

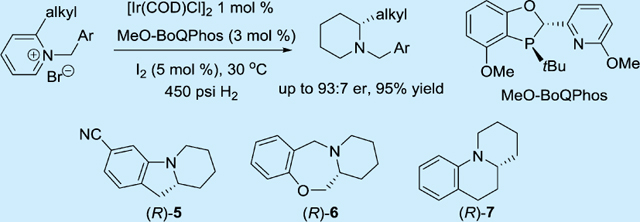
Abstract
An Ir-catalyzed enantioselective hydrogenation of 2-alkylpyridines has been developed using ligand MeO-BoQPhos. High levels of enantioselectivities up to 93:7 er were obtained. The resulting enantioenriched piperidines can be readily converted into biologically interesting molecules such as the fused tricyclic structures 5, 6, and 7 in 99:1 er, providing a novel, concise synthetic route to this family of chiral piperidine-containing compounds.
Graphical Abstract
Chiral 2-alkyl piperidine is a ubiquitous motif found in a wide range of medicinally important compounds including many naturally occurring alkaloids (Figure 1).1,2 2-Alkyl piperidine-containing molecules have exhibited a broad spectrum of interesting biological activities such as potential analgetics,3 antiamnestic agents,4 and for the treatment of neurological diseases and psychiatric disorders.5
Figure 1.

2-Alkyl piperidine-containing alkaloids.
The reported synthetic strategies generally require long synthetic sequences to obtain the target chiral piperidines,6 starting from the chiral pool, use of chiral auxiliaries, or through resolution of racemic mixtures. We are interested in synthesizing a series of chiral 2-alkylpiperidine derivatives and sought to access these molecules through an efficient and enantioselective approach. To this end, we envisioned installing the chiral center directly through asymmetric hydrogenation (AH) of 2-alkylpyridine derivatives. Substituted pyridines are readily available; the successful implementation of this protocol would provide an efficient atom-economical fashion for the preparation of enantiomerically enriched 2-alkyl piperidines.
Recent advances in asymmetric reduction of the heteroarenes have enabled a straightforward synthesis for enantioenriched heterocyclic compounds.7 To facilitate the asymmetric reduction of pyridines, Charette and Legault first demonstrated an activation process for pyridine derivatives by forming N-acylimino-pyridinium ylides; high enantioselectivity was provided for 2-alkylpyridines.8 Nevertheless, safetyconcerns9 related to the use of amination reagents for the preparation of the requisite ylides prevent us from utilizing this elegant method on large scale. Zhou’s10 and Zhang’s11 groups then activated the pyridines by forming N-benzylpyridinium salts. A number of 2-aryl derived pyridines are reduced successfully with high enantioselectivities, but with limited application to 2-alkylpyridines. Herein, we report the development of an Ir-catalyzed enantioselective hydrogenation of 2-alkyl N-benzylpyridinium salts using BI P,N ligand MeO-BoQPhos, and its application toward the synthesis of the fused tricyclic piperidine-containing pharmacophores hexahydro- pyridoindole (R)-5, hexahydrobenzopyridooxazepine (R)-6, and benzoquinolizidine (R)-7 in enantiomerically pure form.
Our investigation started with the readily accessible compound 2-benzyl-N-benzylpyridinium bromide salt 1a. Commercially available chiralphosphineligandswere initially thefocusunderthe conditions of 1 mol % [Ir(COD)Cl]2, 30 °C, and 450 psi H2 pressure (Scheme 1). Disappointing enantioselectivities resulted including Synphos10 and MP2-Segphos,11 which are highly effective for 2-arylpyridinium salt reduction. Iodine is known to activate the iridium catalyst for catalytic hydrogenation reactions.8,12 However, it was found that, for the reduction of 1a with bisphosphine ligands, the presence of iodine was detrimental to both reactivity and enantioselectivity.
Scheme 1. Selected Known Ligands for AH of 1aa.

aTHF/MeOH 9:1, 100% conv to product unless specified, determined by HPLC area % at 220 nm. bIn the presence of 5 mol % I2, ratio of I2/Ir = 2.5:1.
