

Abstract
Some human adenoviruses are tumorigenic in rodents. Subgroup A and B human adenoviruses generally induce sarcomas in both male and female animals, and the gene products encoded within viral early region 1 (E1 region) are both necessary and sufficient for this tumorigenicity. In contrast, subgroup D human adenovirus type 9 (Ad9) induces estrogen-dependent mammary tumors in female rats and requires the E4 region-encoded ORF1 oncoprotein for its tumorigenicity. Considering the established importance of the viral E1 region for tumorigenesis by adenoviruses, we investigated whether this viral transcription unit is also necessary for Ad9 to generate mammary tumors. The nucleotide sequence of the Ad9 E1 region indicated that the gene organization and predicted E1A and E1B polypeptides of Ad9 are closely related to those of other human adenovirus E1 regions. In addition, an Ad9 E1 region plasmid demonstrated focus-forming activity in both low-passage-number and established rat embryo fibroblasts, whereas a large deletion within either the E1A or E1B gene of this plasmid diminished transforming activity. Surprisingly, we found that introducing the same transformation-inactivating E1A and E1B deletions into Ad9 results in mutant viruses that retain the ability to elicit mammary tumors in rats. These results are novel in showing that Ad9 represents a unique oncogenic adenovirus in which the E4 region, rather than the E1 region, encodes the major oncogenic determinant in the rat.
Human adenoviruses cause primarily respiratory, gastrointestinal, and eye infections in people and are divided into six subgroups (A to F) based upon several physical characteristics (25, 48). In rodents, however, the subgroup A and B adenoviruses are tumorigenic, eliciting undifferentiated sarcomas at the site of viral inoculation in both male and female animals (22, 54). Although subgroup D adenoviruses are nononcogenic in hamsters (54), subgroup D human adenovirus type 9 (Ad9) elicits mammary tumors in rats (3, 4, 29). Three months after subcutaneous injection with Ad9, female rats develop exclusively estrogen-dependent mammary tumors, while male rats fail to develop tumors of any kind. Tumors that form in the female rats are predominantly mammary fibroadenomas, the most common type of benign breast tumor found in young women (29, 44).
For the subgroup A and B adenoviruses, the E1A and E1B gene products encoded within the viral early region 1 (E1 region) are both necessary and sufficient for oncogenic transformation of primary rodent cell cultures (22, 49, 51). Individually, E1A is capable of immortalizing cells (26), whereas E1B displays no transforming potential (55). Together, however, these viral genes cooperate to produce transformed cells (22). The mechanism by which E1 region gene products transform cells can be attributed, in part, to their ability to inactivate the cellular tumor suppressor proteins pRB and p53 (48).
Unlike subgroup A and B adenoviruses, subgroup D Ad9 requires the E4 region ORF1 oncoprotein to generate tumors (30, 32). Nevertheless, the facts that (i) E1A mRNA is expressed in Ad9-induced mammary tumors (29) and (ii) the Ad9 E1 and E4 regions together cooperate to induce focus formation in CREF cells (30) suggest that the viral E1 region may also be required for Ad9-induced mammary tumorigenesis. To address this possibility, we constructed Ad9 mutant viruses containing transformation-defective E1A and E1B genes. Despite the critical role of the viral E1 region in oncogenesis by subgroup A and B adenoviruses, we present results here indicating that E1 region transforming functions are dispensable for Ad9 to induce mammary tumors in rats.
MATERIALS AND METHODS
Cell lines.
Rat embryo fibroblasts (REFs) were cultured from 16-day Fisher rat embryos (Harlan Sprague-Dawley, Indianapolis, Ind.) by using standard methods (20). REF cultures, rat CREF (19) and 3Y1 cell lines (37), and human A549 and 293 cell lines (2, 23) were maintained in culture medium (Dulbecco’s modified Eagle medium supplemented with 20 μg of gentamicin per ml and 6 or 10% fetal bovine serum) under a 5% CO2 atmosphere at 37°C.
Nucleotide sequence analyses and plasmid construction.
Plasmids pUC19-Ad9[0-7.5] and pSP72-Ad9[7.5-12.5] containing Ad9 DNA sequences from 0 to 7.5 and 7.5 to 12.5 map units (m.u.), respectively, were used to determine the nucleotide sequence of the Ad9 E1 region.
A DNA fragment (0 to 12.5 m.u.) containing the Ad9 E1 region was inserted into the KpnI and BglII sites of plasmid pSP72 (Promega) to make pAd9E1. Deletions within the Ad9 E1A and E1B genes were first introduced into pUC19-Ad9[0-7.5] by removing the Ad9 SacI-BspEI fragment (nucleotides [nt] 542 to 1049) and the Ad9 NaeI-ClaI fragment (nt 1609 to 2495), respectively. These two deletion mutations were subsequently transferred to pAd9E1 within the Ad9 BamHI-EcoRI fragment (0 to 7.5 m.u.), resulting in pAd9E1(ΔE1A) and pAd9E1(ΔE1B), respectively. The presence of the correct deletion in each mutant plasmid was verified by restriction enzyme and limited sequence analyses.
Construction of adenovirus mutants.
Ad9 mutant viruses having the same E1A and E1B gene deletions described above for plasmids pAd9E1(ΔE1A) and pAd9E1(ΔE1B) were generated. Briefly, the full-length Ad9 genome (0 to 100 m.u.) consists of three EcoRI fragments: A (7.5 to 95 m.u.), B (0 to 7.5 m.u.), and C (95 to 100 m.u.). Deletions were first introduced into the Ad9 EcoRI B fragment of a plasmid, pAd9-EcoRI(B+C), which contains properly oriented terminal Ad9 EcoRI B and C fragments but lacks the intervening Ad9 EcoRI A fragment. Full-length mutant Ad9 genomes were subsequently assembled by inserting a virion-derived Ad9 EcoRI A fragment in the correct orientation at the unique EcoRI site of mutant pAd9-EcoRI(B+C) plasmids. The resulting infectious pAd9-EcoRI(A+B+C) plasmids were digested with SpeI to release intact linear viral genomes, which were transfected into 293 cells to complement expected E1 region deficiencies of the mutant viruses (2, 23). Recovered viruses were amplified and titrated in 293 cells (31, 48).
Isolation of RNA and Northern blot analyses.
Total RNA was isolated from mock-infected or Ad9-infected A549 cells (multiplicity of infection of 10; 9 h postinfection). Cells were washed with ice-cold phosphate-buffered saline (4.3 mM Na2HPO4, 1.4 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl) and lysed in guanidinium solution (4 M guanidinium isothiocyanate, 20 mM sodium acetate [pH 5.2], 0.1 mM dithiothreitol, and 0.5% [wt/vol] Sarkosyl) (12). The resulting lysate was drawn through a 20-gauge needle to shear cellular DNA, layered onto a 5.7 M CsCl cushion, and centrifuged at 150,000 × g for 18 h. The RNA pellet was dissolved in TES buffer (1 mM Tris-HCl [pH 7.5], 2.5 mM EDTA, 1% [wt/vol] sodium dodecyl sulfate [SDS]), precipitated with ethanol, and resuspended in water.
