

Abstract
Abnormal aggregation of the α-synuclein protein is a key molecular feature of Parkinson’s disease and other neurodegenerative diseases. The precise mechanisms that trigger α-synuclein aggregation are unclear, and it is not known what role aggregation plays in disease pathogenesis. Here we use an in vivo zebrafish model to express several different forms of human α-synuclein and measure its aggregation in presynaptic terminals. We show that human α-synuclein tagged with GFP can be expressed in zebrafish neurons, localizing normally to presynaptic terminals and undergoing phosphorylation at serine-129, as in mammalian neurons. The visual advantages of the zebrafish system allow for dynamic in vivo imaging to study α-synuclein, including the use of fluorescence recovery after photobleaching (FRAP) techniques to probe protein mobility. These experiments reveal three distinct terminal pools of α-synuclein with varying mobility, likely representing different subpopulations of aggregated and non-aggregated protein. Human α-synuclein is phosphorylated by an endogenous zebrafish Polo-like kinase activity, and there is a heterogeneous population of neurons containing either very little or extensive phosphorylation throughout the axonal arbor. Both pharmacological and genetic manipulations of serine-129 show that phosphorylation of α-synuclein at this site does not significantly affect its mobility. This suggests that serine-129 phosphorylation alone does not promote α-synuclein aggregation. Together our results show that human α-synuclein can be expressed and measured quantitatively in zebrafish, and that disease-relevant post-translational modifications occur within neurons. The zebrafish model provides a powerful in vivo system for measuring and manipulating α-synuclein function and aggregation, and for developing new treatments for neurodegenerative disease.
Keywords: Alpha-synuclein, Parkinson’s disease, Protein aggregation, Zebrafish, FRAP, in vivo imaging, Phosphorylation, Polo-like kinase
1. Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disease, affecting 1–4% of the US population over the age of 65 (Kowal et al., 2013). Alpha-synuclein, a 140-amino acid protein, forms the major component of Lewy bodies (Spillantini et al., 1997), the intracellular aggregates that characterize both sporadic and familial PD, as well as Dementia with Lewy bodies (DLB). Point mutations in the SNCA gene coding for α-synuclein (e.g., A53T (Polymeropoulos et al., 1997), A30P (Krüger et al., 1998), E46K (Zarranz et al., 2004), H50Q (Proukakis et al., 2013), G51D (Lesage et al., 2013), A53E (Pasanen et al., 2017)), duplication (Chartier-Harlin et al., 2004; Ibáñez et al., 2004), and triplication (Singleton et al., 2003) of the gene all have been identified in families with dominant, heritable PD or DLB, suggesting that α-synuclein is central to the etiology of both Lewy body diseases.
While its precise function is unclear, α-synuclein is enriched in presynaptic terminals (Jakes et al., 1994; Iwai et al., 1995; Withers et al., 1997), can bind membranes, and has particular affinity for highly curved membranes, such as synaptic vesicles (Davidson et al., 1998; Middleton and Rhoades, 2010). Given its interaction with synaptic vesicles and synaptic proteins, α-synuclein has been proposed to play a regulatory role in processes such as synaptic activity, synaptic plasticity, neurotransmitter release, vesicle trafficking, synaptic vesicle pool maintenance, and dopamine metabolism (reviewed in Burré, 2015). Knockout experiments of α-synuclein in mice indicate that it is not an essential protein for neurotransmission, although this may reflect potential compensation by other proteins, including β- and γ-synuclein paralogs (Chandra et al., 2004; Milanese et al., 2012; Toni and Cioni, 2015). Recent data show that α-synuclein can also play a role in modulating DNA repair (Schaser et al., 2019; Weston et al., 2021). Abnormal aggregation of α-synuclein in the cell body is thought to be a key predictor of neuronal loss in disease (Osterberg et al., 2015). Presynaptic terminal α-synuclein aggregation, however, has been suggested to mediate neuronal dysfunction by interfering with synaptic function, even before somatic aggregation begins, and to be a better predictor of clinical disability (Kramer and Schulz-Schaeffer, 2007; Scott et al., 2010; Spinelli et al., 2014). The precise roles that somatic and/or presynaptic aggregation play in neurodegeneration are not known, and the specific form of aggregate/s responsible for neurotoxicity is unclear. Recent hypotheses point toward soluble oligomers as the toxic species (Conway et al., 2000; Winner et al., 2011), while large insoluble aggregates may be neuroprotective (Tanaka et al., 2004; Bodner et al., 2006; reviewed in Chartier and Duyckaerts, 2018). In order to understand the balance between normal, monomeric α-synuclein and toxic oligomeric or aggregated forms that arise in disease, a thorough characterization of the protein’s dynamics and cellular regulation in vivo is essential.
One fundamental question is the relationship between neurodegeneration, aggregation of α-synuclein, and phosphorylation of α-synuclein at serine-129, a modification present in >90% of α-synuclein in Lewy bodies compared to 4% of total brain α-synuclein (Anderson et al., 2006). Past studies in rat, mouse, and Drosophila that have addressed this question using a phosphorylation mimic (S129D) and a non-phosphorylatable (S129A) form of α-synuclein have produced contradicting results. S129D and S129A have each been argued to either enhance or decrease neurotoxicity and protein aggregation (Chen and Feany, 2005; Gorbatyuk et al., 2008; Febbraro et al., 2013), or have been shown to have no effect (McFarland et al., 2009; Escobar et al., 2014). While phosphorylation of α-synuclein may contribute to the development of aggregates, further clarity is necessary regarding the inter-relationship between phosphorylation, aggregation, and neurodegeneration. Alpha-synuclein has been shown to be phosphorylated by members of the Polo-like kinase (PLK) family (Inglis et al., 2009; Mbefo et al., 2010; Oueslati et al., 2013); inhibitors of these kinases present a potentially promising method for determining the impact of phosphorylation on natively phosphorylated α-synuclein in experimental models. An in vivo system that can allow for visualization, manipulation, and measurement of α-synuclein, as well as potentially provide a vertebrate platform for rapidly testing pharmacological treatments would be a useful tool in the study of protein aggregation in PD.
Zebrafish are a powerful model system for studying the nervous system using in vivo fluorescence imaging techniques (Brockway et al., 2019; Cook et al., 2020). Here we present a novel zebrafish model that we have developed for studying α-synuclein aggregation in vivo using transiently expressed human α-synuclein tagged with Enhanced Green Fluorescent Protein (GFP). This allows for subcellular localization and tracking of α-synuclein over time, which was not possible using previous expression systems in zebrafish (Prabhudesai et al., 2012; O’Donnell et al., 2014; Lulla et al., 2016). In our system, human wild-type α-synuclein, as well as α-synuclein containing the A53T point mutation seen in human patients (Polymeropoulos et al., 1997), expresses in zebrafish neurons and localizes to the presynaptic terminal, as in mammals. Using in vivo fluorescence recovery after photobleaching (FRAP), we have measured the sub-cellular mobility of α-synuclein in presynaptic terminals and found that α-synuclein appears to exist in three different states: 1) cytosolic; 2) membrane bound; and 3) aggregated protein. Human α-synuclein is phosphorylated at serine-129 in zebrafish neurons, as it is in humans, likely by an endogenous zebrafish Polo-like kinase (PLK). Interestingly, phosphorylation of α-synuclein results in a bimodal population of neurons containing either very little to no phosphorylation or full phosphorylation throughout the axonal arbor. Neither pharmacologic inhibition of PLKs to reduce serine-129 phosphorylation nor mutation of this serine to the phospho-mimic aspartate or non-phosphorylatable alanine changes the level of presynaptic aggregation in vivo, as measured by FRAP. This suggests that presynaptic aggregation may not be modulated by phosphorylation at serine-129 on its own. Taken together, our results show that human α-synuclein can be expressed and manipulated in zebrafish, and suggest that serine-129 phosphorylation alone does not cause presynaptic aggregation of the α-synuclein protein. This is a powerful new platform for measuring α-synuclein function and toxicity within the living nervous system.
2. Materials and methods
2.1. Generation of DNA constructs
cDNA of 1) EGFP; 2) EGFP-tagged human wild-type (WT) α-synu-clein; 3) EGFP-tagged human A53T α-synuclein; 4) EGFP-tagged human S129A α-synuclein; and 5) EGFP-tagged human S129D α-synuclein (all forms of α-synuclein were gifts from P. McLean, Mayo Clinic) were amplified and flanked with attB1 and attB2 sites for Multisite Gateway Cloning®. PCR products were recombined with Gateway pDONR221 to generate middle entry clones. Entry clones were then recombined with pDestTol2CG (pDESTTol2pA with cmlc2:egfp transgenesis marker; Kwan et al., 2007) with a 5’ neuroD promoter element (p5E containing a ~ 5 kb segment of promoter/enhancer sequence upstream of the neuroD gene; gift from T. Nicolson), and the 3’ SV40 late poly-A element (p3E-polyA, #302; Kwan et al., 2007) to form final expression clones (Table 1). Competent E. coli DH5α (New England Biolabs, Ipswich, MA) or OneShot Top10 (Invitrogen, USA) cells were used to transform and amplify plasmids. Plasmid DNA was collected and purified using the QIAprep Spin Miniprep Kit following kit instructions with minor protocol adjustments for increased yield recommended in Detrich et al., 2011.
Table 1.
Plasmids generated.
| Plasmid name | Referred to in text |
|---|---|
|
| |
| neurod:WTα-synuclein-EGFP | α-synuclein-GFP |
| neurod:A53Tα-synuclein-EGFP | A53T α-synuclein-GFP |
| neurod:S129Dα-synuclein-EGFP | S129D α-synuclein-GFP |
| neurod:S129Aα-synuclein-EGFP | S129A α-synuclein-GFP |
| neurod:EGFP | GFP |
Prior to injection, DNA constructs encoding α-synuclein and GFP were diluted to a concentration of ~10 ng/μL in 0.1 M KCl and 1% phenol red dye (Sigma Aldrich, St. Louis, MO; Alfa Aesar, Ward Hill, MA). All constructs along with their abbreviations are listed in Table 1.
2.2. Zebrafish maintenance and transient expression of DNA constructs
Wild-type (AB/TL strain) zebrafish were reared in a controlled tank facility (Aquaneering Inc., San Diego, CA, USA) held at 28 °C with a 12/12 h light/dark cycle. Fertilized eggs at the one-cell stage were injected with ~0.05 to 0.15 ng DNA through a pulled capillary needle (World Precision Instruments, Sarasota, FL; pulled by Model P-97, Sutter Instrument Co., Novato, CA), under a dissection microscope (SZ61, Olympus, Waltham, MA), using a nitrogen gas-powered microinjection system (World Precision Instruments, Sarasota, FL, USA). Injected embryos were maintained at 28 °C in E3 embryo medium (prepared according to Nusslein-Volhard and Dahm, 2002) and transferred into E3 containing 0.2 mM phenylthiourea (PTU; Alfa Aesar, Ward Hill, MA) at 24 h post fertilization (hpf) to prevent pigmentation. While expression was visualized from 2 to 8 days post fertilization (dpf), experiments were done on fish (larvae) aged 4 dpf, unless noted otherwise, because the motor neuron axonal arbors at this stage were well developed and strongly expressed α-synuclein-GFP.
2.3. Pharmacology
Individual larvae aged 3 dpf were placed in 100 μL of E3/PTU embryo medium containing 100 μM BI 2536 (Boehringer Ingelheim) in 1% DMSO, or 1% DMSO at 28 °C (Sigma Aldrich, St. Louis, MO). The 100 μM concentration was selected after testing a range of doses from 1 to 100 μM (Supplemental Fig. 2), initially suggested by Jeong et al., 2010. After incubating for 10 or 24 h, 4 dpf larvae were rinsed in E3/PTU medium for 15 min, then either imaged live or fixed and processed for phospho-S129 staining.
