

Abstract
Immune regulation plays a crucial role in human health and disease. Inflammatory bowel disease (IBD) is a chronic relapse bowel disease with an increasing incidence worldwide. Clinical treatments for IBD are limited and inefficient. However, the pathogenesis of immune-mediated IBD remains unclear. This review describes the activation of innate and adaptive immune functions by intestinal immune cells to regulate intestinal immune balance and maintain intestinal mucosal integrity. Changes in susceptible genes, autophagy, energy metabolism, and other factors interact in a complex manner with the immune system, eventually leading to intestinal immune imbalance and the onset of IBD. These events indicate that intestinal immune imbalance is an alarm for IBD development, further opening new possibilities for the unprecedented development of immunotherapy for IBD.
1. Introduction
Inflammatory bowel disease (IBD) is a chronic, immune-mediated inflammatory disease characterized by the disruption of the structure and functions of the intestinal epithelial barrier [1]. IBD includes Crohn's disease (CD) and ulcerative colitis (UC), which are chronic of relapsing intestinal inflammation [2–4]. In CD, the inflammation is often transmural, whereas in UC, the inflammation is typically confined to the mucosa [5]. CD and UC differ in the presentation of some symptoms, disease location, and histopathological characteristics, but they share the manifestations of gastrointestinal symptoms such as chronic abdominal pain, intestinal obstruction or diarrhea, mucus, pus, and bloody stools, among other symptoms [6, 7]. IBD complications include strictures, abscesses, fistulas, and colitis-associated cancer [8].
The prevalence of IBD is increasing worldwide, including Europe, North America, and some developing countries [9, 10]. The prevalence of IBD is equal between men and women [11]. Several studies have reported that patients with IBD have an increased risk of colorectal cancer [12, 13] and extraintestinal malignancies (including cholangiocarcinoma, skin cancer, and hematologic malignancies) [14], which is believed to be a consequence of immunosuppressive therapies and an underlying inflammatory state [14]. There is no specific treatment method available for IBD and its related complications. Clinically, supportive treatment is the main treatment strategy [1]. The main goal of IBD treatment is to reduce the side effects during the occurrence of severe episodes, control chronic inflammation, and prevent reactivation of the intestinal inflammatory processes [15].
The etiology of IBD is complex, with a poor prognosis, and few treatment methods exist. Therefore, it is of great significance to clarify its etiology and pathogenesis to determine early diagnostic markers and effective drug targets. An increasing number of studies have revealed that IBD is caused by the interaction of multiple factors, including susceptible genes, the immune system, and host microorganisms [2, 15–17]. Among them, immune dysfunction is receiving increasing attention in the pathogenesis of IBD. Research strongly suggests that IBD is caused by disturbed mucosal immune homeostasis [7, 18, 19]. With the deterioration of the immune system, abnormal responses to the gut microbiota can trigger a series of inflammatory events that damage the intestinal wall, resulting in the disruption of the intestinal mucosal barrier and accelerating the IBD progression [20]. However, the mechanisms underlying immune-mediated IBD have not been systematically and comprehensively clarified.
Here, we reviewed the effect of immune imbalance in IBD from the perspective of immune regulation, with an attempt to provide new insights into the pathogenesis and new immunotherapeutic sites of IBD. In this review, we have provided a framework to understand the changes in the innate immune system, adaptive immune system, autophagy, gene, energy metabolism, and other factors in immune-mediated IBD, as well as to present the available research on how the abnormalities of these factors trigger immune imbalance, which may be the potential pathogenesis of experimental animal colitis and patients with IBD. Meanwhile, we have summarized the physiological and pathophysiological functions of immune cells in the intestine and discussed some of the immune sites that may act as potential therapeutic targets for IBD (Figure 1; Table 1).
Figure 1.
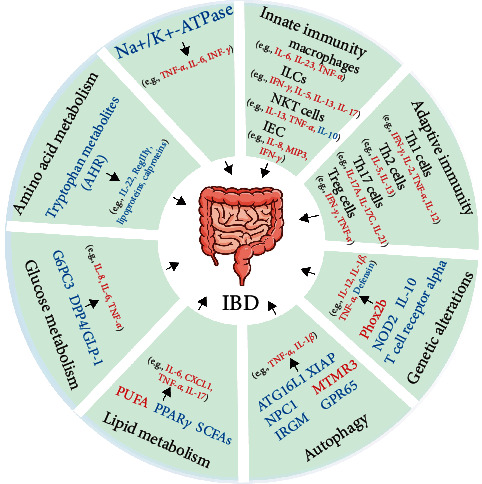
The factors of the innate immune system, adaptive immune system, genetic alterations, impaired autophagy, imbalance of energy metabolism (such as lipid metabolism, glucose metabolism, and amino acid metabolism), and electrolyte disorders interact in complex manners, which ultimately cause intestinal immune imbalance and trigger the onset of IBD. The red font indicates upregulation, while the blue font indicates downregulation. Abbreviations: ILCs, innate lymphocytes; NKT, natural killer T; IEC, intestinal epithelial cell; MIP3, macrophage inflammatory protein 3; NOD2, nucleotide-binding and oligomerization domain 2; Phox2b, paired-like homeobox 2b; Atg16l1, autophagy-related 16-like 1; XIAP, X-linked inhibitor of apoptosis; NPC1, Niemann–Pick disease type C1; MTMR3, myotubularin-related protein 3; IRGM, immunity-related GTPase M; GPR65, G-protein coupled receptor 65; PUFAs, polyunsaturated fatty acids; PPARγ, peroxisome proliferator-activated receptor γ; SCFAs, short-chain fatty acids; G6PC3, glucose-6-phosphatase catalytic subunit 3; DPP-4, dipeptidyl peptidase 4; GLP-1, glucagon-like peptide 1; AhR, aryl hydrocarbon receptor.
Table 1.
Physiological function of immune cells in the intestinal tract and the pathophysiological function of inflammatory bowel disease (IBD).
| Immune cell | Physiological functions in the intestinal tract | The pathophysiological role in IBD |
|---|---|---|
| Macrophages | Assist Treg cells' expansion and production of IL-10 [21, 22]. | Secretion of large amounts of pro-inflammatory cytokines such as IL-23 and TNF-α [29, 30]. |
| ILCs | Controlling tissue homeostasis [34, 35]. | ILC1 can release IFN-γ [36, 37, 41]; LC2 can produce IL-5 and IL-13 cytokines [40]; patients with IBD express ILC3 genes (IL17A, IL22, and IL23R) [44]. |
| NKT cells | Response to infectious pathogens, prevention of autoimmune diseases, and maintenance of self-tolerance [57]. | NKT cells are sources of IL-13 [60]. The expression of IL-13 was significantly increased in UC [60, 61]. |
| IECs | Defend against intestinal luminal bacteria and pathogen-associated molecular patterns [64]. | TLR5 recognizes flagellin, whose activation stimulates the NF-κB, which induces the secretion of chemokines IL-8 and MIP3 [67, 68]. ISG15 expression was increased and ISG15 could enhance IL-12-induced IFN-γ release [71, 73, 74]. |
| Th1 cells | Protective immunity against microbial pathogens and tumors [75]. | In CD, Th1 cell responses are associated with an increased expression of IL-12 [79–81]. |
| Th2 cells | Promote antihelminth immunity, suppress type 1-driven autoimmune disease and maintain metabolic homeostasis [88]. | UC is characterized by Th2-related cytokines (IL-5 and IL-13) [60, 61, 95, 96]. |
| Th17 cells | Maintaining symbiotic populations at important barrier sites [99]. | The mucosa of patients with IBD can detect high levels of IL-17A [78, 105, 106], IL-17C [102], and IL-21 [82, 107]. |
| Treg cells | Tregs act as the key regulators of intestinal homeostasis [108, 109]. | Tregs may differentiate into Th17 cells, resulting in the IL-17+ and FoxP3+ subsets of T cells [48]. |
IL, interleukin; TNF-α, tumor necrosis factor-alpha; ILC, innate lymphocyte; IFN, interferon; NKT, natural killer T; IEC, intestinal epithelial cell; UC, ulcerative colitis; TLR, Toll-like receptor; NF, nuclear factor; ISG15, IFN-stimulated gene 15; MIP3, macrophage inflammatory protein 3; CD, Crohn's disease; Treg, regulatory T.
2. Innate Immunity
2.1. Macrophages
2.1.1. Physiological Roles of Intestinal Macrophages
In intestinal homeostasis, intestinal macrophages produce various cytokines (such as IL-10, IL-6, and iNOS) and soluble mediators (prostaglandin E2 [PGE2], bone morphogenetic protein 2 [BMP2], and WNT ligands) that are involved in the proliferation of epithelial progenitors, intestinal neuronal physiology, and endothelial physiology [21–26]. Tissue macrophages are produced by monocytes in the blood [21]. Past studies have revealed that humans possess two subsets of monocytes based on the cell surface proteins CD14++CD16− and CD14+CD16+ [27]. Monocyte subsets in mice are divided into CX3CR1intLy6Chi and CX3CR1hiLy6Clo [28].
2.1.2. Macrophage Characteristics in Mice Colitis Models and Patients with IBD
A past study reported that Ly6Chi monocytes can be differentiated into CX3CR1intLy6Chi cells, which activated the Toll-like receptor (TLR) and nucleotide-binding and oligomerization domain 2 (NOD2) signal cascade components in both T cell transfer and dextran sodium sulfate (DSS)-induced mouse colitis models, reacted with bacterial products, and led to the secretion of several pro-inflammatory cytokines, including interleukin (IL)-6 and IL-23, thus promoting colitis [28].
Healthy human intestinal macrophages have no anti-inflammatory functions because they have no innate immune receptors (CD14 or TLR) [29]. Interestingly, CD14+ and TLR+ macrophage cells in the lamina propria of patients with CD increased, releasing IL-23 and tumor necrosis factor-alpha (TNF-α) [29, 30]; therefore, the positive innate immune receptor of myeloid cells may be closely related to IBD pathogenesis. Therefore, intestinal macrophages are considered the primary immune cells for establishing and maintaining intestinal homeostasis.
According to the description of the pathogenic role of monocytes/macrophages in experimental colitis models and patients with IBD, this further suggests that regulating monocyte/macrophage differentiation is crucial for the treatment of human IBD.
2.2. Innate Lymphocytes (ILCs)
2.2.1. Physiological Functions of Intestinal ILCs
The ILC family includes ILC1, ILC2, and ILC3, which express lineage-specific transcription factors T-bet, GATA-3, and RORγt, respectively [31–33]. Human mucosal tissues are enriched in ILCs, and they play a key role in controlling tissue homeostasis as effector cells under the conditions of infection, immune response, and inflammation [34, 35].
2.2.2. The Effects of ILCs in Mice Colitis Models and Patients with IBD
ILC1 can release interferon (IFN)-γ to induce inflammation in certain colitis models (Rag1−/− mice treated with anti-CD40) and patients with CD [36, 37]. The depletion of intraepithelial ILC1 can ameliorate symptoms in certain colitis models (Rag1−/− mice treated with anti-CD40) [36]. In addition, a past study demonstrated that IFN-γ produced by ILCs is an inducer of intestinal inflammation in innate colitis and that neutralization of IFN-γ is sufficient to ameliorate disease progression [37]. However, clinical studies have demonstrated that treatment with anti-IFN-γ antibody therapy did not significantly improve the clinical symptoms of patients with CD [37].
Furthermore, ILC2 in patients with UC/CD has been reported to produce IL-5 and IL-13 cytokines, which may be related to early protective immunity on mucosal surfaces [38–40]. Furthermore, IL-13+IFN-γ+ ILC2 was detected in intestinal samples of patients with CD, indicating that ILC2 has plastic functions under inflammatory conditions [40].
The levels of IL-17 produced by ILC3 were significantly increased in the Helicobacter hepaticus-induced mice colitis, the Tbx21−/−Rag2−/− UC, and the T-bet knockout mice models [41–43], suggesting that ILC3 might be involved in the onset of IBD. Furthermore, IL-23 is a key cytokine driving intestinal inflammation, which is mainly related to Th17 cells [44]. ILC3 cells are the innate counterparts of Th17 cells [45]. Research has revealed that IL-23 can act on ILC3, inducing the production of IL-17 and IL-22 [46]. Consistent with this result, after IL-23 stimulation, when compared with the control group, innate immune cells isolated from patients with IBD express ILC3 genes (IL-17A, IL22, and IL23R) [44]. Studies have revealed that the expression of IL-23R on CD4+T cells is increased in T-cell transfer-induced colitis [47]. Therefore, IBD mouse models and patients with IBD are associated with IL23R polymorphisms, which have been reported to induce the T helper type (Th)17 cytokine (IL-17) [42, 46, 48]. Furthermore, according to genome-wide association studies (GWAS) and mouse studies, the IL23/IL-17 axis plays a pivotal role in IBD [49–53]. Therefore, ILCs play an important role in IBD pathogenesis and exhibit plasticity.
2.3. Natural Killer (NK) Cells
2.3.1. Physiological Roles of NK and Natural Killer T (NKT) Cells
According to the cell surface density of CD56, human NK cells can be classified into CD56dimNK cells and CD56brightNK cells [54]. Past research has shown that when compared to CD56dim NK cells, CD56bright NK cells produced high levels of NK-derived cytokines (IFN- γ, IL-10, and IL-13). In the process of innate immune response in the body, the main functions of CD56bright NK cells may be to provide cytokines for macrophages and other antigen-presenting cells (IFN-γ, IL-10, and IL-13), which effectively control infection. In addition, when compared to CD56bright NK cells, CD56dim NK cells are more cytotoxic [54]. NKT cells are primarily detected in the liver, thymus, and intestinal tissues' lamina propria [55, 56]. They are involved in tumor monitoring, immune response to infectious pathogens, autoimmune disease prevention, and maintenance of self-tolerance [57]. NKT cells are a unique subset of lymphocytes that express molecules from conventional T cells and NK cells [57].
