

Summary
The liver is the main site of colorectal cancer (CRC) metastasis. Tumor-associated macrophages (TAMs) play a key role in tumor metastasis. Therefore, modulating the function of tumor-associated macrophages is a potential therapeutic strategy to control tumor metastasis. We found in vivo experiments that the activation of STING inhibited CRC liver metastasis in model mice and affected the macrophage phenotype in the tumor microenvironment. Mechanistically, STING affects TAM polarization and regulates macrophage function through IRG1. And STING activates IRG1 to promote the nuclear translocation of TFEB, affecting the ability of macrophages to suppress tumor metastasis.Therefore, this study highlights the critical role of the STING-IRG1 axis on TAM reprogramming and its role in the process of tumor liver metastasis, which may provide a promising therapeutic strategy for CRC liver metastasis.
Subject areas: Biological sciences, molecular biology, Immunity, cancer
Graphical abstract
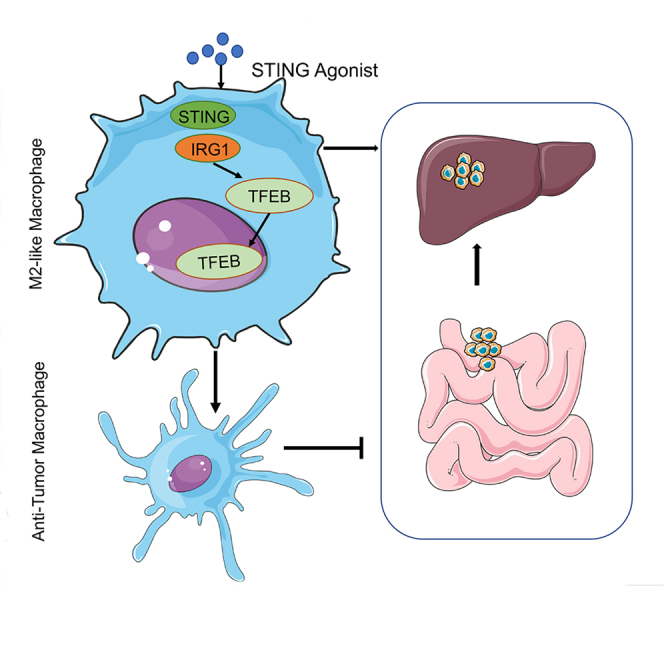
Highlights
-
•
Activation of STING suppresses colon cancer liver metastasis in vivo
-
•
Activation of STING suppresses macrophage M2 polarization
-
•
STING regulates macrophage function and enhances tumor killing ability
-
•
STING suppresses CRC liver metastasis through IRG1-induced TFEB nuclear translocation
Biological sciences; Molecular biology; Immunity; Cancer.
Introduction
Colorectal cancer (CRC) ranks third among the most prevalent malignancies globally and is also identified as the second principal factor contributing to cancer-related mortality worldwide.1,2 Although CRC has a good prognosis, it lacks specific clinical manifestations42. It has an insidious onset, leading to intermediate and advanced disease progression in approximately 30% of patients at the early stage of diagnosis.3 The development of liver metastases in CRC makes treatment more difficult. Liver metastasis of CRC implicates numerous pathobiological pathways regulated by key genes serving as mediators between tumor cells and the surrounding tumor microenvironment (TME).4
The immune cell component within the tumor microenvironment significantly influences tumor progression.5,6,7 Tumor-associated macrophages (TAMs) are the major cell population in different tumor types. TAMs in the tumor microenvironment are a pivotal determinant of the immune response associated with tumors.8 Based on their phenotype, TAMs can either foster or hinder tumor progression. Consequently, TAMs can influence the effectiveness of almost any type of therapy.9 Nevertheless, numerous questions pertaining to the mechanisms that drive the metastatic macrophage phenotype within the tumor environment remain unresolved. Therefore, an investigation into the intricate cellular functions and complexities of TAMs may provide valuable insights into the pathophysiology of the tumor microenvironment.
The pivotal innate immune response molecule called "Stimulator of Interferon Genes" (STING) has been explored in various contexts to accomplish this objective. STING is activated by cytoplasmic DNA from both pathogenic and host sources, which triggers the production of type I interferon and proinflammatory cytokines.10,11,12 Consequently, this mechanism assists in shielding against viral and intracellular bacterial infections and simultaneously modulates the innate anti-tumor immune responses.10,11,12 Correspondingly, STING agonists have shown considerable efficacy in treating pathogenic infections and cancers.13,14
This investigation aimed to analyze the molecular mechanism and function of STING in liver metastasis induced by colorectal cancer cells. In brief, we found that the administration of exogenous STING agonists can inhibit the metastasis of tumor lesions in a mouse model of colorectal cancer liver metastasis. The polarization state of TAMs was altered in metastatic lesions of mice treated with STING agonists. Finally, we demonstrated that STING in macrophages inhibits tumor cell metastasis by mediating TFEB nuclear entry through immune response gene 1 (IRG1).
Results
Expression of stimulator of interferon genes in tumor-associated macrophages correlates with liver metastasis of colorectal cancer
To assess whether STING is involved in liver metastases of colorectal cancer, we analyzed the expression of STING in primary colorectal cancer and metastatic liver foci using immunohistochemistry (IHC). Our study revealed a diminished level of STING in both primary and metastatic tumor foci relative to normal tissue. Notably, we observed that the expression level of STING was also significantly reduced in metastatic foci compared to primary colorectal cancer foci (Figure 1A). Similar results were obtained from CRC patients’ cohorts from the publicly available Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo) (Figure 1D). Previous research indicates that untreated liver metastasis patients show significant enrichment of M2 macrophages, and in patients with colon cancer and liver metastasis, infiltration of these cells is significantly increased. Moreover, the presence of M2 macrophages is also associated with poor prognosis of primary tumors. We also analyzed single-cell RNA sequencing (scRNA-seq) data from patients with colorectal cancer liver metastases (http://www.cancerdiversity.asia/scCRLM).15 Cells were clustered with a particular focus on CCL5+Macrophage(M1) and CCL18+ Macrophage(M2) (Figure 1B). The cell populations were identified using the expression levels of typical marker genes.15 Remarkably, a higher proportion of M2-like macrophages were found in liver metastases of colorectal cancer compared to M1-like macrophages, and STING exhibited a significant reduction in this particular cell population. (Figures 1C and 1E). Furthermore, the correlation of STING with different phenotypes of macrophages was analyzed using the TIMER web tool (https://cistrome.shinyapps.io/timer/) based on the TCGA database,16 which showed that STING in COAD was significantly negatively correlated with M2-like macrophages. In contrast, STING was significantly positively correlated with M1-like macrophages. (Figure 1F). Collectively, our findings suggest that STING is decreased in M2-like macrophages that are linked with liver metastasis in colorectal cancer.
Figure 1.

Expression of STING in TAMs correlates with liver metastasis of colorectal cancer
(A) Representative IHC stained images of STING protein in primary liver metastatic and paracancerous tissue of human colorectal cancers.
(B) Visualization of t-SNE in macrophage clusters based on single-cell transcriptomes.
(C) Expression levels of STING in different macrophage subtypes.
(D) Analysis of STING expression in primary CRC with metastasis tissues in CRC microarray profile.
(E) Boxplots representing the distribution of M1 (CCL5+) or M2 (CCL18+) cells in primary and metastatic tumors.
(F) Correlation between the mRNA expression of STING and M1/M2 macrophage obtained from Tumor Immune Estimation Resource (TIMER2.0, http://timer.cistrome.org/).
