

Abstract
The past 50 years of research in pediatric bone and mineral metabolism have led to remarkable progress in the identification and characterization of disorders that affect the developing skeleton. Progress has been facilitated through advances in both technology and biology and this paper provides a brief description of some but not all of the key findings, including identification of the calcium sensing receptor and the polypeptides parathyroid hormone and parathyroid hormone-related protein as well as their shared receptor and signal generating pathways; the elucidation of vitamin D metabolism and actions; discovery of fibroblast growth factor 23 (FGF23), the sodium-phosphate co-transporters and the other components that regulate phosphate metabolism. Moreover, the past half-century of research has led to the delineation of the molecular bases for genetic forms of hypoparathyroidism, pseudohypoparathyroidism, and primary hyperparathyroidism as well as the determination of the genetic causes of osteogenesis imperfecta, osteopetrosis, hypophosphatasia, and other disorders of mineral/bone homeostasis. During the next decade we expect that many of these fundamental discoveries will lead to the development of innovative treatments that will improve the lives of children with these disorders.
Keywords: bone, bone metabolism, calcium, mineral
This manuscript describes a variety of the extraordinary scientific advances in the biology of bone and mineral metabolism that have occurred in the last half-century and have led to improvements in the evaluation and management of children and adolescents with these disorders. Progress in this field has been facilitated through new knowledge gained by remarkable technological advances that have enabled characterization and measurement of key hormones and metabolites, the identification of novel genes, proteins, steroids, and discovery of biological pathways that affect bone and mineral metabolism. In many instances, new knowledge in these fields has arisen through application of the tools of molecular biology including: genomics/genetics – the identification of the composition and sequence of the units of heredity termed genes which contain the blueprints for synthesizing ribonucleic acid (RNA) and protein molecules based on the precise sequence of four nucleotide bases (adenine, cytosine, guanine and thymine) that are attached to a backbone made of alternating deoxyribose molecules and phosphate groups; transcriptomics – the highly regulated and cell-specific process through which genes are transcribed into messenger RNA molecules that specify the assembly of proteins through the orderly linkage of members of a set of only twenty amino acids, each of which has a unique side chain that can bond with one another to hold a length of protein in a designated shape or configuration; proteomics – the study of all the proteins (proteome) produced by an individual cell or multicellular organism; and metabolomics – the study of the substrates and products and the chemical/physiological reactions (metabolism) within a cell that enable the cell to live and reproduce. Collectively, these fields of study have provided detailed information about the mechanisms that regulate mineral and skeletal homeostasis including the identification of genes, proteins, carbohydrates, and fats specifically associated with disorders of calcium, phosphate, and skeletal homeostasis [1, 2].
The metabolism of calcium and phosphorus/phosphate and skeletal homeostasis are affected by the oral intake and intestinal absorption of these ions, their deposition into and reabsorption from bone, and their renal glomerular filtration and excretion and reabsorption by the renal tubules. These processes are, in turn, regulated by parathyroid hormone (PTH), calcitonin, calcitriol (1,25-dihydroxyvitamin D3 – the biologically active form of vitamin D3), fibroblast growth factor 23 (FGF23), and bone alkaline phosphatase (TNSALP = Tissue non-specific alkaline phosphatase) (Table 1) [2]. Among the most important accomplishments in the fields of mineral and bone metabolism in the past half-century have been:
Table 1:
Genetic regulation of mineral and bone metabolism.
| Gene | Gene product | Gene site | OMIM |
|---|---|---|---|
| Calcium/phosphorus | |||
| CASR | Calcium sensing receptor | 3q13.33-q21.1 | 601199 |
| PHEX | Phosphate-regulating endopeptidase, homolog, X-linked | Xp22.11 | 300550 |
| SLC34A1 | Solute carrier family 34, member 1 (NPT2a) | 5q35.3 | 182309 |
| SLC34A3 | Solute carrier family 34, member 3 (NPT2c) | 9q34.3 | 609826 |
| TNSALP | Tissue non-specific alkaline phosphatase | 1p36.12 | 171760 |
| PTH/PTHRP/calcitonin | |||
| PTH | Parathyroid hormone | 11p15.3 | 168450 |
| PTHLH | Parathyroid-like hormone (PTH related protein - PTHrP) | 12p11.22 | 168470 |
| PTH1R | Parathyroid hormone receptor | 3p21.31 | 168468 |
| CALCA | Calcitonin/calcitonin-related polypeptide, alpha | 11p15.2 | 114130 |
| Vitamin D | |||
| CYP2R1 | Cytochrome P450, subfamily IIR, polypeptide 1 (Vitamin D 25-hydroxylase) | 11p15.2 | 608713 |
| CYP27B1 | Cytochrome P450, subfamily XXVIIB, polypeptide 1 (25-hydroxyvitamin D3–1-alpha hydroxylase) | 12q14.1 | 609506 |
| CYP3A4 | Cytochrome P450, subfamily IIIA, polypeptide 4 (25-hydroxyvitamin D3–23-hydroxylase) | 7q22.1 | 124010 |
| CYP24A1 | subfamily A, polypeptide 1 | 20q13.2 | 126065 |
| VDR | Vitamin D receptor | 12q13.11 | 601764 |
| Fibroblast growth factor 23/receptor | |||
| FGF23 | Fibroblast growth factor 23 | 112p13.2 | 605380 |
| FURIN | Furin, paired basic amino acid cleaving enzyme | 15q26.1 | 136950 |
| FGFR1 | Fibroblast growth factor receptor 1 c | 8p11.23 | 136350 |
| KL | Klotho | 13q13.1 | 604824 |
| PPARGC1A | Peroxisome proliferator-activator receptor-gamma, coactivator, alpha 1 | 4p15.2 | 604517 |
| TRPV | Transient receptor potential cation channel, subfamily V, member 5 | 7q34 | 606679 |
| Bone | |||
| CSF1 | (Macrophage) colony-stimulating factor | 1p13.1 | 120420 |
| BMP1 | Bone morphogenetic protein 1 | 8p21.3 | 112264 |
| SOX9 | SRY-Box 9 | 17q24.3 | 608160 |
| WNT1 | Wingless-type MMTV integration site family, member 1 | 12q13.12 | 164820 |
| CTNNB1 | Catenin, beta 1 | 3p22.1 | 116806 |
| COL1A1 | Collagen, type 1, alpha-1 | 17q21.33 | 120150 |
| COL1A2 | Collagen, type 1, alpha-2 | 7q21.3 | 120160 |
| P4HB | Procollagen-proline, 2-oxoglutarate-4-dioxygenase, beta subunit, Prolyl-4-hydroxylase | 17q25.3 | 176790 |
| PLOD1 | Procollagen-lysine, 2-oxoglutarate-5-dioxygenase, Lysyl hydroxylase | 1p36.2 | 153454 |
| SERPINH1 | Serpin peptidase inhibitor, Clade H, Member 1 Heat shock protein 47 | 11q13.5 | 600493 |
| DMP1 | Dentin matrix acidic phosphoprotein 1 | 600980 | 600980 |
| BGLAP | Gamma-carboxyglutamic acid protein, bone (Osteocalcin) | 1q22 | 112260 |
| TNFRSF11B | Tumor necrosis factor receptor superfamily, member 11B (Osteoprotegerin) | 8q24.12 | 602643 |
| TNFSF11 | Tumor necrosis factor superfamily member 11 (Osteoprotegerin ligand, RANK ligand) | 13q14.11 | 602462 |
| TRPV5 | Transient receptor potential cation channel, subfamily V member 5 | 7q34 | 606679 |
| SP7 | Transcription factor Sp 7 (Osterix) | 12q13.13 | 606633 |
| SPP1 | Secreted phosphoprotein1 (Osteopontin) | 4q22.1 | 166490 |
| Other | |||
| GNAS | Guanine nucleotide-binding protein, alpha-stimulating | 20q13.32 | 139320 |
| GNA11 | Guanine nucleotide binding protein, alpha 11 | 19p13.3 | 139313 |
| STX16 | Syntaxin 16 | 20q13.32 | 603666 |
| GALNT3 | UDP-N-acetyl-alpha- D-galactosamine: Polypepide N-acetylgalactosaminyl-transferase 3 | 2q24.3 | 601756 |
| KDELR2 | KDEL endoplasmic retention protein retention receptor 2 | 7p22.1 | 609024 |
| NFAM1 | NFAT activating protein with ITAM motif 1 [Immunoreceptor tyrosine-based activation motif (ITAM)-mediated co-stimulatory signaling] | 22q13.2 | 608740 |
The discovery of the calcium sensing receptor (CASR) and the development of assays for the measurement of total and ionized calcium and its regulatory components;
Recognition of FGF23, α-Klotho, and Phosphate-regulating endopeptidase, homolog, X-linked (PHEX) as key regulators of phosphorus homeostasis; identification of the roles of FGF23 and α-Klotho in the control of the renal type II sodium-phosphate cotransporter channels (NPT2a – encoded by SLC34A1) and NPT2c (encoded by SLC34A3);
Isolation of PTH, development of assays for its measurement, identification of its cellular receptor, and delineation of the corresponding guanine nucleotide-binding protein G protein signal transduction pathways; identification of PTH-related protein (PTHrP), an important regulator of skeletal development and calcium homeostasis in the fetus as well as postnatally;
Recognition of the molecular bases for genetic forms of hypoparathyroidism, pseudohypoparathyroidism, and primary hyperparathyroidism; analysis of the complexity of GNAS encoding the α subunit of the stimulatory G protein (Gαs);
Elucidation of the metabolism of vitamin D and identification of its bioactive form – 1,25-dihydroxyvitamin D (calcitriol), and of its nuclear receptor (VDR);
Identification of the genetic causes of osteogenesis imperfecta, osteopetrosis, hypophosphatasia, and other disorders of mineral/bone homeostasis.