2.4. Whole mount immunohistochemistry
Immunohistochemistry protocol was adapted from Bae et al., 2009. Larvae were fixed overnight in 4% paraformaldehyde (PFA; Electron Microscopy Sciences, Hatfield, PA) at 4 °C and washed in PBST (PBS + 0.1% TritonX-100; AMRESCO, Solon, OH; Sigma Aldrich, St. Louis, MO) and stored at 4 °C in PBS until use. All rinsing steps were performed with light agitation, and all immunohistochemistry steps were done at room temperature unless otherwise noted. For anti-nitrated α-synuclein staining only, larvae were dissected to optimize antibody penetration by removing the head and yolk following fixation. Fixed larvae were incubated in acetone at −20 °C for 15 min followed by rinsing once with PBST and twice with PBS-DT (PBS, 1% BSA, 1% DMSO, 1% TritonX-100; Sigma Aldrich, St. Louis, MO). Next, larvae were incubated in blocking solution (PBS-DT, 5% NGS; Rockland Immunochemicals, Gilbertsville, PA and Sigma Aldrich, St. Louis, MO) for 60–90 min prior to incubation with primary antibody. Primary antibodies used were mouse monoclonal anti-Syn1 (1:500; anti-α-synuclein; BD Biosciences, San Jose, CA), mouse anti-SV2 anti-synaptic vesicle protein 2 (1:100; DSHB, Iowa City, IA), rabbit monoclonal anti-phospho-S129 (1:200; EP1536Y; Abcam, Cambridge, MA), and mouse monoclonal anti-nitrated α-synuclein (1:100; nSyn505; Thermo Fisher Scientific, Waltham, MA). Secondary antibodies used were anti-mouse Rhodamine Red-X-conjugated secondary antibody (1:500; Jackson ImmunoResearch, West Grove, PA), anti-rabbit Rhodamine Red-X-conjugated secondary antibody (1:400; Jackson ImmunoResearch, West Grove, PA), and anti-mouse Alexa Fluor 633 secondary antibody (1:200–1:400; Thermo Fisher Scientific, Waltham, MA). Antibodies were diluted into antibody buffer (20% blocking solution in PBS, 0.05% sodium azide added for anti-phospho-S129 staining; Sigma Aldrich, St. Louis, MO) and incubated with light agitation. Anti-Syn1 and anti-SV2 primary antibodies were incubated for 48 h, anti-nitrated α-synuclein primary antibody was incubated for 72 h at 4 °C, and anti-phospho-S129 primary antibody was incubated for five days. After removing primary antibodies, larvae were rinsed 30–60 min in PBST then further rinsed in PBST overnight (or rinsed for 4 h total in PBST for anti-nitrated α-synuclein staining). Anti-mouse Rhodamine Red-X-conjugated secondary antibody and anti-rabbit Rhodamine Red-X-conjugated secondary antibody were applied for 1.5–2 h, while anti-mouse Alexa Fluor 633 secondary antibody was applied overnight, followed by rinsing overnight in PBST.
For analysis and quantification of phosphorylation, α-synuclein-GFP expression was used to identify neurons, then anti-phospho-S129 label was identified within each synaptic terminal of GFP-expressing neurons. To control for issues of antibody penetration, larval regions were only analyzed if they showed clear background staining, indicating that antibodies had penetrated the region to be analyzed. Quantification experiments shown in Figs. 4D and 5C were done in 4 dpf fish. In BI 2536 experiments, neurons were simply scored as positive or negative for phosphorylation.
Fig. 4.
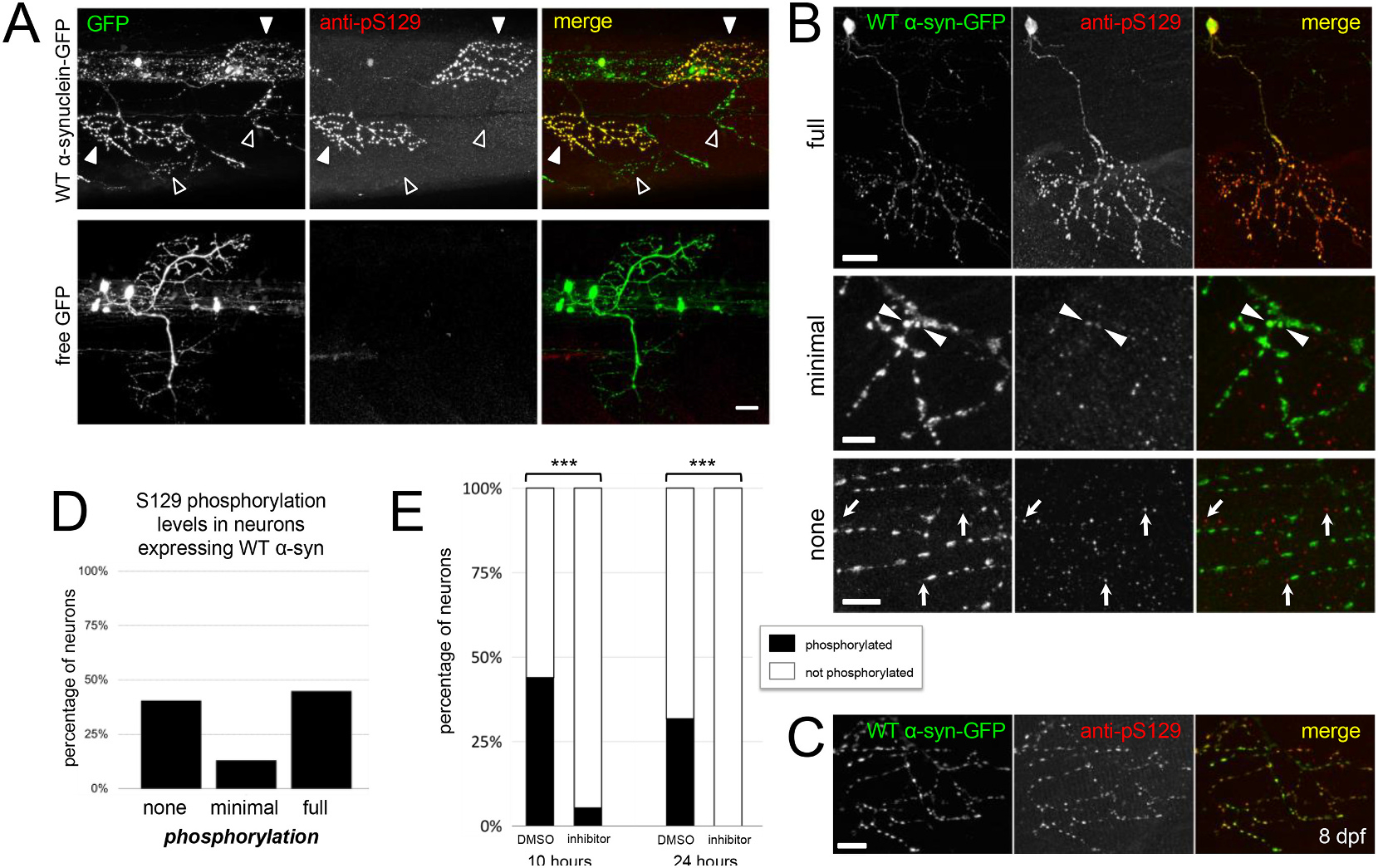
Alpha-synuclein-GFP is phosphorylated by endogenous kinase in a subset of neurons.
A) Immunostaining for anti-phospho-S129 shows that expressed α-synuclein is phosphorylated in 4 dpf larvae (upper row, white arrowheads), but no stain is present when free GFP is expressed (lower row). Open arrowheads show some synuclein-expressing axons with little to no phospho-S129 staining. GFP channel is shown at left, anti-phospho-S129 in the center, and merge at right. Scale bar 20 μm. B) α-synuclein-GFP expressed in primary motor neurons and stained at 4 dpf is either fully phosphorylated throughout the axonal arbor (upper row example), minimally phosphorylated (middle row; a small fraction of terminals throughout arbor, indicated with arrowheads), or not phosphorylated (bottom row). GFP channel is shown at left, anti-phospho-S129 in the center, and merge at right. Arrows in bottom row indicate immunostaining background puncta that are not overlapping with GFP channel. Scale bar 20 μm in upper row, 5 μm in middle row, and 10 μm in bottom row. In panels A-C, caudal is right and dorsal is up. C) Motor axon at 8 dpf shows that expressed α-synuclein is phosphorylated at older ages. GFP channel is shown at left, anti-phospho-S129 in the center, and merge at right. Scale bar 10 μm. All images are maximum intensity projections of stacks with the following depths: A) 28.7 μm top; 118 μm bottom; B) 46.7 μm, 19 μm, and 30.4 μm (top, middle, and bottom respectively); C) 24 μm. D) Quantification of primary motor neurons from 4 dpf zebrafish expressing wild type α-synuclein-GFP shows that 41.2% of neurons do not exhibit phospho-S129 staining, 13.2% show little phosphorylation, and 45.6% are strongly phosphorylated (n = 69 neurons assessed from 8 larvae) E) Following exposure of zebrafish expressing wild type α-synuclein-GFP to 100 μm of PLK inhibitor BI 2536 (beginning at 3 dpf), phosphorylation of α-synuclein at S129 decreased significantly from 43% to 5.4% at 10 h (DMSO: n = 18/41 neurons from 6 larvae; BI 2536: 2/37 neurons from 6 larvae; chi-squared p < 0.001) and from 31.7% to 0% at 24 h (DMSO: n = 13/41 neurons from 5 larvae; BI 2536: 0/53 neurons from 5 larvae; chi-squared p < 0.001) as compared to DMSO control.
Fig. 5.
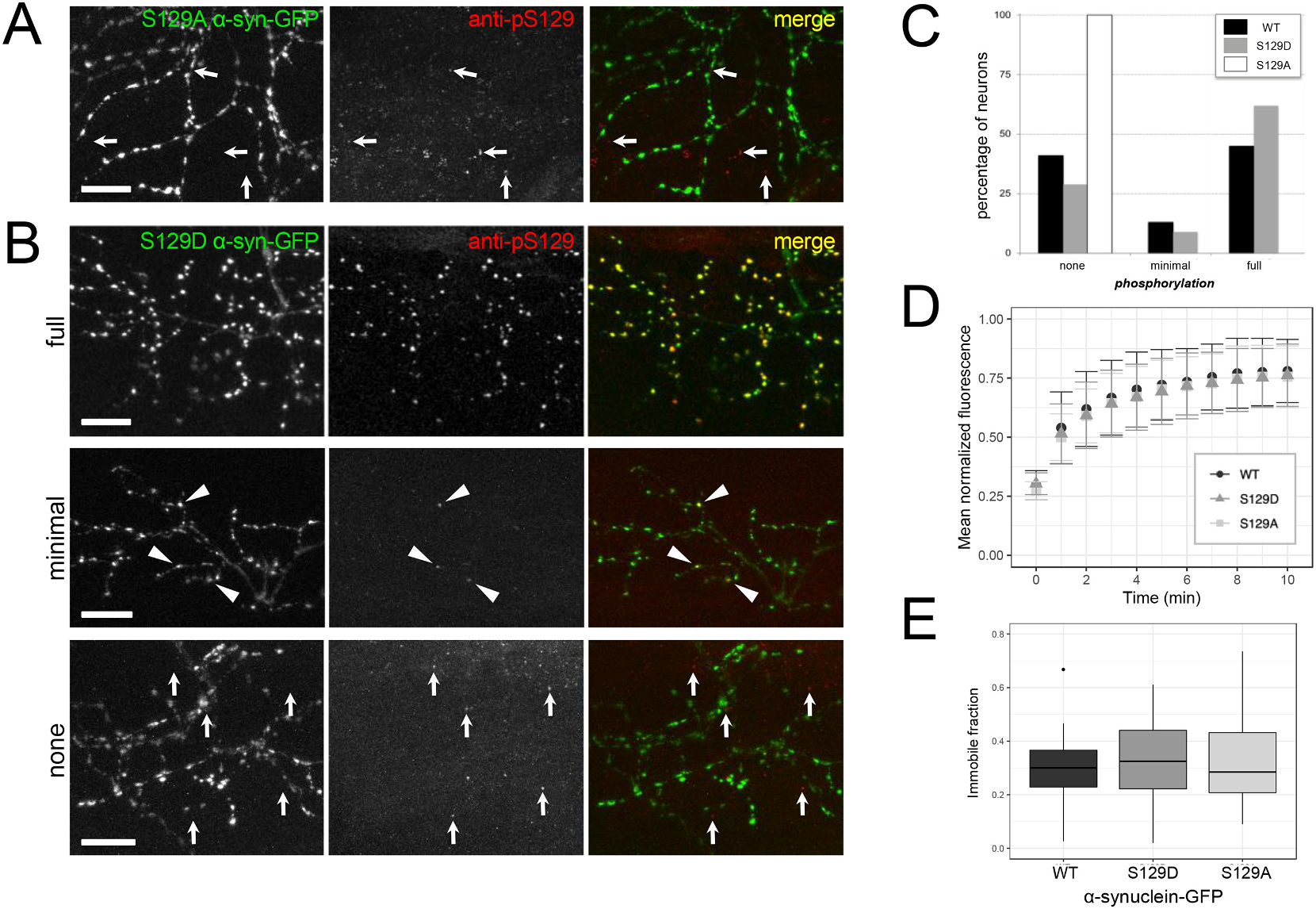
Genetic modification of serine 129 to alanine abolishes phosphorylation, but modification to alanine or aspartate does not affect immobile fraction.