We have recently introduced a family of new phosphorus ligands derived from a modular dihydrobenzooxaphosphole core and demonstrated their unique capability for a wide range of catalytic transformations.13 In particular, the phosphorus-pyridine BoQ-Phos ligands have shown to reduce unfunctionalized alkenes enantioselectively with an iridium catalyst.14 Since 2-alkylpyridines contain largely uncoordinating functionalities, we postulated these ligands could be also effective for the asymmetric reduction of pyridinium salts (Scheme 2). We first tested the unsubstituted ligand L1 for hydrogenation of 1a with [Ir(COD)-Cl]2 at 30 °C and 450 psi H2 pressure; 25% conversion was observed. To enhance the reactivity, electron-donating alkoxy groups were incorporated into the ligand structure. Methoxy substitution on the left-hand side of the phenyl ring improved the conversion slightly to 40%, but again with no enantioselectivity (L2). On the other hand, ligand L3 with a methoxy group on pyridyl generated the desired product 2a in 97% conversion and an 82:18 er. The best enantioselectivity (86:14 er)15 was achieved when methoxy groups were installed on both the phenyl and pyridyl rings as in (S,S)-L4 (MeO-BoQPhos). Further fine-tuning of substitution on the pyridyl group did not significantly improve the selectivity (L5 to L9). Aryl substitution on the phenyl group or phosphorus atom (L10 to L12) decreased the reactivity.
Scheme 2. BoQPhos Ligands for AH of 1aa.

aTHF/MeOH 9:1, 20 h, 100% conv to 2a unless specified, determined by HPLC area % at 220 nm.
We have previously observed that an iridacyclic complex is formed through Ir insertion to the C−H bond of the neighboring OMe group in the presence of a noncoordinating anion such as BArF (Scheme 3).14 This complex is unreactive toward the asymmetric reduction of alkenes. In the presence of a strongly coordinating chloride, the anticipated neutral complex IrCl(MeO-BoQPhos)(COD) 3 was successfully isolated. The single crystal structure of 316 clearly indicated an intact MeO group on pyridine in the molecule.
Scheme 3.

Structure of the Complex [IrCl(MeO-BoQPhos)(COD)] (3)
Complex 3 proved to be effective toward asymmetric hydrogenation of the pyridinium salt 1a and was directly employed for solvent screening to eliminate the uncertainty of incomplete ligation in various solvents (Table 1). A variety of solvents and solvent mixtures were examined, with THF identified as optimal furnishing a 90:10 er (entry 3). This represents the highest enantioselectivity reported for 2-benzyl-N-benzylpyridinium salt 1a compared to those obtained with bisphosphine ligands.10,11
Table 1.
Solvent Screening for AH of 1a Using Complex 3a
| entry | solvent | 2a (%)b | er |
|---|---|---|---|
| 1 | THF/MeOH (9:1) | 100 | 86:14 |
| 2 | THF/MeOH (3:1) | 100 | 82:18 |
| 3c | THF | 100 | 90:10 |
| 4 | DCM | 93 | 75:24 |
| 5 | DCM | 79 | 67:33 |
| 6 | 1,4-dioxane | 92 | 80:20 |
| 7 | toluene | 92 | 82:18 |
| 8 | toluene/DCM (1:1) | 80 | 74:26 |
| 9 | THF/n-BuOH (3:1) | 100 | 82:18 |
| 10 | acetone | 99 | 52:48 |
Reaction conditions: 30 mg of 1a, 2 mol % 3, 5 mol % I2, 30 °C, 450 psi H2 in 0.6 mL of solvent.
HPLC area % at 220 nm.
20 °C.
With the optimized conditions in hand, the substrate scope was then evaluated (Scheme 4). An enantiomeric ratio of 93:7 was observed for the piperidine 2b with 2-bromo-4-cyano benzyl substitution. Most importantly, the catalyst system proved to be chemoselective at reducing the pyridine ring without affecting the reduction sensitive bromo and cyano groups. Hindered substrate 2-diphenylmethylpyridinium salt 1c was reduced as well to produce an 84:16 er. The TBS protected 2-methanol pyridinium salt 1d and 1e are expected to be unfavorable substrates in terms of enantioselectivity; 82:18 er and 84:16 er were obtained, respectively. The catalyst is also effective for 2-alkylpyridinium salts containing long alkyl chains. N-Benzyl-2-phenethylpyridinium 1f was reduced to the corresponding chiral piperidine 2f with an 88:12 er. Pyridinium salts with acetal and ketal functionality on the alkyl group were reduced successfully to yield 89:11 er(2g) and 85:15 er (2h). 2-Methylpyridinium salt 1i was reported to afford product 2i in a 67:33 er upon hydrogenation using MP2-Segphos as a ligand.11 With Ir-MeO-BoQPhos, 2-methylpiperidine 2i was obtained in a much improved 82:18 er. Similarly, 2-propylpiperidine 2j and 2-isopropylpiperidine 2k were obtained in 88:12 er and 91:9 er, respectively. A cyclopropyl substituent is tolerated to generate chiral piperidine 2l in a 92:8 er.
