

Abstract
Purpose:
The BCL-2 family of anti-apoptotic proteins, BCL-2, BCL-XL and MCL-1 can mediate survival of some types of cancer. DT2216 is a PROteolysis-TArgeting Chimera (PROTAC) that degrades BCL-XL specifically and is in phase 1 trials. We sought to define the frequency and mechanism of resistance to DT2216 in T-cell acute lymphoblastic leukemia (T-ALL) cell lines.
Methods:
We measured cell survival and protein levels of BCL-XL, BCL-2, MCL-1 and the pro-apoptotic BIM in 13 distinct T-ALL cell lines after exposure to varying concentrations of DT2216.
Results:
We identified concentrations of DT2216 which were cytotoxic to each T-ALL cell line. These concentrations have no correlation with the initial protein levels of BCL-XL, BCL-2, MCL-1 or BIM in each cell line. However, there was a correlation between survival to DT2216 and the efficiency of degradation of BCL-XL by DT2216. Only one cell line, SUP-T1 had significant resistance to DT2216, defined as an IC50 above what is achievable in murine tumors in vivo.
Conclusion:
Resistance to DT2216 is rare in a wide variety of T-ALL cells but when it occurs is correlated with decreased BCL-XL degradation. Resistance to DT2216 in T-ALL is not predicted by initial BCL-XL or BIM protein levels, or BCL-2 or MCL-1 levels before or after treatment. These data imply that a phase 2 clinical trial of DT2216 in T-ALL should be widely available and not limited to a subset of patients.
INTRODUCTION
BCL-XL, BCL-2 and MCL-1 are the most important members of the anti-apoptotic BCL-2 protein family [1]. These proteins promote neoplastic transformation, tumor progression, and drug resistance by protecting tumor cells from apoptosis [2]. Because of this, there has been intense pharmaceutical interest in developing BCL-2 family protein inhibitors, with the BCL-2 inhibitor venetoclax reaching FDA approval and wide clinical utility in B-cell and myeloid malignancies [3,4]. Unfortunately, clinical development of the dual BCL-2/BCL-XL inhibitor ABT-263 (navitoclax) has been stymied because platelets have an absolute requirement for BCL-XL, which results in unacceptable thrombocytopenia [5]. Thus, BCL-XL is one of the most promising cancer targets yet remains undrugged due to on-target dose-limiting thrombocytopenia.
To overcome this on-target, dose-limiting toxicity, we developed the proteolysis-targeting chimera (PROTAC), DT2216 as a specific BCL-XL protein degrader [6–8]. DT2216 recruits the E3 ligase VHL, commonly expressed in most cancer cells, to ubiquitinate BCL-XL for proteasomal degradation [8]. In pre-clinical studies, DT2216 was highly cytotoxic for MOLT4 T-ALL cells in vitro as well as in two patient-derived xenograft (PDX) models of T-ALL without significant platelet toxicity [8]. In addition, DT2216 enhanced the efficacy of many types of cytotoxic chemotherapy in multiple cancer types in vitro and in murine xenograft or PDX models, including triple negative breast cancer, pancreatic cancer, and small cell lung cancer [8–10].
PROTACs mediate protein degradation via ubiquitination of target proteins for proteasomal degradation [6,11]. PROTACs have many advantages over conventional small molecule inhibitors because they are catalytic and can be recycled within a cell, thus their active cellular half-life exceeds their plasma half-life. In addition, by using ligands to E3 ligases that are preferentially expressed in tumor and not normal tissue they can decrease toxicity and rescue previously discarded drugs. Finally, they can target previously undruggable proteins because the target protein ligands do not have to be a functional inhibitor of the proteins, as long as they can directly bind to or interact with the targets.
Although multiple PROTACs have been developed to efficiently degrade various cancer targets, only a few have reached clinical trials, because they are complex to synthesize and are large and poorly soluble [6,11,12]. Nonetheless, PROTACs are in clinical trials for the androgen receptor, estrogen receptor, IRAK4, BTK, BRD9, and STAT3 [6,11]. DT2216, a BCL-XL degrader received an FDA IND in January of 2021 and is in the midst of a phase 1 trial for dose-finding (NCT04886622). DT2216 demonstrates the power of PROTAC technology in overcoming on-target drug toxicity, in that it selectively degrades BCL-XL without dose-limiting platelet toxicity by using a ligand to VHL for targeting BCL-XL for ubiquitination [8]. VHL is poorly expressed in human platelets and highly expressed in most human tumors, thus differentially targeting BCL-XL for proteasomal degradation in cancer cells [8].