For Northern blot analyses, total RNA was separated on a formaldehyde agarose gel and transferred to a nitrocellulose membrane (13). The membrane was preincubated in hybridization buffer (0.5 M Na2HPO4 [pH 7.2], 1 mM EDTA, 7% [wt/vol] SDS) at 65°C for 4 h and then incubated in hybridization buffer containing a radiolabeled DNA probe (4.3 × 106 cpm/ml) at 65°C for 16 h. E1A and E1B probes, derived from Ad9 E1 region DNA fragments SacI-SphI (nt 542 to 1473) and NaeI-EcoRI (nt 1609 to 2563), respectively, were radiolabeled by the random priming method (17) and purified by gel filtration on NICK columns (Pharmacia). Probed membranes were washed in SSC wash buffer (45 mM NaCl, 4.5 mM sodium citrate, 0.1% [wt/vol] SDS) at 65°C.
Isolation of virion and cellular DNA.
For isolation of adenovirus virion DNA, 293 cells were infected at a multiplicity of infection of 10 and, at 72 h postinfection, were harvested and lysed in lysis buffer (55 mM Tris-HCl [pH 9.0], 0.5 mM EDTA, 0.2% [wt/vol] sodium deoxycholate, 10% [vol/vol] ethanol, 0.5 mM spermine-HCl). Cell lysates were cleared by centrifugation, treated with proteinase K solution (0.75% [wt/vol] SDS, 12.5 mM EDTA, 2.5 mg of proteinase K per ml) at 37°C for 1 h, and extracted with phenol and chloroform. Virion DNA was precipitated with ethanol and resuspended in water.
For isolation of cellular DNA, 400 mg of frozen tumor tissue was ground in a liquid nitrogen-chilled mortar and pestle. The resulting frozen tumor powder was suspended in 4.8 ml of digestion buffer (10 mM Tris-HCl [pH 8.0], 100 mM NaCl, 25 mM EDTA, 0.5% [wt/vol] SDS, 0.1 mg of proteinase K per ml), incubated at 50°C for 16 h, and extracted with phenol (52). Cellular DNA was precipitated with ethanol and resuspended in TE buffer (10 mM Tris-HCl [pH 7.4], 1 mM EDTA).
PCR analyses.
For PCR amplification of cDNAs (reverse transcription-PCR analysis), 2 μg of total RNA was reverse transcribed with Moloney murine leukemia virus reverse transcriptase, using random hexamers, as suggested by the manufacturer (Gibco-BRL). Ad9 E1A cDNAs were PCR amplified with Taq polymerase (Promega) by using E1A primers 1 (nt 551 to 570; 5′ CTC CTG CAG TCC CAG AGA CCG AGA AAA AT 3′) and 2 (nt 1430 to 1411; 5′ CTC AAG CTT AAG CGC ACG TGC GTC TAG TT 3′). PstI and HindIII sites (underlined) engineered within the E1A oligonucleotides allowed PCR products to be inserted at the same sites of plasmid ds56rII6HI (1) for sequencing. Portions of the Ad9 E1A and E1B genes and the entire Ad9 E4 ORF1 gene were PCR amplified from tumor DNAs, using the following oligonucleotide pairs: E1A primers a (nt 487 to 513; 5′ CCA GTC GAG TCC GTC AAG AGG CCA CTC 3′) and b (nt 1487 to 1461; 5′ CCA CAC CTT GCA TGC GTC ACA TAG AC 3′); E1B primers c (nt 1584 to 1609; 5′ ATC CTT GCA GAC TTT AGC AAG ACA CG 3′) and d (nt 2651 to 2628; 5′ CAT GCA GGG TCA TCT GGC TGT TGG 3′); and Ad9 E4 ORF1 primers 1 (5′ ATG GCT GAA TCT CTG TAT GCT TTC 3′) and 2 (5′-CAT GGT TAG TAG AGA TGA GAG TCT GAA 3′). For E1A and E1B nested PCRs, DNA products derived from each of the first PCR amplifications described above were extracted with phenol, precipitated with ethanol, and resuspended in water. One-twentieth of each sample was subjected to a second round of PCR amplification using the following oligonucleotide pairs: E1A primers e (nt 726 to 746; 5′ CCC ATG ATG ACG ACC CTA ACG 3′) and b; and E1B primers c and f (nt 2116 to 2094; 5′ CAA TCC AGC TCC TCT TCC GAC GG 3′).
Immunoprecipitation and immunoblot analyses.
Immunoprecipitations and immunoblot analyses were performed as described previously (32). Briefly, frozen tumor powder, generated as described above for the isolation of cellular DNA, was suspended in ice-cold radioimmunoprecipitation assay buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.1% [wt/vol] SDS, 1% [vol/vol] Nonidet P-40, 0.5% [wt/vol] deoxycholate) containing protease inhibitors (2 μg of aprotinin, 2 μg of leupeptin, and 100 μg of phenylmethylsulfonyl fluoride per ml), sonicated briefly, and cleared by centrifugation (16,000 × g, 10 min). The protein concentration of tumor lysates was determined by the method of Bradford (9). Three milligrams of protein from tumor lysates was subjected to immunoprecipitation with 15 μl of Ad9 E4 ORF1 antiserum prebound to 30 μl of protein A-Sepharose beads (Pharmacia) (32). Beads were washed with ice-cold radioimmunoprecipitation assay buffer and boiled in 2× sample buffer (0.13 M Tris-HCl [pH 6.8], 4% [wt/vol] SDS, 20% [vol/vol] glycerol, 2% [vol/vol] β-mercaptoethanol, 0.003% [wt/vol] bromophenol blue). Proteins were separated by SDS-polyacrylamide gel electrophoresis (40) and electrophoretically transferred to a polyvinylidene difluoride membrane, which was blocked in TBST (50 mM Tris-HCl [pH 7.5], 200 mM NaCl, 0.1% [vol/vol] Tween 20) containing 5% (wt/vol) both nonfat dry milk and bovine serum albumin. In these assays, Ad9 E4 ORF1 antiserum (1:5,000 in TBST) (32) and horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G (1:5,000 in TBST; Southern Biotechnology Associates) were used as primary and secondary antibodies, respectively. After extensive washing with TBST, the membrane was developed by enhanced chemiluminescence (Pierce).
Focus assays.
Plasmid DNA purified by CsCl density gradient centrifugation was transfected onto 50% confluent tertiary REF cultures or CREF cells on 100-mm-diameter dishes, using the calcium phosphate precipitation method with a glycerol shock (38). At 72 h posttransfection, REF and CREF cells were passaged 1:3 and maintained in culture medium containing 10 and 6% filtered fetal bovine serum, respectively. Four to six weeks posttransfection, cells were fixed in methanol and stained with Giemsa to quantify transformed foci (32).
Mammary tumorigenicity of viruses in rats.
