

Abstract
On 15–16 June 2022, the National Institute of Allergy and Infectious Diseases hosted a virtual workshop on the topic of T cell technologies to discuss assays, novel technology development, bench and clinical application of those technologies, and challenges and innovations in the field.
T cells have a central role in adaptive immune responses. However, no accurate assays currently exist that link measurements of ex vivo or in vitro function to effective in vivo T cell responses. Diagnostic detection of T cell function in infectious and immune-mediated diseases also lags in vitro assessments of antibody function. An improved understanding of T cell responses will help researchers and clinicians better predict immune outcomes in response to vaccines, pathogenic infections or immune-mediated diseases.
To address these issues, the National Institute of Allergy and Infectious Diseases (NIAID) convened the ‘T Cell Technologies: Assays, Innovations, Challenges, and Opportunities Workshop’ on 15–16 June 2022. The goals of the workshop were to explore assays and technologic advances that could improve understanding of T cell activation and function in different immune conditions, tissues and infections, and to identify methodologies that best provide an accurate measure of T cell biological relevance.
TCR properties and opportunities
As pointed out by Mark Davis during his overview, the detection and quantification of antigen-specific T cell responses is challenging. One issue addressed was the ability to identify antigen-specific T cells via the T cell receptor (TCR). Although the binding of TCRs with monomeric peptide–major histocompatibility complex (pMHC) multimers is sensitive, it is low affinity and produces a weak and transient signal that is difficult to detect without amplification. pMHC multimer assays, which are based on tetramers, dextramers and spheromers, tackle this issue by triggering simultaneous binding of multiple cell surface TCRs to increase total binding strength and reduce disassociation rates. Recently developed by the Davis group, pMHC spheromers consist of a ferritin structure with 12 pMHC molecules attached; the increased valency of the spheromer further improves detection sensitivity and reduces signal-to-noise ratios1.
Another challenge in T cell detection is the vast diversity of TCR sequence combinations that make it impossible to capture all TCRs specific for a given epitope with a limited number of pMHC multimers. However, by combining TCR sequence information and cluster-based algorithms, such as the ‘grouping of lymphocyte interactions by paratope hotspots’ (GLIPH) algorithm developed by the Davis group and ‘TCR distance measure’ (TCRdist) developed by the Paul Thomas group, we are now able to ‘read’ the TCR repertoire and search for sequence similarities to cluster TCRs into distinct groups.
These analyses can define rules of TCR specificity, predict pathogen history and probable human leukocyte antigen (HLA) types from bulk TCR sequences, and identify major T cell targets in infectious disease, vaccines, autoimmunity or cancer. Together with high-throughput TCR sequencing (TCR-seq) technology, these tools enable the building of TCR-omes to identify meaningful TCR patterns that are associated with immune phenotypes and clinical manifestations, which can eventually be used to establish T cell correlates of protection.
Inbred mouse models are commonly used to validate T cell function in vivo, even though they are only partially predictive of human responses. In recent years, efforts have been made to explore the potential use of human tissue organoids to study immune mechanisms. The Davis group has developed an ex vivo tonsil organoid system that recapitulates key germinal center events, including antigen-specific antibody production, somatic hypermutation and affinity maturation, plasmablast differentiation and class-switch recombination2. Using this system, they have defined the cellular components essential to mount an influenza vaccine response. Human organoid systems can be relatively easy to establish and allow for gene manipulation of different immune components, providing a more accurate analysis of the human immune response compared to some animal models. Efforts are underway to improve the human organoid systems, including co-culturing multiple human tissues to mimic the interactions between different immune niches.