Activation of stimulator of interferon genes in vivo inhibits tumor metastasis
The liver is a primary site of metastasis for colorectal cancer. Therefore, we investigated the effect of STING on liver metastasis in vivo by using spleen injection of colon cancer cells. First, Luciferase-MC38 cells were injected into C57BL/6 mice via the spleen and monitored cell metastasis to the liver by in vivo bioluminescence imaging. The bioluminescence imaging showed a significant reduction in liver metastases after the administration of the STING agonist--DMXAA(i.p.) (Figure 2A). The overall survival of mice treated with the STING agonist (DMXAA) was also prolonged (Figure 2B). We further confirmed our observation by analyzing liver tissue through H&E staining (Figures 2C and 2D). Additionally, we tested two liver function markers, ALT (alanine aminotransferase) and AST (aspartate aminotransferase), in serum samples and found a significant reduction in both markers in the DMXAA group compared to the control group (Figure 2E). We confirmed that the STING downstream proteins, TBK1 and IRF3, were activated in the liver after drug administration (Figure 2F).
Figure 2.

Activation of STING in vivo inhibits tumor metastasis
(A) MC38-luciferase cells were injected to construct a transfer model and treated with DMXAA (i.p.) (10 mg/kg/3 days) or PBS (i.p.) for 3 weeks. Bioluminescence signal imaging of representative mice (left) and quantitation analyses of bioluminescence (right) were done following treatments,n ≥ 5.
(B) Survival curves of liver metastatic mouse model after DMXAA and PBS treatment compared to the non-operated group (mock).
(C) Representative macroscopic photograph of livers of the control model (top panel) and DMXAA (10 mg/kg/3 days)-treated mice (lower panel) with metastatic cancer nodules.
(D) H&E staining of liver from control and DMXAA-treated liver metastasis mouse model.
(E) Levels of ALT and AST in peripheral blood of mice after 3 weeks of DMXAA or PBS treatment.
(F) Western blot to detect the protein levels of STING, TBK1, p-TBK1, IRF3, and p-IRF3 in mouse liver using GAPDH as the loading control.
(G) Flow cytometric analysis of M1 (CD11c+) and M2(CD206+) ratios in F4/80+/CD11b+ macrophage populations isolated from liver metastases.
Stimulator of interferon genes affects macrophage polarization in colorectal cancer liver metastases in vivo
Since TAM polarization affects tumor proliferation, migration, invasion, and angiogenesis, we aimed to elucidate the polarized phenotype of macrophages in metastatic liver lesions. Previous studies have used CD11c and CD206 to classify TAMs into several subpopulations. F4/80+ CD11clow CD206high macrophages were defined as M2 macrophages, whereas F4/80+ CD11chigh CD206low macrophages were defined as M1.17 Using this classification, we observed a higher proportion of F4/80+ CD11chigh CD206low tumor suppressor subtype and a lower proportion of F4/80+ CD11clow CD206high tumor promoter subtype in macrophages from metastatic liver lesions in mice after DMXAA treatment (Figure 2G). Macrophages could recruit effector T cells to kill tumor cells by inducing hemorrhagic necrosis of tumor vessels or by secreting chemokines. Therefore, T cell phenotypes in metastatic liver lesions and peripheral blood were analyzed by flow cytometry. The results showed increased cytotoxic T cell infiltration and significantly decreased regulatory T-cells in liver metastatic lesions after DMXAA treatment (Figure S2).
To explain whether DMXAA plays a role in suppressing tumor metastasis after STING knockout, a colorectal cancer liver metastasis model was established by STINGgt/gt and wild-type mice, which were given DMXAA treatment, respectively. Bioluminescence imaging on the IVIS Lumina II system was used to monitor tumor progression. Our data indicated that treatment with DMXAA after the knockout of STING does not affect tumor metastasis (Figure 3A). This finding was corroborated by an H&E analysis of liver tumor lesions in mice (Figure 3B).
Figure 3.

Inhibition of tumor growth in metastatic liver lesions accompanied by an increase in the proportion of M1-like macrophages after STING agonist treatment
(A) MC38-luciferase cells were injected into WT or STINGgt/gt mice to generate a transfer model, followed by treatments with DMXAA (i.p.) (10 mg/kg/3days) or PBS for three weeks. Bioluminescence signal imaging of representative mice (left) and quantitation analyses of bioluminescence (right) were done after treatments, n ≥ 5.
(B) H&E staining of liver metastatic lesions after the intrasplenic injection of MC38 cells to WT or STINGgt/gt mice.
(C) Immunofluorescence staining of liver tissues with CD206+ (green), CD86+ (red) and DAPI (blue).
(D) Flow cytometry analysis of CD11c+ and CD206+ macrophage subpopulations in tumor tissues of WT or STING gt/gt mice.
Further, we analyzed pro-inflammatory M1 and anti-inflammatory M2 macrophages in liver metastases using the markers CD86 and CD206.18,19 As shown in Figure 3C, the control group showed fewer CD86 pro-inflammatory M1 macrophages and a higher number of CD206 anti-inflammatory M2 macrophages. However, the opposite was confirmed in the DMXAA-treated group. DMXAA treatment after the knockout of STING failed to increase the proportion of M1-like macrophages (Figure 3C). Flow cytometric analysis of tumor lesions also showed that DMXAA treatment after knockout of STING failed to polarize TAM into an anti-tumor subtype (Figure 3D). Clodronate liposomes cleared macrophages in mice, and flow cytometry confirmed that the proportion of macrophages in mice was significantly reduced after clodronate liposome treatment. DMXAA was given to liver metastasis model mice, and the results showed that DMXAA could not inhibit colorectal cancer liver metastasis in mice after macrophage depletion (Figure S5).
Altogether, these data suggested that STING inhibited CRC liver metastasis by affecting macrophage polarization.
Stimulator of interferon genes induces a pro-inflammatory phenotype in macrophages
To investigate whether STING affects macrophage polarization in vitro, bone marrow macrophages(BMDM) were isolated in WT and STINGgt/gt mice. After the induction of M2 polarization by BMDM in vitro, activation of STING increased the expression levels of iNOS and TNF-α, significantly decreased the mRNA levels of Arg-1 and Fizz1, and reprogrammed M2-like macrophages to M1-like macrophages (Figures 4A and 4B). Notably, when STING was eliminated, treatment with DMXAA failed to induce M1 polarization in macrophages (Figures 4A and 4B). In conclusion, our in vitro and in vivo data suggest that STING regulates macrophage polarization.
Figure 4.

STING induces a pro-inflammatory phenotype in macrophages
(A) Flow cytometric analysis of CD11c or CD206 in WT or STINGgt/gt BMDM. The BMDM was induced into M2 phenotype for treatment with DMXAA, followed by flow cytometry.
(B) Bone marrow macrophages were isolated from WT and STINGgt/gt mice, and induced into M2-like macrophages using IL4. The cells were treated with or without DMXAA, and qPCR was performed following total RNA isolation. Each gene was normalized with the housekeeping gene HPRT.
Stimulator of interferon genes activation affects colon cancer cell metastasis via macrophages
To investigate the effects of polarized macrophages on colorectal cancer cell metastasis, we induced M1/M2 polarized macrophages in vitro. We used RAW264.7 and differentiated THP-1 monocyte cell lines, which have been widely studied with regard to the phenotypes mentioned.20 We noticed an increase in the levels of TNF-α and iNOS in M1 macrophages compared to M0 macrophages (Figure S1A). Likewise, Arg-1 and Fizz1 mRNA levels were higher in M2-polarized macrophages. We realized that this observation is similar to the cytokine profile in CRC TAMs.21 After the co-culture of M2 macrophages with CRC cells, M2 macrophages significantly accelerated the migration and invasive potential of DLD1 and MC38 cells (Figures 5A and 5B). Additionally, the STING agonist DMXAA and cGAMP-treated groups significantly inhibited CRC cells' migratory and invasive abilities without any remarkable effect on proliferation (Figures S1B and S1C). A similar phenomenon was observed in the scratch assay for M2-polarized macrophages (Figure 5C). In brief, these data suggest that the STING activation of macrophages inhibits colon cancer cell metastasis.