Normal T-cells require BCL-XL to survive thymic selection [13], and many types of T-cell malignancies re-express BCL-XL to survive neoplastic transformation [7,14]. Thus, T-cell malignancies are an excellent target for demonstrating efficacy of DT2216 in phase 2 trials [7,14]. In this study we systematically surveyed 13 known T-ALL cell lines for expression of the BCL-2 family members and resistance to DT2216. We found that only one cell line, SUP-T1 resisted levels of DT2216 above what is achievable intratumorally in vivo, partially from decreased degradation of BCL-XL [11,12]. Initial levels of BCL-XL, BCL-2, MCL-1 or the pro-apoptotic BIM had no correlation with sensitivity to DT2216.
MATERIALS AND METHODS
Chemicals and antibodies:
DT2216 was synthesized by us as previously described [8] and is 98.0% pure by LC-MS () as described [8]. It is stored at 4° C as a 10 mM stock solution in DMSO and diluted in DMSO before addition to culture media at no less than a 1:1000 dilution. RPMI medium (Hyclone, Logan, UT), fetal bovine serum albumin (Atlanta Biologicals, GA, USA), T-25 and T-75 tissue culture flasks, and 5 ml, 10 ml, and 25 ml pipettes and pipettors were used during the experimental procedure. VHL, BCL-XL, BCL-2, MCL-1, cleaved Caspase 3, and β-actin antibodies were purchased from Cell Signal Technology (USA). Cleaved PARP1 antibody was obtained from Sigma (St. Louis, MO). IL-2 was purchased from Miltenyi Biotech (Auburn, CA, USA) and the Cell Counting Kit 8, was procured from Dojindo (Rockville, MD, USA).
Cell lines and culture:
Human T-ALL cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and Leibniz Institute, DSMZ-German Collection of Microorganisms and Cell Cultures (GmbH, Braunschweig, Germany)(Table S1). Cells were grown in RPMI medium supplemented with 10% or 20% fetal bovine serum albumin (FBS; Atlanta Biologicals, Atlanta, GA, USA) as described by the ATCC or DSMZ at 37°C in a humidified atmosphere of 5% CO2 in a tissue culture incubator. The culture conditions are described in Table S1. TALL-104 required IL-2 as described by the ATCC.
Western blot analysis:
Protein levels of BCL-XL, BCL-2, MCL-1, BIM, and VHL proteins, before and after the treatment of DT2216 in the cells were determined by western blot analysis as we described with β-actin as a loading control [8]. Ten μG of cell lysate was used for each analysis. Each Western was repeated` at least twice with distinct protein preparations.
Cell cytotoxicity assay:
Cell proliferation/cytotoxicity assays were performed on the leukemia cell lines listed in supplemental Table S1. Cell Counting Kit-8 (CCK-8, Dojindo, MD, USA) was used to normalize cell number. Two × 103 to 2 × 104 cells were plated in 96-well plates for each condition in quadruplet, depending on the doubling time of the cell line (Table S1). Varying concentrations of DT2216 were added to each well and cells allowed to grow for 24 h, 48 h, 72 h and 96 h. DMSO was used for dissolving DT2216 and also as the vehicle control. After the treatment, 10 ul of CCK-8 reagent was added to each well and incubated at 37°C for 4 h. After 4 h the intensity of the formazan dye was quantified spectrophotometrically at ʎmax 450 nm on a 96-well plate reader. Absorbance was recorded and % survival was calculated as we described [8].
Statistical analysis:
All survival experiments were repeated at least three times in quadruplicate and data were expressed as Mean ± SE of all experiments. Statistical analysis was performed using Student’s t test. The criterion for statistical significance was p<0.05. For western blotting data, band intensities were measured using ImageJ (NIH) and normalized with β-actin. Each blot was performed twice with different protein preparations each time. The Spearman correlation coefficient, which uses ranks (the nonparametric method) of log-scale values assessed the correlation of the inhibitory concentration of DT2216 that killed 50% (IC50) of T-ALL cells with the concentration that degraded 50% of BCL-XL protein (DC50) and then the relative initial protein expression of BCL-XL.
RESULTS
Because many T-ALL rely on BCL-XL for survival [7,14], they are a key disease target for a DT2216 clinical trial, yet questions remain before embarking on such a trial. Two key questions that would help direct such a trial is whether resistance could be predicted by high initial levels of BCL-XL and whether resistance to DT2216 is due a failure to degrade BCL-XL. It is possible that patients should be selected a priori for DT2216 effectiveness by either immunohistology of tumor BCL-XL or BH3 profiling to assess tumor sensitivity to BCL-XL degradation [7,14].
