

Abstract
Despite an enormous interest in understanding the bioactivity of extracellular vesicles (EV) in physiology and disease for the development of therapeutic applications, the impact of EV preparation methods remains minimally explored. In this study, we implemented density gradient ultracentrifugation combined with size‐exclusion chromatography (DG‐SEC), differential ultracentrifugation (dUC) and/or stand‐alone SEC (sSEC) to fractionate media conditioned by different cancer cells and/or cancer‐associated fibroblasts (CAF). EV‐enriched but protein‐depleted versus EV‐depleted but protein‐enriched DG‐SEC fractions, and EV‐containing dUC and sSEC preparations were quality controlled for particle number, protein concentration, selected protein composition and ultrastructure, characterized for their cytokine content, and dose‐dependently evaluated for monocyte‐derived dendritic cell (MoDC) maturation by measuring surface marker expression and/or cytokine secretion. EV preparations obtained by DG‐SEC from media conditioned by different cancer cell lines or CAF, were depleted from soluble immune suppressive cytokines such as VEGF‐A and MCP‐1 and potently stimulated MoDC maturation. In contrast, EV‐containing dUC or sSEC preparations were not depleted from these soluble cytokines and were unable to mature MoDC. Subsequent processing of dUC EV preparations by SEC dose‐dependently restored the immunomodulatory bioactivity. Overall, our results demonstrate that method‐dependent off‐target enrichment of soluble cytokines has implications for the study of EV immunomodulatory bioactivity and warrants careful consideration.
Keywords: corona, dendritic cells, exosomes, isolation, maturation, microvesicles, separation, vaccines
1. INTRODUCTION
As professional antigen presenting cells, dendritic cells (DC) are essential to elicit sustained anti‐tumour immune responses and targeted by several immunotherapy strategies (Anguille et al., 2014; Wculek et al., 2019). Tumour antigens are used to pulse ex vivo autologous DC, often generated from the differentiation of CD14+ blood monocytes (i.e., monocyte‐derived DC or MoDC), that initiate anti‐tumour immune responses when re‐injected in patients (Dillman et al., 2018; Perez & De Palma, 2019). DC‐based vaccines are safe and well tolerated (Anguille et al., 2014), and started to move to the clinic with the approval of Sipuleucel‐T for the treatment of metastatic castration‐resistant prostate cancer (Kantoff et al., 2010). However, the objective response of patients remains low in clinical trials, paving the way for a new generation of DC vaccines using autologous tumour‐derived extracellular vesicles (EV) as DC maturation and antigen source over commonly used whole tumour lysates that induce variable responses (Wculek et al., 2019; Perez & De Palma, 2019; Squadrito et al., 2018).
EV are nanometer‐sized membrane‐enclosed particles containing proteins, lipids and nucleic acids that are released by cells and mediate local and distant cell‐cell communication between cancer and stromal cells, including cancer‐associated fibroblasts (CAF) and immune cells (Melo et al., 2015). The analysis of EV function requires their separation from other extracellular materials (such as lipoproteins or Ago2‐containing ribonucleoprotein complexes) and soluble factors (such as cytokines) which is challenging and differently achieved by available preparation methods, but introduces biases in downstream functional assays (Whittaker et al., 2020; Van Deun et al., 2014). Recent observations that EV carry a functional biomolecular corona (e.g., heparin‐binding cytokines that interact with proteoglycans on the EV surface) which is differentially maintained during EV preparation further adds to the complexity (Tóth et al., 2021; Wolf et al., 2022). As a consequence, it is not surprising that EV preparations have been attributed both stimulating and inhibiting roles in MoDC maturation and function (Andre et al., 2002; Marton et al., 2012; Ning et al., 2018; Rao et al., 2016; Salimu et al., 2017; Wang et al., 2020; Wolfers et al., 2001; Yang et al., 2011). The presence of soluble cytokines impairing DC maturation such as VEGF, GROα and MCP‐1 (Michielsen et al., 2011; Park et al., 2004; Takahashi et al., 2004; Xu et al., 2022), may confound functional assessment of EV preparations, depending on the specificity of the preparation method implemented. Differential ultracentrifugation (dUC), known to recover EV by high speed pelleting with moderate specificity (Shu et al., 2019; Van Deun et al., 2014), is predominantly used to study the effect of tumour‐derived EV on MoDC maturation and function (Naseri et al., 2020). Polymer‐based precipitation recovers EV preparations with lower specificity containing cytokines (Jung et al., 2020) or Ago2‐containing ribonucleoprotein complexes (Van Deun et al., 2014), introducing biases in downstream functional applications (Jeppesen, 2019; Van Deun et al., 2014). The sequential implementation of density gradient ultracentrifugation (DG) combined with size‐exclusion chromatography (SEC) (combination referred to as DG‐SEC) separates EV with higher specificity from other materials as has been reported for soluble proteins and lipoprotein particles in complex biofluids (blood plasma (Geeurickx et al., 2019; Vergauwen et al., 2021), urine (Dhondt, Geeurickx, et al., 2020; Dhondt, Lumen, et al., 2020), faeces (Tulkens et al., 2018, 2020) and cell culture medium (CM) (Van Deun et al., 2020). Interestingly, the use of SEC and dUC has recently been shown to affect the EV biomolecular corona and abrogate EV bioactivity (Wolf et al., 2022).
In this study we used different methodologies (including DG‐SEC, dUC and stand‐alone SEC (sSEC)) to prepare EV from concentrated CM (CCM) of breast cancer cell lines and CAF. EV separation from other extracellular materials (such as soluble cytokines and Ago2‐containing ribonucleoprotein complexes) was evaluated using complementary characterization methods (including transmission electron microscopy (TEM), nanoparticle tracking analysis (NTA), Qubit protein dosage, western blotting and Luminex® cytokine multi‐analyte profiling). Next, the dose‐dependent potential of EV‐enriched but protein‐depleted versus EV‐depleted but protein‐enriched DG‐SEC fractions, and EV‐containing dUC and sSEC preparations was tested on expression and secretion of MoDC maturation markers. We observe method‐dependent enrichment of soluble cytokines in EV preparations which consequently affect immunomodulatory bioactivity resulting in differential expression and secretion of MoDC maturation markers. Specific depletion of soluble cytokines from EV preparations through the sequential implementation of preparation methods is necessary and sufficient to unlock EV immunomodulatory bioactivity.
2. RESULTS
2.1. DG‐SEC separates EV from soluble cytokines released in medium conditioned by breast cancer cells and CAF
We prepared EV from 300 mL of medium conditioned by MDA‐MB‐231 and MCF‐7 breast cancer cells, and CAF using a sequential implementation of bottom‐up DG followed by SEC (combination referred to as DG‐SEC) (Figure 1a). EV were enriched in fractions with a density of ≈1.085–1.110 g/mL, corresponding to 1 mL fractions 9 and 10 that were pooled as pool (p) 1 as previously described (Dhondt, Geeurickx, et al., 2020; Dhondt, Lumen, et al., 2020; Tulkens et al., 2018, 2020; Van Deun et al., 2020, Vergauwen et al., 2017, 2021) (Figures 1a and S1e). Next, p1 was further processed by SEC . SEC fractions 4‐5‐6‐7 referred to as EV‐enriched SEC fractions , and 8‐9‐10‐11 referred to as protein‐enriched SEC fractions were respectively pooled and concentrated to 100 μL using 10 kDa ultrafiltration units (Figure 1a). To compare the fractions of the DG fractionation process and to ensure that the majority of EV are in p1, we generated a pool (p) 2 with 1 mL fractions 11, 12, 13, (density ≈1.115–1.195 g/mL) and a pool (p) 3 corresponding to 1 mL fractions 14, 15, 16 (density ≈1.200–1.260 g/mL) (Figures 1a and S1e) (Dhondt, Geeurickx, et al., 2020; Vergauwen et al., 2021). For each cell line, p2 and p3 were processed by SEC as done for p1.
FIGURE 1.
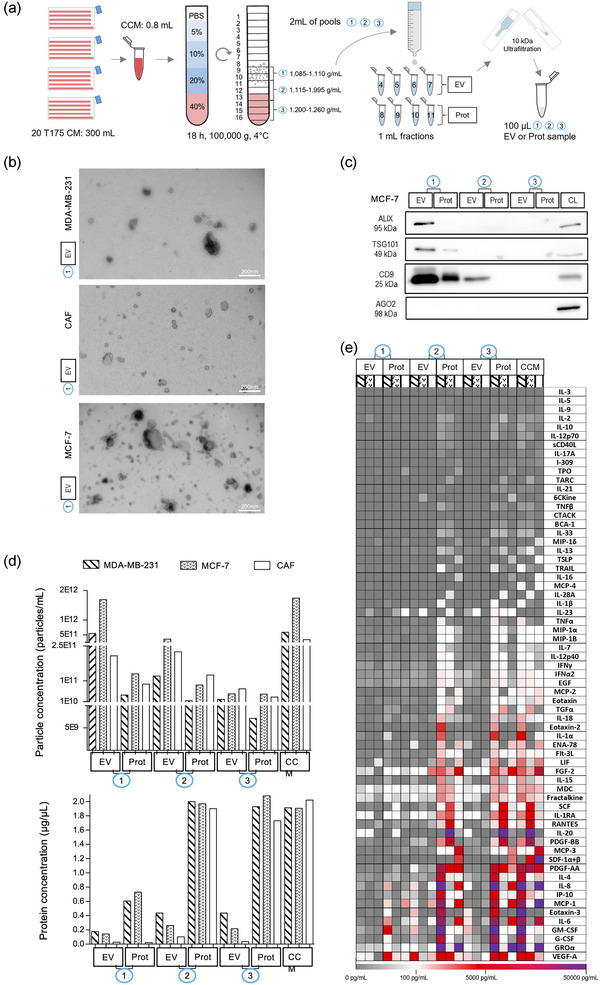
Bottom‐up DG‐SEC efficiently separates EV from soluble cytokines. (a) Schematic representation of the DG‐SEC protocol. CM: conditioned medium, CCM: concentrated CM, EV: EV‐enriched, Prot: protein‐enriched. (b, d, and e) Particle and protein characterization of CCM and pools (p)1, p2 and p3 EV and protein‐enriched samples generated as described in (a) from a representative MDA‐MB‐231, MCF‐7 and CAF harvesting (n = 1) (see Figures S1 c, d,g). (b) Transmission electron microscopy images from MDA‐MB‐231, MCF‐7 or CAF p1 EV‐enriched SEC fractions. Magnification × 30,000, scale bar: 200 nm. (c) Western blot analysis of MCF‐7 p1, p2 and p3 EV and protein‐enriched (Prot) SEC fractions. Thirty microlitres of samples generated by DG‐SEC (a) were denatured as described in the methods section and loaded in each lane. CL: cell lysate (15 μg). (d) Particle and protein concentrations measured in CCM and p1, p2 and p3 EV and Prot samples. Particle and protein concentration were measured by nanoparticle tracking analysis (see (d)) and Qubit protein assay respectively (n = 1). (e) Luminex® cytokine 65‐analytes profiling of CCM and p1, p2 and p3 EV and Prot samples characterized in (b) and (d) (n = 1).
