

Abstract
The development of effective drugs to treat coronavirus infections remains a significant challenge for the scientific community. Recent evidence reports on the sigma-1 receptor (S1R) as a key druggable host protein in the SARS-CoV-1 and SARS-CoV-2 interactomes and shows a potent antiviral activity against SARS-CoV-2 for the S1R antagonist PB28. To improve PB28 activity, we designed and tested a series of its analogues and identified a compound that is fourfold more potent against SARS-CoV-2 than PB28 itself. Interestingly, we found no direct correlation between S1R affinity and SARS-CoV-2 antiviral activity. Building on this, we employed comparative induced fit docking and molecular dynamics simulations to gain insights into the possible mechanism that occurs when specific ligand–protein interactions take place and that may be responsible for the observed antiviral activity. Our findings offer a possible explanation for the experimental observations, provide insights into the S1R conformational changes upon ligand binding and lay the foundation for the rational design of new S1R ligands with potent antiviral activity against SARS-CoV-2 and likely other viruses.
Subject terms: Computational biology and bioinformatics, Drug discovery, Medicinal chemistry
Introduction
Despite the increasingly likely prospect of ending the coronavirus disease 2019 (COVID-19) pandemic with the aid of approved vaccines, we are still experiencing waxing and waning of pandemic patterns, with several viral variants that have followed and will likely continue to emerge. The long-term effects of COVID-19, known as "long COVID," are still under evaluation and include detrimental effects on the central nervous system (CNS), resulting in neurological and psychiatric symptoms. SARS-CoV-2 is the 7th coronavirus to have established respiratory infection between humans. Therefore, developing effective therapies against coronavirus infections remains a critical challenge for the scientific community. In the last two decades, three zoonotic spill overs of coronaviruses to humans have caused significant epidemics. The severe acute respiratory syndrome (SARS) outbreak of 2003 resulted in over 8,000 human infections and approximately 800 deaths1, the Middle East respiratory syndrome (MERS) outbreak of 2012 saw around 2,500 confirmed cases and 858 deaths2. Currently, the ongoing coronavirus disease 2019 (COVID-19) pandemic has resulted in over 760 million confirmed cases and more than 6.8 million deaths3. It is reasonable to postulate that the current pandemic caused by a coronavirus is not the first, nor will it be the last. For this reason, a global effort has been taken to prevent the world from being caught unprepared again. Several in-silico studies4–7 as well as clinical trials8 have been conducted to find effective drugs against COVID-19 through repurposing. The RNA-dependent viral polymerase inhibitor Remdesivir, immunomodulators such as monoclonal antibodies (e.g., Tocilizumab), and kinase inhibitors like Baricitinib have been approved for treating infected patients who are at high risk of progressing to severe COVID-19. Other monoclonal antibodies targeting the spike protein, as well as antiviral drugs like Paxlovid and Molnupiravir, have also received emergency use authorization from the Food and Drug Administration (FDA)9,10. However, the use of monoclonal antibodies is limited by the high frequency of non-susceptible viral variants. Therefore, the FDA has restricted the use of certain monoclonal antibodies in geographic regions characterized by a low frequency of such non-susceptible variants11. The approved antiviral drugs were discovered through drug repurposing approaches that aimed to inhibit viral proteins and counteract replication. However, targeting viral components such as viral enzymes with a drug may lead to the development of resistance due to the high rate of viral mutations12,13. As a result, an ideal broad-spectrum coronavirus antiviral should be able to target host components crucial to the virus's replication cycle or the development of the disease in the host, reducing the likelihood of drug resistance. Based on this evidence, a study published in Nature14 identified the SARS-CoV-2 interactome with 66 druggable host targets, including the endoplasmic sigma receptors. Through affinity-purification mass spectrometry, it was found that the SARS-CoV-2 protein NSP6 interacts with the sigma-1 (S1R) receptor, while ORF9c interacts with the sigma-2 (S2R) receptor. The researchers then tested several drugs already in clinical use for other conditions and known to act on the identified druggable host proteins. Among the tested molecules, PB28, a S1R antagonist and S2R agonist, showed most potent anti-SARS-CoV-2 activity in vitro, with an IC90 value of 0.28 μM. Subsequent studies, using genomic and proteomic approaches, identified S1R as a crucial host dependency factor. Specifically, studies showed that knocking out (KO) or knocking down (KD) S1R prevented the infection of cells (such as Caco-2 and A549-ACE2) by SARS-CoV-2. Conversely, KO and KD of S2R did not have the same effect15. Importantly, S1R displayed the same effect in both SARS-CoV-1 and SARS-CoV-2 infected cells, preventing the infection when knocked out. A retrospective study analysing the outcomes of approximately 800,000 infected patients undergoing typical or atypical antipsychotic treatment provided hints of the in vivo effectiveness of S1R modulation as anti-SARS-CoV-2 therapy16. It is worth noting that typical antipsychotics have an affinity for the S1R, whereas atypical ones do not. The retrospective study showed that a high percentage of infected patients undergoing treatment with typical antipsychotics had better outcomes. Several clinical studies, both retrospective and prospective, have been conducted with fluvoxamine, a serotonin reuptake inhibitor and potent S1R agonist. These studies suggest that fluvoxamine may act as a prophylactic drug for early-stage SARS-CoV-2-infected patients17,18. Furthermore, a S1R polymorphism (rs17775810) has been linked to a reduced death rate associated with COVID-19, with the TT genotype performing better than the CT and CC genotypes, thus providing additional evidence for the S1R involvement in SARS-CoV-2 replication19. It is noteworthy that S1R ligands have been found to exhibit antiviral activity against other viruses as well20. The various findings about the involvement of S1R in coronavirus infections and the positive results obtained from PB28 and other S1R ligands already in clinical use, have sparked interest in further exploring the role of S1R in these infections. This could hopefully lead to the development of novel ligands with the potential to provide therapeutic options against mutant strains that may not respond to existing vaccines and therapies. To develop novel and more potent anti-coronavirus agents, we tested a chemical library of PB28 analogs, focusing on those with high S1R affinity and we identified one agent that is 4-fold more potent against SARS-CoV-2 than PB28. However, we did not observe a direct correlation between S1R affinity and SARS-CoV-2 antiviral activity, suggesting the presence of a mechanism that may only occur if specific ligand–protein interactions take place. To investigate these interactions, we used molecular modelling and analysed the obtained data that will be useful to design new and more potent anti-SARS-CoV-2 S1R ligands21.
Materials and methods
Synthesis
All SR ligands investigated were produced according to the literature reported for each compound in Tables 1, 2, 3, 4. They were obtained with a purity > 95%.
Table 1.