Scheme 4. AH of 2-Alkyl-N-benzylpyridinium Saltsa.

aReaction conditions: pyridinium salt (0.5 g), 20 mL of THF, 5–24 h, isolation yield. b20 °C. c[Ir(COD)Cl]2 2 mol %, ligand 6 mol %, I2 10 mol % at 10 °C and 600 psi H2 for 24 h.
Enantioselective reduction of the more challenging 2,3-disubstituted pyridinium salts including cyclic compound 1m and acyclic compound 1n was also evaluated. Presumably, complete hydrogenation proceeds through a tetrasubstituted enamine intermediate, which has been shown to be the most demanding substrate toward hydrogenation due to a high energy barrier and challenging facial differentiation.17 The resulting fused bicyclic chiral piperidine would be an interesting structure that contains two embedded contiguous stereogenic centers.18 To our delight, the asymmetric reduction of 1m produced the enantioenriched 2m in an exclusive cis-fashion with a 81:19 er and 86% yield. The acyclic 2,3-dimethylpiperidine 2n was obtained in >99.5:0.5 dr and 78:22 er in 82% isolation yield.
Computational studies were conducted on the key enamine intermediate with (S,S)-L4 to understand the enantioinduction process (Figure 2). All calculations were conducted with Gaussian 0919 at the DFT level of theory with the B3LYP20 and LANL2DZ basis set with ECP for iridium21 and D95v for the remaining atoms.22 Aviable reaction pathway was found employing an outer-sphere dissociated mechanism23 similar to the mechanism of iridium catalyzed asymmetric reduction of imines24a and quinolone derivatives.24b This pathway indicates that iodine activated the Ir(I) precatalyst; the resulting Ir(III) iodo complex is maintained as a neutral or charge balanced state throughout the two-step protonation and hydride delivery catalytic cycle. Although the rate-limiting step is the protonation (TS-1), the hydride delivery proceeds through a dissociative mechanism (TS-2) and thus the second transition state would dictate the facial selectivity of the process. Of the three potential hydrides on the iridium(III) intermediate, the one adjacent to the methoxypyridine residue of the ligand is sterically more accessible. The methoxy appendage of the pyridine orients the iminium substrate to expose the si-face for hydride delivery and generating the R-enantiomer of 2a.
Figure 2.

Outer-sphere dissociative catalytic cycle of the Ir-catalyzed reduction of enamine intermediate. DFT calculations: B3LYP/LANL2DZ, CPCM solvation model with THF. Thermal corrections: 298.15 K at 1 atm. Plot: 3D-Model of TS-2.
The resulting enantioenriched 2-alkyl piperidines can be readily converted into naturally occurring alkaloids including Coniine (from 2j) and Pelletierine (from 2h) and pharmaceutically important molecules such as chiral Desoxylpipradrol25 (from 2c). Furthermore, they were demonstrated in the syntheses of several enantiomerically pure fused tricyclic piperidine-containing molecules.
The reaction mixture of 2b was subjected to debenzylation by treatment with α-chloroethyl chloroformate26 in the presence of 0.2 equiv of iPr2NEt in refluxing MeOH (Scheme 5). Recrystallization of the resulted HCl salt enriched the enantiomeric purity of 4−HCl to 99:1 er. The deprotected chiral piperidine was isolated in 76% yield. An intramolecular Hartwig−Buchwald amination furnished (R)-5 in 78% yield upon isolation.
Scheme 5.

Synthesis of Hexahydropyridoindole (R)-5
To our knowledge, this is the first enantioselective synthesis of hexahydropyridoindole derivatives.27
The N-benzyl fragment can also be utilized as an integral part of the molecule for further transformation (Scheme 6). Chiral enrichment on the corresponding alcohol from 2d increased the enantiomeric purityto 96:4 er. Intramolecular ether formation and recrystallization of the HCl salt afforded the enantiomerically pure (R)-65,28 in 99:1 er.