2.3.2. The Effects of NKT Cells in Mice Colitis Models and Patients with IBD
Studies have revealed that UC is characterized by the presence of IL-13Rα2 NKT cells, which can be used as a marker to differentiate UC from CD [58]. DX5+NKT cells can play a role in downregulating the effector immune response in T-cell transfer-induced mice colitis and DSS-induced acute colitis. Mechanistically, DX5+NKT cells require interaction with CD1d (major histocompatibility complex class I-like molecules) to mediate their immune regulatory effects against colitis [59]. NKT cells are sources of IL-13 [60]. Compared with the healthy control group and patients with CD, IL-13 levels significantly increased in patients with UC [60, 61]. The pathogenic effects of IL-13 include the activation of inflammatory colonic mucosal NKT cells and impaired epithelial barrier function [57, 61]. Based on UC pathophysiology, activated NKT cells can strongly produce TNF-α-mediated immune responses [57, 60].
Based on these above-mentioned observations, the pathophysiological functions of NKT cells in experimental colitis mice models and patients with UC/CD are different. Therefore, intestinal NKT cells can secrete various cytokines to suppress (IL-10) [62, 63] or promote (IL-13) the onset of immune-mediated IBD.
2.4. Intestinal Epithelial Cells (IECs)
2.4.1. Physiological Functions of IECs
IECs have tight junctions that defend against intestinal luminal bacteria and pathogen-associated molecular patterns, including lipopolysaccharide (LPS) [64]. In vitro studies have revealed that prolonged exposure of the intestinal epithelium to LPS-enriched conditions increases intestinal epithelial tolerance and cross-tolerance [65, 66].
2.4.2. The Effects of IECs in Mice Colitis Models and Patients with IBD
Studies have shown that the mortality of TLR4 and TLR2 knockdown mice after DSS administration is higher than that of wild-type mice [65]. Furthermore, at the basal lateral of the colon epithelium, TLR5 specifically recognizes flagellin and activates nuclear factor (NF)-κB, which induces the secretion of chemokines IL-8 and macrophage inflammatory protein 3 (MIP3) in model human IECs (prepared by culturing the colonic cell line T84 on collagen-coated permeable supports) [67, 68], the T cell transfer colitis model [69], and the serum of patients with CD [70].
The expression of functional TLR3 in IECs plays a crucial role in the virus-mediated innate intestinal immune response. The activation of this response causes IECs to produce IFN-stimulated gene 15 (ISG15) [70]. ISG15 (which is also known as UCRP, G1P2, IP17, IMD38, IFI15, and IMD38) is expressed at low levels in normal cells and tissues, and its expression is induced by type I IFNs (IFN-α and β) through the binding of IFN regulatory factors to IFN-stimulated response element-containing promoters [71, 72]. The mRNA expression of ISG15 was found to be increased in patients with active IBD and experimental rats with colitis, and ISG15 could enhance the IL-12-induced release of IFN-γ [71, 73, 74].
In short, TLRs participate in the onset of immune-mediated IBD in various ways. The interaction between damage to the IEC barrier function and an innate immune imbalance increases IBD susceptibility. Furthermore, IEC-derived cytokines enhance and prolong the inflammatory response during IBD, which is a potential immunotherapy target.
3. Adaptive Immunity
3.1. Th1 Cells
3.1.1. Physiological Role of Th1 Cells in the Intestine
Type 1 immunity is particularly important for protective immunity against microbial pathogens and tumors. An uncontrolled type 1 cellular immune response leads to tissue damage, immune imbalance, and disease [75].
3.1.2. Pathophysiology Th1 Cells in Mice Colitis Models and Patients with IBD
Studies have shown that during the chronic stage of DSS-induced colitis, Th1 cytokines exhibit no significant expression in the colon mucosa [76]. Earlier studies considered T cell transfer mice colitis to be a Th1-mediated model because it resulted in the development of a large population of T cells producing IFN-γ and TNF-α [77].
Studies have also reported that, compared with the control group and patients with UC, T cells in the tissues of patients with CD produce higher levels of IL-2 and IFN-γ [78]. In patients with CD, Th1 cell responses are associated with increased expression of IL-12 [79–81]. On the one hand, IL-12 can promote the production of IFN-γ by ILC1 in CD [36, 37]. On the other hand, IL-12 positively regulates IL-21 production, further facilitating the synthesis of IFN-γ and IL-17A [82]. Interestingly, IFN-γ and IL-12 can form a feedback loop. IFN-γ can increase macrophage-derived IL-12 production, amplifying the ongoing Th1 immune response in patients with CD [83]. IL-12 expression may be correlated with the chronic, persistent disease course of patients with CD. The CD is believed to be characterized by a Th1 immune response [20, 84–87]. Therefore, Th1 immunity is crucial in the onset of CD and is a potential therapeutic target for CD treatment.
3.2. Th2 Cells
3.2.1. Physiological Function of Th2 Cells in the Intestine
Th2 cells are marked by higher levels of IL-5 and IL-13, along with Gata3 expression [88–90]. Type 2 immune responses promote antihelminth immunity, suppress type 1-driven autoimmune disease, and maintain metabolic homeostasis, whereas the breakdown of these mechanisms may lead to IBD [88, 91, 92].
3.2.2. The Roles of Th2 Cells in Mice Colitis Models and Patients with IBD
Studies have revealed that increased levels of Th2 cytokines were noted in patients with UC and experimental colitis mice models (colitis induced by T cell transfer, 2,4,6-trinitrobenzene sulfonic acid [TNBS], and oxazolone), and the inflammation was alleviated by inhibiting Th2 cytokines [60, 61, 93–97]. UC is characterized by Th2-related cytokines, for example, IL-5 and IL-13 [60, 61, 84, 95–97].
Therefore, the pathogenesis of patients with UC and experimental colitis models is related to Th2 cell-mediated cellular immunity. This provides a new direction for UC diagnosis and immunotherapy.
3.3. Th17 Cells
3.3.1. Physiological Functions of Th17 Cells in the Intestine
Th17 cells play important roles in maintaining symbiotic populations (We live with very large numbers [>1014] of microbes that have taken residence on and in ourselves, termed symbionts [98]) at important barrier sites [99]. They are a subset of T cells characterized by producing high levels of IL-17A, IL-17F, IL-21, and IL-22 [100, 101], among which IL-17A is the marker cytokine for Th17 cells.
3.3.2. The Function of Th17 Cells in Mice Colitis Models and Patients with IBD
Past studies have revealed that when compared with wild-type mice, the DSS-induced mice colitis model showed significantly increased levels of IL-17C, and DSS-treated IL-17C knockout mice exhibit exacerbated intestinal inflammation [102, 103]. Furthermore, carbon monoxide can inhibit the differentiation of Th17 cells and reduce IL-17 secretion in the mouse colitis model induced by T-cell transfer and in IL-10-deficient mice [104].
Compared with normal intestinal mucosa, the mucosa of patients with IBD can detect high levels of IL-17A [78, 105, 106], IL-17C [102], and IL-21 [82, 107]. IL-17 plays a pathogenic role in IBD; however, certain studies have confirmed that blocking IL-17 treatment will exacerbate the symptoms of patients with moderate and severe CD [48].
Briefly, Th17 cells are indispensable in the maintenance of intestinal immune balance. Th17-related cytokines provide immunotherapeutic targets for treating patients with IBD.
3.4. Regulatory T (Treg) Cells
3.4.1. Physiological Functions of Treg Cells in the Intestine
In the adaptive immune system, Treg cells are the key regulators of intestinal homeostasis. They secrete inhibitory cytokines IL-10 and TGF-β, along with the transcription factor fork box P3 (FoxP3), in the intestine primarily via the production of IL-10 to maintain intestinal mucosal homeostasis [108, 109].
3.4.2. The Effect of Treg Cells in Mice Colitis Models and Patients with IBD
Studies have revealed that the adoptive transfer of Treg cells in mice colitis models induced by T cell transfer can inhibit the autoreactive T cell reaction in vivo and prevent intestinal inflammation [110, 111]. Compared with the control group, the levels of FoxP3+Treg cells were significantly reduced in patients with active CD, and the production of TNF-α and IFN-γ in the mucosa of patients with CD decreased after anti-TNF-α treatment [112]. Thus, TNF-α may inhibit Treg cell function in patients with active CD. However, the expressions of Treg cells are increased in patients with active UC, which is involved in inhibiting the proliferation of effector T cells and cytokine production [113]. Treg cells may differentiate into Th17 cells, resulting in IL-17+ and FoxP3+ subsets of T cells, which may explain the reduced inhibition of Treg cells in IBD [48].
Dysfunction in Treg cells is associated with the onset of immune-mediated IBD, along with effector T cells, gene mutations, and other factors. Therefore, Treg cells are crucial in maintaining intestinal homeostasis, and Treg cells are an important therapeutic target for IBD immunotherapy.
4. Effects of Genetic Alterations on Immune-Mediated IBD
4.1. NOD2
NOD2 is a putative intracellular receptor for bacterial peptidoglycans (PGNs) [114]. PGN is a polymer composed of peptide and glycan, with N-acetyl muramic acid and N-acetyl glucosamine linked via the β-1,4 glycosidic bond as the basic skeleton [115]. Its main function is to preserve cell integrity by withstanding turgor. PGN also contributes to the maintenance of bacterial cell shape. In addition, it is intimately involved in the processes of cell growth and cell division [116]). NOD2 is primarily expressed in monocytes, phagocytes [117, 118], and Paneth cells [114]. The NOD2 protein is involved in recognizing muramyl dipeptide (MDP), a PGN found in the cell walls of gram-positive and gram-negative bacteria [119, 120]. MDP binding activates the transcription factor NF-κB, leading to the secretion of proinflammatory cytokines [120–122].
At present, several susceptibility loci (IBD1–IBD8) have been identified for CD [123], and they have the strongest association with the CARD15 gene located within the IBD1 locus, which encodes the cytoplasmic protein [117, 118]. Caspase-activating and recruitment domain-5 (CARD5), also called NOD2, is mainly expressed in the cells of the myeloid lineage and presents with two amino-terminal CARD domains [117]. Some studies have mentioned that the NOD2 mutation increases CD susceptibility. On the one hand, NOD2 mutation or NOD2(CARD15) knockout mice were incapable of recognizing bacterial PGN or MDP [117, 124], increasing TLR2-mediated NF-κB activation and Th1 reaction (IL-12) [117, 125]. On the other hand, studies have shown that because NOD2 and defensin are both located in Paneth cells, the mutation in NOD2 may affect the secretion of defensin in Paneth cells, but the specific underlying mechanism remains unclear [114]. In addition, it has been reported that CD-associated NOD2 mutations result in defective gut responsiveness to LPS, possibly contributing to disease susceptibility [126, 127].
Thus, there is an antagonistic relationship between bacteria and innate immunity, which may affect the ability of the mucosa to eliminate pathogenic bacteria. The pathogenesis remains to be studied. Therefore, NOD2 acts as a protective protein in the intestine and is a potential therapeutic target for IBD treatment.
4.2. IL-10
IL-10 is an anti-inflammatory cytokine secreted by various immune cells (macrophages, Th17 cells, Treg cells), which can downregulate the production of Th1-derived cytokines (IL-12, IL-1β, and TNF-α) and maintain intestinal homeostasis [128–130]. Studies have reported that recombinant IL-10 administration can inhibit IL-1β in a concentration-dependent manner in the mucosal cultures of patients with UC [129]. Studies also revealed that continuous IL-10 administration in models of experimental colitis (IL-10-deficient mice, T cell transfer-induced mice colitis models, and DSS- and TNBS-induced colitis mice) can improve intestinal symptoms. Thus, endogenous IL-10 centrally regulates the mucosal immune response [128, 130]. In addition, the type of IBD diagnosed before six years of age is referred to as very early-onset IBD and presents with a strong family history [131–133]. Furthermore, IBD was diagnosed in two children with mutations in the gene encoding IL-10 [134].
Therefore, changes in the level of the IL-10 gene play an important role in IBD development. The mouse model provides a scientific basis for patients with IBD, and the use of IL-10 as a biological agent for treating IBD requires further studies.
4.3. Relationship between Paired-Like Homeobox 2b (Phox2b), T Cell Receptor Alpha Gene, and IBD
GWAS have identified a single nuclear polymorphism in the enteroendocrine-associated transcription factor Phox2b as a risk factor for CD [135]; however, the specific underlying pathogenesis remains unclear. Studies have shown that T cell receptor alpha knockout mice showed significantly decreased expression of intestinal endocrine cell subpopulations (Neurotensin and Cholecystokinin [CCK] cells) and finally developed UC-like phenotypes [136]. Neurotensin regulates the colon's response to fixed stress by activating mucosal mast cells and regulating the release of colonic goblet mucin [136]. CCK regulation of intestinal growth is also related to intestinal adaptation and tumor growth [136]. Therefore, endocrine cell-related immunoregulatory molecules can maintain intestinal immune balance. However, the specific underlying mechanisms remain unclear.
5. The Role of Autophagy in Immune-Mediated IBD
5.1. Autophagy-Related 16-Like 1 (Atg16l1)
Autophagy is a mechanism for maintaining cellular homeostasis, induced by bactericidal effects and the presentation of endogenous antigens [137]. It has been reported that Ala197Thr in Atg16l1 is associated with CD susceptibility [138, 139]. Moreover, a deficiency of Atg16l1 in mouse spinal cord cells increased DSS-induced colitis and increased proinflammatory cytokine production (IL-1β, TNF-α) [140]. Similar studies revealed that mice with the CD-associated Atg16l1 variant showed decreased autophagy and increased IL-1β production [141]. The expression of the CD-associated Atg16l1T300A risk allele causes defects in plasma membrane damage repair after infection with Listeria monocytogenes and increases cell-to-cell bacterial spread [142, 143]. Furthermore, NOD2 is involved in the autophagic response of invasive bacteria because it induces the recruitment of the autophagy protein Atg16l1 to bacterial entry sites on the plasma membrane [102, 103]; in contrast, this function is lost in cases of NOD2 mutation [144, 145]. Thus, Atg16l1 can maintain intestinal homeostasis and interact with various factors to affect autophagy. This further highlights the importance of Atg16l1 in IBD pathogenesis. Most research is based on animal experiments, so its role in the pathogenesis of patients with IBD remains to be elucidated.
