

Summary
The Sicilian wolf remained isolated in Sicily from the end of the Pleistocene until its extermination in the 1930s–1960s. Given its long-term isolation on the island and distinctive morphology, the genetic origin of the Sicilian wolf remains debated. We sequenced four nuclear genomes and five mitogenomes from the seven existing museum specimens to investigate the Sicilian wolf ancestry, relationships with extant and extinct wolves and dogs, and diversity. Our results show that the Sicilian wolf is most closely related to the Italian wolf but carries ancestry from a lineage related to European Eneolithic and Bronze Age dogs. The average nucleotide diversity of the Sicilian wolf was half of the Italian wolf, with 37–50% of its genome contained in runs of homozygosity. Overall, we show that, by the time it went extinct, the Sicilian wolf had high inbreeding and low-genetic diversity, consistent with a population in an insular environment.
Subject areas: Canine genetics, Evolutionary biology
Graphical abstract
Highlights
-
•
The Sicilian and Italian wolves derive from the same evolutionary lineage
-
•
Sicilian wolf genomes carry Eneolithic dog ancestry
-
•
Nearly half of the Sicilian wolf genome is in runs of homozygosity
Canine genetics; Evolutionary biology
Introduction
The gray wolf (Canis lupus) is one of the most widespread and mobile carnivores in the Holarctic region.1 Genomic studies have grouped modern wolves into two distinct phylogenetic lineages: the Eurasian and North American wolf.1 Recent palaeogenomic studies demonstrated that Eurasian wolf populations were highly connected throughout the Late Pleistocene but experienced a significant turnover after the Last Glacial Maximum (LGM).2,3,4 Today, gray wolf genetic diversity surveys show they form structured populations, which suggests a decline in population connectivity and gene flow potentially due to anthropogenic disturbance of their habitat.5,6
Albeit the deep evolutionary relationship between humans and wolves, their direct competition and human persecution have led wolf populations across their geographical distribution to population decline, genetic bottlenecks, and even local extinctions.7,8 Therefore, changes in population size, admixture between wolves and domestic dogs, as well as recurrent post-divergence gene flow events, are common features of wolves’ evolutionary history.5,7,9
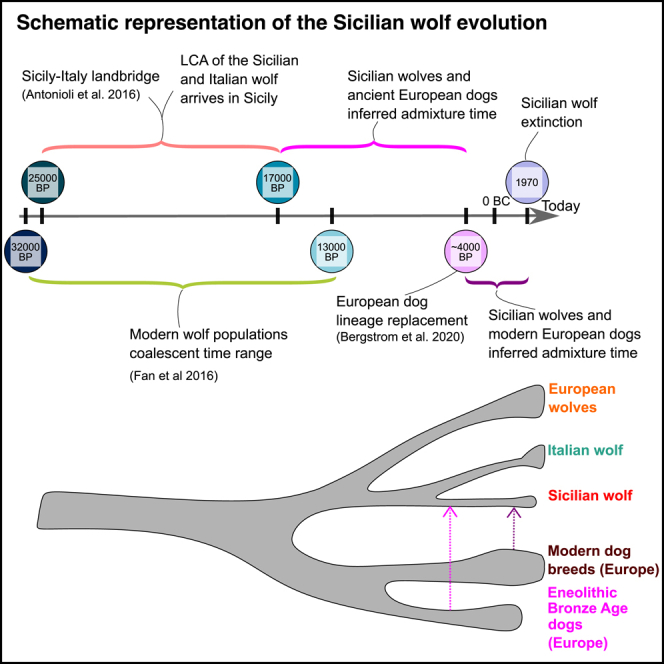
The Italian peninsula was the theater of peculiar population dynamics during LGM. It was a glacial refugium profoundly shaped by sea-level changes. In addition to the recognized local wolf subspecies (C. l. italicus),10,11,12,13 Italy harbored one of the last Mediterranean wolf populations, confined only in Sicily until the mid-1900s.14 The Sicilian wolf remained isolated on the island from the end of the Late Pleistocene (ca. 17kya),15,16 until its human-mediated extermination in the first half of the twentieth century (the 1930s–1960s).17,18,19,20 Today, there are only seven known Sicilian wolf specimens from the 19-20th centuries preserved in museums around Italy. Morphological measurements obtained from the specimens stored in museums and from historical records indicate that this insular population was morphologically distinct from wolves in continental Europe.14,21,22 The Sicilian wolf’s most peculiar features were its small size, the fur tending to light yellow, and the absence of the dark stripes on the forearms, a distinctive character of the peninsular Italic wolf subspecies.23 The minute size of the Sicilian wolf places it among the smallest subspecies of gray wolf, together with the Arab wolf (C. l. arabs), and the extinct Japanese wolf (C. l. hodophilax). Based on these morphometric analyses, the Sicilian wolf has been assigned to the wolf subspecies Canis lupus cristaldii.14 However, despite the modern Italian wolf population being morphologically and genetically well characterized,11,13,24,25,26 few genomes are available from ancient and insular Italian samples27,28 and the genomic identity of the Sicilian wolf remains debated.15,16
To investigate the Sicilian wolf’s genomic ancestry and the potential consequences of the long-term isolation in its genome, we sampled the seven existing museum specimens and were able to recover four nuclear genomes and five mitogenomes (Table S1).
Results and discussion
Reconstructing the genomes of the Sicilian wolves
We sampled the seven existing museum specimens of the Sicilian wolf, from which we recovered four nuclear genomes and five mitogenomes (Table S1). Poor DNA preservation hindered the recovery of genetic material from two of the samples. We achieved an average depth of coverage ranging between 3.8–11.6× for the nuclear genomes and 19.7–1239.2× for the mitochondrial genomes (Table S1). Mapped reads displayed an increase of C to T substitution rates at the 3′ end of the reads, characteristic of sequencing data from historical specimens (Figure S1). For comparative purposes, we sequenced 33 modern wolf genomes (3.6–41.9×) from across Europe and 3 Cirneco dell’Etna dogs (2–2.6×), an old Sicilian hunting breed. We combined our new data with publicly available genomes from dogs and wolves. The final dataset consisted of 57 modern Eurasian and 5 American wolves, 5 Sicilian wolves, 5 Pleistocene wolves, 45 modern dogs, 24 ancient dogs, 4 coyotes (Canis latrans), and 1 Andean fox (Lycalopex culpaeus) (Table S2). The coyote and Andean fox were used as outgroups.
