

Abstract
A method for C(sp3)–C(sp3) cross-coupling of amines is described. Primary amines are converted to 1,2-dialkyldiazenes by treatment with O-nosylhydroxylamines in the presence of atmospheric oxygen. Denitrogenation of the diazenes with an iridium photocatalyst then forges the C–C bond. The substrate scope accommodates a broad latitude of functionality, including heteroaromatics and unprotected alcohols and acids.
Graphical Abstract
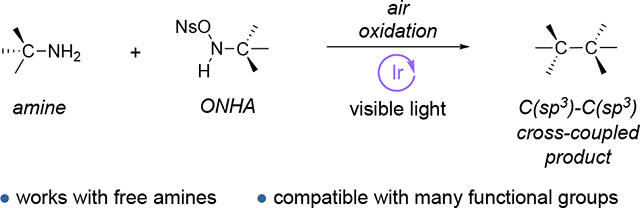
Cross-coupling reactions are highly valued for their ability to construct complex carbon frameworks from simple precursors.1 While conventional cross-couplings utilize coupling partners such as organohalides, organometallics, or boronic acids, recent years have witnessed major efforts to extend the range of functional groups that can be engaged.2 Of particular interest is the cross-coupling of “native” functionalities, such as alcohols3 and carboxylic acids,4 due to their prevalence in natural and synthetic chemical feedstocks. In contrast, amines, which are also broadly available,5 have less commonly been used as cross-coupling partners. This relative paucity is a reflection of the basicity of the amino nitrogen, which tends to complicate transition metal-based reactions, and the strength of the C–N bond.6 Nevertheless, several strategies have been developed to cross-couple amines, most notably through their conversion to Katritzky-type pyridinium salts7 or to redox-active imines,8 which enables the requisite scission of the C–N bond. While these advancements have enabled impressive transformations of amines, C(sp3)–C(sp3) coupling reactions have been limited to radical additions to alkenes,9 coupling with alkyl metal reagents10 and radical couplings with specialized substrates.11 Thus, there remains a strong interest in the development of amine cross-coupling reactions that are simple, versatile, and broadly tolerant of various functional groups for the direct generation of new C(sp3)–C(sp3) coupled products. Here, we report such a method involving the photocatalytic denitrogenation of in situ-generated diazenes.12
It is well known that diazenes can be induced to expel molecular nitrogen, leaving behind carbon-centered radicals that can combine to form C–C bonds.13 Indeed, this reactivity has been employed in wide-ranging applications, including for the total synthesis of highly complex natural products.14 Unfortunately, the current state of the art for this chemistry suffers from several limitations. First, the synthesis of dialkyldiazenes, particularly those with α-C–H bonds, can be challenging because they are prone to isomerize to hydrazones15 which are inactive for denitrogenation. Second, the denitrogenation step often requires elevated temperatures16 or UV radiation,17 which promotes side reactions and limits functional group compatibility. Nevertheless, because the loss of molecular nitrogen provides a powerful driving force that can be leveraged to construct very challenging bond connections, we reasoned that solving the above-mentioned challenges would result in a powerful C–C bond-forming method.
In regard to the denitrogenation problem, we recently reported the electrophotocatalytic decomposition of diazenes to form olefin products (e.g. 2 → 1, Figure 1), using a trisaminocyclopropenium catalyst 4 that enabled the formation of distonic radical cation intermediates.18 As part of that work, we demonstrated that iridium photocatalyst 5 resulted instead in the generation of a diradical intermediate, leading to exclusive C–C bond formation, i.e. 2 → 3. This latter denitrogenation is believed to occur via energy transfer from the photoexcited iridium complex to the diazene group,19 a process that has been employed to great effect for the related decomposition of diazirines to form carbenes.20 In brief, the photocatalyst [Ir] can absorb a photon to access the singlet excited state 1[Ir]*, which can undergo intersystem crossing to the triplet state 3[Ir]* (see Figure 1). Energy transfer from this triplet state to the diazene can then generate the triplet excited state of the diazene. Expulsion of nitrogen from this triplet excited state gives rise to two radical fragments that can recombine to form a new C–C bond. We recognized that this photocatalytic denitrogenation of 1,2-dialkyldiazenes21 could enable an attractive means to cross-couple amines if 1) the process was generally applicable to acyclic diazenes, and 2) a simple synthesis of dialkydiazenes could be developed.
Figure 1.