Figure 5.

STING activation affects colon cancer cell metastasis via macrophages
(A) Determination of migration and invasion ability of MC38 after co-culture with M1 macrophages (RAW264.7) or M2 macrophages (RAW264.7) for 48 h with or without DMXAA or cGAMP treatment.
(B) Determination of migration and invasion ability of DLD1 after co-culture with M1 macrophages (THP1) or M2 macrophages (THP1) for 48 h with or without cGAMP treatment.
(C) With or without DMXAA or cGAMP treatment, MC38 cells were co-cultured with M1 or M2 macrophages and cell migration was determined using a wound healing assay.
Stimulator of interferon genes reprograms tumor-associated macrophages to be tumoricidal
To investigate the effect of STING on the tumor-killing ability of macrophages. Mouse macrophages were cultured in a tumor cell-conditioned medium to induce TAMs, which were then co-cultured with tumor cells. After macrophages were induced as TAMs, the expression level of Arg-1 was significantly increased (Figure 6B). The expression of Arg-1 was suppressed considerably after DMXAA treatment, while the expression levels of TNF-α and iNOS were significantly increased. Furthermore, DMXAA did not affect the viability of CRC cancer cells in the presence of tumor cells alone (Figure S1C). However, DMXAA treatment of macrophages activated STING in them, resulting in a significant reduction in the number of cancer cells within 48 h of co-culture (Figure 6A). Moreover, to determine the key role of STING in the activation of TAM, we isolated BMDM from WT and STINGgt/gt mice. After the generation of TAM and treatment with DMXAA, our data showed that the compound could not induce tumor killing by macrophages when STING was knocked out (Figure 6C).
Figure 6.

STING reprograms TAMs to be tumoricidal
(A) RAW264.7 cells were induced to TAM by tumor cell conditioned medium. Tumor cells (MC38 or CT26) and TAM were then labeled with fluorescence, and the number of tumor cells was counted after co-culture for 48 h.
(B) With or without DMXAA, RAW264.7 cells were induced to TAM using tumor cell-conditioned media. MC38 cells were co-cultured with TAM and qPCR was done for the analysis of the mRNA levels of markers of TAM in RAW264.7.
(C) WT or STING, bone marrow macrophages, were induced to TAM using tumor cell-conditioned media. Tumor cells (MC38 or CT26) and TAM were labeled with fluorescence, and the number of tumor cells was counted after co-culture for 48 h.
Stimulator of interferon genes induces macrophage polarization via IRG1
To investigate the mechanism related to the STING regulation of macrophage polarization, STING binding proteins that may play a key role in macrophage polarization were analyzed by Co-IP and mass spectrometry experiments. IRG1 (ACOD1) has a significant binding effect with STING (Figure 7A). To validate the data obtained by mass spectrometry, RAW264.7 cells were induced by IL4 into M2 macrophages and then the cells were treated with DMXAA or PBS. Co-IP analysis with an anti-STING antibody confirmed that there was indeed an interaction between STING and IRG1 (Figure 7B). In addition, IRG1 was found to be co-localized with STING in the cytoplasm by immunofluorescence(Figure S4).
Figure 7.

STING induces macrophage polarization via IRG1
(A) Mass spectrometry analysis revealed that STING interacts with IRG1.
(B) Co-IP assay confirmed the interaction between STING and IRG1.
(C) RAW264.7 cells were induced to become M2 macrophages using IL4 and then treated at different time points in the presence or absence of DMXAA. qPCR was performed to detect the mRNA level of the indicated markers CXCL10, IFNβ, IRG1 and iNOS.
(D) Flow cytometry analysis of RAW264.7 cells after knockdown of IRG1 in IL4 or LPS/IFN-induced macrophages.
(E) mRNA levels of M1-like macrophages-associated genes measured by qPCR in IRG1 knockdown RAW264.7 cells.
Changes in IRG1 expression during macrophage reprogramming corresponded to the regulation of downstream IFNβ and CXCL10 following STING activation (Figure 7C).
We sought to understand the functional significance of the STING-IRG1 interaction and to elucidate the function of IRG1 in macrophage polarization. For this purpose, IRG1 knockdown RAW264.7 and THP1 cells were generated by RNAi. The effect of IRG1 on the phenotype of M1/M2 macrophages induced in vitro was analyzed using flow cytometry. We noted that IRG1 knockdown significantly inhibited LPS/IFN-γ-induced macrophage M1 polarization compared to control shNC (Figure 7D). In addition, the mRNA levels of M1 macrophage markers TNF-α and iNOS were significantly suppressed upon IRG1 knockdown (Figure 7E).
Activation of IRG1 in macrophages affects colon cancer cell metastasis
To investigate the potential function of IRG1 activation in tumor metastasis, we induced M2-like macrophages in vitro and assessed their effects on CRC through transwell assays. In this co-culture system, macrophages were induced with IL4 to become M2-like and then co-cultured with CRC cells for 48 h to analyze the migration and invasion potential of CRC cells. Treatment with STING agonists (DMXAA\cGAMP) in control shNC significantly suppressed the migration and invasion of MC38 and DLD1 cells. Knockdown of IRG1 in macrophages, DMXAA/cGAMP was not in inhibiting migration and invasion of DLD1 (Figure 8A) and MC38 (Figure 8B) cells. Our data indicate that STING affects macrophage polarization and colorectal cancer cell metastasis via IRG1.
Figure 8.

Activation of IRG1 in macrophages affects colon cancer cell metastasis
(A) THP1 cells were silenced with shRNA against IRG1 or a non-targeting control. Macrophages were seeded into the transwell lower chamber to induce M2 polarization and tumor cells were seeded into transwell upper chambers (size:8 μM). 250 nM cGAMP was added to the transwell lower chamber. Migration and invasion assay of DLD1 after co-culture with M2 macrophages (THP1) for 48 h.
(B) RAW264.7 cells were silenced with shRNA against IRG1 or a non-targeting control. Macrophages were seeded into the transwell lower chamber to induce M2 polarization and tumor cells were seeded into transwell upper chambers (size:8 μM). 50 μg/mL DMXAA was added to the transwell lower chamber. Migration and invasion assay of MC38 after co-culture with M2 macrophages (RAW264.7) for 48 h.
Stimulator of interferon genes induces transcription factor EB into the nucleus after activating IRG1
Transcription factor EB (TFEB) regulates phagocytosis-lysosome-mitochondria crosstalk in macrophages. Activation of TFEB promotes the transcription of aconitate decarboxylase (Acod1, IRG1) and production of its product itaconic acid in response to pathogenic stimuli.22 Activated macrophages express the itaconate synthase IRG1, which converts cis-aconitate to itaconate in the mitochondria. Subsequently, itaconic acid alkylates the transcription factor TFEB in the cytoplasm, prevents TFEB from being phosphorylated by the protein kinase mTOR, destroys the phosphorylated TFEB, and finally transfers TFEB from the cytoplasm.23
To investigate whether TFEB is involved in the STING activation of IRG1, we treated BMDM cells with different concentrations of DMXAA. The results showed that the TFEB protein in the cytoplasm decreased while that in the nucleus increased, but the total protein level remained unchanged (Figure 9A).RAW264.7 cells and BMDM were treated with octyl itaconate (OI), which is a permeable cell substitute for Aconitine salt hydrolyzed by esterase in macrophages.24 OI treatment led to a corresponding increase in itaconic acid concentration within cells.25 Analysis of RAW264.7 cells and BMDM using anti-TFEB antibodies revealed that OI treatment caused concentration-dependent nuclear translocation of TFEB but did not affect the total level of TFEB protein (Figure 9B).