Scheme 6.

Synthesisofhexahydrobenzopyridooxazepine (R)-6
Finally, a benzoquinolizidine derivative 712d,29 was conveniently prepared from 2f (Scheme 7). It was first subjected to chiral enrichment and then converted into compound (R)-7 via debenzylation and an intramolecular amination (Scheme 7).
Scheme 7.

Synthesis of Tricyclic Benzoquinolizidine (R)-7
In summary, an Ir-catalyzed enantioselective hydrogenation of 2-alkyl-pyridinium salts has been developed using MeO-BoQPhos as the ligand. High levels of enantioselectivity up to 93:7 er were obtained, which represents an efficient and practical method for the preparation of enantioenriched piperidines. The resulting piperidines can be readily transformed into biologically interesting molecules such as fused tricyclic hexahydropyridoindole, benzo-quinolizidine, and hexahydrobenzopyridooxazepine derivatives in 99:1 er, providing a concise and novel synthetic route to this family of chiral piperidine-containing compounds.
Supplementary Material
Footnotes
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.6b02401.
Experimental details and characterization data (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1) (a).Ripoche I; Gelas J; Gree D; Gree R; Troin Y. Tetrahedron Lett. 1995, 36, 6675. [Google Scholar]; (b) Arend M; Westermann B; Risch N. Angew. Chem., Int. Ed 1998, 37, 1044. [DOI] [PubMed] [Google Scholar]; (c) Mitchinson A; Nadin AJJ Chem. Soc., Perkin Trans. 1 1999, 1, 2553. [Google Scholar]; (d) Takahashi M; Micalizio GC J. Am. Chem. Soc 2007, 129, 7514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2) (a).Watson PS; Jiang B; Scott B. Org. Lett 2000, 2, 3679. [DOI] [PubMed] [Google Scholar]; (b) Holladay MW; Dart MJ; Lynch JK J. Med. Chem 1997, 40, 4169. [DOI] [PubMed] [Google Scholar]; (c) Lloyd GK; Williams MJ J. Pharm. Exp. Ther 2000, 292, 461. [PubMed] [Google Scholar]; (d) Hong S; Kawaoka AM; Marks TJ J. Am. Chem. Soc 2003, 125, 15878. [DOI] [PubMed] [Google Scholar]; (e) Paŕraga J; Moreno L; Diaz A; El Aouad N; Galań A; Sanz MJ; Caignard D-H; Figader̀e B; Cabedo N; Cortes D. Eur. J. Med. Chem 2014, 86, 700. [DOI] [PubMed] [Google Scholar]
- (3).Beckett AH; Lingard RG; Theobald AE E. J. Med. Chem 1969, 12, 563. [DOI] [PubMed] [Google Scholar]
- (4).Zhao S; Totleben MJ; Freeman JP; Bacon CL; Fox GB; O’Driscoll E; Foley AG; Kelly J; Farrell U; Regan C; Mizsak SA; Szmuszkovicz J. Bioorg. Med. Chem 1999, 7, 1637. [DOI] [PubMed] [Google Scholar]
- (5) (a).Anthony NJ; Gomez RP; Li J; Mercer SP; Roecker AJ; Stump CA; Williams TM PCT Int. Appl 2012, WO 2012061019A2 20120510. [Google Scholar]; (b) Grove SJA; Zhang M; Shahid M. PCTInt. Appl 2002, WO 2002100865A1 20021219. [Google Scholar]
- (6) (a).Bailey PD; Millwood PA; Smith PD Chem. Commun 1998, 633 and references cited therein. [Google Scholar]; (b) Leighty MW; Georg GI ACS Med. Chem. Lett 2011, 2, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Reddy BVS; Ghanty S; Reddy NSS; Reddy YJ; Yadav JS Synth. Commun 2014, 44, 1658. [Google Scholar]