5.2. X-Linked Inhibitor of Apoptosis (XIAP)
During autophagy, the inhibition or loss of XIAP function causes impaired autophagy flux, impaired bacterial clearance, and increased secretion of TNF-α and IL-1β, possibly leading to CD development [146]. XIAP interferes with NOD2-mediated cytokine production [147].
5.3. Niemann–Pick Disease Type C1 (NPC1)
The early onset of CD has been recently associated with NPC, a neurodegenerative lysosomal lipid storage disorder wherein the NPC1 (NPC intracellular cholesterol transporter 1) gene that is involved in lipid transport is mutated [142]. Recent studies have reported that NPC1-related intestinal inflammation and CD-related NOD2 and XIAP gene mutations cause the same functional defects, including impaired antibacterial autophagy, resulting in a similar intestinal phenotype [147]. Changes in NOD2, XIAP, and NPC1 can participate in CD development by affecting autophagy.
5.4. Myotubularin-Related Protein 3 (MTMR3)
The risk allele of MTMR3 associated with IBD indicates increased MTMR3 levels in macrophages of IBD-risk carriers of the rs713875 CC genotype. MTMR3 can reduce the levels of pattern recognition receptor (PRR)-induced phosphatidylinositol 3-phosphate and autophagy levels, further increasing PRR-induced caspase-1 activation and autocrine IL-1β and NF-κB signal transduction, and finally increasing the overall secretion of cytokines [141].
5.5. Immunity-Related GTPase M (IRGM)
IRGM belongs to the p47 immune-related GTPase family. It can resist bacterial invasion via various mechanisms, including the regulation of phagosome processing, cell motility, and autophagy [2]. Paneth cells in IRGM−/− mice showed various changes, including the abnormal development of secretory granules, lowered expression of selective antimicrobial peptides (AMPs) [148], and increased susceptibility to DSS-induced colitis [148]. Furthermore, IRGM has a protective role in the intestine and participates in regulating autophagy. IRGM can maintain intestinal immune balance and can be considered a potential target for IBD therapy.
5.6. G-Protein Coupled Receptor 65 (GPR65)
Recent studies have shown that GPR65 is a susceptible gene for IBD and an H+-sensitive G-protein-coupled receptor that can maintain the pH value of the lysosome, thus maintaining lysosomal energy and contributing to autophagy and pathogen defense [149]. Epithelial cells or lymphoblasts of patients with IBD and GPR65/I231L missense variants show abnormal lysosomal acidification, causing lysosomal dysfunction and impaired autophagy-mediated intracellular bacterial clearance [142, 149]. Compared with wild-type mice, GPR65-deficient mice are more likely to be infected with intestinal diseases caused by Citrobacter. Colitis is also more serious in these mice, along with enhanced T-cell infiltration [142]. Therefore, GPR65 is involved in the intestinal innate immune function to prevent pathogen invasion. It is therefore a potential target for IBD therapy.
Autophagy is closely related to innate immunity and is a response induced by endoplasmic reticulum stress that eventually causes apoptosis and IBD [137]. Based on genetic analyses, changes in NOD2, Atg16l1, and XIAP genes could lead to CD development by affecting autophagy. Therefore, the regulation of autophagy plays an important role in IBD.
6. Metabolic Changes Affecting Immune-Mediated IBD
6.1. The Role of Lipid Metabolism in Immune-Mediated IBD
6.1.1. The Relationship between Polyunsaturated Fatty Acids (PUFAs) and IBD
Lipid metabolites include phospholipids, fatty acids, and cholesterol. Among these, the effects of fatty acid metabolism on immune cells have been extensively studied [150]. Food that is rich in the ω-6 PUFA, such as arachidonic acid (AA), increases IBD risk [151]. The possible mechanisms of ω-6 PUFA involvement in IBD are as follows: (i) nutrients come into direct contact with the colon mucosa, or (ii) nutrients are integrated into the colon cell membrane on the surface of the gastrointestinal tract lumen [152]. IECs from patients with CD exhibit impaired glutathione peroxidase 4 (Gpx4) activity and lipid peroxidation [151]. Furthermore, PUFAs, especially AA, induce the production of IL-6 and chemokine (C-X-C motif) ligand 1 in IECs treated with Gpx4 siRNAs in response to iron availability, lipid peroxidation, and ACSL4, similar to mechanisms of ferroptosis [151, 153]. Animal studies have revealed that mice lacking the Gpx4 allele were administered a PUFA-rich western diet, and it was found that focal granulomatous neutrophilic enteritis was triggered in IECs [151]. Changes in PUFA uptake and GPX4 activity in IECs can cause fatty acid metabolism disorders and damage epithelial barrier functions, which may be closely related to IBD development.
6.1.2. Physiological Functions of Peroxisome Proliferator Activated Receptor γ (PPARγ)
PPARγ belongs to the nuclear receptor family, and it is a lipid-activated transcription factor [154, 155]. PPARγ is primarily expressed in IEC in the intestine. It can be activated by fatty acids and plays a key role in regulating lipid metabolism, inflammation, cancer, insulin sensitization, and cell proliferation [155–157].
6.1.3. Pathological Roles of PPARγ in Immune-Mediated IBD
PPARγ agonists (thiazolidinediones) inhibit the activation of NF-κB through IκBα-dependent mechanisms and further reduce cytokine expression [158]. PPARγ agonists can improve inflammatory symptoms in mice colitis models, including DSS-induced mice colitis and T cell transfer-induced mice colitis models [155, 158, 159]. Interestingly, in these models, the protective effects of thiazolidinediones may be primarily mediated via hematopoietic cells because conditional PPARγ deletion in the intestinal epithelium causes mild spontaneous colitis and increased susceptibility to DSS-induced colitis [160]. In addition, PPARγ ligands (5-ASA) were added to the mucosal culture of patients with IBD, and they were found to increase PPARγ expression [159].
Therefore, PPARγ participates in regulating inflammatory reactions in the intestine and maintaining the integrity of intestinal mucosa. Although PPARγ agonists are widely used in patients with IBD, the specific mechanisms underlying their anti-inflammatory effects on the colon remain unclear.
6.1.4. Physiological Functions of Short-Chain Fatty Acids (SCFAs)
SCFAs include acetate, propionate, and butyrate [161] and are primarily produced by gram-negative bacteria, gram-positive bacteria, and indigestible carbohydrate fermentation [162–165]. The primary SCFA substrate is resistant starch (RS), which is an important source of butyrate [166]. In the intestinal tract, SCFA-mediated immune modulation can be regulated by various specific mechanisms: (i) activation of GPCRs, (ii) stimulation of histone acetyltransferases, (iii) inhibition of histone deacetylases (HDACs), and (iv) stabilization of hypoxia-inducible factor [167–170].
6.1.5. Pathophysiology of SCFAs in Immune-Mediated IBD
In the intestinal inflammatory environment, SCFAs (including butyrate, propionate, and acetate) can bind to GPR41, GPR43, and GPR109A, activating the anti-inflammatory signaling cascade [161, 171–173]. G-protein-coupled receptors (GPCRs, GPRS) are versatile, seven-transmembrane-domain proteins that regulate a diverse array of intracellular signaling cascades [170, 174]. SCFAs are metabolites of intestinal microbiota and promote IEC RegIIIγ in a GPR43-dependent manner along with β-defensins. Therefore, AMP and β-defensin expression were severely impaired in GPR43 knockout mice; the symptoms worsened after DSS administration, and the level of TNF-α and IL-17 proteins increased [175]. Similar studies revealed that inflammation was reduced after administering high-fiber food to mice models with DSS-induced colitis [161]. SCFAs can improve the symptoms of wild-type colitis mice, but the symptoms of GPR43 knockout mice did not improve [167], indicating that the initiation of SCFA anti-inflammatory mechanisms depends on GPR43.
Furthermore, butyrate induces epigenetic modifications that upregulate histone H3 acetylation of FoxP3 and induce Treg differentiation, acting as an anti-inflammatory mediator [176]. SCFAs increased the number of FoxP3+Treg cells in T cell transfer-induced mice model with colitis and depended on the expression of GPR43 in Tregs [176, 177].
SCFAs (including butyrate, propionate, and acetate) can recruit neutrophils, depending on GPR43. In patients with CD, the function of neutrophil recruitment is abnormal, resulting in delayed bacterial clearance and the further onset of granuloma [178]. Increased GPR43+ neutrophil infiltration was found in patients with CD receiving dietary fiber-supplemented (such as resistant starch, non-starch polysaccharides (e.g., celluloses, hemicelluloses, pectins, and gums), non-digestible oligosaccharides, and sugar alcohols [179]) enteral nutrition compared with patients receiving only enteral nutrition [178]. SCFA enemas increase mucosal production, crypt length, and DNA content in colon cells and improve UC symptoms in patients and rats administered with TNBS [180].
Thus, SCFAs produced via the interaction of intestinal bacteria and dietary fiber can activate innate and adaptive immune functions, maintain the intestinal barrier, and provide a new target for IBD treatment.
6.2. Role of Glucose Metabolism in Immune-Mediated IBD
6.2.1. Glucose-6-Phosphatase Catalytic Subunit 3 (G6PC3)
Glucose is an important energy source for immune cells; these cells use glucose to generate ATP and metabolic intermediates [181]. G6PC3 is required for cellular glucose homeostasis and for catalyzing the hydrolysis of glucose 6-phosphate to glucose [182]. Interestingly, neutrophils greatly depend on glycolysis as an energy source [183–185]. Biallelic G6PC3 mutations cause the multisystem autosomal recessive disorder of G6PC3 deficiency (also known as severe congenital neutropenia type 4 [186–189]).
Individuals with severe congenital neutropenia-related IBD are deficient in G6PC3 [186, 190, 191], with CD-like inflammation, frequent stenosis, and severe oral aphthous ulcers [186, 190–193]. Previous studies have reported that G6PC3 deletion may result in the formation of IBD in the following two aspects: (1) deficiencies in the antibacterial activity of the innate immune system in patients with G6PC3-deficiency; and (2) the response of neutrophils with G6PC3 deficiency to LPSs significantly increases the levels of proinflammatory cytokines [185]. The results of similar studies are consistent with these findings. Compared with healthy controls, constitutively activated neutrophils in patients with G6PC3-deficiency released excessive amounts of soluble inflammatory mediators such as IL-8, resulting in IBD [194]. Previous studies have shown that excess IL-8 acts as a potent neutrophil chemotactic factor and activates ligands to drive tissue inflammation, which correlates with the severity of IBD [194–196]. Studies have reported that hematopoietic stem cell transplantation can be used to cure neutropenia and improve IBD caused by G6PC3 deficiency [185, 197].
Taken together, G6PC3 can maintain the homeostasis of human neutrophils. Therefore, normal glucose metabolism is extremely important for maintaining intestinal immune balance.
6.2.2. Role of the Dipeptidyl Peptidase 4 (DPP-4)/Glucagon-Like Peptide 1 (GLP-1) Axis in IBD
GLPs, including GLP-1 and GLP-2, and the peptide YY are secreted by L cells, a type of enteroendocrine cell [198]. GLP-1 enhances glucose-induced insulin responses, promotes pancreatic β-cell survival, and exhibits anti-inflammatory properties [198]. In the present study, we mainly describe the pathophysiological functions of GLP-1 in intestinal inflammation.
Previous studies reported that GLP-1 expression was significantly downregulated in the T cell transfer-induced mice colitis model [199]. Furthermore, another study reported that excess glucose promotes the mitochondria in T cells to produce more reactive oxygen species, which activate some cytokines (TGF-β, IL-17, and IL-6) related to Th17 cells, leading to excessive differentiation and stimulation of Th17 cells, which triggers inflammation in vivo [200]. Therefore, GLP-1 can regulate Th17 differentiation in an inflammatory environment. However, impaired regulation of glucose metabolism caused by high glucose or GLP-1 can result in an intestinal immune imbalance.
Glucagon-like peptide 1 receptor (GLP-1R) is widely expressed in various immune cell populations, particularly in intestinal intraepithelial lymphocytes (IELs) [201, 202]. GLP-1 can bind to GPL-1R in IELs to regulate intestinal immunity. Past studies have demonstrated that the activation of GLP-1R on intestinal IEL in mice can inhibit the expression of inflammatory cytokines (such as IL-2, IL-17a, IFN-γ, and TNF-α). In DSS-induced colitis, when compared with GLP-1R+/+ mice, GLP-1R−/− mice displayed more severe intestinal injury. Therefore, GLP-1R can improve the symptoms of DSS-induced colitis in mice [199, 201]. A similar study found that GLP-1 was significantly increased in mice with DSS-induced colitis treated with DPP-4 inhibitors [199]. Therefore, GPL-1/GPL-1R can maintain the function of the intestinal mucosal barrier.
DPP-4 (also known as CD26) is a type II integrated transmembrane glycoprotein [203]. The major substrates of DPP-4 are incretins, including GLP-1, gastric inhibitory polypeptide (GIP), which is secreted from K cells in the upper small intestine in response to the presence of nutrients, especially fats [204], and GLP-2, which is responsible for glucose metabolism [205, 206]. Apart from its roles as an enzyme, DPP-4 also plays key roles in T cell development, activation, and immune regulation [207, 208]. Functional studies on Jurkat T cell lines transfected with DPP-4 have demonstrated that the cross-linking of DPP-4 and CD3 antigens with their corresponding antibodies enhances intracellular calcium mobilization and IL-2 production, further demonstrating that DPP-4 plays an important role in T cell activation [207].
Interestingly, compared with healthy controls, serum GLP-1 levels were elevated in patients with IBD [209, 210], DPP-4 expression was increased in T cells, and DPP-4 activity was decreased in the circulation. This is considered a counter-regulatory mechanism that is conducive to limiting the occurrence of systemic immune responses [211–213]. Interestingly, DPP4 is expressed as a type II transmembrane protein. Soluble DPP4, which lacks the transmembrane and intracellular segments, is found in the circulation [214, 215].
At present, DPP-4 inhibitors are used to treat experimental mice colitis (DSS-induced mice colitis and TNBS-induced mice colitis), and their mechanism may be inhibiting IL-6 and TNF-α. Myeloperoxidase secretion and IL-10 upregulation ameliorate the inflammatory reaction and promote the healing of the lesions [216–219].