For all cell types, EV were present in EV‐enriched SEC fractions from p1 characterized by a density of ≈1.085–1.110 g/mL (Dhondt, Geeurickx, et al., 2020; Dhondt, Lumen, et al., 2020; Van Deun et al., 2014, 2020, Vergauwen et al., 2017, 2021) as shown by TEM that revealed abundant vesicular structures of 30–200 nm in size (Figures 1b and S1c). Western blot confirmed the enrichment of EV‐associated proteins Alix, TSG101 and CD9 in EV‐enriched SEC fractions from p1 compared to p1 protein‐enriched SEC fractions and p2 and p3‐derived samples for cancer cells (Figures 1c and S1a). Ago2‐containing ribonucleoprotein complexes were depleted in p1 EV‐enriched SEC fractions, in accordance with previous reported results (Van Deun et al., 2014) (Figures 1c and S1a). Western blot analysis of p1, p2 and p3 pellets obtained by 3 h ultracentrifugation at 100,000 g instead of SEC confirmed the enrichment of Alix, TSG101 and CD9 in p1, and the presence of Ago2‐containing ribonucleoprotein complexes in p2 and p3 pellets for CAF (Figure S1b). NTA confirmed particle enrichment in EV‐enriched SEC fractions from p1 compared to p1 protein‐enriched SEC fractions, p2, and p3‐derived samples (Figures 1d and S1d). In compliance with TEM analysis, higher particle concentrations were measured by NTA in p1 EV‐enriched SEC fractions from MCF‐7 cultures (1.36e+12 ± 1.67e+11 particles/mL) compared to MDA‐MB‐231 (6.74e+11 ± 1.21 e+11 particles/mL) and CAF (2.6e+11 ± 3.68 e+10 particles/mL, p = 0.029) (Figure S1f). For all cell lines, protein concentrations measured by the Qubit protein assay were the lowest in EV‐enriched SEC fractions from p1 (<0.2 μg/μL), while p2 and p3 protein‐enriched SEC fractions contained the highest protein concentration (>1.5μg/μL) (Figure 1d).
Luminex® cytokine profiling on a panel of 65 cytokines revealed that EV‐enriched SEC fractions from p1 were efficiently depleted from abundant soluble cytokines present in media conditioned by all cell lines, including VEGF‐A, GROα, IL‐6 and MCP‐1 known to inhibit MoDC maturation (Michielsen et al., 2011; Park et al., 2004; Takahashi et al., 2004; Wculek et al., 2019; Xu et al., 2022) (Figure 1e). In line with the Qubit protein assay, the lowest cytokine concentrations were measured in p1 EV‐enriched SEC fractions while cytokines eluted abundantly in protein‐enriched SEC fractions from p2 and p3 (Figure 1e). Concentrations of the most highly abundant cytokines in media cultured by cancer cells and CAF were all below 1 pg/1e+9 NTA‐measured particles in p1 EV‐enriched SEC fractions and above this threshold in p1 protein‐enriched SEC fractions, p2 and p3‐derived samples (Figure S1g).
Altogether, DG floatation enriched EV at their expected density in p1 and separated EV efficiently from other extracellular materials, including Ago2‐containing ribonucleoprotein complexes and soluble cytokines that remained in EV‐poor higher density pools p2 and p3. SEC processing of p1 further separated EV from soluble cytokines, generating EV‐enriched preparations depleted of cytokines, defined hereafter as DG‐SEC EV preparations. In addition, SEC processing of DG fractions clears iodixanol from the preparations as previously reported (Tulkens et al., 2020; Vergauwen et al., 2017) and confirmed by the detection of iodixanol in protein‐enriched but not EV‐enriched SEC fractions from p1, p2 and p3 (Figure S4a), thus excluding potential confounding effects of density medium on EV immunomodulatory bioactivity.
2.2. DG‐SEC EV from cancer cells stimulate MoDC maturation in a dose‐dependent manner
Maturation of MoDC upon uptake and loading of exogenous antigens involves a significant increased cell‐surface expression of antigen‐presenting major histocompatibility complex MHCII. Co‐stimulatory molecules required for T cell activation such as CD80 and CD86 (Cunningham & Hackstein, 2020) show also increased cell‐surface expression together with PD‐L1 which protect DC from activated T cells following antigen presentation (Oh et al., 2020). We investigated the impact of DG‐SEC EV from MCF‐7 and MDA‐MB‐231 breast cancer cells on MoDC maturation. We generated CD11c+/MHCII+ immature MoDC from healthy volunteers by IL‐4 and GM‐CSF‐induced differentiation of CD14+ blood monocytes (Cunningham & Hackstein, 2020; Tkach, 2017). MoDC were treated for 24 h with DG‐SEC EV from cancer cells, or the TLR4 ligand lipopolysaccharide (LPS) as positive control for MoDC maturation. NTA‐measured particle concentration of DG‐SEC EV preparations (Figure S1f) was used to determine EV treatment dose, and expression of MoDC maturation markers MHCII, CD80, CD86 and PD‐L1 was assessed by flow cytometry (Figure S2a).
First, we compared MHCII, CD80 and CD86 expression in MoDC treated with 4 μg of CCM or EV‐free p3 protein‐enriched SEC fractions (Figure 1), or with DG‐SEC EV at a low dose corresponding to 5,000 NTA‐measured particles per MoDC (part/MoDC) (Figure S2b,c). At this low concentration, DG‐SEC EV did not induce MoDC upregulation of MHCII, CD80 or CD86. In contrast, CCM or p3 protein‐enriched SEC fractions from the different cancer cell lines while not increasing MHCII expression at the tested concentration (40 μg/mL) stimulated CD80 and CD86 expression, underpinning the importance to quality control the specificity of EV preparation prior to functional analysis (Figure S2b,c). This effect was independent of iodixanol present in p3 protein‐enriched SEC fractions (Figure S4a) as treating MoDC with p3 protein‐enriched SEC fractions generated from PBS‐loaded control DG did not induce the expression of MoDC maturation markers (Figure S4b–f).
Next, we evaluated the dose‐response effect of DG‐SEC EV from cancer cells on MoDC maturation using increasing particle concentrations from 12,500 to 75,000 part/MoDC (Figure 2a). DG‐SEC EV from the different cancer cell lines induced MHCII expression in a dose‐dependent manner in MoDC from all tested donors (n = 4 donors per cell line). MHCII increase was statistically significant from 75,000 part/MoDC for MDA‐MB‐231 (p < 0.05) and MCF‐7 DG‐SEC EV (p < 0.05), and associated with an increase in the percentage of CD80, CD86 and PD‐L1 positive populations as well as an increase in CD80, CD86 and PD‐L1 expression (Figure 2a). The average percentage of MoDC expressing CD80 more than doubled following treatment with DG‐SEC EV at 75,000 part/MoDC (Figure 2a). While less than 30% of non‐treated MoDC expressed CD86, the CD86 positive population increased in a dose‐dependent manner to over respectively 65% and 80% following treatment with DG‐SEC EV from MDA‐MB‐231 and MCF‐7 (p < 0.05). While PD‐L1 is expressed by most MoDC, the PD‐L1 positive population and PD‐L1 expression significantly increased in response to DG‐SEC EV from the different cancer cell lines (Figure 2a). PD‐L1 mean fluorescence intensity more than doubled in MoDC treated with DG‐SEC EV at 75,000 part/MoDC from the different cancer cell lines compared to PBS‐treated cells (Figure 2a). As expected MoDC matured by LPS exhibited the strongest MHCII and T cell costimulatory CD80, CD86 and PD‐L1 expression (Figure 2a). In addition, DG‐SEC EV prepared from PBS‐loaded empty DG did not induce expression of MoDC maturation markers (Figure S4b–f).
FIGURE 2.
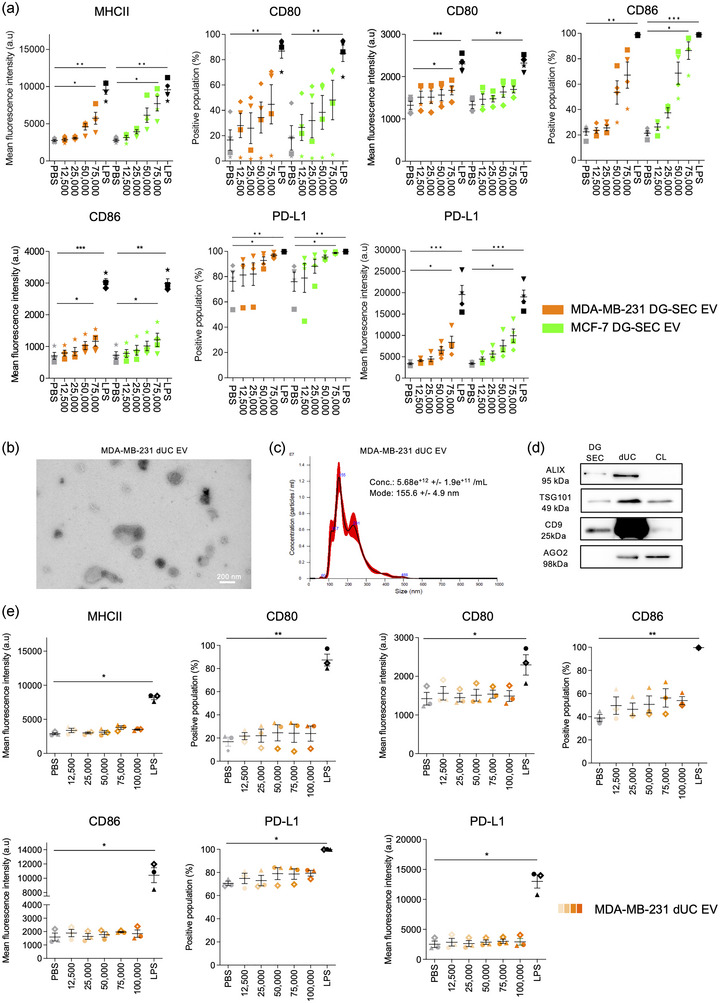
EV separated by DG‐SEC induce a dose‐dependent increase in MoDC maturation markers. a. Flow cytometry analysis after 24 h treatment of 200,000 immature MoDC from healthy volunteers with DG‐SEC EV from MDA‐MB‐231 or MCF‐7 (n = 4 MoDC donors per cell line). Treatment is indicated as corresponding number of NTA‐measured particles per MoDC. LPS (100 ng/mL) was used as a positive control to induce MoDC maturation. PBS was used as a negative control. Data represent mean and standard error. Statistical analysis: Friedman test with Dunn's multiple comparisons test (*p < 0.05, **p < 0.01, ***p < 0.001). (b) Transmission electron microscopy images from MDA‐MB‐231 dUC EV. Magnification x30,000, scale bar: 200 nm. (c) NTA size distribution profile of MDA‐MB‐231 dUC EV represented as mean (black line) and standard error (red shaded area). Conc.: particle concentration. Particle concentration and mode are expressed as mean and standard error (n = 3 videos). (d) Western blot analysis of MDA‐MB‐231 EV DG‐SEC and dUC EV preparations. Thirty microliters of EV preparations were denatured as described in the methods section and loaded in each lane. CL: cell lysate (15 μg). (e) Flow cytometry analysis after 24 h treatment of 200,000 immature MoDC from healthy volunteers with dUC EV from MDA‐MB‐231 (n = 3 MoDC donors). LPS (100 ng/mL) was used as a positive control to induce MoDC maturation. PBS was used as a negative control. Data represent mean and standard error. Statistical analysis: Friedman test with Dunn's multiple comparisons test (*p < 0.05, **p < 0.01, ***p < 0.001).