S1R/S2R affinities and anti-SARS-CoV-2 activity of a first set of PB28 analogs.
| General structure of PB28 and its analogs | ||||||
|---|---|---|---|---|---|---|
 | ||||||
| Compound | A or B | R | n | AKi nM S1R | BKi nM S2R | CIC50 (μM) SARS-CoV-2 |
| PB2839 | A | 5-OCH3 | 3 | 0.38 | 0.68 | 0.299 |
| (-)-R-PB2839 | A | 5-OCH3 | 3 | 0.63 | 0.49 | 0.309 |
| ( +)-S-PB2839 | A | 5-OCH3 | 3 | 0.13 | 1.18 | 0.455 |
| 140 | A | 5-OCH3 | 0 | 0.40 | 7.90 | NOT ACTIVE |
| 240 | A | 5-OCH3 | 1 | 0.31 | 16.4 | 5.90 |
| 340 | A | 5-OCH3 | 2 | 1.57 | 21.1 | 12.57 |
| 440 | A | 5-OCH3 | 4 | 1.54 | 3.58 | 0.320 |
| 540 | A | 5-OCH3 | 5 | 1.52 | 0.35 | 0.19 |
| 6 | A | 5-OCH3 | 6 | 3.07 | 103 | 1.32 |
| 740 | A | H | 3 | 0.61 | 0.68 | 0.438 |
| 840 | A | H | 4 | 0.036 | 14.6 | 0.317 |
| 940 | A | H | 5 | 1.45 | 7.85 | 0.19 |
| 1040 | A | 6-OCH3 | 3 | 0.36 | 5.42 | 0.47 |
| 1140 | B | H | 3 | 2.16 | 0.69 | 0.94 |
| 1240 | B | H | 5 | 2.40 | 0.57 | 0.52 |
| 1340 | B | 5-OCH3 | 3 | 1.57 | 9.24 | 0.306 |
| 1441 | B | 6-OCH3 | 3 | 3.16 | 9.02 | NOT ACTIVE |
| 1541 | A | 5-OH | 3 | 5.40 | 2.66 | NOT ACTIVE |
| 1641 | A | 6-OH | 3 | 0.69 | 1.12 | NOT ACTIVE |
| 1741 | B | 6-OH | 3 | 6.78 | 26.4 | NOT ACTIVE |
| 1841 | B | 7-OH | 3 | 5.48 | 11.8 | NOT ACTIVE |
A,BKi values represent the mean of three experiments in duplicate, ± SEM is reported in the cited manuscript; AKi values from radioligand binding assay: [3H]-( +)-Pentazocine on guinea pig brain according to the generally accepted protocol; BKi values from radioligand binding assay: [3H]-DTG on rat liver masking with (+)-Pentazocine according to the generally accepted protocol; CDetection of viral genomes by RT-qPCR on Vero E6 cells infected with SARS-CoV-2 virus14. Listed values are one experiment that is representative of experiments performed at least three times.
Table 2.
SR ligands with chemotype different from PB28.
| Compound | AKi nM S1R | BKi nM S2R | CIC50 (μM) SARS-CoV-2 | |
|---|---|---|---|---|
| 1942 |  |
2435 | 4.24 | NOT ACTIVE |
| 2042 |  |
311 | 3.56 | NOT ACTIVE |
| 2142 |  |
709 | 4.84 | NOT ACTIVE |
| 2242 | 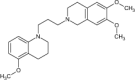 |
3987 | 1.42 | NOT ACTIVE |
A,BKi values represent the mean of three experiments in duplicate, ± SEM is reported in the cited manuscript; AKi values from radioligand binding assay: [3H]-( +)-Pentazocine on guinea pig brain according to the generally accepted protocol; BKi values from radioligand binding assay: [3H]-DTG on rat liver masking with ( +)-Pentazocine according to the generally accepted protocol; CDetection of viral genomes by RT-qPCR on Vero E6 cells infected with SARS-CoV-2 virus14. Listed values are one experiment that is representative of experiments performed at least three times.
Table 3.
S1R/S2R affinities and anti-SARS-CoV-2 activity of a second set of PB28 analogs.
| A. Replacement of the cyclohexyl ring | |||||
|---|---|---|---|---|---|
| General structure | |||||
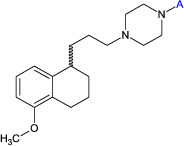 | |||||
| Compound | A | AKi nM S1R | BKi nM S2R | CIC50 (μM) SARS-CoV-2 | |
| 2343 | CH3- | 17.8 | 663 | 5.78 | |
| 2443 | CH3CH2CH3- | 9.6 | 42.5 | 2.26 | |
| 2543 | (CH3CH2)2CH- | 12.0 | 12.7 | NOT ACTIVE | |
| B. Insertion of Heteroatoms | |||||
|---|---|---|---|---|---|
| General structure | |||||
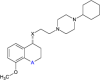 | |||||
| A | X | ||||
| 2644 | CH2 | N | 23.9 | 8.49 | 0.30 |
| ( +)-R-2644 | CH2 | N | 13.6 | 14.4 | 0.08 |
| (-)-S-2644 | CH2 | N | 23.3 | 8.52 | 0.41 |
| 2744 | CH2 | NCH3 | 4.17 | 15.3 | 1.27 |
| 2844 | CH2 | O | 3.90 | 9.03 | 5.83 |
| 2944 | O | CH2 | 12.5 | 9.72 | 10.49 |
| 3044 | 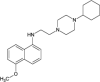 |
5.44 | 18.5 | 2.1 | |
| 3144 | 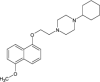 |
65%** | 2.40 | NOT ACTIVE | |
| C. Replacement of the tetralin core | |||||
|---|---|---|---|---|---|
| General structure | |||||
 | |||||
| 3245 | 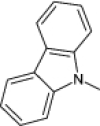 |
3450 | 12.6 | 1.18 | |
| 3346 | 0.25 | 4.70 | NOT ACTIVE | ||
| 3447 |  |
24.7 | 26.6 | NOT ACTIVE | |
| 3548 | 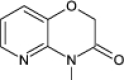 |
12.0 | 16.0 | NOT ACTIVE | |
| 36cis49 | 0.042 | 3.66 | NOT ACTIVE | ||
| 36trans49 | 0.045 | 4.77 | NOT ACTIVE | ||
| 37cis49 | 0.81 | 1.59 | 2.349 | ||
| 37trans49 | 0.23 | 1.21 | 2.313 | ||
| 38cis49 | 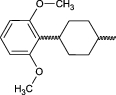 |
0.26 | 7.92 | NA | |
| 38trans49 | 0.32 | 0.21 | NA | ||
A,BKi values represent the mean of three experiments in duplicate, ± SEM is reported in the cited manuscript; AKi values from radioligand binding assay: [3H]-( +)-Pentazocine on guinea pig brain according to the generally accepted protocol;BKi values from radioligand binding assay: [3H]-DTG on rat liver masking with ( +)-Pentazocine according to the generally accepted protocol; CDetection of viral genomes by RT-qPCR on Vero E6 cells infected with SARS-CoV-2 virus14. Listed values are one experiment that is representative of experiments performed at least three times.