In this manuscript, the authors have selected a limited number of topics concerning advances in mineral and bone metabolism over the past 50 years upon which to focus.
Calcium and the calcium-sensing receptor
Among the most important accomplishments in the fields of mineral and bone metabolism in the past half-century has been the development of accessible and accurate methods for the measurement of both total and ionized serum calcium concentrations. Calcium is present in the serum in three fractions: that linked to proteins – primarily albumin (45 percent); that complexed to anions such as phosphate or citrate (7 percent); and that in the free or ionized state (Ca2+) (48 percent), the latter being the biologically active form of this cation [3, 4]. The total serum calcium concentration is measured colorimetrically; it may also be determined by atomic absorption spectrophotometry. Measurement of total serum calcium serves as a useful proxy for Ca2+ determination, if total serum calcium levels are not extreme and concentrations of serum proteins are normal. Although a variety of mathematical algorithms have been developed to “adjust” the total serum calcium value when serum levels of proteins, particularly albumin, diverge from normal, none of these corrections is entirely satisfactory. One of the most welcome technological advances for this field was the development of ion-selective electrodes that enabled the direct measurement of Ca2+. The accuracy of this method requires strict adherence to the technique of sample acquisition and preparation and rapid transport of the chilled, anerobic serum sample to the analytical laboratory.
A significant advance in our field was the identification and cloning of the biological receptor that enables cells to detect (sense) extracellular concentrations of Ca2+ (CASR). The CASR is a heptahelical transmembrane protein that is expressed on the surface of parathyroid, bone, and renal tubular cells as well as many other cells throughout the body. The CASR is a member of a large family of receptor proteins that transduce extracellular signals by interacting with heterotrimeric (α, β, and γ subunits) GTP-binding proteins (G-proteins). Ligand-binding to receptor leads to activation of GTP and dissociation of the α subunit from the βγ complex. The free α subunit regulates downstream effector targets. G protein-coupled signal transduction offers a number of advantages over other forms of transmembrane signal transduction. Firstly, heptahelical receptors often activate multiple classes of G proteins, thereby eliciting diverse signals from one ligand. Secondly, G protein-coupled signal transduction amplifies receptor signals as one activated Gα molecule can interact with multiple downstream effectors, thereby achieving exquisite sensitivity. Thirdly, there is endogenous control of the duration of signaling through the rate of GTP to GDP (guanosine diphosphate) hydrolysis which provides a molecular timer that limits the duration of signaling action. Hydrolysis of GTP to GDP facilitates reassociation of the α subunit with the βγ complex - resetting the G protein for a new cycle of signal transduction events. Thus, G protein-coupled signaling enables precise control of enzymes, ion channels, and transporter proteins that regulate intracellular messaging that controls diverse cellular processes.
The CASR binds Ca2+ with high affinity but can also bind other divalent cations such as magnesium. In the parathyroid cell, increasing concentrations of extracellular Ca2+ bind to the CASR leading to dissociation of G-protein α subunits from G11 and Gq with consequent activation of phospholipase Cβ (PLCβ). PLCβ hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and 1,4,5-trisphosphate (IP3) which serve as second messengers that activate pathways that suppress PTH synthesis and release. In the kidney, activation of CASR signaling reduces tubular reabsorption of calcium and thus increases its urinary excretion, thereby lowering serum Ca2+ concentrations. Heterozygous variants of CASR that result in reduced receptor activity cause type 1 familial hypocalciuric hypercalcemia (FHH1) (OMIM 145980) – which is often clinically silent and manifested primarily by mild elevations in serum PTH and calcium with low urine calcium excretion. Patients are generally asymptomatic, although pancreatitis or chondrocalcinosis may develop in some affected adults. By contrast, biallelic variants cause the more severe disorder neonatal severe hyperparathyroidism (OMIM 239200) – typified by asymptomatic, and often life-threatening hypercalcemia (i.e., failure to thrive, skeletal demineralization). CASR variants that result in gain-of-function cause autosomal dominant hypocalcemia type 1 (OMIM 601198) – a form of hypoparathyroidism that is characterized by markedly increased renal excretion of calcium. Mutations in genes encoding other components of the CASR signaling pathway, with loss-of-function variants in GNA11 and AP2S1 causing FHH type 2 and type 3, respectively, while gain-of-function variants in GNA11 cause autosomal dominant hypocalcemia type 2 (OMIM 615361).
Phosphorus/phosphate metabolism
Phosphorus is an abundant element that is widespread in its distribution and is a major intracellular anion. In vivo, phosphorus is complexed with oxygen to form phosphate. Approximately 85 percent of total body phosphorus is in the skeleton in the form of the bone mineral hydroxyapatite – Ca5(PO4)3(OH); the remaining 15 percent is present in soft tissues. Most of intracellular phosphorus in the body is in the organic form as a complex with carbohydrates, lipids, and proteins, while nearly all the phosphorus found in the extracellular fluid space is in the form of inorganic phosphate [5]. Serum inorganic phosphate reflects only a very minor percentage of total body phosphorus, but this form is easily measurable and provides an indication of total body phosphorus stores. Phosphorus plays a critical biochemical role through its involvement in cellular and extracellular metabolism, as an integral component of nucleic acids, cell membranes, and high-energy compounds [e.g., adenosine triphosphate (ATP)] utilized in metabolism, and through regulating the activity of many enzymes. Phosphorus is also an important component of the hydroxyapatite crystal, which provides mechanical strength to mineralized tissues and participates in maintaining the proper pH of extracellular fluids. Phosphorus is readily absorbed by the intestinal tract and reabsorbed from glomerular filtrate in the proximal renal tubule. Exploration of the mechanisms that regulate renal phosphate reabsorption has revealed the essential roles played in this process by PTH as well as FGF23, the latter synthesized primarily by bone osteoblasts and osteocytes, and α-Klotho (KL, OMIM 604824), a molecule whose extracellular domain serves with FGF receptor 1c (FGFR1, OMIM 136350) as a co-receptor for FGF23 [6–10]. FGF23 (OMIM 605380) expression is stimulated by Ca2+, phosphate, calcitriol, and PTH. Transcription of FGF23 in osteocytes is inhibited by dentin matrix protein 1 (DMP1, OMIM 600980) interacting with the X-linked phosphate regulating endopeptidase (PHEX, OMIM 300550) (vide infra) [11, 12].
The FGF23 gene encodes the 251 amino acid polypeptide which includes a 24-amino acid signal peptide. FGF23 is stabilized by N-acetyl glycosylation of amino acid THr 178 by UDP-Gal-NAc transferase 3, a member of the GalNAc-transferases family that is encoded by GALNT3 (OMIM 601756). After cleavage of the signal peptide, biologically active “intact” FGF23 is secreted. Intact FGF23 protein contains the arginine-X-X-arginine (RXXR) motif that is a protease recognition site for furin (FURIN, OMIM 136950), a preprotein convertase. When proteolytically cleaved between Arg179 and Ser180 the intact FGF23 (~23 kDa) forms biologically inactive – and C-terminal (~12 kDa) fragments.