A) S129A α-synuclein-GFP expressed in motor neurons and stained at 4 dpf does not exhibit phospho-S129 staining. GFP channel is shown at left, anti-phospho-S129 in the center, and merge at right. Arrows indicate immunostaining background puncta that are not overlapping with GFP channel. B) Similar to WT α-synuclein-GFP, S129D α-synuclein-GFP expressed in motor neurons and stained at 4 dpf is either fully phosphorylated throughout the axonal arbor (upper row example), minimally phosphorylated (middle row; a small fraction of terminals throughout arbor, indicated with arrowheads), or not phosphorylated (bottom row). In bottom row, arrows indicate immunostaining background puncta that are not overlapping with GFP channel. GFP channel is shown at left, anti-phospho-S129 in the center, and merge at right. Scale bars 10 μm; caudal is right and dorsal is up. All images are maximum intensity projections of stacks with the following depths: A) 39.2 μm B) 65.2 μm, 30.9 μm, and 66.6 μm (top, middle, and bottom respectively). C) Quantification of phosphorylation in motor neurons expressing each form of α-synuclein shows that neurons expressing wild type α-synuclein-GFP are distributed as 41.2% not phosphorylated, 13.2% minimally phosphorylated, and 45.6% fully phosphorylated (n = 69 neurons from 8 larvae). Neurons expressing S129D α-synuclein-GFP show a similar distribution with 28.6% not phosphorylated, 9.5% minimally, and 61.9% fully phosphorylated (n = 103 neurons from 14 larvae). In contrast, S129A α-synuclein-GFP expressed in motor neurons is not phosphorylated (100%; n = 130 neurons from 10 larvae). D) Group data of fluorescence recovery over time. Error bars represent standard deviation. Average tau for WT α-synuclein-GFP was 2.8 min (n = 26 terminals from 11 larvae); S129D was 2.7 min (n = 39 terminals from 13 larvae), and S129A was 2.3 min (n = 29 terminals from 7 larvae; one-way ANOVA, p = 0.638). E) The average immobile fraction for WT α-synuclein-GFP was 29.3% (n = 26 terminals from 11 larvae), S129D was 32.5% (n = 39 terminals from 13 larvae), and S129A was 32.8%; (n = 29 terminals from 7 larvae; one-way ANOVA, p = 0.641). In E, bars indicate range of data within 1.5 IQR; outlier outside this range shown as a point.
2.5. Imaging of fixed larvae
Stained larval zebrafish were positioned onto glass slides in Vectashield® mounting medium (Vector Laboratories, Burlingame, CA) and sealed under a glass coverslip. To prevent larvae from damage, the coverslip was elevated slightly using droplets of vacuum grease on its four corners. Slides were sealed and stored at 4 °C in a dark chamber. Larvae were imaged using a laser scanning confocal microscope (LSM; Carl Zeiss LSM 710, Oberkochen, Germany) under a 40× oil (1.3 NA) objective. Motor neurons were identified based on their classic morphology (Myers, 1985; Myers et al., 1986; Westerfield et al., 1986). GFP was excited using a 488 nm laser, and rhodamine was excited using a 561 nm laser, and Alexa Fluor 633 was excited using a 633 nm laser. Following acquisition, linear adjustments were made to some images for visual clarification on screen and for use in figures. All images shown are maximum intensity projections unless noted in figure legend.
2.6. Live imaging and FRAP
In preparation for live imaging, zebrafish aged 3–4 dpf were anesthetized in a solution of ~0.2 mM MS-222 tricaine (Sigma Aldrich, St. Louis, MO). Larvae were mounted onto glass slides in 1–2% low-melt agarose (AMRESCO Agarose SFR™; AMRESCO, Solon, OH) and imaged using either a confocal (LSM 710 Carl Zeiss, Oberkochen, Germany) or two-photon (7 MP Carl Zeiss, Oberkochen, Germany) LSM, both under a 20× water immersion (1.0NA) objective. Presumptive presynaptic terminals in spinal cord axons or in motor neurons were imaged at 8× magnification under 20×.
Fast FRAP (line scan) experiments (Fig. 2) were done on the 7 MP using 860 nm excitation for GFP, as described in Spinelli et al. (2014). Line scan bleaches were performed at 20–60% laser power and images were collected at 7.5 ms intervals for a period of 750 ms (150 ms at baseline and 600 ms post bleach). Slower FRAP experiments (on whole synaptic terminals; Figs. 3 and 5) were done on the LSM 710, using an argon laser to excite GFP at 488 nm. In both types of FRAP experiments, bleaches were performed at 100% laser power, a measure that was found to bleach terminals to at least ~40% of the baseline fluorescence intensity. To detect the rate of recovery, single Z-plane images mid-terminal (Fig. 3) or small, ~5 μm Z-stacks encompassing the entire terminal (Fig. 5) were collected every minute for up to 15 min post-bleach in synuclein-GFP experiments, and every 10 s for 5 min post-bleach in free GFP experiments.
Fig. 2.
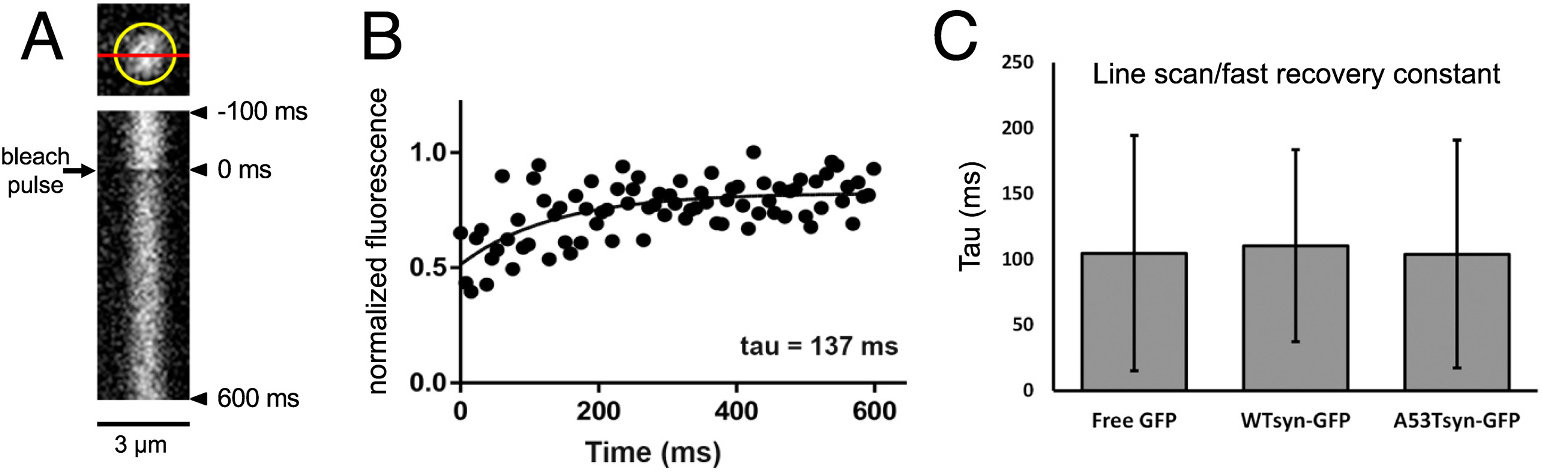
Fast synaptic terminal in vivo FRAP imaging reveals a pool of α-synuclein-GFP with identical recovery to free GFP, likely representing freely diffusing α-synuclein.
A) (Top) Schematic of a line-bleached terminal (circled in yellow; single plane). The red line indicates the area bleached and measured over a recovery period of 600 ms. (Bottom) representative x-t plot of a line-bleached terminal expressing WT α-synuclein-GFP. The horizontal axis represents microns, aligned with the red line drawn above. The vertical axis represents time (increasing downward). Line through terminal was scanned every 7.5 ms, thus each y-axis pixel represents 7.5 ms. Bleach pulse occurs at 0 ms. B) Representative graph of individual terminal recovery in (A), plotting presynaptic terminal fluorescence intensity (normalized to baseline fluorescence) over time, fit with a single exponential function. C) Group data of recovery tau (mean) for presynaptic terminals expressing free GFP (105 ± 90 ms; n = 13 terminals from 4 larvae), wild-type α-synuclein-GFP (110 ± 73 ms; n = 15 terminals from 3 larvae), or A53T α-synuclein-GFP (104 ± 87 ms; n = 22 terminals from 3 larvae).
Fig. 3.
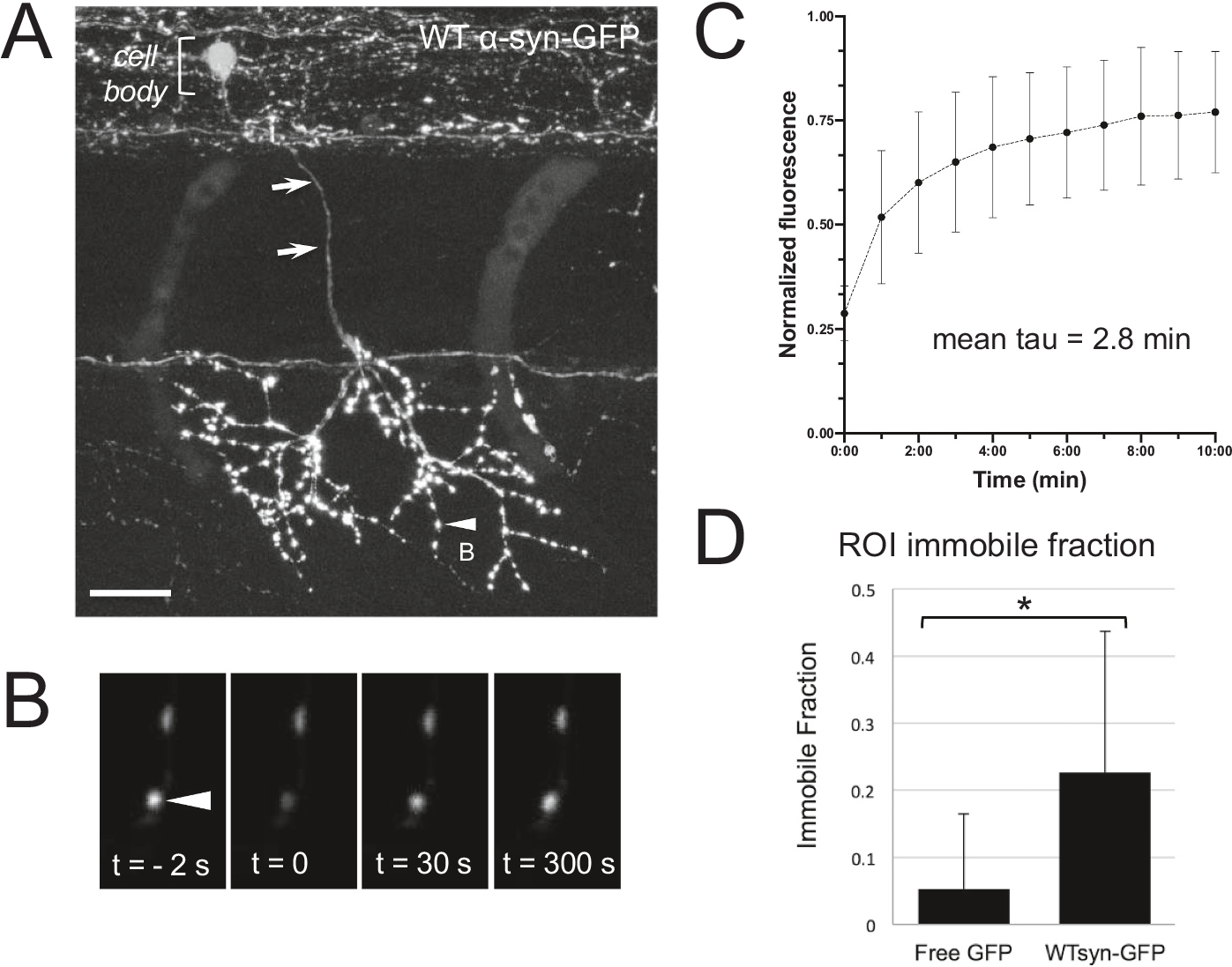
Slow synaptic terminal in vivo FRAP imaging reveals a more slowly recovering pool and a non-recovering pool of α-synuclein-GFP.