Table 4.
Reference subnanomolar S1R ligands.
| AKi Nm S1R | BKi nM S2R | CIC50 (μM) SARS-CoV-2 | ||
|---|---|---|---|---|
| PB19050 | 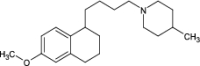 |
0.42 | 36.3 | NOT ACTIVE |
| PB21250 |  |
0.030 | 17.9 | NOT ACTIVE |
| 3950 |  |
0.35 | 238 | NOT ACTIVE |
A,BKi values represent the mean of three experiments in duplicate, ± SEM is reported in the cited manuscript; AKi values from radioligand binding assay: [3H]-( +)-Pentazocine on guinea pig brain according to the generally accepted protocol; BKi values from radioligand binding assay: [3H]-DTG on rat liver masking with ( +)-Pentazocine according to the generally accepted protocol; CDetection of viral genomes by RT-qPCR on Vero E6 cells infected with SARS-CoV-2 virus14. Listed values are one experiment that is representative of experiments performed at least three times.
Biology
Materials for antiviral activity assays
VeroE6 (ATCC #CRL-1586) and A549-ACE2, a human lung epithelial cell line provided by the lab of Olivier Schwartz that is overexpressing the SARS-CoV-2 receptor, were cultured in DMEM (Gibco #31,966,021) supplemented with 10% FBS (Gibco #A3160801) and penicillin/streptomycin (100 U/mL and 100 µg/mL, Gibco #15,140,122) at 37 °C in a 5% CO2 atmosphere. In order to maintain the selection of the ACE2 clones, Blasticidin (10 µg/mL – Sigma Aldrich #SBR00022-10ML) was added to the media.
SARS-CoV-2 BA.1 strain (GISAID ID: EPI_ISL_6794907) was kindly provided by the Virus and Immunity Unit (Institut Pasteur, PMID: 35,322,239). Viral stocks were generated by infecting VeroE6 cells at a multiplicity of infection (MOI) of 0.01 in DMEM supplemented with 2% of FBS and 1 µg/mL of TPCK-Tryspin (Sigma-Aldrich #1426-100MG). Supernatant was harvested 3 days post-infection (p.i.) and stored at − 80 °C.
Antiviral activity assays
A549-ACE2 cells were seeded in 96 well plates at a concentration of 1.5E4 cells per well in DMEM supplemented with 10% of FBS and incubated overnight at 37 °C, 5% CO2.
Two hours prior to infection, the supernatant was replaced with 100 uL of DMEM – 2% FBS containing the compound of interest at a range of concentrations (100 µM, 50 µM, 20 µM, 10 µM, 1 µM or 0,1 µM); or an equivalent volume of DMSO (Sigma Aldrich—#D2650), vehicle used as a control. At the time of infection, the media was replaced with virus inoculum (MOI = 0.1 PFU/cell).
Following a one-hour adsorption at 37 °C, the virus inoculum was replaced by 200 µL of drug- (or vehicle-) containing media and cells were incubated for an additional 72 h at 37 °C, 5% CO2. The supernatants were then collected for RT-qPCR and cell viability assay.
Virus quantification by RT-qPCR
72 hpi, supernatants were collected and inactivated 10 min at 95 °C to further be used for RT-qPCR. SARS-CoV-2 specific primers targeting the N gene region: 5’TAATCAGACAAGGAACTGATTA-3’ (Forward) and 5’CGAAGGTGTGACTTCCATG-3’ (Reverse) were used with the Luna Universal One-Step RT-qPCR kit (New England Biolabs, #E3005) in an Applied Biosystems QuantStudio 6 thermocycler with the following cycling conditions: 55 °C for 10 min, 95 °C for 1 min, and 40 cycles of 95 °C for 10 s followed by 60 °C for 1 min. The number of viral genomes is expressed as PFU equivalents/mL and calculated by performing a standard curve with RNA derived from a viral stock with a known viral titer. Datas was fit using nonlinear regression and IC50s for each experiment were determined using GraphPad Prism version 8.1.0 (San Diego, CA).
Cell toxicity assay
Cell viability was assessed in drug treated cells by using Cell TiterGlo following the manufacturer’s instructions (Promega #G7570). Luminescence was measured in a Tecan Infinity 2000 plate reader.
Percentage of viability was calculated relative to untreated cells and cells lysed with 20% of ethanol.
Computational details
Docking studies
PB28, 10, 14 and 16 were docked on the recently published X-ray structure of S1R in complex with haloperidol (co-x) (resolution 3.08 Å – pdb code: 6DJZ)22. The retrieved .pdb file was prepared using the Protein Preparation Wizard tool, available from the Schrodinger Suite 2021–223 for adding missing hydrogen atoms, reconstructing incomplete side chains, assigning favourable protonation states at physiological pH and performing a force field based minimization of the 3D protein structures. All ligands were prepared using the LigPrep tool for generating all the possible ionization states and tautomers at a pH value of 7.0 ± 2.024. The obtained files were employed for docking simulations performed by Grid-based ligand docking with energetics25. In particular, we performed Induced Fit Docking (IFD)26 simulations in order to properly take into account putative conformational rearrangements of the protein binding site during molecular recognition. Docking simulations were performed using the SP mode and all the default settings, building a cubic grid centered on the co-x and having an inner box of 10 Å × 10 Å × 10 Å and an outer box of 30 Å × 30 Å × 30 Å. In particular, these steps were followed: i) docking of each ligand using a softened potential and retaining only those poses (maximum 20) that meet specific criteria based on Coulomb-vdW (less than 100) and H-bond (less than –0.05) thresholds; ii) prediction of the best orientation of that residues within a certain distance (5 Å) of any ligand pose; iii) minimization of the obtained complexes to accommodate the ligand structure in the induced-fit protein conformation; iv) redocking using the standard precision (SP) protocol of each protein–ligand complex structure within a specified energy of the lowest-energy structure (30 kcal/mol), v) computation of the binding energy (IFDScore) for each output pose. The top-scored docking complexes were subjected to molecular mechanics/generalized Born surface area calculations (MM-GBSA) using the OPLS2005 force field27. Notice that during this calculation, no flexibility was allowed for the residues of the binding site since a conformational rearrangement of the binding site had already been considered during the IFD simulations.