BCL-XL, BCL-2, MCL-1, BIM and VHL levels in T-ALL cells
Using western blot analysis, we first asked whether there was an inverse relationship between BCL-XL, BCL-2, and MCL-2 protein levels in 13 T-ALL cell lines, since these proteins can trade off providing survival signals to cancer cells [1,2]. Figure 1A shows that there is not an inverse correlation between initial levels of these three anti-apoptotic proteins in the 13 T-ALL cell lines tested here. High BIM levels have been reported to promote sensitivity to chemotherapy in the T-ALL cell line SUP-T1 [15]. Therefore, we measured BIM levels in the 13 T-ALL cell lines using western blot analysis normalized to actin (Fig. 1B). We found that all three BIM isoforms, EL, L and S were lower in SUP-T1 cells than 8 other T-ALL lines, but there were 4 T-ALL lines that had far more sensitivity to DT2216 and yet lower BIM levels than SUP-T1, suggesting that BIM expression is not correlated with T-ALL cell sensitivity to DT2216. There was no correlation between protein levels of VHL, the E3 ligase recruited by DT2216 to BCL-XL and sensitivity to DT2216 in the 13 cell lines (Fig. S1).
Figure 1. Pre-treatment protein levels of BCL-XL, BCL-2 and MCL-1.
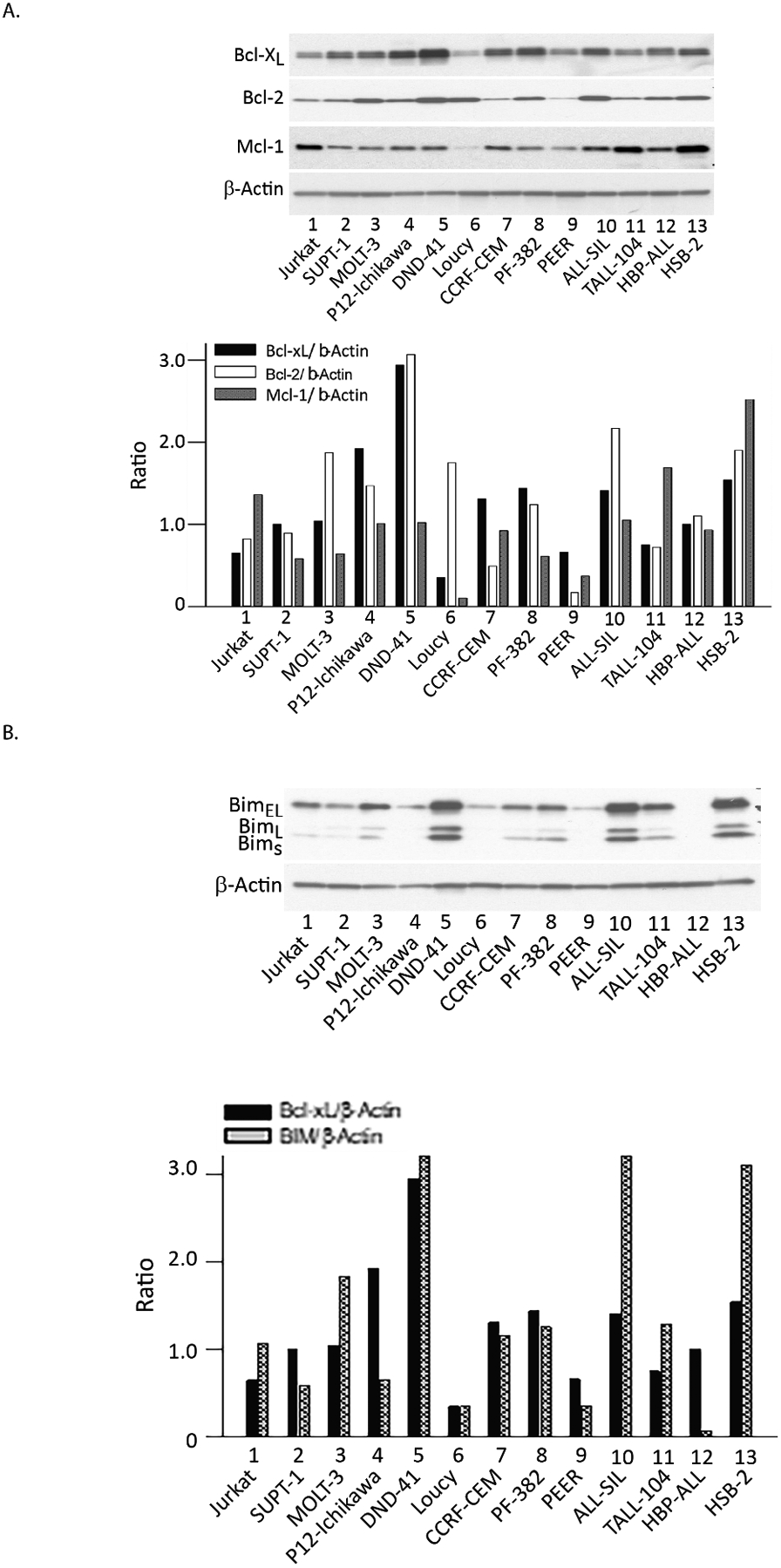
A- Prior to DT2216 exposure western analysis of the 13 T-ALL cell lines defined BCL-XL, BCL-2 and MCL-1 protein levels relative to actin using densitometry. B- Prior to DT2216 exposure, the protein levels of BIM were assessed by western blot analysis and compared with actin in each cell line.