A) Motor neuron expressing WT α-synuclein-GFP in 3 dpf zebrafish, with cell body in spinal cord (upper left bracket) and axon (arrows) descending ventrally to innervate muscle. Image in (A) is a maximum projection of a stack (70.5 μm thick) that was acquired after animal was fixed, following live imaging on highlighted synaptic terminal (arrowhead) shown enlarged in (B). Nonlinear adjustments were done in the larger panel only (A), in order to highlight thin axon descending from spinal cord (arrows). Scale bar in A 20 μm. B) Time series of in vivo ROI bleach experiment on presynaptic terminal indicated in (A). Only a single plane of section is shown. Arrowhead indicates the terminal measured before and after the bleach pulse and during the recovery phase. C) Group data of fluorescence recovery over time for WT α-synuclein-GFP. Average tau is 2.8 min (n = 26 terminals from 11 larvae). D) Group data of mean immobile fraction in terminals expressing free GFP (5 ± 11%; n = 22 terminals, 3 larvae) or wild-type (21 ± 25%; n = 27 terminals, 4 larvae). * represents p < 0.05, post-hoc Tukey test.
2.7. FRAP analysis
Analysis of FRAP image series was performed using Fiji (Schindelin et al., 2012). Single plane images or maximum intensity projections from a single bleached terminal were condensed into a time series and drift-corrected using the StackReg plugin (Biomedical Imaging Group, Swiss Federal Institute of Technology, Lausanne, Switzerland). Average fluorescence intensity within the bleached ROI as well as a neighboring, unbleached GFP-labeled control terminal in the field was collected for all time points. ROI fluorescence intensity was then normalized to baseline fluorescence of each terminal pre-bleach; the normalized intensity of the reference terminal was used to correct the normalized intensity of the bleach terminal at each timepoint to account for inadvertent photobleaching. For Fig. 5, fluorescence of the bleach terminal was not corrected for photobleaching; on average, the reference terminals in these experiments retained ~99% of fluorescence (n = 99 terminals) over 10 min of imaging, indicating photobleaching was not a significant concern. The immobile fraction (IF) of synuclein-GFP or free GFP was calculated as follows:
Graphs of fluorescence intensity with respect to time were generated using Prism5 or Prism8 software (GraphPad, La Jolla, CA) and fit with a single exponential association curve. Time constants (tau), representing the time at which fluorescence had recovered to (1–1/e), or ~ 63%, of the final fluorescence value were measured from the association curves. For Fig. 5, the value of the fluorescence plateau was also taken from this curve and used to calculate the immobile fraction.
2.8. Colocalization analysis
Colocalization analysis was performed on confocal stacks using the JaCoP plugin (Bolte and Cordelières, 2006) in Fiji. Stacks were cropped in x, y, and z to include only relevant GFP fluorescence in the spinal cord and/or motor neurons, while autofluorescent structures, such as blood vessels, were cropped from the images when possible. For the anti-Syn1 and anti-phospho-S129 colocalization analysis, Pearson’s correlation coefficients were calculated. The Pearson’s correlation coefficient is a measure of the covariance of each channel over all pixels of the image and ranges from −1 (perfect negative correlation) to 1 (perfect positive correlation). Alternately, the SV2 and nitrated α-synuclein analysis required that the analysis be limited to only pixels in motor neurons with GFP fluorescence, making a Mander’s colocalization analysis more appropriate. Mander’s colocalization coefficients for GFP were calculated after setting intensity thresholds in each channel. As acquisition settings and the intensity of both the GFP fluorescence and staining were variable, it was not possible to use a set threshold for all images. Instead, thresholds were set manually for each channel in each image. The threshold was set at one intensity level above the level where individual pixels in background areas were no longer detected. To control for the effect of thresholding on the Mander’s coefficient generated, for nitrated α-synuclein analysis, each image was independently thresholded and analyzed three times and the mean Mander’s coefficient was used.
2.9. Statistics
All numbers are represented as the mean ± SD unless otherwise noted. Numbers and statistical tests are reported in each section and/or figure legend. For each FRAP experiment analysis, larvae were pooled from 2 to 7 different clutches injected on different days. For all staining data shown in figures (with the exception of Fig. 4E), larvae were pooled from 2 to 5 different clutches injected on different days. Statistical analysis was performed in R: A language and environment for statistical computing (R Core Team, 2017), Microsoft Excel (Redmond, WA), or StatPro (AnalystSoft, USA). For Fig. 4E, Yate’s correction for continuity was applied to correct for chi-squared test bias on 2 × 2 contingency tables.
3. Results
3.1. Human α-synuclein is exogenously expressed in zebrafish motor neurons
To model the behavior of α-synuclein in vivo, we stochastically expressed human α-synuclein with a C-terminal Green Fluorescent Protein tag (α-synuclein-GFP) in zebrafish larvae (Fig. 1). We used the zebrafish neuroD promoter in order to drive expression in a subset of neurons. We focused our analysis on α-synuclein activity within motor neurons because they provide a relatively homogeneous population that is morphologically distinct, and their axonal arbors can be spatially isolated from other neurons (Myers, 1985; Myers et al., 1986; Westerfield et al., 1986). Furthermore, presynaptic terminals can be identified clearly along the axons of motor neurons, subclasses of which project stereotypically into the dorsal and/or ventral trunk. At 2–4 days post fertilization (dpf) in living zebrafish, α-synuclein-GFP appeared in the soma, axon, and presynaptic terminals of a small percentage of neurons throughout the central and peripheral nervous system, including some motor neurons projecting from the spinal cord (Fig. 1A–C). Robust expression persisted and neurons appeared healthy until at least 8 dpf (Fig. 1D). Alpha-synuclein-GFP within motor axons appeared punctate and did not appreciably label axonal segments between varicosities, in contrast to free GFP, suggesting that α-synuclein was enriched at presynaptic terminals (Fig. 1B, arrowheads).
Fig. 1.
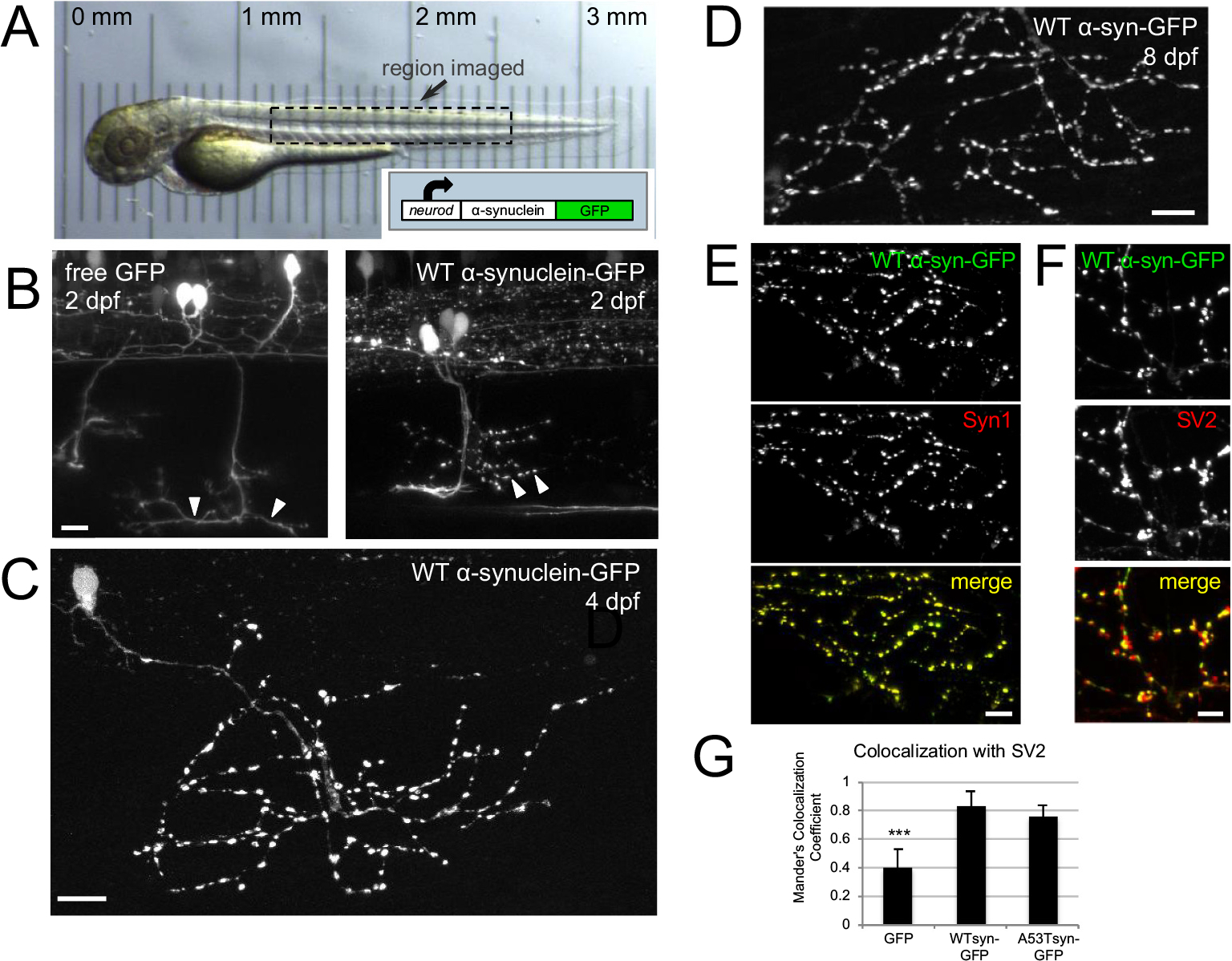
Human α-synuclein-GFP expresses in zebrafish neurons and localizes to presynaptic terminals.