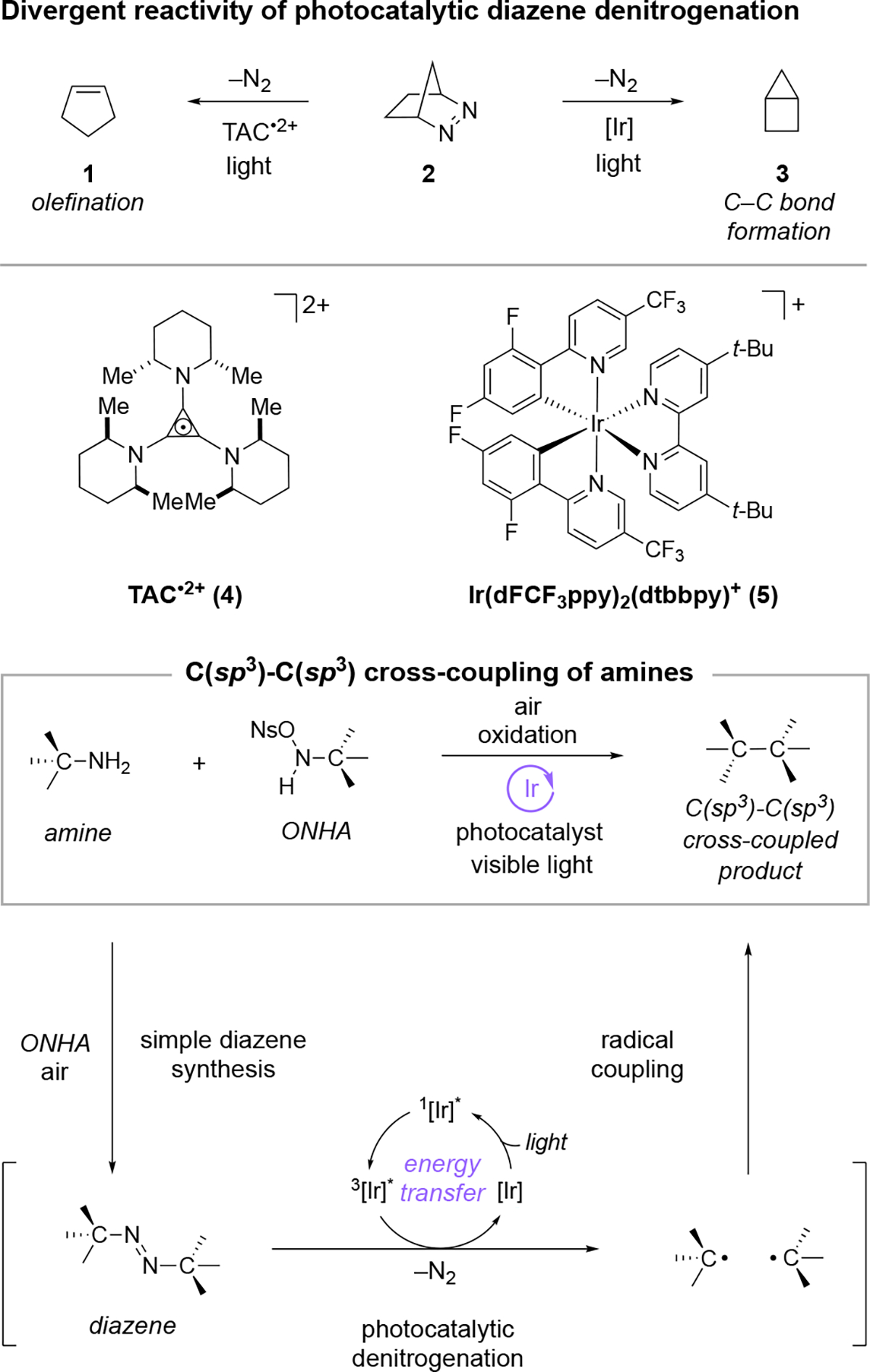
Cross-coupling of amines via photocatalytic denitrogenation of in situ-generated diazenes.
In regard to the second question, although several methods to prepare dialkyldiazenes are known, they tend to be inefficient,22 and in our hands proved unserviceable for the development of a useful cross-coupling procedure. We envisioned making use of the facile autooxidation of hydrazines23 as a way to prepare diazenes under mild conditions; our goal thus became to convert a primary amine substrate to a hydrazine intermediate. While it is known that treatment of an amine with an N-chloroamine can forge the requisite N–N bond to form diaziridines,24 N-chloroamines are problematic and did not suit our purposes. Inspired by the diaziridine work and limited examples utilizing tosyl-substituted hydroxylamines,25 we found that reaction with O-nosylhydroxyamines (ONHAs) provided the desired efficiency.