α-Klotho is a single-pass transmembrane glycoprotein of 1,012 amino acids that is highly expressed in the kidney and brain and is also present in the parathyroid glands. There are three primary isoforms of the α-Klotho protein – a full length transmembrane form (mKl), a shed soluble form consisting of the extracellular domain of the parent protein (sKl), and a secreted truncated form that is generated by alternative splicing of KL mRNA. Membrane-bound α-Klotho associates with FGFR1c forming a pocket that serves as the receptor for FGF23 [9]. This complex then dimerizes initiating intracellular transduction of the FGF23 signal. Synthesis of α-Klotho is enhanced by calcitriol and ligands for peroxisome proliferator-activated receptor-γ (PPARγ, PPARGC1A, OMIM 604517) and inhibited by FGF23.
In the proximal renal tubule, FGF23 reduces transcellular reabsorption of phosphate by increasing the rates of internalization and degradation of the apically-sited sodium phosphate co-transporters - NPT2a and NPT2c [encoded by SLC34A1 (OMIM 182309) and SCL34A3 (OMIM 609826), respectively] [7]. FGF23 increases reabsorption of calcium in the renal distal convoluted tubule via α-Klotho, which stabilizes the calcium transporter TPRV5 (encoded by TRPV5, OMIM 606679). FGF23 antagonizes some of the actions of vitamin D and PTH by repressing expression of CYP27B1 (OMIM 609506, encoding 25-hydroxyvitamin D-1-α-hydroxylase) thus decreasing synthesis of calcitriol in proximal renal tubules and by inactivating vitamin D metabolites via induction of CYP24A1 (OMIM 126065, encoding cholesterol-24-hydroxylase). In bone, FGF23 depresses mineralization of hydroxyapatite. In the rat parathyroid gland, FGF23 inhibits the synthesis of PTH.
Circulating levels of FGF23 are increased in patients with autosomal dominant hypophosphatemic rickets (OMIM 193100), X-linked hypophosphatemic rickets (OMIM 307800), and the autosomal recessive forms of hypophosphatemic rickets 1 and 2 (OMIM 241520 and 613312, respectively), as well as fibrous dysplasia and tumor induced osteomalacia – the latter due to excessive synthesis of FGF23 by abnormal cells in the bone lesion or tumor, respectively. The secretion of FGF23 is also increased in patients with chronic renal disease and may be pathogenetically associated with some of the adverse consequences of this state such as myocardial hypertrophy. Serum concentrations of intact FGF23 are decreased in patients with familial hyperphosphatemic tumoral calcinosis type 1 (OMIM 211900) due to mutations in GALNT3 that impair activity of the encoded glycosyl-transferase needed for stabilization of FGF23 via O-glycosylation of Thr178, as well as in subjects with familial hyperphosphatemic tumoral calcinosis type 2 (OMIM 617993) due to loss of function mutations in FGF23 itself.
X-linked hypophosphatemic rickets (XLHR, OMIM 307800) and other forms of FGF23-dependent hypophosphatemic rickets are characterized by suboptimal mineralization of bone that, in the growing child, leads to deformation of the long cartilagenous bones (resulting in genu varum and/or genu valgus) and decreased resistance to external pressure of membranous bones (craniotabes) as well as craniosynostosis. XLHR is due to inactivating variants of phosphate-regulating endopeptidase homolog, X-linked (PHEX, OMIM 300550); the PHEX protein is an enzyme that normally degrades osteopontin (SPP1, OMIM 166490), a protein that inhibits bone mineralization [13].
XLHR is also associated with high circulating levels of FGF23 resulting in hyperphosphaturia and hypophosphatemia and decreased synthesis of calcitriol, thus impairing intestinal absorption of calcium. The conventional management of this disorder has consisted of administration of active forms of vitamin D plus multiple daily doses of phosphate; lack of compliance with dosing regimens complicates management of the disease. The treatment of XLHR has changed dramatically with the development of burosumab, a humanized FGF23-neutralizing monoclonal antibody [14–16]. Subcutaneous administration of burosumab every 14 days to children with XLHR and every 28 days to adults with this disease results in normalization of serum concentrations of phosphate, calcitriol, and alkaline phosphatase with clinical and radiological improvement in rickets and bone deformities but without amelioration of the associated dental defects [17, 18]. Burosumab has also been effective in patients with ARHR1 due to inactivating variants of dentin matrix acidic phosphoprotein 1 (DMP1, OMIM 600980) with normalization of serum phosphate concentrations, amelioration of bone pain, and healing of pseudo-fractures [19].
Parathyroid hormone and PTH-related protein (PTHrP)
PTH is synthesized and secreted by the parathyroid glands via a precise mechanism that is based on the ability of the CASR to sense extracellular concentrations of Ca2+; as Ca2+ values decline, synthesis and secretion of PTH increase. PTH increases serum levels of calcium and decreases serum levels of phosphate via interaction with the type 1 PTH receptor (PTH1R, OMIM 168468) which is coupled via the G protein – Gs – to activation of adenylyl cyclase [20]. When administered intermittently, PTH enhances bone formation, in part by inhibiting apoptosis of osteoblasts. PTH-related protein (PTHLH, OMIM 168470) is a cytokine essential in utero where it increases the linear growth of endochondral bone and postnatally as it contributes to bone remodeling; PTHrP is synthesized by keratinocytes, endothelial and smooth muscle cells, and pancreatic islet cells. The common receptor for PTH and PTHrP – PTH1R (PTH1R, OMIM 168468) – acts principally through Gs to stimulate adenyl cyclase and to generate the second messenger cyclic AMP which activates specific protein kinase A signal transduction pathways that ultimately produce the physiologic actions of PTH or PTHrP. Because PTH and PTHrP share significant amino acid sequence homology within the first 13 amino acids of their protein sequences, they can bind to the common PTH1R and effect similar actions.
The stimulatory guanine nucleotide-binding protein, Gs
The discovery that cyclic AMP was the principal second messenger for PTH action was a landmark observation and quickly led to studies to determine the basis for PTH resistance in patients with pseudohypoparathyroidism type 1A (PHP1A, OMIM 103580) in which biochemical hypoparathyroidism (i.e., hypocalcemia and hyperphosphatemia) is due to resistance to PTH rather than its absence. Most of these patients also manifest features of Albright’s hereditary osteodystrophy (AHO, OMIM 103580), a constellation of developmental defects characterized by short stature, brachydactyly, and subcutaneous ossifications. Early studies had shown that these patients failed to manifest the normal phosphaturic response to PTH administration, while subsequent studies demonstrated that PTH infusion failed to elicit an increase in urinary excretion of nephrogenous cyclic AMP. The recognition that PTH1R action required the heterotrimeric Gs protein for activation of adenylyl cyclase led to studies in accessible cells that demonstrated a 50 percent reduction in the levels of its alpha subunit (Gαs) in patients with PHP1A. These patients also demonstrated resistance to a variety of hormones whose receptors require Gαs for activation of adenylyl cyclase as well as early-onset obesity and neurocognitive delays. [PHP1A has also been designated “inactivating PTH/PTH Related Protein (PTHrP) signaling disorder (iPPSD)”]. By contrast, other patients with PHP1 who lacked major features of AHO and in whom hormone resistance was principally to PTH had normal levels of Gαs in accessible cells, a disorder now termed PTH1B (OMIM 603223). Patients with pseudopseudohypoparathyroidism (PPHP, OMIM 612463) manifest clinical features of AHO but do not have hormone resistance.
GNAS is a complex, reciprocally imprinted gene with alternative first exons that generate a variety of transcripts in addition to Gαs (Figure 1) [21, 22]. Gαs is expressed biallelically (i.e., from both the maternal and paternal alleles) in most tissues. However, specific cells in the proximal renal tubule, pituitary, thyroid, gonads, hypothalamus, and brown adipose tissue express Gαs transcripts preferentially from the maternal allele. Hormone resistance in patients with PHP1A is the consequence of mutations that affect exons 1–13 of the maternally derived GNAS allele and which reduce expression or function of Gαs protein. By contrast, the features of AHO arise as a consequence of a 50 percent reduction of Gαs in non-imprinted tissues. PPHP patients carry a mutation on the paternal GNAS allele that do not affect Gαs expression in cells in which GNAS is imprinted, because the paternal allele is not expressed in these cells; hence, these patients have AHO but do not manifest hormone resistance [21–23].