Our results are strongly indicative that cancer cell derived EV‐enriched but cytokine depleted DG‐SEC fractions stimulate MoDC maturation. To explore the impact of different EV preparation methods on MoDC maturation, we repeated these experiments with EV‐containing preparations obtained from MDA‐MB‐231 cultures by differential ultracentrifugation (dUC) pelleting at 100,000 g (Théry et al., 2006) (Figure 3a), and assessed MoDC MHCII, CD80, CD86 and PD‐L1 expression by flow cytometry (Figure 2b–e). Abundant vesicular structures of 30–300 nm in size were recovered in dUC pellets as shown by TEM (Figure 2b, S2d), NTA (Figure 2c) and western blot analysis for Alix, TSG101 and CD9 (Figure 2d). More EV were recovered from MDA‐MB‐231 cultures with dUC compared to DG‐SEC as shown by increased abundance of EV‐associated proteins by western blot analysis (Figure 2d), and vesicular structures by TEM (Figures 1b, S1c, 2b). However, western blot analysis identified a substantial amount of Ago2‐containing ribonucleoprotein complexes in dUC pellets but not in DG‐SEC EV preparations (Van Deun et al., 2014), confirming the presence of other extracellular materials and previously reported lower specificity of dUC EV preparations (Cocozza et al., 2020; Van Deun et al., 2014) (Figure 2d). Treatment of MoDC with dUC EV up to 100,000 part/MoDC did not significantly increase MHCII and CD80 expression compared to PBS‐treated MoDC while PD‐L1 expression and CD86 positive population only moderately increased in MoDC treated with 100,000 part/MoDC (Figure 2e). Interestingly, subsequent processing of dUC EV preparations by SEC dose‐dependently restored EV immunomodulatory bioactivity (Figure s3). Treatment with 12,500 to 75,000 part/MoDC dUC‐SEC EV (Figure S3a,b) but not dUC EV dose‐dependently increased MHCII and PD‐L1 expression, and the percentage of CD80 and CD86 positive populations reaching statistical significance at 75,000 part/MoDC (p < 0.05, Friedman test with Dunn's multiple comparisons test) (Figure S3c).
FIGURE 3.
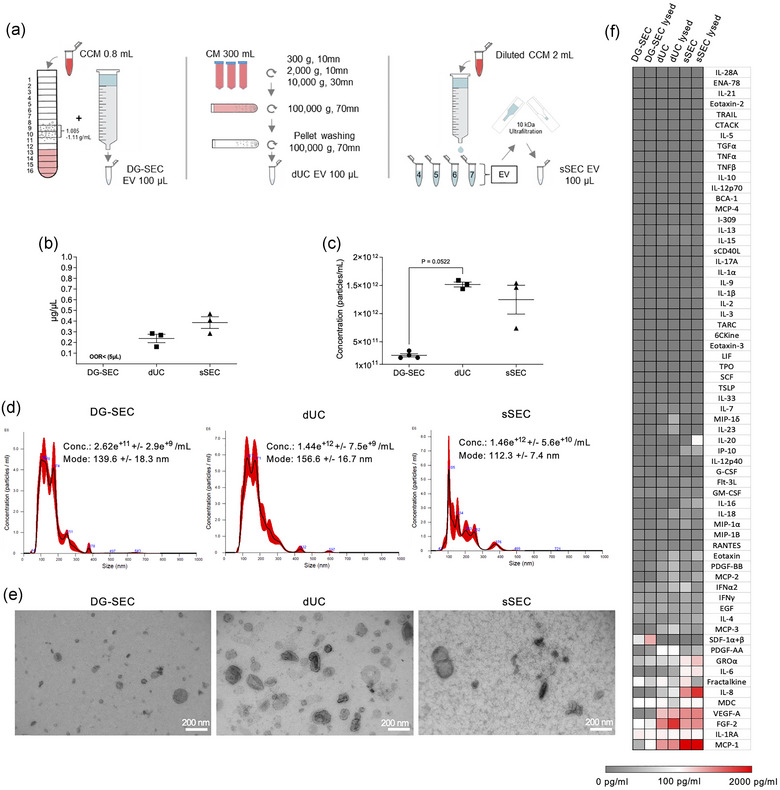
Impact of separation method on breast CAF EV particle and protein content. (a) Schematic view of CAF EV separation using different methods. (b) Protein concentration measured by Qubit using 5 μL of each preparation (DG‐SEC [n = 4], dUC [n = 3], sSEC [n = 3]). (c) Particle concentration measured by NTA in DG‐SEC (n = 4), dUC (n = 3), sSEC (n = 3). Three videos of 30 s were acquired for each replicate. (b–d) Data represent mean and standard error. Statistical analysis: Kruskal Wallis test with Dunn's multiple comparisons test. (d) Representative NTA size distribution profile of DG‐SEC, dUC and sSEC EV represented as mean (black line) and standard error (red shaded area). Conc.: particle concentration. Particle concentration and mode are expressed as mean and standard error. (e) Transmission EM of DG‐SEC, dUC, and sSEC CAF EV, magnification × 30,000, scale bar 200 nm. (f) Cytokine profiling in one representative DG‐SEC, dUC and sSEC EV preparation lysed or not with 0.2% Triton X‐100. Signal from PBS 0.2% Triton X100 control was subtracted.
To further confirm that cytokines present in EV preparations impair EV immunomodulatory bioactivity, we compared MoDC response to 75,000 part/MoDC DG‐SEC EV prepared from media conditioned by MDA‐MB‐231 cells, alone, or in combination with 4 μg of p1 protein‐enriched SEC fraction or VEGF‐A (1 ng/mL or 10 ng/mL) known to inhibit MoDC maturation (Takahashi et al., 2004) and separated from EV by DG‐SEC (Figure S5). While MoDC treated with DG‐SEC EV increased the percentage of CD80, CD86 and PDL1 positive populations, MoDC treatment with DG‐SEC EV supplemented with 4 μg of p1 protein‐enriched SEC fractions or VEGF impaired MoDC maturation (Figure S5).
Our results support that cancer cell‐derived EV preparations specifically separated by DG‐SEC from other extracellular materials induce a dose‐dependent maturation of MoDC (Figure 2a). This bioactivity may however depend on the specificity of the EV preparation method as MDA‐MB‐231 dUC EV showed a reduced potency to stimulate expression of MoDC maturation markers compared to DG‐SEC EV (Figure 2b–e). Notably, SEC processing of dUC EV restored EV immunomodulatory bioactivity with significantly upregulated MoDC maturation markers at 75,000 part/MoDC (Figure S3c). Addition of VEGF‐A or p1 protein‐enriched SEC fractions impaired EV‐induced surface remodelling of CD80/86 costimulatory molecules during MoDC maturation further underpinning the need to deplete EV preparations from cytokines and soluble proteins to enable the study of EV immunomodulatory bioactivity (Figure S5).
2.3. EV separation specificity determines EV bioactivity in MoDC maturation
To further study the impact of EV separation on MoDC maturation, we used EV preparations obtained by DG‐SEC, dUC (Théry et al., 2006), or sSEC from medium conditioned by CAF (Figure 3a). We selected CAF to study the impact of EV separation on MoDC maturation from technical perspectives: (1) CCM evaluation revealed a high protein concentration (Figure 1d) and high abundance of immunosuppressive cytokines such as VEGF‐A, MCP‐1 and GRO‐α (Figure 1e) or Wnt2 (Huang et al., 2021) potentially interfering with EV immunomodulatory bioactivity; and (2) in contrast to cancer cell lines, CAF show contact inhibition of growth, allowing sequential harvesting of confluent monolayers of a same cell batch limiting pre‐analytical variables.
Due to the relative higher protein concentration of CAF CCM (Figure 1d), abundant proteins started to elute already in EV‐enriched fraction n°7 by sSEC, generating EV preparations with a defined protein contamination (Figure s6a). Accordingly, protein concentration measurements identified the highest concentrations in EV‐containing sSEC preparations in contrast to EV‐enriched DG‐SEC preparations which were below the detection limit of the instrument (Figure 3b). Western blot revealed that EV‐associated proteins TSG101 and CD9 were detected in dUC and sSEC EV‐containing preparations while Ago2 was not substantially depleted in dUC EV preparations (Figure S6b). NTA measurements identified that particle concentrations obtained by dUC (1.52 e+12 ± 4.3e+9 part/mL, n = 3) and sSEC (1.25e+12 ± 2.5e+11 part/mL, n = 3) were respectively over 5, and 4‐fold higher to DG‐SEC (2.74e+11 ± 3.6e+9 part/mL, n = 4) (Figure 3c,d). TEM identified 30–200 nm sized vesicular structures in the three preparations, but also proteinaceous background in sSEC preparation (Figure 3e, S6c). Thus, despite lower yield, DG‐SEC recovered EV with higher specificity compared to dUC and sSEC as previously reported (Van Deun et al., 2014).
Cytokine profiling of DG‐SEC, dUC or sSEC EV preparations reflected the results from the protein concentration measurements, with the lowest concentrations for GROα, IL‐6, IL‐8, VEGF‐A, FGF‐2 and MCP‐1 identified in DG‐SEC EV compared to dUC and sSEC EV (Figure 3f). Cytokine concentrations reported as pg per 1e+9 particles confirmed that DG‐SEC improved EV separation from cytokines including VEGF‐A, FGF2 and MCP‐1 compared to dUC while GROα, IL‐6 and IL‐8 relative amounts were similarly low in DG‐SEC and dUC (Figure 3f). In accordance with protein concentration measurements, cytokines were increased in sSEC preparations with a relative concentration regularly over 1 pg/1e+9 particles (Figure S6d). EV lysis with 0.2% Triton X‐100 initiated a consistent increase in the detection of most cytokines except fractalkine (CX3CL1) that decreased in all lysed preparations in line with its transmembrane nature (Figures 3f and S6d). Overall, cytokines such as MCP‐1 and VEGF‐A, known to inhibit MoDC maturation (Alfaro et al., 2009; Omata et al., 2002; Oyama et al., 1998), were mainly identified in dUC and sSEC EV preparations. In addition, GROα, IL‐6, IL‐8 were only identified in sSEC EV preparations in line with its higher protein concentration.
Pooled EV preparations from multiple harvestings of a same cell batch (Figure 3a) were used to perform NTA particle‐based dose response‐treatment (12,500–75,000 part/MoDC) of MoDC from healthy volunteers. Maturation was monitored by flow cytometry measuring MHCII, CD80, CD86 and PD‐L1 expression and percentages of positive populations (Figure 4), and by Luminex‐based MoDC cytokine measurements (Figure 5). From 25,000 part/MoDC, DG‐SEC EV significantly increased expression of all MoDC membrane markers and the percentage of positive populations compared to PBS‐treated cells (Figure 4). While DG‐SEC EV at 25,000 to 75,000 part/MoDC significantly increased MHCII expression compared to PBS‐treated MoDC, dUC and sSEC EV induced a moderate but not significant MHCII upregulation at highest particle concentrations (75,000 part/MoDC) (Figure 4a). MHCII expression following treatment with DG‐SEC EV at 50,000 part/MoDC doubled compared to PBS‐treated MoDC and was higher than after dUC (p < 0.05, Friedman test with Dunn's multiple comparisons test) and sSEC EV treatment at 75,000 part/MoDC. The percentage of CD80 positive cells increased significantly following DG‐SEC EV treatment reaching a maximum of 71.9 ± 10.1% (p < 0.01) with 50,000 part/MoDC compared to PBS‐treated MoDC (45.3 ± 9.1%) (Figure 4b,c). CD86 positive population significantly increased after treatment with DG‐SEC EV over 25,000 part/MoDC compared to PBS‐treated MoDC (34 ± 7.5%), reaching a maximum of 85.9 ± 3.4% (p < 0.001) with 50,000 part/MoDC. In addition to DG‐SEC EV and LPS positive control, only sSEC EV preparations resulted in a significant increase of CD86 positive population from 50,000 part/MoDC, with a maximum of (61.4 ± 8%) at 75,000 part/MoDC (p < 0.05) while dUC EV induced a maximum of 49.4 ± 6% (p = 0.4991) at the highest particle concentration (Figure 4d,e). PD‐L1 expression increased significantly in response to DG‐SEC EV preparations from 25,000 to 75,000 part/MoDC and sSEC EV preparations at 75,000 part/MoDC compared to PBS‐treated MoDC. In line with MHCII, CD86 and CD80, dUC EV preparations did not induce a significant upregulation of PD‐L1 expression compared to PBS‐treated MoDC (Figure 4f,g). Interestingly DG‐SEC EV‐induced maturation markers expression was higher at a concentration of 50,000 than 75,000 part/MoDC, suggesting MoDC response regulation to prevent overactivation (Figure 4). These results indicate that EV separated by DG‐SEC from other extracellular materials potentially stimulate MoDC antigen presentation and confirm T cell co‐stimulator expression in a dose dependent manner. While sSEC EV preparations were unable to stimulate MoDC antigen presentation they could increase MoDC CD80, CD86 and PD‐L1 markers only at highest particle amounts and at the lowest significance levels. In contrast, dUC EV were unable to induce a significant increase of MoDC markers at all tested concentrations, highlighting that despite the presence of EV in the dUC/sSEC preparations, they have a reduced potential in stimulating MoDC maturation.