Figure 9.

STING induces TFEB into the nucleus after activating IRG1
(A) Western Blot analysis of BMDM treated with different concentrations of DMXAA (0 μg,25 μg,50 μg,75 μg), the expression difference of TFEB in the cytoplasm and nucleus (β-Actin is a cytoplasmic internal reference protein, LaminB1 is a nuclear internal reference protein).
(B) Western Blot analysis of TFEB expression differences in cytoplasm and nucleus after RAW264.7 and BMDM cells were treated with 150 μM OI (4-Octyl Itaconate) for 4 h (β-Actin is a cytoplasmic internal reference protein, LaminB1 is a nuclear internal reference protein), Whole: total cell protein, Cyto: cytoplasmic protein, Nuc: nuclear protein.
(C) Western Blot analysis of TFEB protein expression difference in cytoplasm and nucleus after knockdown of IRG1 in RAW264.7 cells after 50 μg DMXAA treatment.
(D) Western Blot detection of TFEB protein expression in RAW264.7 cells after IRG1 knockdown.
(E) Immunofluorescence detection of nuclear translocation of TFEB protein after RAW264 cells were treated with 50 μg/mL DMXAA or RAW264.7 cells were treated with 150 μM OI (4-Octyl Itaconate).
(F) After RAW264.7 knocked down IRG1, polarization was induced by IL4, and the migration and invasion of MC38 cells were detected after being treated with OI for 4 h and co-cultured with MC38 for 48 h in a transwell chamber.
(G) After spleen injection of MC38-Luc cells to construct a colorectal cancer liver metastasis model, the liver metastasis model mice were treated with OI (i.p.) (50 mg/kg/2 day) or PBS for 3 weeks. Bioluminescence signal imaging (left) and quantitative analysis of bioluminescence (right) in representative mice, n ≥ 5.
(H) ALT and AST levels in peripheral blood of mice with colorectal cancer liver metastases after 3 weeks of OI or PBS treatment.
Itaconic acid is produced by IRG1 through cis-aconitic acid and its gene expression is induced by LPS treatment in macrophages.24 In fact, itaconic acid expression can be detected after stimulating IRG1 in macrophages.25 To further investigate the role of IRG1 in TFEB activation, RAW264.7cells with IRG1 knockdown were used. It was found that DMXAA stimulation induced TFEB nuclear translocation in wild-type RAW264.7 cells, but this effect was significantly weakened in IRG1 knockdown RAW264.7 cells (Figure 9C). However, OI treatment restored DMXAA-induced TFEB nuclear translocation in IRG1 knockdown RAW264.7 cells (Figure 9D). In addition, immunofluorescence experiments were used to detect TFEB nuclear translocation after DMXAA and OI treatment of RAW264.7 cells, and the results showed that both drug treatments induced TFEB nuclear entry (Figure 9E). By co-culturing RAW264.7 with MC38, we found that the OI treatment of RAW264.7 could inhibit the migration and invasion of tumor cells MC38, while OI treatment could also inhibit the migration and invasion of tumor cells after knocking down IRG1 (Figure 9F). These results indicate that under STING stimulation, IRG1 can induce TFEB nuclear translocation, thereby inhibiting tumor migration and invasion.
Activation of IRG1 inhibits colorectal cancer liver metastasis in vivo
After being activated, IRG1 catalyzes cis-aconitate in the tricarboxylic acid cycle to produce a large amount of itaconic acid. Itaconic acid then participates in the nuclear translocation of TFEB. Mice with liver metastases of colorectal cancer were treated with OI (OI can be converted into intracellular itaconic acid by macrophages in vivo) for three consecutive weeks. The status of tumor cell metastasis to the liver was monitored by in vivo bioluminescence imaging. The results showed that tumor metastases in the liver were significantly reduced after the intraperitoneal injection of OI (i.p.) (50 mg/kg) for three weeks (Figure 9G). Compared with the control group, ALT and AST in the serum of mice in the OI group were significantly reduced (Figure 9H), and liver damage was also alleviated after OI treatment. Furthermore, pro-inflammatory M1 and anti-inflammatory M2 macrophages in liver metastases were analyzed using the markers CD86 and CD206. The results showed that the infiltrating M1 macrophages in the tumor tissues increased after OI treatment (Figure S3). These data suggest that IRG1 can affect colorectal cancer liver metastasis in vivo.
Discussion
STING (Stimulator of Interferon Genes) is an important immune regulatory molecule that regulates the immune response of cells to DNA viruses and cancer. In recent years, more and more studies have shown that STING regulates the immune response of macrophages to tumors, thereby affecting the growth and metastasis of tumors.26,27 However, its specific function and mechanism in colorectal cancer metastasis are still to be resolved. This study discovered the role of STING in regulating tumor cell migration and invasion and that STING affects tumor cell migration and invasion by inducing IRG1-mediated TFEB nuclear translocation.
In this study, we found that the expression level of STING was lower in colorectal cancer liver metastases than in the primary lesions, and it was also significantly lower in M2-type macrophages associated with liver metastases. Therefore, STING may play an important role in colorectal cancer liver metastasis, and its downregulation may be related to M2-type macrophages.
Tumor cells, owing to their unrestrained proliferative capacity, often experience flawed DNA replication resulting in the leakage of genetic material into the cytoplasm. This accumulation triggers the activation of STING, instigating a pro-inflammatory response and thus inducing apoptosis as a tumor suppressor mechanism.28,29,30 However, melanoma and telomerase-deficient cancer cells have exhibited inhibited STING activation.31 Conversely, low expression of STING has been strongly associated with poor prognosis in patients with gastric cancer.32 In patients with advanced colon cancer, STING or cGAS protein expression deletion has been observed.33 Notably, our data demonstrated significant downregulation of STING expression in colorectal cancer cell lines, highlighting the pivotal role of the cGAS-STING signaling pathway in the progression of colorectal cancer.
In vivo experiments, we established a mouse model of colorectal cancer liver metastasis to confirm that STING can induce the polarization of macrophages in the tumor and significantly inhibit the liver metastasis of colorectal cancer after activation in vivo.
Through co-culturing macrophages and colorectal cancer cells, it was found in vitro that STING mediates the reprogramming of tumor-associated macrophages. These results suggest that STING may regulate the function of macrophages, alter the immune cell status in the liver metastasis microenvironment, and impact the treatment of colorectal cancer liver metastasis. In the present study, DMXAA was found to produce antitumor effects in colorectal cancer liver metastases by acting on macrophages. However, tissue-resident macrophages (kupper cells) are also present in the liver, and whether they play a role in this process has not been fully determined. Therefore, further studies are needed to validate the role of macrophages from different sources (e.g., BMDMs and kupper cells) in this process.
Analysis of the tumor microenvironment of colorectal cancer liver metastases in mice by flow cytometry revealed that after treatment with STING agonists, tumor-associated macrophages were polarized into a pro-inflammatory phenotype, contributing to the inhibition of tumor growth and diffusion.
Further exploring the molecular mechanism of STING affecting colorectal cancer liver metastasis, we found that STING induces TFEB into the nucleus through IRG1, thereby affecting the migration and invasion of tumor cells. This study provides clues for in-depth exploration of the mechanism of STING in the tumor microenvironment. IRG1 is an immunoregulatory gene and a critical metabolic regulatory molecule involved in various metabolic pathways.34 Recent studies have shown that IRG1 also plays a vital role in tumor cells, it can promote the metabolic transformation of tumor cells, and participate in the growth, migration and metastasis of tumors. Through its metabolic regulation, IRG1 affects the metabolic transformation in tumor cells, such as lactate production and glucose uptake, and can affect the proliferation, survival and invasion ability of tumor cells.35 In addition, the expression of IRG1 also regulates the immune escape of tumor cells, which is one of the crucial processes in tumorigenesis and progression.36 Therefore, IRG1 is considered as a potential therapeutic target, which can be used to control the metabolism and immune escape of tumor cells, thereby inhibiting tumor growth and metastasis.