Model system preparation
The S1R-(S)-PB28 and S1R-(R)-16 complexes returned by IFD simulations were subjected to Molecular Dynamics (MD) simulations to get insights into putative effects of the ligand binding on S1R conformation. Using the system builder28, the complexes were inserted into a pre-equilibrated (T = 310 K) 1-palmitolyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) lipid bilayer and fully solvated into a minimized, orthorhombic TIP3P water-box (10 × 10 × 10 Å3). Na+ and Cl− ions were added generating a 150 mM ionic concentration. In doing that, we obtained two electrically neutral systems including ~ 90,000 atoms. The default OPLS4 force field29 was used for both protein and ligands.
MD simulation protocol
Simulations were performed on GPUs by using Desmond 4.2, implemented in the Schrodinger Suite 2022–428, as software program. A non-bonded cut-off of 9 Å was used. All the prepared systems were minimized, equilibrated and simulated using an isothermalisobaric ensemble (NPγT, P = 1 atm, T = 300 K) with a Nosè–Hoover thermostat30 and a Martyna-Tobias-Klein barostat31. We performed a 200 ns-long MD simulation for each investigated complex using a time step equal to 2 fs and storing the coordinates with a recording interval of 100 ps. In doing that, 2001 frames were generated and analysed for each system.
MD simulation analysis
The obtained trajectories were analysed using the trajectory player available in the Schrodinger Suite 2022–428. More specifically, the Root Mean Square Deviation (RMSD) and Root Mean Square Fluctuations (RMSF) of the alpha-carbon atoms of the protein were computed using the 'Compute Properties Over Trajectory' tool. All the results were saved in a .csv file used as input file for an in-house R script written to generate the corresponding 2D plots.
Molecular properties calculation
We utilized QikProp32 and Epik 733 to predict the octanol/water partition coefficient (referred to as logP) and the macro-pKa of all investigated compounds in this study. Specifically, Epik 7 employs a reliable machine-learning model to predict the pKa for all the reasonable ionization sites within each molecule33.
Results and discussion
PB28 derivatives and anti-SARS-CoV-2 in vitro activity
To investigate a range of biological features that affect anti-SARS-CoV-2 activity, we constructed a library of SR binders consisting of several S1R and S2R ligands closely related to PB28. This library included ligands with an affinity for SR subtypes ranging from subnanomolar to micromolar Ki values, with varying degrees of S1R vs S2R selectivity. We also investigated S1R or S2R ligands with different chemotypes than PB28 to determine whether the anti-SARS-CoV-2 activity was specifically associated with a particular structure type. While the literature has extensively discussed the structure-affinity relationship studies of the S1R and S2R ligands reported in Tables 1, 2, 3, 4, as recently reviewed34, in this study we present their activity as IC50 values against SARS-CoV-2 virus in Vero E6 cells and correlate the obtained values with the known affinities of SRs. As PB28 is a chiral compound, we investigated the corresponding pure enantiomers, namely (-)-(R)-PB28 and ( +)-(S)-PB28. Notably, we did not observe a substantial change in antiviral activity compared to the racemate, which is in line with the measured S1R and S2R affinities. It is worth noting that the activity of the compounds is basically unaffected when the alkyl chain is elongated from propyl to hexyl, with hexyl being only threefold less potent (6). However, lower homologs exhibit reduced activity, with a 20- or 40-fold reduction observed for compounds 2 and 3, respectively, which feature a methylene or ethylene linker, respectively. Furthermore, despite the subnanomolar affinity at the S1R returned by compound 1, the absence of a linker between the tetralin nucleus and the piperazine moiety results in a loss of activity. The change in position of the methoxy group (6–OCH3, 10), on the other hand, did not affect the activity. Additionally, the replacement of the tetralin ring with a naphthalene nucleus, along with the presence (5–OCH3, 13) or absence (11, 12) of the methoxy group, did not alter the activity. A notable finding was that changing the position of the methoxy group in the naphthalene series resulted in a complete loss of activity, despite the compound's 1-digit nanomolar affinity for the S1R (for example, compound 14 with a methoxy group at position 6). Interestingly, replacing the methoxy group with a hydroxyl group in either the 5- or 6-position also led to a complete loss of antiviral activity, despite high S1R affinity observed for both the tetralin (16, 15) and naphthalene (17, 18) series. The above compounds generally exhibit subnanomolar or 1-digit nanomolar affinity at the S1R and slightly lower affinity at the S2R, except for the hexyl-bearing compound 6. We also tested a small series of S2R ligands that have no or negligible affinity at the S1R (Table 2). These compounds feature the 6,7-dimethoxytetrahydroisoquinoline as the basic moiety instead of the cyclohexylpiperazine, and a dihydroisoquinolinone (19, 20, 21) or tetrahydroquinoline (22) as the hydrophobic moiety. They were all inactive in the anti-viral assay, which supports the conclusion that S2R is not involved in the anti-SARS-CoV-2 activity. However, based on the collected data, it is not possible to establish a correlation between the anti-viral activity and the S1R affinity of these compounds, as ligands with high affinity for S1R may result in highly active or inactive compounds. Confounding factors in the anti-SARS-CoV-2 assays have been reported due to phospholipidosis35 induced by cationic amphiphilic drugs, such as some SR ligands. However, given the structural similarity of the compounds analysed, it would be expected that all of them have the potential to induce phospholipidosis and show antiviral activity in vitro, since this phenomenon is a common side effect of many cationic amphiphilic drugs and is mainly related to their lipophilicity36. To investigate this further, we have expanded our analysis to include other derivatives of PB28 with varying lipophilicities and chemical structures different from PB28 (Tables 3 and 4). Replacing the cyclohexyl ring with shorter alkyl chains, such as methyl (23) or n-propyl (24), results in a 20- or 8-fold reduction in anti-SARS-CoV-2 activity, respectively (Table 3A). However, the compound (25) with a 3-pentyl group in place of the cyclohexyl one shows no anti-SARS-CoV-2 activity, despite its better match with the PB28 physicochemical properties (Table 3A). Compounds with heteroatoms in the linker or a tetralin portion with lower lipophilicity than PB28 were also evaluated (Table 3B and Table 3C). Various modifications were made to the structure of the compounds, and as a result, their anti-SARS-CoV-2 activity was affected differently. Among less lipophilic compounds, those with N-heteroatoms displayed activity comparable to PB28, with