In summary, dysregulation of the DPP-4/GLP axis can lead to glucose metabolism disorders and immune imbalances, which further lead to IBD formation. Therefore, normal glucose metabolism can maintain the function of the intestinal mucosal barrier.
6.3. Role of Amino Acid Metabolism in Immune-Mediated IBD
Amino acids are essential for synthesizing various specific proteins, including cytokines and antibodies [220]. They are not only involved in the development of immune organs and the proliferation and differentiation of immune cells but also affect the secretion of cytokines and the regulation of immune responses [150]. Amino acid deficiency leads to atrophy of immune organs and dysfunction of immune cells [221].
Intestinal tryptophan (Trp) can be metabolized by the intestinal flora, Trp hydroxylase, and indoleamine 2,3-dioxygenase 1 to produce metabolites endogenous aryl hydrocarbon receptor (AhR) ligands (such as indole-3-aldehyde [IAld] and 6-formylindolo [3,2-b] carbazole) [222–225]. AhR is a cytoplasmic transcription factor present in many types of immune cells [226]. AhR ligands obtained from diet induce IL-22 secretion, which, in turn, facilitates intestinal production of mucins and AMP that are responsible for pathogen resistance and mucosal protection [223, 227, 228].
A study revealed that IAld activates AhR to enhance IL-22 expression in ILC3 and then stimulates epithelial cells to produce antimicrobial proteins, including REGIIIγ, lipoproteins, and calprotectin [223]. Animal studies reported that the Th17 cell response in the intestine of AhR−/− mice was significantly upregulated [223]. These studies further prove that AhR is involved in regulating the balance between ILC3 and Th17 cells and maintaining intestinal immune homeostasis.
Studies have shown that the Trp metabolism in the intestinal flora is impaired in patients with IBD; this results in AhR activation deficiency [224]. Furthermore, animal studies have reported that AhR deficiency aggravates inflammation in T cell transfer- and DSS-induced mice colitis and that it induces colitis by reducing IL-22 release [229]. Interestingly, AhR agonists and lactobacillus strains that can metabolize Trp can improve intestinal inflammation in mice [230]. These studies further indicate that AhR and AhR ligands are potential therapeutic targets for IBD.
Intestinal chromaffin cells can convert Trp into 5-hydroxytryptamine via Trp hydroxylase. There was an increase in 5-hydroxytryptamine in DSS- and TNBS-induced mouse colitis models [231, 232]. The use of a specific tryptophan hydroxylase 1 (TpH1, which is responsible for serotonin synthesis in the peripheral tissues and is mainly expressed in the enterochromaffin cells of the gut and the pineal gland [233]) enzyme inhibitor can inhibit the synthesis of 5-hydroxytryptamine and further improve the inflammatory symptoms of experimental colitis in mice [234]. In contrast, 5-HT tissue levels are significantly reduced in patients with CD and UC, and the intestinal chromaffin cells may be affected by the relevant inflammatory process [235]. However, the specific mechanism remains unelucidated.
Taken together, these results suggest that amino acid metabolites, intestinal microorganisms, and the host interact to maintain the intestinal mucosal barrier. Therefore, changes in amino acid metabolism have a potential role in IBD pathogenesis and could be new therapeutic targets for IBD.
6.4. Relationship between Na+/K+-ATPase and Immune-Mediated IBD
Na+/K+-ATPase is present in the basolateral membrane of villus and crypt intestinal cells. Cl− secretion and Na+ absorption depend on the function of Na+/K+-ATPase in the basolateral membrane [236]. Therefore, Na+/K+-ATPase can maintain the electrolyte balance in the intestine.
Previous studies reported that, compared with healthy controls, the activity of Na+/K+-ATPase was decreased in patients with active UC-associated diarrhea, and its activity was found to be related to inflammatory cell infiltration [237]. Furthermore, anti-CD3mAb mice can activate T cells to produce proinflammatory cytokines, such as TNF-α, IFN-γ, and IL-6, increase mucosal permeability, and decrease Na+/K+-ATPase activity in epithelial cells, further causing transient watery diarrhea [236]. Interestingly, several studies have reported that TNF-α plays an important role in T cell-induced diarrhea [238–240]. Similarly, IBD-induced diarrhea is associated with enhanced T cell activation and reduced Na+/K+-ATPase activity and sodium and water reabsorption [236, 237]. In adults, approximately 8–10 l of fluid enter the intestinal cavity every day. These liquids are derived from dietary intake and intestinal contribution, and are secreted from exocrine glands. However, only 0.1–0.2 l of the liquid are excreted from the feces. Therefore, the normal gut has a strong reabsorption function [241, 242]. In addition, studies on IBD-related diarrhea have demonstrated that the colon's ability to absorb water is significantly impaired [241] and that the disruption of the intestinal barrier increases permeability, leading to an increase in water secretion [243]. In clinical treatment, TNF-α inhibitors, such as infliximab, have been used to prevent diarrheal symptoms in patients with IBD [244, 245]. Taken together, electrolyte balance can help maintain intestinal immune balance and protect the integrity of the intestinal mucosa.
7. Conclusions and Outlook
Previous studies have revealed that immune balance is important for protecting the intestinal mucosal barrier and provided convincing evidence. However, the involvement of immune imbalance in IBD pathogenesis remains completely unclear. In this field, systematic reviews highlighting the potential mechanisms in experimental mouse colitis models (T cell transfer-induced colitis, DSS-induced colitis, and TNBS-induced colitis) and patients with IBD after immune imbalance caused by multiple factors are lacking. Therefore, we hope to provide a basic and systematic review of immune regulation and IBD, which can promote researchers to pay attention to the immune regulation process from the normal intestinal mucosa to IBD. This will provide a new perspective not only for treatment but also for prevention.
Acknowledgments
This work was supported by the National Natural Science Foundation of China under grant numbers: 82103206, 82173360; General Project of Chongqing Natural Science Foundation under grant number: cstc2021jcyj-msxmX0018; Special Support for Chongqing Postdoctoral Research Project under grant number: 2021XM3027; Kuanren Talents Program of the second affiliated hospital of Chongqing Medical University under grant numbers: kryc-yq-2224, 13-003-023; Senior Medical Talents Program of Chongqing for Young and Middle-aged under grant number: 11-020; and Chongqing medical scientific research project (Joint project of Chongqing Health Commission and Science and Technology Bureau) under grant number: 2020GDRC014. No external funding was received for this study.
Contributor Information
Lin Lv, Email: lin-miaomiao@cqmu.edu.cn.
Chuanfei Li, Email: leechuanfei@cqmu.edu.cn.
Zhechuan Mei, Email: meizhechuan@cqmu.edu.cn.
Data Availability
The data are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Authors' Contributions
Chunli Hu conceptualized and drafted the manuscript. Shengtao Liao assisted with the initial drafting and instructive layout of figures and tables. Zhechuan Mei, Chuanfei Li, and Lin Lv gave constructive comments. Zhechuan Mei, Chuanfei Li, and Lin Lv critically reviewed and revised the final version of the manuscript. All authors read and approved the final manuscript.
References
- 1.Mao Q., Pan H., Zhang Y., et al. GelNB molecular coating as a biophysical barrier to isolate intestinal irritating metabolites and regulate intestinal microbial homeostasis in the treatment of inflammatory bowel disease. Bioactive Materials . 2023;19:251–267. doi: 10.1016/j.bioactmat.2022.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Y.-Z., Li Y.-Y. Inflammatory bowel disease: pathogenesis. World Journal of Gastroenterology . 2014;20(1):91–99. doi: 10.3748/wjg.v20.i1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marafini I., Sedda S., Dinallo V., Monteleone G. Inflammatory cytokines: from discoveries to therapies in IBD. Expert Opinion on Biological Therapy . 2019;19(11):1207–1217. doi: 10.1080/14712598.2019.1652267. [DOI] [PubMed] [Google Scholar]
- 4.Reza Lahimchi M., Eslami M., Yousefi B. Interleukin-35 and Interleukin-37 anti-inflammatory effect on inflammatory bowel disease: application of non-coding RNAs in IBD therapy. International Immunopharmacology . 2023;117 doi: 10.1016/j.intimp.2023.109932.109932 [DOI] [PubMed] [Google Scholar]
- 5.Abraham C., Cho J. H. Inflammatory bowel disease. New England Journal of Medicine . 2009;361(21):2066–2078. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baumgart D. C., Sandborn W. J. Crohn’s disease. The Lancet . 2012;380(9853):1590–1605. doi: 10.1016/S0140-6736(12)60026-9. [DOI] [PubMed] [Google Scholar]
- 7.Gomez-Bris R., Saez A., Herrero-Fernandez B., Rius C., Sanchez-Martinez H., Gonzalez-Granado J. M. CD4 T-cell subsets and the pathophysiology of inflammatory bowel disease. International Journal of Molecular Sciences . 2023;24(3) doi: 10.3390/ijms24032696.2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gecse K. B., Vermeire S. Differential diagnosis of inflammatory bowel disease: imitations and complications. The Lancet Gastroenterology & Hepatology . 2018;3(9):644–653. doi: 10.1016/S2468-1253(18)30159-6. [DOI] [PubMed] [Google Scholar]
- 9.Kaplan G. G. The global burden of IBD: from 2015 to 2025. Nature Reviews Gastroenterology & Hepatology . 2015;12(12):720–727. doi: 10.1038/nrgastro.2015.150. [DOI] [PubMed] [Google Scholar]
- 10.Ananthakrishnan A. N., Kaplan G. G., Ng S. C. Changing global epidemiology of inflammatory bowel diseases: sustaining health care delivery into the 21st century. Clinical Gastroenterology and Hepatology . 2020;18(6):1252–1260. doi: 10.1016/j.cgh.2020.01.028. [DOI] [PubMed] [Google Scholar]
- 11.Euers L., Abughazaleh S., Glassner K., et al. Risk factors for and frequency of CT scans, steroid use, and repeat visits in inflammatory bowel disease patients seen at a single-center emergency department: a retrospective cohort study. Journal of Clinical Medicine . 2021;10(12) doi: 10.3390/jcm10122679.2679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pan X., Zhu Q., Pan L.-L., Sun J. Macrophage immunometabolism in inflammatory bowel diseases: from pathogenesis to therapy. Pharmacology & Therapeutics . 2022;238 doi: 10.1016/j.pharmthera.2022.108176.108176 [DOI] [PubMed] [Google Scholar]
- 13.Shteingart S., Rapoport M., Grodzovski I., et al. Therapeutic potency of IL2-caspase 3 targeted treatment in a murine experimental model of inflammatory bowel disease. Gut . 2009;58(6):790–798. doi: 10.1136/gut.2008.153981. [DOI] [PubMed] [Google Scholar]
- 14.Faye A. S., Holmer A. K., Axelrad J. E. Cancer in inflammatory bowel disease. Gastroenterology Clinics of North America . 2022;51(3):649–666. doi: 10.1016/j.gtc.2022.05.003. [DOI] [PubMed] [Google Scholar]
- 15.Gowd V., Kanika, Jori C., et al. Resveratrol and resveratrol nano-delivery systems in the treatment of inflammatory bowel disease. The Journal of Nutritional Biochemistry . 2022;109 doi: 10.1016/j.jnutbio.2022.109101.109101 [DOI] [PubMed] [Google Scholar]
- 16.Zhang H., Wang L., Li C., et al. Exosome-induced regulation in inflammatory bowel disease. Frontiers in Immunology . 2019;10 doi: 10.3389/fimmu.2019.01464.1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaya D. R., Russell R. K., Nimmo E. R., Satsangi J. New genes in inflammatory bowel disease: lessons for complex diseases? The Lancet . 2006;367(9518):1271–1284. doi: 10.1016/S0140-6736(06)68345-1. [DOI] [PubMed] [Google Scholar]
- 18.Jiang P., Zheng C., Xiang Y., et al. The involvement of TH17 cells in the pathogenesis of IBD. Cytokine & Growth Factor Reviews . 2023;69:28–42. doi: 10.1016/j.cytogfr.2022.07.005. [DOI] [PubMed] [Google Scholar]
- 19.Kudo T., Shimizu T. Mucosal immune systems of pediatric inflammatory bowel disease: a review. Pediatrics International . 2023;65(1) doi: 10.1111/ped.15511.e15511 [DOI] [PubMed] [Google Scholar]
- 20.Xavier R. J., Podolsky D. K. Unravelling the pathogenesis of inflammatory bowel disease. Nature . 2007;448(7152):427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 21.Na Y. R., Stakenborg M., Seok S. H., Matteoli G. Macrophages in intestinal inflammation and resolution: a potential therapeutic target in IBD. Nature Reviews Gastroenterology & Hepatology . 2019;16(9):531–543. doi: 10.1038/s41575-019-0172-4. [DOI] [PubMed] [Google Scholar]
- 22.Bain C. C., Mowat A. M. Macrophages in intestinal homeostasis and inflammation. Immunological Reviews . 2014;260(1):102–117. doi: 10.1111/imr.12192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mowat A. M. To respond or not to respond — a personal perspective of intestinal tolerance. Nature Reviews Immunology . 2018;18(6):405–415. doi: 10.1038/s41577-018-0002-x. [DOI] [PubMed] [Google Scholar]
- 24.Koelink P. J., Bloemendaal F. M., Li B., et al. Anti-TNF therapy in IBD exerts its therapeutic effect through macrophage IL-10 signalling. Gut . 2020;69(6):1053–1063. doi: 10.1136/gutjnl-2019-318264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zigmond E., Bernshtein B., Friedlander G., et al. Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity . 2014;40(5):720–733. doi: 10.1016/j.immuni.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 26.Shouval D. S., Biswas A., Goettel J. A., et al. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity . 2014;40(5):706–719. doi: 10.1016/j.immuni.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ingersoll M. A., Spanbroek R., Lottaz C., et al. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood . 2010;115(3):e10–e19. doi: 10.1182/blood-2009-07-235028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zigmond E., Varol C., Farache J., et al. Ly6C hi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity . 2012;37(6):1076–1090. doi: 10.1016/j.immuni.2012.08.026. [DOI] [PubMed] [Google Scholar]