Figure 1:

Functional organization of the GNAS locus on chromosome 20q13.32 and variations associated with pseudohypoparathyroidism (PHP) and pseudopseudohypoparathyroidism (PPHP). Exons 1–13 of GNAS encode the α subunit of the G-stimulatory protein (Gαs) requisite for intracellular signal transduction. In most tissues, GNAS is expressed by both maternal and paternal alleles; however, in the proximal renal tubule, brown adipose tissue, anterior pituitary, thyroid, and gonads, GNAS is expressed only by the maternal allele. Deleterious variants of maternal GNAS result in pseudohypoparathyroidism (PHP), while variants occurring in the paternal GNAS allele lead to pseudopseudohypoparathyroidism (PPHP). Centromeric to exon 1 of the GNAS chromosomal site are: the A/B transcript, a very large form of Gαs – XLαs, the neuroendocrine secretory protein 55 (NESP55), and the antisense (AS) transcript. The A/B, XLαs, and NESP transcripts are first exons/promoters that bypass exon 1 of GNAS and attached directly to exons 2–13. Maternal variants of GNAS exons 1–13 result in Albright’s hereditary osteodystrophy and PHP1A; while paternal variants of GNAS exons 1–13 lead to PPHP or progressive osseous heteroplasia (POH). Further centromeric of GNAS is STX16 – variants of which lead to PHP1B (isolated renal tubular resistance to PTH). [Reproduced with permission from [21] Juppner H. Molecular definition of pseudohypoparathyroidism variants. J Clin Endocrinol Metab 2021;106:1541–1552. doi: 10.1210/cline/dgab060].
Patients with PHP1B lack typical features of AHO except for mild brachydactyly. PHP1B (OMIM 603223; also termed iPPSD3) is primarily a disorder of imprinting [24]. All subjects with PHP1B described to date show complete loss of methylation (LOM) of the maternal exon A/B differentially methylated region (DMR) which leads to biallelic expression of exon A/B transcripts and suppression of Gαs transcription in those tissues in which Gαs is normally expressed preferentially from the maternal allele. Approximately 15–20 percent of patients with PHP1B demonstrate autosomal dominant transmission through a maternal GNAS allele. Subjects with familial PHP1B usually have loss of methylation that is limited to the A/B differentially methylated region and carry microdeletions within the maternal STX16 allele approximately 220 kb upstream of GNAS that correspond to 3 kb (missing exons 4–6) or 4.4 kb (missing exons 2–4), although larger deletions have been rarely identified. Other mutations that lead to loss of methylation that is limited to exon A/B include a few duplications/triplications involving the region centromeric of exon A/B. Some patients with familial PHP1B have additional methylation defects that affect other differentially methylated regions (DMR) and that are associated with microdeletions that include sequences within NESP and/or the paternally expressed antisense transcript of AS of the GNAS complex locus. The genetic bases for most cases of sporadic PHP1B remain unknown, however. These patients often have global epigenetic defects in methylation that affect all four GNAS DMRs, perhaps related to failure of remethylation of GNAS during oogenesis [25]. In some patients, partial or complete paternal uniparental disomy for chromosome 20 has been identified.
The McCune-Albright syndrome (OMIM 174800) of polyostotic fibrous dysplasia, irregularly bordered café-au-lait macules (CALM) of the skin, and multiple endocrinopathies – the most frequent of which is peripheral isosexual precocious puberty – is the consequence of post-conceptual, somatic, gain-of-function variants of GNAS [26, 27]. The most common activating variants of GNAS are Arg201Cys or His in exon 8 and result in increased activity of Gαs and hormone-independent, cell-autonomous activation of adenylyl cyclase. Mutation-bearing cells are distributed in a mosaic pattern that in the skin can be seen to follow the developmental lines of Blashko (e.g., the unusual pattern of CALM lesions) and lead to phenotypic consequences only in cells that are affected by excess cyclic AMP production. Further variability in the phenotype arises as a consequence of the parental origin of the GNAS allele bearing the post-zygotic mutation, as paternal imprinting of Gαs leads to the milder effect of mutations that occur on the paternal allele. The proportion of cells with an activating variant of GNAS within a tissue will determine the extent of the phenotype. For example, the magnitude of fibrous dysplasia may range from a single, asymptomatic lesion to multiple, diffuse, polyostotic lesions and substantial impairment of skeletal shape and strength. Patients with extensive fibrous dysplasia often have increased serum concentrations of FGF23 which lead to hypophosphatemic rickets and osteomalacia. Endocrinopathies associated with the McCune–Albright syndrome include: gonadotropin-independent isosexual precocious puberty and recurrent ovarian cysts in affected females; Leydig and Sertoli cell hyperplasia with macro-orchidism in males, hyperthyroidism associated with thyroid nodularity, neonatal hypercortisolism, hypersomatotropism, and hyperprolactinemia. Many of these endocrinopathies occur as isolated disorders due to identical somatic Arg201 GNAS mutations that occur much later in development (and even postnatally) and are present only in a single tissue.
Abnormalities of vitamin D metabolism or function
Vitamin D-dependent rickets (VDDR) is distinct from nutritional vitamin D deficiency and is the consequence of abnormalities in the metabolism of this vitamin or in the tissue responsiveness to its active metabolite – 1,25-dihydroxyvitamin D/calcitriol. Cholecalciferol is first 25-hydroxylated by the hepatic 25-hydroxylase enzyme (CYP2R1, OMIM 608717) to form calcifidiol (25-hydroxyvitamin D) and then by renal 25-hydroxyvitamin D3-1α-hydroxylase (CYP27B1, OMIM 609506) to form calcitriol, the active metabolite of vitamin D that binds to the vitamin D receptor (VDR, OMIM 601769) and modulates target gene expression (Figure 2). There are five forms of VDDR [28, 29]. VDDR1A (OMIM 264700) is the result of inactivating variants of CYP27B1 – thus impairing conversion of calcifidiol to calcitriol. VDDR1B (OMIM 600081) is a consequence of biallelic mutations in CYP2R1 – thereby depressing synthesis of calcidiol. VDDR2A (OMIM 277440) is due to a loss-of-function variant of VDR resulting in a functionally abnormal vitamin D receptor. VDDR2B (OMIM 600785) is associated with normal or increased synthesis of calcidiol and calcitriol and a normal vitamin D receptor whose pathogenesis is as yet unidentified, but may be related to an abnormality of the interaction between the vitamin D receptor and its target hormone response element. VDDR1 and VDDR2 are autosomal recessive in transmission while VDDR3 (OMIM 619073) is transmitted as an autosomal dominant disorder; VDDR3 is due to a recurrent, gain-of-function variant of CYP3A4 (OMIM 124010) that leads to rapid inactivation of vitamin D metabolites [30]. In contrast to genetic disorders that impair vitamin D action, hypercalcemia of infancy, type 1 (OMIM 143880) is associated with excessive vitamin D action as a consequence of biallelic inactivating variants of CYP24A1 (OMIM 126065), encoding the enzyme – 25-hydroxyvitamin D-24-hydroxylase – that inactivates calcifediol and calcitriol. Patients with this disorder have hypercalcemia and hypercalciuria with markedly elevated serum concentrations of calcitriol and very low levels of PTH. Hypercalcemia of infancy, type 2 (OMIM 616963) results from inactivating variants of SLC34A1 (OMIM 182309) encoding the renal sodium-phosphate co-transporter NPT2a, the functional loss of which results in hypophosphatemia, suppressed levels of FGF23, and consequent increased production of calcitriol [31].
