

Abstract
The aggregation of amyloid fibrils can lead to various diseases including Alzheimer’s, Parkinson’s disease, and transmissible spongiform encephalopathy. Amyloid fibrils can develop from a variety of proteins in the body as they misfold into a primarily β sheet structure and aggregate. Human lysozyme has been shown to have far reaching effects in human health—a homologous enzyme, hen egg white lysozyme (HEWL), has been shown to denature to a primarily β sheet structure at low pH and high alcohol content solution. We have studied these systems in atomic-level detail with a combination of constant pH and μ-second long molecular dynamics simulations in explicit solvent, which cumulatively total over 10 μs of simulation time. These studies have allowed us to determine two potential unfolding pathways depending on the protonation state of a key glutamic acid residue, as well as the effect of solution dynamics and pH on the unfolding process.
Graphical Abstract
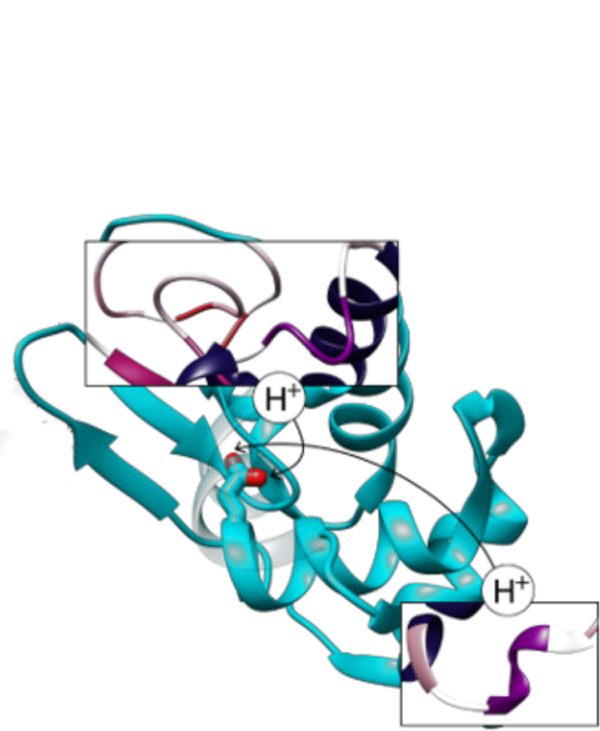
Introduction
Protein structural conformations and folding pathways are of great interest for their role in proper protein function and, often, malfunction in disease states. The reformation and aggregation of proteins in the body can potentially cause large-scale physiological consequences, and have been implicated in Alzheimer’s, Parkinson’s disease, and transmissible spongiform encephalopathy.1, 2 These diseases have been connected with the formation of large plaques structurally dominated by β sheets, such as amyloid fibrils in Alzheimer’s disease. Amyloid fibrils can develop from a variety of proteins in the body, including human lysozyme, as they misfold into a primarily β sheet structure, which has been linked to amyloidosis in particular.3, 4 While this phenomenon and the aggregation of amyloid plaques in the body is well known5, there is still a great deal of interest in the unfolding mechanism and refolding process due to the large number of proteins that can display this morphology. Additionally, further insight into partially unfolded and mostly native protein states has been of interest due to the possibility of protein-rich clusters as precursors for amyloid fibrils, among other aggregates.6
Hen egg white lysozyme (HEWL) is homologous to human lysozyme, and provides a convenient proxy to study the folding processes of amyloid plaque-forming enzymes.7 Lysozyme in particular has been implicated in genetic amyloidosis and has been shown to form amyloid fibrils in vivo under a variety of experimental conditions, including heat, pH adjustment and type of solvent.8–11 These results have shown evidence for multiple types of unfolding pathways for lysozyme, including two-step folding, passing through an intermediate transition state, and molten globule intermediates, among others.12–14
The unfolding/refolding/misfolding of proteins involve processes that can span a large range of time-scales, from microseconds to hours. These time-scales are not achievable by conventional all-atom classical molecular dynamics (MD) simulations. However, MD simulations can provide insights on possible states and likely pathways.15–19
Here, we present a theoretical study of HEWL unfolding using constant pH and multi-microsecond (μs) long MD simulations to probe the initial steps of the unfolding transition. We particularly focus on a low pH system with a high concentration of ethanol, which has been shown by circular dichroism spectroscopy to favor a β sheet-rich structure at these conditions (90% ethanol, pH 4, disulfide bonds reduced). 20 We have explored the initial unfolding landscape of lysozyme with MD by investigating a set of HEWL systems at constant pH, determining the most likely protonation, and using those states as a starting point for 5–6 μs MD simulations on Anton.21 Our simulations indicate that the protonation state of Glu35, while rotationally degenerate, results in two different unfolding pathways. The importance of proton exchange via constant pH simulations has been shown several times in the literature for unfolding and dynamics in MD, though we are not aware of other work that shows differences between rotationally degenerate protonation states with regard to unfolding, likely because constant pH simulations are a relatively new avenue of research.18, 22–24 Our results are consistent with previous literature reports, and help provide insights on the potential role of pH on the initial unfolding process of HEWL.