- (7) (a).Wang D-S; Chen Q-A; Li W; Yu C-B; Zhou Y-G; Zhang XJ Am. Chem. Soc 2010, 132, 8909. [DOI] [PubMed] [Google Scholar]; (b) Dobereiner GE; Nova A; Schley ND; Hazari N; Miller SJ; Eisenstein O; Crabtree RH J. Am. Chem. Soc 2011, 133, 7547. [DOI] [PubMed] [Google Scholar]; (c) Woodmansee DH; Pfaltz A. Top. Organomet. Chem 2011, 34, 31. [Google Scholar]; (d) Wang D-S; Chen Q-A; Lu S-M; Zhou Y-G Chem. Rev 2012, 112, 2557. [DOI] [PubMed] [Google Scholar]; (e) Huang W-X; Liu L-J; Wu B; Feng G-S; Wang B; Zhou Y-G Org. Lett 2016, 18, 3082. [DOI] [PubMed] [Google Scholar]; (f) Gualandi A; Savoia D. RSC Adv. 2016, 6, 18419. [Google Scholar]
- (8).Legault CY; Charette AB J. Am. Chem. Soc 2005, 127, 8966. [DOI] [PubMed] [Google Scholar]
- (9).Compounds o-(2,4-dinitrophenyl)hydroxylamine and N-Boc-hydroxylamine show high energy decomposition at a relatively low onset temperature. Handling, shipment, and storage of these materials pose significant safety risks on large scales. See the Supporting Information for the Differential Scanning Calorimetry (DSC) plots. [Google Scholar]
- (10).Ye Z-S; Chen M-W; Chen Q-A; Shi L; Duan Y; Zhou Y-G Angew. Chem., Int. Ed 2012, 51, 10181. [DOI] [PubMed] [Google Scholar]
- (11).Chang M; Huang Y; Liu S; Chen Y; Krska SW; Davies IW; Zhang X. Angew. Chem., Int. Ed 2014, 53, 12761. [DOI] [PubMed] [Google Scholar]
- (12) (a).Chan YNC; Osborn JA J. Am. Chem. Soc 1990, 112, 9400. [Google Scholar]; (b) Xiao D; Zhang X. Angew. Chem., Int. Ed 2001, 40, 3425. [DOI] [PubMed] [Google Scholar]; (c) Blaser H-U Adv. Synth. Catal 2002, 344, 17. [Google Scholar]; (d) Wang D-W; Wang X-B; Wang D-S; Lu S-M; Zhou Y-G; Li Y-XJ Org. Chem 2009, 74, 2780. [DOI] [PubMed] [Google Scholar]; (e) Tang W-J; Tan J; Xu L-J; Lam KH; Fan Q-H; Chan AS C. Adv. Synth. Catal 2010, 352, 1055. [Google Scholar]
- (13) (a).Tang W; Qu B; Capacci AG; Rodriguez S; Wei X; Haddad N; Narayanan B; Ma S; Grinberg N; Yee NK; Krishnamurthy D; Senanayake CH Org. Lett 2010, 12, 176. [DOI] [PubMed] [Google Scholar]; (b) Tang W; Capacci AG; Wei X; Li W; White A; Patel ND; Savoie J; Gao JJ; Rodriguez S; Qu B; Haddad N; Lu BZ; Krishnamurthy D; Yee NK; Senanayake CH Angew. Chem., Int. Ed 2010, 49, 5879. [DOI] [PubMed] [Google Scholar]; (c) Qu B; Samankumara LP; Ma S; Fandrick KR; Desrosiers J-N; Rodriguez S; Li Z; Haddad N; Han ZS; McKellop K; Pennino S; Grinberg N; Gonnella NC; Song JJ; Senanayake CH Angew. Chem., Int. Ed 2014, 53, 14428. [DOI] [PubMed] [Google Scholar]; (d) Du K; Guo P; Chen Y; Cao Z; Wang Z; Tang W. Angew. Chem., Int. Ed 2015, 54, 3033. [DOI] [PubMed] [Google Scholar]; (e) Fu W; Nie M; Wang A; Cao Z; Tang W. Angew. Chem., Int. Ed 2015, 54, 2520. [DOI] [PubMed] [Google Scholar]; (f) Qu B; Haddad N; Rodriguez S; Sieber JD; Desrosiers J-N; Patel ND; Zhang Y; Grinberg N; Lee H; Ma S; Ries UJ; Yee NY; Senanayake CH J. Org. Chem 2016, 81, 745. [DOI] [PubMed] [Google Scholar]
- (14).Qu B; Samankumara LP; Savoie J; Fandrick DR; Haddad N; Wei X; Ma S; Lee H; Rodriguez S; Busacca CA; Yee NK; Song JJ; Senanayake CH J. Org. Chem 2014, 79, 993. [DOI] [PubMed] [Google Scholar]
- (15).The major enantiomer is (R)-configuration, derived from DFT calculation and the X-ray single crystal structure of (R)-7-HCl salt. [Google Scholar]