Figure 2:
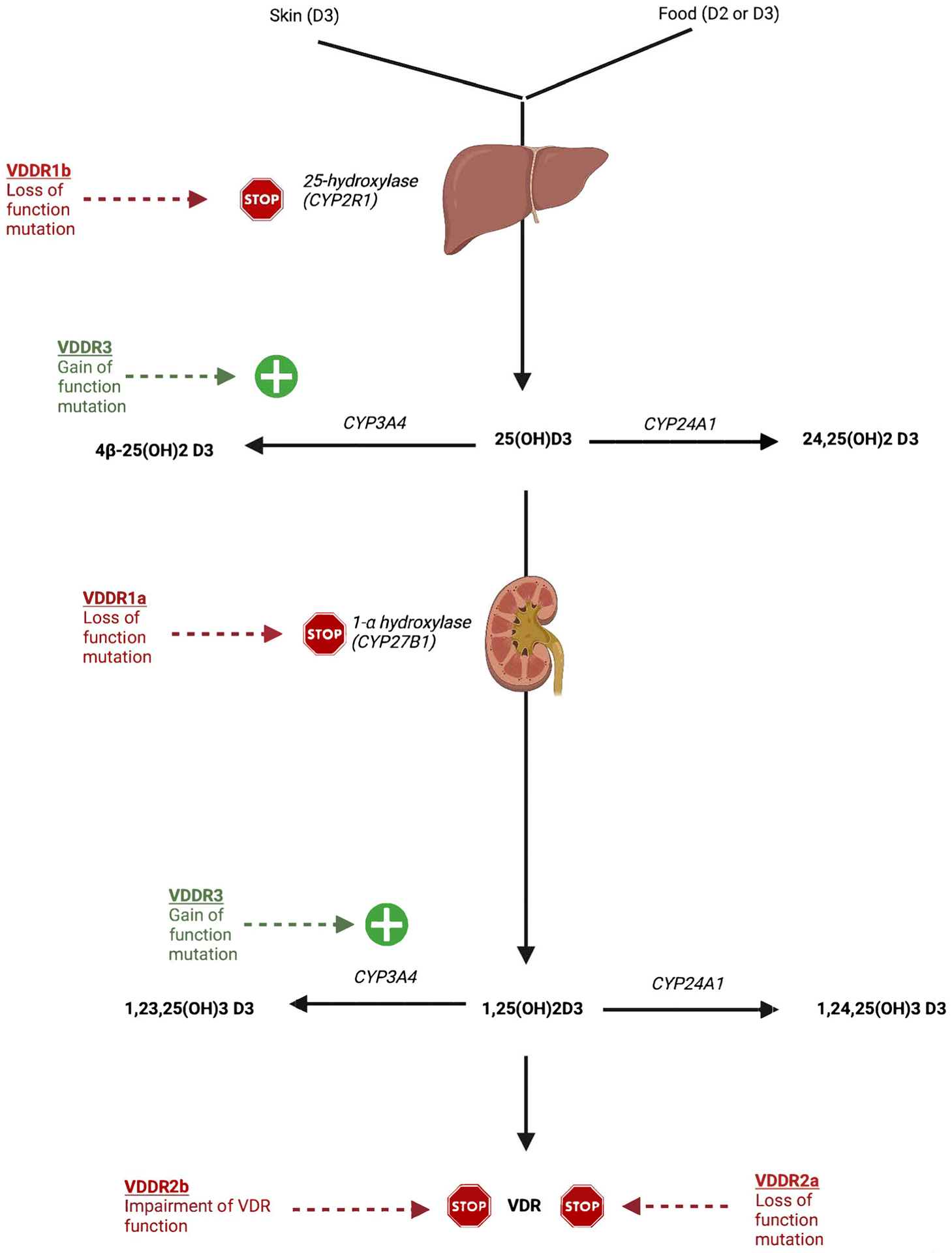
Metabolism of vitamin D – causes of non-nutritional vitamin D deficiency: cholecalciferol (vitamin D3) or ergocalciferol (vitamin D2) is initially hydroxylated at carbon 25 by 25-hydroxylase encoded by CYP2R1 (Table 1) to form calcidiol and then by 25-hydroxyvitamin D3–1-alpha hydroxylase (CYP27B1) to form calcitriol. Hydroxylation at carbon 24 by 25-hydroxyvitamin D3–24-hydroxylase (CYP24A1) leads to water soluble calcitroic acid (1,24,25-trihydroxycholecalciferol) and its excretion in urine or bile. Hydroxylation at carbon 23 by the enzyme encoded by CYP3A4 results in water soluble 1,23,25-trihydroxycholecalciferol. Loss of function variants of CYP2R1 and CYP27B1 interfere with synthesis of calcitriol, while gain–of-function variants of CYP3A4 increase its rate of excretion (see text). [Reproduced with permission from [29] Stoffers AJ, Weber DR, Levine MA. An update on vitamin D deficiency in the twenty-first century: nature and nurture. Curr Opin Endocrinol Diabetes Obes 2022;29:36–43. DOI: 10.1097/MED.0000000000000691].
Hypophosphatasia
Hypophosphatasia (OMIM 241500) is a disorder of bone mineralization due to loss-of-function variants of TNSALP encoding tissue nonspecific (liver, bone and kidney) alkaline phosphatase (OMIM 171760) resulting in clinical manifestations of rickets. Alkaline phosphatase is a phosphohydrolase ectoenzyme linked to the outer membrane of osteoblasts. Depending on the mutant site within TNSALP and whether the mutant site is monoallelic or biallelic, variable degrees of loss of bone alkaline phosphatase activity result in clinical manifestations of rickets of increasing severity [32, 33]. Thus, there are perinatal, infantile, childhood, and adult forms of hypophosphatasia as well as types designated odontohypo-phosphatasia (clinical manifestations related to abnormal dentinogenesis and premature shedding of teeth) and pseudohypophosphatasia (a disorder in which alkaline phosphatase activity toward artificial substrates is intact, but biologic alkaline phosphatase activity toward endogenous substrates is compromised). In patients with hypophosphatasia, inability to free protein-bound phosphate leads to extracellular accumulation of inorganic pyrophosphate that impairs normal growth of hydroxyapatite crystals and hence results in subnormal skeletal mineralization. With the development of a recombinant bone-targeted human TNSALP termed asfotase alpha, management of neonates, infants, and children with hypophosphatasia has changed dramatically; in the perinatal and infantile forms of hypophosphatasia administration of asfotase alpha increases skeletal mineralization leading to improvement in respiratory function and improved survival rate [34].
Hyperparathyroidism – familial hypocalciuric hypercalcemia
A single autonomously functioning parathyroid adenoma is present in most patients with sporadic primary hyperparathyroidism. By contrast, familial forms of primary hyperparathyroidism are associated with enlargement and excess function of all four parathyroid glands. Familial hyperparathyroidism is usually inherited as an autosomal dominant disorder and most often occurs as a component of complex syndromes that involve multiple tissues: multiple endocrine neoplasia types: 1 – due to deleterious variants of MEN1 (OMIM 613733) encoding the tumor suppressor menin; 2A – related to inactivating mutations in RET (OMIM 164761) specifying the RET protooncogene, a receptor tyrosine kinase; and 4 – attributable to variants of CDKN1B (OMIM 600778) encoding cyclin-dependent kinase inhibitor 1B, a molecule that impairs the normal transfer of a phosphate group from cyclic AMP to a recipient protein and hence interrupts progression of the normal cell cycle [35]. Familial isolated hyperparathyroidism type 1 (OMIM 145000) has been ascribed to mutations in CDC73 (OMIM 607393), variants of which are also associated with the hyperparathyroidism-ossifying jaw tumor syndrome (OMIM 145001). CDC73 (OMIM 607393) encodes parafibromin, a tumor suppressor that is involved in transcriptional and post-transcriptional pathways that control cell cycling. Patients with CDC73 mutations usually have single-gland parathyroid disease and have a high risk of developing parathyroid carcinoma. Familial isolated hyperparathyroidism type 2 (OMIM 618883) has been related to activating variants of the transcription factor glial cells missing-2 (GCM2, OMIM 603716) [36]. Variants of hyperparathyroidism include three forms of familial hypocalciuric hypercalcemia (FHH) which differ from other forms of familial primary hyperparathyroidism due to the very low fractional excretion of calcium characteristic of these conditions, onset of mild hypercalcemia within the first days of life, and their generally benign natural history. FHH1 is the consequence of heterozygous loss-of-function mutations in CASR (OMIM 601199) encoding the plasma membrane CASR; FHH2 is due to loss-of-function mutations in GNA11 (OMIM 139313) encoding the α subunit of G11 that interacts with the CASR, and FHH3 is due to loss-of-function mutations in AP2S1 (OMIM 602242) encoding the adaptor-related protein complex 2, sigma 1 subunit, a component of a large protein complex required for proper insertion of the CASR into the plasma membrane [37]. Conversely, activating variants of CASR result in autosomal dominant hypocalcemia type 1 (OMIM 601198), while activating mutations in GNA11 are responsible for autosomal dominant hypocalcemia type 2 (OMIM 615361).
Abnormalities of bone strength: osteogenesis imperfecta and osteopetrosis
Cartilage is formed by chondroblasts that are derived from mesenchymal stem cells in response to the product of SOX9 (OMIM 608160) [38, 39]. Bone is formed by osteoblasts that have differentiated from mesenchymal cells – either directly as membranous bone (e.g., skull, clavicles) or after antecedent cartilage formation (e.g., long bones, vertebrae) under the direction of bone morphogenetic protein (BMP1, OMIM 112264) or follow the WNT-βCatenin pathway involving the low density lipoprotein receptor-related proteins 5 and 6 and epigenetic factors including a number of miRNAs, lncRNAs, and circRNAs [40]. Osteoclasts arise from bone marrow hematopoietic monocyte/macrophage precursor cells that undergo differentiation – initially into mononuclear osteoclasts – in response to the osteoblast – derived ligand for receptor activator of nuclear factor kβ (NFkβ) (RANK-ligand) (TNFSF11, OMIM 602642), macrophage colony-stimulating factor (CSF1; 120420), and immunoreceptor tyrosine-based activation motif (ITAM)-mediated co-stimulatory signaling (NFAM1, OMIM 608740) [40]. RANK-ligand links to RANK expressed on the cell membrane of the osteoclast precursor cell.