Methods
An initial crystal structure (pdbid 1YKZ) was chosen to be consistent with the high alcohol content of the experimental work.25 The structure was checked and hydrogenated using MolProbity.26 Six initial systems were created to study the impact and potential order of disulfide bond breakage—one system with all the disulfide bonds present, one with none, and four more with one disulfide bond present in each. These systems were employed to perform constant pH simulations in pure water for 100 ns to determine likely protonation sites. Three pH values were considered: 4, 6.8, and 9. The results at pH 4 were used to create two systems to investigate the two different protonation states for Glu35. The surrounding solvent was replaced with a 90% ethanol/10% TIP3P water mixture.27 Ethanol parameters were generated using R.E.S.P28 fitting for the partial charges and GAFF for the geometric parameters.29 These two systems were simulated for 4.5 μs each on the Anton server.21 In total, about ~13 μs of production simulation was performed.
All systems were run with the AMBER ff99SB force field using a 2 fs timestep.30 The constant pH and equilibration MD simulations were run using AMBER’s pmemd.cuda program.31, 32 sPME was used for long-range electrostatics, with an 8 Å cutoff for non-bonded interactions.33 All covalent hydrogen bonds were restrained using SHAKE.34 These simulations were performed in the NVT ensemble at 300 K with Langevin dynamics, after a slow heating at constant volume and solvent equilibration at constant pressure.35 The collision frequency for Langevin dynamics was set to 5 ps−1. For the constant pH simulations in water, AMBER’s constant pH in explicit solvent Monte Carlo module was used with one protonation state change attempted every 100 steps and 100 solvent relaxation steps performed following successful pH changes.
Anton requires slightly different parameters from the initial production due to limitations and speed requirements. For the two Anton simulations, the long range electrostatics were treated with Gaussian split Ewald.36 As recommended for Anton simulations, the multigrator was used with the NoseHoover thermostat and a relaxation time of 1 fs, with the MTK barostat and a relaxation time of 10 fs.37
Results and Discussion
Molecular dynamics (MD) simulations were performed for a series of six systems in explicit solvent. A representative structure of HEWL can be seen in Figure 1.
Figure 1.
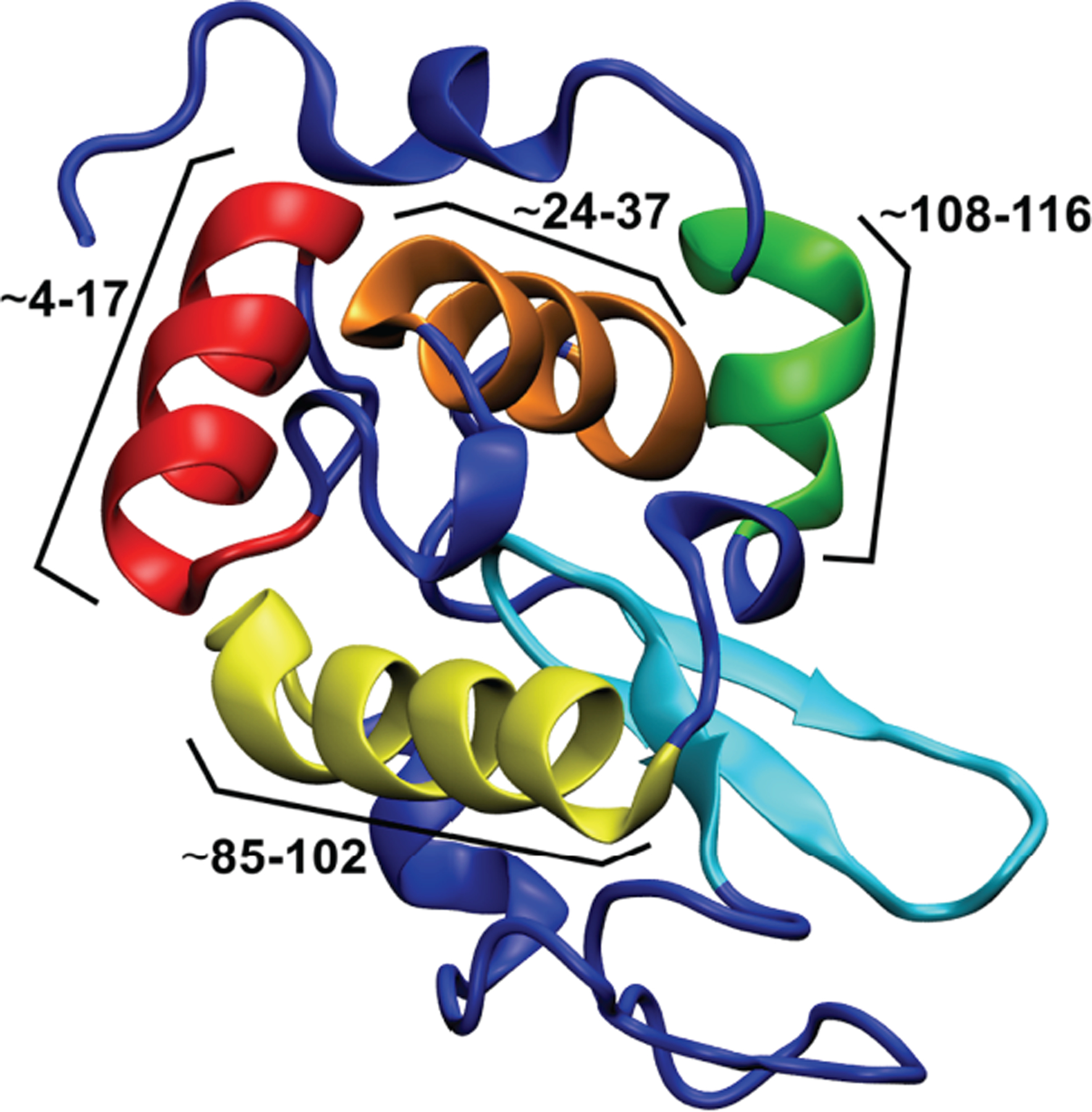
Representative structure for HEWL in its native conformation, with unstructured loops and α turns colored in dark blue, the β sheet section colored in cyan, and the large α helices colored and labeled according to their residue ranges in red (~4–17), orange (~24–37), yellow (~85–102), and green (~108–116), respectively.