Under the influence of external stimuli, macrophages can elevate the expression of IRG1 metabolic enzyme and produce itaconic acid. This compound is one of the most significantly upregulated metabolites in activated macrophages and exhibits anti-inflammatory properties that facilitate inflammation reduction.23,37,38 The results of our study differ from previous reports. However, recent studies have shown that itaconic acid exhibits a dual role in macrophages through interaction with IRG1. Specifically, low doses of itaconic acid hinder inflammation, while high doses promote inflammation.39 In in vivo experiments with OI, the survival cycle of mice treated with relatively low doses of OI in a colorectal cancer liver metastasis model was significantly reduced, thus preventing the simultaneous collection of the required experimental data. These studies seem to be somewhat contradictory. In fact, this is not necessarily contradictory; the role of IRG1 is multifaceted, and it may exhibit different effects in different settings. In some cases, IRG1 may promote the inflammatory response, while in other cases it may suppress it. Therefore, in future studies, the mechanism of IRG1’s role in tumor development and treatment and its interactions with other molecules and pathways need to be further explored to better understand the function and role of IRG1 in tumors.
Our study showed that IRG1 plays a crucial role in colorectal cancer liver metastasis and is essential for STING in regulating the conversion of macrophages to a tumor suppressor phenotype. Earlier research has reported that itaconic acid promotes the repositioning of TFEB from the cytoplasm to the nucleus, which consequently stimulates lysosome biosynthesis and heightens macrophage invasion. TFEB ordinarily resides in the cytoplasm during homeostasis, but its activation and movement toward the nucleus can be induced by different stimuli.40 TFEB was also found to be a promising therapeutic target for breast cancer and other possible solid malignancies, and TFEB activation was able to inhibit a variety of effector molecules that exert TAM pro-tumor functions, including Arg-1, IL-10, and PGE2.41
In the study, STING activation was found to significantly affect the nucleation of TFEB in a concentration-dependent manner. Upon being treated with the itaconic acid analog OI, macrophages have the capability to uptake and process OI into intracellular itaconic acid. It has been observed that OI treatment triggers the transportation of TFEB into the nucleus of macrophages. In addition, inhibition of IRG1 activation by knocking down IRG1 could inhibit DMXAA-induced TFEB activation. These results strongly support the ability of STING to affect the nuclear translocation of TFEB upon the activation of IRG1.
We should also acknowledge some limitations of this study: the liver is a highly immune cell-infiltrated organ containing various immune cells, including macrophages, B cells, T cells, natural killer cells, and others. Various types of immune cells have distinct roles in regulating the immune response in the liver and participate in a range of physiological and pathological processes. Therefore, we need to investigate in more depth the possible therapeutic impact of other types of immune cells and the macrophages that play a role in colorectal cancer liver metastasis. By analyzing these intricate immune regulatory networks, we can enhance our comprehension of the immune response in the liver and accelerate the progress of developing effective therapeutic interventions.
Taken together, our data suggest that the regulation of the STING-IRG1-TFEB axis in macrophages may be a potential therapeutic target for mitigating liver metastasis and may be a promising therapeutic strategy for liver metastasis in colorectal cancer.
Limitations of the study
The liver is an organ highly infiltrated by immune cells, containing a variety of immune cells, such as macrophages, B cells, T cells, natural killer cells, and so forth., each of which has a unique role in regulating the immune response of the liver and participates in a series of physiological and pathological processes process. Therefore, we need to delve deeper into the role of other types of immune cells in the treatment of colorectal cancer liver metastases, not just focus on macrophages. By analyzing these intricate immune regulatory networks, we can better understand the immune response in the liver and rapidly improve the development of effective therapeutic interventions. At the same time, our study failed to further elucidate the interaction mode between STING-IRG1-TFEB, and further exploration is needed.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TMEM173/STING Polyclonal antibody | Proteintech | Cat#19851-1-AP; RRID: AB_10665370 |
| STING (D2P2F) Rabbit mAb | Cell Signaling Technology | Cat#13647; RRID: AB_2732796 |
| TBK1/NAK (E8I3G) Rabbit mAb | Cell Signaling Technology | Cat#38066; RRID: AB_2827657 |
| Phospho-TBK1/NAK (Ser172) (D52C2) Rabbit mAb | Cell Signaling Technology | Cat#5483; RRID: AB_10693472 |
| GAPDH Monoclonal antibody | Proteintech | Cat#60004-1-Ig; RRID: AB_2107436 |
| IRG1 Polyclonal Antibody | Thermo Fisher Scientific | Cat#PA5-102893; RRID: AB_2815944 |
| IRF3 Polyclonal antibody | Proteintech | Cat#11312-1-AP; RRID: AB_2127004 |
| Phospho-IRF-3 (Ser396) (D6O1M) Rabbit mAb | Cell Signaling Technology | Cat#29047; RRID: AB_2773013 |
| TFEB Polyclonal antibody | Proteintech | Cat#13372-1-AP; RRID: AB_2199611 |
| Lamin B1 Polyclonal antibody | Proteintech | Cat#12987-1-AP; RRID: AB_2136290 |
| β-Actin Rabbit mAb | Abclonal | Cat#AC026; RRID: AB_2768234 |
| Zombie NIR™ Fixable Viability Kit | Biolegend | Cat#423105 |
| Brilliant Violet 510™ anti-mouse CD45 Antibody | Biolegend | Cat#103137; RRID: AB_256139 |
| FITC anti-mouse/human CD11b | Biolegend | Cat#101206; RRID: AB_312789 |
| APC anti-mouse F4/80 | Biolegend | Cat#123116; RRID: AB_893481 |
| PE anti-mouse CD206 (MMR) Antibody | Biolegend | Cat#141706; RRID: AB_10895754 |
| PE/Cyanine7 anti-mouse CD11c Antibody | Biolegend | Cat#117318; RRID: AB_493568 |
| FITC anti-mouse CD3 Antibody | Biolegend | Cat#100204; RRID: AB_312661 |
| PerCP/Cyanine5.5 anti-mouse CD4 Antibody | Biolegend | Cat#100434; RRID: AB_893324 |
| PE/Cyanine7 anti-human CD8 Antibody | Biolegend | Cat#344750; RRID: AB_2687201 |
| APC anti-mouse CD69 Antibody | Biolegend | Cat#104514; RRID: AB_492843 |
| PE anti-mouse CD25 Antibody | Biolegend | Cat#113704; RRID: AB_2927943 |
| Brilliant Violet 421™ anti-mouse CD127 (IL-7Rα) Antibody | Biolegend | Cat#135027; RRID: AB_2563103 |
| Bacterial and virus strains | ||
| sh-mouseIRG1-1 (5′ to 3′): TGAAGTACAGGGACGATTAAT | Sangon Biotech | N/A |
| sh-mouseSTING-2 (5′ to 3′): CCTGACGTCCAGTACGTAAAC | Sangon Biotech | N/A |
