

Introduction and importance:
Vanishing white matter (VWM) is a neurological disorder that has an autosomal recessive mode of inheritance. VWM is caused due to a mutation in in any of the five genes of eukaryotic translation initiation factor 2B (eIF2B). The etiology is unknown.
Case presentation:
The authors report two cases of VWM disease. In the first case, an 8-month-old female child, brought to the pediatric clinic with seizure and loss of consciousness. The second case was a 24-month-old girl, presented with weakness, a disability to walk and swallow, and poor feeding. Her brain MRI demonstrated cystic changes (white matter rarefaction) in supratentorial peri-ventricular white matter and genetic testing result showed an EIF2B3 gene mutation.
Clinical discussion:
Leukoencephalopathy with VWM, also known as Cree encephalopathy is caused by mutations in the EIF2B gene. The disease is inherited in an autosomal recessive fashion. There are various agents leading to symptoms and signs of VWM disease. Physical stress like head trauma even in a mild degree, infections, and febrile diseases can be mentioned as causes of VWM. The eIF2B complex, plays a role as an important factor in the regulation of protein synthesis in cells under different conditions.
Conclusion:
As a conclusion, genetic counseling could be recommended to all individuals with VWM disease and their family members for next pregnancies and possible precautions for consanguineous marriages.
Keywords: exome sequencing, magnetic resonance imaging, point mutation
Introduction
Highlights
The result of this study demonstrated that:
Vanishing white matter (VWM) Leukoencephalopathy can be diagnosed by means of clinical and molecular genetic tests.
We had access to both of diagnostic aspects and confirmed the disease. When MRI findings, signs and symptoms are consistent with VWM disease, genetic testing can be performed as a confirmatory test.
Therefore, genetic counseling could be recommended to all individuals with VWM disease and their family members for their next pregnancies and possible precautions for consanguineous marriages.
Leukoencephalopathy with vanishing white matter (VWM), is an autosomal recessive neurological disease. It is also known as childhood ataxia with central hypomyelination disease1. It was first described in infantile form in 1988 in Canada. VWM can affect people of all ages and the infantile form is the most severe type. It commonly appears in children of ages 1–5 years2. The incidence is estimated 1 in 80 000 live births. In 42 of 210 families, there was parental consanguinity3.
VWM disease is characterized by chronic and progressive childhood ataxia, spasticity, and optic atrophy4. There are few biochemical markers for VWMD. The first marker found was an increase of cerebrospinal fluid glycine concentrations. Recently identified biomarker is the reduction of asialotransferrin in cerebrospinal fluid concentration5. It is caused by mutations in any of the five genes encoding the subunits of the eukaryotic translation initiation factor 2B (eIF2B). eIF2B is a significant factor for the translation initiation of mRNA into proteins6. Given that, at the moment, VWM has no specific treatment5. We would like to mention that although VWM has been known as a rare disease, we identified two cases in the same region in Iran. In addition, it is important to state that both patients were born of consanguineous marriage.
Case presentation
We report two cases of VWM diseases in Gorgan, Iran. Our first case is an 8-month-old female child, born in the 35th week of gestational age by Cesarean Section (C-section) due to preterm rupture of membrane. The initial neurological assessment was normal at birth. Her APGAR score at 1 and 5 min after birth was 8 and 9, respectively. The patient’s birth weight was 1900 g (10%).The weight of the hospitalization time was 4800 g (below 5th percentile), head circumference was 41 cm (10th percentile), and supine length was 61 cm (below 5th percentile). Vaccination report was up to date. There was developmental deterioration as the patient was not able to sit and grab objects. She was the first issue of consanguineous parents (cousins). The patient was also born with maternal hypothyroidism. A history of neurological and familial disorders were not reported by parents.
Her first hospital admission was at the age of 4-month-old, by complaint of seizure. The patient has been on antiseizure medication since then. In her 8 months, she was presented with seizure and loss of consciousness. She has suffered from poor feeding since the last day. No history of head trauma was mentioned.