There are four major α helix secondary structure areas and one β sheet in the native conformation, along with four disulfide bonds that stabilize the native conformation. Constant pH simulations were performed for each system in pure water to determine the most likely protonation states at the experimental pHs. We make this approximation because, currently, there is not a straightforward way of simulating an ethanol-water mixture at constant pH. All standard MD simulations were performed in a solution of 90% ethanol. All of the simulations were performed using AMBER (see Methods section).32 The systems considered include one native system with all four disulfide bonds, four with one disulfide bond each (three reduced disulfides), and one with no disulfide bonds. These were chosen to reflect the experimental result that, during the α/β transition, 0–1 disulfide bonds were observed in the mass spectra.38 All six systems are represented schematically in Figure 2.
Figure 2.
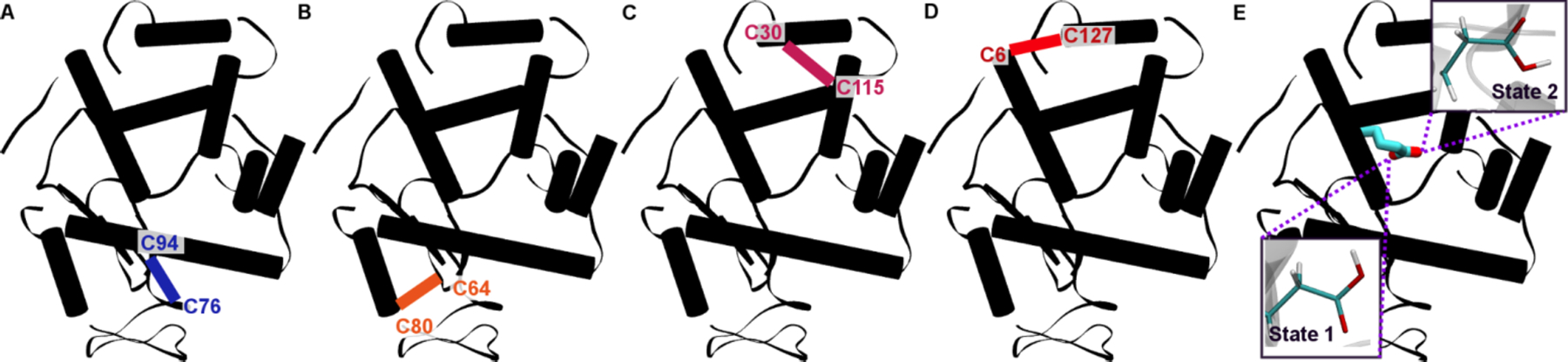
A schematic representation of the non-native subsystems created for the MD simulations. (A-D) show the relevant single disulfide bond maintained for each subsystem colored in blue (C94-C76), orange (C80-C64), pink (C30-C115), and red (C6-C127), respectively, with the labels denoting the cysteine residues involved in the disulfide bond. (E) shows the two sub-states based on pH and with all reduced disulfide bonds, with the rotationally degenerate GLU35 highlighted in purple for State 1 and State 2.
After an initial relaxation and 100 ns of constant pH simulation, the systems were examined to determine potential protonation states at a pH of 4, 6.8, and 9 as per experimental conditions. At a pH of 4, corresponding to the pH of the solution to induce unfolding of the protein, a rotationally degenerate glutamic acid that can be protonated on either of the two oxygens was observed (Figure 2E, Table S1). Lysozyme’s titratable residues are well-established in the literature at low pH, and most of the titratable residues were static in all pH 4 systems. One exception was the rotationally degenerate Glu35, for which protonation was present on one side or the other for ~50% of the simulation time (Table S1).39 This Glu is both a known active site residue for hen egg white lysozyme and a participant in the catalytic mechanism by activating proton transfer.40, 41 Thus, we hypothesize that it is also protonated in 90% ethanol at low pH. Asp18 and Asp101 were the only other titrated residues that showed dynamic protonation changes, though less so than Glu35 (Table S1). They were not considered further since their sidechains extend into solvent and are not involved in hydrogen bonding networks within the protein. We also determined that HEWL at pH 4 has a charge state of +11.