the (R)-enantiomer exhibiting a fourfold increased activity (26), while methylation of the NH group (27) or aromatization of the tetralin core to naphthalene (30) led to a 4- to 7- fold reduction in activity (Table 3B). However, the insertion of an oxygen atom in the linker or the tetralin core caused a drop-in activity, with a significant reduction for the tetralin-bearing compounds (28 and 29) and a complete loss for the naphthalene counterpart (31) (Table 3B). Furthermore, replacement of the tetralin nucleus with other structures, such as carbazole, cyclohexane, dihydro-isoquinolinone, pyrido-ozaxinone, or differently substituted benzene-cyclohexyl ring, led to inactive compounds in the anti-SARS-CoV-2 assay, except for 37cis and 37trans couple, whose IC50 value is in the 2 µM range (Table 3C). Again, all these compounds display affinity at the SRs spanning from subnanomolar to micromolar. It is interesting to note that PB190 and PB212, which are known to have a subnanomolar affinity as a S1R agonist and antagonist, respectively, did not demonstrate any antiviral effect in the performed assay, nor did their congener 39 (Table 4). LogP and pKa values were calculated for all the compounds (Tables 1, 2, 3, 4) and reported for comparison (Table S1). The former values ranged from 3.30 to 5.51 while the latter from 4.03 to 13.31. Overall, the data obtained suggest that: (i) S1R ligands with Ki values ranging from subnanomolar to two-digit nanomolar have anti-SARS-CoV-2 activity, (ii) pure S2R ligands do not exhibit antiviral activity, and (iii) some high-affinity S1R ligands may not possess anti-SARS-CoV-2 activity. Notably, hydroxy-derivatives of PB28 (15, 16, 17 and 18, Table 1), despite having low nanomolar S1R affinity, are inactive in the anti-SARS-CoV-2 assay. Additionally, the results obtained from PB190 and PB212 suggest that the measured antiviral activities cannot be justified solely by the affinity or functional activity, when available, of S1R ligands. S1R has been defined as a pluripotent modulator that mainly resides at the interface between mitochondria and endoplasmic reticulum. It is able to translocate and interact with several proteins modulating their functions thus resulting in multiple functions and therapeutic applications37. It should be noted that the currently available functional assays may not account for all possible S1R protein interactions, making it difficult to assign the terms "agonist" or "antagonist" to S1R ligands38. Based on the obtained data and reported conformational changes described in the literature, we have postulated that the S1R ligands may induce a conformational rearrangement of the S1R upon binding to produce the antiviral effect, possibly by interacting with NSP6.
Computational studies
Induced fit docking (IFD) simulations
Based on the experimental data obtained, which suggests the presence of a conformational change responsible for the observed antiviral activities, we conducted IFD simulations to gain new molecular insights into potential conformational crosstalks between S1R and the ligands in our series. Additionally, we focused our attention on three ligands from our series that were selected as molecular probes due to their high structural similarity with the reference compound PB28 (Ki (S1R) = 0.69 nM) and strong affinity for S1R. Specifically, we investigated the effect of: (i) a methoxy shift from the 5- to 6-position (10, Ki (S1R) = 0.38 nM); (ii) the substitution of the tetralin core with a naphthalene one (14, Ki (S1R) = 3.16 nM); and (iii) the replacement of the methoxy substituent in position 6 with a hydroxy group (16, Ki (S1R) = 0.69 nM). Although these compounds share a high structural similarity, only PB28 and 10 were found to be responsible for antiviral activities (Table 1). Figure 1 displays the top-scored docking poses returned by the IFD protocol. As expected, the predicted binding mode for each investigated compound closely resembles that observed experimentally for the co-crystallized ligand, based on the crystal structure. Furthermore, all investigated ligands produced high MM-GBSA scores, with the best ΔG being returned by (S)-PB28 (− 145.7 kcal/mol), followed by (S)-10 (− 143.61 kcal/mol), (R)-16 (− 142.9 kcal/mol), and 14 (− 138.46 kcal/mol), in full agreement with their measured sub-nanomolar (PB28, 16, 10) and 1-digit nanomolar (14) S1R affinities. Additionally, IFD data suggests the presence of several interactions shared by all investigated ligands, including (i) an ionic interaction between a positively charged nitrogen atom and E172, a negatively charged residue, (ii) a cation-pi interaction involving the same nitrogen atom and F107, an aromatic residue, and (iii) several hydrophobic interactions with L182, L186, and, except for (R)-16, Y206. Although most of the interactions are shared among the simulated compounds, a distinct orientation can be observed when comparing the top-scored conformations of (S)-PB28 and (S)-10 with those returned by 14 and (R)-16. Specifically, (S)-PB28 and (S)-10 are predicted to position their methoxy group towards L95, thus forming a hydrophobic interaction. On the other hand, 14 and (R)-16 direct their R-group (see Fig. 1) away from this residue. Interestingly, visual inspection suggests that a slight variation in terms of posing could potentially be responsible for a different conformation of the protein cavity. Indeed, an induced-fit rearrangement involving the α4 helix of the protein is observed upon binding with (S)-PB28 and (S)-10 only. Figure 2, showing the superposition of the S1R-(S)-PB28 and S1R-(R)-16 complexes returned by IFD simulations, provides a concrete idea of the observed slight α4 displacement. Notably, Schmidt et al.22 indicate that the conformation adopted by the α4 helix of S1R upon ligand binding is associated with its response (agonism vs. antagonism). More specifically, the authors report on the differences between the S1R crystal structure complexed with haloperidol (antagonist) and that complexed with pentazocine (agonist), highlighting that these two compounds induce two distinct α4 conformations, namely one closer to the binding site (haloperidol) and the other away from it (pentazocine). Importantly, PB28 is herein predicted to induce a further α4 helix approach, consistently with its known S1R antagonist activity.
Figure 1.

Top-scored docking poses within the binding pocket of S1R of: (A) (S)-PB28; (B) (S)-10, (C) 14, (D) (R)-16. For the sake of clarity, only polar hydrogen atoms are shown. Important residues are rendered as sticks while the proteins are represented as cartoons. Salt-bridge and cation-pi interactions are depicted by a red and green line, respectively.
Figure 2.

Superposition of the S1R (cyan)-(S)-PB28 (orange) and S1R (gray)-(R)-16 (sand) complexes. The observed α4-helix shift is indicated by a bidirectional black arrow. For the sake of clarity, only polar hydrogen atoms are shown. Important residues are rendered as sticks while the proteins are represented as cartoons.