Assays to assess T cell activation and function
A variety of methodologies are available to measure T cell responses, including interferon-γ release assay (IGRA), enzyme-linked immunosorbent spot (ELISpot) assay, intracellular cytokine staining (ICS), activation-induced marker (AIM) assay, multimers, TCR profiling, single-cell mRNA sequencing (scRNA-seq) and many others (Fig. 1). Overall, there is fundamental tension between comprehensiveness, complexity, depth of analysis and granularity of information with ease of use, robustness, throughput and affordability. IGRAs are the simplest but least informative tests. ELISpot assays are robust and relatively high throughput but have limited sensitivity and do not provide information on the phenotype of responding cells (CD4 and/or CD8) or other surface markers. ICS provides an opportunity to detect functional responses combined with phenotypic information. The AIM assay is the most sensitive, being agnostic of a particular cytokine functionality and allowing for the isolation of single cells for downstream analysis. AIM assays also can be combined with ICS to include functional measurements3.
Fig. 1 |. Measuring T cell responses using various technologies.
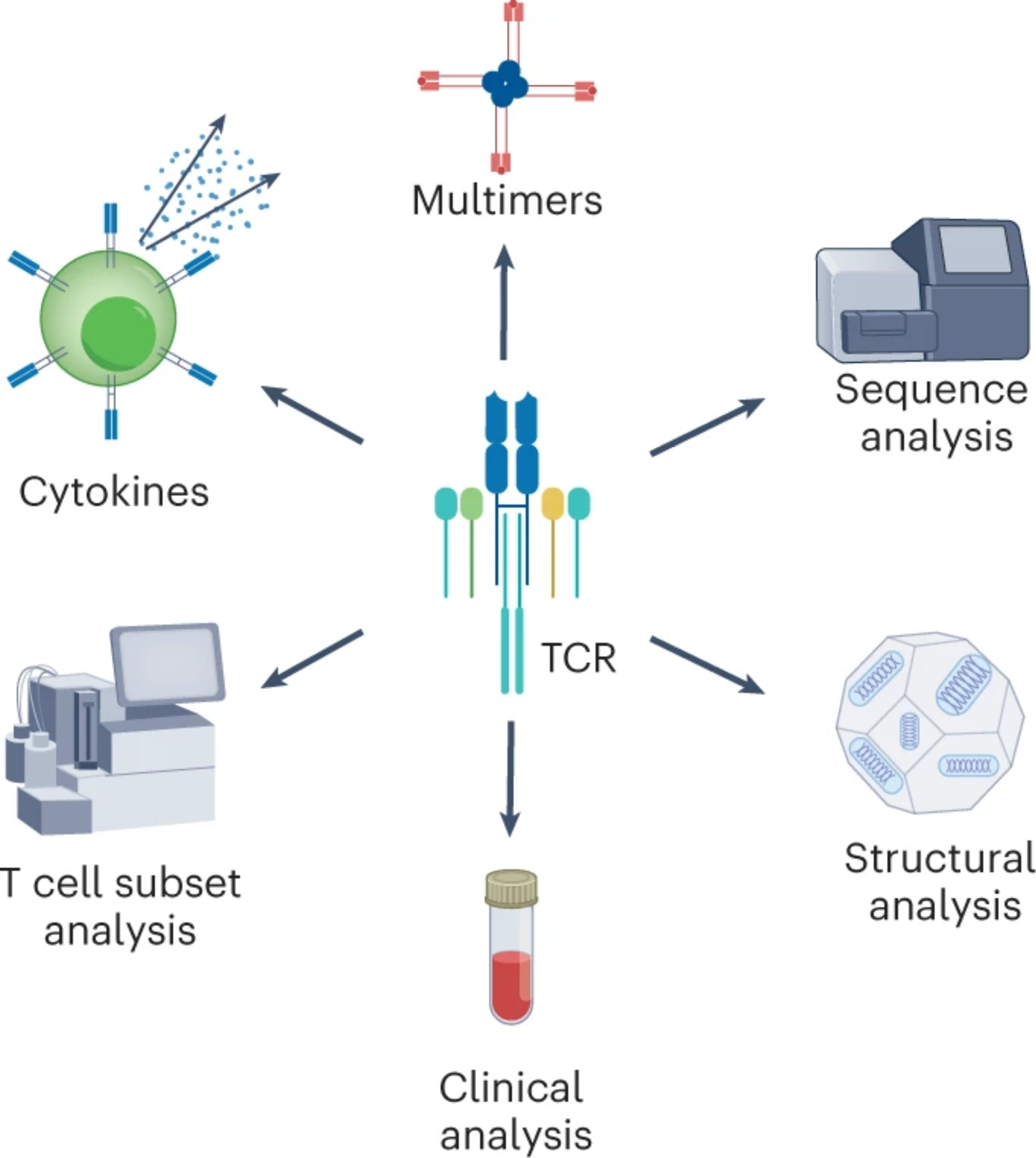
Assays have been developed and continuously evolved to detect, quantify and characterize T cell populations, defining antigen-specificity, activation status and functional significance. Incorporated with innovative computational tools, high-throughput, high-resolution and high-dimensional profiling of T cells has become a trend in basic research and large cohort clinical studies.