A) In vivo transmitted light image of zebrafish at 2 dpf; box indicates general region targeted for imaging. Translucency of larva is demonstrated by micrometer placed below; reprinted from Brockway et al., 2019. Inset shows schematic of neurod:α-synuclein-GFP DNA. B) In vivo imaging at 2 dpf shows that free GFP fills axons smoothly, while α-synuclein-GFP labeling appears more punctate at presumptive synaptic terminals. Arrowheads indicate regions of axons containing synaptic terminals at 2 dpf, which become more mature by 4 dpf (C). C) WT α-synuclein-GFP expression shown at 4 dpf in fixed larvae. D) α-synuclein-GFP expression persists until at least 8 dpf (image from fixed fish). E) Expression of α-synuclein-GFP colocalizes with synuclein protein. Ventrally projecting axons of motor neuron expressing wild-type α-synuclein-GFP (top), anti-Syn1 staining (middle), and merge (bottom). Scale bars 10 μm in B-E. F-G) α-synuclein-GFP expression in motor axons correlates more strongly with presynaptic terminal marker SV2 than does free GFP. F) High magnification view of motor axons expressing wild-type α-synuclein-GFP (top), SV2 antibody staining (middle), and two-channel merge (bottom). Scale bar 5 μm. G) Average Mander’s colocalization coefficient calculated on confocal stacks shows that WT α-synuclein-GFP and A53T α-synuclein-GFP colocalize significantly more strongly with SV2 (0.83 ± 0.11, n = 4 regions from 3 larvae; and 0.77 ± 0.07, n = 5 regions from 4 larvae, respectively) than does free GFP (0.41 ± 0.12, n = 9 regions from 4 larvae; one-way ANOVA, post-hoc Tukey tests p < 0.002 and p < 0.004). In all panels, caudal is right and dorsal is up. Panels B–F are maximum intensity projections of stacks with the following depths: B) left 45 μm; B) right 36 μm; C) 56.9 μm; D) 24 μm; E) 80 μm; F) 80 μm.
To verify that the GFP signal was a clear indication of α-synuclein protein, we performed whole-mount antibody stains on 4 dpf zebrafish using the Syn1 antibody (Perrin et al., 2003), which recognizes human α-synuclein (Fig. 1E). Zebrafish express members of the synuclein family (Sun and Gitler, 2008; Milanese et al., 2012), some of which have been suggested to aggregate (Lulla et al., 2016), but do not contain an ortholog to α-synuclein (Toni and Cioni, 2015). We thus expected the Syn1 antibody to colocalize completely and exclusively with α-synuclein-GFP expression. As expected, Syn1 staining revealed a high degree of colocalization with both human wild-type α-synuclein-GFP and a modified version containing the A53T mutation (McLean et al., 2001) in zebrafish neurons (A53T α-synuclein-GFP). Pearson’s correlation coefficients of 0.89 ± 0.03 for α-synuclein-GFP and 0.81 ± 0.05 for A53T α-synuclein-GFP confirmed this, while the Pearson’s correlation coefficient for stacks containing only free GFP were expectedly low, 0.038 ± 0.06 (α-syn n = 4 regions from 2 larvae; A53T α-syn n = 6 regions from 4 larvae; GFP n = 5 regions from 3 larvae); one-way ANOVA p < 0.001; post-hoc Tukey WT/GFP p < 0.0001, A53T/GFP p < 0.0001). These results confirm that GFP fluorescence is an accurate representation of α-synuclein expression in zebrafish expressing α-synuclein-GFP, and that the differences observed between α-synuclein-GFP and free GFP expression are attributable to the α-synuclein protein.
3.2. Alpha-synuclein is enriched at presynaptic terminals
We next tested whether the axonal varicosities observed in α-synuclein-GFP-expressing motor neurons corresponded to mature presynaptic terminals. To accomplish this, we labeled presynaptic terminals in 4 dpf zebrafish expressing free GFP or α-synuclein-GFP using an antibody recognizing a common marker of zebrafish presynaptic terminals in the peripheral nervous system: synaptic vesicle protein 2 (SV2; Panzer et al., 2005; Fig. 1F, G). To quantify the differences in synaptic localization between free GFP and α-synuclein-GFP, we calculated a Mander’s colocalization coefficient for GFP overlap with SV2, indicating the proportion of total pixels in the GFP channel that colocalize with pixels in the SV2 channel. Using this analysis of colocalization, we hypothesized that if the axonal varicosities observed in α-synuclein-GFP expression correspond to synapses, a larger proportion of total α-synuclein-GFP would be concentrated in synapses compared to free GFP, which is also present at comparable levels in the cytoplasm. Thus, α-synuclein-GFP conditions should generate a Mander’s colocalization coefficient closer to 1. Indeed, analysis of motor axons expressing wild-type α-synuclein-GFP and A53T α-synuclein-GFP produced Mander’s colocalization coefficients of 0.83 ± 0.11 and 0.77 ± 0.07, respectively, significantly larger than that of free GFP, which was 0.41 ± 0.12 (one-way ANOVA p < 0.002; post-hoc Tukey WT/GFP p < 0.002, A53T/GFP p < 0.004; Fig. 1G). These results demonstrate that α-synuclein-GFP is enriched at presynaptic terminals, suggesting that human α-synuclein expressed in zebrafish localizes normally within neurons.
Presynaptic terminal α-synuclein functions to regulate synaptic transmission (Burré, 2015), and it has been suggested that abnormal aggregation of this protein in the terminal can cause toxicity, leading to dysfunctional synapses (Scott et al., 2010; Spinelli et al., 2014) and clinical symptoms in humans (Kramer and Schulz-Schaeffer, 2007). To begin to explore whether enriched α-synuclein might be aggregating in zebrafish synaptic terminals, we first used an antibody that recognizes nitrated forms of the protein. This antibody reacts with disease-related α-synuclein aggregates in human tissue (Duda et al., 2002) by preferentially binding to misfolded forms of the protein (Waxman et al., 2008; Tsika et al., 2010). Indeed, we detected nitrated forms of α-synuclein in axonal terminals of 4 dpf zebrafish transiently expressing WT α-synuclein-GFP (Supplemental Fig. 1), raising the possibility that aggregation could be occurring. A Mander’s colocalization analysis showed that nitrated synuclein colocalizes with the α-synuclein-GFP signal (0.4193 ± 0.195; n = 9 images from 5 larvae), significantly more than with secondary antibody signal alone (0.005167 ± 0.00397; n = 8 images from 5 larvae; t-test p < 0.0005).
3.3. Presynaptic α-synuclein exists in three pools with differing mobility
To test further whether α-synuclein may be aggregating in presynaptic terminals of zebrafish neurons, we utilized fluorescence recovery after photobleaching (FRAP) imaging techniques to detect the mobility of the α-synuclein protein in vivo. Measurements of protein mobility are an established method for in vivo detection of aggregated protein, because aggregated proteins have limited mobility compared to their non-aggregated counterparts (Lippincott-Schwartz et al., 2001). In the mouse cortex, our previous in vivo FRAP imaging of α-synuclein has revealed three pools of presynaptic α-synuclein with distinct mobility, likely corresponding to freely diffusing, synaptic vesicle-bound, and aggregated α-synuclein (Spinelli et al., 2014, 2015). To test whether α-synuclein acts similarly in the zebrafish nervous system, we used in vivo imaging to perform FRAP on zebrafish presynaptic terminals in 4 dpf larvae (Figs. 2–3).
In all conditions, we expected that at least some α-synuclein-GFP should exist in a freely diffusing state. To detect this fast, freely diffusing pool of α-synuclein, we performed FRAP in line-scan mode (Fig. 2), which enables detection of fluorescence recovery on the millisecond scale. To quantify the rate of recovery, we calculated the recovery constant, tau, which measures the time to recover to (1–1/e), or ~ 63%, of the final intensity value for each terminal bleached. As expected, we found that the rate of recovery of the fastest pool of α-synuclein-GFP and A53T α-synuclein-GFP in zebrafish presynaptic terminals is indistinguishable from free GFP, with average tau of 110 ± 73 ms, 104 ± 87 ms, 105 ± 90 ms, respectively (Fig. 2C; n = 15 terminals/3 larvae; 22 terminals/3 larvae; 13 terminals/4 larvae; one-way ANOVA p = 0.976). These results suggest that a pool of presynaptic α-synuclein-GFP in zebrafish neurons is freely diffusing, as is found in mice (Spinelli et al., 2014).
Next, we performed longer-term FRAP experiments that would enable detection of additional, more slowly recovering pools. Our previous work in mouse cortex has suggested the existence of two additional pools: vesicle-bound and aggregated pools of α-synuclein (Spinelli et al., 2014). We bleached circular regions of interest encompassing individual presynaptic terminals, and measured α-synuclein-GFP recovery for up to 15 min (Fig. 3). In vivo FRAP on this timescale revealed a slower recovering portion of synuclein-GFP. The average tau for wild-type α-synuclein-GFP was 2.8 min (Fig. 3C; n = 26 terminals from 11 larvae).
In addition to distinguishing a pool of α-synuclein that recovers more slowly than freely diffusible GFP, the slow recovery FRAP experiments also revealed a third pool of α-synuclein that did not recover fully within the 15 min post-bleach. This immobile fraction (IF) was quantified by measuring the amount of fluorescence that did not return to baseline as a fraction of the total bleached (Lippincott-Schwartz et al., 2001). In presynaptic terminals of 4 dpf zebrafish expressing α-synuclein-GFP, the immobile fraction comprised 21 ± 25% of total α-synuclein-GFP (Fig. 3D), significantly greater than the immobile fraction in terminals expressing free GFP, which was only 5 ± 11% (one-way ANOVA across WT/A53T/GFP conditions, post-hoc Tukey WTsyn vs. free GFP p < 0.05). In terminals expressing A53T α-synuclein-GFP, the immobile fraction was 15 ± 18% (n = 24 terminals from six larvae). Interestingly, this did not differ significantly from either α-synuclein-GFP or free GFP. This may be explained by the fact that the A53T α-synuclein-GFP construct had lower expression levels than WT, since in other systems there is a correlation between expression level and presynaptic aggregation as measured by FRAP (Unni et al., 2010; Spinelli et al., 2014).
3.4. Alpha synuclein is phosphorylated within a subset of neurons
Complementary to our in vivo FRAP imaging experiments, we also assessed whether α-synuclein in the zebrafish nervous system is phosphorylated at serine-129. This specific modification has been associated with aggregated α-synuclein, since the majority of aggregated α-synuclein in human disease is phosphorylated at this site (Anderson et al., 2006), although our previous work suggests that a dissociation between serine-129 phosphorylation and presynaptic α-synuclein aggregation may exist in mouse presynaptic terminals (Spinelli et al., 2015; Weston et al., 2021). We performed whole mount immunohistochemistry using an antibody directed specifically against human α-synuclein that is phosphorylated at serine-129 (anti-phospho-S129). Indeed, we found that α-synuclein expressed in zebrafish undergoes this same modification seen in human Lewy bodies. As shown in Fig. 4A, presynaptic terminals in motor neurons contain phosphorylated α-synuclein, while this staining was not observed in motor neurons expressing free GFP, suggesting that human α-synuclein expressed in zebrafish is phosphorylated by an endogenous zebrafish kinase. Interestingly, the phosphorylation of α-synuclein appeared to occur in a bimodal pattern within neurons, whereby whole axonal arbors expressing the protein revealed either robust phosphorylation throughout their extent (45.6% of imaged neurons), or very little (13.2%) to none (41.2% of imaged neurons; n = 69 neurons from 8 larvae; Fig. 4B–D). Phosphorylation persisted until at least 8 dpf (Fig. 4C).
3.5. Polo-like kinase inhibitor, BI 2536, decreases phosphorylation of α-synuclein with no significant effect on immobile fraction
In mammals, α-synuclein can be phosphorylated by a class of kinases known as the Polo-like kinase (PLK) family (Inglis et al., 2009; Mbefo et al., 2010; Oueslati et al., 2013). To test whether zebrafish PLKs phosphorylate human α-synuclein tagged with GFP in vivo, we used the specific PLK inhibitor BI 2536, which is known to decrease serine-129 phosphorylation of α-synuclein in mammals (Inglis et al., 2009; Bergeron et al., 2014; Fig. 4E). We added 100 μM BI 2536 to fish water for 10 or 24 h followed by immunohistochemistry with phospho-S129 antibody, and compared phosphorylation to larvae exposed only to equivalent DMSO vehicle concentrations. We found that 24-h exposure to BI 2536 essentially abolished phosphorylation of α-synuclein in zebrafish neurons, as compared to a vehicle (Fig. 4E; n = 41 neurons from 5 larvae in DMSO condition and 53 neurons from 5 larvae in BI 2536; chi-squared test yielded p < 0.001; also see Supplemental Fig. 2). This decrease in α-synuclein phosphorylation by BI 2536 suggests that endogenous zebrafish PLKs (Elling et al., 2008) phosphorylate exogenously expressed human α-synuclein in vivo. To test whether the BI 2536-induced reduction in serine-129 phosphorylation also changed presynaptic α-synuclein aggregation as measured by in vivo FRAP, we measured the terminal immobile fraction in fish treated with BI 2536 or vehicle. We did not detect any significant change in immobile fraction between these two groups (BI 2536: 41%, n = 7 terminals from 2 larvae; DMSO: 57%, n = 9 terminals from 3 larvae; p = 0.43, t-test), arguing that serine-129 phosphorylation alone does not directly modulate levels of presynaptic α-synuclein aggregation.