- 29.Kamada N., Hisamatsu T., Okamoto S., et al. Unique CD14+ intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-γ axis. The Journal of Clinical Investigation . 2008;118(6):2269–2280. doi: 10.1172/JCI34610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bernardo D., Marin A. C., Fernández-Tomé S., et al. Human intestinal pro-inflammatory CD11chighCCR2+CX3CR1+ macrophages, but not their tolerogenic CD11c−CCR2−CX3CR1− counterparts, are expanded in inflammatory bowel disease. Mucosal Immunology . 2018;11(4):1114–1126. doi: 10.1038/s41385-018-0030-7. [DOI] [PubMed] [Google Scholar]
- 31.Klose C. S. N., Flach M., Möhle L., et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell . 2014;157(2):340–356. doi: 10.1016/j.cell.2014.03.030. [DOI] [PubMed] [Google Scholar]
- 32.Fallon P. G., Ballantyne S. J., Mangan N. E., et al. Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. The Journal of Experimental Medicine . 2006;203(4):1105–1116. doi: 10.1084/jem.20051615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reynders A., Yessaad N., Vu Manh T. P., et al. Identity, regulation and in vivo function of gut NKp46+RORγt+ and NKp46+RORγt− lymphoid cells. The EMBO Journal . 2011;30(14):2934–2947. doi: 10.1038/emboj.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Forkel M., Mjösberg J. Dysregulation of group 3 innate lymphoid cells in the pathogenesis of inflammatory bowel disease. Current Allergy and Asthma Reports . 2016;16(10) doi: 10.1007/s11882-016-0652-3.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Artis D., Spits H. The biology of innate lymphoid cells. Nature . 2015;517(7534):293–301. doi: 10.1038/nature14189. [DOI] [PubMed] [Google Scholar]
- 36.Fuchs A., Vermi W., Lee J. S., et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-γ-producing cells. Immunity . 2013;38(4):769–781. doi: 10.1016/j.immuni.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bernink J. H., Peters C. P., Munneke M., et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nature Immunology . 2013;14(3):221–229. doi: 10.1038/ni.2534. [DOI] [PubMed] [Google Scholar]
- 38.Forkel M., van Tol S., Höög C., Michaëlsson J., Almer S., Mjösberg J. Distinct alterations in the composition of mucosal innate lymphoid cells in newly diagnosed and established Crohn’s disease and ulcerative colitis. Journal of Crohn’s and Colitis . 2019;13(1):67–78. doi: 10.1093/ecco-jcc/jjy119. [DOI] [PubMed] [Google Scholar]
- 39.De Salvo C., Buela K. A., Creyns B., et al. NOD2 drives early IL-33-dependent expansion of group 2 innate lymphoid cells during Crohn’s disease-like ileitis. Journal of Clinical Investigation . 2021;131(5) doi: 10.1172/JCI140624.e140624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lim A. I., Menegatti S., Bustamante J., et al. IL-12 drives functional plasticity of human group 2 innate lymphoid cells. Journal of Experimental Medicine . 2016;213(4):569–583. doi: 10.1084/jem.20151750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buonocore S., Ahern P. P., Uhlig H. H., et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature . 2010;464(7293):1371–1375. doi: 10.1038/nature08949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Powell N., Walker A. W., Stolarczyk E., et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity . 2012;37(4):674–684. doi: 10.1016/j.immuni.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garrett W. S., Lord G. M., Punit S., et al. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell . 2007;131(1):33–45. doi: 10.1016/j.cell.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Geremia A., Arancibia-Cárcamo C. V., Fleming M. P. P., et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. Journal of Experimental Medicine . 2011;208(6):1127–1133. doi: 10.1084/jem.20101712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clottu A. S., Humbel M., Fluder N., Karampetsou M. P., Comte D. Innate lymphoid cells in autoimmune diseases. Frontiers in Immunology . 2021;12 doi: 10.3389/fimmu.2021.789788.789788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takatori H., Kanno Y., Watford W. T., et al. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. The Journal of Experimental Medicine . 2009;206(1):35–41. doi: 10.1084/jem.20072713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eken A., Singh A. K., Treuting P. M., Oukka M. IL-23R+ innate lymphoid cells induce colitis via interleukin-22-dependent mechanism. Mucosal Immunology . 2014;7(1):143–154. doi: 10.1038/mi.2013.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Verstockt B., Van Assche G., Vermeire S., Ferrante M. Biological therapy targeting the IL-23/IL-17 axis in inflammatory bowel disease. Expert Opinion on Biological Therapy . 2017;17(1):31–47. doi: 10.1080/14712598.2017.1258399. [DOI] [PubMed] [Google Scholar]
- 49.Duerr R. H., Taylor K. D., Brant S. R., et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science . 2006;314(5804):1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hue S., Ahern P., Buonocore S., et al. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. The Journal of Experimental Medicine . 2006;203(11):2473–2483. doi: 10.1084/jem.20061099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kullberg M. C., Jankovic D., Feng C. G., et al. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. The Journal of Experimental Medicine . 2006;203(11):2485–2494. doi: 10.1084/jem.20061082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maloy K. J. The Interleukin-23/Interleukin-17 axis in intestinal inflammation. Journal of Internal Medicine . 2008;263(6):584–590. doi: 10.1111/j.1365-2796.2008.01950.x. [DOI] [PubMed] [Google Scholar]
- 53.Abraham C., Cho J. Interleukin-23/Th17 pathways and inflammatory bowel disease. Inflammatory Bowel Diseases . 2009;15(7):1090–1100. doi: 10.1002/ibd.20894. [DOI] [PubMed] [Google Scholar]
- 54.Cooper M. A., Fehniger T. A., Caligiuri M. A. The biology of human natural killer-cell subsets. Trends in Immunology . 2001;22(11):633–640. doi: 10.1016/S1471-4906(01)02060-9. [DOI] [PubMed] [Google Scholar]
- 55.Liao C.-M., Zimmer M. I., Shanmuganad S., Yu H.-T., Cardell S. L., Wang C.-R. Dysregulation of CD1d-restricted type II natural killer T cells leads to spontaneous development of colitis in mice. Gastroenterology . 2012;142(2):326–334.e2. doi: 10.1053/j.gastro.2011.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bendelac A., Savage P. B., Teyton L. The biology of NKT cells. Annual Review of Immunology . 2007;25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711. [DOI] [PubMed] [Google Scholar]
- 57.Lai L. J., Shen J., Ran Z. H. Natural killer T cells and ulcerative colitis. Cellular Immunology . 2019;335:1–5. doi: 10.1016/j.cellimm.2018.08.010. [DOI] [PubMed] [Google Scholar]
- 58.Fuss I. J., Joshi B., Yang Z., et al. IL-13Rα2-bearing, type II NKT cells reactive to sulfatide self-antigen populate the mucosa of ulcerative colitis. Gut . 2014;63(11):1728–1736. doi: 10.1136/gutjnl-2013-305671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hornung M., Farkas S. A., Sattler C., Schlitt H. J., Geissler E. K. DX5+ NKT cells induce the death of colitis-associated cells: involvement of programmed death ligand-1. European Journal of Immunology . 2006;36(5):1210–1221. doi: 10.1002/eji.200535332. [DOI] [PubMed] [Google Scholar]
- 60.Fuss I. J., Heller F., Boirivant M., et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. The Journal of Clinical Investigation . 2004;113(10):1490–1497. doi: 10.1172/JCI19836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heller F., Florian P., Bojarski C., et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology . 2005;129(2):550–564. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 62.Saubermann L. J., Beck P., De Jong Y. P., et al. Activation of natural killer T cells by alpha-galactosylceramide in the presence of CD1d provides protection against colitis in mice. Gastroenterology . 2000;119(1):119–128. doi: 10.1053/gast.2000.9114. [DOI] [PubMed] [Google Scholar]
- 63.Burdin N., Brossay L., Kronenberg M. Immunization with alpha-galactosylceramide polarizes CD1-reactive NK T cells towards Th2 cytokine synthesis. European Journal of Immunology . 1999;29(6):2014–2025. doi: 10.1002/(SICI)1521-4141(199906)29:06<2014::AID-IMMU2014>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 64.Hershberg R. M. The epithelial cell cytoskeleton and intracellular trafficking. V. Polarized compartmentalization of antigen processing and toll-like receptor signaling in intestinal epithelial cells. American Journal of Physiology-Gastrointestinal and Liver Physiology . 2002;283(4):G833–G839. doi: 10.1152/ajpgi.00208.2002. [DOI] [PubMed] [Google Scholar]
- 65.Abreu M. T., Fukata M., Arditi M. TLR signaling in the gut in health and disease. The Journal of Immunology . 2005;174(8):4453–4460. doi: 10.4049/jimmunol.174.8.4453. [DOI] [PubMed] [Google Scholar]
- 66.Otte J.-M., Cario E., Podolsky D. K. Mechanisms of cross hyporesponsiveness to toll-like receptor bacterial ligands in intestinal epithelial cells. Gastroenterology . 2004;126(4):1054–1070. doi: 10.1053/j.gastro.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 67.Gewirtz A. T., Navas T. A., Lyons S., Godowski P. J., Madara J. L. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. The Journal of Immunology . 2001;167(4):1882–1885. doi: 10.4049/jimmunol.167.4.1882. [DOI] [PubMed] [Google Scholar]
- 68.Rhee S. H., Keates A. C., Moyer M. P., Pothoulakis C. MEK is a key modulator for TLR5-induced interleukin-8 and MIP3alpha gene expression in non-transformed human colonic epithelial cells. Journal of Biological Chemistry . 2004;279(24):25179–25188. doi: 10.1074/jbc.M400967200. [DOI] [PubMed] [Google Scholar]
- 69.Lodes M. J., Cong Y., Elson C. O., et al. Bacterial flagellin is a dominant antigen in Crohn disease. Journal of Clinical Investigation . 2004;113(9):1296–1306. doi: 10.1172/JCI200420295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guo L., Xu X.-Q., Zhou L., et al. Human intestinal epithelial cells release antiviral factors that inhibit HIV infection of macrophages. Frontiers in Immunology . 2018;9 doi: 10.3389/fimmu.2018.00247.247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bogunovic D., Byun M., Durfee L. A., et al. Mycobacterial disease and impaired IFN-γ immunity in humans with inherited ISG15 deficiency. Science . 2012;337(6102):1684–1688. doi: 10.1126/science.1224026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mirzalieva O., Juncker M., Schwartzenburg J., Desai S. ISG15 and ISGylation in human diseases. Cells . 2022;11(3) doi: 10.3390/cells11030538.538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.D’Cunha J., Knight E., Jr, Haas A. L., Truitt R. L., Borden E. C. Immunoregulatory properties of ISG15, an interferon-induced cytokine. Proceedings of the National Academy of Sciences of the United States of America . 1996;93(1):211–215. doi: 10.1073/pnas.93.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Østvik A. E., Svendsen T. D., Granlund A. B., et al. Intestinal epithelial cells express immunomodulatory ISG15 during active ulcerative colitis and Crohn’s disease. Journal of Crohn’s and Colitis . 2020;14(7):920–934. doi: 10.1093/ecco-jcc/jjaa022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hoeve M. A., Savage N. D., de Boer T., et al. Divergent effects of IL-12 and IL-23 on the production of IL-17 by human T cells. European Journal of Immunology . 2006;36(3):661–670. doi: 10.1002/(ISSN)1521-4141. [DOI] [PubMed] [Google Scholar]
- 76.Dieleman L. A., Palmen M. J., Akol H., et al. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clinical and Experimental Immunology . 1998;114(3):385–391. doi: 10.1046/j.1365-2249.1998.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kiesler P., Fuss I. J., Strober W. Experimental models of inflammatory bowel diseases. Cellular and Molecular Gastroenterology and Hepatology . 2015;1(2):154–170. doi: 10.1016/j.jcmgh.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Breese E., Braegger C. P., Corrigan C. J., Walker-Smith J. A., MacDonald T. T. Interleukin-2- and interferon-gamma-secreting T cells in normal and diseased human intestinal mucosa. Immunology . 1993;78(1):127–131. [PMC free article] [PubMed] [Google Scholar]
- 79.Monteleone G., Biancone L., Marasco R., et al. Interleukin 12 is expressed and actively released by Crohn’s disease intestinal lamina propria mononuclear cells. Gastroenterology . 1997;112(4):1169–1178. doi: 10.1016/S0016-5085(97)70128-8. [DOI] [PubMed] [Google Scholar]
- 80.Monteleone G., Parrello T., Luzza F., Pallone F. Response of human intestinal lamina propria T lymphocytes to interleukin 12: additive effects of interleukin 15 and 7. Gut . 1998;43(5):620–628. doi: 10.1136/gut.43.5.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Parronchi P., Romagnani P., Annunziato F., et al. Type 1 T-helper cell predominance and interleukin-12 expression in the gut of patients with Crohn’s disease. The American Journal of Pathology . 1997;150(3):823–832. [PMC free article] [PubMed] [Google Scholar]
- 82.Sarra M., Monteleone I., Stolfi C., et al. Interferon-gamma-expressing cells are a major source of interleukin-21 in inflammatory bowel diseases. Inflammatory Bowel Diseases . 2010;16(8):1332–1339. doi: 10.1002/ibd.21238. [DOI] [PubMed] [Google Scholar]