Genetic affinities of the Sicilian wolf
We investigated the placement of the Sicilian wolves in the context of global dog and wolf genomic diversity by performing a multidimensional scaling (MDS) analysis including the Sicilian wolves, modern Eurasian and North American wolves, modern and ancient dogs, and Pleistocene Siberian wolves (Figure 1A).1,3,29 In the resulting plot, the first dimension (Dim1 - 15.42%) separated dogs from wolves, while the second dimension (Dim2 - 3.62%) separated different groups of wolves, with Asian Highland and Italian wolves at each end of the distribution. In the MDS, the Sicilian wolves are placed within the European wolf diversity in proximity to the Italian wolves but shifted toward the cluster of dogs (Figure 1A). An MDS analysis restricting to Eurasian wolves separated the Sicilian wolf from the rest of the wolves in dimension 1, while separating the Italian and Sicilian wolves from the Highland wolf in dimension 2 (Figure S2A). The clear separation of the Sicilian wolves from all other groups in the MDS is consistent with an island population with a history of long-term isolation.
Figure 1.

Population structure of the Sicilian wolves retrieved from genomic data
(A) Multidimensional scaling plot showing the genomic diversity of the Sicilian wolf, ancient and modern wolves, and dogs. Dimensions one (Dim1 - 15.42%) and two (Dim2 - 3.62%) are represented on the x and y axes. Each point indicates a different sample, and colors indicate the broad geographic categories in the legend.
(B) ADMIXTURE plots assuming two (top) and five (bottom) ancestry components. Vertical bars correspond to different samples, and the colors represent the estimated ancestry components and proportions. Horizontal bars in different colors below the ADMIXTURE results indicate the main geographical and temporal groups.
To further explore the ancestry and potential admixture in the Sicilian wolf genomes, we used the model-based clustering approach implemented in ADMIXTURE v1.330 (Figures 1B, S2B and S2C). When assuming two ancestry components (Ks), the samples are mostly separated into dogs and wolves, with the Sicilian wolves being modeled as a mixture of both components. This dual admixture pattern was not exclusive to the Sicilian wolves. However, the Sicilian wolf displayed the largest proportion of the dog-related component, only after the Sierra Morena wolf,31 which derives one-third of its genome from dog ancestry. The dog ancestry component remained in both the Sicilian and Sierra Morena wolves after increasing the number of components to 3 and 4 (Figure S2B). This admixture pattern is consistent with the MDS analysis (Figure 1A), where the Sicilian and Sierra Morena wolves are shifted toward the cluster of dogs, suggesting the Sicilian wolf might also carry dog ancestry. When assuming five ancestry components, we identified two components mostly present among dogs, two mostly present among wolves and a fifth component shared by the Italian and Sicilian wolves (Figure 1B). This similarity between the Sicilian and Italian wolves is in agreement with our mitochondrial phylogeny and previous mitochondrial DNA haplogroup studies, which showed the Sicilian wolves are placed in the same clade as the Italian and Eastern European wolves (Figure S2E; see also mitochondrial phylogeny methods section).15,16 As we increased the number of ancestry components (K = 7–20), the Sicilian wolves were assigned their own cluster (Figure S2B). Overall, the MDS and ADMIXTURE analyses suggested that the Sicilian wolf is a genetically differentiated population most similar to the Italian wolf but has a genetic affinity to dogs.
The dual wolf-dog ancestry of the Sicilian wolf
We further explored whether the patterns observed in the MDS and ADMIXTURE analyses were due to the Sicilian wolf being a highly drifted population or dog admixture. We used outgroup f3-statistics, which measure the shared genetic drift between pairs of samples, to identify the closest populations to the Sicilian wolf. We found that the Sicilian wolf was most closely related to ancient and modern European dogs, followed by modern Italian wolves (Figure 2A). Among modern dogs, the Italian dogs, such as Spinone, Cane Corso, and Cirneco, had the highest genetic affinity to the Sicilian wolf (Figures 2A and S3A–S3D). Recent introgression of stray dog ancestry present on the island could explain the high affinity between the Sicilian wolf and modern Italian dogs. However, the high affinity between the Sicilian wolf and ancient Eneolithic and Bronze Age dogs (∼5000–3000 BP; e.g., Croatian ALPO01, Italian AL2397, Irish Dog-1PU, and Newgrange) suggests a more ancient origin for this link. We observed a similar pattern in all four Sicilian wolf genomes, with the Sicilian wolf 1 being particularly close to ancient European dogs (Figures 2A and S3A–S3D). In contrast, outgroup f3-statistics for the Italian and Iberian wolves, which are the closest to the Sicilian wolf (Figure 1) and share a similar history of recent isolation and bottlenecks, did not show the same pattern (Figures S3E and S3F).
Figure 2.

Ancestry composition of the Sicilian wolf
(A) Outgroup f3-statistics of the form f3(Andean fox; Sic1, X), showing the genetic affinity between the Sicilian wolf (Sic1) and other dogs and wolves (X). Names are indicated for the samples with the highest affinity to the Sicilian wolves (highest f3). Error bars correspond to 3.3 standard errors estimated through a block jackknife approach. Corresponding plots for the other Sicilian wolf genomes are shown in Figure S3.
(B) D-statistic tests of the form D(Sic1, Italian wolf, H3, Andean fox) (x axis), and D(Sic7, Italian wolf, H3, Andean fox) (y axis), where H3 corresponds to different groups of dogs and wolves. Each point corresponds to a different test where H3 varies. Error bars correspond to 3.3 standard errors estimated through a block jackknife approach. All tests yielded significant results (Z score >3.3) suggesting gene flow between the Sicilian wolf and the corresponding dog in each test. A dotted line indicates the identity line, with points above the line showing tests where the Sicilian wolf 7 had higher affinity to H3 compared to Sicilian wolf 1.
(C) TreeMix admixture graph incorporating 3 migration edges (arrows): from ancient southern European dogs into the Sicilian wolf, from a Mesolithic Swedish dog (C89) into the Alaskan Malamute, and from the Middle eastern dog into ancient southern European dogs. The corresponding TreeMix residuals are shown in Figure S5.