FIGURE 4.
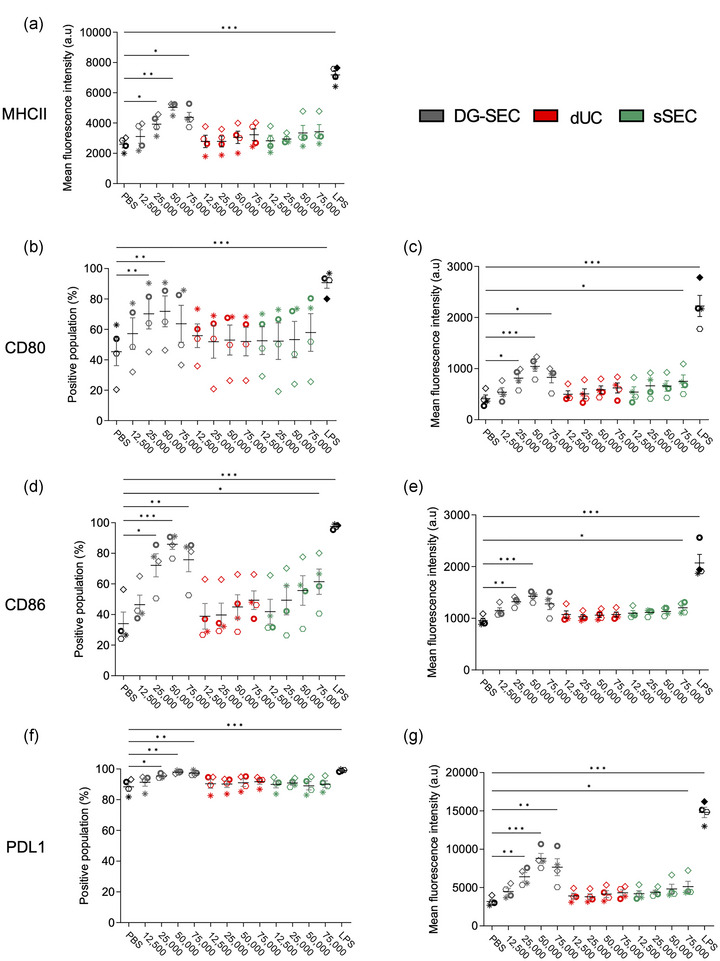
Impact of separation method on CAF EV potential to stimulate MoDC maturation markers expression. (a–g) Flow cytometry analysis after 24 h treatment of 200,000 immature MoDC from healthy volunteers with DG‐SEC, dUC or sSEC EV breast CAF (n = 4 MoDC donors). Treatment is indicated as corresponding number of NTA‐measured particles per MoDC. LPS (100 ng/mL) was used as a positive control to induce MoDC maturation. PBS was used as a negative control. Data represent mean and standard error. Statistical analysis: Friedman test with Dunn's multiple comparisons test (*p < 0.05, **p < 0.01, ***p < 0.001).
FIGURE 5.
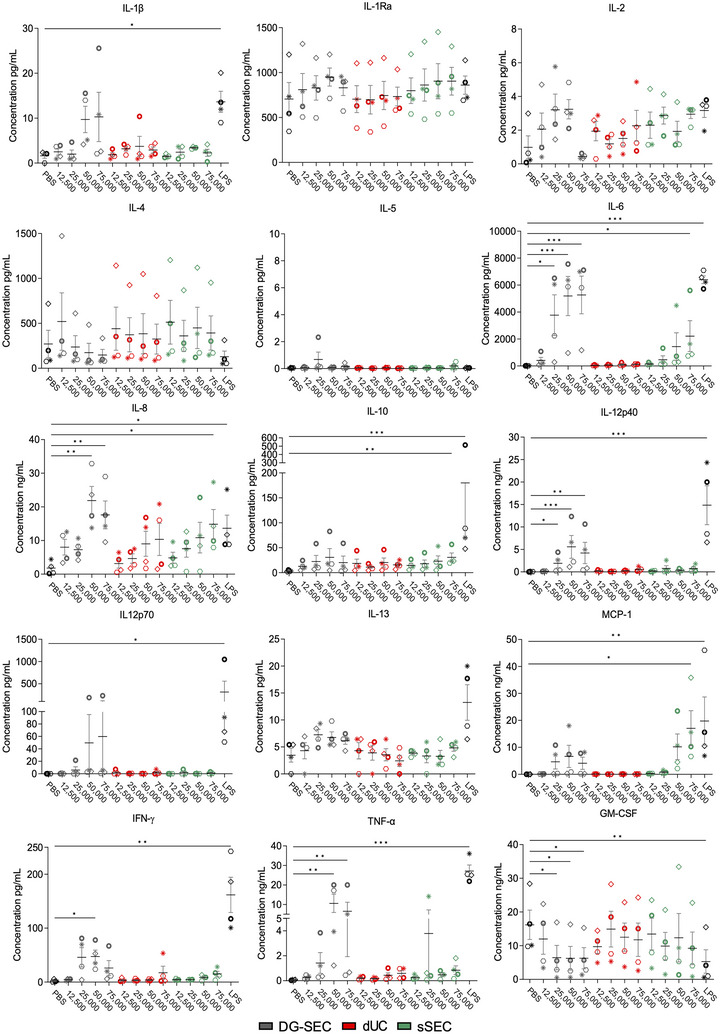
Impact of separation method on CAF EV‐induced cytokine release by MoDC. Luminex 15‐plex cytokine profiling in MoDC culture medium following treatment with breast CAF DG‐SEC, dUC and sSEC EV preparations (Figure 4). LPS (100 ng/mL) was used as a positive control to induce MoDC maturation. PBS was used as a negative control. Data represent mean and standard error. Statistical analysis: Friedman test with Dunn's multiple comparisons test (*p < 0.05, **p < 0.01, ***p < 0.001).
T cell activation requires antigen presentation and presence of co‐stimulatory molecules but also the release by DC of stimulatory cytokines, such as type I interferons, and IL‐12 secreted as a functional 75 kDa heterodimer (p70) composed of p35 and p40 subunits (Curtsinger & Mescher, 2010; Tugues et al., 2014; Wculek et al., 2019). We used Luminex® multi‐analyte profiling to measure cytokine released by MoDC in the culture medium in response to DG‐SEC, dUC and sSEC EV preparations (Figure 5). In line with increased surface expression of MHCII, CD80 and CD86, MoDC treated with DG‐SEC EV exhibited a secretory phenotype of mature MoDC with increased IL‐1β, IL‐6, IL‐8, IL‐12 (p40 and p70), MCP‐1, TNFα, IFN‐γ and a reduced release of IL‐4 and GM‐CSF compared to PBS‐treated MoDC (Figure 5). This effect is in agreement with MoDC maturation induced by LPS. In response to DG‐SEC EV, MoDC secreted high amounts of IL‐6 (5.2 ± 1.5 ng/mL), IL‐12p40 (5.6 ± 2.6 ng/mL), TNF‐a (10.6 ± 4.7 ng/mL), IL‐8 (21.9 ± 4.2 ng/mL) and MCP‐1 (6.6 ± 4.1 ng/mL). With the exception of IL‐8, a key chemoattractant of neutrophils, myeloid‐derived suppressor cells, and blood monocytes (Ha et al., 2017), dUC EV did not strongly induce MoDC cytokine release at tested particle concentrations, inducing a maximum of 0.12 ± 0.02 ng/mL IL‐6, 0.54 ± 0.25 ng/mL IL‐12p40, 0.59 ± 0.22 ng/mL TNF‐a, 10.3 ± 4.8 ng/mL IL‐8 and 0.13 ± 0.02 ng/mL MCP‐1 when treated with 75,000 part/MoDC. sSEC preparations of 75,000 part/MoDC were only capable to induce higher IL‐6 (2.2 ± 1.2 ng/mL but not significantly) and IL‐8 (14.9 ± 4.4 ng/mL, p < 0.05). Only sSEC EV were able to induce statistically significant MCP‐1 (17.1 ± 6.5 ng/mL, p < 0.05) release compared to PBS‐treated MoDC again strongly indicating the difference in MoDC activation potential compared to the other EV preparations. IL‐6 released by activated DC is an essential contributor to effector T cell proliferation and activation, inhibiting regulatory T cell differentiation and function, and participating in the differentiation of T helper 17 and T helper 2 cells (Xu et al., 2022). IFN‐γ is the main mediator of IL‐12 potent antitumor immunity functions and its signalling promotes DC maturation and IL‐12 secretion in a positive feedback loop (Subbiah et al., 2018; Trinchieri, 2003; Tugues et al., 2014). Accordingly, MoDC released more IFNγ in response to DG‐SEC EV at 50,000 part/MoDC (47.5 ± 11.8 pg/mL, p < 0.05) than dUC EV (17.3 ± 12.2 pg/mL) and sSEC EV at 75,000 part/MoDC (15 ± 5.2 pg/mL).
In conclusion, cytokine profiling confirmed that EV separated with high specificity from other extracellular materials by DG‐SEC stimulate MoDC maturation in a dose‐dependent manner, with after 24 h a maximal response at a concentration of 50,000 part/MoDC that was reduced at 75,000 part/MoDC, suggesting a mechanism of autocrine loop regulating MoDC response to excessive EV concentrations. This could be mediated by regulators as IL‐10 or IL‐6 at high concentrations. IL‐6 was notably released at similar concentration in MoDC medium following treatment with 50,000 and 75,000 part/MoDC (Figure 5) (Wculek et al., 2019). MoDC maturation response was not recapitulated by EV‐containing preparations obtained by dUC or sSEC at tested particle concentrations, demonstrating that EV separation specificity is essential to identify and characterize EV bioactivity.
3. DISCUSSION
EV separation from other extracellular materials is a challenge achieved with differential specificity by currently available methodologies. Assessment of the impact of EV preparation methods is however critical to our understanding of EV bioactivity in physiology and disease, and to the development of EV‐based therapeutic applications and their manufacturing (De Wever & Hendrix, 2019; Van Deun et al., 2014; Whittaker et al., 2020). In addition, appropriate controls, including assessment of the bioactivity of EV‐depleted samples and dose response experiments, are other methodological aspects that require careful consideration.
DC are the most sensitive sensors of the innate immune system, detecting molecular changes in the extracellular microenvironment. EV are a potential source of maturation signals and tumour antigens with the capacity to improve the efficacy of DC‐based cancer immunotherapy (Perez & De Palma, 2019; Wculek et al., 2019). Indeed, cancer cell EV engineered to express chimeric receptors notably improve EV uptake and tumour antigen presentation by DC (Squadrito et al., 2018). Considering this therapeutic potential, we implemented MoDC as a model system in this study to evaluate their ability to respond to EV obtained from different cancer cell and CAF cultures using diverse methods that prepare EV with variable specificity.