Female rats with 1- or 2-day-old litters were obtained from Harlan Sprague-Dawley; 12 to 24 h after arrival, newborn rats were injected subcutaneously with 0.4 ml of virus solution on their anterior flanks, using a 26-gauge needle. Beginning 2 months postinfection, animals were examined weekly by palpation for the presence of tumors, until the experiment was terminated at 8 months postinfection. At this time, animals were euthanized, and portions of tumors were removed and either fixed in 10% formalin for histological examination or frozen at −80°C for isolation of DNA or protein. Animals were cared for and handled according to institutional guidelines.
Protein sequence alignments.
Sequences of Ad12, Ad7, Ad5, and Ad40 E1 region polypeptides were obtained from GenBank. Alignments were made by using the Pairwise Sequence Alignment program (ALIGN) of the BCM Search Launcher (50).
Nucleotide sequence accession number.
The nucleotide and polypeptide sequences reported in this paper were submitted to GenBank (accession no. AF099665).
RESULTS
Gene organization and predicted polypeptides of the Ad9 E1 region.
To initiate our characterization of the subgroup D Ad9 E1 region, we determined the sequence of the left 4006 nt of the Ad9 genome. From this analysis, we found that the gene organization of the Ad9 E1 region closely resembles that of other human adenovirus E1 regions (Fig. 1A) (48). In addition, the predicted Ad9 13S E1A, 19K and 55K E1B, and pIX proteins displayed significant sequence similarity with the corresponding proteins from other human adenoviruses, although they were most closely related to the E1 region polypeptides of subgroup B adenoviruses (Table 1).
FIG. 1.



(A) Nucleotide sequence of the Ad9 E1 region. The locations of E1A, E1B, and IX gene promoter TATA boxes, known and putative splice donor (SD) and splice acceptor (SA) sites, and poly(A) signal sequences are shown. Relevant restriction enzyme sites are indicated (underlined) on the nucleotide sequence. E1A and E1B mRNA splice variants result from the use of the following SD and SA sites: 10S E1A, SD1 and SA1 plus SD2 and SA2; 12S E1A, SD2 and SA2; 13S E1A, SD3 and SA2; 13S E1B, SD4A or SD4B and SA3; and 22S E1B, SD5 and SA3. The predicted amino acid sequences of the 13S E1A, 19K and 55K E1B, and pIX polypeptides are shown beneath their coding sequences. (B) Comparison of the Ad9 13S, 12S, and 10S E1A polypeptide sequences. CR1, CR2, spacer region, and CR3 are indicated (22, 33, 48, 53).
TABLE 1.
Amino acid sequence identities between subgroups A to D and F adenovirus E1A, E1B, and pIX proteinsa
| Virus | E1A 13S
|
E1B 19K
|
E1B 55K
|
pIX
|
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ad12 | Ad7 | Ad5 | Ad9 | Ad40 | Ad12 | Ad7 | Ad5 | Ad9 | Ad40 | Ad12 | Ad7 | Ad5 | Ad9 | Ad40 | Ad12 | Ad7 | Ad5 | Ad9 | Ad40 | |
| Ad12 | 100 | 100 | 100 | 100 | ||||||||||||||||
| Ad7 | 41.9 | 100 | 41.6 | 100 | 47.9 | 100 | 53.7 | 100 | ||||||||||||
| Ad5 | 39.9 | 37.5 | 100 | 43.2 | 48.1 | 100 | 48.6 | 53.8 | 100 | 49.3 | 49.6 | 100 | ||||||||
| Ad9 | 39.3 | 43.1 | 38.1 | 100 | 43.2 | 52.4 | 47.6 | 100 | 45.3 | 56.2 | 52.2 | 100 | 50.0 | 58.2 | 46.5 | 100 | ||||
| Ad40 | 38.4 | 38.5 | 33.7 | 40.1 | 100 | 48.5 | 45.5 | 43.6 | 41.8 | 100 | 55.9 | 48.3 | 48.6 | 44.5 | 100 | 62.5 | 51.7 | 49.3 | 49.6 | 100 |
Sequence identities were determined with the full-length sequence of each polypeptide by using the ALIGN program (50). Ad12, subgroup A; Ad7, subgroup B; Ad5, subgroup C; Ad9, subgroup D; Ad40, subgroup F. Boldface values indicate amino acid sequence comparisons between Ad9 E1 region polypeptides and other E1 region polypeptides; values in italics show that all Ad9 E1 region polypeptides are most closely related to those of subgroup B virus Ad7.
Northern blot analyses of total cellular RNA isolated from Ad9-infected A549 cells were also performed to detect Ad9 E1A and E1B mRNAs. In these assays, a diffuse E1A mRNA band migrating at approximately 1 kb and distinct 1.2- and 2.2-kb E1B mRNA bands were observed (Fig. 2). The size of the E1A mRNA band was consistent with that predicted for the 12S and 13S transcripts (see below), and the two E1B mRNAs corresponded well with the sizes predicted for 13S and 22S transcripts (47).
FIG. 2.

Northern blot analyses of Ad9 E1A and E1B mRNAs. Total RNA (23 μg), isolated from Ad9-infected (9 h postinfection) or mock-infected A549 cells, was separated on a formaldehyde agarose gel, transferred to a nitrocellulose membrane, and hybridized to either an E1A or E1B 32P-labeled DNA probe. RNA bands were visualized by autoradiography. Locations of 28S and 18S rRNAs are indicated. The indicated Ad9 mRNA species are predicted from their sizes.
Because E1A but not E1B mRNA is detected in Ad9-induced rat mammary tumors (29), we determined the structures of Ad9 E1A transcripts by using reverse transcription-PCR techniques on total RNA from Ad9-infected A549 cells. Sequencing of PCR products obtained from these analyses revealed three Ad9 E1A splice-variant transcripts resembling 13S, 12S, and 10S mRNAs from other human adenoviruses (Fig. 1A) (47). Ad9 E1A mRNA having a size consistent with that expected for the 10S mRNA was not detected by Northern blot analyses (Fig. 2), presumably due to its low abundance at early times after infection. The 13S, 12S, and 10S Ad9 E1A cDNAs are predicted to encode 251-, 189-, and 133-amino-acid-residue polypeptides, respectively (Fig. 1B). Between conserved regions 2 (CR2) and 3 (CR3), the 13S E1A protein of subgroup A virus Ad12 possesses an alanine-rich spacer region which is, in part, responsible for the highly oncogenic phenotype of this virus (33, 53). In contrast, the Ad9 13S E1A protein was found to contain a non-alanine-rich spacer region similar to the one present in 13S E1A proteins of weakly oncogenic subgroup B adenoviruses (Fig. 1B).
A large deletion within the E1A or E1B gene abolishes focus-forming activity by the Ad9 E1 region.