We then used D-statistics to formally test for gene flow between the Sicilian wolves and modern and ancient dogs. We tested if the Sicilian wolf shared more alleles with dogs compared to its closest wolf (the Italian wolf) (Figures 2A and 2C). Our results showed that the Sicilian wolf shares significantly more alleles with dogs compared to the Italian wolf (Z score >3.33). Furthermore, ancient European dogs yielded the largest D values (Figure 2B), confirming that ancient European dogs are the closest to the Sicilian wolf among dogs. A similar result was obtained for all four Sicilian wolf genomes (Figures S4A and S4B). However, when comparing the D-statistics obtained for different pairs of Sicilian wolves, we found each has a different degree of affinity to dogs suggesting they have varying dog ancestry proportions. Two scenarios could explain these observations: gene flow (ancient, recent, or both) from dogs into the Sicilian wolf, or the Sicilian wolf being closer to a now extinct (and unsampled) wolf lineage that contributed to ancient European dogs. While the second scenario is supported by mtDNA based phylogenetic studies,27,28 which suggest genetic continuity between Late Pleistocene wolves in Italy and dogs, our results support the first scenario. The D-statistics showed significant gene flow between the Sicilian wolf and dogs, regardless of the dog used in the test (Figures 2B and S4A). If the gene flow direction were from the Sicilian wolf into specific dog breeds, we would expect to observe significant values only for those dog breeds carrying Sicilian wolf ancestry. Furthermore, these results were consistent when using the Portuguese wolf instead of the Italian wolf in the D-statistic test (Figure S4E). Stray dogs have been common in Sicily, potentially since their introduction to the island, making it possible that the dog ancestry in the Sicilian wolf could derive from recent admixture. We used D-statistics to determine whether an ancient or modern dog was a better source for the dog ancestry in the Sicilian wolf. We computed D (Eneolithic Croatian dog, X; Sicilian wolf, Andean fox) to estimate the amount of shared derived alleles between the Sicilian wolf and the Eneolithic Croatian dog (ALPO01) compared to all other dogs in the dataset (X). Then, we computed the same test substituting the Eneolithic Croatian dog for the Cirneco dog and compared the results of both tests (Figure S4B). We found that in most of the tests, the D-statistic obtained with the Eneolithic Croatian dog is larger than that obtained with the Cirneco dog, suggesting the former is a better source for the dog ancestry of the Sicilian wolf.
To place the Sicilian wolves in the broader phylogenetic context of modern and ancient wolves and dogs while allowing for potential admixture events, we used TreeMix.32 The obtained graph without migration edges was consistent with a nuclear phylogeny estimated using RAxML33 (Figure S2D), placing the Sicilian wolves as intermediate between the dog and wolf clades (Figure S5A). Once migrations were incorporated, the Sicilian wolves were modeled as a sister clade to the Italian wolves with ancestry contribution from the Croatian Eneolithic dog lineage into the base of the Sicilian wolf clade (Figures 2C and S5). These results were consistent with f-statistics-based admixture graphs that showed the Sicilian wolf can be modeled as a mixture of ancient dog (∼31–49%) and Italian wolf (∼51–69%) ancestries (Figures S4C and S4D).
To further validate our results and to measure the extent of the dog ancestry in the Sicilian wolves, we performed local ancestry inference using PCAdmix.34 We modeled each Sicilian wolf genome as a mixture of dog and wolf ancestry using nine dogs and ten wolves as sources. Our results showed that the Sicilian wolves carry between ∼50-60% wolf and ∼50-40% dog ancestry (Figure S6), in agreement with the admixture graph modeling (Figures S4C and S4D). Altogether, these results showed that the Sicilian wolf is a mixture of two ancestries: wolf (most similar to the modern Italian wolf) and dog (most similar to ancient European dogs).
In light of our results, we hypothesize that geographic isolation, a small population size, and the introduction of dogs to Sicily, provided a fertile ground for hybridization and retention of ancient dog ancestry in the Sicilian wolf. A previous study on Sierra Morena wolves (C. l. signatus) found that one-third of their genomes were of dog ancestry.31 The Sierra Morena and the Sicilian wolves have similar demographic histories, being a small, isolated, and declining population in a human-modified landscape. A similar pattern of admixture with modern dogs was documented in one of the last specimens of Japanese wolves (C. l. hodophilax), which became extinct at the same time as the Sicilian wolf.35,36 Thus, our findings further show that admixture with dogs might be favored in low-density isolated populations in anthropogenic contexts. The affinity between Sicilian wolves and ancient European dogs represents the most striking finding, as it shows that ancient dog ancestry prevailed until recently in the Sicilian wolf genome. Overall, our results showed that the dual ancestry of the Sicilian wolf genome resulted from a long-term multi-factor evolutionary process involving geographic isolation and admixture with dogs, potentially influenced by a drastic population reduction due to resource competition and direct conflict with humans.
The effects of island isolation on the Sicilian wolf
We explored the insular effect and the consequences of the population decline in the last few hundred years on the genomic diversity of the Sicilian wolf genome. We estimated the average nucleotide diversity of the Sicilian wolf and European wolves for comparative purposes. The Sicilian wolf displayed low-average nucleotide diversity compared to the Italian, Iberian, and Scandinavian wolves (Figure 3A). Similarly, heterozygosity estimates for the two highest coverage Sicilian wolf genomes were low (Figure 3B). A decrease in diversity is a characteristic of island populations. Therefore the Sicilian wolf genomic diversity is consistent with the expectations from a population that was shaped by a strong founder effect, long-term isolation, and small effective population size.37,38,39,40
Figure 3.

Island isolation impact on the Sicilian wolf genomes
(A) Population level nucleotide diversity of Sicilian, Italian, Iberian, and Scandinavian wolves. The chromosomes and the nucleotide diversity values are shown on the x and y axis respectively. The red line represents the mean diversity value.
(B) Heterozygosity in sliding windows (excluding repetitive regions) for different wolf samples show reduced levels of heterozygosity in the Sicilian wolves Sic1 and Sic2.
(C) Individual runs of homozygosity represented as the number of ROH segments on the y axis and fraction of the genome contained in ROH (Mbp) on the x axis.
(D) Histogram showing the distribution of ROH segments of different size categories. The Sicilian wolves (Sic1 and Sic2) show a high number of short ROHs.