The optimized amine cross-couping conditions we identified involved treatment of a primary amine substrate, such as leelamine, with one equivalent of the isopropyl ONHA•TFA26 salt 6 and 2,6-lutidine in MeCN at room temperature with exposure to air (Table 1). After 12 h, 2 mol% [Ir(dFCF3ppy)2(dtbbpy)]PF6 was added, and the mixture was irradiated by blue LEDs for 24 h. This procedure resulted in an 85% isolated yield of the cross-coupled product 7.27 In addition to this example, products derived from t-Bu glycinate 8 and leucine p-nitroanilide 9 were generated in high yields. Reactions to form products 7 and 8 were also conducted on a preparative scale (1 mmol) with reasonable yields. Meanwhile, we found that unprotected 1,2-aminoalcohols were viable substrates, with aminoindanol furnishing 10 and threonine benzyl ester giving rise to 11, both as single diastereomers. In the case of 10, both cis and trans aminoindanol substrates led to the same trans product. Unprotected amino acids could also be directly coupled, as with products 12-14 derived from asparagine, methionine, and methyldopa respectively. Peptide starting materials aspartame and lisinopril were converted to adducts 15 and 16 in good and modest yields, respectively. The benzazapinone derivative 17 was prepared in 76% yield, and reaction of aminoethyl biotinamide led to 18 in 75% yield.
Table 1.
Scope of photocatalytic cross-coupling of amines with N-isopropyl-O-nosylhydroxylaminea
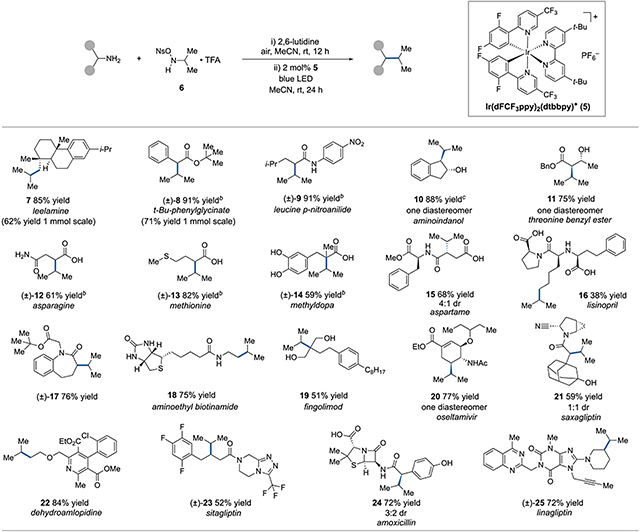
|
See SI for detailed procedures. Reaction Conditions: Amine (1.0 equiv), 6 (1.0 equiv), 2,6-lutidine (2.0 equiv) under ambient air in dry MeCN at room temperature for 12 h, followed by addition of 5 (2 mol%) and irradiation with blue LEDs under N2 at room temperature for 24 h. Yields determined on purified products. Diastereomeric ratios (dr) determined by 1H NMR spectroscopy.
Enantiopure starting materials were used. Products were obtained in racemic form.
(1R,2R)-1-aminoindan-2-ol used as substrate. The (1S,2R) substrate furnished the same product diastereomer. Ns = 4-nitrobenzenesulfonyl.
To further explore the applicability of this reaction to complex molecular settings, we examined the cross-coupling of several amine-containing drug molecules. For example, the reactions of fingolimod, oseltamivir, saxagliptin, dehydroamlopidine, sitagliptin, amoxicillin, and linagliptin to furnish adducts 19-25 in good yields highlight the compatibility of this procedure with a range of functionality and complex architectures. Due to the nature of the radical intermediates, stereoselectivity for these reactions relies exclusively on substrate control. Thus, while 20 was formed as a single diastereomer; 21 and 24, derived from substrates with more isolated amino groups, were generated as mixtures of diastereomers.
Next, we examined the scope of the ONHA component, using threonine benzyl ester 26 as the amine coupling partner (Table 2). Carbocycles of various sizes could be engaged to furnish adducts 27-29 with complete diastereoselectivity for the trans product. We also found that products incorporating tetrahydropyran 30 and piperidine 31 rings could be formed in good yields and as single stereoisomers. When non-α-branched ONHA partners were employed, products 32-34 were obtained, albeit as 1:1 mixtures of diastereomers. In the case of product 35, some measure of stereocontrol was exerted by the existing stereocenter on the ONHA coupling partner.
Table 2.