Molecular dynamics simulations
Encouraged by these preliminary results and in an effort to support this hypothesis, two complexes returned by IFD simulations were used as the starting point for 200-ns long MD simulations, specifically, S1R-(S)-PB28 and S1R-(R)-16. Notice that these complexes were selected since (S)-PB28 and (R)-16 represent ideal molecular probes. Indeed, despite their high S1R affinity and structural similarity (see Table 1) only one of them (i.e., (S)-PB28) acts as antiviral. As an initial step of the study, we computed the time-dependent Root Mean Square Deviation (RMSD) for all alpha-carbon atoms of the protein during the simulation. Figure S1 in the supporting information indicates that equilibration of the complexes required 70 ns (S1R-(R)-16) and 150 ns (S1R-(S)-PB28), and consequently, the last 130 ns and 50 ns were analyzed for S1R-16 and S1R-PB28, respectively. Notably, average RMSD values of 0.97 Å ± 0.38 and 2.06 Å ± 0.38 were computed for S1R-(S)-PB28 and S1R-(R)-16, respectively, indicating the reliability of the performed IFD simulations. Specifically, the interactions with E172 and F107 remained intact throughout the simulations of both S1R-(S)-PB28 and S1R-(R)-16. Moreover, additional interactions involving the –OH substituent were predicted to be established with T202 and Y206, as shown in Figure S2 of the supporting information. Figure 3A reports the Root Mean Square Fluctuation (RMSF) values computed for the alpha-carbon atoms of each residue within the investigated systems, a higher level of flexibility can be observed in both α4 (residues 177–192) and α1 (residues 8–32) helixes of S1R-(R)-16, in comparison to S1R-(S)-PB28. Expanding on these findings, we computed the time-dependent RMSD values returned by the alpha-carbon atoms of the α4 helix (as shown in Fig. 3B). It is worth noting that our starting IFD complexes exhibited, between them, an initial shift of the α4 helix by 0.56 Å. The values obtained from our MD simulations indicate that this shift becomes more pronounced in time, and this protein portion can reach a different state depending on the bound ligand. This data again supports the robustness of the IFD docking data. Significantly, the observation that the α4 helix undergoes a conformational change, resulting in a different state induced by ligand-binding, is consistent with a recent study that reported results from MD simulations51.
Figure 3.

(A) RMSF of the alpha-carbon atoms belonging to each residue (Å) of the α4 helix of S1R-(R)-16 (blue) and S1R-(S)-PB28 (orange); (B) Time dependence of RMSD (Å) computed for the α4 helix of S1R-(R)-16 (blue) and S1R-(S)-PB28 (orange); (C) Superposition of selected snapshots of S1R-(R)-16 (gray) and S1R-(S)-PB28 (cyan) extracted from the performed MD simulations. The α1 and α4 helix shift is highlighted by a bidirectional black arrow; (D) Time dependence of the dihedral angle (dα) defined by the alpha-carbon atoms of the first (R8 and V177) and the last (T32 and S192) residues of α1 and α4. The initial equilibration trajectories were not included in the analysis.
In particular, Figure S3 (available in the supporting information) displays the superposition of the average conformations of S1R-(S)-PB28 and S1R-(R)-16 complexes as returned by the performed MD simulations. Remarkably, a shift of 1.8 Å involving the alpha-carbon atom belonging to A185 is observed after comparing the two complexes. This observation is in full agreement with the paper by Schmidt et al. (see Fig. 2C of reference 22) reporting the same conformational switch after comparing the x-ray crystal structures in complex with haloperidol and pentazocine. Furthermore, we computed the time-dependence of the dihedral angle defined by the alpha-carbon atoms of the first and the last residues of α1 and α4 (residues R8, T32, V177 and S192, hereinafter referred to as dα). As evident in Fig. 3C and 3D, during the simulation S1R-(S)-PB28 reaches a very different conformational state (dα ≈ − 140°) with respect to S1R-(R)-16 (dα ≈ − 30°), hence suggesting the presence of a conformational coupling involving these two helices. Noteworthy the visual inspection performed on the obtained trajectories indicates that such a difference is mostly due to a different orientation of the protein with respect to the membrane, rather than to a movement of α1 within the membrane. Such a significant conformation effect might represent a mechanical switch triggered by ligand binding. Whether this switch affects the oligomerization of S1R or its interactions with other membrane proteins or cytoplasmic biomolecules remains to be ascertained. Notably, our data support the physiological relevance of α1 whose conformation was recently hypothesized to be crucial for the closed-to-open transition of the protein51.
Conclusions
As recently reviewed, S1R may play a critical role in early-stage virus replication17. In addition to the evidence already described, further support for S1R as a potential target for antiviral action is provided by the following observations: i) S1R is enriched in lipid rafts where it colocalizes with viral replicative proteins, such as NSP6; ii) viruses use stress response mechanisms to promote their replication, and S1R regulates stress response20,52,53; iii) Activation of S1R by its ligands induces autophagy, a mechanism used by some viruses including coronaviruses to evade the host immune system52,54; iv) several drugs intended for different uses were screened against viruses (e.g.; EBV, HCV, Ebola, Influenza H5N1), and drugs associated with antiviral activity were also S1R17. Based on our findings and the evidence that pan-viral disease mechanisms were revealed through the comparison of host-coronavirus protein interaction networks, we have tested a chemical library of S1R ligands. Some returned anti-SARS-CoV-2 effects, but we have not found a clear correlation between their S1R affinity and antiviral activity. The phospholipidosis effect has been proposed as an explanation for the lack of in vivo activity of some S1R ligands in preclinical studies, and we cannot rule it out. However, the correlation of the chemical properties of the compounds tested with the antiviral activity suggests that the phospholipidosis effect is not responsible for the observed effects. Very similar compounds in terms of structure, pKa and logP (see Table S1 in the Supporting Information) would exert the same effect, which is not the case. Compounds 13 and 14 give one representative example with identical physico-chemical properties and opposite anti-SARS-CoV-2 activity. Additionally, the phospholipidosis hypothesis contrasts with the genomic data and retrospective/prospective clinical studies mentioned above. Even though the effect of S1R ligands on phospholipidosis remains to be tested, our investigation has focused on the conformational changes these ligands may induce in the S1R protein, which may affect its interactions with client proteins. Recent bioluminescence resonance energy transfer (BRET) assays have shown that different S1R responses occur based on receptor multimerization, depending on the binding of structurally diverse ligands38. Our preliminary data support the hypothesis that shedding light on the conformational effects induced by ligand binding is crucial for the rational design of S1R ligands. Therefore, despite the complexity of the framework, our findings suggest that modulation of S1R could still be a viable approach to combat SARS-CoV-2 and SARS-CoV-1 infections as well as other highly pathogenic viruses.