On her physical examination, the child was ill-looking and pale. Her vital signs were: BP: 73/30 mmHg, HR: 180/min, RR=28/min, T: 37.2°C and with SPO2: 95%, blood sugar: 75 mg/dl. Her neurological examination included spasticity, a reduced tone and power, a neutral plantar reflex, and an increased deep tendon reflex. Co-ordination and gait could not be assessed. Sensory function and cranial nerve examination was intact. Other systems physical examination was normal. Her lab results showed respiratory alkalosis and metabolic acidosis. A high performance liquid chromatography plasma amino acid profile test was done. The lab results showed: lowering of the levels of Tyrosine and Leucine, an elevated level of Glycine, and a normal range of other Amino acids. EEG demonstrated generalized slow wave. Brain MRI was obtained and reported by an expert radiologist. Imaging findings represented: (a) axial T2 (b) sagittal T2 (c) axial fluid attenuated inversion recovery (FLAIR) at the level of cerebellum (d) coronal FLAIR images of the brain showing diffuse supratentorial white matter high signal abnormalities in T2 images, with associated FLAIR high signal abnormalities in the deep white matter of both cerebellar hemispheres. In axial FLAIR image and subcortical U fiber high signal intensity changes were seen in coronal FLAIR images. In addition, there are some early cystic changes in supratentorial peri-ventricular white matter for this child, all considered important MR imaging features of VWM syndrome (Figure 1). Also, routine blood examination performed presented in Table 1.
Figure 1.
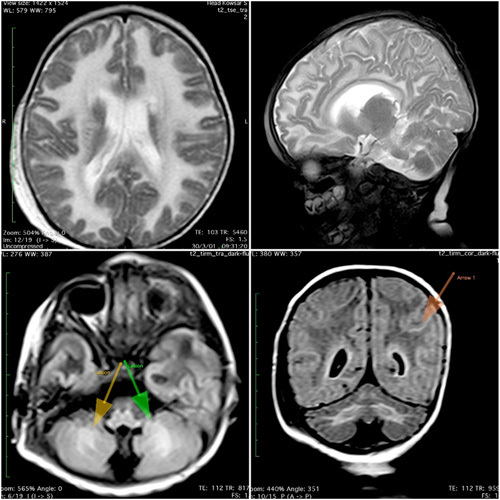
Brain MRI of case one.
Table 1.
Routine blood examination performed.
| Biochemistry | Case 1 | Case 2 | Unit |
|---|---|---|---|
| WBC | 13 900 | 23 900 | mm |
| RBC | 4.79 | 4.92 | mil/cumm |
| Hb | 12.3 | 12.4 | gr/dl |
| Hct | 38.3 | 41.0 | % |
| MCV | 79.96 | 83.33 | fl |
| MCH | 25.68 | 25.2 | Pg |
| Plt | 252 000 | 251 000 | µL |
| Poly | 70 | 89 | % |
| Lymph | 30 | 11 | % |
| BUN | 8 | 35 | mg/dl |
| Cr | 0.5 | 0.9 | mg/dl |
| Na | 137 | 140 | mEq/L |
| K | 4.3 | 4.1 | mEq/L |
| Calcium | 8 | 11.5 | mg/dl |
| Phosphorus | 4.5 | 2.6 | mg/dl |
| Mg | 2.6 | 1.8 | mg/dl |
| Albumin | 3.3 | 2.8 | mg/dl |
| TG | 136 | – | mg/dl |
| Cholesterol | 210 | – | mg/dl |
| Blood Sugar | 119 | 154 | mg/dl |
| AST | 10 | 20 | U/L |
| ALT | 12 | 10 | U/L |
| ALP | 539 | 299 | U/L |
| ESR | 4 | 35 | – |
| CRP | Negative | 1+ | – |
| Blood group , Rh | O positive | O positive | |
| VBG | |||
| PH | 7.44 | 7.45 | |
| P02 | 97.9 | 50 | mmHg |
| PCO2 | 52.3 | 40.6 | mmHg |
| HCO3A | 34.8 | mmol/L | |
| HCO3S | 31.5 | mmol/L | |
| SEROLOGY | |||
| Amoniac | 68 | 103 | mg/dl |
| Lactat | 26 | 11 | mg/dl |
| Blood Homocysteine | 5.92 | – | µmol/L |
| Urine Homocysteine | 0.5 | – | µmol/L |
µL, microliter; µmol/L, micromoles per liter; ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BUN, blood urea nitrogen; Cr, creatinine; CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; fl, femtoliters; gr/dl, grams per deciliter; Hb, hemoglobin; Hct, hematocrit; K, potassium; MCH, mean corpuscular hemoglobin; MCV, mean corpuscular volume; mg/dl, milligram per deciliter; Mg, magnesium; mil/cumm, million per cubic millimeter; mm, millimeter; mmHg, millimeters of mercury; mmol/L, millimoles per liter; Na, sodium; pg, picograms; Plt, platelet; RBC, red blood cell count; Rh, rhesus; TG, triglyceride; U/L, unit per liter; VBG, venous blood gas; WBC, white blood cell count.