| sh-humanIRG1-1 (5′ to 3′): GCTTCCAACTGACTACATTAA | Sangon Biotech | N/A |
| sh-mouseSTING-2 (5′ to 3′): GCAAGGCCGATTACTGCATTT | Sangon Biotech | N/A |
| Biological samples | ||
| TRIzol | YI FEI XUE BIO TECH | Cat#YFXM0011P |
| Percoll | GE healthcare | Cat#17-0891-09 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat#11668019 |
| Ceturegel® Matrix High Concentration, LDEV-Free Matrigel Matrix | Yasen | Cat#40187ES10 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Mouse IL-4 Protein | R&D Systems | Cat#404-ML |
| Lipopolysaccharides (LPS) | Cell Signaling Technology | Cat#14011 |
| Recombinant Mouse IFN-gamma Protein | R&D Systems | Cat#485-MI |
| DMXAA | Aladdin | Cat#D129922 |
| Critical commercial assays | ||
| AceQ Universal SYBR qPCR Master Mix | Vazyme | Cat#Q511-02 |
| HiScript III 1st Strand cDNA Synthesis Kit | Vazyme | Cat#R312-01 |
| Deposited data | ||
| GSE41568 | GEO | https://www.ncbi.nlm.nih.gov/geo/ |
| GSE41258 | GEO | https://www.ncbi.nlm.nih.gov/geo/ |
| scRNA-seq data | http://www.cancerdiversity.asia/scCRLM | N/A |
| Experimental models: Cell lines | ||
| CT26 | ATCC | Cat#CRL-2638 |
| MC38 | Coweldgen | Cat#KMCC-001-0304 |
| THP1 | ATCC | Cat#TIB-202 |
| Raw264.7 | ATCC | Cat#TIB-71 |
| DLD1 | ATCC | Cat#CCL-221 |
| Experimental models: Organisms/strains | ||
| C57BL/6 | Animal Core Facility of Nanjing Medical University | N/A |
| STINGgt/gt | FJNU Research Center of Southern Biomedical | N/A |
| Oligonucleotides | ||
| IRG1 forward: GGTATCATTCGGAGGAGCAAGAG | This paper | N/A |
| IRG1 reverse: ACAGTGCTGGAGGTGTTGGAAC | This paper | N/A |
| Fizz1 forward: CCAATCCAGCTAACTATCCCTCC | This paper | N/A |
| Fizz1 reverse: ACCCAGTAGCAGTCATCCCA | This paper | N/A |
| IFNβ forward: ACAGTGCTGGAGGTGTTGGAAC | This paper | N/A |
| IFNβ reverse: ACACTGTCTGCTGGTGGAGTTC | This paper | N/A |
| CXCL10 forward: ATCATCCCTGCGAGCCTATCCT | This paper | N/A |
| CXCL10 reverse: GACCTTTTTTGGCTAAACGCTTTC | This paper | N/A |
| HPRT forward: CTGGTGAAAAGGACCTCTCGAAG | This paper | N/A |
| HPRT reverse: CCAGTTTCACTAATGACACAAACG | This paper | N/A |
| Arg1 forward: CTCCAAGCCAAAGTCCTTAGAG | This paper | N/A |
| Arg1 reverse: AGGAGCTGTCATTAGGGACATC | This paper | N/A |
| iNOS forward: CGGAGAACAGCAGAGTTGGT | This paper | N/A |
| iNOS reverse: TTGTGGTGAAGGGTGTCGTG | This paper | N/A |
| TNF-α forward: AAGCAAGCAGCCAACCAG | This paper | N/A |
| TNF-α reverse: CCACAAGCAGGAATGAGAAGA | This paper | N/A |
| Recombinant DNA | ||
| pMDLg/pRRE | Addgen | Cat#12251 |
| pRSV-Rev | Addgen | Cat#12253 |
| pMD2.G | Addgen | Cat#12259 |
| pLKO.1 puro | Addgen | Cat#8453 |
| Software and algorithms | ||
| ImageJ | NIH | N/A |
| FlowJo | Treestar | N/A |
| GraphPad Prism 8 | GraphPad | N/A |
| Other | ||
| RPMI 1640 medium | Gibco | Cat#C11875500BT |
| DMEM medium | Gibco | Cat#C11995500BT |
| Fetal bovine serum | Gibco | Cat#10099141 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Hongping Xia (101013473@seu.edu.cn).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
The datasets used and/or analyzed during the current study are available within the manuscript and its supplementary information files.
-
•
This paper does not report the original code. Two CRC liver metastasis datasets were downloaded from GEO datasets (GSE41568 and GSE41258). The scRNA-seq data of patients with colorectal cancer liver metastases were obtained from Single-Cell Colorectal Cancer Liver Metastases (CRLM) Atlas (http://www.cancerdiversity.asia/scCRLM)
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
Experimental model and study participant details
Cell culture
THP1 cells were cultured in RPIM (Gibco) with 10%FBS (Gibco) and 1% penicillin-streptomycin. DLD1, MC38, CT26 and RAW264.7 cell lines were cultured in DMEM (Gibco) with 10% FBS (Gibco) and 1% penicillin-streptomycin. All cells were cultured at 37°C in an incubator with 5% CO2.
Animals and tumor mouse models
STINGgt/gt mice (C57BL/6 background) were generated in Dr. Qi Chen’s laboratory (Fujian Normal University, Biomedical Research Center of South China). 4-6-week-old male C57BL/6 mice were obtained from the Laboratory Animal Center of Nanjing Medical University. Nanjing Medical University’s Institutional Animal Care and Use Committee approved all animal studies(ID number 2020-461), and the Ethical Committee approved protocols for the studies. For the colorectal cancer liver metastasis model, after anesthetizing the mice, a scalpel was used to make a transverse incision on the left side to expose the spleen. Next, the spleen was injected with 100 μL of 3 × 105 MC38-luciferase cells in PBS and then returned to the abdominal cavity for wound closure.
Patient sample collection
Colorectal cancer tissues, normal paracancerous tissues, and colorectal cancer liver metastases were obtained from Nanjing Drum Tower Hospital. All experimental procedures were approved by the Medical(ID number 2020-467) Ethics Committee of Nanjing Medical University and performed in accordance with the relevant guidelines and regulations. Written informed consents were obtained from all patients.
Method details
Macrophage exhaustion
One week before the construction of the mouse liver metastasis model, macrophages were depleted in mice using clodronate liposomes (CLO), and CLO was injected 150 μL into the tail vein for the first time, followed by 100 μL per mouse every 3 days, while the control group was treated with PBS-Liposomes.
Macrophage induction and transwell co-culture assay
For M1-like or M2-like macrophages, BMDMs or RAW264.7 cells were polarized to M1 using 100 ng/mL LPS (CST) and 20 ng/mL IFN-γ (R&D) or M2 with 20 ng/mL IL-4 (R&D) for 24 h. For THP-1 cells, THP-1-derived macrophages were obtained by incubation in 10% FBS RPMI with 100 ng/mL PMA for 24. Furthermore, the cells were incubated with LPS and IFN for 48 h to induce M1-like macrophages and 20 ng/mL IL4 for 48 h to induce M2-like macrophages. A conditioned medium for CT26 or MC38 cells was used to induce RAW264.7 cells or BMDMs to generate TAMs in vitro.
Macrophages were seeded into 24-well plates to induce M1/M2 polarization and tumor cells were seeded into small chambers (size:8 μM). Two hours later, 50 μg/mL DMXAA or 250 nM cGAMP (PFO pretreatment for 30 min) was added to the transwell lower chamber. Forty-eight hours later, the cells were stained with 0.1% crystalline violet and photographed under a microscope.
shRNA knockdown
The packaging plasmids (pMDLg/pRRE, pRSV-Rev and pMD2.G) were co-transfected with the core plasmids PLKO.1 into HEK293T to produce recombinant shRNA virus particles. THP1 cells (1×106 cells per well in a six-well plate), and RAW264.7 cells (3×105 cells per well in a six-well plate) were stained with lentivirus in serum-free medium and replaced with normal medium after 8 h. Screening of cells using puromycin after 24 h of culture.