DT2216 effectiveness in a wide variety of T-ALL cells
We next studied the effects of DT2216 on cell survival in 13 T-ALL cell lines: Jurkat, SUP-T1, CCRF-CEM, DND-41, P12-Ichikawa, MOLT-3 (same patient but distinct clone from MOLT-4, similar response to DT2216, 8), Loucy, PEER, PF-382, ALL-SIL, TALL-104, HSB-2, and HBP-ALL. The concentrations required to inhibit growth by 50% in the cells (IC50) after 48 hrs of exposure are reported in Figure 2. Full dose-dependent survival curves for all cell lines at 24, 48, 72 and 96 hrs are described in Supplemental Figure S2. Time course IC50 values for all 13 T-ALL cell lines to DT2216 are in Supplemental Figure S3. Among of these T-ALL cell lines, only SUP-T1 required levels of DT2216 that exceeded what we reported could be achieved within a T-ALL xenografted tumor in vivo in immunodeficient mice, 800 nM [8]. All 12 other T-ALL cell lines had IC50 values well below this, implying they would be sensitive to DT2216, at least in this in vivo model. The most sensitive T-ALL cell line was Loucy, with an IC50 of 2 nM.
Figure 2. DT2216 concentrations required for cell death and BCL-XL degradation.
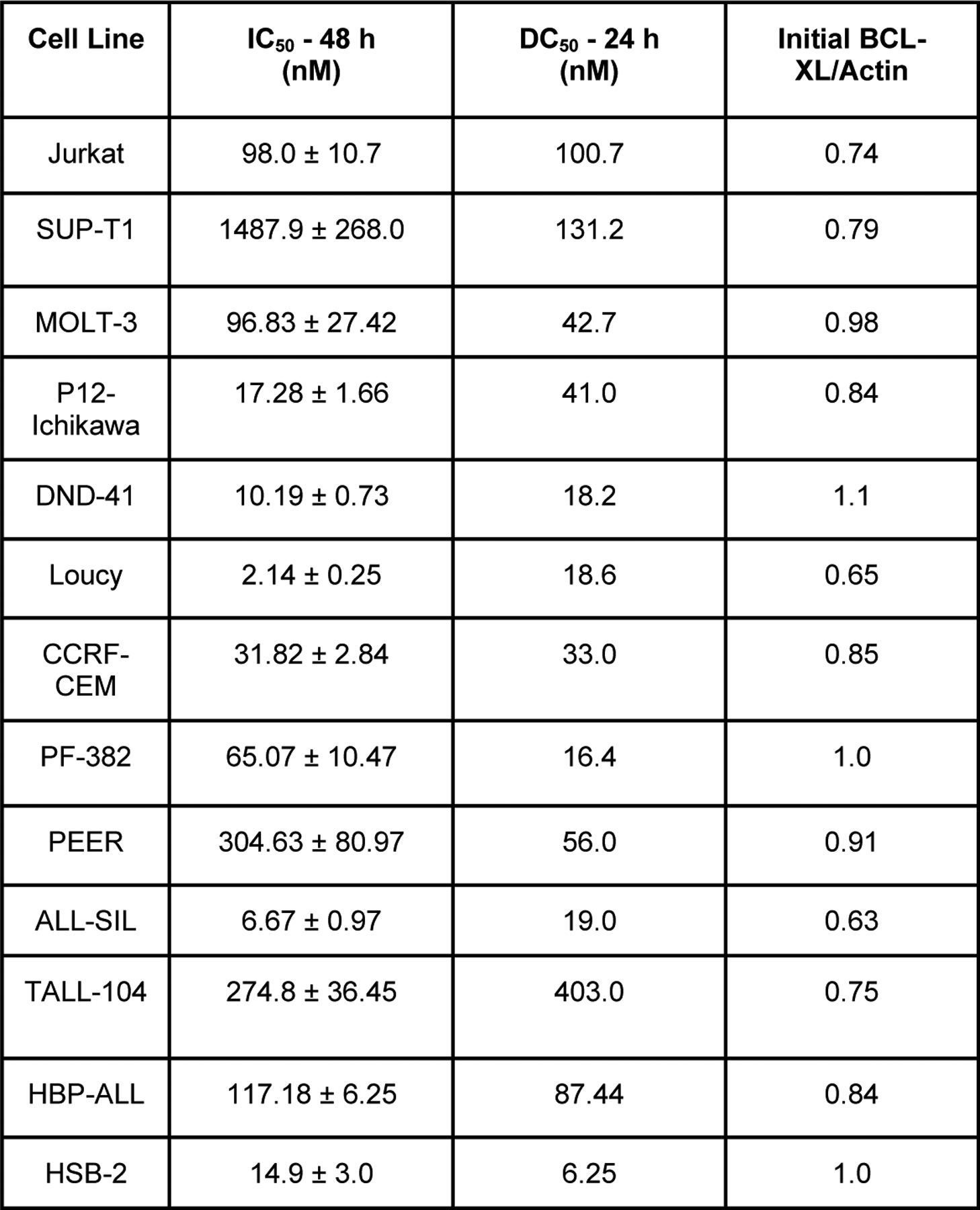
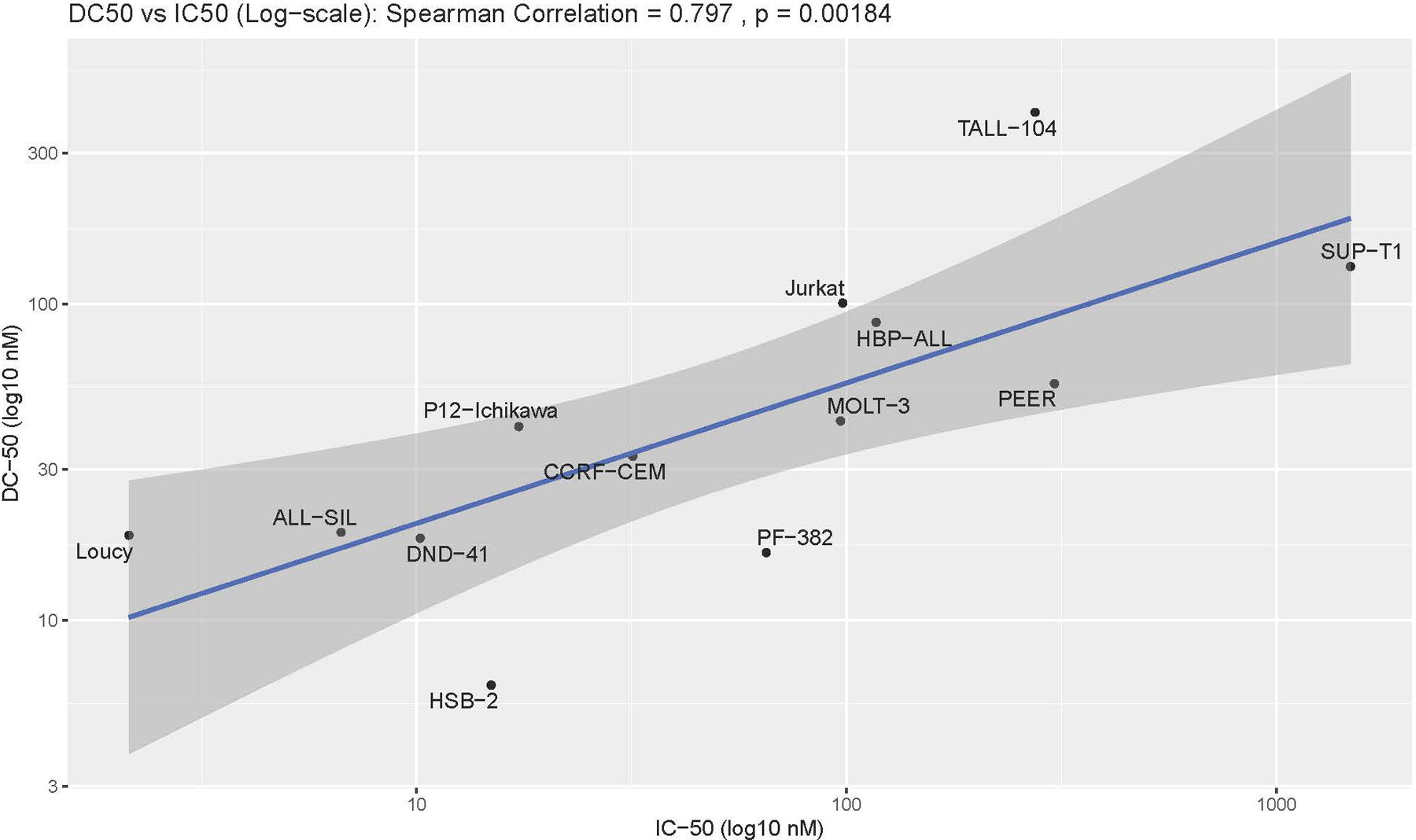
A- The left column has the DT2216 inhibitory concentration for 50% of cells (IC50) calculated using MTS assays of the 13 T-ALL cell lines exposed to varying concentrations of DT2216 for 48 hrs. The middle column is the DT2216 concentration for degrading 50% of BCL-XL (DC50) calculated using densitometry on western blots after 24 hrs of DT2216. The right column is the ratio of initial BCL-XL protein levels compared to actin using densitometry of western blot analyses. Full survival curves and western analyses of BCL-XL protein degradation are in the supplemental figures. B- Graphical representation of the Spearman Coefficient correlation between the IC50 and the DC50, P=0.0018.