Ex vivo assays of fresh or frozen blood or peripheral blood mononuclear cells (PBMCs) are the most physiologic and are suited to combination with transcriptomics. In vitro restimulation formats involve expansion of antigen-specific T cells and detection of less abundant cell types. However, the longer assay time makes this format more laborious. In addition, certain cell types (for example, regulatory T cells) may not expand well, phenotypes may be altered and certain T cell specificities might preferentially expand, leading to underestimations of repertoire complexity. Alessandro Sette’s group has shown particular interest in capturing the full breadth of human T cell responses in an unbiased manner. With the recent SARS-CoV-2 pandemic, the ancestorial strain of SARS-CoV-2 and specific amino acid mutations that arose were used to generate peptides associated with the different variants. More than 2,000 SARS-CoV-2 human T cell epitopes have been experimentally validated4, providing a striking example of the complexity of human T cell responses. This complexity explains why, thus far, the majority of T cell responses are preserved in SARS-CoV-2 variants3.
The Sette laboratory has also exhibited interest in the impact of HLA polygeny and polymorphism on T cell detection. Because of heterozygosity, an individual can express up to 14 different HLA molecules (8 HLA class II and 6 HLA class I). The 10 HLA molecules most frequently expressed offer 95.3% coverage, when coverage is defined as the percentage of individuals expressing at least one of these HLAs. However, if coverage is defined as the percentage of total HLA genes expressed in the population, then the top 10 HLAs only account for 35.4% of the expressed HLAs. Furthermore, few allelic variants expressed in non-European or European Americans are often considered5. To address these issues, the Sette group developed detection methods based on pools of large numbers of peptides, produced by sequential lyophilization (MegaPool approach), that were selected as overlapping peptides spanning the antigen of interest, predicted epitopes from the comprehensive collection of HLA variants or experimentally defined epitopes. The use of overlapping peptides is most comprehensive but becomes expensive or unwieldy when large antigens or genomes are being analyzed. In those cases, use of predicted or experimentally defined epitopes allows for a reduction in the complexity of the antigen pools tested.
Also interested in understanding pMHC and TCR interactions, Michael Birnbaum focused on building new tools with high-throughput capacity. His group developed a yeast display platform that directly assesses pMHC binding in large collections of samples to identify peptide repertoires6. Birnbaum’s team showed that yeast-display-trained models improve the prediction of peptide-binding affinity for pathogen- and tumor-associated peptides. To enable high-throughput screening of antigen-specific TCRs, they developed ‘receptor–antigen pairing by targeted retroviruses’ (RAPTR), which match TCRs with their cognate antigens based on specific infection of TCR-expressing cells by antigen-displaying viruses7. This technology enables the screening of single or polyclonal TCRs and further identification by bulk or single-cell sequencing.