- 83.Monteleone G., Parrello T., Monteleone I., Tammaro S., Luzza F., Pallone F. Interferon-gamma (IFN-gamma) and prostaglandin E2 (PGE2) regulate differently IL-12 production in human intestinal lamina propria mononuclear cells (LPMC) Clinical and Experimental Immunology . 1999;117(3):469–475. doi: 10.1046/j.1365-2249.1999.00991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Di Sabatino A., Biancheri P., Rovedatti L., MacDonald T. T., Corazza G. R. New pathogenic paradigms in inflammatory bowel disease. Inflammatory Bowel Diseases . 2012;18(2):368–371. doi: 10.1002/ibd.21735. [DOI] [PubMed] [Google Scholar]
- 85.Cobrin G. M., Abreu M. T. Defects in mucosal immunity leading to Crohn’s disease. Immunological Reviews . 2005;206(1):277–295. doi: 10.1111/j.0105-2896.2005.00293.x. [DOI] [PubMed] [Google Scholar]
- 86.Neurath M. F., Weigmann B., Finotto S., et al. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn’s disease. The Journal of Experimental Medicine . 2002;195(9):1129–1143. doi: 10.1084/jem.20011956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fuss I. J., Neurath M., Boirivant M., et al. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. The Journal of Immunology . 1996;157(3):1261–1270. [PubMed] [Google Scholar]
- 88.Wynn T. A. Type 2 cytokines: mechanisms and therapeutic strategies. Nature Reviews Immunology . 2015;15(5):271–282. doi: 10.1038/nri3831. [DOI] [PubMed] [Google Scholar]
- 89.Ohtani K., Ohtsuka Y., Ikuse T., et al. Increased mucosal expression of GATA-3 and STAT-4 in pediatric ulcerative colitis. Pediatrics International: Official Journal of the Japan Pediatric Society . 2010;52(4):584–589. doi: 10.1111/j.1442-200X.2009.03019.x. [DOI] [PubMed] [Google Scholar]
- 90.Li J., Ueno A., Fort Gasia M., et al. Profiles of lamina propria T helper cell subsets discriminate between ulcerative colitis and Crohn’s disease. Inflammatory Bowel Diseases . 2016;22(8):1779–1792. doi: 10.1097/MIB.0000000000000811. [DOI] [PubMed] [Google Scholar]
- 91.Howitt M. R., Lavoie S., Michaud M., et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science . 2016;351(6279):1329–1333. doi: 10.1126/science.aaf1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Luo X., Villablanca E. J. Type 2 immunity in intestinal homeostasis and inflammatory bowel disease. Biochemical Society Transactions . 2021;49(5):2371–2380. doi: 10.1042/BST20210535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Popp V., Gerlach K., Mott S., et al. Rectal delivery of a DNAzyme that specifically blocks the transcription factor GATA3 and reduces colitis in mice. Gastroenterology . 2017;152(1):176–192.e5. doi: 10.1053/j.gastro.2016.09.005. [DOI] [PubMed] [Google Scholar]
- 94.Boirivant M., Fuss I. J., Chu A., Strober W. Oxazolone colitis: a murine model of T helper cell type 2 colitis treatable with antibodies to interleukin 4. Journal of Experimental Medicine . 1998;188(10):1929–1939. doi: 10.1084/jem.188.10.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rosen M. J., Karns R., Vallance J. E., et al. Mucosal expression of type 2 and type 17 immune response genes distinguishes ulcerative colitis from colon-only Crohn’s disease in treatment-naive pediatric patients. Gastroenterology . 2017;152(6):1345–1357.e7. doi: 10.1053/j.gastro.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nalleweg N., Chiriac M. T., Podstawa E., et al. IL-9 and its receptor are predominantly involved in the pathogenesis of UC. Gut . 2015;64(5):743–755. doi: 10.1136/gutjnl-2013-305947. [DOI] [PubMed] [Google Scholar]
- 97.Targan S. R., Karp L. C. Defects in mucosal immunity leading to ulcerative colitis. Immunological Reviews . 2005;206(1):296–305. doi: 10.1111/j.0105-2896.2005.00286.x. [DOI] [PubMed] [Google Scholar]
- 98.Ohnmacht C., Marques R., Presley L., Sawa S., Lochner M., Eberl G. Intestinal microbiota, evolution of the immune system and the bad reputation of pro-inflammatory immunity. Cellular Microbiology . 2011;13(5):653–659. doi: 10.1111/j.1462-5822.2011.01577.x. [DOI] [PubMed] [Google Scholar]
- 99.Weaver C. T., Elson C. O., Fouser L. A., Kolls J. K. The Th17 pathway and inflammatory diseases of the intestines, lungs, and skin. Annual Review of Pathology . 2013;8(1):477–512. doi: 10.1146/annurev-pathol-011110-130318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Maxwell J. R., Zhang Y., Brown W. A., et al. Differential roles for interleukin-23 and interleukin-17 in intestinal immunoregulation. Immunity . 2015;43(4):739–750. doi: 10.1016/j.immuni.2015.08.019. [DOI] [PubMed] [Google Scholar]
- 101.Ramesh R., Kozhaya L., McKevitt K., et al. Pro-inflammatory human Th17 cells selectively express P-glycoprotein and are refractory to glucocorticoids. Journal of Experimental Medicine . 2014;211(1):89–104. doi: 10.1084/jem.20130301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Friedrich M., Diegelmann J., Schauber J., Auernhammer C. J., Brand S. Intestinal neuroendocrine cells and goblet cells are mediators of IL-17A-amplified epithelial IL-17C production in human inflammatory bowel disease. Mucosal Immunology . 2015;8(4):943–958. doi: 10.1038/mi.2014.124. [DOI] [PubMed] [Google Scholar]
- 103.Reynolds J. M., Martinez G. J., Nallaparaju K. C., Chang S. H., Wang Y. H., Dong C. Cutting edge: regulation of intestinal inflammation and barrier function by IL-17C. The Journal of Immunology . 2012;189(9):4226–4230. doi: 10.4049/jimmunol.1103014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Takagi T., Naito Y., Tanaka M., et al. Carbon monoxide ameliorates murine T-cell-dependent colitis through the inhibition of Th17 differentiation. Free Radical Research . 2018;52(11-12):1328–1335. doi: 10.1080/10715762.2018.1470327. [DOI] [PubMed] [Google Scholar]
- 105.Kobayashi T., Okamoto S., Hisamatsu T., et al. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn’s disease. Gut . 2008;57(12):1682–1689. doi: 10.1136/gut.2007.135053. [DOI] [PubMed] [Google Scholar]
- 106.Sugihara T., Kobori A., Imaeda H., et al. The increased mucosal mRNA expressions of complement C3 and interleukin-17 in inflammatory bowel disease. Clinical and Experimental Immunology . 2010;160(3):386–393. doi: 10.1111/j.1365-2249.2010.04093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Monteleone G., Monteleone I., Fina D., et al. Interleukin-21 enhances T-helper cell type I signaling and interferon-gamma production in Crohn’s disease. Gastroenterology . 2005;128(3):687–694. doi: 10.1053/j.gastro.2004.12.042. [DOI] [PubMed] [Google Scholar]
- 108.Kmieć Z., Cyman M., Ślebioda T. J. Cells of the innate and adaptive immunity and their interactions in inflammatory bowel disease. Advances in Medical Sciences . 2017;62(1):1–16. doi: 10.1016/j.advms.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 109.Roncarolo M. G., Gregori S., Bacchetta R., Battaglia M., Gagliani N. The biology of T regulatory type 1 cells and their therapeutic application in immune-mediated diseases. Immunity . 2018;49(6):1004–1019. doi: 10.1016/j.immuni.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 110.Saruta M., Yu Q. T., Fleshner P. R., et al. Characterization of FoxP3+CD4+ regulatory T cells in Crohn’s disease. Clinical Immunology (Orlando, Fla) . 2007;125(3):281–290. doi: 10.1016/j.clim.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 111.Makita S., Kanai T., Oshima S., et al. CD4+CD25bright T cells in human intestinal lamina propria as regulatory cells. Journal of Immunology . 2004;173(5):3119–3130. doi: 10.4049/jimmunol.173.5.3119. [DOI] [PubMed] [Google Scholar]
- 112.Ricciardelli I., Lindley K. J., Londei M., Quaratino S. Anti tumour necrosis-alpha therapy increases the number of FOXP3 regulatory T cells in children affected by Crohn’s disease. Immunology . 2008;125(2):178–183. doi: 10.1111/j.1365-2567.2008.02839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Holmén N., Lundgren A., Lundin S., et al. Functional CD4+CD25high regulatory T cells are enriched in the colonic mucosa of patients with active ulcerative colitis and increase with disease activity. Inflammatory Bowel Diseases . 2006;12(6):447–456. doi: 10.1097/00054725-200606000-00003. [DOI] [PubMed] [Google Scholar]
- 114.Wehkamp J., Harder J., Weichenthal M., et al. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut . 2004;53(11):1658–1664. doi: 10.1136/gut.2003.032805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sun Q., Liu X., Li X. Peptidoglycan-based immunomodulation. Applied Microbiology and Biotechnology . 2022;106(3):981–993. doi: 10.1007/s00253-022-11795-4. [DOI] [PubMed] [Google Scholar]
- 116.Vollmer W., Blanot D., De Pedro M. A. Peptidoglycan structure and architecture. FEMS Microbiology Reviews . 2008;32(2):149–167. doi: 10.1111/j.1574-6976.2007.00094.x. [DOI] [PubMed] [Google Scholar]
- 117.Athman R., Philpott D. Innate immunity via toll-like receptors and Nod proteins. Current Opinion in Microbiology . 2004;7(1):25–32. doi: 10.1016/j.mib.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 118.Chamaillard M., Girardin S. E., Viala J., Philpott D. J. Nods, Nalps and Naip: intracellular regulators of bacterial-induced inflammation. Cellular Microbiology . 2003;5(9):581–592. doi: 10.1046/j.1462-5822.2003.00304.x. [DOI] [PubMed] [Google Scholar]
- 119.Inohara N., Ogura Y., Fontalba A., et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. Journal of Biological Chemistry . 2003;278(8):5509–5512. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 120.Girardin S. E., Boneca I. G., Viala J., et al. NOD2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. Journal of Biological Chemistry . 2003;278(11):8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 121.Saez A., Gomez-Bris R., Herrero-Fernandez B., Mingorance C., Rius C., Gonzalez-Granado J. M. Innate lymphoid cells in intestinal homeostasis and inflammatory bowel disease. International Journal of Molecular Sciences . 2021;22(14) doi: 10.3390/ijms22147618.7618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hugot J.-P., Chamaillard M., Zouali H., et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature . 2001;411(6837):599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 123.Brant S. R., Shugart Y. Y. Inflammatory bowel disease gene hunting by linkage analysis: rationale, methodology, and present status of the field. Inflammatory Bowel Diseases . 2004;10(3):300–311. doi: 10.1097/00054725-200405000-00019. [DOI] [PubMed] [Google Scholar]
- 124.Hisamatsu T., Suzuki M., Reinecker H.-C., Nadeau W. J., McCormick B. A., Podolsky D. K. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology . 2003;124(4):993–1000. doi: 10.1053/gast.2003.50153. [DOI] [PubMed] [Google Scholar]
- 125.Watanabe T., Kitani A., Murray P. J., Strober W. NOD2 is a negative regulator of toll-like receptor 2-mediated T helper type 1 responses. Nature Immunology . 2004;5(8):800–808. doi: 10.1038/ni1092. [DOI] [PubMed] [Google Scholar]
- 126.Bonen D. K., Ogura Y., Nicolae D. L., et al. Crohn’s disease-associated NOD2 variants share a signaling defect in response to lipopolysaccharide and peptidoglycan. Gastroenterology . 2003;124(1):140–146. doi: 10.1053/gast.2003.50019. [DOI] [PubMed] [Google Scholar]
- 127.Cuthbert A. P., Fisher S. A., Mirza M. M., et al. The contribution of NOD2 gene mutations to the risk and site of disease in inflammatory bowel disease. Gastroenterology . 2002;122(4):867–874. doi: 10.1053/gast.2002.32415. [DOI] [PubMed] [Google Scholar]
- 128.Li M.-C., He S.-H. IL-10 and its related cytokines for treatment of inflammatory bowel disease. World Journal of Gastroenterology . 2004;10(5):620–625. doi: 10.3748/wjg.v10.i5.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ishizuka K., Sugimura K., Homma T., et al. Influence of interleukin-10 on the interleukin-1 receptor antagonist/interleukin-1 beta ratio in the colonic mucosa of ulcerative colitis. Digestion . 2001;63(Suppl 1):22–27. doi: 10.1159/000051906. [DOI] [PubMed] [Google Scholar]
- 130.Rennick D. M., Fort M. M. Lessons from genetically engineered animal models. XII. IL-10-deficient (IL-10−/−) mice and intestinal inflammation. American Journal of Physiology-Gastrointestinal and Liver Physiology . 2000;278(6):G829–G833. doi: 10.1152/ajpgi.2000.278.6.G829. [DOI] [PubMed] [Google Scholar]
- 131.Uhlig H. H., Schwerd T., Koletzko S., et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology . 2014;147(5):990–1007.e3. doi: 10.1053/j.gastro.2014.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kelsen J. R., Baldassano R. N., Artis D., Sonnenberg G. F. Maintaining intestinal health: the genetics and immunology of very early onset inflammatory bowel disease. Cellular and Molecular Gastroenterology and Hepatology . 2015;1(5):462–476. doi: 10.1016/j.jcmgh.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Muise A. M., Snapper S. B., Kugathasan S. The age of gene discovery in very early onset inflammatory bowel disease. Gastroenterology . 2012;143(2):285–288. doi: 10.1053/j.gastro.2012.06.025. [DOI] [PubMed] [Google Scholar]
- 134.Glocker E.-O., Frede N., Perro M., et al. Infant colitis—it’s in the genes. The Lancet . 2010;376(9748) doi: 10.1016/S0140-6736(10)61008-2.1272 [DOI] [PubMed] [Google Scholar]
- 135.Rioux J. D., Xavier R. J., Taylor K. D., et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nature Genetics . 2007;39(5):596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rubin D. C., Zhang H., Qian P., Lorenz R. G., Hutton K., Peters M. G. Altered enteroendocrine cell expression in T cell receptor alpha chain knock-out mice. Microscopy Research and Technique . 2000;51(2):112–120. doi: 10.1002/(ISSN)1097-0029. [DOI] [PubMed] [Google Scholar]