(E) Genetic load represented as the number of transversion sites that are homozygous is shown on the y axis compared to the proportion of the genome in ROH (FROH). The Sicilian wolves display a higher transversion load compared to other wolf populations.
To get insights into the distribution of low-heterozygosity regions in the Sicilian wolf genome we estimated runs of homozygosity (ROHs) for the two highest coverage samples. ROHs abundance and their length can be informative about population sizes and inbreeding.41 Our results showed that 37–50% of the Sicilian wolf genome can be found in ROHs with average ROHs lengths of 9.5Mb and 6.6Mb for Sicilian wolf 1 (Sic1) and 2 (Sic2), respectively (Figures 3C–3E). While long ROHs (>10Mb) were still quite prevalent in the Sicilian wolves (15 and 30% from total ROHs), the number of short and medium ROHs was higher than those observed in recently inbred populations such as the Mexican wolf (C. l. baileyi) (Figures 3C and 3D). We found the Sic2 had more ROHs compared to the Sic1. Notably, our D-statistic tests showed the Sic1 had a higher proportion of dog ancestry compared to Sic2. Admixture between two distinct populations leads to an increase in heterozygosity, thus it is possible that the smaller number of ROHs in Sic1 is due to its higher proportion of dog ancestry. Compared to the Italian wolf, its genetically closest wolf, the Sicilian wolf had a larger number of ROHs (Figure 3C) but their size distribution was similar (Figure 3D). A larger proportion of shorter ROHs could be the result of long-term reduction in population size of the Sicilian wolf, combined with influx of ancestry from dogs, which would break the larger ROH blocks.
To measure the potential impact of the small population size in the fitness of the Sicilian wolf compared to mainland populations we estimated their homozygous transversion load.42 The Sicilian wolf had a relatively higher mutation load compared to other wolves (particularly Sic2), higher than the mutation load in the inbred Mexican wolf (Figure 3E). Combined, these results indicate that the Sicilian wolf population was suffering from the effects of small population size already at the end of the 19th century. Finally, our results showing the Sicilian wolf carried ancient dog ancestry together with the short ROH segments, suggest the admixture happened in the past, rather than toward the time of extinction.
Conclusion
The genomes of some of the last surviving Sicilian wolves showed that their evolutionary history was profoundly shaped by admixture with dogs. Our results showed they derive from the same lineage as modern Italian wolves but carry ancient Eneolithic and Bronze Age dog ancestry. Such ancient dog ancestry was shown to be widespread in Europe until 3-4kya.43 Our results suggest this ancient dog lineage survived in the Sicilian wolf until the 20th century. In this respect, the Sicilian wolf is unique among all other extant and extinct European wolf populations genetically characterized to date. The phylogenetic placement of the Sicilian wolf placed together with the Italian wolf (Figure 2C), coupled with the evidence for long-term isolation, supports that by 25-17 kya (the time of the colonization of Sicily), the Italian and Sicilian wolves’ ancestor had already split from other European wolf lineages.44 Previous genomic studies estimated that contemporary wolf populations share a common ancestor between 32 and 13 kya.1,29,45 Our results provide a lower limit for the split of wolves from their last common ancestor, suggesting that contemporary populations were already differentiated by the time of the colonization of Sicily (25kya). Finally, these results clarified previous hypotheses based on mitochondrial haplogroups, suggesting the Sicilian and Italian wolves form a clade with the Pleistocene Siberian wolves.15 Instead, our f3-statistic and MDS analyses showed that Siberian Pleistocene wolves were not closely related to the Sicilian wolf (Figures 2A and S3A–S3D).
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| 5 Sicilian wolves (historical remains - bones and skin) | This paper | Sic1 (C11875), Sic2 (AN/855), Sic3 (M/18), Sic4 (TSA / 9), Sic7 |
| 3 Cirneco dell'Etna dogs (swabs) | This paper | MW1085_CIR1, MW1095_CIR11,MW1098_CIR14 |
| 4 Italian modern wolf (tissue) | This paper | MW303 (14b), MW305 (16b), MW307 (19a), MW310 (TS86a) |
| 2 Norwegian modern wolf | This paper | MW005 (M406281), MW011 (M495008) |
| 2 Finnish modern wolf | This paper | MW347 (13172), MW423 (SF1695) |
| 2 Lithuanian modern wolf | This paper | MW020 (CL9), MW366 (LT3) |
| 2 modern wolf from Czech Republic | This paper | MW045 (CK), MW333 (KRNAP) |
| 2 Slovakian modern wolf | This paper | MW339 (20/0047_2), MW049 (CL693) |
| 2 German modern wolf | This paper | MW055 (W150470), MW098 (W181390) |
| 2 Romanian modern wolf | This paper | MW062 (W151402), MW066 (W151413) |
| 2 Portuguese modern wolf | This paper | MW119 (L474), MW107 (L090) |
| 2 Spanish modern wolf | This paper | MW122 (L546), MW127 (L590) |
| 1 Austrian modern wolf | This paper | MW515 (1032) |
| 1 modern wolf from Bosnia and Herzegovina | This paper | MW703A (5755 - CL231) |
| 1 Bulgarian modern wolf | This paper | MW706A (7386 - CL422) |
| 2 Polish modern wolf | This paper | MW259 (BIE31), MW295 (PSN05) |
| 3 Croatian modern wolf | This paper | MW1118 (1231), MW1120 (2736), MW1126 (7201) |
| 1 modern wolf from Greece | This paper | MW1135 (8571) |
| 2 modern wolf from Latvia | This paper | MW1151 (10686), MW1152 (10687) |
| Chemicals, peptides, and recombinant proteins | ||
| Proteinase K | Sigma-Aldrich | Cat#3115844001 |
| Critical commercial assays | ||
| MinElute PCR Purification Kit | QIAGEN | Cat#28006 |
| PfuTurbo Cx Hotstart DNA Polymerase | Agilent | Cat#600414 |