Although cell–cell interactions between T cells and other immune cells are essential for mounting an optimal immune response, these interactions are mostly transient, and monitoring their dynamics in vivo remains challenging. Gabriel Victora described the ‘labelling immune partnerships by sortagging intercellular contacts’ (LIPSTIC) technology, which allows direct measurement of T follicular helper (TFH) cell interactions with dendritic cells during T cell priming in vivo8. LIPSTIC is complementary to intravital microscopy, and it has the advantage of being able to isolate cell populations for downstream transcriptomic analysis. Victora’s group also recently developed a receptor-agnostic version of LIPSTIC to extend cell–cell labeling to non-hematopoietic lineages for broader applications.
The measurement of T cell responses has a variety of applications, including basic investigations of T cell biology, study of host–pathogen interactions and evaluation of different vaccine platforms. The specific application can inform the choice of assay, antigen and modality to be used. Donna Farber has adapted integrated T cell assays to study human T cell subsets in peripheral tissues using organ donor tissues. Phenotype profiling revealed that human T cell composition, tissue-resident memory T (TRM) cell frequency and age-associated changes are tissue site specific. Assessed by scRNA-seq, human TRM cells exhibit transcriptional changes and site-specific adaptation that enable their long-term maintenance in tissues. T cell clonal analysis showed that TRM cell clones are segregated in barrier sites and exhibit site-specific clonal expansion shared mostly with associated lymph node, but not blood9. These studies support the notion of site-specific targeting of TRM cells for monitoring and promoting immunity.
In another application, Cecilia Berin discussed food-allergen-specific T cell analyses in pediatric cohorts. Owing to the low blood volume and weak direct ex vivo T cell response in pediatric samples, allergen-responsive T cells are assessed mainly using conventional activation-marker-based detection following ex vivo stimulation of PBMCs. Frequencies of allergen-responsive CD154+CD4+ cells co-expressing interleukin-4 (IL-4) and/or IL-13 have been associated with allergen-specific immunoglobulin E (IgE) levels. Although these cells can be used to monitor the therapeutic efficacy of food allergy immunotherapy, a higher frequency of the cells at baseline was also associated with treatment failure. Several clinical studies (with smaller cohort sizes) also have used pMHC dextramers and scRNA-seq paired with TCR-seq to provide in-depth profiling of peanut-reactive T cells10. As emphasized by Berin, these high-dimensional analysis methods are a trend for future large cohort studies in food allergy research.
Evolving T cell technologies
Beginning with flow cytometry, immunologists have continuously innovated methods to enhance single-cell resolution methods. For T cells, the critical determinants of cell identity include the specificity of the TCR and the fate or functional state of the cell itself. Recent advances in scRNA-seq and high-dimensional cytometric profiling allow simultaneous assessment of TCR sequence, gene expression and expression of combinations of surface and intracellular proteins. These ever-larger datasets have necessitated the development of analytical approaches to define cell states in high-dimensional space and to translate the expansive diversity of TCR sequences into measures of specificity.
Alex Shalek addressed the problem of characterizing T cell states and their functional significance. Using natural and vaccine-induced control of tuberculosis as an example, he described how high-resolution profiling, such as scRNA-seq and single-cell TCR-seq, can be used to define distinct cell states and their molecular circuitry, as well as strategies to link these features with distinct disease outcomes and mechanisms underlying pathology11. Previous approaches had relied on relatively coarse-grained cell descriptions, often obscuring underlying variation due to an inability to simultaneously measure the necessary number of parameters. The work demonstrates that these ostensibly ‘subtle’ variations can describe fundamentally distinct pathophysiological processes that may be key to disease course and to the rational design of preventions and cures for acute and chronic conditions, including cancer, allergy, and pathogenic infections. Shalek’s talk also highlighted outstanding challenges in the field, such as clinical sample collection and hypothesis testing at scale, as well as emerging methods to tackle the challenges. Gaurav Gaiha addressed a similar question, describing a method to expand rare cell populations prior to the simultaneous assessment of specificity and function. The cytolytic function of CD8+ T cells, which had been expanded by stimulation with the dominant HIV response peptide identified by Gaiha and his group, was assessed to measure the ability of the cells to eliminate peptide-pulsed autologous target cells, thus allowing for the longitudinal examination of durable HIV control12. Maintaining the stability of the cell differentiation state throughout expansion allowed the group to measure killing and other key features of cell function. The expanded cell populations were also more tractably analyzed for specificity via stimulation or multimer staining.