The sequential combination of density‐based and size‐based preparation methods can be used to process EV from the most complex biofluids (Dhondt, Geeurickx, et al., 2020; Dhondt, Lumen, et al., 2020; Tulkens et al., 2020, 2018; Simonsen, 2017; Vergauwen et al., 2021) and to study EV bioactivity in pre‐clinical research. Our results show that EV separated with high specificity by DG‐SEC from other extracellular materials, including soluble cytokines known to inhibit DC maturation, induce, without any additional stimulatory cytokine cocktail, a dose‐dependent increase of MoDC maturation markers such as MHCII CD80, CD86 and PD‐L1. Given that EV uptake is a low yield process (∼1% spontaneous rate at 1 h (Bonsergent et al., 2021)) it is not surprising to observe a dose‐dependency of DG‐SEC EV induced effects. At a given number of EV corresponding in our set‐up to final concentration a maximum bioactivity is observed most probably due to saturation of binding, uptake, and cytosolic release mechanisms. Interestingly, while these effects are observed for EV‐enriched preparations obtained by DG‐SEC they are not observed with EV‐containing preparations obtained by dUC or sSEC from the same batches of CCM and using the same batches of MoDC. Cytokines such as MCP‐1, GROα and growth factors such as VEGF‐A are known to inhibit DC maturation but are not efficiently depleted from EV preparations obtained by dUC or sSEC. Subsequent processing of dUC EV preparations by SEC dose‐dependently restored the immunomodulatory bioactivity, while addition of VEGF‐A impaired DG‐SEC EV induced EV immunomodulatory bioactivity.
Our experimental set‐up demonstrates that co‐separated extracellular materials, including soluble cytokines, obscure EV bioactivity which is only revealed using highly specific separation. Other groups have shown that bioactivity is attributed to co‐separated soluble cytokines or growth factors rather than EV (Whittaker et al., 2020; Wolf et al., 2022). Indeed, the field is still reminiscent to claimed pro‐angiogenic functions misattributed to MSC‐derived EV with detailed investigations concluding that VEGF co‐separated with EV was responsible for the observed regenerative effects (Whittaker et al., 2020). Recent breakthrough research (Tóth et al., 2021; Wolf et al., 2022) postulates the intriguing possibility that soluble cytokines or growth factors can be part of a biomolecular corona on the surface of EV which may be affected by particular EV preparation methods, especially when separating EV with higher specificity. Two independent research groups reported that EV bioactivity was, at least partly, attributed to the biomolecular corona (Tóth et al., 2021; Wolf et al., 2022). Intriguingly, although increasing specificity by combining tangential flow filtration with dUC or SEC has been shown to abrogate EV bioactivity, presumably by affecting the biomolecular corona (Wolf et al., 2022), in our experimental EV bioactivity was only obtained after highly specific separation with DG‐SEC or dUC‐SEC. Although the contribution of the biomolecular corona in our experimental set‐up remains to be defined, our study highlights the importance of higher specificity methods, especially in relation to EV‐based therapeutic manufacturing and application. Indeed, recent clinical evidence demonstrated that ex vivo secretion of IL‐6, IL‐8, Il‐12p40 and TNF‐α from cancer patient‐derived MoDC, partially matured ex vivo with IFNγ and inactivated Bacillus Calmette‐Guerin, positively correlated with survival or stable disease following MoDC intratumor injection (Subbiah et al., 2018). This ex vivo secretion of IL‐6, IL‐8, Il‐12p40 and TNF‐α was only achieved upon incubation of MoDC with DG‐SEC EV, but not dUC or sSEC EV in our experimental set‐up.
Although demonstrated here with in vitro cell culture‐derived EV and allogenic healthy volunteer‐derived MoDC, our results strongly indicate that specificity of preparation methodology is also critical to uncover therapeutic applications using autologous MoDC and tumour EV abundantly present in more complex biofluids such as blood plasma (Vergauwen et al., 2021) or supernatants from ex vivo patient‐derived tissue fragments. Uncovering the full therapeutic potential of EV immunomodulatory bioactivity therefore requires implementing community guidelines to ascertain the EV‐associated nature of results, through appropriate controls, transparent reporting, and cross validation with higher specificity methods (De Wever & Hendrix, 2019; Théry et al., 2018; Van Deun et al., 2017; Whittaker et al., 2020). It must be emphasized that the methodological threats identified in this study are most probably also relevant to evaluation of EV bioactivity in general.
4. MATERIALS AND METHODS
4.1. Cell culture
Human breast cancer cells oestrogen receptor‐positive MCF‐7 (ATCC, Manassas, VA), and triple‐negative MDA‐MB‐231 (ATCC, Manassas, VA) were cultured in Dulbecco's Modified Eagle's Medium high glucose (DMEM) supplemented with 10% heat‐inactivated foetal bovine serum (FBS) at 37°C, 10% CO2. Human breast cancer associated fibroblasts (CAF) were isolated from a breast tumour resection immortalized by transduction of a human telomerase reverse transcriptase construct and cultured as previously described (De Vlieghere et al., 2015; De Wever et al., 2004). All Cell cultures were regularly tested and confirmed negative for mycoplasma contamination using the MycoAlert Mycoplasma Detection Kit (Lonza, Verviers, Belgium).
4.2. Antibodies and reagents
Primary antibodies used for western blotting included anti‐human Alix (1:1000, mouse monoclonal, Cell Signalling, #2171S), anti‐human TSG101 (C‐2) (1:1000, mouse monoclonal, Santa Cruz Biotechnology, #sc‐7964), anti‐human CD9 (1:1000, rabbit monoclonal, Cell Signalling, #13403), anti‐human Ago2 (1:1000, rabbit polyclonal, Abcam, #ab32381). Secondary antibodies used for western blot included: anti‐mouse horseradish peroxidase‐linked antibody (1:3000, sheep, GE Healthcare Life Sciences, #NA931V), and anti‐rabbit horseradish peroxidase‐linked antibody (1:4000, donkey, GE Healthcare Life Sciences, #NA934V). Antibodies used for flow cytometry experiments included PE/Cyanine 7 anti‐human CD11c (1:200, BioLegend, #337216), PE anti‐human CD80 (1:100, BioLegend, #305208), PerCP/Cyanine5.5 anti‐human CD86 (1:200, BioLegend, #305420), Brilliant Violet 421™ anti‐human CD274 (B7‐H1, PD‐L1) (1:50, BioLegend, #329714), FITC anti‐human HLA‐DR (MHCII) clone: REA805 (1:200, Miltenyi, #130‐111‐788). In all experiments particle‐free sterile commercial Phosphate‐buffered saline (PBS) pH 7.2 was used (Thermo Fisher Scientific, Gibco™ #20012027).
4.3. Preparation of conditioned medium
MCF‐7, MDA‐MB‐231 and CAF were expanded to 20 T175 culture flasks in 875 cm2 multilayer culture flasks (Corning, #353144) for each cell line as previously described (Geeurickx et al., 2021). An additional T175 was plated for each cell line and trypsinized at the moment of harvesting to determine cell number and viability using a Countess Automatic Cell Counter and 0.1% trypan blue exclusion test (Thermo Fisher Scientific, Erembodegem, Belgium). Cell viability was always greater than 95%. When 70% confluence was reached for MCF‐7 and MDA‐MB‐231 (approximately 2e+8 cells), or 100% confluence for CAF that have contact inhibition of growth (approximately 2.5e+8 cells), cells were washed three times with pre‐warmed serum‐free DMEM using first 10 mL per T175 for 1 min, replaced by 15 mL per T175 for 10 min, and lastly 15 mL for 30 min at 37°C. After the last washing, cells were incubated with 15 mL of pre‐warmed EV‐harvesting medium per T175 and kept at 37°C, 10% CO2 for 24 h (Geeurickx et al., 2021; Van Deun et al., 2014, 2020). EV‐harvesting medium consisted for CAF of DMEM and Ham's F‐12 nutrient mixture (1:1), 100 U/mL penicillin, and 100 U/mL streptomycin, and for MCF‐7 and MDA‐MB‐231 of DMEM supplemented with 0.5% EV‐depleted serum (EDS), 100 U/mL penicillin, and 100 U/mL streptomycin. EDS was obtained through 18 h centrifugation of FBS at 100,000 g, 4°C (SW 32.1Ti rotor) and subsequent 0.2 μm filtration of the EV‐depleted supernatant (Van Deun et al., 2020).
4.4. Conditioned medium collection and concentration
After 24 h, 300 mL conditioned medium (CM) was collected for each cell line in 50 mL tubes and centrifuged for 10 min at 300 g, 4°C, followed by 0.45 μm filtration through a cellulose acetate bottle top filter (Corning, Amsterdam, The Netherlands). Filtered CM was concentrated to 800 μL using Centricon® Plus‐70 centrifugal filter units with a 10 kDa nominal molecular weight limit at 3200 g, 4°C (Merck Millipore, Burlington, MA). Concentrated conditioned medium (CCM) was recovered after inversion of centrifugal filter units and 10 min centrifugation at 1000 g, 4°C, and stored at −80°C until EV separation. Cancer cells were discarded after one CM collection. For CAF, 15 mL of culture medium (DMEM F‐12 (1:1), 10% FBS, 100 U/mL penicillin, 100 U/mL streptomycin) was added per T175 after collection and CAF confluent monolayers were kept at 37°C, 5% CO2 at least 24 h before a next washing and collection.
4.5. EV separation by bottom‐up density gradient and size‐exclusion chromatography (DG‐SEC)
Total 5%, 10%, and 20% OptiprepTM iodixanol gradient solutions were prepared by mixing appropriate amounts of a 50% iodixanol working solution (WS) with homogenisation medium (0.25 M sucrose,1mM EDTA, 10 mM Tris‐HCL, pH 7.4) as previously described (Dhondt, Geeurickx, et al., 2020; Dhondt, Lumen, et al., 2020; Geeurickx et al., 2021; Van Deun et al., 2020). The 50% iodixanol WS was prepared by mixing OptiPrepTM (60% (w/v) aqueous iodixanol solution, Axis‐Shield, Oslo, Norway) with appropriate amounts of buffer (0.25 M sucrose, 6 mM EDTA, 60 mM Tris‐HCl, pH 7.4). The sample‐containing 40% solution was prepared by mixing 800 μL of CCM with 3.2 mL of WS. The gradient was built in a sterile 16.8 mL polyallomer open top centrifuge tube using the Biomek 4000 automated workstation (Beckman Coulter) as previously described (Tulkens et al., 2020). A total of 4 mL of the 40% sample‐containing solution was first gently added to the bottom of the tube, followed by 4 mL of 20% solution, 4 mL 10% solution, 3.5 mL 5% solution and 1 mL of sterile commercial PBS as previously detailed (Dhondt, Geeurickx, et al., 2020; Dhondt, Lumen, et al., 2020). Gradients were centrifuged 18 h at 100,000 g, 4°C (SW 32.1 Ti rotor, Beckman Coulter). After the centrifugation, 16 fractions of 1 mL were collected in sterile 1.5 mL Eppendorf tubes using the Biomek 4000 workstation and kept at 4°C. Density of collected fractions was determined by measuring 340 nm absorbance of individual fractions collected from of blank PBS‐loaded bottom‐up density gradient as previously described (Dhondt, Geeurickx, et al., 2020).