- 137.Kaser A., Blumberg R. S. Autophagy, microbial sensing, endoplasmic reticulum stress, and epithelial function in inflammatory bowel disease. Gastroenterology . 2011;140(6):1738–1747.e2. doi: 10.1053/j.gastro.2011.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hampe J., Franke A., Rosenstiel P., et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nature Genetics . 2007;39(2):207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 139.Libioulle C., Louis E., Hansoul S., et al. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genetics . 2007;3(4) doi: 10.1371/journal.pgen.0030058.e58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhang H., Zheng L., McGovern D. P., et al. Myeloid ATG16L1 facilitates host-bacteria interactions in maintaining intestinal homeostasis. The Journal of Immunology . 2017;198(5):2133–2146. doi: 10.4049/jimmunol.1601293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lahiri A., Hedl M., Abraham C. MTMR3 risk allele enhances innate receptor-induced signaling and cytokines by decreasing autophagy and increasing caspase-1 activation. Proceedings of the National Academy of Sciences of the United States of America . 2015;112(33):10461–10466. doi: 10.1073/pnas.1501752112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Larabi A., Barnich N., Nguyen H. T. T. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy . 2020;16(1):38–51. doi: 10.1080/15548627.2019.1635384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Tan J. M. J., Mellouk N., Osborne S. E., et al. An ATG16L1-dependent pathway promotes plasma membrane repair and limits Listeria monocytogenes cell-to-cell spread. Nature Microbiology . 2018;3(12):1472–1485. doi: 10.1038/s41564-018-0293-5. [DOI] [PubMed] [Google Scholar]
- 144.Travassos L. H., Carneiro L. A. M., Ramjeet M., et al. NOD1 and NOD2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nature Immunology . 2010;11(1):55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 145.Cooney R., Baker J., Brain O., et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nature Medicine . 2010;16(1):90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 146.Gradzka S., Thomas O. S., Kretz O., et al. Inhibitor of apoptosis proteins are required for effective fusion of autophagosomes with lysosomes. Cell Death & Disease . 2018;9(5) doi: 10.1038/s41419-018-0508-y.529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Schwerd T., Pandey S., Yang H.-T., et al. Impaired antibacterial autophagy links granulomatous intestinal inflammation in Niemann–Pick disease type C1 and XIAP deficiency with NOD2 variants in Crohn’s disease. Gut . 2017;66(6):1060–1073. doi: 10.1136/gutjnl-2015-310382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Liu B., Gulati A. S., Cantillana V., et al. Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. American Journal of Physiology-Gastrointestinal and Liver Physiology . 2013;305(8):G573–G584. doi: 10.1152/ajpgi.00071.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lassen K. G., McKenzie C. I., Mari M., et al. Genetic coding variant in GPR65 alters lysosomal pH and links lysosomal dysfunction with colitis risk. Immunity . 2016;44(6):1392–1405. doi: 10.1016/j.immuni.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Xia L., Oyang L., Lin J., et al. The cancer metabolic reprogramming and immune response. Molecular Cancer . 2021;20(1) doi: 10.1186/s12943-021-01316-8.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mayr L., Grabherr F., Schwärzler J., et al. Dietary lipids fuel GPX4-restricted enteritis resembling Crohn’s disease. Nature Communications . 2020;11(1) doi: 10.1038/s41467-020-15646-6.1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.de Silva P. S. A., Olsen A., Christensen J., et al. An association between dietary arachidonic acid, measured in adipose tissue, and ulcerative colitis. Gastroenterology . 2010;139(6):1912–1917. doi: 10.1053/j.gastro.2010.07.065. [DOI] [PubMed] [Google Scholar]
- 153.Xu S., He Y., Lin L., Chen P., Chen M., Zhang S. The emerging role of ferroptosis in intestinal disease. Cell Death & Disease . 2021;12(4) doi: 10.1038/s41419-021-03559-1.289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bensinger S. J., Tontonoz P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature . 2008;454(7203):470–477. doi: 10.1038/nature07202. [DOI] [PubMed] [Google Scholar]
- 155.Dubuquoy L., Rousseaux C., Thuru X., et al. PPARγ as a new therapeutic target in inflammatory bowel diseases. Gut . 2006;55(9):1341–1349. doi: 10.1136/gut.2006.093484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Debril M.-B., Renaud J.-P., Fajas L., Auwerx J. The pleiotropic functions of peroxisome proliferator-activated receptor gamma. Journal of Molecular Medicine . 2001;79(1):30–47. doi: 10.1007/s001090000145. [DOI] [PubMed] [Google Scholar]
- 157.Fajas L., Debril M. B., Auwerx J. Peroxisome proliferator-activated receptor-gamma: from adipogenesis to carcinogenesis. Journal of Molecular Endocrinology . 2001;27(1):1–9. doi: 10.1677/jme.0.0270001. [DOI] [PubMed] [Google Scholar]
- 158.Su C. G., Wen X., Bailey S. T., et al. A novel therapy for colitis utilizing PPAR-γ ligands to inhibit the epithelial inflammatory response. Journal of Clinical Investigation . 1999;104(4):383–389. doi: 10.1172/JCI7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rousseaux C., Lefebvre B., Dubuquoy L., et al. Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator-activated receptor- Journal of Experimental Medicine . 2005;201(8):1205–1215. doi: 10.1084/jem.20041948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bassaganya-Riera J., Reynolds K., Martino-Catt S., et al. Activation of PPAR γ and δ by conjugated linoleic acid mediates protection from experimental inflammatory bowel disease. Gastroenterology . 2004;127(3):777–791. doi: 10.1053/j.gastro.2004.06.049. [DOI] [PubMed] [Google Scholar]
- 161.Parada Venegas D., De la Fuente M. K., Landskron G., et al. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Frontiers in Immunology . 2019;10 doi: 10.3389/fimmu.2019.00277.277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Puddu A., Sanguineti R., Montecucco F., Viviani G. L. Evidence for the gut microbiota short-chain fatty acids as key pathophysiological molecules improving diabetes. Mediators of Inflammation . 2014;2014:9. doi: 10.1155/2014/162021.162021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Deleu S., Machiels K., Raes J., Verbeke K., Vermeire S. Short chain fatty acids and its producing organisms: an overlooked therapy for IBD? EBioMedicine . 2021;66 doi: 10.1016/j.ebiom.2021.103293.103293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Louis P., Flint H. J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiology Letters . 2009;294(1):1–8. doi: 10.1111/j.1574-6968.2009.01514.x. [DOI] [PubMed] [Google Scholar]
- 165.Louis P., Flint H. J. Formation of propionate and butyrate by the human colonic microbiota. Environmental Microbiology . 2017;19(1):29–41. doi: 10.1111/1462-2920.13589. [DOI] [PubMed] [Google Scholar]
- 166.Champ M. M.-J. Physiological aspects of resistant starch and in vivo measurements. Journal of AOAC International . 2004;87(3):749–755. doi: 10.1093/jaoac/87.3.749. [DOI] [PubMed] [Google Scholar]
- 167.Licciardi P. V., Ververis K., Karagiannis T. C. Histone deacetylase inhibition and dietary short-chain fatty acids. ISRN Allergy . 2011;2011 doi: 10.5402/2011/869647.869647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kelly C. J., Zheng L., Campbell E. L., et al. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host & Microbe . 2015;17(5):662–671. doi: 10.1016/j.chom.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Donohoe D. R., Collins L. B., Wali A., Bigler R., Sun W., Bultman S. J. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Molecular Cell . 2012;48(4):612–626. doi: 10.1016/j.molcel.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Vinolo M. A. R., Hirabara S. M., Curi R. G-protein-coupled receptors as fat sensors. Current Opinion in Clinical Nutrition and Metabolic Care . 2012;15(2):112–116. doi: 10.1097/MCO.0b013e32834f4598. [DOI] [PubMed] [Google Scholar]
- 171.Brown A. J., Goldsworthy S. M., Barnes A. A., et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. Journal of Biological Chemistry . 2003;278(13):11312–11319. doi: 10.1074/jbc.M211609200. [DOI] [PubMed] [Google Scholar]
- 172.Le Poul E., Loison C., Struyf S., et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. Journal of Biological Chemistry . 2003;278(28):25481–25489. doi: 10.1074/jbc.M301403200. [DOI] [PubMed] [Google Scholar]
- 173.Taggart A. K. P., Kero J., Gan X., et al. (d)-β-Hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. Journal of Biological Chemistry . 2005;280(29):26649–26652. doi: 10.1074/jbc.C500213200. [DOI] [PubMed] [Google Scholar]
- 174.Hilger D., Masureel M., Kobilka B. K. Structure and dynamics of GPCR signaling complexes. Nature Structural & Molecular Biology . 2018;25(1):4–12. doi: 10.1038/s41594-017-0011-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhao Y., Chen F., Wu W., et al. GPR43 mediates microbiota metabolite SCFA regulation of antimicrobial peptide expression in intestinal epithelial cells via activation of mTOR and STAT3. Mucosal Immunology . 2018;11(3):752–762. doi: 10.1038/mi.2017.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Furusawa Y., Obata Y., Fukuda S., et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature . 2013;504(7480):446–450. doi: 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- 177.Smith P. M., Howitt M. R., Panikov N., et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science . 2013;341(6145):569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zhao M., Zhu W., Gong J., et al. Dietary fiber intake is associated with increased colonic mucosal GPR43+ polymorphonuclear infiltration in active Crohn’s disease. Nutrients . 2015;7(7):5327–5346. doi: 10.3390/nu7075223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Slavin J. Fiber and prebiotics: mechanisms and health benefits. Nutrients . 2013;5(4):1417–1435. doi: 10.3390/nu5041417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Jacobasch G., Schmiedl D., Kruschewski M., Schmehl K. Dietary resistant starch and chronic inflammatory bowel diseases. International Journal of Colorectal Disease . 1999;14(4-5):201–211. doi: 10.1007/s003840050212. [DOI] [PubMed] [Google Scholar]
- 181.Palmer C. S., Ostrowski M., Balderson B., Christian N., Crowe S. M. Glucose metabolism regulates T cell activation, differentiation, and functions. Frontiers in Immunology . 2015;6 doi: 10.3389/fimmu.2015.00001.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Jun H. S., Lee Y. M., Cheung Y. Y., et al. Lack of glucose recycling between endoplasmic reticulum and cytoplasm underlies cellular dysfunction in glucose-6-phosphatase-β-deficient neutrophils in a congenital neutropenia syndrome. Blood . 2010;116(15):2783–2792. doi: 10.1182/blood-2009-12-258491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Chacko B. K., Kramer P. A., Ravi S., et al. Methods for defining distinct bioenergetic profiles in platelets, lymphocytes, monocytes, and neutrophils, and the oxidative burst from human blood. Laboratory Investigation . 2013;93(6):690–700. doi: 10.1038/labinvest.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kramer P. A., Ravi S., Chacko B., Johnson M. S., Darley-Usmar V. M. A review of the mitochondrial and glycolytic metabolism in human platelets and leukocytes: implications for their use as bioenergetic biomarkers. Redox Biology . 2014;2:206–210. doi: 10.1016/j.redox.2013.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bolton C., Burch N., Morgan J., et al. Remission of inflammatory bowel disease in glucose-6-phosphatase 3 deficiency by allogeneic haematopoietic stem cell transplantation. Journal of Crohn’s and Colitis . 2020;14(1):142–147. doi: 10.1093/ecco-jcc/jjz112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Banka S., Newman W. G. A clinical and molecular review of ubiquitous glucose-6-phosphatase deficiency caused by G6PC3 mutations. Orphanet Journal of Rare Diseases . 2013;8(1) doi: 10.1186/1750-1172-8-84.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Smith B. N., Evans C., Ali A., et al. Phenotypic heterogeneity and evidence of a founder effect associated with G6PC3 mutations in patients with severe congenital neutropenia. British Journal of Haematology . 2012;158(1):146–149. doi: 10.1111/j.1365-2141.2012.09110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Boztug K., Appaswamy G., Ashikov A., et al. A syndrome with congenital neutropenia and mutations in G6PC3. New England Journal of Medicine . 2009;360(1):32–43. doi: 10.1056/NEJMoa0805051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.McDermott D. H., De Ravin S. S., Jun H. S., et al. Severe congenital neutropenia resulting from G6PC3 deficiency with increased neutrophil CXCR4 expression and myelokathexis. Blood . 2010;116(15):2793–2802. doi: 10.1182/blood-2010-01-265942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Bégin P., Patey N., Mueller P., et al. Inflammatory bowel disease and T cell lymphopenia in G6PC3 deficiency. Journal of Clinical Immunology . 2013;33(3):520–525. doi: 10.1007/s10875-012-9833-6. [DOI] [PubMed] [Google Scholar]
- 191.Cullinane A. R., Vilboux T., O’Brien K., et al. Homozygosity mapping and whole-exome sequencing to detect SLC45A2 and G6PC3 mutations in a single patient with oculocutaneous albinism and neutropenia. Journal of Investigative Dermatology . 2011;131(10):2017–2025. doi: 10.1038/jid.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Desplantes C., Fremond M. L., Beaupain B., et al. Clinical spectrum and long-term follow-up of 14 cases with G6PC3 mutations from the French Severe Congenital Neutropenia Registry. Orphanet Journal of Rare Diseases . 2014;9(1) doi: 10.1186/s13023-014-0183-8.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Mistry A., Scambler T., Parry D., et al. Glucose-6-phosphatase catalytic subunit 3 (G6PC3) deficiency associated with autoinflammatory complications. Frontiers in Immunology . 2017;8 doi: 10.3389/fimmu.2017.01485.1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Goenka A., Doherty J. A., Al-Farsi T., et al. Neutrophil dysfunction triggers inflammatory bowel disease in G6PC3 deficiency. Journal of Leukocyte Biology . 2021;109(6):1147–1154. doi: 10.1002/JLB.5AB1219-699RR. [DOI] [PubMed] [Google Scholar]