BMDM isolation
Cells were flushed from the bone marrow of wild-type (WT) C57BL/6 and STINGgt/gt mice by using serum-free DMEM. Bone marrow cells were cultured in a macrophage medium (10% FBS and 20 ng/mL M-CSF) for 7 days. Cells attached to the bottom of the culture dish after 7 days were identified as unpolarized macrophages.
Tumor cell and macrophage co-culture experiments
For direct contact culture conditions, macrophages were seeded in 24-well plates and induced into TAMs with a tumor cell-conditioned medium. Cells were subsequently labeled using Celltracker CM-DiI (YEASEN). Tumor cells were labeled using CMFDA (YEASEN) and seeded in the same culture dish with macrophages. Next, the medium was replaced with a tumor cell medium. DMXAA (50 μg/mL) was added to the medium (0 h), co-cultured for 48 h, and then photographed and recorded to determine the number of cancer cells by counting the cells in 3–5 fields of view.
Flow cytometry
After harvesting mouse tumor tissue, tumors were dissociated, and red blood cells were removed using erythrocyte lysate. The dissociated cells were filtered using a 70 μM cell sieve and lymphocytes were isolated from the cell population using Percoll(life). The macrophage and T cell subsets were subsequently detected using flow cytometry.
Immunoprecipitation-mass spectrometry (IP-MS)
After the treatment of IL-4-induced M2-type macrophages with or without DMXAA (50 μg/mL) for 24 h, cells were lysed in protein lysis buffer. The supernatant was incubated with antibodies against STING overnight at 4°C, washed three times with protein lysis buffer and incubated with Protein G agarose beads overnight at 4°C. The samples were then boiled in an SDS loading buffer for 10 min to separate the proteins. The collected protein samples were further separated from the extracts by SDS-PAGE, and the protein fractions in the two lanes were analyzed by mass spectrometry after staining with Komas Brilliant Blue staining solution.
Quantification and statistical analysis
All data were analyzed using GraphPad Prism (GraphPad). All experiments were repeated at least three times. Unless otherwise stated in the legend, all results were shown as mean ± SD/SEM. p values ∗ ≤0.05, ∗∗ ≤0.01, ∗∗∗ ≤0.001 and ∗∗∗∗ ≤0.0001 were used to ascertain significance between groups.
Acknowledgments
The authors would like to thank Prof. Qi Chen from Fujian Normal University Biomedical Research Center of South China for kind sharing the STINGgt/gt mice (C57BL/6 background).This study was supported by grants from the National Natural Science Foundation of China (No. 82072739 to Xia H, 82101849 to Zhang C, 81871988 to Cheng Z), The Recruitment Program of Overseas High-Level Young Talents, Jiangsu Provincial Health and Health Commission scientific research project key project (ZD2021019); Xisike Clinical Oncology Research Foundation; Nanjing Health Science and Technology Development Special Project (YKK22204); Funding of “Peak” Training Program for Scientific Research and “Climbing Peak” Training Program for Innovative Technology team of Yijishan Hospital, Wannan Medical College, and starting up funding of Southeast University, Nanjing Medical University and the First Affiliated Hospital (Yijishan Hospital), Wannan Medical College.
Author contributions
YXL, and HPX designed the research and supervised the project. YXL, QS, CFZ, and MD were responsible for conducting experiments. QZ and ZJM supported the study and performed the statistical analysis. QS, GRZ, XMW, ZJC, and HPX were responsible for the clinical sample collection, funding support and critical discussion. HJX and CFZ provided technical support for the experiments. All authors read and approved the final article.
Declaration of interests
The authors declare no competing interests.
Published: July 13, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107376.
Contributor Information
Guoren Zhou, Email: zhouguoren888@njmu.edu.cn.
Xiaoming Wang, Email: wnmcxiaomingwang@163.com.
Zhangjun Cheng, Email: chengzhangjun@seu.edu.cn.
Hongping Xia, Email: 101013473@seu.edu.cn.
Supplemental information
References
- 1.Dekker E., Tanis P.J., Vleugels J.L.A., Kasi P.M., Wallace M.B. Colorectal cancer. Lancet. 2019;394:1467–1480. doi: 10.1016/S0140-6736(19)32319-0. [DOI] [PubMed] [Google Scholar]
- 2.Baidoun F., Elshiwy K., Elkeraie Y., Merjaneh Z., Khoudari G., Sarmini M.T., Gad M., Al-Husseini M., Saad A. Colorectal Cancer Epidemiology: Recent Trends and Impact on Outcomes. Curr. Drug Targets. 2021;22:998–1009. doi: 10.2174/1389450121999201117115717. [DOI] [PubMed] [Google Scholar]
- 3.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 4.Liu Y., Zhang Q., Xing B., Luo N., Gao R., Yu K., Hu X., Bu Z., Peng J., Ren X., Zhang Z. Immune phenotypic linkage between colorectal cancer and liver metastasis. Cancer Cell. 2022;40:424–437.e5. doi: 10.1016/j.ccell.2022.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Vitale I., Manic G., Coussens L.M., Kroemer G., Galluzzi L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metabol. 2019;30:36–50. doi: 10.1016/j.cmet.2019.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Meurette O., Mehlen P. Notch Signaling in the Tumor Microenvironment. Cancer Cell. 2018;34:536–548. doi: 10.1016/j.ccell.2018.07.009. [DOI] [PubMed] [Google Scholar]
- 7.Xiao Y., Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol. Ther. 2021;221 doi: 10.1016/j.pharmthera.2020.107753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen D., Zhang X., Li Z., Zhu B. Metabolic regulatory crosstalk between tumor microenvironment and tumor-associated macrophages. Theranostics. 2021;11:1016–1030. doi: 10.7150/thno.51777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cassetta L., Pollard J.W. Tumor-associated macrophages. Curr. Biol. 2020;30:R246–R248. doi: 10.1016/j.cub.2020.01.031. [DOI] [PubMed] [Google Scholar]
- 10.Ishikawa H., Barber G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun L., Wu J., Du F., Chen X., Chen Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang X., Shi H., Wu J., Zhang X., Sun L., Chen C., Chen Z.J. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell. 2013;51:226–235. doi: 10.1016/j.molcel.2013.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kwon J., Bakhoum S.F. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov. 2020;10:26–39. doi: 10.1158/2159-8290.CD-19-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McLaughlin M., Patin E.C., Pedersen M., Wilkins A., Dillon M.T., Melcher A.A., Harrington K.J. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat. Rev. Cancer. 2020;20:203–217. doi: 10.1038/s41568-020-0246-1. [DOI] [PubMed] [Google Scholar]
- 15.Wu Y., Yang S., Ma J., Chen Z., Song G., Rao D., Cheng Y., Huang S., Liu Y., Jiang S., et al. Spatiotemporal Immune Landscape of Colorectal Cancer Liver Metastasis at Single-Cell Level. Cancer Discov. 2022;12:134–153. doi: 10.1158/2159-8290.CD-21-0316. [DOI] [PubMed] [Google Scholar]