3.6. Point mutation of α-synuclein serine-129 to aspartate or alanine does not affect immobile fraction
Pharmacologic PLK inhibition is likely to have multiple effects on neurons in addition to changing levels of α-synuclein phosphorylation. In order to specifically modulate serine-129 phosphorylation without interfering with other PLK targets, we next generated two different point mutation constructs (McFarland et al., 2009) that express either non-phosphorylatable alanine (S129A) or the phospho-mimic aspartate (S129D) at the 129 position in α-synuclein. Immunohistochemical analysis with the serine-129 phospho-antibody showed no staining of the S129A α-synuclein-GFP protein when expressed in vivo, as predicted (Fig. 5A, C). Interestingly, expression of the S129D mutant showed variable staining with the phospho-synuclein antibody, whereby some motor axons showed strong staining and others showed little to no staining (Fig. 5B, C), similar to our data with the wild-type serine at this position (quantified in Fig. 5C). This may suggest that the epitope recognized by the anti-phospho-S129 antibody in vivo is more complicated than simple phosphorylation/charge difference at serine-129, and may require other changes in protein structure before antibody binding will occur. We next performed in vivo FRAP experiments of terminal α-synuclein-GFP in larvae expressing wild-type, S129D, or S129A α-synuclein, and did not detect any significant difference in immobile fraction (29.3%, 32.5%, and 32.8% respectively; n = 26, 39, and 29 terminals from 11, 13, and 7 larvae; one-way ANOVA, p = 0.641; Fig. 5E) or recovery tau (2.8 min, 2.7 min, and 2.3 min respectively; one-way ANOVA, p = 0.638; Fig. 5D). Taken together, two parallel approaches for decreasing phosphorylation of α-synuclein at serine-129 – one pharmacological and one using genetic manipulation – both demonstrate that phosphorylation of α-synuclein at serine-129 does not directly alter presynaptic aggregation as measured by FRAP.
4. Discussion
Here we demonstrate that human α-synuclein can be expressed in zebrafish, where it localizes to presynaptic terminals and undergoes disease-relevant post-translational modifications. In vivo FRAP measurements suggest that this presynaptic protein exists in three distinct pools, with one pool having very low mobility, as seen in mouse cortex (Spinelli et al., 2014), indicating the presence of presynaptic aggregates. Endogenous zebrafish PLKs phosphorylate human α-synuclein in our system, suggesting that the heterologous protein can undergo similar posttranslational modifications by the same kinase family as in mammalian neurons. Following both pharmacologic inhibition of endogenous zebrafish PLKs, and mutation of the serine-129 site to mimic or prevent α-synuclein’s phosphorylation, we did not detect any significant change in the amount of presynaptic aggregation as measured by in vivo FRAP. This suggests that serine-129 phosphorylation alone does not promote α-synuclein aggregation.
4.1. Alpha-synuclein expression and aggregation in zebrafish
We have generated and characterized a new zebrafish model for expressing and manipulating human α-synuclein. In this study, we focused our analysis on α-synuclein activity within motor neurons because it allowed us to isolate a relatively homogeneous neuronal population that is morphologically distinct, with axonal arbors that can be spatially separated from other neurons (Myers, 1985; Myers et al., 1986; Westerfield et al., 1986). Combining in vivo imaging and immunohistochemistry with a synaptic vesicle marker, we found that α-synuclein expression, while present in the soma and axons, is enriched at presynaptic terminals. This finding is consistent with reports of α-synuclein’s presynaptic localization in other fish, rodent, and human neurons (Maroteaux et al., 1988; Maroteaux and Scheller, 1991; Jakes et al., 1994), and suggests that α-synuclein localizes normally in zebrafish. In axonal branches, approximately 80% of both wild-type and A53T α-synuclein-GFP localized to the synapse, compared to only 40% of free GFP, indicating that α-synuclein preferentially localizes to presynaptic terminals, and that a smaller fraction of α-synuclein is freely diffusing through the axon compared to free GFP. As punctate synaptic enrichment of α-synuclein has been widely observed in neurons expressing α-synuclein (Withers et al., 1997; Spinelli et al., 2014; Taguchi et al., 2014), our results suggest that human α-synuclein-GFP is trafficked normally in zebrafish and could be assuming normal synaptic function by binding to synaptic vesicles (Maroteaux et al., 1988; reviewed in Sulzer and Edwards, 2019). In other studies, expression of α-synuclein in zebrafish led to neuronal toxicity by 3 dpf (Prabhudesai et al., 2012; O’Donnell et al., 2014). Here, α-synuclein-GFP persisted in healthy motor neurons until at least 8 dpf. This difference may be attributable to expression levels and/or our use of the neuroD promoter. In the future it will be interesting to use different promoters, including those that express selectively in other important populations of brain and spinal cord neurons – like midbrain dopamine neurons – to study presynaptic terminal α-synuclein-GFP aggregation with similar approaches.
Our work complements previous zebrafish expression models in that we used a tagged form of α-synuclein specifically so that we can measure and quantify protein mobility. Our measurements of the sub-cellular mobility of α-synuclein using FRAP provide further evidence that human α-synuclein expressed in zebrafish neurons is assuming a role similar to that seen in mammals (Spinelli et al., 2014). First, through rapid measurements of sub-cellular mobility, we detected a population of α-synuclein-GFP with recovery indistinguishable from free GFP, indicating that a pool of α-synuclein-GFP is freely diffusing through the cell and that the GFP tag does not by itself promote protein aggregation. Although the α-synuclein-GFP hybrid protein is larger than GFP, the 1.5-fold mass increase introduced by α-synuclein is unlikely to be detected, since FRAP resolution appears to be limited to about ten-fold differences in molecular weight (Lippincott-Schwartz et al., 2001). The recovery constant, tau, for this fast recovering pool was approximately 100 ms, which is essentially identical to the recovery we previously observed in a mouse model of α-synuclein-GFP (102 msec; Spinelli et al., 2014). Second, using longer term FRAP experiments we detected a slower pool of α-synuclein that was mobile on the minute time scale (2.8 min). The mobility of this second pool of α-synuclein-GFP likely corresponds to the binding and unbinding of the protein to synaptic vesicles; α-synuclein has been shown in in vivo systems to undergo binding events on this time scale (Spinelli et al., 2014, 2015). The corresponding intermediate pool of slowly recovering α-synuclein-GFP detected in in vivo FRAP experiments in mouse has a very similar recovery constant, tau, of 2.6 ± 2.5 min (Spinelli et al., 2014). This suggests that in the zebrafish and mouse models α-synuclein has a similar binding affinity for the highly curved lipid membranes found in fish and mammalian synaptic vesicles.
Our findings that human α-synuclein is trafficked normally to the synapse and binds synaptic vesicles are important, as zebrafish lack an ortholog to human α-synuclein, although they do express orthologs to β- and γ-synucleins (Sun and Gitler, 2008). The β and γ forms of synuclein may aggregate in vivo (Lulla et al., 2016; Prabhudesai et al., 2016), and exogenous α-synuclein expressed in zebrafish has been associated with cellular toxicity (Prabhudesai et al., 2012; O’Donnell et al., 2014; reviewed in Barnhill et al., 2020). Reports that addition of human α-synuclein mRNA can rescue hypokinesia induced by double knockdown of zebrafish β- and γ-synucleins (Milanese et al., 2012) further support our assessment that the zebrafish nervous system is a viable platform to study both the normal and disease-relevant behavior of α-synuclein. The model system that we introduce here specifically allows for quantification of α-synuclein aggregation using in vivo imaging approaches. In order to perform these in vivo approaches, our experiments necessarily needed to occur at a larval stage in development when zebrafish are optically transparent. It will be interesting in the future to develop approaches that can image zebrafish in the adult to determine whether similar effects on presynaptic terminal α-synuclein aggregation are also present at later developmental stages.
Beyond demonstrating the ability of human α-synuclein-GFP to function in zebrafish neurons, our experiments in the zebrafish nervous system help clarify how human α-synuclein may be acting pathologically. Our in vivo FRAP experiments measuring sub-cellular α-synuclein-GFP mobility detected a third pool of α-synuclein-GFP at presynaptic terminals that did not recover fully within 15 min post-bleach, suggesting a pool of aggregated α-synuclein. On average, presynaptic terminals contained an immobile α-synuclein fraction of around 25% at 4 dpf, although there was significant variability from terminal to terminal, possibly due to varying levels of protein expression. In experiments performed in the in vivo mouse brain, a similar immobile fraction of α-synuclein is also observed (Spinelli et al., 2014). Additionally, in mouse cortical neurons the fraction of total synaptic α-synuclein that was resistant to proteinase K digestion, a biochemical property of aggregated α-synuclein (Miake et al., 2002), also falls within this range (Spinelli et al., 2014). Varying protein levels across neurons that result from transient expression via microinjection in zebrafish embryos (Williams et al., 1996) may account in part for the range of immobile fraction that we measured here. Taken together, our results suggest that the immobile fraction of α-synuclein-GFP that we detected in zebrafish likely corresponds to a pool of aggregated α-synuclein species, and that steady-state fractions of aggregated α-synuclein in zebrafish are similar to what has been observed in the mammalian brain (Spinelli et al., 2014). Interestingly, we detected the formation of nitrated α-synuclein species within the presynaptic terminal. Nitration of proteins is often due to oxidative stress and our results suggest that terminal mitochondria may be affected by α-synuclein aggregation in such a way as to promote an oxidizing environment, as has been shown to occur in other α-synuclein-expressing systems (Parihar et al., 2008; Ganjam et al., 2019). This could cause a feedforward cycle, leading to α-synuclein nitration, misfolding and further mitochondrial impairments.
We also investigated the mobility of α-synuclein containing the A53T mutation seen in human patients (Polymeropoulos et al., 1997). In our experiments, the average immobile fraction of pooled A53T α-synuclein-GFP was not significantly different from either WT α-synuclein-GFP or free GFP). It is possible that subtle effects of the A53T mutation were not resolved here, however. A53T α-synuclein-GFP expression levels were, on the whole, lower than that of wild-type α-synuclein-GFP and, in light of the relationship between protein levels and aggregation, it is possible that the low immobile fraction of A53T α-synuclein was due to an insufficient protein level to induce substantial aggregation.
4.2. Alpha-synuclein phosphorylation at serine-129
Although zebrafish do not contain an ortholog of α-synuclein, our immunohistochemical data show that heterologous expression of the human protein in this system leads to phosphorylation at serine-129 in presynaptic terminals, as occurs in mammals. The specific PLK inhibitor, BI 2536, abolished serine-129 phosphorylation. These data suggest that the PLK kinase family, strongly implicated as being important for mammalian synucleins (Inglis et al., 2009; Mbefo et al., 2010; Oueslati et al., 2013), is also active within zebrafish neurons and can phosphorylate α-synuclein at serine-129. PLK may also phosphorylate β-synuclein in zebrafish as it does in mammals (Mbefo et al., 2010). Since the transparency of zebrafish enables visualization and spatial distinction of individual neurons throughout the nervous system, we were able to detect an interesting multimodal pattern to the phosphorylation of α-synuclein, with whole axonal arbors showing either robust serine-129 phosphorylation, very little or none. This type of variability could reflect differences in zebrafish PLK expression or some other form of protein modulation that affects axonal arbors in motor neurons. It may also reveal a progressive, stepwise pattern to phosphorylation within a given neuron. In vivo experiments in zebrafish will be particularly useful for exploring this possibility; the view of whole axonal arbors will readily allow for quantifying spatial dynamics within neurons as well as across neighboring neurons over time.