Correlation of IC50 with degradation of BCL-XL
We had postulated that resistance to DT2216 could be due to decreased BCL-XL degradation by DT2216. We found that there was indeed a statistical correlation between the concentration of DT2216 needed to degrade 50% of the initial BCL-XL protein level (DC50) and the IC50 (Fig. 2A, Supplemental Fig. S4).
No increase of BCL-2 and MCL-1 levels after DT2216 treatment
We then asked whether DT2216 could induce the expression of BCL-2 or MCL-1, and they could replace BCL-XL as a survival signal (Supplemental Fig. S4). Their expression was not induced in any cell line by DT2216. Indeed, in some instances BCL-2 or MCL was also degraded at the higher DT2216 concentrations. This is likely due to cleavage of these proteins by activated caspases, as we previously reported [8]. Notably, the resistant SUP-T1 cells had no change in their BCL-2 or MCL-1 protein levels after exposure to DT2216. In the DT2216-resistant SUP-T1 cells BCL-XL was effectively degraded and still generated markers of apoptosis such as cleaved PARP1 and Caspase 3 well below the IC50 (Supplemental Fig. S5), which implies that there is likely another mechanism of resistance for this line.
DISCUSSION
T-ALL is an uncommon malignancy that can be cured with aggressive chemotherapy [14–20]. However, when it becomes resistant and relapses, most patients will succumb to their disease; thus, new therapies for relapsed T-ALL are needed, and one promising alternative is DT2216 [6–8]. Indeed, the wide sensitivity of 12 of 13 cell lines, all derived from relapsed patients implies that perhaps adding DT2216 to maintenance T-ALL chemotherapy would prevent relapse [20].
Double positive CD4+/CD8+ T-cells that do not react with self-antigen express BCL-XL to survive thymic selection, and after thymic selection most normal T-cells no longer require BCL-XL [13], with the notable exception of CD25+ regulatory T-cells [16]. Interestingly, many but not all types of T-cell malignancies rely on BCL-XL for survival, implying that these malignancies utilize the same pathway for survival as normal thymic T-cells [7,14]. Using BH3 profiling, Weinstock and team found that the majority of cutaneous T-cell lymphomas and a minority fraction of peripheral T-cell lymphomas rely on BCL-XL for survival [17]. We then demonstrated that DT2216 was an effective therapeutic agent against T-cell lymphomas in vitro and in vivo [7].
Letai and colleagues used BH3 profiling to assess the dependency of T-ALL cells on the BCL-2 family members [14]. They found that the majority of T-ALL were dependent on BCL-XL for survival, similar to the double positive T-cells undergoing thymic selection [13]. However, they noted that a minority of T-ALL, those with an immature phenotype and poor prognosis, termed early T-cell progenitor (ETP) subgroup, did not require BCL-XL but rather BCL-2 [14]. ETP T-ALL has a unique immunophenotype (CD1a−, CD8−, CD5weak with stem-cell/myeloid markers), high expression of ERG, and lack of biallelic TCRγ locus deletion, which means that the T-ALL has differentiation arrest prior to V(D)J recombination of this locus. The model cell line cited as an example for an ETP T-ALL was Loucy; However, in our hands Loucy cells were highly sensitive to DT2216 (Fig. 2A, supplemental Fig. S2). The cell line PEER has also been suggested as an ETP-ALL [21], and it was less sensitive to DT2216 than Loucy (48 hr IC50 of 305 versus 2 nM for Loucy), indicating diversity of response to DT2216 in ETP-ALL.
There are several potential reasons for the finding that DT2216 could kill Loucy cells at a low IC50. First, it is likely that DT2216 is far more potent than BH3 profiling, since as noted above it degrades BCL-XL and does not just inhibit it. Second, the cell line could have drifted in culture, and resulted in a greater reliance on BCL-XL than BCL-2. Third, it is possible that while DT2216 does not degrade BCL-2 it could at high concentrations inhibit it. However, the IC50 of DT2216 in Loucy cells makes this unlikely. Nonetheless, DT2216 could be useful in T-ALL with the ETP phenotype, where it would be needed, since this group is far more likely to relapse.