To investigate the transforming potential of the Ad9 E1 region, we constructed an Ad9 E1 region (0 to 12.5 m.u.) plasmid, pAd9E1, and examined its ability to induce transformed foci on low-passage-number REF cultures. Unlike other adenovirus E1 regions, the Ad9 E1 region is unable to transform primary REF or baby rat kidney cell cultures (28). Consistent with these previous findings, pAd9E1 alone failed to generate transformed foci on REFs (Table 2). Nevertheless, whereas an activated ras plasmid alone also lacked detectable focus-forming activity on REFs, pAd9E1 and the activated ras plasmid together cooperated to produce transformed foci on these cells (Table 2). To determine whether Ad9 E1A and E1B gene functions were required for this cooperation, we introduced a large deletion into each of these genes within pAd9E1. A segment of the E1A gene coding for the initiation codon, conserved region 1 (CR1), CR2, and half of CR3 (48) was removed in plasmid pAd9E1(ΔE1A), and E1B gene coding sequences downstream of E1B-19K amino acid residue 14, as well as the first 208 amino acid residues of the 495-residue E1B-55K protein, were removed in plasmid pAd9E1(ΔE1B) (Fig. 3). Each deletion would be anticipated to inactivate the transforming potential of the relevant gene (21, 22, 57). When cotransfected with the activated ras plasmid, pAd9E1(ΔE1A) failed to generate any foci on REFs, whereas pAd9E1(ΔE1B) retained significant focus-forming activity, albeit at a reduced efficiency compared to wild-type pAd9E1 (Table 2). These results are concordant with previous results showing that activated ras cooperates with the Ad5 E1A but not the E1B gene (18). Therefore, our findings provided evidence that the transforming potential of the E1A gene is inactivated in pAd9E1(ΔE1A); however, it was unclear from these REF assays whether the deletion in pAd9E1(ΔE1B) similarly affects the transforming potential of the E1B gene.
TABLE 2.
Focus formation by wild-type and mutant Ad9 E1 region plasmids on low-passage-number REF cultures and the CREF cell linea
| Plasmid(s) | No. of transformed foci/ 2 100-mm-diam dishes
|
|||||
|---|---|---|---|---|---|---|
| REF culturesb
|
CREF cell linec
|
|||||
| −ras
|
+ras
|
Expt 1 | Expt 2 | |||
| Expt 1 | Expt 2 | Expt 1 | Expt 2 | |||
| pSP72 | 0 | 0 | 0 | 0 | 0 | 1 |
| pAd9E1 | 0 | 0 | 24 | 53 | 71 | 116 |
| pAd9E1(ΔE1A) | 0 | 0 | 0 | 0 | 2 | 3 |
| pAd9E1(ΔE1B) | 0 | 0 | 12 | 14 | 0 | 0 |
| pAd9E1(ΔE1A) + pAd9E1(ΔE1B) | NDd | ND | ND | ND | 36 | 45 |
50% confluent tertiary REF or CREF cells on 100-mm-diameter dishes were transfected with the indicated plasmid(s). At 72 h posttransfection, cells were passaged 1:3 and then maintained in culture medium. REF and CREF cells were fixed in methanol and stained with Giemsa at 4 and 6 weeks posttransfection, respectively, to quantify the number of transformed foci.
15 μg of the indicated Ad9 E1 region plasmid plus 5 μg of empty pSP72 (−ras) or 5 μg of pSP72-ras (+ras) plasmid (18) were transfected into REF cells.
10 μg of the indicated Ad9 E1 region plasmid plus 10 μg of empty pSP72 plasmid were transfected into CREF cells. For the pAd9E1(ΔE1A)-plus-pAd9E1(ΔE1B) cotransfection, 10 μg of each plasmid was used.
ND, not determined.
FIG. 3.

Illustration of E1A and E1B deletion mutations introduced into the Ad9 E1 region of plasmid pAd9E1. Wild-type Ad9 sequences are represented by a black line; deleted sequences are represented by a hatched line. The restriction enzyme sites used to generate the deletions are shown. The locations of E1A CR1, CR2, and CR3 (22, 48) are also indicated. ITR, inverted terminal repeat.
In an attempt to reveal more striking transforming deficiencies for pAd9E1(ΔE1B), we next performed focus assays in the established REF cell line CREF (19). Contrary to results obtained in REFs, transfection of pAd9E1 alone into CREF cells led to the formation of numerous transformed foci (Table 2). The fact that a plasmid containing Ad9 sequences from 0 to 17.5 m.u. exhibits weaker transforming activity in CREF cells (30) may indicate that Ad9 sequences from 12.5 to 17.5 m.u. interfere with focus formation in these cells. More important, when transfected individually into CREF cells, both pAd9E1(ΔE1A) and pAd9E1(ΔE1B) displayed significantly impaired focus-forming activity compared to wild-type pAd9E1 (Table 2). Cotransfection of pAd9E1(ΔE1A) and pAd9E1(ΔE1B) into CREF cells, however, resulted in a moderate number of transformed foci, revealing cooperation between the functional E1A and E1B genes retained collectively in the two plasmids. Taken together, the results obtained for low-passage-number REFs and the cell line CREF showed that the deletions within pAd9E1(ΔE1A) and pAd9E1(ΔE1B) greatly diminish the transforming activity of the Ad9 E1A and E1B genes, respectively.
Isolation of Ad9 E1A or E1B deletion mutant viruses.
The importance of the Ad9 E1 region in mammary oncogenesis was assessed by introducing the same E1A and E1B deletion mutations of pAd9E1(ΔE1A) and pAd9E1(ΔE1B) into infectious Ad9 plasmids for recovery of mutant viruses. To complement their E1 region deficiencies, we transfected each of the mutant viral DNAs into human 293 cells, which stably express Ad5 E1 region proteins (2, 23). In 293 cells, the E1A mutant virus Ad9ΔE1A replicated to titers comparable to those of wild-type Ad9, whereas the E1B mutant virus Ad9ΔE1B replicated to titers approximately 10-fold lower. Because wild-type Ad9 fails to complement the replication defects of the Ad5 E1B-55K mutant dl252 (28), the reduced replication of Ad9ΔE1B conversely may be due to it being poorly complemented by Ad5 E1B proteins expressed in 293 cells. Restriction enzyme analyses of virion DNA verified that Ad9ΔE1A and Ad9ΔE1B contained the expected deletions and further showed that these viruses had not acquired Ad5 E1 region sequences from the 293 cells (Fig. 4).
FIG. 4.