The calculated +11 states were used to create two sub-systems, State 1 and State 2 (Figure 2E), with all disulfide bonds broken and Glu35 protonated either cis (State 1) or trans (State 2). State 1 and State 2 were simulated with long timescale conventional MD simulations on the supercomputer Anton,21 totaling over 10 μs of simulation time (~5 μs per system). These systems, along with the single disulfide bond systems and the original wildtype structure, were compared to investigate the structural aspects of HEWL unfolding. The sulfur-sulfur distances of the reduced disulfides show similar distances in each of the systems studied. This was also consistent with the measured disulfide distances for the long timescale simulations on Anton, which are shown for both State 1 and State 2 in Figure 3. Regardless of whether or not the relevant disulfide bonds are present, the C76-C94 sulfurs remain structurally very close (less than 4Å), followed by the C64-C80 distance, which fluctuates slightly more but remains fairly constant at 5–6 Å. This result does not account for the possibility of reformation and cleavage, since these are classical MD simulations. However, it does correlate with the previous literature reports indicating that 0–1 disulfide bonds remain after unfolding42, and indicate that the C76-C94 disulfide bond breakage is most likely to reform easily. The C30-C115 distance shows relatively large fluctuations over the course of the simulation and distances of ~10–15 Å, and the C6-C127 shows both the highest fluctuation and distance reaching from 20–25 Å. This suggests that the majority of the unfolding from the reduction of the disulfide bonds comes from these two bonds in particular.
Figure 3.
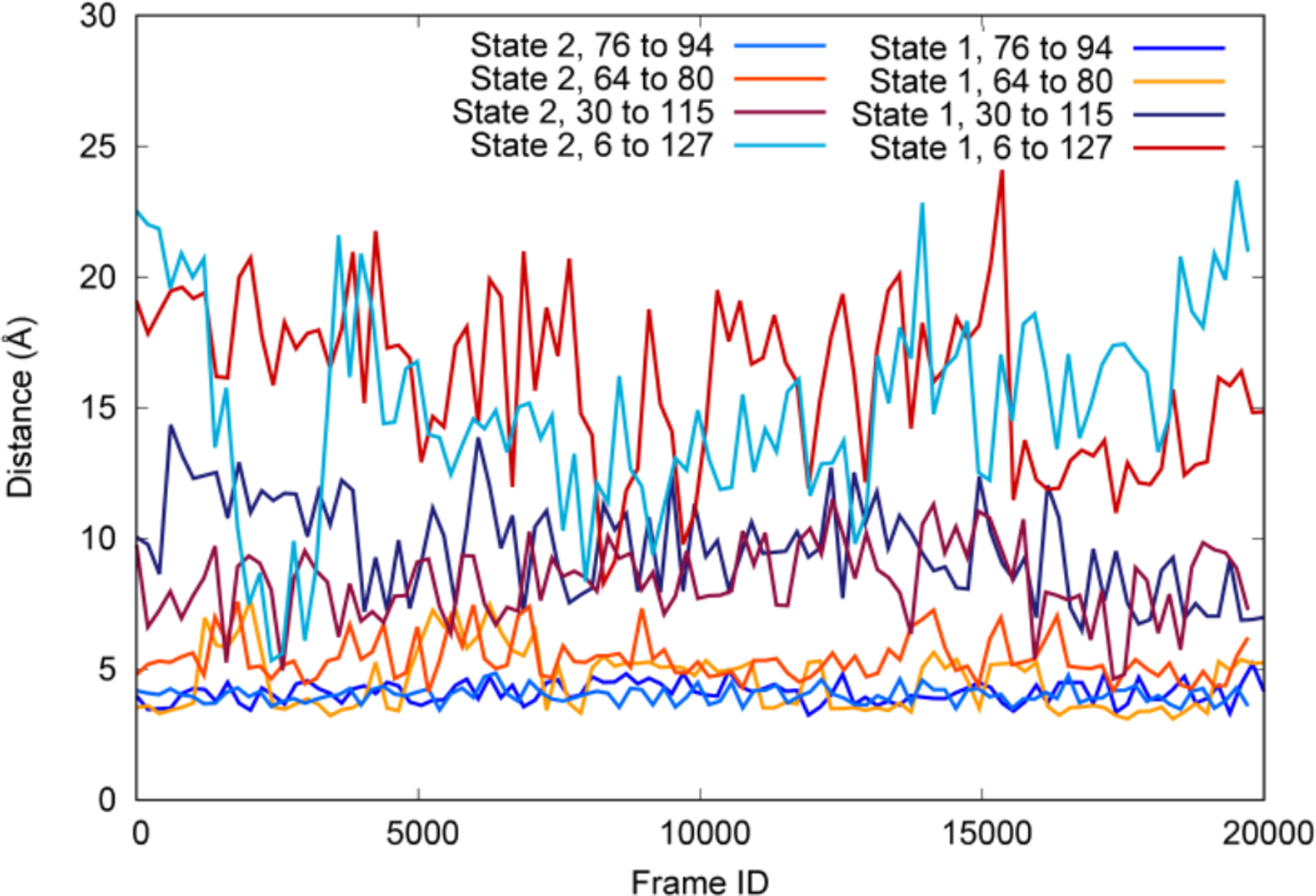
Distance between sulfur atoms for the four reduced disulfide bonds in the long timescale MD simulations. These distances are relatively consistent over time and between the two systems despite other structural differences between them, indicating relative importance to the unfolding process.