The expansion of discrimination in cell fate has coincided with the development of numerous methods for sequencing the TCR itself, either in bulk samples or from single cells13. Bulk sequencing has the advantage of enormous depth and comprehensive characterization of even relatively large samples but is unable to provide paired αβ-chain information. By contrast, single-cell sequencing provides paired αβ-chain data but with substantially lower throughputs. Both approaches have massively expanded our TCR catalog, but currently no algorithm can directly translate sequence into specificity. Paul Thomas described a computational and empirical workflow called ‘reverse epitope discovery’ that relates condition-associated paired TCRs from single-cell data to HLA and antigen associations based on various bulk and single-cell functional assays. These combinatorial deconvolutions allow for rapid epitope specificity assignment of immunodominant public TCR clusters14. Harlan Robins presented a complementary approach based on bulk TCRαβ-seq to define condition-associated TCRs that can be used to generate exquisitely precise classifiers that distinguish infection history and pathologic states15. Individuals could be reliably diagnosed as having a history of SARS-CoV-2 infection based on TCR sequence alone, building on previous work from Robin’s group showing that such classifiers also could be constructed for chronic infections such as cytomegalovirus.
The ultimate goal of these approaches is the empirical deconvolution of the TCR repertoire, which remains elusive. The combination of ever-growing empirical datasets with advanced analytical techniques offers optimism that a hybrid empirical–analytical translation from sequence to specificity is possible, as described in Ramy Arnaout’s talk. Arnaout compared TCR-repertoire-wide measures of diversity, which are normally divorced from epitope-specific features of the repertoire, to empirically determined specificities. He identified features of repertoire-wide ‘class’ diversity and further exploited this analysis to identify divergent features of otherwise similarly diverse repertoires16. The diagnostic implications of these analyses have great potential for the generation of future correlates of risk and protection after infection or vaccination. Historically, such correlates have primarily been antibody based for infectious disease, but interrogating the specific contributions of cellular immunity to clinical outcomes will be key for improved therapeutic intervention.
To this end, Jun Huang discussed his work on engineered chimeric antigen receptor T (CAR T) cells17. These cells have engineered specificity but must still be identified and isolated for single-cell analyses, requiring the use of novel multimer-based reagents for isolation and analysis. These multimer-based reagents include dodecamers that have higher sensitivity and better specificity compared to other reagents. They can be utilized for in vivo detection and activation of CAR T cells in individuals with cancers17. Furthermore, the engineered receptor generates a new range of potential cell states that may not always map effectively onto the endogenous cell states defined in infectious or even tumor-associated responses.
Identifying pathogenic T cell populations in autoimmune disease
Assumptions made about the contributions of T helper cell subsets to the pathogenesis of autoimmune disease were challenged by Michael Brenner in his talk, which highlighted the need for an unbiased approach to assess the clinical significance of T cells in autoimmune and infectious diseases. Owing to MHC-II associations, autoimmune diseases such as rheumatoid arthritis were thought of as T helper type 1 (TH1)- and/or TH17-driven conditions. Further investigation has identified two major new populations of T cells that may serve as major contributors to inflammation seen in rheumatoid arthritis and several other autoimmune diseases.