According to the history, physical examination, and MRI reports, the probable diagnosis could be VWM disease. Molecular analysis confirmed the eIF2B5 gene mutation Table 2.
Table 2.
Whole Exome Sequencing (WES).
| Patient | Gene and transcript | Variant | Disease | exon | Zygosity | Variant classification |
|---|---|---|---|---|---|---|
| Case 1 | eIF2B5 (NM-003907.3) |
Chr3:183855548 A>G: c.461A>G: p.Asp154Gly. |
Leukoencephalopathy with vanishing white matter (MIM:603896). |
3 | Homozygote AR | Likely pathogenic |
| Case 2 | eIF2B3 | c.937G>A p.Va1313Met NM-020365.5 |
Leukoencephalopathy with vanishing white matter. | – | – | Likely pathogenic |
The other case we report is a 28-month-old girl born in 37th week of gestational age, an uncomplicated pregnancy, by C-Section arising from repeated C-Section with normal initial neurological assessment. Her APGAR score at 1 and 5 min after birth was 7 and 9, respectively. The growth details were as the following: Birth weight: 3500 g (50–75% normal percentile). At the time of admission: Weight: 8800 g (<5th percentile), height:47 cm (<5th percentile), head circumference: 86 cm (<5th percentile). Her developmental status gradually had faced progressive delay since 12 months without any other complaints. Vaccination state was done according to the national immunization program in Iran.
She was the second issue of third-degree consanguineous marriage (parents were cousins). Parents and the older sibling were normal and healthy and there was not any remarkable history of familial genetic disorders. At the age of 24 months, she was presented with weakness, a disability to walk and swallow, poor feeding, and difficulty at standing without support. Head trauma was not reported. On her physical examination, the child appeared ill. Her vital signs were: BP=95/50 mmHg, HR=122/min, RR=24/min, T=37°C. On central nervous system examination, upper motor neuron signs were detected in her limbs with intact sensory functions and cranial nerves. Her other general physical examination was normal. EEG results showed generalized slow wave. Brain imaging revealed: axial T2 in fronto-temporal lobes; axial T2 in mid cerebellum; axial FLAIR at the level of fronto-temporal lobes; and axial FLAIR images in mid cerebellum, all of brain, showing diffuse supratentorial white matter high signal abnormalities in T2 and FLAIR images, with associated T2/FLAIR high signal abnormalities in deep white matter of both cerebellar hemispheres (arrows in b and d) and cystic changes (white matter rarefaction) in supratentorial peri-ventricular white matter for this child (arrows in c), all considered important MR imaging features of VWM syndrome (Figure 2). Also, routine blood examination performed presented in Table 1.
Figure 2.

Brain MRI of case two.
The patient was suspicious to VWM. Thus, whole exome sequencing was performed, and the results showed an EIF2B3 gene mutation (Table 2).
Discussion
Leukoencephalopathy with VWM, also known as Cree encephalopathy is caused by mutations in EIF2B gene. The disease is inherited in an autosomal recessive fashion syndrome (Figure 1)2. In this case report study, the cases carry this mutations despite they are from Iranian nationality. Patients with onset at less than 1 year consistently have a rapidly progressive disease and might even die within several months. Many of cases show epilepsy3. Our study confirms that VWM has an extremely broad phenotypic range and that age at onset is an important determinant of prognosis. The basic defect of this disease is associated with one of the five subunits of eukaryotic translation initiation factor eIF2B which is essential for protein synthesis and its regulation under the stress conditions7.
There are various agents leading symptoms and signs of VWM disease. Physical stress like head trauma even in a mild degree, infections and febrile diseases can be mentioned as causes of VWM. The eIF2B complex, plays role as an important factor in the regulation of protein synthesis in cells under different conditions, in the condition of different stress such as fever, etc.8.
Pathological characteristics of VWM include increasing white matter rarefaction and cystic degeneration, highly characteristic foamy oligodendrocytes with oligodendrocytosis, meager astrogliosis with dysmorphic astrocytes, and loss of oligodendrocytes because of apoptosis, despite their exact pathophysiology would be poorly discovered9 and also in our case series’ MRI all features are evaluated and cystic changes and such others could be seen.
MRI feature of VWM disease include white matter abnormalities and has been used for years as a critical step in diagnosis. Different MRI sequences including T1W, T2W, and FLAIR signals vary in their sensitivity to different structural components of the brain matter10.