| T4 DNA ligase | New England Biolabs Inc. | Cat#M0202L |
| T4 Polynucleotide Kinase | New England Biolabs Inc. | Cat#M0201L |
| T4 DNA Polymerase | New England Biolabs Inc. | Cat#M0203S |
| BSt 2,0 warmstart polymerase | New England Biolabs Inc. | Cat#M0538S |
| Phusion® High-Fidelity PCR Master Mix with HF buffer | New England Biolabs Inc. | Cat#M0531S |
| DNeasy® Blood and Tissue Kit | Qiagen | Cat#69504 |
| Deposited data | ||
| 5 Sicilian wolves (historical remains - bones and skin) | This study | Table S2; ENA accession number: PRJEB57290 |
| 3 Cirneco dell'Etna dogs | This study | Table S2; ENA accession number: PRJEB57290 |
| 33 modern european wolves | This study | Table S2; ENA accession number: PRJEB57290 |
| 1 modern Andean fox (Lcu2_Pastora) | Auton et al.46 | Table S2 |
| 4 Coyotes | vonHoldt et al.47 Gopalakrishnan et al.48 |
Table S2 |
| 10 Tibetan and Chinese modern wolves | Zhang et al.49; Wang et al.50 | Table S2 |
| 1 Greenlandic wolf | Gopalakrishnan et al.48 | Table S2 |
| 4 North American and Mexican wolves | Fan et al.1; Sinding et al.51 Gopalakrishnan et al.48 |
Table S2 |
| 3 Iberian wolves | Gómez-Sánchez et al.31; Fan et al.1 | Table S2 |
| 1 Croatian wolf | Freedman et al.29 | Table S2 |
| 2 Italian wolves | Fan et al.1 | Table S2 |
| 3 Russian wolves | Wang et al.52 | Table S2 |
| 1 Syrian wolf | Gopalakrishnan et al.48 | Table S2 |
| 1 Saudi Arabian wolf | Gopalakrishnan et al.48 | Table S2 |
| 1 Israeli wolf | Freedman et al.29 | Table S2 |
| 1 Iranian wolf | Fan et al.1 | Table S2 |
| 1 Indian wolf | Fan et al.1 | Table S2 |
| 5 Pleistocene wolves | Sinding et al.53; Ramos-Madrigal et al.3 | Table S2 |
| 24 ancient dogs | Frantz et al.54; Botigué et al.55; Ní Leathlobhair et al.56; Bergström et al.43; Feuerborn et al.57 | Table S2 |
| 42 modern dogs | Wiedmer et al.58; Decker et al.59; Metzger et al.60; Auton et al.46; Wang et al.52; Wang et al.50; Sinding et al.53; Freedman et al.29; Marsden et al.61; Lindblad et al.62 | Table S2 |
| Oligonucleotides | ||
| Illumina-compatible adapters | Meyer and Kircher63 | N/A |
| BGI-compatible adapters | Mak et al.64 | N/A |
| Software and algorithms | ||
| PALEOMIX v1.2.13.1 | Schubert et al.65 | https://github.com/MikkelSchubert/paleomix |
| AdapterRemoval2 | Schubert et al.66 | https://github.com/MikkelSchubert/adapterremoval |
| bwa-backtrack v0.7.15 | Li and Durbin67 | http://bio-bwa.sourceforge.net/ |
| Picard v2.9.1 | N/A | https://broadinstitute.github.io/picard |
| GATK v4.1 | McKenna et al.68 | https://software.broadinstitute.org/gatk/ |
| ANGSD 0.931 | Korneliussen et al.69 | https://github.com/ANGSD/angsd |
| samtools v1.9 | Li et al.70 | http://samtools.sourceforge.net/ |
| ADMIXTOOLS v.5.1 | Patterson et al.71 | https://github.com/DReichLab/AdmixTools |
| TreeMix v.1.13 | Pickrell and Pritchard32 | https://bitbucket.org/nygcresearch/treemix/wiki/Home |
| mapDamage 2.0 | Jónsson et al.72 | https://github.com/ginolhac/mapDamage |
| MUSCLE | Edgar73 | https://github.com/rcedgar/muscle |
| Plink 1.9 | Purcell et al.74 | https://www.cog-genomics.org/plink/2.0/ |
| ADMIXTURE v.1.3 | Alexander et al.30 | http://dalexander.github.io/admixture/download.html |
| RAxML-ng | Kozlov et al.33 | https://github.com/amkozlov/raxml-ng |
| FigTree v.1.4.4 | Rambaut75 | http://tree.bio.ed.ac.uk/software/figtree/ |
| BEDtools | Quinlan and Hall76 | https://github.com/arq5x/bedtools2 |
| IQ-TREE v.2.1.2 | Minh et al.77 | http://www.iqtree.org/ |
| Astral III | Zhang et al.78 | https://github.com/smirarab/ASTRAL |
| qpBrute | Leathlobhair et al.56, Liu et al.79 | https://github.com/ekirving/qpbrute |
| PCAdmix | Brisbin et al.34 | N/A |
| VCFtools v.0.1.16 | Danecek et al.80 | https://vcftools.sourceforge.net/ |
| Rstudio | Allaire81 | https://github.com/rstudio/rstudio |
| Beagle v4.1 | Browning and Browning82 | https://faculty.washington.edu/browning/beagle/b4_1.html |
| Beagle v5.4 | Browning et al.83 | http://faculty.washington.edu/browning/beagle/beagle.html |
| ROHan | Renaud et al.84 | https://github.com/grenaud/ROHan |
| Variant Effect Predictor | Watterson85 | https://www.ensembl.org/info/docs/tools/vep/index.html |
| all_genotype_probability_v3.py | Greer42 | https://github.com/sagitaninta/mutationLoad and https://github.com/DebGreer/MSc_Project |
Resource availability
Lead contact
Further information and requests of resources and reagents should be directed to and will be fulfilled by the lead contact, Marta Maria Ciucani (ciucani@palaeome.org).
Materials availability
This study did not generate new unique reagents.
Method details
Description of the historical Sicilian samples
The sample Sic1 (cat number C11875) was a petrous bone from a wolf skull preserved at the Museum of Natural History, Section of Zoology ‘La Specola’, University of Florence, Florence, Italy. The sample still has the original tag from the 19th century, and it belongs to a wolf killed in 1883 at Vicari (Palermo, PA). The sample Sic2 (cat number AN/855) is represented by a petrous bone from a wolf skull belonging to a juvenile individual from 1879 preserved at the Museum of Zoology ‘Pietro Doderlein’ of the University of Palermo, Palermo, Italy. The sample Sic3 (cat number M/18) is a mounted specimen of an adult male. The individual Sic2 was killed in Sicily, presumably around 1870-1880, as documented by an old picture in the museum records. The specimen is preserved in the Museum of Zoology ‘Pietro Doderlein’ of the University of Palermo, Palermo, Italy. The individual Sic4 (cat number 9) is represented by a hide labelled as an adult wolf that was shot in 1924 at Bellalampo (Palermo, PA), and it is preserved in the Regional Museum of Terrasini (Palermo, PA), Italy. The last sample, Sic7 (cat number NA), is a hide dated around 1880-1920 and is preserved at the Museum of Termini Imerese (Palermo, PA), Italy.