Examination of the rheumatoid synovium by high-dimensional cytometry by time of flight (CyTOF) identified an expanded population of PD-1hi CXCR5neg CD4 cells that were transcriptionally distinct from TFH cells. These cells were designated as peripheral helper T (TPH) cells18. TPH cells produced cytokines that provided B cell help but were distinct from TFH cells in their lack of BCL-6 expression and high expression of Blimp-1 and peripheral homing receptors18. TPH cells also were found to be a dominant species in systemic lupus erythematous (SLE), comprising approximately 5% of the CD4+ cells in the blood and correlating with disease severity as determined by the SLE disease activity index — something that was not attributed to TFH cells. Since their identification in 2017, T cells with a TPH cell phenotype have been identified in a number of autoimmune-related diseases, including autoimmune hepatitis, celiac disease, primary biliary cholangitis, Sjögren’s syndrome, type 1 diabetes, ulcerative colitis, IgA nephropathy and juvenile arthritis. These observations demonstrate that TPH cells may be important across many different autoimmune conditions where autoantibodies have an important role in disease progression or overall pathogenesis.
Another surprising clinical finding from the analysis of T cells in the synovium of rheumatoid arthritis patients is the identification of a high abundance of CD8+ T cells. These cells produce interferon-γ and express granzyme K, suggesting that rheumatoid arthritis may also be a disease of CD8+ cells19. These unique CD8+ cells are also a dominant species in Crohn’s disease and ulcerative colitis, and represent a significant population in SLE19. Importantly, this novel CD8+ T cell population is also seen in infectious disease and was recently identified in the bronchoalveolar lavage of patients with COVID pneumonia19.
Clinical T cell functional analysis
Three presentations on clinical T cell analyses covered topics on local immunity, vaccination, transplantation and transgenic T cell therapy, and described the use of TCR-seq to discover or track specific T cells populations. David Koelle discussed the characterization of TCRs recovered from biopsies after resolution of infection using co-cultivated autologous herpes-simplex-virus-2-fed monocyte-derived dendritic cells, with the goal of understanding the polyclonal swarm of CD8+ T cell TCRs used to recognize the proteome encoded by a large-genome viral pathogen. Koelle also employed an in vitro method to delete viral HLA class-I immune-evasion genes and boost signals from TCR-transduced reporter cells. The fine specificity of reactive TCRs were then defined using virus-covering open reading frame libraries and panels of artificial antigen-presenting cells expressing subject-specific HLA20.
Aude Chapuis reviewed the differential proteasome subunit expression seen in tumors and how these changes can modulate tumor antigen processing, leading to suppressed peptide presentation and immune escape from tumor-specific TCR recognition. Tumor-associated proteins often include an array of peptides for a given HLA restriction. Therefore, careful consideration of target peptides that are not influenced, or are less influenced, by modulations of the peptide-processing machinery should be taken in account before selecting TCRs for clinical translation21. Most naturally occurring tumor-targeting TCRs are HLA class-I restricted and confer functionality to CD8+ T cells, but only a few bind peptide-HLA at an affinity high enough to be independent of CD8 co-receptor engagement to induce functional CD4+ T cell responses. Co-delivery of CD8+ T cells and therapeutic TCRs is currently being pursued as a way to provide the beneficial proliferative and survival advantages of antigen-specific CD4+ T cells to transgenic TCR cell products21. Despite these enhancements, it is anticipated that transgenic T cell infiltration of solid tumors will remain a challenge. A variety of strategies are being developed to overcome the negative factors associated with the tumor environment. These strategies include synthetic constructs that convert negative tumor-associated extracellular signals into positive intracellular T cell signals, abrogating the effects of the negative signal or directly triggering a positive co-receptor signal following TCR engagement.
Research advances in CAR T cell optimization were described by Joseph Fraietta. The use of unbiased high-dimensional flow and RNA-seq systems approaches to examine the expression profile of CAR-T-cell-responding patients showed that those expressing higher frequencies of CD8+PD-1−CD27+ T cells at the pre-manufacture or apheresis stage of CAR T cell generation were associated with improved responses to this adoptive cell therapy22. TCR tracking of polyclonal CAR T cell products revealed a dramatic repertoire change over time that was associated with clinical response. Current research focuses on solid tumor CAR T cell treatment to reduce TGFβ sensitivity of the infused cell23.