Cluster analysis was performed on the long timescale simulations for State 1 and State 2. Further details of this analysis can be found in the Supporting Information (Figure S1). The cluster analysis indicates that while the disulfide distances are similar between systems, their relationship to the root mean square deviation (RMSD) of the overall structure is quite different. This further supports that States 1 and 2 proceed by different unfolding pathways, despite having similar structures.
Additionally, the MD structures remain in largely the same shape compared to the native structure. Rather than the α helices completely unwinding and then reforming, small parts of the α helices denature and begin extending the originally small β sheet structure in native HEWL. The secondary structure nature of each residue was calculated over time for each MD trajectory, the full results of which can be seen in Figure S2, showing that the α helices unwind and denature slowly into structures with a higher β sheet character. Interestingly, the different protonation states lead to similar overall levels of transition over time but in entirely different parts of the lysozyme’s structure sequence depending on the presence or absence of particular hydrogen bonds to this GLU residue, as can be seen in Table 1 and Figure 4. In State 1, the hydrogen bonding between GLU35 and ARG114 is absent, thus causing a central portion of the ~108–116 helix to denature into an unstructured loop and a turn, as can be seen in Figure 4A, highlighted in orange. Our results suggest that this effect also seems to destabilize the subsequent ~85–102 helix and cause it to unravel by a turn at either end and feed into the β sheet structure. By contrast, the ~108–116 helix is highly stable in State 2, but there is a great deal more transition from α helix to unstructured loop, bend and turn character in the ~4–17 and ~24–37 helices (Figure 4B). This is consistent with the loss of hydrogen bonding with ALA31 in State 2 as compared to State 1. Taken together, these results indicate that degenerate protonation states may have a strong effect on the unfolding process and support the observation of two unfolding pathways based on the protonation of this particular Glu35 residue.
Table 1:
Calculated Hydrogen bonding for GLU 35
| Acceptor | Donor | State 1 % present | State 2 % present |
|---|---|---|---|
| Ala31 | Glu35 | 76.4% | 50.0% |
| Glu35 | Gln57 | 4.4% | 21.46% |
| Glu35 | Arg114 | -- | 100.0% |
| Phe34 | Glu35 | -- | 32.3% |
| Lys33 | Glu35 | -- | 3.5% |
| Ala32 | Glu35 | -- | 0.5% |
| Leu56 | Glu35 | 2.62% | 0.5% |
Figure 4.
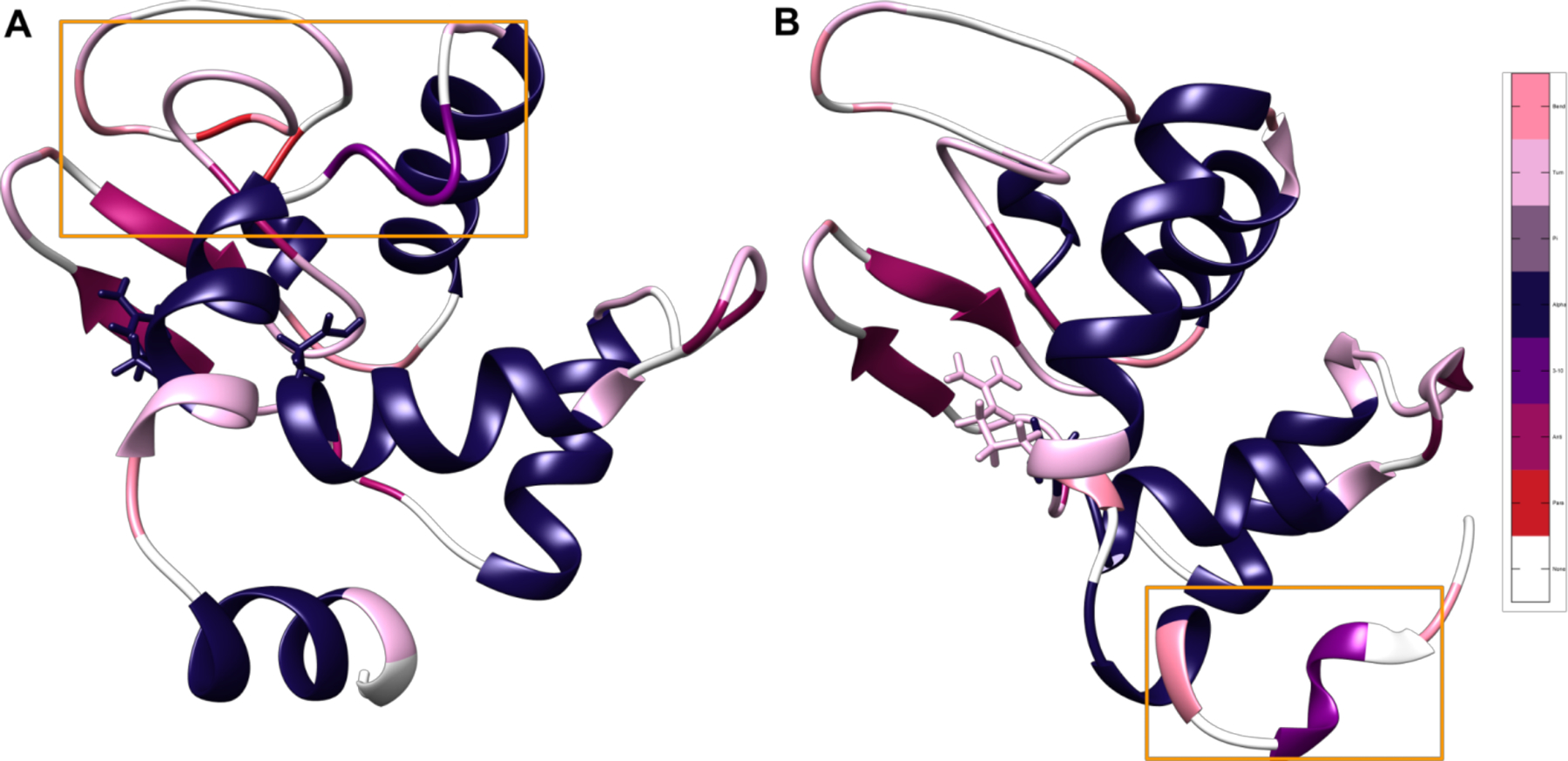