Significantly, there is no correlation between BCL-XL DC50 values and the initial levels of BCL-XL. This indicates that DT2216 is effective at BCL-XL degradation no matter the initial BCL-XL protein levels. Thus, in subsequent clinical trials T-ALL patients should not be excluded based on initial high levels of BCL-XL in the marrow blasts. However, there was a correlation between IC50 and DC50 levels, with SUP-T1 cells having the highest IC50 and DC50, despite its initial BCL-XL levels being less than the majority of other T-ALL lines (Fig. 1A, Fig. 2A). Thus, one pathway towards resistance to DT2216 is a decrease in its degradation of BCL-XL, but there must also be other resistance mechanisms since DT2216 had an IC50 at 48 hrs (1487.9 nM) against SUP-T1 cells that is almost 10-fold higher than its DC50 (131.2 nM) (Fig. 2A, Supplemental Fig. S1, S2).
Levels of BCL-XL, BCL-2, MCL-1, VHL or BIM in SUPT1 cells did not predict its resistance and levels of these proteins in the sensitive cell lines did not predict their sensitivity to DT2216. These data indicate that levels of BCL-XL, BCL-2, MCL-1, VHL or BIM in an initial marrow biopsy in T-ALL should not dictate exclusion of any patient from therapy with DT2216. In addition, we recently developed a dual BCL-XL/BCL-2 degrader that would prevent shifting reliance for survival to BCL-2, although this may add toxicity without added benefit since 12 of 13 T-ALL lines are sensitive to DT2216 [22].
In summary, this study found that DT2216 is a highly potent agent for T-ALL cells regardless of their expression levels of BCL-XL, BCL-2, MCL-1, or BIM. Many of the sensitive cell lines, including CCRF-CEM, ALL-AIL and Jurkat are reported to be highly drug resistant (steroids, 6TG, asparaginase, etoposide), which is not surprising since they were derived from relapsed samples [18,19]. This implies that no patient should be excluded from a trial of DT2216 based on a history of multi-drug resistance. The finding that DT2216 is highly active in these resistant cell lines as well as the ETP cell line Loucy indicates that it is a promising agent in a disease where relapse is most often fatal [20].
Supplementary Material
Acknowledgements-
Funding from US National Institutes of Health (NIH) grant R01 CA242003 (D.Z. and G.Z.), RO1 CA205224 (R.H.) and from the Cancer Prevention of Research Institute of Texas Individual Investigator grant RP220269.
Footnotes
Competing interests- G.Z. and D.Z. hold patents for use of BCL-XL PROTACs as senolytic and antitumor agents. G.Z., D.Z., and R.H. have equity in Dialectic Therapeutics, which owns the license for DT2216.
Data Availability-
All data generated or analyzed during this study are included in this published article and its supplementary information files.
REFERENCES
- 1.Kale J, Osterlund EJ, & Andrews DW (2017). Bcl-2 family proteins: Changing partners in the dance towards death. Cell Death & Differentiation, 25(1), 65–80. 10.1038/cdd.2017.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shamas-Din A, Kale J, Leber B, & Andrews DW (2013). Mechanisms of action of bcl-2 family proteins. Cold Spring Harbor Perspectives in Biology, 5(4). 10.1101/cshperspect.a008714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Elmore SW (2013) ABT-199, a potent and selective bcl-2 inhibitor, achieves antitumor activity while sparing platelets. Nature Medicine 19(2), 202–208. 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 4.Zhang X, Liu X, Zhou D, Zheng G. Targeting anti-apoptotic BCL-2 family proteins for cancer treatment (2020). Future Med Chem. Apr;12(7):563–565. doi: 10.4155/fmc-2020-0004. [DOI] [PubMed] [Google Scholar]
- 5.Gandhi L, Camidge DR, Ribeiro de Oliveira M, Bonomi P, Gandara D, Khaira D, Hann CL, McKeegan EM, Litvinovich E, Hemken PM, Dive C, Enschede SH, Nolan C, Chiu YL, Busman T, Xiong H, Krivoshik AP, Humerickhouse R, Shapiro GI, Rudin CM. (2011) Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol. Mar 1;29(7):909–16. doi: 10.1200/JCO.2010.31.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He Y, Khan S, Huo Z, Lv D, Zhang X, Liu X, Yuan Y, Hromas R, Xu M, Zheng G, & Zhou D (2020). Proteolysis targeting chimeras (protacs) are emerging therapeutics for hematologic malignancies. Journal of Hematology & Oncology, 13(1). 10.1186/s13045-020-00924-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He Y, Koch R, Budamagunta V, Zhang P, Zhang X, Khan S, Thummuri D, Ortiz YT, Zhang X, Lv D, Wiegand JS, Li W, Palmer AC, Zheng G, Weinstock DM, & Zhou D (2020). DT2216—a bcl-xl-specific degrader is highly active against bcl-xl-dependent T cell lymphomas. Journal of Hematology & Oncology, 13(1). 