The potentially harmful roles of donor-specific alloreactive T cells in solid organ transplantation highlight the need to monitor both the magnitude and character of alloreactive T cell responses during transplantation. One critical unmet need is the ability to better identify the risk of sensitization, or the presence of donor-reactive memory T cell responses, prior to transplantation to improve survival of HLA-mismatched transplanted organs. Current practice avoids organ transplantation for which the recipient possesses donor-specific antibodies, but the magnitude and quality of the donor-specific T cell repertoire is not measured as part of clinical practice and is likely to impact the risk of rejection as well as the requirement for immunosuppression following transplantation. Mandy Ford reviewed specific T cell phenotypes associated with increased risk of transplant rejection, including highly differentiated CD4+CD57+ PD-1 cells and reduced CD8+ T cell expression of the FcγRIIB co-inhibitory receptor that results in reduced T cell apoptosis24. Megan Sykes discussed recent advances in the development of tools to identify the donor-reactive TCR repertoire via a pre-transplant in vitro mixed lymphocyte reaction followed by TCR-seq of the responding cells. Findings from these studies provide new insights, including demonstration that: (1) the alloimmune repertoire is highly specific for a given donor–recipient pair; (2) most alloreactive T cell clones are present at low frequencies; and (3) many TCRs are capable of recognizing alloantigens. Her team also used TCR-seq to identify alloreactive TCRs among single cells in intestinal allografts and recipient bone marrow, enabling determination of the functional phenotype of defined alloreactive T cell clones in situ. Moreover, combined use of TCR-seq and scRNA-seq has enabled the discovery of TCR sequences and clonotype enrichment and/or deletion associated with clinical outcomes such as transplant rejection and tolerance25. Such strategies could potentially be used to modulate the amount of immunosuppression in individual transplant recipients.
Conclusion and recommendations
There have been substantial advances in our ability to understand T cell sensitivity and specificity. These advances have largely been technology-driven with the ability to combine high-throughput scRNA-seq, paired TCRαβ-seq and high-dimensional flow cytometric profiling. Analytic tools and approaches have created pathways to translate these expansive datasets into TCR repertoires that are associated with infectious diseases, vaccination, allergy, autoimmunity and cancer.
High-quality data paired with metainformation on the clinical state of the individual from whom the sample was obtained may be critical in driving the T-cell-sequencing space to its next phase of being predictive of clinical diagnoses and clinical outcomes. Large-scale collection of T cell sequences has proven somewhat effective at predicting T cell function but has not led to an understanding of the ternary structures of the TCRs. An in-depth analysis of TCR structure across the full structural space, combined with TCR sequence and immune response data, is needed. This will help define what structures are biologically possible in the context of an immune response and will enable scientists to look at algorithm-predicted structures and create linkages with immunogen structures. A TCR structure project where biologically validated high-quality structures are assembled and made accessible could address this need. Incorporation of data across the immunological space is also critical. A TCR structure project would ideally incorporate user- and academically friendly informatics tools to help generate hypotheses that could be validated and fed back into a growing database.
Novel technological innovations in the epitope discovery space will increase our understanding of T cell specificity and antigen immunogenicity. There is need for enhanced prediction tools and more experimental epitope data for large genome parasites, bacteria and allergens. Additional data in these areas will allow for tetramer and multimer generation for the identification of antigen-specific T cells and increased understanding of T cell correlates of protection.
To address gaps identified in this workshop, an intentional strategy must be adopted to incorporate subject sample acquisition at important timepoints and from clinically relevant sites, with immunologically validated structure and sequence data. The scientific community will require access to user-friendly tools and datasets, with the ability to add their own validated data to a comprehensive and curated resource. Use of new technologies, foundational methods, cross-disciplinary ideas and data across the immune space will lead to the generation of novel hypotheses and discoveries and produce essential correlates of T cell immune protection.