Scope studies for the ONHA coupling partner.a
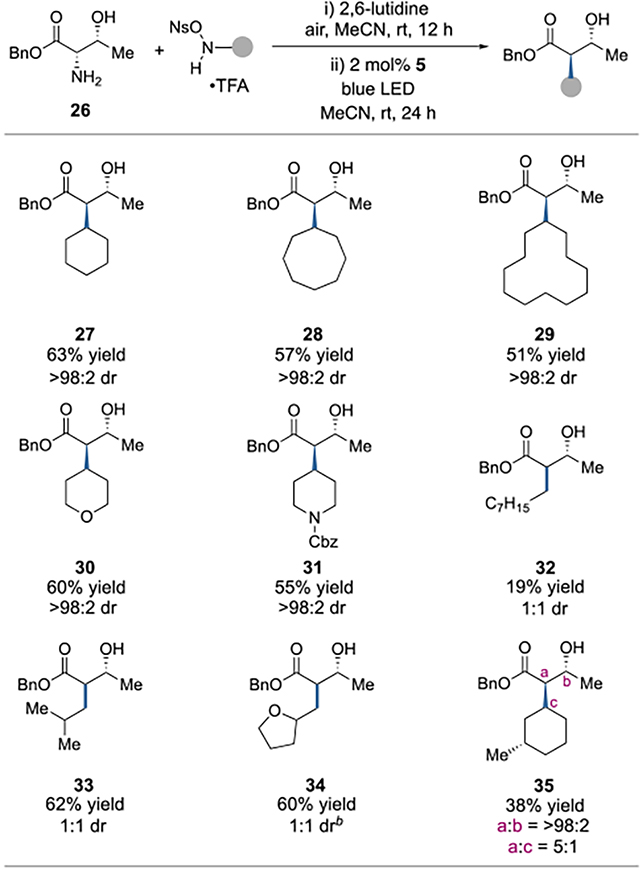
|
See SI for detailed procedures. Reaction Conditions: 26 (1.0 equiv), ONHA (1.0 equiv), 2,6-lutidine (2.0 equiv) under ambient air in MeCN at room temperature for 12 h, followed by addition of 5 (2 mol%) and irradiation with blue LEDs under N2 at room temperature for 24 h. Yields determined on purified products. Diastereomeric ratios (dr) determined by 1H NMR spectroscopy.
Diastereomeric ratio refers to the α and β stereocenters. Racemic ONHA was used, so all diastereomers were obtained.
Under the assumption that the hydrazine formation step proceeds by nucleophilic attack of the amine on the nitrogen of the ONHA, we anticipated that selective cross-coupling of only one amino group of a diamine substrate based on steric differences might be possible. Indeed, treatment of lysine (36) with one equivalent of ONHA 6 under the standard conditions led to the selective formation of 37 in 68% yield (Figure 2, eq 1). No products derived from reaction of the α-amino group were detected. Finally, we examined the applicability of this amine cross-coupling to forge C–C bonds in the context of a highly complex natural product, namely the antibiotic natamycin (38, Figure 2, eq 2). This macrocyclic compound bears a dense array of potentially sensitive functionality, including multiple hydroxyl groups, an allylic epoxide, a carboxylic acid, and a conjugated tetraene, in addition to the aminoglycan appendage. Even within this formidable setting, this coupling method enabled the formation of isopropyl derivative 39 in 48% yield as a single diastereomer, highlighting the potential for late-stage editing of complex molecules.
Figure 2.
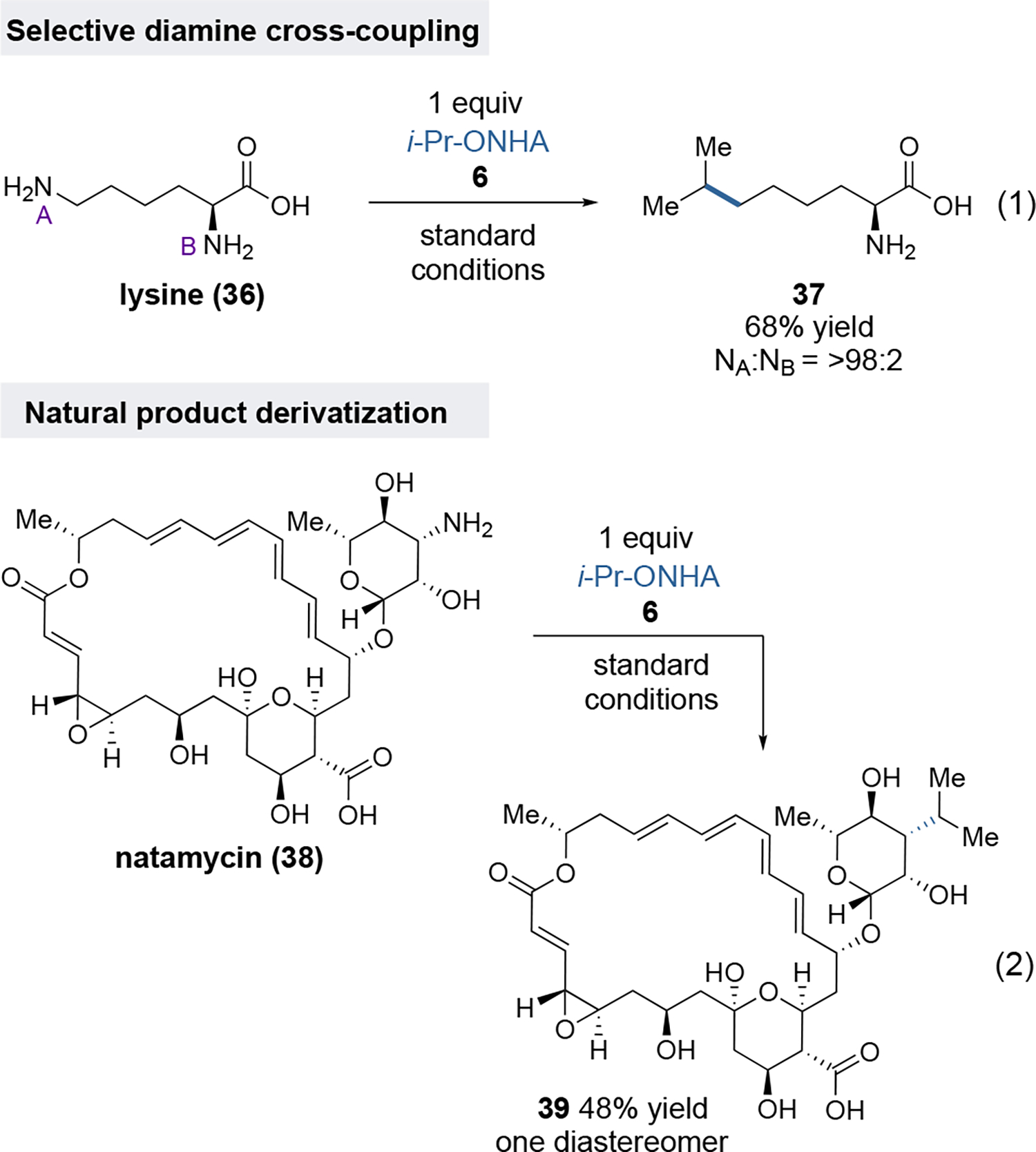
Selective cross-coupling of lysine (eq. 1) and cross-coupling of natamycin (eq. 2).
In summary, we have developed a new method to convert primary amines to C(sp3)–C(sp3) cross-coupled products by employing a simple yet effective 1,2-dialkyldiazene synthesis followed by the use of visible light photocatalysis to trigger their in situ denitrogenation. Because of the mild nature of these conditions, a wide range of functional groups were found to be compatible, including unprotected alcohols, carboxylic acids, and even other amino groups. This work thus expands the ability to utilize widely available amines as building blocks for the construction of complex carbon frameworks.
Supplementary Material
Acknowledgments:
Financial support for this work was provided by NIGMS (R35 GM127135).
Footnotes
Supporting Information Available: Experimental procedures and product characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).(a) Johnston CP; Smith RT; Allmendinger S; MacMillan DWC Metallaphotoredox-Catalysed Sp3–Sp3 Cross-Coupling of Carboxylic Acids with Alkyl Halides. Nature 2016, 536, 322–325. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Corbet J-P; Mignani G Selected Patented Cross-Coupling Reaction Technologies. Chem. Rev. 2006, 106, 2651–2710. [DOI] [PubMed] [Google Scholar]; (c) Choi J; Fu GC Transition Metal–Catalyzed Alkyl-Alkyl Bond Formation: Another Dimension in Cross-Coupling Chemistry. Science 2017, 356, eaaf7230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Li C-J Cross-Dehydrogenative Coupling (CDC): Exploring C–C Bond Formations beyond Functional Group Transformations. Acc. Chem. Res. 2009, 42, 335–344. [DOI] [PubMed] [Google Scholar]; (b) Scheuermann CJ Beyond Traditional Cross Couplings: The Scope of the Cross Dehydrogenative Coupling Reaction. Chem. Asian J. 2010, 5, 436–451. [DOI] [PubMed] [Google Scholar]