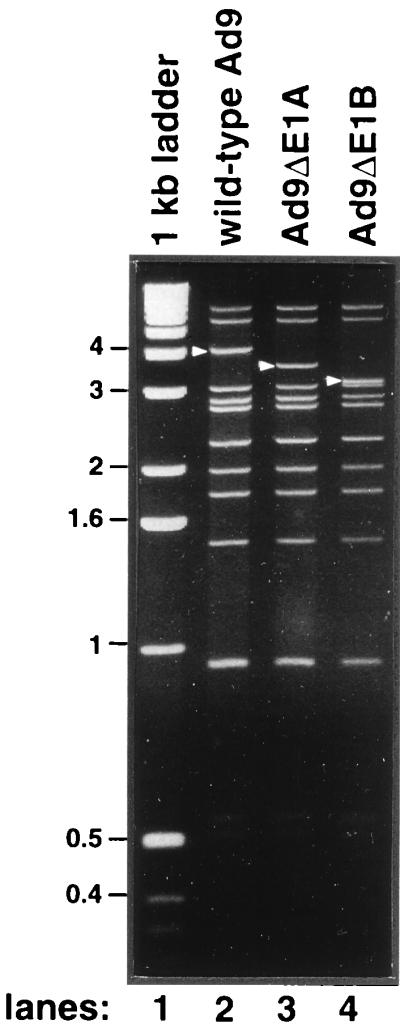
SmaI digestion pattern of wild-type and E1A and E1B mutant Ad9 virion DNAs. The wild-type Ad9 E1 region-containing SmaI DNA band migrating at approximately 4 kb, as well as the corresponding faster-migrating DNA bands of the Ad9 E1A mutant virus Ad9ΔE1A and of the Ad9 E1B mutant virus Ad9ΔE1B, are indicated with arrows. A 1-kb ladder (Gibco-BRL) was used as a DNA size marker (lane 1).
Ad9 E1A and E1B mutant viruses retain the ability to elicit mammary tumors in rats.
We next tested the ability of mutant viruses Ad9ΔE1A and Ad9ΔE1B to generate mammary tumors in Wistar-Furth rats. In accordance with our previous results (29, 30), wild-type Ad9 elicited mammary tumors in all of the female rats but none of the male rats, whereas subgroup D Ad26 failed to elicit tumors in any animals (Table 3). Significantly, we found that both Ad9ΔE1A and Ad9ΔE1B retained the ability to generate mammary tumors in female rats, despite the fact that Ad9ΔE1B-infected animals received a ninefold-lower dose of virus than did animals infected with either wild-type Ad9 or Ad9ΔE1A (Table 3). The tumorigenic phenotype of Ad9ΔE1B may not be surprising, considering that, unlike E1A mRNA, E1B mRNA is not detected in Ad9-induced mammary tumors (29). Furthermore, although mammary tumors elicited by all of the viruses were histologically identical (Table 4), the tumors produced by Ad9ΔE1A were generally smaller than those induced by either wild-type Ad9 or Ad9ΔE1B, both of which generated tumors of similar size (data not shown).
TABLE 3.
Tumorigenicities of wild-type and mutant Ad9 viruses in Wistar-Furth ratsa
| Virus | No. of rats that developed tumors/ no. infected with virus
|
|
|---|---|---|
| Females | Males | |
| Ad9 | 3/3 | 0/2 |
| Ad9ΔE1A | 8/8 | 0/2 |
| Ad9ΔE1Bb | 3/3 | 0/3 |
| Ad26c | 0/3 | 0/3 |
Two- to three-day-old Wistar-Furth rats were injected subcutaneously with 7 × 107 PFU of virus. Animals were monitored by palpation for tumor development over an 8-month period.
Due to replication deficiencies of virus Ad9ΔE1B in 293 cells, the dose used to infect rats with this virus (8 × 106 PFU) was approximately ninefold lower than that used for the other viruses.
Ad26, a nononcogenic subgroup D human adenovirus closely related to Ad9 (31), served as a negative control in this experiment.
TABLE 4.
Histologies of wild-type and mutant Ad9-induced tumorsa
| Virus | Tumor sample | Histology |
|---|---|---|
| Wild-type Ad9 | 1 | Fibroadenoma |
| 2 | Fibroadenoma | |
| 3 | Fibroadenoma | |
| Ad9ΔE1A | 1 | Fibroadenoma, one area of increased cellularity |
| 2 | Focally cellular fibroadenoma | |
| 3 | Fibroadenoma | |
| 4 | Cellular fibroadenoma, focally increased mitoses, and focal phyllodes-like tumorb | |
| 5 | Fibroadenoma | |
| 6 | Fibroadenoma | |
| 7 | Fibroadenoma | |
| 8 | Fibroadenoma | |
| Ad9ΔE1B | 1 | Fibroadenoma |
| 2 | Fibroadenoma | |
| 3 | Fibroadenoma |
For histological examination, tumor samples were fixed in 10% formalin, and sections were stained with hematoxylin and eosin.
A section of this tumor contained an area resembling a phyllodes-like tumor, a type of mammary tumor occasionally observed in rats infected by wild-type Ad9 (29).
Mutant Ad9 virus-induced tumors do not contain wild-type Ad9 E1 region sequences.
Because retention of tumorigenicity by both Ad9ΔE1A and Ad9ΔE1B was unanticipated, it was important to demonstrate that the mammary tumors caused by these viruses do not contain wild-type Ad9 DNA. For this analysis, we subjected tumor DNAs to a two-step nested PCR procedure (Fig. 5). In the first step, DNAs were PCR amplified with E1A primers (a plus b) or E1B primers (c plus d) flanking the deleted regions (Fig. 5A). From these reactions, wild-type Ad9-induced tumors yielded the expected 1,001-bp E1A product and 1,068-bp E1B product, but Ad9ΔE1A-induced tumors and Ad9ΔE1B-induced tumors yielded only the expected smaller 550-bp E1A product and 180-bp E1B product, respectively (Fig. 5B). To rule out the possibility of low-level contamination by wild-type Ad9 genomes in these mutant virus-induced tumors, we next used a nested set of E1A primers (e plus b) or E1B primers (c plus f) to subject the DNA products of the first PCRs described above to a second PCR (Fig. 5A). Using these nested primers in control PCRs, we were able to amplify the expected 762-bp E1A and 533-bp E1B products directly from the DNA of a wild-type Ad9-induced tumor (Fig. 5C). In contrast, we failed to amplify any such wild-type Ad9 DNA products from the first E1A and E1B PCRs of mutant virus-induced tumor DNAs (Fig. 5C). These results indicated that wild-type Ad9 E1 region sequences are absent from the mutant virus-induced mammary tumors and, consequently, that Ad9ΔE1A and Ad9ΔE1B are able to produce mammary tumors in rats.
FIG. 5.

Tumors induced by viruses Ad9ΔE1A and Ad9ΔE1B do not contain wild-type Ad9 E1 region sequences. (A) Locations of Ad9 E1 region primers used in PCRs. ITR, inverted terminal repeat. (B) PCR 1 utilized either E1A primers a and b or E1B primers c and d. Genomic DNA from rat 3Y1 cells or water (no DNA) represented negative controls in these reactions; 1.5 μg of genomic DNA or 10 ng of virion DNA was used as a template. (C) Nested PCR 2 utilized E1A primers e and b or E1B primers c and f. In these reactions, DNA from a wild-type Ad9-induced tumor and 3Y1 genomic DNA represented positive and negative controls, respectively; 1/20 of the DNA products from PCR (B) was used as a template for PCR 2. PCR conditions: 30 cycles of denaturation at 94°C for 1 min, annealing at 65°C (E1A reaction 1), 58°C (E1A reaction 2), or 60°C (E1B reactions 1 and 2) for 1 min, and extension at 72°C for 1 min, followed by a final 72°C extension for 15 min. DNA products were separated by agarose gel electrophoresis and visualized with ethidium bromide.