An important, additional factor that could be influencing whether phosphorylation (and possibly aggregation) may initiate in a particular neuron is the intracellular concentration of α-synuclein. While not always the case, some neurons with the most pervasive phosphorylation also appeared to have high levels of α-synuclein-GFP expression. This observation is consistent with findings from the mouse studies of α-synuclein pathology, which found that the immobile fraction and extent of proteinase-K resistant α-synuclein (an alternate marker of aggregates) were both found to be correlated with synaptic concentration of α-synuclein (Unni et al., 2010; Spinelli et al., 2014). Moreover, α-synuclein concentration has been highly implicated in initiating human PD, as duplication or triplication of the α-synuclein gene can lead to dominant, heritable forms of early-onset PD (Singleton et al., 2003, 2004; Chartier-Harlin et al., 2004; Ibáñez et al., 2004). In transient zebrafish expression, protein levels can vary among neurons within a given fish. This is a potential explanation for some of the variability we observed in both our measurements of the immobile fraction of α-synuclein and in the levels of phosphorylation. Generation of a stable transgenic zebrafish line expressing constant levels of α-synuclein would help to investigate further the origins of the variability that we have measured. A stable transgenic line should also lead to long-term expression of α-synuclein, allowing for investigations of its function beyond the minutes to days timescale highlighted here, potentially extending to the effects of α-synuclein aggregation on adult behavior.
4.3. Alpha-synuclein phosphorylation and aggregation
Since phosphorylation at serine-129 has been associated with aggregation of α-synuclein in Lewy bodies (Fujiwara et al., 2002; Anderson et al., 2006), we set out to test whether phosphorylation correlated with presynaptic aggregation of α-synuclein as measured by in vivo FRAP. Two different and complementary strategies – inhibiting phosphorylation either pharmacologically using BI 2536 or genetically via point mutation of the 129 site to aspartate or alanine – both showed the same result: that phosphorylation of this residue does not directly alter presynaptic aggregation as measured by FRAP. This complements our previous work in the mouse nervous system, which also showed that modulating serine-129 phosphorylation alone could be dissociated from presynaptic aggregation in vivo (Spinelli et al., 2015; Weston et al., 2021). Our findings suggest that aggregation of α-synuclein at presynaptic terminals may be independent of this posttranslational modification at serine-129, or may be governed by multiple factors, including other potential posttranslational modifications that are known to occur, such as additional phosphorylation events (e.g. a phosphorylation “code”; Lippens et al., 2012), nitration (He et al., 2019) and/or C-terminal truncation (Li et al., 2005). The importance of these and potentially other posttranslational modifications on the normal and pathological functions of α-synuclein are still to be determined. It is clear that modifications can alter α-synuclein interactions with other proteins present within the presynaptic terminal (El Turk et al., 2018), which could affect mobility as measured by FRAP or other techniques. It will be interesting in future work to dissect out the importance of relevant posttranslational modifications to α-synuclein’s interactions with both lipid membrane and protein components within the terminal.
5. Conclusions
In summary, our work shows that human α-synuclein can be expressed in zebrafish and that phosphorylation at serine-129 alone does not directly drive α-synuclein aggregation. The zebrafish system is a powerful in vivo model for quantifying and manipulating α-synuclein function in neurons (Barnhill et al., 2020), and specifically for dissecting the mechanisms involved in protein aggregation. The ability to visualize identifiable whole neurons at subcellular resolution throughout the nervous system allows us to investigate regional specificity and progression over time with high temporal resolution. Furthermore, this in vivo system can facilitate testing of specific biochemical and cell biological pathways to help determine potential treatments for neurodegenerative disease.
Supplementary Material
Acknowledgements
We would like to thank: Pamela McLean for α-synuclein DNA; Teresa Nicolson for p5E-neurod; Alex Nechiporuk and Kara Cerveny for sharing reagents and embryos; K. Maggie Carey for zebrafish maintenance and mating; and Greg Hermann, Nikolaus Loening, Yung-Pin Chen, Margaret Metz, Janis Lochner, Pamela Zobel-Thropp, Eve Lowenstein, Mako Gedi, and Molly Marra for helpful discussions.
Funding
This work was supported by the National Institutes of Health (Award NS069625, NS102227), the National Science Foundation (Awards 1553764, 1338188), the Fischer Family Foundation, the M.J. Murdock Charitable Trust (Awards 2011279, 2015256), and the John S. Rogers Research Program at Lewis & Clark College, Portland, OR.
Footnotes
Appendix A. Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2021.105291.
References
- Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J, Kling K, Lee M, Diep L, Keim PS, Shen X, Chataway T, Schlossmacher MG, Seubert P, Schenk D, Sinha S, Gai WP, Chilcote TJ, 2006. Phosphorylation of Ser-129 is the dominant pathological modification of α-Synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 281, 29739–29752. [DOI] [PubMed] [Google Scholar]
- Bae Y-K, Kani S, Shimizu T, Tanabe K, Nojima H, Kimura Y, Higashijima S, Hibi M, 2009. Anatomy of zebrafish cerebellum and screen for mutations affecting its development. Dev. Biol. 330, 406–426. [DOI] [PubMed] [Google Scholar]
- Barnhill L, Murata H, Bronstein JM, 2020. Studying the pathophysiology of Parkinson’s disease using Zebrafish. Biomedicines 8, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron M, Motter R, Tanaka P, Fauss D, Babcock M, Chiou SS, Nelson S, San Pablo F, Anderson JP, 2014. In vivo modulation of polo-like kinases supports a key role for PLK2 in Ser129 α-synuclein phosphorylation in mouse brain. Neuroscience 256, 72–82. [DOI] [PubMed] [Google Scholar]
- Bodner RA, Outeiro F, Altmann S, Maxwell MM, Cho SH, Hyman BT, Mclean PJ, Young AB, Housman DE, Kazantsev AG, 2006. Pharmacological promotion of inclusion formation: a therapeutic approach for Huntington’s and Parkinson’s diseases. PNAS 103 (11), 4246–4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolte S, Cordelières FP, 2006. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232. [DOI] [PubMed] [Google Scholar]
- Brockway NL, Cook ZT, O’Gallagher MJ, Tobias ZJC, Gedi M, Carey KM, Unni VK, Pan YA, Metz MR, Weissman TA, 2019. Multicolor lineage tracing using in vivo time-lapse imaging reveals coordinated death of clonally related cells in the developing vertebrate brain. Dev. Biol. 453 (2), 130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burré J, 2015. The synaptic function of α-Synuclein. J. Parkinsons Dis. 5, 699–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Fornai F, Kwon H-B, Yazdani U, Atasoy D, Liu X, Hammer RE, Battaglia G, German DC, Castillo PE, Südhof TC, 2004. Double-knockout mice for α–and β-synucleins: effect on synaptic functions. PNAS 101 (41), 14966–14971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier S, Duyckaerts C, 2018. Is Lewy pathology in the human nervous system chiefly an indicator of neuronal protection or of toxicity? Cell Tissue Res. 373, 149–160. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destée A, 2004. α-Synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169. [DOI] [PubMed] [Google Scholar]
- Chen L, Feany MB, 2005. α-Synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 8 (5), 657–663. [DOI] [PubMed] [Google Scholar]
- Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT, 2000. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. PNAS 97, 571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook ZT, Brockway NL, Weissman TA, 2020. Visualizing the developing brain in living Zebrafish using Brainbow and time-lapse confocal imaging. J. Vis. Exp. 157. [DOI] [PubMed] [Google Scholar]
- Core Team, R., 2017. R: A language and environment for statistical computing. In: R Foundation for Statistical Computing. Austria. URL, Vienna. https://www.R-project.org/. [Google Scholar]
- Davidson WS, Jonas A, Clayton DF, George JM, 1998. Stabilization of α-Synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 273 (16), 9443–9449. [DOI] [PubMed] [Google Scholar]
- The Zebrafish: genetics, genomics and informatics. In: Detrich HW III, Westerfield M, Zon LI (Eds.), 2011. Methods in Cell Biology, 3rd ddVol. 104. Elsevier, Boston. [Google Scholar]
- Duda JE, Giasson BI, Mabon ME, Lee VM, Trojanowski JQ, 2002. Novel antibodies to synuclein show abundant striatal pathology in Lewy body diseases. Ann. Neurol. 52 (2), 205–210. [DOI] [PubMed] [Google Scholar]
- El Turk F, De Genst E, Guilliams T, Fauvet B, Hejjaoui M, Di Trani J, Chiki A, Mittermaier A, Vendruscolo M, Lashuel HA, Dobson CM, 2018. Exploring the role of post-translational modifications in regulating α-synuclein interactions by studying the effects of phosphorylation on nanobody binding. Protein Sci. 27 (7), 1262–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elling RA, Fucini RV, Hanan EJ, Barr KJ, Zhu J, Paulvannan K, Yang W, Romanowski MJ, 2008. Structure of the Brachydanio rerio polo-like kinase 1 (Plk1) catalytic domain in complex with an extended inhibitor targeting the adaptive pocket of the enzyme. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 64, 686–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar VD, Kuo Y-M, Orrison BM, Giasson BI, Nussbaum RL, 2014. Transgenic mice expressing S129 phosphorylation mutations in α-synuclein. Neurosci. Lett. 563, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Febbraro F, Sahin G, Farran A, Soares S, Jensen PH, Kirik D, Romero-Ramos M, 2013. Ser129D mutant alpha-synuclein induces earlier motor dysfunction while S129A results in distinctive pathology in a rat model of Parkinson’s disease. Neurobiol. Dis. 56, 47–58. [DOI] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T, 2002. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 4, 160–164. [DOI] [PubMed] [Google Scholar]
- Ganjam GK, Bolte K, Matschke LA, Neitemeier S, Dolga AM, Höllerhage M, Höglinger GU, Adamczyk A, Decher N, Oertel WH, Culmsee C, 2019. Mitochondrial damage by α-synuclein causes cell death in human dopaminergic neurons. Cell Death Dis. 10 (11), 865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbatyuk OS, Li S, Sullivan LF, Chen W, Kondrikova G, Manfredsson FP, Mandel RJ, Muzyczka N, 2008. The phosphorylation state of Ser-129 in human alpha-synuclein determines neurodegeneration in a rat model of Parkinson disease. PNAS 105, 763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Yu Z, Chen S, 2019. Alpha-synuclein nitration and its implications in Parkinson’s disease. ACS Chem. Neurosci. 10 (2), 777–782. [DOI] [PubMed] [Google Scholar]
- Ibáñez P, Bonnet A-M, Débarges B, Lohmann E, Tison F, Agid Y, Dürr A, Brice A, Pollak P, 2004. Causal relation between α-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1169–1171. [DOI] [PubMed] [Google Scholar]
- Inglis KJ, Chereau D, Brigham EF, Chiou SS, Schöbel S, Frigon NL, Yu M, Caccavello RJ, Nelson S, Motter R, Wright S, Chian D, Santiago P, Soriano F, Ramos C, Powell K, Goldstein JM, Babcock M, Yednock T, Bard F, Basi GS, Sham H, Chilcote TJ, McConlogue L, Griswold-Prenner I, Anderson JP, 2009. Polo-like kinase 2 (PLK2) phosphorylates α-Synuclein at serine 129 in central nervous system. J. Biol. Chem. 284, 2598–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, Rohan de Silva H, Kittel A, Saitoh T, 1995. The precursor protein of non-Aβ component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 14, 467–475. [DOI] [PubMed] [Google Scholar]