- (3).(a) Zhang X; MacMillan DWC Alcohols as Latent Coupling Fragments for Metallaphotoredox Catalysis: Sp3–Sp2 Cross-Coupling of Oxalates with Aryl Halides. J. Am. Chem. Soc. 2016, 138, 13862– 13865. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lackner GL; Quasdorf KW; Overman LE Direct Construction of Quaternary Carbons from Tertiary Alcohols via Photoredox-Catalyzed Fragmentation of tert-Alkyl N-Phthalimidoyl Oxalates. J. Am. Chem. Soc. 2013, 135, 15342– 15345. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nawrat CC; Jamison CR; Slutskyy Y; MacMillan DWC; Overman LE Oxalates as Activating Groups for Alcohols in Visible Light Photoredox Catalysis: Formation of Quaternary Centers by Redox-Neutral Fragment Coupling. J. Am. Chem. Soc. 2015, 137, 11270– 11273. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zheng Y-L; Newman SG Cross-Coupling Reactions with Esters, Aldehydes, and Alcohols. Chem. Commun. 2021, 57, 2591–2604. [DOI] [PubMed] [Google Scholar]; (e) Dong Z; MacMillan DWC Metallaphotoredox-Enabled Deoxygenative Arylation of Alcohols. Nature 2021, 598, 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Wei Y; Ben-zvi B; Diao T Diastereoselective Synthesis of Aryl C-Glycosides from Glycosyl Esters via C–O Bond Homolysis. Angew. Chem. Int. Ed. 2021, 60, 9433– 9438. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Chi BK; Widness JK; Gilbert MM; Salgueiro DC; Garcia KJ; Weix DJ In-Situ Bromination Enables Formal Cross-Electrophile Coupling of Alcohols with Aryl and Alkenyl Halides. ACS Catal. 2022, 12, 580–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Rodriguez N; Gooβen LJ Decarboxylative coupling reactions: a modern strategy for C–C bond formation. Chem. Soc. Rev. 2011, 40, 5030–5048. [DOI] [PubMed] [Google Scholar]; (b) Zuo Z; Ahneman DT; Chu L; Terrett JA; Doyle AG; MacMillan DWC Merging Photoredox with Nickel Catalysis: Coupling of α-Carboxyl sp3-Carbons with Aryl Halides. Science 2014, 345, 437– 440. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dong Z; MacMillan DWC Metallaphotoredox-Enabled Deoxygenative Arylation of Alcohols. Nature 2021, 598, 451– 456. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Sakai HA; MacMillan DWC Nontraditional Fragment Couplings of Alcohols and Carboxylic Acids: C(Sp3)–C(Sp3) Cross-Coupling via Radical Sorting. J. Am. Chem. Soc. 2022, 144, 6185–6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) McGrath NA; Brichacek M; Njardarson JT A Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives. J. Chem. Educ. 2010, 87, 1348–1349. [Google Scholar]; (b) Sanderson K Amino Acid Provides Shortcut to Drugs. Nature 2012, 488, 266. [DOI] [PubMed] [Google Scholar]