Mammary tumors contain and express the Ad9 E4 ORF1 gene.
As E4 ORF1 is an essential viral determinant for tumorigenesis by Ad9 (32), we next sought to confirm that Ad9 mutant virus-induced mammary tumors retain this gene and express the protein. By PCR amplification or immunoblot analysis, we detected the Ad9 E4 ORF1 gene (Fig. 6A) or its protein expression (Fig. 6B), respectively, in all mammary tumors, including those elicited by viruses Ad9ΔE1A and Ad9ΔE1B. As smaller tumors had arisen in Ad9ΔE1A virus-infected animals, it was noteworthy that the levels of Ad9 E4 ORF1 protein in Ad9ΔE1A-induced tumors were lower than those in both wild-type Ad9-induced and Ad9ΔE1B-induced tumors (Fig. 6B).
FIG. 6.

(A) PCR amplification of the Ad9 E4 ORF1 gene from tumor DNAs. PCRs were performed with Ad9 E4 ORF1 primers as described for Fig. 5 except that a 55°C annealing temperature was used. Genomic DNA from rat 3Y1 cells represented a negative control in these reactions. (B) Detection of Ad9 E4 ORF1 protein in tumors. Tumor lysates containing 3 mg of protein were subjected to immunoprecipitation followed by immunoblot analysis using Ad9 E4 ORF1 polyclonal antiserum. Preimmune serum served as a negative control for immunoprecipitations (lane 2).
DISCUSSION
In this study, we determined the nucleotide sequence of the subgroup D Ad9 E1 region and showed that its gene organization and predicted protein products are highly related to those of E1 regions from other human adenoviruses. Additionally, to investigate the role of the Ad9 E1 region in Ad9-induced mammary oncogenesis, we engineered the same E1A and E1B deletion mutations into both Ad9 E1 region plasmids and Ad9 viruses. We found that while E1A and E1B mutant Ad9 E1 region plasmids displayed significantly impaired focus-forming activity in vitro, the corresponding E1A and E1B mutant Ad9 viruses retained the ability to generate mammary tumors in rats. These results indicate that although the Ad9 E1 region alone or in cooperation with activated ras exhibits transforming activity in vitro, this activity is not required for mammary tumorigenesis by Ad9 in vivo. Similar examples in which transformation in vitro fails to predict tumorigenicity in vivo are also known for other viral and cellular transforming proteins (6, 8, 35, 45, 48, 56).
In addition to showing that Ad9 E1 region transforming functions are dispensable for mammary tumorigenesis by Ad9, our results further argue that the Ad9 E4 region-encoded ORF1 transforming gene represents the major oncogenic determinant of this virus. In this respect, Ad9 represents the first example of an oncogenic adenovirus for which the E1 region is not the major oncogenic determinant. The fact that the oncogenic avian adenovirus CELO lacks genes related to the human adenovirus E1A and E1B oncogenes (11) further suggests that additional examples non-E1 region oncogenic determinants for adenoviruses will be found.
Although the mechanism by which Ad9 reaches the mammary glands of rats after subcutaneous inoculation has not been established, we hypothesize that the inoculated Ad9 virions are able to directly infect mammary cells to cause tumors in the animals. This idea is based on the fact that rodent cells are generally nonpermissive for replication of human adenoviruses (48), a property that would limit spread of the virus by successive rounds of viral replication in tissues of rats. Moreover, in this study, we found that Ad9 E1A and E1B mutant viruses (Ad9ΔE1A and Ad9ΔE1B, respectively) retained the capacity to generate mammary tumors in these animals. Because E1A and E1B genes encode critical functions needed for efficient replication of adenoviruses (48), these new results with E1A and E1B mutant viruses provide additional support for the idea that viral replication in rats is not required for Ad9 to produce mammary tumors.
Although tumors elicited by wild-type and E1 region mutant Ad9 viruses in this study were found to be histologically identical, the tumors induced by the E1A mutant Ad9 virus were generally smaller than those generated by both the wild-type and E1B mutant Ad9 viruses. This finding suggests that E1A transforming functions may, in fact, enhance the growth of Ad9-induced mammary tumors. Nevertheless, it must also be considered that, separate from its transforming functions, E1A also serves an important role in the viral life cycle by transcriptionally activating other viral gene regions, including the E4 region (7, 34, 42). In the E1A mutant virus Ad9ΔE1A, we introduced a large deletion extending from the E1A initiation codon through half of CR3, a mutation which in addition to abolishing the transforming potential of E1A would also be expected to block transcriptional activation mediated by this gene. With regard to such a lack of E1A transcriptional activity in virus Ad9ΔE1A, it may be relevant that mammary tumors generated by this virus expressed reduced levels of the E4 ORF1 protein (Fig. 6B). This finding may indicate that E1A plays an accessory role in Ad9 mammary tumorigenesis by transcriptionally activating the viral E4 region and, thereby, elevating expression of the Ad9 E4 ORF1 oncogenic determinant. Similar indirect roles in viral oncogenesis have been ascribed to the bovine papillomavirus type 1 E2 and the Epstein-Barr herpesvirus EBNA2 transactivators, which participate in tumor formation by increasing expression of the transforming genes of their respective viruses (14, 16, 24, 36, 43).
In addition to promoting tumorigenesis, the oncoproteins of DNA tumor viruses may also contribute to determining which particular tissues are targeted for neoplasia. Comparisons of two related families of viruses, the papillomaviruses (PVs) and fibropapillomaviruses (FPVs), can be used to illustrate this idea. Although members of both families of viruses encode three different, structurally conserved transforming proteins, E5, E6, and E7 (5, 10, 15, 27, 39, 46), PVs and FPVs target distinct tissues in vivo, with PVs causing papillomas in epithelial keratinocytes and FPVs causing fibropapillomas in dermal fibroblasts (27). It has been established that E6 and E7 represent the major transforming proteins of PVs, whereas the E5 gene product is the major transforming protein of FPVs (27). Such observations have led to the hypothesis that the use of functionally different oncogenic determinants contributes to the unique tumorigenic tissue tropisms of PVs and FPVs (27). Likewise, Ad9 causes estrogen-dependent mammary tumors, whereas other oncogenic adenoviruses induce sarcomas in rodents. Therefore, one intriguing possibility is that novel molecular mechanisms which underly the transforming activity of Ad9 E4 ORF1 (41) permit Ad9 to selectively target mammary cells for tumorigenesis.
ACKNOWLEDGMENTS
We thank Stephen Hoang for technical assistance and Sylvia Lee for generously supplying REF cultures. We also thank Sylvia Lee, Britt Glaunsinger, Ezequiel Fuentes, and Nader Ghebranious for helpful suggestions.
D.L.T. was supported by National Research Service Award CA09197 from the National Cancer Institute and by the Federal Work-Study program of the U.S. Department of Education. This work was funded by grants from the NIH (NCI R01 CA/AI58541), ACS (RP6-97-068-01-VM), and Department of the Army (DAMD17-97-1-7082) to R.T.J. and by an NIH grant (POI CA41086) to T.E.S. T.E.S. is an American Cancer Society Professor and an Investigator of the Howard Hughes Medical Institute.