Data generation for the historical Sicilian samples
The Sicilian wolf samples were processed under strict clean laboratory conditions at the Globe Institute, University of Copenhagen. DNA extractions were performed following the silica-based protocol described in Dabney et al. (2013)86 for the bone samples and Campos & Gilbert (2009)87 for keratin samples (hides and claws). Both bone and keratin samples were digested using 1 mL of digestion buffer. The extracts were purified using modified PB buffer (Qiagen), washed twice with PE buffer (Qiagen) and eluted twice in 20 μL of buffer EB - with 10 minutes of incubation time at 37°C. The concentration of each extract was checked on a Qubit Fluorometer (ThermoFisher Scientific) in ng/μL and on the Tapestation (Agilent Technologies) for concentration and fragment size.
BGI libraries for the samples Sic1 and Sic4 were constructed following Carøe et al. (2018)88 and Mak et al. (2017)64,89 while BEST Illumina libraries for Sic2, Sic3 and Sic7 were built following Carøe et al. (2018) using Illumina adapters.88 Libraries were prepared using up to 32 μL of DNA in a final reaction volume of 80 μL.
The appropriate number of cycles for the amplification was determined using Mx3005 qPCR (Agilent Technologies) in which 1 μL of SYBRgreen fluorescent dye (Invitrogen, Carlsbad, CA, USA) was loaded in 20 μL indexing reaction volume also using 1 μL of template, 0.2 mM dNTPs (Invitrogen), 0.04 U/μL AmpliTaq Gold DNA polymerase (Applied Biosystems, Foster City, CA, USA), 2.5 mM MgCl2 (Applied Biosystems), 1X GeneAmp® 10X PCR Buffer II (Applied Biosystems), 0.2 μM forward and reverse primers mixture,64 and 16.68 μL AccuGene molecular biology water (Lonza, Basel, CH). qPCR cycling conditions were 95°C for 10 minutes, followed by 40 cycles of 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 45 seconds.
The libraries were amplified using PfuTurbo Cx HotStart DNA Polymerase (Agilent Technologies) and Phusion® High-Fidelity PCR Master Mix with HF buffer (New England Biolabs Inc). The amplification was performed in 50 μL PCR reactions that contained:
-
-
14 μL of purified library, 0.1 μM of each forward (BGI 2.0) and custom made reverse BGI primers, 2x Phusion® High-Fidelity PCR Master Mix with HF buffer and 8.6 μL AccuGene molecular biology water.
-
-
10 μL of purified library, forward and reverse primers, 2.5U/L PfuTurbo Cx HotStart DNA Polymerase, 0.4 BSA, 1 μL of dNTPs (25 μM), 1 μL of Buffer 10X and 30.6 μL AccuGene molecular biology water.
PCR cycling conditions for libraries amplified using Phusion® High-Fidelity PCR Master Mix with HF buffer were: initial denaturation at 98°C for 45 seconds followed by 18 to 20 cycles of 98°C for 20 seconds, 60°C for 30 seconds, and 72°C for 20 seconds, and a final elongation step at 72°C for 5 minutes. Amplified libraries were then purified using 1.5x ratio of SPRI beads to remove adapter dimers and fragments smaller than 90-100bp and eluted in 50 μL of EB (Qiagen) buffer after 10 minutes of incubation at 37°C. The samples Sic1 and Sic4 were sequenced using BGIseq platform, while the samples Sic2, Sic3 and Sic7 were pooled together and sequenced on Novaseq6000 Illumina platform, S2 flow-cell PE50.
Data generation for modern samples
Cirneco dell’Etna buccal swab samples - DNA sample collection, storage and extraction
SK-1S Isohelix buccal swabs for non-invasive DNA collection were used to sample three Cirneco dell’Etna individuals (MW1085_CIR1, MW1095_CIR11, MW1098_CIR14). The dogs were sampled by their owners at least 30 minutes after eating, and to avoid contamination, gloves and face masks were used. The swabs were inserted into the dogs’ mouths, rubbed on their cheek for ca. 1 minute and placed back into the collector tube with the SGC-50 Isohelix Dri-capsule to preserve and stabilise the DNA at room temperature during shipping. Once received in the Modern DNA labs of the Globe Institute (University of Copenhagen), the samples were stored at -20°C.
Buccal swab samples were placed in 2 mL Eppendorf tubes and were extracted using a modified version of the DNeasy® Blood and Tissue Kit (Qiagen). In each tube were added 380 μL of ATL Buffer and 20 μL of Proteinase K (Roche). The samples were placed in a thermomixer for 1 hour at 56°C. The lysate was transferred in a different tube and 400 μL of AL Buffer, and 400 μL of 96% Ethanol were added. The extraction reaction was then spun down in a DNeasy Mini column, and the filter was washed with 500 μL of Buffer AW1 and 500 μL of Buffer AW2. The DNA was eluted twice using 50 μL of AE Buffer directly onto the DNeasy Mini column membrane.
Modern Italian wolves tissue samples - DNA sample collection, storage and extraction
Four Italian wolf samples were collected around 2010 and 2012 from road-killed individuals populating the Simbruini Mountain Range Regional Park and National Park of Abruzzo in the Central Apennines (See Table S2). The muscle samples were stored in ethanol at -20°C and subsequently processed in the “Modern DNA laboratory” at the Fondazione Edmund Mach (FEM). Small pieces of tissue of around 25 mg were extracted using the DNeasy Blood and Tissue Kit (Qiagen) with overnight digestion at 56°C. The elution was conducted at the GLOBE Institute (University of Copenhagen) using two washes of 50 μL of AE buffer, with 10 minutes of incubation at 37°C. Until the elution, samples were stored at -20°C inside the DNeasy Mini spin columns.