- 16.Li T., Fan J., Wang B., Traugh N., Chen Q., Liu J.S., Li B., Liu X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017;77:e108–e110. doi: 10.1158/0008-5472.CAN-17-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jung S.B., Choi M.J., Ryu D., Yi H.S., Lee S.E., Chang J.Y., Chung H.K., Kim Y.K., Kang S.G., Lee J.H., et al. Reduced oxidative capacity in macrophages results in systemic insulin resistance. Nat. Commun. 2018;9:1551. doi: 10.1038/s41467-018-03998-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolf M.T., Dearth C.L., Ranallo C.A., LoPresti S.T., Carey L.E., Daly K.A., Brown B.N., Badylak S.F. Macrophage polarization in response to ECM coated polypropylene mesh. Biomaterials. 2014;35:6838–6849. doi: 10.1016/j.biomaterials.2014.04.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rao S.S., Hu Y., Xie P.L., Cao J., Wang Z.X., Liu J.H., Yin H., Huang J., Tan Y.J., Luo J., et al. Omentin-1 prevents inflammation-induced osteoporosis by downregulating the pro-inflammatory cytokines. Bone Res. 2018;6:9. doi: 10.1038/s41413-018-0012-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hallmann R., Zhang X., Di Russo J., Li L., Song J., Hannocks M.J., Sorokin L. The regulation of immune cell trafficking by the extracellular matrix. Curr. Opin. Cell Biol. 2015;36:54–61. doi: 10.1016/j.ceb.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 21.Liu H., Liang Z., Zhou C., Zeng Z., Wang F., Hu T., He X., Wu X., Wu X., Lan P. Mutant KRAS triggers functional reprogramming of tumor-associated macrophages in colorectal cancer. Signal Transduct. Targeted Ther. 2021;6:144. doi: 10.1038/s41392-021-00534-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schuster E.M., Epple M.W., Glaser K.M., Mihlan M., Lucht K., Zimmermann J.A., Bremser A., Polyzou A., Obier N., Cabezas-Wallscheid N., et al. TFEB induces mitochondrial itaconate synthesis to suppress bacterial growth in macrophages. Nat. Metab. 2022;4:856–866. doi: 10.1038/s42255-022-00605-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lampropoulou V., Sergushichev A., Bambouskova M., Nair S., Vincent E.E., Loginicheva E., Cervantes-Barragan L., Ma X., Huang S.C.C., Griss T., et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metabol. 2016;24:158–166. doi: 10.1016/j.cmet.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mills E.L., Ryan D.G., Prag H.A., Dikovskaya D., Menon D., Zaslona Z., Jedrychowski M.P., Costa A.S.H., Higgins M., Hams E., et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. 2018;556:113–117. doi: 10.1038/nature25986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Z., Chen C., Yang F., Zeng Y.X., Sun P., Liu P., Li X. Itaconate is a lysosomal inducer that promotes antibacterial innate immunity. Mol. Cell. 2022;82:2844–2857.e10. doi: 10.1016/j.molcel.2022.05.009. [DOI] [PubMed] [Google Scholar]
- 26.Guerriero J.L., Sotayo A., Ponichtera H.E., Castrillon J.A., Pourzia A.L., Schad S., Johnson S.F., Carrasco R.D., Lazo S., Bronson R.T., et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature. 2017;543:428–432. doi: 10.1038/nature21409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaneda M.M., Messer K.S., Ralainirina N., Li H., Leem C.J., Gorjestani S., Woo G., Nguyen A.V., Figueiredo C.C., Foubert P., et al. Corrigendum: PI3Kgamma is a molecular switch that controls immune suppression. Nature. 2017;542:124. doi: 10.1038/nature21026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mackenzie K.J., Carroll P., Martin C.A., Murina O., Fluteau A., Simpson D.J., Olova N., Sutcliffe H., Rainger J.K., Leitch A., et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548:461–465. doi: 10.1038/nature23449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harding S.M., Benci J.L., Irianto J., Discher D.E., Minn A.J., Greenberg R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548:466–470. doi: 10.1038/nature23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanada K., Budzowska M., Davies S.L., van Drunen E., Onizawa H., Beverloo H.B., Maas A., Essers J., Hickson I.D., Kanaar R. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct. Mol. Biol. 2007;14:1096–1104. doi: 10.1038/nsmb1313. [DOI] [PubMed] [Google Scholar]
- 31.Xia T., Konno H., Barber G.N. Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res. 2016;76:6747–6759. doi: 10.1158/0008-5472.CAN-16-1404. [DOI] [PubMed] [Google Scholar]
- 32.Song S., Peng P., Tang Z., Zhao J., Wu W., Li H., Shao M., Li L., Yang C., Duan F., et al. Decreased expression of STING predicts poor prognosis in patients with gastric cancer. Sci. Rep. 2017;7 doi: 10.1038/srep39858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhong G., Peng C., Chen Y., Li J., Yang R., Wu M., Lu P. Expression of STING and PD-L1 in colorectal cancer and their correlation with clinical prognosis. Int. J. Clin. Exp. Pathol. 2018;11:1256–1264. [PMC free article] [PubMed] [Google Scholar]
- 34.Zeng Y.R., Song J.B., Wang D., Huang Z.X., Zhang C., Sun Y.P., Shu G., Xiong Y., Guan K.L., Ye D., Wang P. The immunometabolite itaconate stimulates OXGR1 to promote mucociliary clearance during the pulmonary innate immune response. J. Clin. Invest. 2023;133 doi: 10.1172/JCI160463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riquelme S.A., Lozano C., Moustafa A.M., Liimatta K., Tomlinson K.L., Britto C., Khanal S., Gill S.K., Narechania A., Azcona-Gutiérrez J.M., et al. CFTR-PTEN-dependent mitochondrial metabolic dysfunction promotes Pseudomonas aeruginosa airway infection. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aav4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu R., Chen F., Wang N., Tang D., Kang R. ACOD1 in immunometabolism and disease. Cell. Mol. Immunol. 2020;17:822–833. doi: 10.1038/s41423-020-0489-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weiss J.M., Davies L.C., Karwan M., Ileva L., Ozaki M.K., Cheng R.Y., Ridnour L.A., Annunziata C.M., Wink D.A., McVicar D.W. Itaconic acid mediates crosstalk between macrophage metabolism and peritoneal tumors. J. Clin. Invest. 2018;128:3794–3805. doi: 10.1172/JCI99169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu R., Liu J., Wang N., Zeng L., Yu C., Chen F., Wang H., Billiar T.R., Jiang J., Tang D., Kang R. Aconitate decarboxylase 1 is a mediator of polymicrobial sepsis. Sci. Transl. Med. 2022;14 doi: 10.1126/scitranslmed.abo2028. [DOI] [PubMed] [Google Scholar]
- 39.Muri J., Wolleb H., Broz P., Carreira E.M., Kopf M. Electrophilic Nrf2 activators and itaconate inhibit inflammation at low dose and promote IL-1beta production and inflammatory apoptosis at high dose. Redox Biol. 2020;36 doi: 10.1016/j.redox.2020.101647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He R., Wang M., Zhao C., Shen M., Yu Y., He L., Zhao Y., Chen H., Shi X., Zhou M., et al. TFEB-driven autophagy potentiates TGF-beta induced migration in pancreatic cancer cells. J. Exp. Clin. Cancer Res. 2019;38:340. doi: 10.1186/s13046-019-1343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Y., Hodge J., Liu Q., Wang J., Wang Y., Evans T.D., Altomare D., Yao Y., Murphy E.A., Razani B., Fan D. TFEB is a master regulator of tumor-associated macrophages in breast cancer. J. Immunother. Cancer. 2020;8 doi: 10.1136/jitc-2020-000543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu S., Jin J. Radiotherapy guidelines for rectal cancer in China (2020 edition) Prec Radiat Oncol. 2022;6:4–31. doi: 10.1002/pro6.1141. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The datasets used and/or analyzed during the current study are available within the manuscript and its supplementary information files.
-
•
This paper does not report the original code. Two CRC liver metastasis datasets were downloaded from GEO datasets (GSE41568 and GSE41258). The scRNA-seq data of patients with colorectal cancer liver metastases were obtained from Single-Cell Colorectal Cancer Liver Metastases (CRLM) Atlas (http://www.cancerdiversity.asia/scCRLM)
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request