Acknowledgements
We thank the organizing committee, workshop participants and keynote speakers for their expertise and insights on T cell technologies. Workshop organizers, participants and keynote speakers include: R. Arnaout, M. Cecilia Berin, M. Birnbaum, M. Brenner, A. Chapuis, W. Davidson, M. Davis, D. Farber, J. A. Fraietta, G. Gaiha, J. Huang, M. Jenkins, P. Kehn, M. Morsheimer, J. Peyman, J. Rice, H. Robins, M. Sykes and G. Victora.
Footnotes
Competing interests
D.M.K. has received research funding from Sanofi Pasteur and Sensei; is on the scientific advisory boards of two companies with candidate vesicular stomatitis virus vaccines, Curevo (US) and MaxHealth LLC (China); and is co-inventor on University of Washington or University of Washington–Fred Hutchinson co-managed awarded patents on candidate herpes simplex virus vaccines. A.S. is a consultant for Gritstone Bio, Flow Pharma, Moderna, AstraZeneca, Qiagen, Fortress, Gilead, Sanofi, Merck, RiverVest, MedaCorp, Turnstone, NA Vaccine Institute, Emervax, Gerson Lehrman Group and Guggenheim. L.J.I. has filed for patent protection on various aspects of T cell epitope and vaccine design work. A.K.S. reports compensation for consulting and/or scientific advisory board membership from Merck, Honeycomb Biotechnologies, Cellarity, Repertoire Immune Medicines, Hovione, Third Rock Ventures, Ochre Bio, FL82, Empress Therapeutics, Relation Therapeutics, Senda Biosciences, Santa Ana Bio, IntrECate biotherapeutics and Dahlia Biosciences unrelated to this work. A.K.S. has received research support from Merck, Novartis, Leo Pharma, Janssen, the Bill and Melinda Gates Foundation, the Moore Foundation, the National Institutes of Health, the Moore Foundation, Wellcome Leap, the Pew-Stewart Trust, Foundation MIT, the Chan Zuckerberg Initiative, Novo Nordisk and the Food and Drug Administration unrelated to this work. P.G.T. has consulted and/or received honoraria and travel support from Illumina, Johnson and Johnson and 10X Genomics; serves on the scientific advisory boards of Immunoscape and Cytoagents; and has been awarded or applied for patents related to these topics.
References
- 1.Mallajosyula V et al. Sci. Immunol 6, eabg5669 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wagar LE et al. Nat. Med 27, 125–135 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tarke A et al. Cell 185, 847–859 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grifoni A et al. Cell Host Microbe 29, 1076–1092 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hensen L et al. Front. Immunol 13, 812393 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rappazzo CG, Huisman BD & Birnbaum ME Nat. Commun 11, 4414 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dobson CS et al. Nat. Methods 19, 449–460 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pasqual G et al. Nature 553, 496–500 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miron M et al. Genome Med 13, 100 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monian B et al. J. Clin. Invest 132, e150634 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gideon HP et al. Immunity 55, 827–846 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collins DR et al. Immunity 54, 2372–2384 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pai JA & Satpathy AT Nat. Methods 18, 881–892 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pogorelyy MV et al. Cell Rep. Med 3, 100697 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Snyder TM et al. Preprint at medRxiv 10.1101/2020.07.31.20165647 (2020). [DOI] [Google Scholar]
- 16.Arora R & Arnaout R Proc. Natl Acad. Sci. USA 119, e2203505119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu Y et al. Matter 4, 3917–3940 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rao DA et al. Nature 542, 110–114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jonsson AH et al. Sci. Transl. Med 14, eabo0686 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng T et al. Front. Immunol 12, 735643 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lahman MC et al. Sci. Transl. Med 14, eabg8070 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fraietta JA et al. Nat. Med 24, 563–571 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Narayan V et al. Nat. Med 28, 724–734 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morris AB et al. Immunity 52, 136–150 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zuber J et al. Sci. Immunol 1, eaah3732 (2016).28239678 [Google Scholar]