Fractions 1–8 of the DG were discarded, while the other fractions were pooled in 5 mL Eppendorf tubes as follows: 9–10 (2 mL), 11–13 (3 mL), 14–16 (3 mL). Two millilitres of each pool was loaded on top of a SEC column. Columns were prepared as previously described (Dhondt, Geeurickx, et al., 2020) at least 1 day before performing SEC. Briefly, for each SEC column ±20 mL of Sepharose CL‐2B (GE Healthcare, Machelen, Belgium) was washed three times with commercial PBS. A nylon net with 20 μm pore size (Merck Millipore, NY2002500) was placed on the bottom of a 10 mL syringe (Romed, 3SYR‐10ML), followed by stacking of 10 mL washed Sepharose CL‐2B. EV‐rich 1 mL SEC fractions (4–7) (Dhondt, Geeurickx, et al., 2020; Dhondt, Lumen, et al., 2020; Van Deun et al., 2020; Vergauwen et al., 2021) and protein‐rich SEC fractions (8–11) were collected and concentrated to 100 μL with Amicon® ultra 2 mL centrifugal filters (10 kDa NMWL). Concentrated DG‐SEC samples were stored at −80°C.
4.6. Stand‐alone size exclusion chromatography (sSEC)
CAF CCM was prepared as described above and diluted with sterile commercial PBS to obtain a 2 mL sample. The sample was loaded on top of a SEC column and EV‐rich fractions (4–7) were pooled and concentrated to 100 μL as previously described.
4.7. EV separation by differential ultracentrifugation pelleting (dUC)
EV recovery by dUC from MDA‐MB‐231 or CAF CM (300 mL) was carried out according to the protocol from Théry et al. (2006). Briefly, CM was collected in 50 mL sterile conical polypropylene centrifuge tubes and centrifuged at 300 g, 4°C for 10 min, after which the supernatant was transferred to 6 new 50 mL centrifuge tubes and centrifuged at 2000 g, 4°C for 10 min. Next, the supernatant was transferred to 38.5 mL open‐top thin wall polypropylene tubes and centrifuged at 4°C, 10,000 g for 30 min (SW 32.1 Ti rotor, Beckman Coulter). Again, the supernatant was collected and transferred to new 38.5 mL open‐top thin wall polypropylene tubes and centrifuged in the same ultracentrifuge at 4°C and 100,000 g (SW 32.1 Ti rotor, Beckman Coulter) for 70 min. As much medium as possible was removed above the pellets that were resuspended in 1 mL of PBS, pooled in a single 17 mL open‐top thin wall ultra‐clear tube, and centrifuged again at 4°C, 100,000 g for 70 min (SW 32.1 Ti, Beckman Coulter). The final EV‐enriched pellet was resuspended in 100 μL PBS and stored at −80°C.
4.8. Nanoparticle tracking analysis (NTA)
NTA was performed with NanoSight LM10‐HS equipped with an automated syringe pump, a temperature sensor and 488 nm laser (NanoSight Ltd.), and software 3.0. Samples were diluted in commercial PBS to a particle concentration within the linear range of the instrument: 2e+8 – 1 e+9 particles/mL and a final volume of 1 mL which was taken up with a 1 mL syringe and introduced in the chamber. For every sample, the pump flow was set at 20 and 3 videos of 30 s were recorded with screen gain set at 1.0 and camera level at 13. For processing the videos, the screen gain was set at 1.0 and the detection threshold at 3.0.
Transmission electron microscopy: Samples were deposited on a formvar coated grid, stabilised with evaporated carbon film and glow discharged before sample application. Sample staining with neutral uranyl acetate (2% in aqua distilled (AD)) was done after which grids were coated with 2% methyl cellulose/uranyl acetate (0.4%) solution. These grids were analysed using a Tecnai G2 Spirit transmission electron microscope (FEI) operated at 100 kV and images were acquired with a Quemesa charge‐coupled device camera (Olympus Soft Imaging Solutions GMBH).
4.9. Protein analysis
Protein concentration of cell lysates, DG‐SEC, and EV samples was measured using the Qubit Protein assay kit (Thermo Fisher Scientific) and Qubit fluorometer 3.0 following manufacturer's instructions. Cell lysates were obtained in Laemmli lysis buffer (0.125 M Tris–HCl [pH 6.8], 10% glycerol, 2.3% SDS) and proteins were lysed in reducing sample buffer (0.5 M Tris‐HCl (pH 6.8), 43% glycerol, 9.2% SDS, 5% 2‐mercaptoethanol, 5% bromophenol blue) and boiled for 5 min at 95°C. DG‐SEC sample and EV preparation lysates were obtained by mixing 30μL of sample with 5 μL of reducing sample buffer (0.5 M Tris‐HCl (pH 6.8), 43% glycerol, 9.2% SDS, 5% 2‐mercaptoethanol, 5% bromophenol blue) and boiled for 5 min at 95°C. Proteins were separated by SDS–PAGE (SDS polyacrylamide gel electrophoresis), transferred to nitrocellulose membranes, blocked in 5% non‐fat milk in PBS 0.5% Tween‐20, and incubated overnight at 4°C with primary antibodies described in the reagents section. Secondary antibodies were added for 60 min at room temperature after extensive washing with blocking buffer. After final washing, Blots were developed using the WesternBright Sirius reagent (Advansta, Menlo Park, CA) and visualized on a Proxima 2850 Imager (IsoGen Life Sciences) and images were analysed using ProXima AQ‐4 software.
4.10. Cytokine assays
Cytokine assays were conducted at Eve Technologies (Calgary, Canada) using the Luminex® xMAP® technology. Cytokine profiling in DG‐SEC samples and EV preparations were assessed by the human cytokine array/chemokine array 65‐Plex Panel (HD65). Twenty‐five microliters of each sample was diluted to 100 μL with commercial PBS and stored at −80°C until analysis. Cytokines released by MoDC in the culture medium were assessed by the human Cytokine Array Proinflammatory Focused 15‐plex (HDF15). Following MoDC treatment, cells were pelleted by centrifugation 2 min at 2000 rpm, 4°C and 75 μL of MoDC culture medium was collected per well and stored at −80°C until analysis.
4.11. Human monocyte‐derived dendritic (MoDC) cell generation
Blood samples were obtained from 22‐ to 35‐year‐old voluntary male (n = 5) and female (n = 3) healthy donors that gave written informed consent. Fifty millilitres of blood was collected from each donor in 9 mL Vacuette® citrate tubes (Greiner Bio One, Kremsmünster, Austria) and diluted to 90 mL with commercial sterile PBS. Thirty millilitres of diluted blood was layered on 16 mL of Ficoll‐Paque™ PLUS density gradient media (Cytiva, Marlborough) in 50 mL sterile centrifugation tubes and centrifuged 30 min at 400 g at room temperature without brake. The ring of peripheral blood mononuclear cells (PBMCs) was collected in a new tube, diluted to 50 mL with RPMI medium and centrifuged 10 min at 400 g at room temperature. The PBMCs pellet was washed two more times with RMPI and centrifuged for 10 min at 400 g and room temperature. The final PBMCs pellet was resuspended in PBS, 2% ultra‐low endotoxin FBS (Biosera, Nuaillé, France), 1mM EDTA at a concentration of 1e+8 cells/mL and CD14+ cells were separated using the EasySepTM Human CD14 Positive Selection Kit II (STEMCELL Technologies, Saint‐Egrève, France, #17858) with the EasySepTM Magnet (STEMCELL Technologies, #18000) following manufacturer instructions. Separated CD14+ cells were cultured at 2 million cells per ml for 5 days in DC medium (RPMI 1640, 10 mM Hepes, 100 U/mL penicillin,100 U/mL streptomycin (Fisher Scientific, Merelbeke, Belgium), 10% ultra‐low endotoxin FBS) supplemented with IL‐4 and GM‐CSF (50 and 100 ng/mL, respectively; Miltenyi Biotec, Bergisch Gladbach, Germany) to generate immature MoDC as previously described (Tkach, 2017).
4.12. MoDC treatment, staining and fixation
For each donor, MoDC were centrifuged 10 min at 400 g at room temperature, resuspended in fresh DC medium, counted, and viability controlled by 0.1% trypan blue stain exclusion (Life Technologies, Merelbeke, Belgium) was greater than 95%. Cells were resuspended at 4 million cells per mL in DC medium and 50 μL of cell suspension (200,000 cells) was placed per well in 96‐well plates and kept at 37°C, 5% CO2. For treatment, particle concentration in EV samples was first measured by NTA and particles corresponding to a treatment of 5,000–100,000 particles per MoDC were resuspended in DC medium and adjusted to final volume of 50 μL. For treatment with CCM or p3 protein‐enriched samples (Figure S2), 4 μg of protein measured with Qubit protein assay were resuspended in 50 μL of DC medium. Fifty microlitres of particle or protein suspensions, LPS‐EB Ultrapure 200 ng/mL (Invivogen, San Diego, #tlrl‐3pelps), or particle‐free sterile PBS (Thermo Fisher Scientific, Gibco™ #20012027) were then added to the 96‐well plates and gently mixed with 50 μL of MoDC cell suspension. Plates were incubated 24 h at 37°C, 5% CO2. Plates were then centrifuged for 2 min at 2000 rpm, 4°C, and the supernatant was discarded. Fifty microlitres of eBioscienceTM Fixable Viability Dye eFluorTM 780 (Invitrogen, Waltham MA, USA) was added to the cell pellets and incubated 15 min at 4°C protected from light following manufacturer's instructions. Pellets were then washed with staining buffer (PBS, 0.5% BSA, EDTA 2 mM) and centrifuged 2 min at 2000 rpm at 4°C. Supernatant was discarded and human FcR blocking reagent (Miltenyi Biotec) was added and incubated 15 min at 4°C protected from light. After washing with staining buffer and 2 min centrifugation at 2000 rpm, 4°C, supernatant was discarded and 25 μL of staining antibodies diluted in staining buffer (see antibodies and reagents) was added and incubated 30 min at 4°C protected from light. After washing with staining buffer and 2 min centrifugation at 2000 rpm, 4°C, the supernatant was discarded and 50 μL of 3% formaldehyde was added for fixation and incubated for 15 min at 4°C protected from light. After a last washing supernatant was discarded, and cells resuspended in 100 μL of staining buffer. Plates were kept protected from light at 4°C until flow cytometry analysis.
4.13. Flow cytometry
Flow cytometry was performed on a BD LSRII cytometer equipped with a high throughput sampler and using BD FACS Diva™ Software. Cells were gated on forward (FSC‐A) and side scatter area (SSC‐A) to exclude cells debris, followed by FSC‐A/FSC‐H gating to define single cell populations (Figure S2a). Exclusion of Fixable Viability Dye eFluorTM 780 (Invitrogen, Waltham MA) was used to gate living cells following manufacturer's instructions. Fluorescence minus one (FMO) control samples established for each donor with LPS‐treated MoDC were used to define positive population gates of each staining antibody (Figure S2a). CD11c+/MHCII+ positive cells were defined as MoDC and a minimum of 10,000 events within the MoDC population were acquired for each treatment condition. Flow cytometry data were analysed using the FlowJo V10 software.
4.14. Statistical analysis
All statistical analyses performed in this manuscript were done using GraphPad Prism v9. Particle concentration measurements were not assumed to be normally distributed and to have equal variance; they were analysed using Kruskal Wallis test with Dunn's multiple comparisons test. Flow cytometry and Luminex data from human MoDC were not assumed to be normally distributed and to have equal variance; they were analysed using Friedman test (pairing within donors) with Dunn's multiple comparisons test or using Mann‐Whitney test (no pairing within donors, Figure S2c).
AUTHOR CONTRIBUTIONS
Quentin Roux: Conceptualization; Data curation; Formal analysis; Investigation; Validation; Visualization; Writing — original draft. Robin Boiy: Conceptualization; Data curation; Formal analysis; Investigation; Validation; Visualization; Writing — review & editing. Felix De Vuyst: Investigation; Writing — review & editing. Mercedes Tkach: Methodology; Resources; Validation; Writing — review & editing. Claudio Pinheiro: Investigation. Sofie De Geyter: Investigation. Ilkka Miinalainen: Investigation. Clotilde Théry: Resources; Validation; Writing—review & editing. Olivier De Wever: Supervision; Writing — review & editing. An Hendrix: Conceptualization; Funding acquisition; Supervision; Writing — review & editing.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
EV‐TRACK
We have submitted all relevant data of our experiments to the EV‐TRACK knowledgebase (EV‐TRACK ID: EV220024) (Van Deun J, et al. EV‐TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nature methods. 2017;14(3):228‐32).