Most frequently occurring structures in the cluster corresponding to the distance between the broken disulfide bond between residues 6 and 127. Representative structures for the two protonation states, colored by the most common secondary structure after the 5 μs mark, with salmon representing bend character, pink representing turn character, gray representing pi helix character, navy blue representing α helix character, purple representing 3–10 helix character, mauve representing antiparallel β sheet, red representing parallel β sheet, and white representing unstructured loops. Glu35 and its corresponding Arg114 hydrogen bonding partner are displayed in licorice. Areas that are particularly different between states are highlighted with orange boxes. The difference in location of unfolding indicates the presence of two divergent unfolding pathways based on protonation state and subsequent hydrogen bonding. (A) State 1 with all disulfide bonds broken and the Glu35 hydrogen on OE1. (B) State 2 with all disulfide bonds broken and the Glu35 hydrogen on OE2.
CONCLUSION
A MD study has been performed on HEWL to investigate the initial states of misfolding. The constant-pH MD simulations suggest that GLU35 has different ionization states. Subsequent multi-μs MD simulations on the two possible rotationally degenerate ionization states indicate that, although the overall structure exhibits similar shapes experimentally and theoretically, the α/β transition begins on different sites. This difference depends on the relevant hydrogen bonding network from the protonation sites. These results have uncovered strong evidence for multiple unfolding pathways related to degenerate protonation states, and further explored the relationship between the reduction of disulfide bonds and relevant unfolding pathways.
Supplementary Material
ACKNOWLEDGMENT
Anton computer time was provided by the Pittsburgh Supercomputing center (PSC) through Grant R01GM116961 from the National Institutes of Health and the Anton machine at PSC was generously made available by D.E. Shaw Research.
ABBREVIATIONS
- HEWL
hen egg white lysozyme
- MD
molecular dynamics
- EtOH
ethanol
- RMSD
root mean square deviation
- WT
wild type
Footnotes
Publisher's Disclaimer: “Just Accepted” manuscripts have been peer-reviewed and accepted for publication. They are posted online prior to technical editing, formatting for publication and author proofing. The American Chemical Society provides “Just Accepted” as a service to the research community to expedite the dissemination of scientific material as soon as possible after acceptance. “Just Accepted” manuscripts appear in full in PDF format accompanied by an HTML abstract. “Just Accepted” manuscripts have been fully peer reviewed, but should not be considered the official version of record. They are citable by the Digital Object Identifier (DOI®). “Just Accepted” is an optional service offered to authors. Therefore, the “Just Accepted” Web site may not include all articles that will be published in the journal. After a manuscript is technically edited and formatted, it will be removed from the “Just Accepted” Web site and published as an ASAP article. Note that technical editing may introduce minor changes to the manuscript text and/or graphics which could affect content, and all legal disclaimers and ethical guidelines that apply to the journal pertain. ACS cannot be held responsible for errors or consequences arising from the use of information contained in these “Just Accepted” manuscripts.