Modern wolves - DNA sample collection, storage and extraction
Twenty-nine modern wolf (tissue or blood) samples from several locations in Europe (Table S2) were extracted using the Thermo Scientific KingFisher instrument and following the manufacturer’s protocol. The concentration of the samples was measured with a Qubit Fluorometer (ng/μL). All the extracts, with the exception of the modern Italian wolves and the Cirneco dell’Etna, were sent to BGI Copenhagen for library build and sequenced on ⅛ of a lane each on DNBSEQ PE150.
Library build, amplification and sequencing of modern Italian wolves and Cirneco dogs
Extracts were fragmented in the Covaris LE220 plus Focused-ultrasonicator with the parameters set for getting 350-bp fragment length. The extracts were diluted to obtain 100 ng concentration and BGI libraries for the Italian wolves and Cirneco dogs were constructed following Carøe et al. (2018)88 and Mak et al. (2017)64 using 10 μM adaptors. Libraries were purified using MinElute columns using PE buffer (Qiagen) and eluted in 60 μL of EB buffer.
The PCR mixture for the Italian wolf libraries consisted of: 20 μL of purified library, 0.2 μM of forward and reverse BGI primers, 2.5 U/μL PfuTurbo Cx HotStart DNA Polymerase, 10 μL of Buffer 10X, 0.08 mg/mL BSA, 0.5 mM of dNTPs (25 μM) and 61.2 μL AccuGene molecular biology water (Lonza, Basel, CH). The Cirneco dog libraries were amplified using 20 μL of purified library, 0.2 μM of forward and reverse BGI primers, 0.05 U/μL of AmpliTaq Gold (Thermo Fisher Scientific, USA), 0.4 mg/mL BSA, 0.2 mM of dNTPs (25 μM), 10 μL of Buffer 10X, 2.5mM of MgCl2 and 50.2 μL AccuGene molecular biology water in a total volume of 100 μL reaction.
The amplification of the Italian wolves was performed as follows: initial denaturation at 95°C for 2 minutes followed by 10 to 12 cycles of 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 110 seconds, and a final elongation step at 72°C for 10 minutes. PCR cycling conditions for Cirneco dog libraries were: initial denaturation at 95°C for 10 minutes followed by 12 cycles of 95°C for 20 seconds, 60°C for 30 seconds, and 72°C for 45 seconds, and a final elongation step at 72°C for 5 minutes. The Italian wolves and Cirneco dogs were sequenced on ⅛ of a lane each on MGIseq2000 PE150 and DNBSEQ PE150, respectively.
Quantification and statistical analysis
Data processing and historical DNA authentication
Short sequencing reads from modern, historical, and ancient samples were mapped to the dog reference genome (CanFam3.1)62 using Paleomix v.1.2.13 pipeline.65 First, adapter sequences were removed using AdapterRemoval 2.066 with default settings, and BWA v0.7.12 backtrack67 was used to perform the alignment of reads to the dog genome (bwa seed was disabled) setting the minimum base mapping quality to 0 to retain all the reads in this step. In further computational steps, reads with mapping quality lower than 20 or 30 (as specified in each analysis) were discarded. Picard MarkDuplicates v2.9.190 was used to identify and remove PCR duplicates and GATK v4.1.091,68 was used to perform the indel-realignment step. For the historical Sicilian samples in this study the post-mortem DNA profiles and misincorporation patterns were estimated through mapDamage2.072 (Figure S1).
Sex determination of the Sicilian wolves
We determined the genetic sex of four Sicilian wolves, by comparing the average genomic coverage with the coverage of the X chromosome. The samples Sic1, Sic2 and Sic3 were identified as males (XY) while Sic7 was shown to be female (XX) (Table S1).
Mitochondrial phylogeny
We investigated maternal lineage of the Sicilian wolves generating a mitochondrial phylogeny using RAxML-ng33 including all the wolves and dogs in our dataset and using the coyote as the outgroup. The Maximum-Likelihood mitochondrial phylogeny is consistent with previous publications15,16 and four out of the five Sicilian wolves (Sic1, Sic2, Sic3 and Sic7) cluster together in the phylogeny as a sister clade to wolves from Romania and Slovakia (Figure S2D). They also fall within the same clade as wolves from Italy, Bulgaria, and Poland. The last sample, Sic4, falls within the dog mitochondrial diversity, as shown in a previous work based on the d-loop region.15 Unfortunately, we did not recover sufficient nuclear data from Sic4 to further investigate its ancestry. However, the sample was labelled as Canis lupus, but morphological analysis identified dog features, suggesting a hybrid origin. Our mitochondrial phylogenetic analysis placing Sic4 within dogs, is in agreement with the hybrid origin or, alternatively, it might suggest a potential mislabelling.
Dataset
The dataset compiled for this study is represented by 146 samples, including one Andean fox (Lycalopex culpaeus),46 4 coyotes (Canis latrans),5,47 62 modern wolves (Canis lupus) from Eurasia and North America,1,5,29,31,49,50,51,52,53 45 modern dogs (Canis lupus familiaris),29,62,46,50,52,53,59,60,61,92,93 24 ancient dogs (Canis lupus familiaris),43,53,54,55,56,57 5 Pleistocene wolves3,53 and 5 newly sequenced Sicilian wolves (Canis lupus cristaldii) (Table S2). In this dataset, 33 modern wolves and 3 Cirneco dogs were newly sequenced as part of this study.
Variant calling
In order to account for the varying depth of coverage of the samples and to incorporate the low-coverage ancient samples, we used a pseudo-haploid dataset instead of performing genotype calling. For each modern sample at each genomic site, we sampled a random read using ANGSD v0.93169 (-doHaploCall 1) from the reads with a minimum mapping quality of 30 (-minMapQ 30) and bases with a minimum quality of 20 (-minQ 20). Additionally, the following parameters were used: -minMinor 1 -maxMis 10 -skipTriallelic 1 -doMajorMinor 2 -C 50 -baq 1 -remove_bads 1 -only_proper_pairs 1 -rmTrans 1. Transitions were discarded (-rmTrans 1) to reduce the aDNA-derived error in the historical samples included in our dataset. The output file was converted into Plink files using the haploToPlink tool from ANGSD. Once the SNPs were chosen based on the modern genomes, we used ANGSD with -doHaploCall (option -s) to obtain pseudo-haploid calls for the ancient samples, historical samples, and outgroups (Coyote and Andean fox). The pseudo-haploid calls for the modern, ancient, and historical genomes were merged using Plink. The resulting dataset consisted of 74.5 million SNPs.