- (16).CCDC-1477152 and CCDC-1478240 contain supplementary crystallographic data for compounds 3 and 7, respectively. This material is free of charge from The Cambridge Crystallographic Data Centre via; www.ccdc.cam.ac.uk/data_request/cif. [Google Scholar]
- (17) (a).Wang Q; Huang W; Yuan H; Cai Q; Chen L; Lv H; Zhang XJ Am. Chem. Soc 2014, 136, 16120. [DOI] [PubMed] [Google Scholar]; (b) Molinaro C; Scott JP; Shevlin M; Wise C; Ménard, A.; Gibb, A.; Junker, E. M.; Lieberman, D. J. Am. Chem. Soc 2015, 137, 999. [DOI] [PubMed] [Google Scholar]
- (18) (a).Burns NZ; Krylova IN; Hannoush RN; Baran PS J. Am. Chem. Soc 2009, 131, 9172. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ito T; Overman LE; Wang JJ Am. Chem. Soc 2010, 132, 3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Gaussian 09, Revision C.01, Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas Ö; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian, Inc., Wallingford CT, 2010. [Google Scholar]
- (20)(a).Becke AD J. Chem. Phys 1993, 98, 5648. [Google Scholar]; (b) Lee C; Yang W; Parr RG Phys. Rev. B: Condens. Matter Mater. Phys 1988, 37, 785. [DOI] [PubMed] [Google Scholar]
- (21)(a).Hay PJ; Wadt WR J. Chem. Phys 1985, 82, 270. [Google Scholar]; (b) Wadt WR; Hay PJ J. Chem. Phys 1985, 82, 299. [Google Scholar]
- (22).Dunning TH Jr.; Hay PJ In Modern Theoretical Chemistry, Vol. 3; Methods of Electronic Structure Theory; Schaefer F III, Ed.; Plenum: New York, 1977; pp 1–28. [Google Scholar]
- (23) (a).Martín M; Sola E; Tejero S; Andrés JL.; Oro LA Chem. -Eur. J 2006, 12, 4043. [DOI] [PubMed] [Google Scholar]; (b) Martín M; Sola E; Tejero S; Andrés JL; Oro LA Chem. - Eur. J 2006, 12, 4057. [DOI] [PubMed] [Google Scholar]
- (24) (a).Hopmann KH; Bayer A. Organometallics 2011, 30, 2483. [Google Scholar]; (b) Dobereiner GE; Nova A; Schley ND; Hazari N; Miller SJ; Eisenstein O; Crabtree RH J. Am. Chem. Soc 2011, 133, 7547. [DOI] [PubMed] [Google Scholar]
- (25).Corkery JM; Elliott S; Schifano F; Corazza O; Ghodse AH Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2012, 39, 253. [DOI] [PubMed] [Google Scholar]
- (26) (a).Olofson RA; Martz JT; Senet J-P; Piteau M; Malfroot TJ Org. Chem 1984, 49, 2081. [Google Scholar]; (b) Yang BV; O’Rourke D; Li J. Synlett 1993, 1993, 195. [Google Scholar]
- (27).For nonenantioselective syntheses, see: [Google Scholar]; (a) Ozaki S; Mitoh S; Ohmori H. Chem. Pharm. Bull 1996, 44, 2020. [Google Scholar]; (b) Krogsgaard-Larsen N; Begtrup M; Herth MM; Kehler J. Synthesis 2010, 2010, 4287. [Google Scholar]; (c) Mahoney SJ; Fillion E. Chem. - Eur. J 2012, 18, 68. [DOI] [PubMed] [Google Scholar]; (d) Kaldas SJ; Cannillo A; McCallum T; Barriault L. Org. Lett 2015, 17, 2864. [DOI] [PubMed] [Google Scholar]
- (28).For nonchiral analogues: Schultz AG; Macielag M; Sundararaman P; Taveras AG; Welch MJ Am. Chem. Soc 1988, 110, 7828. [Google Scholar]
- (29) (a).Ito Y; Miyata S; Nakatsuka M; Saegusa TJ Am. Chem. Soc 1981, 103, 5250. [Google Scholar]; (b) Rueping M.; Hubener L. Synlett 2011, 2011, 1243. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.