Osteogenesis imperfecta is characterized by bone fragility and increased risk/frequency of fracture with minimal or no apparent injury. It is due to variants of genes whose malfunction impairs bone strength by interfering with formation or modeling of bone collagen or its proper mineralization (Table 2) [41–43]. It may be due to deleterious variants of genes required for osteoblast differentiation and maturation or impairing osteoblast function; e.g., those encoding osteoblast synthesis of procollagens types I-IV or those necessary for the normal processing, structure, modifications, and calcification/mineralization of bone collagen [38–43]. As of October 2022, 22 “types” of osteogenesis imperfecta have been formally categorized by OMIM. Osteogenesis imperfecta type 22 is due to deleterious variants of CCDC134 (OMIM 618788) encoding a secreted protein that regulates intracellular signal transduction through the mitogen-activated protein kinase pathway. Inactivating variants of CCDC134 have been identified in patients with extreme bone fragility and multiple bone fractures – at times beginning in utero, pseudoarthroses, scoliosis, and impaired growth but normal dentinogenesis; in these patients the expression of COL1A1 is decreased [44–46]. Additionally, there are other gene variants listed in Table 2 that are associated with decreased bone strength. For example, PLOD2 encodes lysyl hydroxylase 2, an enzyme required for cross-linking of collagen fibers that is mutated in patients with Bruck syndrome 2 (OMIM 609220), manifested by the association of congenital contractures, pterygia, and extreme bone fragility [47]. P4HB encodes the beta subunit of prolyl-4-hydroxylase that is mutated in patients with one form of the Cole-Carpenter syndrome 1 (OMIM 112240) of craniosynostosis and bone fragility. Zoledronic acid, a modified bisphosphonate, has proven effective in the treatment of patients with osteogenesis imperfecta; by decreasing bone resorption, zoledronic acid increases bone mineral content [48, 49].
Table 2:
Osteogenesis imperfecta.
| Type | OMIM phenotype | Defective gene | Protein | OMIM | Locus gene | Inheritance, associated features |
|---|---|---|---|---|---|---|
| Defects in procollagen/collagen synthesis and processing | ||||||
| I | 166200 | COL1A1 | Collagen, type I, alpha −1 | 120150 | 17q21.33 | AD, variant structure of the collagen propeptide, modifications, helical formation; multiple bone fractures with minimal trauma, blue sclerae, normal teeth, near normal height |
| II | 166219 | COL1A1, COL1A2 | Collagen, type I, alpha-2 | 120160 | 7q21.3 | AD, in utero and perinatal fractures, death often in perinatal period |
| III | 259420 | COL1A1, COL1A2 | AD, fading blue sclerae, progressive deformities in late childhood, dentinogenesis imperfecta of primary teeth | |||
| IV | 166220 | COL1A1, COL1A2 | AD, Prepubertal bone fragility nearly white sclerae | |||
| XIII | 614856 | BMP1 | Bone morphogenetic protein | 112264 | 8p21.3 | AR, deficiency of C-propeptidase, 1 severe skeletal deformity |
| XXII | 618788 | CCDC134 | Coiled-coil domain-containing protein 134 | 618788 | 22q13.2 | AR, impacts transcription, intracellular signal transduction, decreases expression of COL1A1 |
| Ehlers-Danlos syndrome/Osteogenesis imperfecta | ||||||
| 619115 | COL1A1 | Collagen, type I, alpha 1 | 120150 | 17q21.33 | AD, Bone fragility with joint hyperextensibility | |
| Defects in collagen modification | ||||||
| VII | 610682 | CRTAP | Cartilage-associated protein | 605497 | 3p22.3 | AR, scafolding protein forming complex with prolyl 3-hydroxylase; severe to lethal |
| VIII | 610915 | P3H1 | Prolyl 3-hydroxylase 1 | 610339 | 1p34.2 | AR, Hydroxylates carbon 3 of proline at position 986 of collagen type 1 alpha 1- essential for collagen cross-linking, severe to lethal |
| IX | 259440 | PPIB | Peptidyl-prolyl isomerase B | 12384 | 15q22.31 | AR, catalyzes isomerization of proline, an amino acid essential for synthesis of bone collagen, gray sclerae, bowing of femur, tibia |
| XIV | 615066 | TMEM38B | Transmembrane protein 38B | 611236 | 9q31.2 | AR, monovalent cation channel in endoplasmic reticulum important for intracellular collagen movement, normal to blue sclerae, severe |
| Abnormalities of collagen folding and cross-linking | ||||||
| X | 613848 | SERPINH1 | Serpin peptidase inhibitor | 600943 | 11q13.5 | AR, collagen binding protein, also termed heat shock protein 47; monitors rate of movement of type I procollagen from endoplasmic reticulum to the Golgi apparatus, severe, dentinogenesis imperfecta |
| XI | 610968 | FKBP10 | FK506-binding protein | 607063 | 17q21.2 | AR, collagen chaperone assisting in collagen folding; interacts with SERPINH1, joint contractures, Bruck syndrome #1 |
| 259450 | FKBP10 | AR, bone fragility, congenital contractures, pterygia, Bruck syndrome #2 | ||||
| NAa | 609220 | PLOD2 | Procollagen-lysine, 2-oxoglutarate, 5-dioxygenase 2 | 601865 | 3q24 | AR, lysyl hydroxylase 2 – enzyme that hydroxylates lysine residues in telopeptides of domains enabling cross-linking of collagen strands; associated with bone fragility, joint contractures, pterygia Bruck syndrome #2/osteogenesis imperfecta |
| XXI | 619131 | KDELR2 | KDEL endoplasmic reticulum protein retention receptor 2 | 609024 | 7p22.1 | AR, receptor that cycles between the endoplasmic reticulum and Golgi apparatus transferring proteins with a carboxyl-terminal sequence of Lys-Asp-Glu-Leu (KDEL) |
| Abnormalities of osteoblast differentiation and function | ||||||
| XII | 613849 | SP7 | Transcription factor SP7 | 606633 | 12q13.13 | AR, controls osteoblastic bone formation, moderately severe |
| XV | 615220 | WNT1 | Wingless-type MMTV integrative site family, member 1 | 164820 | 12q13.12 | AR/AD, stimulates osteoblast differentiation, severe skeletal deformities, neurological defects |
| XVI | 616229 | CREB3L1 | cAMP Response element binding protein 3-like 1 | 616215 | 11 p11.2 | AR, affects the unfolded protein response resulting in alteration of protein processing in the osteoblast, developmental delay |
| XVII | 616507 | SPARC | Secreted protein, acidic, cysteine-rich | 182120 | 5q33.1 | AR, glycoprotein that binds to type 1 collagen in the extracellular matrix, pectus deformity |
| XVIII | 617952 | TENT5A | Terminal nucleotidyl-transferase 5A | 611357 | 6q14.1 | AR, increases expression of COL1A1 and COL1A2, joint contractures |
| XIX | 301014 | MBTPS2 | Membrane-bound transcription factor protease, site 2 | 300294 | Xp22.12 | XLR, acts within Golgi apparatus to release membrane-bound proteins |
| XX | 618644 | MSED | Mesoderm development LRP chaperone LRP 5 and 6 | 607783 | 15q25.1 | AR, essential for osteoblast differentiation as a chaperone for defects in mineralization of bone |
| V | 610967 | IFITM5 | Interferon-induced transmembrane protein 5 | 614757 | 11 p15.5 | AD, ossification of forearm interosseous membrane, hyperplastic callus formation |
| VIa | IFITM5 | Fish-scale pattern in lamellar bone, increased osteoid | ||||
| VI | 613982 | SERPINF1 | Serpin peptidase inhibitor, Clade F, Member 1 | 172860 | 17p13.3 | AR, fish-scale pattern in lamellar bone, increased osteoid |
| NAa - Cole-Carpenter syndrome type 1 | 112240 | P4HB | Prolyl 4-hydroxylase, beta subunit | 176790 | 17q25.3 | AD, bone fragility, craniosynostosis; hydroxylates prolyl residues in preprocollagen |
| NAa - Cole-Carpenter syndrome type 2 | 616294 | SEC24D | SEC24-related gene family, member D | 607186 | 17q25.3 | AR, bone fragility, craniosynostosis; mediates protein transport from endoplasmic reticulum |
NA, not assigned; AD, autosomal dominant; AR, autosomal recessive; XLR, X-linked recessive. [Modified from 41. Jovanovic M, Guterman-Ram G, Marini JC. Osteogenesis imperfecta: Mechanisms and signaling pathways connecting classical and rare OI types. Endocrine Rev 2022;43:61–90. doi:10.1210/endrev/bnab017. 43. Etich J, Leβmeier L, Rehberg M, et al. Osteogenesis imperfecta – pathophysiology and therapeutic options. Mol Cell Pediatr 2020;7:9. https://doi.org/10.1186/s40348-020-00101-9].