Supplementary Information
Abbreviations
- (S1R)
Sigma-1 receptor
- (CNS)
Central nervous system
- (SARS)
Severe acute respiratory syndrome
- (MERS)
Middle East respiratory syndrome
- (COVID-19)
Coronavirus disease 2019
- (FDA)
Food and Drug Administration
- (S2R)
Sigma-2 receptor
- (KO)
Knocking out
- (KD)
Knocking down
- (IFD)
Induced Fit Docking
- (MD)
Molecular Dynamics
- (RMSD)
Root Mean Square Deviation
Author contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by C.A. and G.F.M. Computational studies and analyses were performed by P.D., I.M., G.L. and G.F.M. Anti-SARS-CoV-2 assays were designed and performed by V.R., S.B. and M.V. Synthesis of S1R ligands was performed by F.S.A. The first draft of the manuscript was written by C.A. and G.F.M. and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Funding
This work was funded by the National Research Council (CNR—Italy) under the program “PROGETTI DI RICERCA @CNR” (acronym DATIAMO).
Data availability
All data are reported in the manuscript and the SI provided.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Giuseppe Felice Mangiatordi, Email: giuseppe.mangiatordi@ic.cnr.it.
Carmen Abate, Email: carmen.abate@uniba.it.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-023-39662-w.
References
- 1.SARS | Basics Factsheet | CDC. https://www.cdc.gov/sars/about/fs-sars.html.
- 2.Memish ZA, Perlman S, Van Kerkhove MD, Zumla A. Middle East respiratory syndrome. Lancet. 2020;395:1063–1077. doi: 10.1016/S0140-6736(19)33221-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coronavirus Disease (COVID-19) Situation Reports. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports.
- 4.Delre P, Caporuscio F, Saviano M, Mangiatordi GF. Repurposing known drugs as covalent and non-covalent inhibitors of the SARS-CoV-2 papain-like protease. Front. Chem. 2020 doi: 10.3389/fchem.2020.594009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ali F, et al. Implication of in silico studies in the search for novel inhibitors against SARS-CoV-2. Arch. Pharm. (Weinheim) 2022;355:e2100360. doi: 10.1002/ardp.202100360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou Y, Wang F, Tang J, Nussinov R, Cheng F. Artificial intelligence in COVID-19 drug repurposing. Lancet Digital Health. 2020;2:e667–e676. doi: 10.1016/S2589-7500(20)30192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Onyango H, Odhiambo P, Angwenyi D, Okoth P. In silico identification of new anti-SARS-CoV-2 main protease (Mpro) molecules with pharmacokinetic properties from natural sources using molecular dynamics (MD) simulations and hierarchical virtual screening. J. Trop. Med. 2022;2022:3697498. doi: 10.1155/2022/3697498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chakraborty C, Sharma AR, Bhattacharya M, Agoramoorthy G, Lee S-S. The drug repurposing for COVID-19 clinical trials provide very effective therapeutic combinations: lessons learned from major clinical studies. Front. Pharmacol. 2021 doi: 10.3389/fphar.2021.704205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Commissioner, O. of the. Coronavirus (COVID-19) Update: FDA Authorizes First Oral Antiviral for Treatment of COVID-19. FDAhttps://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-first-oral-antiviral-treatment-covid-19 (2021).
- 10.Commissioner, O. of the. Coronavirus (COVID-19) Update: FDA Authorizes Additional Oral Antiviral for Treatment of COVID-19 in Certain Adults. FDAhttps://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-additional-oral-antiviral-treatment-covid-19-certain (2021).
- 11.Commissioner, O. of the. Coronavirus (COVID-19) Update: FDA Limits Use of Certain Monoclonal Antibodies to Treat COVID-19 Due to the Omicron Variant. FDAhttps://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-limits-use-certain-monoclonal-antibodies-treat-covid-19-due-omicron (2022).
- 12.Sanjuán R, Domingo-Calap P. Mechanisms of viral mutation. Cell Mol Life Sci. 2016;73:4433–4448. doi: 10.1007/s00018-016-2299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Callaway E. The coronavirus is mutating — Does it matter? Nature. 2020;585:174–177. doi: 10.1038/d41586-020-02544-6. [DOI] [PubMed] [Google Scholar]
- 14.Gordon DE, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gordon DE, et al. Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science. 2020;370:eabe9403. doi: 10.1126/science.abe9403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Canal-Rivero M, et al. Lower risk of SARS-CoV2 infection in individuals with severe mental disorders on antipsychotic treatment: A retrospective epidemiological study in a representative Spanish population. Schizophr. Res. 2021;229:53–54. doi: 10.1016/j.schres.2021.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hashimoto Y, Suzuki T, Hashimoto K. Mechanisms of action of fluvoxamine for COVID-19: A historical review. Mol. Psychiatry. 2022;27:1898–1907. doi: 10.1038/s41380-021-01432-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khani E, Entezari-Maleki T. Fluvoxamine and long COVID-19; a new role for sigma-1 receptor (S1R) agonists. Mol. Psychiatry. 2022;27:3562–3562. doi: 10.1038/s41380-022-01545-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lehrer S, Rheinstein PH. Homozygosity for rs17775810 minor allele associated with reduced mortality of COVID-19 in the UK biobank cohort. In Vivo. 2021;35:965–968. doi: 10.21873/invivo.12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vela JM. Repurposing Sigma-1 receptor ligands for COVID-19 therapy? Front. Pharmacol. 2020 doi: 10.3389/fphar.2020.582310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Research, C. for D. E. and. Coronavirus (COVID-19) | Drugs. FDAhttps://www.fda.gov/drugs/emergency-preparedness-drugs/coronavirus-covid-19-drugs (2023).
- 22.Schmidt HR, Betz RM, Dror RO, Kruse AC. Structural basis for σ1 receptor ligand recognition. Nat Struct Mol Biol. 2018;25:981–987. doi: 10.1038/s41594-018-0137-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schrödinger Release 2021–2: Protein Preparation Wizard; Epik, Schrödinger, LLC, (2021).
- 24.Schrödinger Release 2021–2: LigPrep, Schrödinger, LLC, (2021).
- 25.Schrödinger Release 2021–2: Glide, Schrödinger, LLC, New York, NY, (2021).
- 26.Schrödinger Release 2021–2: Induced Fit Docking protocol, Schrödinger, LLC, New York, NY, (2021).
- 27.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B. 2001;105:6474–6487. doi: 10.1021/jp003919d. [DOI] [Google Scholar]
- 28.Schrödinger Release 2022–4: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2021. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, (2021).
- 29.Lu C, et al. OPLS4: improving force field accuracy on challenging regimes of chemical space. J. Chem. Theory Comput. 2021;17:4291–4300. doi: 10.1021/acs.jctc.1c00302. [DOI] [PubMed] [Google Scholar]
- 30.Feller SE, Zhang Y, Pastor RW, Brooks BR. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995;103:4613–4621. doi: 10.1063/1.470648. [DOI] [Google Scholar]
- 31.Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994;101:4177–4189. doi: 10.1063/1.467468. [DOI] [Google Scholar]
- 32.Schrödinger Release 2022–4: QikProp, Schrödinger, LLC, New York, NY, (2021).