- (6).(a) Ouyang K; Hao W; Zhang W-X; Xi Z Transition-Metal-Catalyzed Cleavage of C–N Single Bonds. Chem. Rev. 2015, 115, 12045–12090. [DOI] [PubMed] [Google Scholar]; (b) Ruiz-Castillo P; Buchwald SL Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Reviews on Katritsky Salts as Radical Precursors: Li Y-N; Xiao F; Guo Y; Zeng Y-F Recent Developments in Deaminative Functionalization of Alkyl Amines. Eur. J. Org. Chem. 2021, 2021 (8), 1215–1228. [Google Scholar]; (b) Correia JTM; Fernandes VA; Matsuo BT; Delgado JAC; de Souza WC; Paixão MW Photoinduced Deaminative Strategies: Katritzky Salts as Alkyl Radical Precursors. Chem. Commun. 2020, 56, 503–514. [DOI] [PubMed] [Google Scholar]; (a) Selected Examples: Plunkett S; Basch CH; Santana SO; Watson MP Harnessing Alkylpyridinium Salts as Electrophiles in Deaminative Alkyl–Alkyl Cross-Couplings. J. Am. Chem. Soc. 2019, 141, 2257–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Basch CH; Liao J; Xu J; Piane JJ; Watson MP Harnessing Alkyl Amines as Electrophiles for Nickel-Catalyzed Cross Couplings via C–N Bond Activation. J. Am. Chem. Soc. 2017, 139, 5313–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Klauck FJR; James MJ; Glorius F Deaminative Strategy for the Visible-Light-Mediated Generation of Alkyl Radicals. Angew. Chem. Int. Ed. 2017, 56, 12336–12339. [DOI] [PubMed] [Google Scholar]
- (8).(a) Dorsheimer JR; Ashley MA; Rovis T Dual Nickel/Photoredox-Catalyzed Deaminative Cross-Coupling of Sterically Hindered Primary Amines. J. Am. Chem. Soc. 2021, 143, 19294–19299. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ashley MA; Rovis T Photoredox-Catalyzed Deaminative Alkylation via C–N Bond Activation of Primary Amines. J. Am. Chem. Soc. 2020, 142, 18310–18316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Liu Y; Tao X; Mao Y; Yuan X; Qiu J; Kong L; Ni S; Guo K; Wang Y; Pan Y Electrochemical C–N Bond Activation for Deaminative Reductive Coupling of Katritzky Salts. Nat Commun 2021, 12, 6745. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Baker KM; Baca DL; Plunkett S; Daneker ME; Watson MP Engaging Alkenes and Alkynes in Deaminative Alkyl–Alkyl and Alkyl–Vinyl Cross-Couplings of Alkylpyridinium Salts. Org. Lett. 2019, 21, 9738–9741. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jin Y; Wu J; Lin Z; Lan Y; Wang C Merger of C–F and C–N Bond Cleavage in Cross-Electrophile Coupling for the Synthesis of Gem-Difluoroalkenes. Org. Lett. 2020, 22, 5347–5352. [DOI] [PubMed] [Google Scholar]
- (10).Ni S; Li C-X; Mao Y; Han J; Wang Y; Yan H; Pan Y Ni-Catalyzed Deaminative Cross-Electrophile Coupling of Katritzky Salts with Halides via C─N Bond Activation. Sci. Adv. 2019, 5, eaaw9516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Xu Y; Xu Z-J; Liu Z-P; Lou H Visible-Light-Mediated de-Aminative Alkylation of N-Arylamines with Alkyl Katritzky Salts. Org. Chem. Front. 2019, 6, 3902–3905. [Google Scholar]; (b) Wang C; Qi R; Xue H; Shen Y; Chang M; Chen Y; Wang R; Xu Z Visible-Light-Promoted C(Sp3)–H Alkylation by Intermolecular Charge Transfer: Preparation of Unnatural α-Amino Acids and Late-Stage Modification of Peptides. Angew. Chem. Int. Ed. 2020, 59, 7461–7466. [DOI] [PubMed] [Google Scholar]
- (12).During the course of this project, we became aware of related work by Prof. Quentin Michaudel at Texas A&M University, which has just appeared as a preprint. Chattapadhyay D; Aydogan A; Doktor K; Maity A; Wu JW; Michaudel Q Harnessing Sulfur(VI) Fluoride Exchange Click Chemistry and Photocatalysis for Deaminative Benzylic Arylation. ChemRxiv (Catalysis). Cambridge: Cambridge Open Engage; Submission date: May 02, 2023; Doi. 10.26434/chemrxiv-2023-9r9w1. (accessed 2023-05-08). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Hui C; Wang S; Xu C Dinitrogen Extrusion from Diazene in Organic Synthesis. Chinese Chemical Letters 2022, 33, 3695–3700. [Google Scholar]; (b) Adam W; Doerr M Wagner-Meerwein Rearrangements of Radical Cations Generated by Triphenylpyrylium Tetrafluoroborate Photosensitized Electron Transfer of Azoalkanes. J. Am. Chem. Soc. 1987, 109, 1570–1572. [Google Scholar]