REFERENCES
- 1.Abate C, Luk D, Gentz R, Rauscher F, Curran T. Expression and purification of the leucine zipper and DNA-binding domains of fos and jun: both fos and jun contact DNA directly. Proc Natl Acad Sci USA. 1990;87:1032–1036. doi: 10.1073/pnas.87.3.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aiello L, Guilfoyle R, Huebner K, Weinmann R. Adenovirus 5 DNA sequences present and RNA sequences transcribed in transformed human embryo kidney cells. Virology. 1979;94:460–469. doi: 10.1016/0042-6822(79)90476-8. [DOI] [PubMed] [Google Scholar]
- 3.Ankerst J, Jonsson N. Adenovirus type 9-induced tumorigenesis in the rat mammary gland related to sex hormonal state. J Natl Cancer Inst. 1989;81:294–298. doi: 10.1093/jnci/81.4.294. [DOI] [PubMed] [Google Scholar]
- 4.Ankerst J, Jonsson N, Kjellen L, Norrby E, Sjogren H O. Induction of mammary fibroadenomas in rats by adenovirus type 9. Int J Cancer. 1974;13:286–290. doi: 10.1002/ijc.2910130303. [DOI] [PubMed] [Google Scholar]
- 5.Baker C C. Sequence analysis of papillomavirus genomes. In: Salzman N, Howley P M, editors. The Papovaviridae. Vol. 2. New York, N.Y: Plenum Publishing Corp.; 1987. pp. 321–385. [Google Scholar]
- 6.Barbosa M S, Schlegel R. The E6 and E7 genes of HPV-18 are sufficient for inducing two-stage in vitro transformation of human keratinocytes. Oncogene. 1989;4:1529–1532. [PubMed] [Google Scholar]
- 7.Berk A J, Lee F, Harrison T, Williams J, Sharp P A. Pre-early adenovirus 5 gene product regulates synthesis of early viral messenger RNAs. Cell. 1979;17:935–944. doi: 10.1016/0092-8674(79)90333-7. [DOI] [PubMed] [Google Scholar]
- 8.Bos J L, Jochemsen A G, Bernards R, Schrier P I, van Ormondt H, van der Eb A J. Deletion mutants of region E1a of AD12 E1 plasmids: effect on oncogenic transformation. Virology. 1983;129:393–400. doi: 10.1016/0042-6822(83)90178-2. [DOI] [PubMed] [Google Scholar]
- 9.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 10.Bubb V, McCance D J, Schlegel R. DNA sequence of the HPV-16 E5 ORF and the structural conservation of its encoded protein. Virology. 1988;163:243–246. doi: 10.1016/0042-6822(88)90259-0. [DOI] [PubMed] [Google Scholar]
- 11.Chiocca S, Kurzbauer R, Schaffner G, Baker A, Mautner V, Cotten M. The complete DNA sequence and genomic organization of the avian adenovirus CELO. J Virol. 1996;70:2939–2949. doi: 10.1128/jvi.70.5.2939-2949.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 13.Chomczynski R. One-hour downward alkaline capillary transfer for blotting of DNA and RNA. Anal Biochem. 1992;201:134–139. doi: 10.1016/0003-2697(92)90185-a. [DOI] [PubMed] [Google Scholar]
- 14.Cohen J I, Wang F, Kieff E. Epstein-Barr virus nuclear protein 2 mutations define essential domains for transformation and transactivation. J Virol. 1991;65:2545–2554. doi: 10.1128/jvi.65.5.2545-2554.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Danos O, Engel L W, Chen E Y, Yaniv M, Howley P M. Comparative analysis of the human type 1a and bovine type 1 papillomavirus genomes. J Virol. 1983;46:557–566. doi: 10.1128/jvi.46.2.557-566.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DiMaio D, Metherall J, Neary K. Nonsense mutation in open reading frame E2 of bovine papillomavirus DNA. J Virol. 1986;57:475–480. doi: 10.1128/jvi.57.2.475-480.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feinberg A P, Vogelstein B. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal Biochem. 1983;132:6–13. doi: 10.1016/0003-2697(83)90418-9. [DOI] [PubMed] [Google Scholar]
- 18.Finlay C A, Hinds P W, Levine A J. The p53 proto-oncogene can act as a suppressor of transformation. Cell. 1989;57:1083–1093. doi: 10.1016/0092-8674(89)90045-7. [DOI] [PubMed] [Google Scholar]
- 19.Fisher P B, Babiss L E, Weinstein I B, Ginsberg H S. Analysis of type 5 adenovirus transformation with a cloned rat embryo cell line (CREF) Proc Natl Acad Sci USA. 1982;79:3527–3531. doi: 10.1073/pnas.79.11.3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freshney R I. Culture of animal cells: a manual of basic technique. 2nd ed. New York, N.Y: Alan R. Liss, Inc.; 1987. pp. 107–126. [Google Scholar]
- 21.Fukui Y, Saito I, Shiroki K, Shimojo H. Isolation of transformation-defective, replication-nondefective early region 1B mutants of adenovirus type 12. J Virol. 1984;49:154–161. doi: 10.1128/jvi.49.1.154-161.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Graham F. Transformation by and oncogenicity of human adenoviruses. In: Ginsberg H, editor. The adenoviruses. New York, N.Y: Plenum Press; 1984. pp. 339–398. [Google Scholar]
- 23.Graham F L, Smiley J, Russell W C, Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol. 1977;36:59–72. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 24.Groff D E, Lancaster W D. Genetic analysis of the 3′ early region transformation and replication functions of bovine papillomavirus type 1. Virology. 1986;150:221–230. doi: 10.1016/0042-6822(86)90281-3. [DOI] [PubMed] [Google Scholar]
- 25.Horwitz M S. Adenoviruses. In: Fields B N, Knipe D M, Howley P M, editors. Fields virology. Vol. 2. Philadelphia, Pa: Lippincott; 1996. pp. 2149–2171. [Google Scholar]
- 26.Houweling A, van den Elsen P, van der Eb A. Partial transformation of primary rat cells by the leftmost 4.5% fragment of adenovirus 5 DNA. Virology. 1980;105:537–550. doi: 10.1016/0042-6822(80)90054-9. [DOI] [PubMed] [Google Scholar]
- 27.Howley P M. Papillomavirinae: the viruses and their replication. In: Fields B N, Knipe D M, Howley P M, editors. Fields virology. Vol. 2. Philadelphia, Pa: Lippincott; 1996. pp. 2045–2076. [Google Scholar]
- 28.Jannun R, Chinnadurai G. Functional relatedness between the E1a and E1b regions of group C and group D human adenoviruses. Virus Res. 1987;7:33–48. doi: 10.1016/0168-1702(87)90056-6. [DOI] [PubMed] [Google Scholar]
- 29.Javier R, Raska K, Jr, Macdonald G J, Shenk T. Human adenovirus type 9-induced rat mammary tumors. J Virol. 1991;65:3192–3202. doi: 10.1128/jvi.65.6.3192-3202.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Javier R, Raska K, Jr, Shenk T. Requirement for the adenovirus type 9 E4 region in production of mammary tumors. Science. 1992;257:1267–1271. doi: 10.1126/science.1519063. [DOI] [PubMed] [Google Scholar]