ASSOCIATED CONTENT
Theoretical cluster analysis, theoretical time resolved secondary structure character, input files for molecular dynamics simulations. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Rambaran RN; Serpell LC Amyloid fibrils: Abnormal protein assembly. Prion 2008, 2 (3), 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nazabal A; Maddelein M-L; Bonneu M; Saupe SJ; Schmitter J-M Probing the structure of the infectious amyloid form of the prion-forming domain of HET-s using high resolution hydrogen/deuterium exchange monitored by mass spectrometry. J. Biol. Chem 2005, 280 (14), 13220–13228. [DOI] [PubMed] [Google Scholar]
- 3.Booth DR; Sunde M; Bellotti V; Robinson CV; Hutchinson WL; Fraser PE; Hawkins PN; Dobson CM; Radford SE; Blake CCF; Pepys MB Instability, unfolding and aggregation of human lysozyme variants underlying amyloid fibrillogenesis. Nature 1997, 385 (6619), 787–793. [DOI] [PubMed] [Google Scholar]
- 4.Buell AK; Dhulesia A; Mossuto MF; Cremades N; Kumita JR; Dumoulin M; Welland ME; Knowles TPJ; Salvatella X; Dobson CM Population of nonnative states of lysozyme variants drives amyloid fibril formation. J. Am. Chem. Soc 2011, 133 (20), 7737–7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dobson CM Principles of protein folding, misfolding and aggregation. Semin. Cell Dev. Biol 2004, 15 (1), 3–16. [DOI] [PubMed] [Google Scholar]
- 6.Safari MS; Byington MC; Conrad JC; Vekilov PG Polymorphism of lysozyme condensates. J. Phys. Chem. B 2017, 121 (39), 9091–9101. [DOI] [PubMed] [Google Scholar]
- 7.Burnett LC; Burnett BJ; Li B; Durrance ST; Xu S, A. Lysozyme concentration pH, and time-dependent isothermal transformation diagram reveals fibrous amyloid and non-fibrous, amorphous aggregate species. Open J. Biophys 2014, 04 (02), 39–50. [Google Scholar]
- 8.Meersman F; Atilgan C; Miles AJ; Bader R; Shang W; Matagne A; Wallace BA; Koch MHJ Consistent picture of the reversible thermal unfolding of hen egg-white lysozyme from experiment and molecular dynamics. Biophys. J 2015, 99 (7), 2255–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sabulai B Thermal unfolding of hen egg-white lysozyme in the presence of 4-chlorobutan. Pure & Appl. Chem 1998, 70 (3), 665–670. [Google Scholar]
- 10.Haezebrouck P; Joniau M; Van Dael H; Hooke SD; Woodruff ND; Dobson CM An equilibrium partially folded state of human lysozyme at low pH. J. Mol. Bio 1995, 246 (3), 382–387. [DOI] [PubMed] [Google Scholar]
- 11.Bonincontro A; Cinelli S; Comaschi T; Onori G Influence of urea on thermal denaturation of lysozyme investigated by optical and dielectric spectroscopies. Phys. Chem. Chem. Phys 2004, 6 (5), 1039–1042. [Google Scholar]
- 12.Ibarra-Molero B; Sanchez-Ruiz JM Are there equilibrium intermediate states in the urea-induced unfolding of hen egg-white lysozyme? J. Phys. Chem. B 1997, 36 (31), 9616–9624. [DOI] [PubMed] [Google Scholar]
- 13.Hameed M; Ahmad B; Fazili KM; Andrabi K; Hasan Khan R Different molten globule-like folding intermediates of hen egg white lysozyme induced by high pH and tertiary butanol. J. Biochem 2007, 141 (4), 573–583. [DOI] [PubMed] [Google Scholar]
- 14.Carra JH; Murphy EC; Privalov PL Thermodynamic effects of mutations on the denaturation of T4 Lysozyme. Biophys. J 1996, 71 (4), 1994–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Babin V; Roland C; Darden TA; Sagui C The free energy landscape of small peptides as obtained from metadynamics with umbrella sampling corrections. J. Chem. Phys 2006, 125, 204909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krishna KA; Rao GV; Rao K Chaperonin GroEL: structure and reaction cycle. Curr. Protein Pep. Sci 2007, 8 (5), 418–425. [DOI] [PubMed] [Google Scholar]
- 17.Williams SL; de Oliveira CAF; McCammon JA Coupling constant pH molecular dynamics with accelerated molecular dynamics. J. Chem. Theory Comput 2010, 6 (2), 560–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goh GB; Laricheva EN; Brooks CL Uncovering pH-dependent transient states of proteins with buried ionizable residues. J. Am. Chem. Soc 2014, 136 (24), 8496–8499.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garrison BJ; Postawa Z Computational view of surface based organic mass spectrometry. Mass Spectrom. Rev 2008, 27 (4), 289–315. [DOI] [PubMed] [Google Scholar]
- 20.Goda S; Takano K; Yamagata Y; Nagata R; Akutsu H; Maki S; Namba K; Yutani K Amyloid protofilament formation of hen egg lysozyme in highly concentrated ethanol solution. Protein Sci.. 2000, 9(2), 369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shaw DE; Dror RO; Salmon JK; Grossman JP; Mackenzie KM; Bank JA; Young C; Deneroff MM; Batson B; Bowers KJ, et al. Millisecond-scale molecular dynamics simulations on Anton. A Proceedings of the Conference on High Performance Computing Networking, Storage and Analysis November 14–20, 2009, Portland, Oregon.