- Jakes R, Spillantini MG, Goedert M, 1994. Identification of two distinct synucleins from human brain. FEBS Lett. 345, 27–32. [DOI] [PubMed] [Google Scholar]
- Jeong K, Jeong JY, Lee HO, Choi E, Lee H, 2010. Inhibition of PLK1 induces mitotic infidelity and embryonic growth defects in developing zebrafish embryos. Dev. Biol. 345 (1), 34–48. [DOI] [PubMed] [Google Scholar]
- Kowal SL, Dall TM, Chakrabarti R, Storm MV, Jain A, 2013. The current and projected economic burden of Parkinson’s disease in the United States. Mov. Disord. 28, 311–318. [DOI] [PubMed] [Google Scholar]
- Kramer ML, Schulz-Schaeffer WJ, 2007. Presynaptic α-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J. Neurosci. 27, 1405–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schols L, Riess O, 1998. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat. Genet. 18, 106–108. [DOI] [PubMed] [Google Scholar]
- Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, Campbell DS, Parant JM, Yost HJ, Kanki JP, Chien C-B, 2007. The Tol2kit: a multisite gateway-based construction kit forTol2 transposon transgenesis constructs. Dev. Dyn. 236, 3088–3099. [DOI] [PubMed] [Google Scholar]
- Lesage S, Anheim M, Letournel F, Bousset L, Honoré A, Rozas N, Pieri L, Madiona K, Dürr A, Melki R, Verny C, Brice A, 2013. G51D α-synuclein mutation causes a novel Parkinsonian-pyramidal syndrome. Ann. Neurol. 73, 459–471. [DOI] [PubMed] [Google Scholar]
- Li W, West N, Colla E, Pletnikova O, Troncoso JC, Marsh L, Dawson TM, Jäkälä P, Hartmann T, Price DL, Lee MK, 2005. Aggregation promoting C-terminal truncation of α-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. PNAS 102, 2162–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippens G, Amniai L, Wieruszeski J-M, Sillen A, Leroy A, Landrieu I, 2012. Towards understanding the phosphorylation code of tau. Biochem. Soc. Trans. 40 (4), 698–703. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Snapp E, Kenworthy A, 2001. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2, 444–456. [DOI] [PubMed] [Google Scholar]
- Lulla A, Barnhill L, Bitan G, Ivanova MI, Nguyen B, O’Donnell K, Stahl MC, Yamashiro C, Klärner FG, Schrader T, Sagasti A, Bronstein JM, 2016. Neurotoxicity of the parkinson disease-associated pesticide ziram is synuclein-dependent in zebrafish embryos. Environ. Health Perspect. 124, 1766–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroteaux L, Scheller RH, 1991. The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Mol. Brain Res. 11, 335–343. [DOI] [PubMed] [Google Scholar]
- Maroteaux L, Campanelli JT, Scheller RH, 1988. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 8 (8), 2804–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbefo MK, Paleologou KE, Boucharaba A, Oueslati A, Schell H, Fournier M, Olschewski D, Yin G, Zweckstetter M, Masliah E, Kahle PJ, Hirling H, Lashuel HA, 2010. Phosphorylation of Synucleins by members of the polo-like kinase family. J. Biol. Chem. 285, 2807–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland NR, Fan Z, Xu K, Schwarzschild MA, Feany MB, Hyman BT, McLean PJ, 2009. α-Synuclein S129 phosphorylation mutants do not Alter nigrostriatal toxicity in a rat model of Parkinson disease. J. Neuropathol. Exp. Neurol. 68, 515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean PJ, Kawamata H, Hyman BT, 2001. α-Synuclein-enhanced green fluorescent protein fusion proteins form proteasome sensitive inclusions in primary neurons. Neuroscience 104, 901–912. [DOI] [PubMed] [Google Scholar]
- Miake H, Mizusawa H, Iwatsubo T, Hasegawa M, 2002. Biochemical characterization of the Core structure of α-Synuclein filaments. J. Biol. Chem. 277 (21), 19213–19219. [DOI] [PubMed] [Google Scholar]
- Middleton ER, Rhoades E, 2010. Effects of curvature and composition on α-synuclein binding to lipid vesicles. Biophys. J. 99, 2279–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanese C, Sager JJ, Bai Q, Farrell TC, Cannon JR, Greenamyre JT, Burton EA, 2012. Hypokinesia and reduced dopamine levels in zebrafish lacking β- and γ1-synucleins. J. Biol. Chem. 287, 2971–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers PZ, 1985. Spinal motoneurons of the larval zebrafish. J. Comp. Neurol. 236, 555–561. [DOI] [PubMed] [Google Scholar]
- Myers PZ, Eisen JS, Westerfield M, 1986. Development and axonal outgrowth of identified Motoneurons in the Zebrafish. J. Neurosci. 6 (8), 2278–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusslein-Volhard C, Dahm R, 2002. Zebrafish; A Practical Approach. Oxford University Press. [Google Scholar]
- O’Donnell KC, Lulla A, Stahl MC, Wheat ND, Bronstein JM, Sagasti A, 2014. Axon degeneration and PGC-1α-mediated protection in a zebrafish model of α-synuclein toxicity. Dis. Model. Mech. 7, 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterberg VR, Spinelli KJ, Weston LJ, Luk KC, Woltjer RL, Unni VK, 2015. Progressive aggregation of alpha-synuclein and selective degeneration of lewy inclusion-bearing neurons in a mouse model of parkinsonism. Cell Rep. 10, 1252–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oueslati A, Schneider BL, Aebischer P, Lashuel HA, 2013. Polo-like kinase 2 regulates selective autophagic α-synuclein clearance and suppresses its toxicity in vivo. PNAS 110 (41), E3945–E3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panzer JA, Gibbs SM, Dosch R, Wagner D, Mullins MC, Granato M, Balice-Gordon RJ, 2005. Neuromuscular synaptogenesis in wild-type and mutant zebrafish. Dev. Biol. 285, 340–357. [DOI] [PubMed] [Google Scholar]
- Parihar MS, Parihar A, Fujita M, Hashimoto M, Ghafourifar P, 2008. Mitochondrial association of alpha-synuclein causes oxidative stress. Cell. Mol. Life Sci. 65 (7–8), 1272–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasanen P, Palin E, Pohjolan-Pirhonen R, Pöyhönen M, Rinne JO, Päivärinta M, Martikainen MH, Kaasinen V, Hietala M, Gardberg M, Saukkonen AM, Eerola-Rautio J, Kaakkola S, Lyytinen J, Tienari PJ, Paetau A, Suomalainen A, Myllykangas L, 2017. SNCA mutation p.Ala53Glu is derived from a common founder in the Finnish population. Neurobiol. Aging 50, 168.e5–168.e8. [DOI] [PubMed] [Google Scholar]
- Perrin RJ, Payton JE, Barnett DH, Wraight CL, Woods WS, Ye L, George JM, 2003. Epitope mapping and specificity of the anti-α-synuclein monoclonal antibody Syn-1 in mouse brain and cultured cell lines. Neurosci. Lett. 349, 133–135. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL, 1997. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- Prabhudesai S, Sinha S, Attar A, Kotagiri A, Fitzmaurice AG, Lakshmanan R, Ivanova MI, Loo JA, Klärner FG, Schrader T, Stahl M, Bitan G, Bronstein JM, 2012. A novel “molecular tweezer” inhibitor of α-synuclein neurotoxicity in vitro and in vivo. Neurotherapeutics 9, 464–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhudesai S, Bensabeur FZ, Abdullah R, Basak I, Baez S, Alves G, Holtzman NG, Larsen JP, Møller SG, 2016. LRRK2 knockdown in zebrafish causes developmental defects, neuronal loss, and synuclein aggregation. J. Neurosci. Res. 94, 717–735. [DOI] [PubMed] [Google Scholar]
- Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL, Houlden H, Schapira AH, 2013. A novel α-synuclein missense mutation in Parkinson disease. Neurology 80, 1062–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaser AJ, Osterberg VR, Dent SE, Stackhouse TL, Wakeham CM, Boutros SW, Weston LJ, Owen N, Weissman TA, Luna E, Raber J, Luk KC, McCullough AK, Woltjer RL, Unni VK, 2019. Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci. Rep. 9, 10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A, 2012. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9 (7), 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DA, Tabarean I, Tang Y, Cartier A, Masliah E, Roy S, 2010. A pathologic cascade leading to synaptic dysfunction in α-synuclein-induced neurodegeneration. J. Neurosci. 30, 8083–8095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K, 2003. Alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302 (5646), 841. [DOI] [PubMed] [Google Scholar]
- Singleton A, Gwinn-Hardy K, Sharabi Y, Li S-T, Holmes C, Dendi R, Hardy J, Singleton A, Crawley A, Goldstein DS, 2004. Association between cardiac denervation and parkinsonism caused by a-synuclein gene triplication. Brain 127, 768–772. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM-Y, Trojanowski JQ, Jakes R, Goedert M, 1997. α-Synuclein in Lewy bodies. Nature 388, 839–840. [DOI] [PubMed] [Google Scholar]
- Spinelli KJ, Taylor JK, Osterberg VR, Churchill MJ, Pollock E, Moore C, Meshul CK, Unni VK, 2014. Presynaptic alpha-Synuclein aggregation in a mouse model of Parkinson’s disease. J. Neurosci. 34 (6), 2037–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinelli KJ, Osterberg VR, Meshul CK, Soumyanath A, Unni VK, 2015. Curcumin treatment improves motor behavior in α-Synuclein transgenic mice. PLoS One 10, e0128510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Edwards RH, 2019. The physiological role of α-synuclein and its relationship to Parkinson’s disease. J. Neurochem. 150, 475–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Gitler AD, 2008. Discovery and characterization of three novel synuclein genes in zebrafish. Dev. Dyn. 237, 2490–2495. [DOI] [PubMed] [Google Scholar]
- Taguchi K, Watanabe Y, Tsujimura A, Tatebe H, Miyata S, Tokuda T, Mizuno T, Tanaka M, 2014. Differential expression of alpha-synuclein in hippocampal neurons. PLoS One 9 (2), e89327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Kim YM, Lee G, Junn E, Iwatsubo T, Mouradian MM, 2004. Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. J. Biol. Chem. 279, 4625–4631. [DOI] [PubMed] [Google Scholar]
- Toni M, Cioni C, 2015. Fish synucleins: an update. Mar. Drugs. 13 (11), 6665–6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsika E, Moysidou M, Guo J, Cushman M, Gannon P, Sandaltzopoulos R, Giasson BI, Krainc D, Ischiropoulos H, Mazzulli JR, 2010. Distinct region-specific alpha-synuclein oligomers in A53T transgenic mice: implications for neurodegeneration. J. Neurosci. 30 (9), 3409–3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unni VK, Weissman TA, Rockenstein E, Masliah E, McLean PJ, Hyman BT, 2010. In vivo imaging of α-synuclein in mouse cortex demonstrates stable expression and differential subcellular compartment mobility. PLoS One 5 (5), e10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman EA, Duda JE, Giasson BI, 2008. Characterization of antibodies that selectively detect alpha-synuclein in pathological inclusions. Acta Neuropathol. 116 (1), 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M, McMurray JV, Eisen JS, 1986. Identified motoneurons and their innervation of axial muscles in the zebrafish. J. Neurosci. 6, 2267–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston LJ, Stackhouse TL, Spinelli KJ, Boutros SW, Rose EP, Osterberg VR, Luk KC, Raber J, Weissman TA, Unni VK, 2021. Polo-like kinase 2 genetic deletion reduces alpha-synuclein serine-129 phosphorylation in presynaptic terminals but not Lewy bodies. J. Biol. Chem. 10.1016/j.jbc.2021.100273. In press; PMID: 33428941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DW, Müller F, Lavender FL, Orbán L, Maclean N, 1996. High transgene activity in the yolk syncytial layer affects quantitative transient expression assays in zebrafish (Danio rerio) embryos. Transgenic Res. 5, 433–442. [DOI] [PubMed] [Google Scholar]
- Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, Hetzer C, Loher T, Vilar M, Campioni S, Tzitzilonis C, Soragni A, Jessberger S, Mira H, Consiglio A, Pham E, Masliah E, Gage FH, Riek R, 2011. In vivo demonstration that α-synuclein oligomers are toxic. PNAS 108, 4194–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withers GS, George JM, Banker GA, Clayton DF, 1997. Delayed localization of synelfin (synuclein, NACP) to presynaptic terminals in cultured rat hippocampal neurons. Dev. Brain Res. 99, 87–94. [DOI] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Tortosa EG, del Ser T, Muñoz DG, de Yebenes JG, 2004. The new mutation, E46K, of α-synuclein causes parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.