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGEMENTS
This research was funded by European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska‐Curie grant agreement No [722148] (Q.R., A.H., and C.T.), Ghent University (Q.R. and A.H.), Kom op tegen Kanker (Stand up to Cancer), the Flemish cancer society grant agreement No [13050] (R.B.), Fund for Scientific Research Flanders (FWO).
Roux, Q. , Boiy, R. , De Vuyst, F. , Tkach, M. , Pinheiro, C. , de Geyter, S. , Miinalainen, I. , Théry, C. , De Wever, O. , & Hendrix, A. (2023). Depletion of soluble cytokines unlocks the immunomodulatory bioactivity of extracellular vesicles. Journal of Extracellular Vesicles, 12, e12339. 10.1002/jev2.12339
Quentin Roux and Robin Boiy equally contributed to this work.
REFERENCES
- Alfaro, C. , Suarez, N. , Gonzalez, A. , Solano, S. , Erro, L. , Dubrot, J. , Palazon, A. , Hervas‐Stubbs, S. , Gurpide, A. , Lopez‐Picazo, J. M. , Grande‐Pulido, E. , Melero, I. , & Perez‐Gracia, J. L. (2009). Influence of bevacizumab, sunitinib and sorafenib as single agents or in combination on the inhibitory effects of VEGF on human dendritic cell differentiation from monocytes. British Journal of Cancer, 100(7), 1111–1119. 10.1038/sj.bjc.6604965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre, F. , Schartz, N. E. , Movassagh, M. , Flament, C. , Pautier, P. , Morice, P. , Pomel, C. , Lhomme, C. , Escudier, B. , Le Chevalier, T. , Tursz, T. , Amigorena, S. , Raposo, G. , Angevin, E. , & Zitvogel, L. (2002). Malignant effusions and immunogenic tumour‐derived exosomes. The Lancet, 360(9329), 295–305. 10.1016/s0140-6736(02)09552-1 [DOI] [PubMed] [Google Scholar]
- Anguille, S. , Smits, E. L. , Lion, E. , van Tendeloo, V. F. , & Berneman, Z. N. (2014). Clinical use of dendritic cells for cancer therapy. The Lancet Oncology, 15, e257–267. [DOI] [PubMed] [Google Scholar]
- Bonsergent, E. , Grisard, E. , Buchrieser, J. , Schwartz, O. , Théry, C. , & Lavieu, G. (2021). Quantitative characterization of extracellular vesicle uptake and content delivery within mammalian cells. Nature Communications, 12(1), 1864. 10.1038/s41467-021-22126-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocozza, F. , Grisard, E. , Martin‐Jaular, L. , Mathieu, M. , & Théry, C. (2020). SnapShot: Extracellular vesicles. Cell, 182, 262–262.e1. [DOI] [PubMed] [Google Scholar]
- Cunningham, S. , & Hackstein, H. (2020). Recent advances in good manufacturing practice‐grade generation of dendritic cells. Transfusion Medicine and Hemotherapy, 47, 454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtsinger, J. M. , & Mescher, M. F. (2010). Inflammatory cytokines as a third signal for T cell activation. Current Opinion in Immunology, 22, 333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vlieghere, E. , Gremonprez, F. , Verset, L. , Mariën, L. , Jones, C. J. , De Craene, B. , Berx, G. , Descamps, B. , Vanhove, C. , Remon, J.‐P. , Ceelen, W. , Demetter, P. , Bracke, M. , De Geest, B. G. , & De Wever, O. (2015). Tumor‐environment biomimetics delay peritoneal metastasis formation by deceiving and redirecting disseminated cancer cells. Biomaterials, 54, 148–157. 10.1016/j.biomaterials.2015.03.012 [DOI] [PubMed] [Google Scholar]
- De Wever, O. , & Hendrix, A. (2019). A supporting ecosystem to mature extracellular vesicles into clinical application. The EMBO Journal, 38, e101412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Wever, O. , Nguyen, Q. , Van Hoorde, L. , Bracke, M. , Bruyneel, E. , Gespach, C. , & Mareel, M. (2004). Tenascin‐C and SF/HGF produced by myofibroblasts in vitro provide convergent proinvasive signals to human colon cancer cells through RhoA and Rac. The FASEB Journal, 18(9), 1016–1018. Portico. 10.1096/fj.03-1110fje [DOI] [PubMed] [Google Scholar]
- Dhondt, B. , Geeurickx, E. , Tulkens, J. , Van Deun, J. , Vergauwen, G. , Lippens, L. , Miinalainen, I. , Rappu, P. , Heino, J. , Ost, P. , Lumen, N. , De Wever, O. , & Hendrix, A. (2020). Unravelling the proteomic landscape of extracellular vesicles in prostate cancer by density‐based fractionation of urine. Journal of Extracellular Vesicles, 9(1), 1736935. Portico. 10.1080/20013078.2020.1736935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhondt, B. , Lumen, N. , De Wever, O. , & Hendrix, A. (2020). Preparation of multi‐omics grade extracellular vesicles by density‐based fractionation of urine. STAR Protocols, 1, 100073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillman, R. O. , Cornforth, A. N. , Nistor, G. I. , McClay, E. F. , Amatruda, T. T. , & Depriest, C. (2018). Randomized phase II trial of autologous dendritic cell vaccines versus autologous tumor cell vaccines in metastatic melanoma: 5‐year follow up and additional analyses. Journal for ImmunoTherapy of Cancer, 6(1), 19. 10.1186/s40425-018-0330-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geeurickx, E. , Lippens, L. , Rappu, P. , De Geest, B. G. , De Wever, O. , & Hendrix, A. (2021). Recombinant extracellular vesicles as biological reference material for method development, data normalization and assessment of (pre‐)analytical variables. Nature Protocols, 16(2), 603–633. 10.1038/s41596-020-00446-5 [DOI] [PubMed] [Google Scholar]
- Geeurickx, E. , Tulkens, J. , Dhondt, B. , Van Deun, J. , Lippens, L. , Vergauwen, G. , Heyrman, E. , De Sutter, D. , Gevaert, K. , Impens, F. , Miinalainen, I. , Van Bockstal, P.‐J. , De Beer, T. , Wauben, M. H. M. , Nolte‐‘t‐Hoen, E. N. M. , Bloch, K. , Swinnen, J. V. , van der Pol, E. , Nieuwland, R. , … Hendrix, A. (2019). The generation and use of recombinant extracellular vesicles as biological reference material. Nature Communications, 10(1), 1–12. 10.1038/s41467-019-11182-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha, H. , Debnath, B. , & Neamati, N. (2017). Role of the CXCL8‐CXCR1/2 axis in cancer and inflammatory diseases. Theranostics, 7, 1543–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, T.‐X. , Tan, X.‐Y. , Huang, H.‐S. , Li, Y.‐T. , Liu, B.‐L. , Liu, K.‐S. , Chen, X. , Chen, Z. , Guan, X.‐Y. , Zou, C. , & Fu, L. (2021). Targeting cancer‐associated fibroblast‐secreted WNT2 restores dendritic cell‐mediated antitumour immunity. Gut, 71(2), 333–344. 10.1136/gutjnl-2020-322924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen, D. K. , Fenix, A. M. , Franklin, J. L. , Higginbotham, J. N. , Zhang, Q. , Zimmerman, L. J. , Liebler, D. C. , Ping, J. , Liu, Q. , Evans, R. , Fissell, W. H. , Patton, J. G. , Rome, L. H. , Burnette, D. T. , & Coffey, R. J. (2019). Reassessment of exosome composition. Cell, 177(2), 428–445.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, H. H. , Kim, J.‐Y. , Lim, J. E. , & Im, Y.‐H. (2020). Cytokine profiling in serum‐derived exosomes isolated by different methods. Scientific Reports, 10, 14069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantoff, P. W. , Higano, C. S. , Shore, N. D. , Berger, E. R. , Small, E. J. , Penson, D. F. , Redfern, C. H. , Ferrari, A. C. , Dreicer, R. , Sims, R. B. , Xu, Y. , Frohlich, M. W. , & Schellhammer, P. F. (2010). Sipuleucel‐T immunotherapy for castration‐resistant prostate cancer. New England Journal of Medicine, 363(5), 411–422. 10.1056/nejmoa1001294 [DOI] [PubMed] [Google Scholar]
- La Shu, S. , Yang, Y. , Allen, C. L. , Hurley, E. , Tung, K. H. , Minderman, H. , Wu, Y. , & Ernstoff, M. S. (2019). Purity and yield of melanoma exosomes are dependent on isolation method. Journal of Extracellular Vesicles, 9(1), 1692401. Portico. 10.1080/20013078.2019.1692401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marton, A. , Vizler, C. , Kusz, E. , Temesfoi, V. , Szathmary, Z. , Nagy, K. , Szegletes, Z. , Varo, G. , Siklos, L. , Katona, R. L. , Tubak, V. , Howard, O. M. Z. , Duda, E. , Minarovits, J. , Nagy, K. , & Buzas, K. (2012). Melanoma cell‐derived exosomes alter macrophage and dendritic cell functions in vitro. Immunology Letters, 148(1), 34–38. 10.1016/j.imlet.2012.07.006 [DOI] [PubMed] [Google Scholar]
- Melo, S. A. , Luecke, L. B. , Kahlert, C. , Fernandez, A. F. , Gammon, S. T. , Kaye, J. , LeBleu, V. S. , Mittendorf, E. A. , Weitz, J. , Rahbari, N. , Reissfelder, C. , Pilarsky, C. , Fraga, M. F. , Piwnica‐Worms, D. , & Kalluri, R. (2015). Glypican‐1 identifies cancer exosomes and detects early pancreatic cancer. Nature, 523(7559), 177–182. 10.1038/nature14581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michielsen, A. J. , Hogan, A. E. , Marry, J. , Tosetto, M. , Cox, F. , Hyland, J. M. , Sheahan, K. D. , O'Donoghue, D. P. , Mulcahy, H. E. , Ryan, E. J. , & O'Sullivan, J. N. (2011). Tumour tissue microenvironment can inhibit dendritic cell maturation in colorectal cancer. PLoS ONE, 6(11), e27944. 10.1371/journal.pone.0027944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naseri, M. , Bozorgmehr, M. , Zöller, M. , Ranaei Pirmardan, E. , & Madjd, Z. (2020). Tumor‐derived exosomes: The next generation of promising cell‐free vaccines in cancer immunotherapy. Oncoimmunology, 9, 1779991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning, Y. , Shen, K. , Wu, Q. , Sun, X. , Bai, Y. , Xie, Y. , Pan, J. , & Qi, C. (2018). Tumor exosomes block dendritic cells maturation to decrease the T cell immune response. Immunology Letters, 199, 36–43. 10.1016/j.imlet.2018.05.002 [DOI] [PubMed] [Google Scholar]
- Oh, S. A. , Wu, D.‐C. , Cheung, J. , Navarro, A. , Xiong, H. , Cubas, R. , Totpal, K. , Chiu, H. , Wu, Y. , Comps‐Agrar, L. , Leader, A. M. , Merad, M. , Roose‐Germa, M. , Warming, S. , Yan, M. , Kim, J. M. , Rutz, S. , & Mellman, I. (2020). PD‐L1 expression by dendritic cells is a key regulator of T‐cell immunity in cancer. Nature Cancer, 1(7), 681–691. 10.1038/s43018-020-0075-x [DOI] [PubMed] [Google Scholar]