The present study reports the two cases of VWM in Iran. It had strengths and limitations. We had full access to the medical profile of patients, MRI center, and medical genetic laboratory. One of the limitations was that the genetic tests were expensive and costly (in comparison with the routine blood tests and hospitalization) and also time-consuming. Moreover, convincing parents about the necessity of genetic study on their children was another issue. Although, we have illustrated two pediatric cases of VWM in Iran, in the preceding discussion, there are a number of special situations warranting further study like assessing acquired (trauma, febrile conditions) and hereditary (consanguinity of parents ) causes of the disease.
Conclusion
As a conclusion VWM Leukoencephalopathy can be diagnosed by means of clinical and molecular genetic tests. We had access to both of diagnostic aspects and confirmed the disease. When MRI findings, signs and symptoms are consistent with VWM disease, genetic testing can be performed as a confirmatory test. Therefore, genetic counseling could be recommended to all individuals with VWM disease and their family members for next pregnancies and possible precautions for consanguineous marriages. Also, given that causes of VWM that is not hereditary (e.g. trauma, infections, etc.). This distinction between hereditary and ‘acquired’ (i.e. secondary to other pathological conditions) VWM useful for more study and follow.
Ethical approval
NA.
Consent to participate
Written informed consent was obtained from the patient for the publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request. The whole research was done under the permission of the Ethics committee of GolestanUniversity of Medical Sciences and also Genetic Testing for parents was done because of their.
Sources of funding
None.
Author contribution
S.A.H. and M.G.G.: diagnosed, and managed this patient and interpretation; A.K., A.L., and P.S.H.: writing the first manuscript draft; M.G.G. revised the manuscript and finalized the draft.
Conflicts of interest disclosure
The authors declare that they have no financial conflict of interest with regard to the content of this report.
Research registration unique identifying number (UIN)
Name of the registry:
Unique Identifying number or registration ID:
Hyperlink to your specific registration (must be publicly accessible and will be checked):
Guarantor
Hossein-Ali Nikbakht, Babol University of Medical Sciences, Babol 47176-47745, Iran. Tel: +98 9119122546, fax: +98 1132363857, E-mail: ep.nikbakht@gmail.com.
Data availability statement
The datasets are available from the corresponding author on reasonable request.
Provenance and peer review
Not commissioned, externally peer-reviewed.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Published online 28 June 2023
Contributor Information
Atefe Kami, Email: atefeh.kami@gmail.com.
Alale Langari, Email: alale.langari@gmail.com.
Mohammad H. Gharib, Email: drgharib@goums.ac.ir.
Mousa Ghelichi-Ghojogh, Email: m.ghelichi97@gmail.com.
Parmis S. Hosseini, Email: mghelichi2000@yahoo.com.
Seyed A. Hosseini, Email: sahmadhosseini2023@gmail.com.
References
- 1.Filareto I, Cinelli G, Scalabrini I, et al. EIF2B2 gene mutation causing early onset vanishing white matter disease: a case report. Ital J Pediatr 2022;48:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robbins K, Arraj P, Sanchez LD, et al. CT and MRI findings in infantile vanishing white matter. Radiol Case Rep 2021;16:116–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hamilton EMC, van der Lei HDW, Vermeulen G, et al. Natural history of vanishing white matter. Ann Neurol 2018;84:274–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Navya B, Kondle VK, Uppal K. A case report of leukoencephalopathy with vanishing white matter disease. Int J Contemp Pediatr 2020;7:1438. [Google Scholar]
- 5.Deginet E, Tilahun R, Bishaw S, et al. Probable vanishing white matter disease: a case report and literature review. Ethiop J Health Sci 2021;31:1307–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Man JH, van Gelder CA, Breur M, et al. Cortical pathology in vanishing white matter. Cells 2022;11:3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.vander Knaap MS, Pronk JC, Scheper GC. Vanishing white matter disease. Lancet Neurol 2006;5:413–423. [DOI] [PubMed] [Google Scholar]
- 8.Kaczorowska M, Kuczynski D, Jurkiewicz E, et al. Acute fright induces onset of symptoms in vanishing white matter disease—Case report. Eur J Paediatr Neurol 2006;10:192–193. [DOI] [PubMed] [Google Scholar]
- 9.Hettiaracchchi D, Neththikumara N, Pathirana B, et al. A novel mutation in the EIF2B4 gene associated with leukoencephalopathy with vanishing white matter. Case Rep Pediatr 2018;2018:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ashrafi MR, Amanat M, Garshasbi M, et al. An update on clinical, pathological, diagnostic, and therapeutic perspectives of childhood leukodystrophies. Expert Rev Neurother 2020;20:65–84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets are available from the corresponding author on reasonable request.