Osteopetrosis is a disorder of bone formation characterized by increased bone mass/density due to abnormalities in osteoclast differentiation or function and hence decreased bone mineral reabsorption [50]. Paradoxically, however, osteopetrosis is associated with increased “brittleness” of bone and predisposition to fracture [50–52]. The forms of osteopetrosis may be classified according to the site of abnormality of osteoclast differentiation (osteoclastogenesis), osteoclast function, or interaction with matrix (Table 3). Thus, FERMT3 (OMIM 607901) and ITGB3 (OMIM 173470) encode integrins – adhesion factors that facilitate cell-to-cell-to-matrix communication [53].
Table 3:
Osteopetrosis.
| Type | OMIM | Defective gene | Protein | OMIM | Locus | Inheritance, associated features |
|---|---|---|---|---|---|---|
| Osteoclast differentiation | ||||||
| AR2 | 259710 | TNFSF11 | Tumor necrosis factor ligand superfamily, member 11 | 602642 | 13q14.11 | AR, receptor activator of NFkB – ligand (RANKL) |
| AR7 | 612301 | TNFRSF11A | Tumor necrosis factor receptor superfamily, member 11A | 603499 | 18q21.33 | AR, receptor activator of NFkB – (RANK) |
| AD1 | 607634 | LRP5 | Low density lipoprotein-receptor-related protein 5 | 603506 | 11 q13.2 | AD, transduces WNT signaling enabling osteoclastogenesis |
| NA | 300248 | IKBKG | IKBKG Inhibitor of nuclear factor kappa-B kinase, Regulatory subunit gamma | 300248 | Xq28 | XLR, enables NFkB- mediated osteoclast differentiation, associated with ectodermal dysplasia |
| NA | IGSF23 | Immunoglobulin superfamily member 23 | NA | 19q13.31 | AR, receptor promoting M-CSF and RANK ligand – mediated differentiation of osteoclasts by activating MAPK signaling | |
| NA | 617306 | MITF | Microphthalmia -associated transcription factor | 156845 | 3p13 | AR, developmental protein, variant associated with syndrome of coloboma, osteopetrosis, microphthalmia, albinism |
| Osteoclast function | ||||||
| NA | FERMT3 | Fermitin family, member 3 | 607901 | 11q13.1 | AR. transmembrane adhesion receptor facilitating cell-to-cell-to-extracellular matrix protein interaction | |
| NA | ITGB3 | Integrin, beta 3 | 173470 | 17q21.32 | AR, transmembrane adhesion receptor that facilitates cell-to-cell-to-extracellular matrix protein interaction | |
| AR22 | CCDC134 | Coil-coiled domain-containing protein 134 | 618788 | 22q13 | AR, regulator of transcription, signal transduction | |
| Synthesis of hydrochloric acid | ||||||
| AR3 | 259730 | CA2 | Carbonic anhydrase II | 611492 | 8q21.2 | AR, catalyzes dissociation of carbonic acid, associated with renal tubular acidosis |
| Acid transport/secretion/mineral dissolution | ||||||
| AR8 | 615085 | SNX10 | Sorting nexin 10 | 614780 | 7p15.2 | AR, necessary for vacuole formation and trafficking severe neonatal form |
| AR6 | 611497 | PLEKHM1 | Pleckstrin homology domain containing protein,family M, member 1 | 611466 | 17q21.31 | AR/AD, enables vesicular transport in osteoclasts, intermediate severity |
| AD3 | 618107 | PLEKHM1 | ||||
| AR4 | 611490 | CLCN7 | Chloride channel 7 | 602727 | 16p13.3 | AR/AD, chloride channel that interacts with OSTM1, variable severity |
| AD2 | 166600 | CLCN7 | ||||
| AR5 | 259720 | OSTM1 | Osteopetrosis-associated transmembrane protein1 | 607649 | 6q21 | AR, forms stable molecular complex with CLCN7, infantile form with involvement of nervous system |
| AR1 | 259700 | TCIRG1 | T Cell immune regulator 1 | 604592 | 11 q13.2 | AR, osteoclast proton pump, severe neonatal form |
| Other | ||||||
| Pycknodysostosis | 265800 | CTSK | Cathepsin K | 601105 | 1q21.3 | AR, cysteine proteinase that degrades bone collagen matrix; bone fragility despite osteosclerosis |
| Sclerosteosis 1 | 269500 | SOST | Sclerostin | 605740 | 17q21.31 | AR, bone morphogenic protein antagonist secreted by osteocytes; interacts with LRP5, LRP6 |
NA, not assigned; AD, autosomal dominant; AR, autosomal recessive; XLR, X-linked recessive.
Conclusions
In this brief overview we have presented some highlights and discoveries that have been made over the past fifty years in pediatric bone and mineral metabolism. These remarkable achievements have led to the generation of new diagnostic algorithms and stimulated the development of novel treatments that address the fundamental biological defect that is responsible for the clinical disorder. It is clear that the pace of discovery has accelerated with the resolution of the genome at the single base level, and we anticipate that many new and important advances in understanding physiology and disease will soon be disclosed through enhanced understanding of the mechanisms that regulate gene transcription and by further definition of the physical interactions of proteins with genes and chromatin. Further, new knowledge from the emerging field of epigenetics also promises to extend our understanding of human disease. We predict that over the next ten years we will see the harnessing of these discoveries to develop even more bold approaches to therapeutics, including gene therapy.
Research funding:
None declared.
Footnotes
Competing interests: Authors state no conflict of interest.
Contributor Information
Allen W. Root, Johns Hopkins All Children’s Hospital, St. Petersburg, FL, USA.
Michael A. Levine, The Center for Bone Health at the Children’s Hospital of Philadelphia, Philadelphia, PA, USA.