- 33.Johnston, R. C. et al. Epik: pKa and Protonation State Prediction through Machine Learning. Preprint at 10.26434/chemrxiv-2023-c6z8t (2023). [DOI] [PubMed]
- 34.Abate C, et al. PB28, the Sigma-1 and Sigma-2 receptors modulator with potent anti–SARS-CoV-2 activity: a review about its pharmacological properties and structure affinity Relationships. Front. Pharmacol. 2020 doi: 10.3389/fphar.2020.589810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tummino TA, et al. Drug-induced phospholipidosis confounds drug repurposing for SARS-CoV-2. Science. 2021;373:541–547. doi: 10.1126/science.abi4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hanumegowda UM, et al. Phospholipidosis as a function of basicity, lipophilicity, and volume of distribution of compounds. Chem. Res. Toxicol. 2010;23:749–755. doi: 10.1021/tx9003825. [DOI] [PubMed] [Google Scholar]
- 37.Su T-P, Su T-C, Nakamura Y, Tsai S-Y. The sigma-1 receptor as a pluripotent modulator in living systems. Trends Pharmacol. Sci. 2016;37:262–278. doi: 10.1016/j.tips.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yano H, et al. Pharmacological profiling of sigma 1 receptor ligands by novel receptor homomer assays. Neuropharmacology. 2018;133:264–275. doi: 10.1016/j.neuropharm.2018.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abate C, Mosier PD, Berardi F, Glennon RA. A structure-affinity and comparative molecular field analysis of sigma-2 (sigma2) receptor ligands. Cent. Nerv. Syst. Agents Med. Chem. 2009;9:246–257. doi: 10.2174/1871524910909030246. [DOI] [PubMed] [Google Scholar]
- 40.Berardi F, et al. 4-(Tetralin-1-yl)- and 4-(Naphthalen-1-yl)alkyl derivatives of 1-cyclohexylpiperazine as σ receptor ligands with agonist σ2 activity. J. Med. Chem. 2004;47:2308–2317. doi: 10.1021/jm031026e. [DOI] [PubMed] [Google Scholar]
- 41.Ferorelli S, et al. Design and evaluation of Naphthol- and Carbazole-containing fluorescent σ ligands as potential probes for receptor binding studies. J. Med. Chem. 2007;50:4648–4655. doi: 10.1021/jm070373b. [DOI] [PubMed] [Google Scholar]
- 42.Abate C, et al. Development of 3,4-dihydroisoquinolin-1(2H)-one derivatives for the positron emission tomography (PET) imaging of σ2 receptors. Eur. J. Med. Chem. 2013;69:920–930. doi: 10.1016/j.ejmech.2013.09.018. [DOI] [PubMed] [Google Scholar]
- 43.Perrone R, et al. N-aryl- or N-alkylpiperazine derivatives: The role of N-substituent on σ1, σ2, 5-HT1A and D2 receptor affinity. Med. Chem. Res. 2000;10:201–207. [Google Scholar]
- 44.Abate C, et al. Analogues of σ receptor ligand 1-Cyclohexyl-4-[3-(5-methoxy-1,2,3,4-tetrahydronaphthalen-1-yl)propyl]piperazine (PB28) with added polar functionality and reduced lipophilicity for potential use as positron emission tomography radiotracers. J. Med. Chem. 2011;54:1022–1032. doi: 10.1021/jm1013133. [DOI] [PubMed] [Google Scholar]
- 45.Niso M, et al. Sigma-2 receptor agonists as possible antitumor agents in resistant tumors: hints for collateral sensitivity. ChemMedChem. 2013;8:2026–2035. doi: 10.1002/cmdc.201300291. [DOI] [PubMed] [Google Scholar]
- 46.Berardi F, et al. Exploring the importance of piperazine N-Atoms for σ2 receptor affinity and activity in a series of analogs of 1-cyclohexyl-4-[3-(5-methoxy-1,2,3,4-tetrahydronaphthalen-1-yl)propyl]piperazine (PB28) J. Med. Chem. 2009;52:7817–7828. doi: 10.1021/jm9007505. [DOI] [PubMed] [Google Scholar]
- 47.Abate C, et al. Arylamides hybrids of two high-affinity σ2 receptor ligands as tools for the development of PET radiotracers. Eur J Med Chem. 2011;46:4733–4741. doi: 10.1016/j.ejmech.2011.05.057. [DOI] [PubMed] [Google Scholar]
- 48.Abate C, et al. 2-Aminopyridine derivatives as potential σ2 receptor antagonists. ChemMedChem. 2012;7:1847–1857. doi: 10.1002/cmdc.201200246. [DOI] [PubMed] [Google Scholar]
- 49.Abate C, et al. 1-Cyclohexyl-4-(4-arylcyclohexyl)piperazines: Mixed σ and Human Δ8–Δ7 Sterol Isomerase Ligands with Antiproliferative and P-glycoprotein inhibitory activity. ChemMedChem. 2011;6:73–80. doi: 10.1002/cmdc.201000371. [DOI] [PubMed] [Google Scholar]
- 50.Berardi F, et al. Methyl substitution on the piperidine ring of N-[ω-(6-methoxynaphthalen-1-yl)alkyl] derivatives as a probe for selective binding and activity at the σ1 receptor. J. Med. Chem. 2005;48:8237–8244. doi: 10.1021/jm050654o. [DOI] [PubMed] [Google Scholar]
- 51.Meng F, Xiao Y, Ji Y, Sun Z, Zhou X. An open-like conformation of the sigma-1 receptor reveals its ligand entry pathway. Nat Commun. 2022;13:1267. doi: 10.1038/s41467-022-28946-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gouda AS, Mégarbane B. Molecular bases of serotonin reuptake inhibitor antidepressant-attributed effects in COVID-19: A new insight on the role of bradykinins. J. Personaliz. Med. 2022;12:1487. doi: 10.3390/jpm12091487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsai S-YA, Pokrass MJ, Klauer NR, De Credico NE, Su T-P. Sigma-1 receptor chaperones in neurodegenerative and psychiatric disorders. Expert Opin Ther Targets. 2014;18:1461–1476. doi: 10.1517/14728222.2014.972939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brimson JM, et al. Drugs that offer the potential to reduce hospitalization and mortality from SARS-CoV-2 infection: The possible role of the sigma-1 receptor and autophagy. Expert Opin. Ther. Targets. 2021;25:435–449. doi: 10.1080/14728222.2021.1952987. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are reported in the manuscript and the SI provided.