10.1186/s13045-020-00928-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khan S, Zhang X, Lv D, Zhang Q, Zhang P, Liu X, Thummuri D, Yuan Y, Wiegand J, Pei J, Zhang W, Sharma A, Mccurdy C, Kuruvilla V, Baran N, Ferrando A, Kim Y, Rogojina A, & Zhou D (2019). A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nature Medicine 25. 10.1038/s41591-019-0668-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thummuri D, Khan S, Underwood PW, Zhang P, Wiegand J, Zhang X, Budamagunta V, Sobh A, Tagmount A, Loguinov A, Riner AN, Akki AS, Williamson E, Hromas R, Vulpe CD, Zheng G, Trevino JG, Zhou D. (2022) Overcoming Gemcitabine Resistance in Pancreatic Cancer Using the BCL-XL-Specific Degrader DT2216. Mol Cancer Ther. Jan;21(1):184–192. doi: 10.1158/1535-7163.MCT-21-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khan S, Wiegand J, Zhang P, Hu W, Thummuri D, Budamagunta V, Hua N, Jin L, Allegra CJ, Kopetz SE, Zajac-Kaye M, Kaye FJ, Zheng G, Zhou D. (2022) BCL-XL PROTAC degrader DT2216 synergizes with sotorasib in preclinical models of KRASG12C-mutated cancers. J Hematol Oncol. Mar 9;15(1):23. doi: 10.1186/s13045-022-01241-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Békés M, Langley DR & Crews CM (2022). PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discov. 21: 181–200. 10.1038/s41573-021-00371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao h, Sun X, & Rao Y. (2020) PROTAC Technology: Opportunities and Challenges. ACS Med. Chem. Lett 11 (30): 237–240. 10.1021/acsmedchemlett.9b00597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma A, Pena JC, Chang B, Margosian E, Davidson L, Alt FW, Thompson CB. (1995) Bclx regulates the survival of double-positive thymocytes. Proc Natl Acad Sci U S A. 1995 May 23;92(11):4763–7. doi: 10.1073/pnas.92.11.4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chonghaile TN, Roderick JE, Glenfield C, Ryan J, Sallan SE, Silverman LB, Loh ML, Hunger SP, Wood B, DeAngelo DJ, Stone R, Harris M, Gutierrez A, Kelliher MA, Letai A. (2014) Maturation stage of T-cell acute lymphoblastic leukemia determines BCL-2 versus BCL-XL dependence and sensitivity to ABT-199. Cancer Discov. Sep;4(9):1074–87. doi: 10.1158/2159-8290.CD-14-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leung KT, Li KK, Sun SS, Chan PK, Ooi VE, Chiu LC. (2008) Activation of the JNK pathway promotes phosphorylation and degradation of BimEL--a novel mechanism of chemoresistance in T-cell acute lymphoblastic leukemia. Carcinogenesis. Mar;29(3):544–51. doi: 10.1093/carcin/bgm294. [DOI] [PubMed] [Google Scholar]
- 16.Kolb R, De U, Khan S et al. (2021) Proteolysis-targeting chimera against BCL-XL destroys tumor-infiltrating regulatory T cells. Nat Commun. 12:1281. 10.1038/s41467-021-21573-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koch R, Christie AL, Crombie JL, Palmer AC, Plana D, Shigemori K, Morrow SN, Van Scoyk A, Wu W, Brem EA, Secrist JP, Drew L, Schuller AG, Cidado J, Letai A, Weinstock DM. (2019) Biomarker-driven strategy for MCL1 inhibition in T-cell lymphomas. Blood. Feb 7;133(6):566–575. doi: 10.1182/blood-2018-07-865527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Follini E, Marchesini M, Roti G. (2019) Strategies to Overcome Resistance Mechanisms in T-Cell Acute Lymphoblastic Leukemia. Int J Mol Sci. Jun 20;20(12):3021. doi: 10.3390/ijms20123021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beesley A, Palmer ML., Ford J et al. (2016) Authenticity and drug resistance in a panel of acute lymphoblastic leukaemia cell lines. Br J Cancer 95: 1537–1544. 10.1038/sj.bjc.6603447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raetz EA, & Teachey DT (2016). T-cell acute lymphoblastic leukemia. Hematology, 2016(1), 580–588. 10.1182/asheducation-2016.1.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goossens S, Radaelli E, Blanchet O et al. (2015) ZEB2 drives immature T-cell lymphoblastic leukaemia development via enhanced tumour-initiating potential and IL-7 receptor signalling. Nat Commun. Jan 7; 6:5794. 10.1038/ncomms6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lv D, Pal P, Liu X et al. (2021) Development of a BCL-xL and BCL-2 dual degrader with improved anti-leukemic activity. Nat Commun. 12:6896. 10.1038/s41467-021-27210-x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files.