- Omata, N. , Yasutomi, M. , Yamada, A. , Iwasaki, H. , Mayumi, M. , & Ohshima, Y. (2002). Monocyte chemoattractant protein‐1 selectively inhibits the acquisition of CD40 ligand‐dependent IL‐12‐producing capacity of monocyte‐derived dendritic cells and modulates Th1 immune response. The Journal of Immunology, 169(9), 4861–4866. 10.4049/jimmunol.169.9.4861 [DOI] [PubMed] [Google Scholar]
- Oyama, T. , Ran, S. , Ishida, T. , Nadaf, S. , Kerr, L. , Carbone, D. P. , & Gabrilovich, D. I. (1998). Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor‐κB activation in hemopoietic progenitor cells. The Journal of Immunology, 160(3), 1224–1232. 10.4049/jimmunol.160.3.1224 [DOI] [PubMed] [Google Scholar]
- Park, S.‐J. , Nakagawa, T. , Kitamura, H. , Atsumi, T. , Kamon, H. , Sawa, S. , Kamimura, D. , Ueda, N. , Iwakura, Y. , Ishihara, K. , Murakami, M. , & Hirano, T. (2004). IL‐6 regulates in vivo dendritic cell differentiation through STAT3 activation. The Journal of Immunology, 173(6), 3844–3854. 10.4049/jimmunol.173.6.3844 [DOI] [PubMed] [Google Scholar]
- Perez, C. R. , & De Palma, M. (2019). Engineering dendritic cell vaccines to improve cancer immunotherapy. Nature Communications, 10, 5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao, Q. , Zuo, B. , Lu, Z. , Gao, X. , You, A. , Wu, C. , Du, Z. , & Yin, H. (2016). Tumor‐derived exosomes elicit tumor suppression in murine hepatocellular carcinoma models and humans in vitro. Hepatology, 64(2), 456–472. 10.1002/hep.28549 [DOI] [PubMed] [Google Scholar]
- Salimu, J. , Webber, J. , Gurney, M. , Al‐Taei, S. , Clayton, A. , & Tabi, Z. (2017). Dominant immunosuppression of dendritic cell function by prostate‐cancer‐derived exosomes. Journal of Extracellular Vesicles, 6(1), 1368823. 10.1080/20013078.2017.1368823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen, J. B. (2017). What are we looking at? Extracellular vesicles, lipoproteins, or both? Circulation Research, 121, 920–922. [DOI] [PubMed] [Google Scholar]
- Squadrito, M. L. , Cianciaruso, C. , Hansen, S. K. , & Palma, M. (2018). EVIR: Chimeric receptors that enhance dendritic cell cross‐dressing with tumor antigens. Nature Methods, 15(3), 183–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah, V. , Murthy, R. , Hong, D. S. , Prins, R. M. , Hosing, C. , Hendricks, K. , Kolli, D. , Noffsinger, L. , Brown, R. , McGuire, M. , Fu, S. , Piha‐Paul, S. , Naing, A. , Conley, A. P. , Benjamin, R. S. , Kaur, I. , & Bosch, M. L. (2018). Cytokines produced by dendritic cells administered intratumorally correlate with clinical outcome in patients with diverse cancers. Clinical Cancer Research, 24(16), 3845–3856. 10.1158/1078-0432.ccr-17-2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, A. , Kono, K. , Ichihara, F. , Sugai, H. , Fujii, H. , & Matsumoto, Y. (2004). Vascular endothelial growth factor inhibits maturation of dendritic cells induced by lipopolysaccharide, but not by proinflammatory cytokines. Cancer Immunology, Immunotherapy, 53(6), 543–550. 10.1007/s00262-003-0466-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry, C. , Amigorena, S. , Raposo, G. , & Clayton, A. (2006). Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Current Protocols in Cell Biology, Chapter 3, Unit 3.22. [DOI] [PubMed] [Google Scholar]
- Théry, C. , Witwer, K. W. , Aikawa, E. , Alcaraz, M. J. , Anderson, J. D. , Andriantsitohaina, R. , Antoniou, A. , Arab, T. , Archer, F. , Atkin‐Smith, G. K. , Ayre, D. C. , Bach, J.‐M. , Bachurski, D. , Baharvand, H. , Balaj, L. , Baldacchino, S. , Bauer, N. N. , Baxter, A. A. , Bebawy, M. , … Zuba‐Surma, E. K. (2018). Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of Extracellular Vesicles, 7(1), 1535750. 10.1080/20013078.2018.1535750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkach, M. (2017). Qualitative differences in T‐cell activation by dendritic cell‐derived extracellular vesicle subtypes. The EMBO Journal, 36(20), 3012–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tóth, E. Á. , Turiák, L. , Visnovitz, T. , Cserép, C. , Mázló, A. , Sódar, B. W. , Försönits, A. I. , Petővári, G. , Sebestyén, A. , Komlósi, Z. , Drahos, L. , Kittel, Á. , Nagy, G. , Bácsi, A. , Dénes, Á. , Gho, Y. S. , Szabó‐Taylor, K. É. , & Buzás, E. I. (2021). Formation of a protein corona on the surface of extracellular vesicles in blood plasma. Journal of Extracellular Vesicles, 10(11), e12140.. 10.1002/jev2.12140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchieri, G. (2003). Interleukin‐12 and the regulation of innate resistance and adaptive immunity. Nature Reviews Immunology, 3, 133–146. [DOI] [PubMed] [Google Scholar]
- Tugues, S. , Burkhard, S. H. , Ohs, I. , Vrohlings, M. , Nussbaum, K. , vom Berg, J. , Kulig, P. , & Becher, B. (2014). New insights into IL‐12‐mediated tumor suppression. Cell Death & Differentiation, 22(2), 237–246. 10.1038/cdd.2014.134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulkens, J. , De Wever, O. , & Hendrix, A. (2020). Analyzing bacterial extracellular vesicles in human body fluids by orthogonal biophysical separation and biochemical characterization. Nature Protocols, 15, 40–67. [DOI] [PubMed] [Google Scholar]
- Tulkens, J. , Vergauwen, G. , Van Deun, J. , Geeurickx, E. , Dhondt, B. , Lippens, L. , De Scheerder, M.‐A. , Miinalainen, I. , Rappu, P. , De Geest, B. G. , Vandecasteele, K. , Laukens, D. , Vandekerckhove, L. , Denys, H. , Vandesompele, J. , De Wever, O. , & Hendrix, A. (2018). Increased levels of systemic LPS‐positive bacterial extracellular vesicles in patients with intestinal barrier dysfunction. Gut, 69(1), 191–193. 10.1136/gutjnl-2018-317726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Deun, J. , Mestdagh, P. , Agostinis, P. , Akay, Ö. , Anand, S. , Anckaert, J. , Martinez, Z. A. , Baetens, T. , Beghein, E. , Bertier, L. , Berx, G. , Boere, J. , Boukouris, S. , Bremer, M. , Buschmann, D. , Byrd, J. B. , Casert, C. , Cheng, L. , … Hendrix, A. (2017). EV‐TRACK: Transparent reporting and centralizing knowledge in extracellular vesicle research. Nature Methods, 14(3), 228–232. 10.1038/nmeth.4185 [DOI] [PubMed] [Google Scholar]
- Van Deun, J. , Mestdagh, P. , Sormunen, R. , Cocquyt, V. , Vermaelen, K. , Vandesompele, J. , Bracke, M. , De Wever, O. , & Hendrix, A. (2014). The impact of disparate isolation methods for extracellular vesicles on downstream RNA profiling. Journal of Extracellular Vesicles, 3(1), 24858. 10.3402/jev.v3.24858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Deun, J. , Roux, Q. , Deville, S. , Van Acker, T. , Rappu, P. , Miinalainen, I. , Heino, J. , Vanhaecke, F. , De Geest, B. G. , De Wever, O. , & Hendrix, A. (2020). Feasibility of mechanical extrusion to coat nanoparticles with extracellular vesicle membranes. Cells, 9(8), 1797. 10.3390/cells9081797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergauwen, G. , Dhondt, B. , Van Deun, J. , De Smedt, E. , Berx, G. , Timmerman, E. , Gevaert, K. , Miinalainen, I. , Cocquyt, V. , Braems, G. , Van den Broecke, R. , Denys, H. , De Wever, O. , & Hendrix, A. (2017). Confounding factors of ultrafiltration and protein analysis in extracellular vesicle research. Scientific Reports, 7(1), 2704. 10.1038/s41598-017-02599-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergauwen, G. , Tulkens, J. , Pinheiro, C. , Avila Cobos, F. , Dedeyne, S. , De Scheerder, M. , Vandekerckhove, L. , Impens, F. , Miinalainen, I. , Braems, G. , Gevaert, K. , Mestdagh, P. , Vandesompele, J. , Denys, H. , De Wever, O. , & Hendrix, A. (2021). Robust sequential biophysical fractionation of blood plasma to study variations in the biomolecular landscape of systemically circulating extracellular vesicles across clinical conditions. Journal of Extracellular Vesicles, 10(10), e12122. 10.1002/jev2.12122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C. , Huang, X. , Wu, Y. , Wang, J. , Li, F. , & Guo, G. (2020). Tumor cell‐associated exosomes robustly elicit anti‐tumor immune responses through modulating dendritic cell vaccines in lung tumor. International Journal of Biological Sciences, 16(4), 633–643. 10.7150/ijbs.38414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wculek, S. K. , Cueto, F. J. , Mujal, A. M. , Melero, I. , Krummel, M. F. , & Sancho, D. (2019). Dendritic cells in cancer immunology and immunotherapy. Nature Reviews Immunology, 20(1), 7–24. 10.1038/s41577-019-0210-z [DOI] [PubMed] [Google Scholar]
- Whittaker, T. E. , Nagelkerke, A. , Nele, V. , Kauscher, U. , & Stevens, M. M. (2020). Experimental artefacts can lead to misattribution of bioactivity from soluble mesenchymal stem cell paracrine factors to extracellular vesicles. Journal of Extracellular Vesicles, 9, 1807674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf, M. , Poupardin, R. W. , Ebner‐Peking, P. , Andrade, A. C. , Blöchl, C. , Obermayer, A. , Gomes, F. G. , Vari, B. , Maeding, N. , Eminger, E. , Binder, H. , Raninger, A. M. , Hochmann, S. , Brachtl, G. , Spittler, A. , Heuser, T. , Ofir, R. , Huber, C. G. , Aberman, Z. , … Strunk, D. (2022). A functional corona around extracellular vesicles enhances angiogenesis, skin regeneration and immunomodulation. Journal of Extracellular Vesicles, 11(4), e12207. 10.1002/jev2.12207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfers, J. , Lozier, A. , Raposo, G. , Regnault, A. , Théry, C. , Masurier, C. , Flament, C. , Pouzieux, S. , Faure, F. , Tursz, T. , Angevin, E. , Amigorena, S. , & Zitvogel, L. (2001). Tumor‐derived exosomes are a source of shared tumor rejection antigens for CTL cross‐priming. Nature Medicine, 7(3), 297–303. 10.1038/85438 [DOI] [PubMed] [Google Scholar]
- Xu, Y.‐D. , Cheng, M. , Shang, P.‐P. , & Yang, Y.‐Q. (2022). Role of IL‐6 in dendritic cell functions. Journal of Leukocyte Biology, 111, 695–709. [DOI] [PubMed] [Google Scholar]
- Yang, C. , Kim, S.‐H. , Bianco, N. R. , & Robbins, P. D. (2011). Tumor‐derived exosomes confer antigen‐specific immunosuppression in a murine delayed‐type hypersensitivity model. PLoS ONE, 6(8), e22517. 10.1371/journal.pone.0022517 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information