- (14).(a) Lindovska P; Movassaghi M Concise Synthesis of (−)-Hodgkinsine, (−)-Calycosidine, (−)-Hodgkinsine B, (−)-Quadrigemine C, and (−)-Psycholeine via Convergent and Directed Modular Assembly of Cyclotryptamines. J. Am. Chem. Soc. 2017, 139, 17590–17596. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lathrop SP; Pompeo M; Chang W-TT; Movassaghi M Convergent and Biomimetic Enantioselective Total Synthesis of (−)-Communesin F. J. Am. Chem. Soc. 2016, 138, 7763–7769. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Movassaghi M; Ahmad OK; Lathrop SP Directed Heterodimerization: Stereocontrolled Assembly via Solvent-Caged Unsymmetrical Diazene Fragmentation. J. Am. Chem. Soc. 2011, 133, 13002–13005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Pompeo MM; Cheah JH; Movassaghi M Total Synthesis and Anti-Cancer Activity of All Known Communesin Alkaloids and Related Derivatives. J. Am. Chem. Soc. 2019, 141, 14411–14420. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wender PA; Kee J-M; Warrington JM Practical Synthesis of Prostratin, DPP, and Their Analogs, Adjuvant Leads Against Latent HIV. Science 2008, 320, 649–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Nicolas RC; Wilén C-E; Roth M; Pfaendner R; King III RE Azoalkanes: A Novel Class of Flame Retardants. Macromolecular Rapid Communications 2006, 27, 976–981. [Google Scholar]
- (16).Dannenberg JJ; Rocklin D A Theoretical Study of the Mechanism of the Thermal Decomposition of Azoalkanes and 1,1-Diazenes. J. Org. Chem. 1982, 47, 4529–4534. [Google Scholar]
- (17).Engel PS Mechanism of the Thermal and Photochemical Decomposition of Azoalkanes. Chem. Rev. 1980, 80, 99–150. [Google Scholar]
- (18).Steiniger KA; Lambert TH Olefination of Carbonyls with Alkenes Enabled by Electrophotocatalytic Generation of Distonic Radical Cations. Sci. Adv. 2023, 9, eadg3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Orłowska K; Santiago JV; Krajewski P; Kisiel K; Deperasińska I; Zawada K; Chaładaj W; Gryko D UV Light Is No Longer Required for the Photoactivation of 1,3,4-Oxadiazolines. ACS Catal. 2023, 13, 1964–1973. [Google Scholar]
- (20).Geri JB; Oakley JV; Reyes-Robles T; Wang T; McCarver SJ; White CH; Rodriguez-Rivera FP; Parker DL; Hett EC; Fadeyi OO; Oslund RC; MacMillan DWC Microenvironment Mapping via Dexter Energy Transfer on Immune Cells. Science 2020, 367, 1091–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).(a) 1,1-diazenes can also be denitrogenated to form C–C bonds. For impressive recent examples, see: Zou X; Zou J; Yang L; Li G; Lu H Thermal Rearrangement of Sulfamoyl Azides: Reactivity and Mechanistic Study. J. Org. Chem. 2017, 82, 4677–4688. [DOI] [PubMed] [Google Scholar]; (b) Qin H; Cai W; Wang S; Guo T; Li G; Lu H N-Atom Deletion in Nitrogen Heterocycles. Angew. Chem. Int. Ed. 2021, 60, 20678–20683. [DOI] [PubMed] [Google Scholar]; (c) Kennedy SH; Dherange BD; Berger KJ; Levin MD Skeletal Editing Through Direct Nitrogen Deletion of Secondary Amines. Nature 2021, 593, 223–227. [DOI] [PubMed] [Google Scholar]; (d) Hui C; Brieger L; Strohmann C; Antonchick AP Stereoselective Synthesis of Cyclobutanes by Contraction of Pyrrolidines. J. Am. Chem. Soc. 2021, 143, 18864–18870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).