References
- (1).Burley SK, Arap W, Pasqualini R. Predicting proteome-scale protein structure with artificial intelligence. N Engl J Med 2021;385:2191–4. [DOI] [PubMed] [Google Scholar]
- (2).Sun M, Wu X, Yu Y, Wang L, Xie D, Zhang Z, et al. Disorders of calcium and phosphorus metabolism and the proteomics/metabolomics-based research. Front Cell Dev Biol 2020;8:576110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ong GSY, Walsh JP, Stuckey BGA, Brown SJ, Rossi E, Ng JL, et al. The importance of measuring ionized calcium in characterizing calcium status and diagnosing primary hyperparathyroidism. J Clin Endocrinol Metab 2012;97:3138–45. [DOI] [PubMed] [Google Scholar]
- (4).Tinawi M Disorders of calcium metabolism: hypocalcemia and hypercalcemia. Cureus 2021;13. 10.7759/cureus.12420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Tinawi M Disorders of phosphate metabolism: hypophosphatemia and hyperphosphatemia. Arch Clin Biomed Res 2021;5:538–55. [Google Scholar]
- (6).Chen G, Liu Y, Goetz R, Fu L,Jayataman S, Hu MC, et al. aKlotho is a non-enzymatic molecular scaffold for FGF23 hormone signaling. Nature 2018;553:461–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Erben RG. Physiologic actions of fibroblast growth factor-23. Front Endocrinol 2018;9:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kurpas A, Supel K, Idzikowska K, Zielinska M. FGF23: a review of its role in mineral metabolism and renal and cardiovascular disease. Dis Markers 2021:8821292. 10.1155/2021/8821292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kuro-o M The Klotho proteins in health and disease. Nat Rev Endocrinol 2019;15:27–44. [DOI] [PubMed] [Google Scholar]
- (10).Rausch S, Foller M. The regulation of FGF23 under physiological and pathophysiological conditions. EurJ Physiol 2022;474:281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Dussold C, Gerber C, White S, Wang X, Qi L, Francis C, et al. DMP1 prevents osteocyte alterations, FGF23 elevation and left ventricular hypertrophy in mice with chronic kidney disease. Bone Res 2019;7:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Rowe PSN. Regulation of bone-renal mineral and energy metabolism: the PHEX, FGF23, DMP1, MEPE ASARM pathway. Crit Rev Eukaryot Gene Expr 2012;22:61–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Boukpessi T, Hoac B, Coyac BR, Leger T, Garcia C, Wicart P, et al. Osteopontin and the dento-osseous pathobiology of X-linked hypophosphatemia. Bone 2017;95:151–61. [DOI] [PubMed] [Google Scholar]
- (14).Dahir K, Roberts MS, Krolczyk S,Simmons JH. X-linked hypophosphatemia: a new era in management. J Endocr Soc 2020;4:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Imel EA, Glorieux FH, Whyte MP, Munns CF, Ward L, Nilsson O, et al. Burosumab versus conventional therapy in children with X-linked hypophosphatemia: a randomised, active-controlled, open-label, phase 3 trial. Lancet 2019;393:2416–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Gordon RJ, Levine MA. Burosumab treatment of children with X-linked hypophosphatemic rickets. Lancet 2019;393:2364–6. [DOI] [PubMed] [Google Scholar]
- (17).Linglart A, Imel EA, Whyte MP, Portale AA, Hogler W, Boot AM, et al. Sustained efficacy and safety of burosumab, a monoclonal antibody to FGF23, in children with X-linked hypophosphatemia. J Clin Endocrinol Metab 2022;107:813–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Schindeler A, Biggin A, Munns CF. Clinical evidence for the benefits of burosumab therapy for X-linked hypophosphatemia (XLH) and other conditions in adults and children. Front Endocrinol 2020;11: 338–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Bai X, Levental M, Karaplis AC. Burosumab treatment for autosomal recessive hypophosphatemic rickets type 1 (ARHR1). J Clin Endocrinol Metab 2022;107:2777–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Martin TJ, Sims NA, Seeman E. Physiological and pharmacological roles of PTH and PTHrP in bone using their shared receptor, PTH1R. Endocr Rev 2021;42:383–406. [DOI] [PubMed] [Google Scholar]
- (21).Juppner H Molecular definition of pseudohypoparathyroidism variants. J Clin Endocrinol Metab 2021;106:1541–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Juppner J. Obesity and Gas variants. N Engl J Med 2021;385:1619–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lemos MC, Thakker RV. GNAS mutations in pseudohypoparathyroidism type 1a and related disorders. Hum Mutat 2015;36:11–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hanna P, Francou B, Delemer B, Juppner H, Linglart A. A novel familial PHP1B variant with incomplete loss of methylation at GNAS-AB and enhanced methylation at GNAS-AS2.J Clin Endocrinol Metab 2021;106: 2779–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Milioto A, Reyes M, Hanna P, Kiuchi Z, Turan S, Zeve D, et al. Lack of GNAS remethylation during oogenesis may be a cause of sporadic pseudohypoparathyroidism type 1b. J Clin Endocrinol Metab 2022;107: e1610–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Hartley I, Zhadina M, Collins MT, Boyce AM. Fibrous dysplasia of bone and McCune-Albright syndrome: a bench to bedside review. Calcif Tissue Int 2019;104:517–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Spencer T, Pan KS, Collins MT, Boyce AM. The clinical spectrum of McCune-Albright syndrome and its management. Horm Res Paediatr 2019;92:347–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Levine MA. Diagnosis and management of vitamin D dependent rickets. Front Pediatr 2020;8:315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Stoffers AJ, Weber DR, Levine MA. An update on vitamin D deficiency in the twenty-first century: nature and nurture. Curr Opin Endocrinol Diabetes Obes 2022;29:36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Roizen JD, Li D, O’Lear L,Javaid MK, Shaw NJ, Ebeling PR, et al. CYP34A mutation causes vitamin D-dependent rickets type 3. J Clin Invest 2018; 128:1913–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lenherr-Taube N, Furman M, Assor E, Elia Y, Collins C, Thummel K, et al. Mild idiopathic infantile hypercalcemia – part 2: a longitudinal observational study. J Clin Endocrinol Metab 2021;106:2938–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Whyte MP. Hypophosphatasia: aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol 2016;12:233–46. [DOI] [PubMed] [Google Scholar]
- (33).Whyte MP. Hypophosphatasia: enzyme replacement therapy brings new opportunities and new challenges (perspective). J Bone Miner Res 2017;32:667–75. [DOI] [PubMed] [Google Scholar]
- (34).Tournis S, Yavropoulou MP, Polyzos SA, Doulgeraki A. Hypophosphatasia. J Clin Med 2021;10:5676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Newey PJ. Hereditary primary hyperparathyroidism. Endocrinol Metab Clin N Am 2021;50:663–81. [DOI] [PubMed] [Google Scholar]
- (36).Canaff L, Guarnieri V, Kim Y, Wong BYL, Nolin-Lapalme A, Cole DEC, et al. Novel glial cell missing-2 (GCM2) variants in parathyroid disorders. Eur J Endocrinol 2022;186:351–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).LeeJY Shoback DM. Familial hypocalciuric hypercalcemia and related disorders. Best Pract Res Clin Endocrinol Metabol 2018;32:609–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Rutkovskiy A, Stenslokken KO, Vaage IJ. Osteoblast differentiation at a glance. Med Sci Monit Basic Res 2016;22:95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ponzetti M, Rucci N. Osteoblast differentiation and signaling: established concepts and emerging topics. Int J Mol Sci 2021;22:6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Inoue K, Ng, Xia Y, Zhao B. Regulation of osteoclastogenesis and bone resorption by miRNAs. Front Cell Dev Biol 2021;9:651161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Jovanovic M, Guterman-Ram G, Marini JC. Osteogenesis imperfecta: mechanisms and signaling pathways connecting classical and rare OI types. Endocr Rev 2022;43:61–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Charoenngam N, Cevik MB, Holick MF. Diagnosis and management of pediatric metabolic bone diseases associated with skeletal fragility. Curr Opin Pediatr 2020;32:560–73. [DOI] [PubMed] [Google Scholar]
- (43).Etich J, LeBmeier L, Rehberg M, Sill H, Zaucke F, Netzer C, et al. Osteogenesis imperfecta – pathophysiology and therapeutic options. Mol Cell Pediatr 2020;7:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Dubail J, Brunnelle P, Baujat G, Huber C, Doyard M, Michot C, et al. Homozygous loss-of-function mutations in CCDC134 are responsible for a severe form of osteogenesis imperfecta. J Bone Miner Res 2020;35: 1470–80. [DOI] [PubMed] [Google Scholar]
- (45).Holick MF, Shirvani A, Charoenngam N. Fetal fractures in an infant with maternal Ehlers–Danlos syndrome, CCDC134 pathogenic mutation and a negative genetic test for osteogenesis imperfecta. Children 2021;8:512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Ali TM, Linnenkamp BDW, Yamamoto GI, Honjo RS, Filho HCM, Kim CA, et al. The recurrent homozygous translation start site variant in CCDC134 in an individual with severe osteogenesis imperfecta of non-Moroccan ancestry. Am J Med Genet 2022;188:1545–9. [DOI] [PubMed] [Google Scholar]
- (47).Gistelinck C, Weis MA, Rai J, Schwarze U, Niyazov D, Song KM, et al. Abnormal bone collagen cross-linking in osteogenesis imperfecta/Bruck syndrome caused by compound heterozygous PLOD2 mutations. J Bone Miner Res 2021;5:e10454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Sun L, Hu J, Liu J, Zhang Q, Wang O, Jiang Y, et al. Relationship of pathogenetic mutations and responses to zolendronic acid in a cohort of osteogenesis imperfecta children. J Clin Endocrinol Metab 2022;107:2571–9. [DOI] [PubMed] [Google Scholar]
- (49).Lim DBN, Moon RJ, Davies JH. Advances in diagnosis and management of childhood osteoporosis. J Clin Res Pediatric Endocrinol 2022;14:370–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Wu CC, Econs MJ, DiMeglio LA,Insogna KL, Levine MA, Orchard PJ, et al. Diagnosis and management of osteopetrosis: consensus guidelines from the osteopetrosis working group. J Clin Endocrinol Metab 2017; 102:3111–23. [DOI] [PubMed] [Google Scholar]
- (51).Penna S, Capo V, Palagano E, Sobacchi C, Villa A. One disease, many genes: implications for the treatment of osteopetroses. Front Endocrinol 2019;10:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Povoroznyuk VV, Dedukh MV, Bystrytska MA, Musilienko AS. Osteopetrosis: classification, pathomorphology, genetic disorders, clinical manifestations (literature review and case report). Pract Med 2019;9:135–42. [Google Scholar]
- (53).Sendon C, Esquibies AE. ITGB (integrin subunit beta) 3 mutation in pulmonary hemorrhage and osteopetrosis. Respir Med Case Rep 2019; 26:270–2. [DOI] [PMC free article] [PubMed] [Google Scholar]