- 195.Daig R., Andus T., Aschenbrenner E., Falk W., Schölmerich J., Gross V. Increased interleukin 8 expression in the colon mucosa of patients with inflammatory bowel disease. Gut . 1996;38(2):216–222. doi: 10.1136/gut.38.2.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Arnold R., König W. Interleukin-8 release from human neutrophils after phagocytosis of Listeria monocytogenes and Yersinia enterocolitica. Journal of Medical Microbiology . 1998;47(1):55–62. doi: 10.1099/00222615-47-1-55. [DOI] [PubMed] [Google Scholar]
- 197.Fioredda F., Iacobelli S., van Biezen A., et al. Stem cell transplantation in severe congenital neutropenia: an analysis from the European Society for Blood and Marrow Transplantation. Blood . 2015;126(16):1885–1892. doi: 10.1182/blood-2015-02-628859. [DOI] [PubMed] [Google Scholar]
- 198.Zietek T., Rath E. Inflammation meets metabolic disease: gut feeling mediated by GLP-1. Frontiers in Immunology . 2016;7 doi: 10.3389/fimmu.2016.00154.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Duan L., Rao X., Braunstein Z., Toomey A. C., Zhong J. Role of incretin axis in inflammatory bowel disease. Frontiers in Immunology . 2017;8 doi: 10.3389/fimmu.2017.01734.1734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Zhang D., Jin W., Wu R., et al. High glucose intake exacerbates autoimmunity through reactive-oxygen-species-mediated TGF-β cytokine activation. Immunity . 2019;51(4):671–681.e5. doi: 10.1016/j.immuni.2019.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Yusta B., Baggio L. L., Koehler J., et al. GLP-1R agonists modulate enteric immune responses through the intestinal intraepithelial lymphocyte GLP-1R. Diabetes . 2015;64(7):2537–2549. doi: 10.2337/db14-1577. [DOI] [PubMed] [Google Scholar]
- 202.Hadjiyanni I., Siminovitch K. A., Danska J. S., Drucker D. J. Glucagon-like peptide-1 receptor signalling selectively regulates murine lymphocyte proliferation and maintenance of peripheral regulatory T cells. Diabetologia . 2010;53(4):730–740. doi: 10.1007/s00125-009-1643-x. [DOI] [PubMed] [Google Scholar]
- 203.Zhong J., Maiseyeu A., Davis S. N., Rajagopalan S. DPP4 in cardiometabolic disease: recent insights from the laboratory and clinical trials of DPP4 inhibition. Circulation Research . 2015;116(8):1491–1504. doi: 10.1161/CIRCRESAHA.116.305665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Deacon C. F., Nauck M. A., Meier J., Hücking K., Holst J. J. Degradation of endogenous and exogenous gastric inhibitory polypeptide in healthy and in type 2 diabetic subjects as revealed using a new assay for the intact peptide. The Journal of Clinical Endocrinology and Metabolism . 2000;85(10):3575–3581. doi: 10.1210/jcem.85.10.6855. [DOI] [PubMed] [Google Scholar]
- 205.Drucker D. J. Biological actions and therapeutic potential of the glucagon-like peptides. Gastroenterology . 2002;122(2):531–544. doi: 10.1053/gast.2002.31068. [DOI] [PubMed] [Google Scholar]
- 206.Zhong J., Rao X., Rajagopalan S. An emerging role of dipeptidyl peptidase 4 (DPP4) beyond glucose control: potential implications in cardiovascular disease. Atherosclerosis . 2013;226(2):305–314. doi: 10.1016/j.atherosclerosis.2012.09.012. [DOI] [PubMed] [Google Scholar]
- 207.Kameoka J., Tanaka T., Nojima Y., Schlossman S. F., Morimoto C. Direct association of adenosine deaminase with a T cell activation antigen, CD26. Science . 1993;261(5120):466–469. doi: 10.1126/science.8101391. [DOI] [PubMed] [Google Scholar]
- 208.Morimoto C., Schlossman S. F. The structure and function of CD26 in the T-cell immune response. Immunological Reviews . 1998;161(1):55–70. doi: 10.1111/j.1600-065X.1998.tb01571.x. [DOI] [PubMed] [Google Scholar]
- 209.Keller J., Beglinger C., Holst J. J., Andresen V., Layer P. Mechanisms of gastric emptying disturbances in chronic and acute inflammation of the distal gastrointestinal tract. American Journal of Physiology-Gastrointestinal and Liver Physiology . 2009;297(5):G861–G868. doi: 10.1152/ajpgi.00145.2009. [DOI] [PubMed] [Google Scholar]
- 210.Keller J., Binnewies U., Rösch M., et al. Gastric emptying and disease activity in inflammatory bowel disease. European Journal of Clinical Investigation . 2015;45(12):1234–1242. doi: 10.1111/eci.12542. [DOI] [PubMed] [Google Scholar]
- 211.Hildebrandt M., Rose M., Rüter J., Salama A., Mönnikes H., Klapp B. F. Dipeptidyl peptidase IV (DP IV, CD26) in patients with inflammatory bowel disease. Scandinavian Journal of Gastroenterology . 2001;36(10):1067–1072. doi: 10.1080/003655201750422675. [DOI] [PubMed] [Google Scholar]
- 212.Xiao Q., Boushey R. P., Cino M., Drucker D. J., Brubaker P. L. Circulating levels of glucagon-like peptide-2 in human subjects with inflammatory bowel disease. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology . 2000;278(4):R1057–R1063. doi: 10.1152/ajpregu.2000.278.4.R1057. [DOI] [PubMed] [Google Scholar]
- 213.Buljevic S., Detel D., Pugel E. P., Varljen J. The effect of CD26-deficiency on dipeptidyl peptidase 8 and 9 expression profiles in a mouse model of Crohn’s disease. Journal of Cellular Biochemistry . 2018;119(8):6743–6755. doi: 10.1002/jcb.26867. [DOI] [PubMed] [Google Scholar]
- 214.Enz N., Vliegen G., De Meester I., Jungraithmayr W. CD26/DPP4 - a potential biomarker and target for cancer therapy. Pharmacology & Therapeutics . 2019;198:135–159. doi: 10.1016/j.pharmthera.2019.02.015. [DOI] [PubMed] [Google Scholar]
- 215.De Meester I., Durinx C., Bal G., et al. Natural substrates of dipeptidyl peptidase IV. Advances in Experimental Medicine and Biology . 2000;477:67–87. doi: 10.1007/0-306-46826-3_7. [DOI] [PubMed] [Google Scholar]
- 216.Abbott C. A., Yazbeck R., Geier M. S., Demuth H.-U., Howarth G. S. Dipeptidyl peptidases and inflammatory bowel disease. Advances in Experimental Medicine and Biology . 2006;575:155–162. doi: 10.1007/0-387-32824-6_16. [DOI] [PubMed] [Google Scholar]
- 217.Hu X., Wang X., Xue X. Therapeutic perspectives of CD26 inhibitors in imune-mediated diseases. Molecules . 2022;27(14) doi: 10.3390/molecules27144498.4498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Ning M.-M., Yang W.-J., Guan W.-B., Gu Y.-P., Feng Y., Leng Y. Dipeptidyl peptidase 4 inhibitor sitagliptin protected against dextran sulfate sodium-induced experimental colitis by potentiating the action of GLP-2. Acta pharmacologica Sinica . 2020;41(11):1446–1456. doi: 10.1038/s41401-020-0413-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Mimura S., Ando T., Ishiguro K., et al. Dipeptidyl peptidase-4 inhibitor anagliptin facilitates restoration of dextran sulfate sodium-induced colitis. Scandinavian Journal of Gastroenterology . 2013;48(10):1152–1159. doi: 10.3109/00365521.2013.832366. [DOI] [PubMed] [Google Scholar]
- 220.Li P., Yin Y.-L., Li D., Kim S. W., Wu G. Amino acids and immune function. British Journal of Nutrition . 2007;98(2):237–252. doi: 10.1017/S000711450769936X. [DOI] [PubMed] [Google Scholar]
- 221.Nielsen J. P., Foged N. T., Sørensen V., Barfod K., Bording A., Petersen S. K. Vaccination against progressive atrophic rhinitis with a recombinant Pasteurella multocida toxin derivative. Canadian Journal of Veterinary Research . 1991;55(2):128–138. [PMC free article] [PubMed] [Google Scholar]
- 222.Hubbard T. D., Murray I. A., Perdew G. H. Indole and tryptophan metabolism: endogenous and dietary routes to ah receptor activation. Drug Metabolism and Disposition . 2015;43(10):1522–1535. doi: 10.1124/dmd.115.064246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Kawajiri K., Fujii-Kuriyama Y. The aryl hydrocarbon receptor: a multifunctional chemical sensor for host defense and homeostatic maintenance. Experimental Animals . 2017;66(2):75–89. doi: 10.1538/expanim.16-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Lamas B., Richard M. L., Leducq V., et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nature Medicine . 2016;22(6):598–605. doi: 10.1038/nm.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Agus A., Planchais J., Sokol H. Gut microbiota regulation of tryptophan metabolism in health and disease. Cell Host & Microbe . 2018;23(6):716–724. doi: 10.1016/j.chom.2018.05.003. [DOI] [PubMed] [Google Scholar]
- 226.Monteleone I., Pallone F., Monteleone G. Aryl hydrocarbon receptor and colitis. Seminars in Immunopathology . 2013;35(6):671–675. doi: 10.1007/s00281-013-0396-2. [DOI] [PubMed] [Google Scholar]
- 227.Lee J. S., Cella M., McDonald K. G., et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of notch. Nature Immunology . 2011;13(2):144–151. doi: 10.1038/ni.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Quintana F. J., Sherr D. H. Aryl hydrocarbon receptor control of adaptive immunity. Pharmacological Reviews . 2013;65(4):1148–1161. doi: 10.1124/pr.113.007823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Ji T., Xu C., Sun L., et al. Aryl hydrocarbon receptor activation down-regulates IL-7 and reduces inflammation in a mouse model of DSS-induced colitis. Digestive Diseases and Sciences . 2015;60(7):1958–1966. doi: 10.1007/s10620-015-3632-x. [DOI] [PubMed] [Google Scholar]
- 230.Russo E., Giudici F., Fiorindi C., Ficari F., Scaringi S., Amedei A. Immunomodulating activity and therapeutic effects of short chain fatty acids and tryptophan post-biotics in inflammatory bowel disease. Frontiers in Immunology . 2019;10 doi: 10.3389/fimmu.2019.02754.2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Oshima S., Fujimura M., Fukimiya M. Changes in number of serotonin-containing cells and serotonin levels in the intestinal mucosa of rats with colitis induced by dextran sodium sulfate. Histochemistry and Cell Biology . 1999;112(4):257–263. doi: 10.1007/s004180050445. [DOI] [PubMed] [Google Scholar]
- 232.Linden D. R., Chen J.-X., Gershon M. D., Sharkey K. A., Mawe G. M. Serotonin availability is increased in mucosa of guinea pigs with TNBS-induced colitis. American Journal of Physiology-Gastrointestinal and Liver Physiology . 2003;285(1):G207–G216. doi: 10.1152/ajpgi.00488.2002. [DOI] [PubMed] [Google Scholar]
- 233.Maffei M. E. 5-Hydroxytryptophan (5-HTP): natural occurrence, analysis, biosynthesis, biotechnology, physiology and toxicology. International Journal of Molecular Sciences . 2020;22(1) doi: 10.3390/ijms22010181.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Manocha M., Khan W. I. Serotonin and GI disorders: an update on clinical and experimental studies. Clinical and Translational Gastroenterology . 2012;3(4) doi: 10.1038/ctg.2012.8.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Magro F., Vieira-Coelho M. A., Fraga S., et al. Impaired synthesis or cellular storage of norepinephrine, dopamine, and 5-hydroxytryptamine in human inflammatory bowel disease. Digestive Diseases and Sciences . 2002;47(1):216–224. doi: 10.1023/a:1013256629600. [DOI] [PubMed] [Google Scholar]
- 236.Musch M. W., Clarke L. L., Mamah D., et al. T cell activation causes diarrhea by increasing intestinal permeability and inhibiting epithelial Na+/K+-ATPase. The Journal of Clinical Investigation . 2002;110(11):1739–1747. doi: 10.1172/JCI15695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Allgayer H., Kruis W., Paumgartner G., Wiebecke B., Brown L., Erdmann E. Inverse relationship between colonic (Na+ + K+)-ATPase activity and degree of mucosal inflammation in inflammatory bowel disease. Digestive Diseases and Sciences . 1988;33(4):417–422. doi: 10.1007/BF01536025. [DOI] [PubMed] [Google Scholar]
- 238.Maher M. M., Gontarek J. D., Bess R. S., Donowitz M., Yeo C. J. The Na+/H+ exchange isoform NHE3 regulates basal canine ileal Na+ absorption in vivo. Gastroenterology . 1997;112(1):174–183. doi: 10.1016/s0016-5085(97)70232-4. [DOI] [PubMed] [Google Scholar]
- 239.Schmitz H., Fromm M., Bode H., Scholz P., Riecken E. O., Schulzke J. D. Tumor necrosis factor-alpha induces Cl− and K+ secretion in human distal colon driven by prostaglandin E2. The American Journal of Physiology . 1996;271(4 Pt 1):G669–G674. doi: 10.1152/ajpgi.1996.271.4.G669. [DOI] [PubMed] [Google Scholar]
- 240.Yu L. C., Perdue M. H. Immunologically mediated transport of ions and macromolecules. Annals of the New York Academy of Sciences . 2000;915:247–259. doi: 10.1111/j.1749-6632.2000.tb05248.x. [DOI] [PubMed] [Google Scholar]
- 241.Anbazhagan A. N., Priyamvada S., Alrefai W. A., Dudeja P. K. Pathophysiology of IBD associated diarrhea. Tissue Barriers . 2018;6(2) doi: 10.1080/21688370.2018.1463897.e1463897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Rao M. C. Physiology of electrolyte transport in the Gut: implications for disease. Comprehensive Physiology . 2011;9(3):947–1023. doi: 10.1002/cphy.c180011. [DOI] [PubMed] [Google Scholar]
- 243.Salim S. Y., Söderholm J. D. Importance of disrupted intestinal barrier in inflammatory bowel diseases. Inflammatory Bowel Diseases . 2011;17(1):362–381. doi: 10.1002/ibd.21403. [DOI] [PubMed] [Google Scholar]
- 244.Evans R. C., Clarke L., Heath P., Stephens S., Morris A. I., Rhodes J. M. Treatment of ulcerative colitis with an engineered human anti-TNFalpha antibody CDP571. Alimentary Pharmacology and Therapeutics . 1997;11(6):1031–1035. doi: 10.1046/j.1365-2036.1997.00251.x. [DOI] [PubMed] [Google Scholar]
- 245.Baert F. J., D’Haens G. R., Peeters M., et al. Tumor necrosis factor alpha antibody (infliximab) therapy profoundly down-regulates the inflammation in Crohn’s ileocolitis. Gastroenterology . 1999;116(1):22–28. doi: 10.1016/S0016-5085(99)70224-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data are available from the corresponding author upon reasonable request.