(a) Ohme R; Preuschhof H; Heyne H-U Azoethane: Diazene, Diethyl. In Organic Syntheses; John Wiley & Sons, Inc., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2003; pp 11. [Google Scholar]; (b) Kempa S; Wallach L; Rück-Braun K Product Class 6: Aliphatic Azo Compounds. In Category 5, Compounds with One Saturated Carbon Heteroatom Bond; Science of Synthesis; Georg Thieme Verlag KG: Stuttgart, 2015; Vol. 41. [Google Scholar]; (c) Rademacher P Product Class 7: Hydrazines and Hydrazinium Salts. In Category 5, Compounds with One Saturated Carbon Heteroatom Bond; Science of Synthesis; Georg Thieme Verlag KG: Stuttgart, 2015; Vol. 40b. [Google Scholar]
- (23).(a) Overberger CG; Merkel TF Syntheses of Seven-Membered Cyclic Azo Compounds. J. Org. Chem. 1981, 46, 442–446. [Google Scholar]; (b) Marković D; Varela-Álvarez A; Sordo JA; Vogel P Mechanism of the Diphenyldisulfone-Catalyzed Isomerization of Alkenes. Origin of the Chemoselectivity: Experimental and Quantum Chemistry Studies. J. Am. Chem. Soc. 2006, 128, 7782–7795. [DOI] [PubMed] [Google Scholar]
- (24).(a) Schmitz E Diaziridine. In Dreiringe mit Zwei Heteroatomen: Oxaziridine · Diaziridine Cyclische Diazoverbindungen; Schmitz E, Ed.; Organische Chemie in Einzeldarstellungen; Springer: Berlin, Heidelberg, 1967; pp 67–113. [Google Scholar]; (b) Shevtsov AV; Kuznetsov VV; Molotov SI; Lyssenko KA; Makhova NN Synthesis of 4-Aroyl-1,2,4-Triazolidin-3-Ones via Ring Extension in Reactions of 1,2-Di-and 1,2,3,3-Tetraalkyldiaziridines with Aroyl Isocyanates. Russ Chem Bull 2006, 55, 554–558. [Google Scholar]; (c) Ohme R; Schmitz E; Dolge P Diaziridine, VII. Diaziridine aus Aminalen des Formaldehyds. Chem. Ber. 1966, 99, 2104–2109. [Google Scholar]
- (25).(a) Zhao RY; Zhuo X; Yang Q; Zhao L; Huang Y; Ye H; Yang C; Lei J; Gai S; Guo H; Jia J; Bai L; Xie H; Zhou X; Guo Z; Li W; Cao M; Zheng J; Ye Z; Yang Y Cross-Linked Pyrrolobenzodiazepine Dimer (Pbd) Derivative and Its Conjugates. WO2020006722A1, January 9, 2020. [Google Scholar]; (b) Okawara T; Kanazawa Y; Yamasaki T; Furukawa M Convenient Syntheses of 1-Acyl-2-Alkylhydrazines. Synthesis 1987, 1987, 183–184. [Google Scholar]
- (26). N-Alkyl-O-arenesulfonylhydroxylamines are known to decompose by elimination or rearrangement: Hoffman RV; Belfoure EL The Preparation of N-Alkyl-O-Arenesulfonylhydroxylamines. Synthesis 1983, 1983, 34–35. They can be stored in the solid state but decompose in solution, making characterization of pure materials challenging. We have not observed any issues of them acting as high-energy materials; nevertheless, caution may be warranted, especially for lower molecular weight compounds.
- (27).We have not observed products of homodimerization, suggesting the generated radicals do not escape the solvent cage under these conditions. Gould IR; Zimmt MB; Turro NJ; Baretz BH; Lehr GF Dynamics of Radical Pair Reactions in Micelles. J. Am. Chem. Soc. 1985, 107, 4607–4612. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.