- 22.Khandogin J; Chen J; Brooks CL Exploring atomistic details of pH-dependent peptide folding. Proc. Natl. Acad. Sci. 2006, 103 (49), 18546–18550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soares RO; Torres PHM; da Silva ML; Pascutti PG Unraveling HIV protease flaps dynamics by constant pH molecular dynamics simulations. J. Struct. Bio 2016, 195 (2), 216–226. [DOI] [PubMed] [Google Scholar]
- 24.Liu H; Tan Q; Han L; Huo S Observations on AMBER force field performance under the conditions of low pH and high salt concentrations. J. Phys. Chem. B 2017, 121(42), 9838–9847. [DOI] [PubMed] [Google Scholar]
- 25.Deshpande A; Nimsadkar S; Mande SC Effect of alcohols on protein hydration: crystallographic analysis of hen egg-white lysozyme in the presence of alcohols. Acta Cryst. D 2005, 61 (7), 1005–1008. [DOI] [PubMed] [Google Scholar]
- 26.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS; Richardson DC MolProbity: all-atom structure validation for macromolecular crystallography. Acta Cryst. D 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79 (2), 926–935. [Google Scholar]
- 28.Fox T; Kollman PA Application of the RESP methodology in the parametrization of organic solvents. J. Phys. Chem. B 1998, 102 (41), 8070–8079. [Google Scholar]
- 29.Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Development and testing of a general Amber force field. J. Comp. Chem 2004, 25 (9), 1157–1174. [DOI] [PubMed] [Google Scholar]
- 30.Hornak V; Abel R; Okur A; Strockbine B; Roitberg A; Simmerling C Comparison of multiple AMBER force fields and development of improved protein backbone parameters. Proteins 2006, 65 (3), 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salomon-Ferrer R; Götz AW; Poole D; Le Grand S; Walker RC Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. J. Chem. Theory Comput 2013, 9 (9), 3878–3888. [DOI] [PubMed] [Google Scholar]
- 32.Swails JM; York DM; Roitberg AE Constant pH replica exchange molecular dynamics in explicit solvent using discrete protonation states: Implementation, testing, and validation. J. Chem. Theory Comput 2014, 10 (3), 1341–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG A smooth particle mesh Ewald method. J. Chem. Phys 1995, 103 (19), 8577–8593. [Google Scholar]
- 34.Ryckaert J-P; Ciccotti G; Berendsen HJC Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Chem. Phys 1977, 23 (3), 327–341. [Google Scholar]
- 35.Loncharich RJ; Brooks BR; Pastor RW Langevin dynamics of peptides: The frictional dependence of isomerization rates of N-acetylalanyl-N′-methylamide. Biopolymers 1992, 32 (5), 523–535. [DOI] [PubMed] [Google Scholar]
- 36.Shan Y; Klepeis JL; Eastwood MP; Dror RO; Shaw DE Gaussian split Ewald: A fast Ewald mesh method for molecular simulation. J. Chem. Phys 2005, 122 (5), 54101. [DOI] [PubMed] [Google Scholar]
- 37.Lippert RA; Predescu C; Ierardi DJ; Mackenzie KM; Eastwood MP; Dror RO; Shaw DE Accurate and efficient integration for molecular dynamics simulations at constant temperature and pressure. J. Chem. Phys 2013, 139 (16), 164106. [DOI] [PubMed] [Google Scholar]
- 38.Valentine SJ; Anderson JG; Ellington AD; Clemmer DE; Disulfide-intact and reduced lysozyme in the gas phase: conformations and pathways of folding and unfolding. J. Phys. Chem. B 1997, 101, 3891–3900. [Google Scholar]
- 39.Swails JM; Roitberg AE Enhancing conformation and protonation state sampling of hen egg white lysozyme using pH replica exchange molecular dynamics. J. Chem. Theory Comput 2012, 8 (11), 4393–4404. [DOI] [PubMed] [Google Scholar]
- 40.Vocadlo DJ; Davies GJ; Laine R; Withers SG Catalysis by hen egg-white lysozyme proceeds via a covalent intermediate. Nature 2001, 412 (6849), 835–838. [DOI] [PubMed] [Google Scholar]
- 41.Held J; van Smaalen S The active site of hen egg-white lysozyme: flexibility and chemical bonding. Acta Cryst. D 2014, 70 (Pt 4), 1136–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi H; Pierson NA; Valentine SJ; Clemmer DE Conformation types of ubiquitin [M+8H]8+ ions from water:methanol solutions: evidence for the N and A States in aqueous solution. J. Phys. Chem. B 2012, 116 (10), 3344–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.