- 31.Javier R, Shenk T. Mammary tumors induced by human adenovirus type 9: a role for the viral early region 4 gene. Breast Cancer Res Treat. 1996;39:57–67. doi: 10.1007/BF01806078. [DOI] [PubMed] [Google Scholar]
- 32.Javier R T. Adenovirus type 9 E4 open reading frame 1 encodes a transforming protein required for the production of mammary tumors in rats. J Virol. 1994;68:3917–3924. doi: 10.1128/jvi.68.6.3917-3924.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jelinek T, Pereira D S, Graham F L. Tumorigenicity of adenovirus-transformed rodent cells is influenced by at least two regions of adenovirus type 12 early region 1A. J Virol. 1994;68:888–896. doi: 10.1128/jvi.68.2.888-896.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones N, Shenk T. An adenovirus type 5 early gene function regulates expression of other early viral genes. Proc Natl Acad Sci USA. 1979;76:3665–3669. doi: 10.1073/pnas.76.8.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaur P, McDougall J K. Characterization of primary human keratinocytes transformed by human papillomavirus type 18. J Virol. 1988;62:1917–1924. doi: 10.1128/jvi.62.6.1917-1924.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kieff E. Epstein-Barr Virus and its replication. In: Fields B N, Knipe D M, Howley P M, editors. Fields virology. Vol. 2. Philadelphia, Pa: Lippincott; 1996. pp. 2343–2396. [Google Scholar]
- 37.Kimura G, Itagaki A, Summers J. Rat cell line 3Y1 and its virogenic polyoma and SV40 transformed derivatives. Int J Cancer. 1975;15:694–706. doi: 10.1002/ijc.2910150419. [DOI] [PubMed] [Google Scholar]
- 38.Kingston R E, Chen C A, Okayama H. Calcium phosphate transfection. In: Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current protocols in molecular biology. New York, N.Y: Greene Publishing Associates and Wiley-Interscience; 1990. pp. 9.1.1–9.1.9. [Google Scholar]
- 39.Kulke R, DiMaio D. Biological activities of the E5 protein of the deer papillomavirus in mouse C127 cells: morphologic transformation, induction of cellular DNA synthesis, and activation of the platelet-derived growth factor receptor. J Virol. 1991;65:4943–4949. doi: 10.1128/jvi.65.9.4943-4949.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 41.Lee S S, Weiss R S, Javier R T. Binding of human virus oncoproteins to hDlg/SAP97, a mammalian homolog of the Drosophila discs large tumor suppressor protein. Proc Natl Acad Sci USA. 1997;94:6670–6675. doi: 10.1073/pnas.94.13.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nevins J. Mechanism of activation of early viral transcription by the adenovirus E1A gene product. Cell. 1981;26:213–220. doi: 10.1016/0092-8674(81)90304-4. [DOI] [PubMed] [Google Scholar]
- 43.Rabson M S, Yee C, Yang Y-C, Howley P M. Bovine papillomavirus type 1 3′ early region transformation and plasmid maintenance functions. J Virol. 1986;60:626–634. doi: 10.1128/jvi.60.2.626-634.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robbins S L, Angell M, Kumar V. Basic pathology. Philadelphia, Pa: The W. B. Saunders Co.; 1981. pp. 564–595. [Google Scholar]
- 45.Schlegel R, Phelps W C, Zhang Y-L, Barbosa M. Quantitative keratinocyte assay detects two biological activities of human papillomavirus DNA and identifies viral types associated with cervical carcinoma. EMBO J. 1988;7:3181–3187. doi: 10.1002/j.1460-2075.1988.tb03185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schwarz E, Durst M, Demankowski C, Lattermann O, Zech R, Wolfsperger E, Suhai S, zur Hausen H. DNA sequence and genome organization of genital human papillomavirus type 6b. EMBO J. 1983;2:2341–2348. doi: 10.1002/j.1460-2075.1983.tb01744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sharp P A. Adenovirus transcription. In: Ginsberg H, editor. The adenoviruses. New York, N.Y: Plenum Press; 1984. pp. 173–204. [Google Scholar]
- 48.Shenk T. Adenoviridae: the viruses and their replication. In: Fields B N, Knipe D M, Howley P M, editors. Fields virology. Vol. 2. Philadelphia, Pa: Lippincott; 1996. pp. 2111–2148. [Google Scholar]
- 49.Shenk T, Flint J. Transcriptional and transforming activities of the adenovirus E1A proteins. Adv Cancer Res. 1991;57:47–85. doi: 10.1016/s0065-230x(08)60995-1. [DOI] [PubMed] [Google Scholar]
- 50.Smith R F, Wiese B A, Wojzynski M K, Davison D B, Worley K C. BCM Search Launcher—an integrated interface to molecular biology database search and analysis services available on the World Wide Web. Genome Res. 1996;6:454–462. doi: 10.1101/gr.6.5.454. [DOI] [PubMed] [Google Scholar]
- 51.Stillman B. Functions of the adenovirus E1B tumor antigens. Cancer Surv. 1986;5:389–404. [PubMed] [Google Scholar]
- 52.Strauss W M. Preparation of genomic DNA from mammalian tissue. In: Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current protocols in molecular biology. Vol. 1. New York, N.Y: Greene Publishing Associates and Wiley-Interscience; 1994. pp. 2.2.1–2.2.3. [Google Scholar]
- 53.Telling G C, Williams J. Constructing chimeric type 12/type 5 adenovirus E1A genes and using them to identify an oncogenic determinant of adenovirus type 12. J Virol. 1994;68:877–887. doi: 10.1128/jvi.68.2.877-887.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trentin J, Yabe Y, Taylor G. The quest for human cancer viruses: a new approach to an old problem reveals cancer induction in hamster by human adenovirus. Science. 1962;137:835–841. doi: 10.1126/science.137.3533.835. [DOI] [PubMed] [Google Scholar]
- 55.van den Elsen P, Houweling A, van der Eb A. Expression of region E1b of human adenoviruses in the absence of region E1a is not sufficient for complete transformation. Virology. 1983;128:377–390. doi: 10.1016/0042-6822(83)90264-7. [DOI] [PubMed] [Google Scholar]
- 56.van Leeuwen F N, van der Kammen R A, Habets G G M, Collard J G. Oncogenic activity of Tiam 1 and Rac1 in NIH3T3 cells. Oncogene. 1995;11:2215–2221. [PubMed] [Google Scholar]
- 57.Yew P R, Kao C C, Berk A J. Dissection of functional domains in the adenovirus 2 early 1B 55K polypeptide by suppressor-linker insertional mutagenesis. Virology. 1990;179:795–805. doi: 10.1016/0042-6822(90)90147-j. [DOI] [PubMed] [Google Scholar]