Plink v1.974 was used to further filter the pseudo-haploid panel. We kept sites with a minimum allele frequency of 5% (-maf 0.05) and minimum genotyping rates of 25% (-geno 0.25). Finally, we performed a linkage disequilibrium (LD) pruning in windows of size 10kb (--indep-pairwise 10kb 2 0.5). After filtering, the pseudo-haploid panel consisted of 2.3 million. This dataset was used in the following analyses: ADMIXTURE, MDS, f3-statistics, D-statistics, TreeMix and f4-statistics-based admixture graphs. From here on, we refer to this dataset as “pseudo-haploid SNP panel”.
MDS
The pseudo-haploid SNP panel described in the previous section (146 samples and a total of 2.3 million transversion sites) and Plink v1.9 –mds-plot74 option were used to perform a multidimensional scaling (MDS) analysis. We used Plink v1.9 –mds-plot option on two subsets of the samples: a subset including all modern and ancient wolf and dog samples (Figure 1A) and a second subset restricting to only modern and ancient wolves (Figure S2A).
Admixture
The pseudo-haploid SNP panel, restricted to modern and ancient wolves and dogs, was used to run ADMIXTURE v.1.330 in order to estimate their ancestry components. The outgroups were not included in this analysis. ADMIXTURE was run assuming 2 to 20 ancestry components (K2 to K20). We ran 50 independent replicates for each value of K keeping the results from the replicate with the best likelihood. To explore which K best fitted out data, we performed cross-validation (--cv) for the best replicate for each K (Figure S2C). The results from the cross-validation procedure showed K=2 is the K that best fits our dataset. Therefore, we show the results from K=2 in Figure 1. Additionally, we show the results obtained with K=5 to show that the Sicilian wolf clusters with the Italian wolf, which is consistent with MDS and f3-statistic analyses (Figures 1A and 2A). Pong94 was used to visualise the admixture plots.
Mitochondrial and nuclear phylogenies
We built a mitochondrial phylogeny including 131 the samples in the dataset and using the coyote as the outgroup. First, we extracted the reads mapping to the chrMT from the genomic alignments using samtools.70 Then, we built a majority-count consensus sequence using ANGSD (-dofasta 2). The mitochondrial sequences for each sample were combined and aligned using MUSCLE.73 Finally, a ML tree was built using RAxML-ng,33 setting the evolutionary model to GTR+G and running 500 bootstrap replicates. The resulting tree was visualised using FigTree v.1.4.475 (Figure S2E).
To build the nuclear phylogeny on all the modern and ancient individuals in the dataset, we first used ANGSD to generate a random-read sequence for each genome using the dog reference genome (CanFam3.1)62 as reference and sampling a random reads at each genomic site (-dofasta1). Bases with base quality lower than 20 and reads with mapping quality lower than 20 were discarded (-minQ 20 -minmapq 20). The minimum coverage for each individual was set to 3x (-setminDepthInd 3), and the following additional filters were used: -doCounts 1 -remove_bads 1 -uniqueOnly 1 -baq 1 -C 50. Then, from autosomal chromosomes, we selected 1000 random regions, each 5000bp long, from the dog reference genome using BEDTools76 random and the following parameters: -l 5000 -n 1000. We used samtools70 to select from each sample the regions of interest and combined them into a multi-sequence alignment (MSA) fasta file. Each MSA file was used in IQ-TREE77 v.2.1.2 to reconstruct a phylogeny using 1000 bootstrap replicates (-B 1000) with UFBoot2,95 1000 bootstrap replicates for SH-aLRT (-alrt 1000) and ModelFinder Plus96 to identify the best evolutionary model for each region. The resulting 1000 trees were concatenated using Astral-III78 to estimate the nuclear phylogeny. FigTree75 v.1.4.4 tool was used to visualise the species tree estimated with Astral-III (Figure S2D).
Outgroup f3-statistics
We calculated outgroup f3-statistics using the qp3pop tool from Admixtools71 v.5.1 and the pseudo-haploid SNP panel to estimate the shared genetic drift between pairs of samples since their separation from an outgroup. For each of the Sicilian wolf genomes, we estimated an f3-statistic of the form f3(Andean Fox: Sicilian wolf, X), where X represents all remaining samples in the dataset (Figures 2A and S3). The more recently a pair of samples split the larger is the expected f3-statistic.
D-statistics
We computed D-statistics using the qpDstats tool from Admixtools71 v.5.1 and the pseudo-haploid SNP panel, to study how modern and ancient wolves and dogs are related to the Sicilian wolves.
We estimated the following D-statistics tests:
D(Sicilian wolf, Italian wolf; X, Andean fox)
Our treemix admixture graphs showed the most likely position in the tree for the Sicilian wolf is forming a clade with the Italian wolf. Therefore, we used this D-statistic to test if the Sicilian wolf shared more alleles with dogs (X) in comparison to the Italian wolf, which would be indicative of gene-flow. We performed this test for each of the four Sicilian wolf genomes independently (Figures 2B and S4A). Additionally, this test allowed us to determine the direction of the gene flow between dogs and the Sicilian wolf. If the gene flow was from the dogs into the Sicilian wolf, we would expect significant results regardless of the dog in X. Conversely, if the geno flow was from the Sicilian wolf into specific dog breeds we would expect significant results only when those breeds are placed in X. We find significant positive results for all dogs suggesting there is dog ancestry in the Sicilian wolf (Figure S4E).
D(Sicilian wolf, Portuguese wolf; X, Andean fox)
We confirmed the result obtained in the previous test changing the Italian wolf for the Portuguese wolf (Figure S4E).
D(X, Cirneco dog; Sicilian wolf, Andean fox) and D(X, Croatian Eneolithic dog (ALP01); Sicilian wolf, Andean fox)
Where X corresponds to all dogs in the dataset. We performed these two tests in order to identify which of the Cirneco dogs and the Croatian Eneolitic dog (ALP01) was a better source for the dog ancestry in the Sicilian wolf. For every X, we expect the D-statistic to be larger for the dog that is a better source of ancestry (Cirneco or Croatian Eneolitic dog). Overall, we obtain larger values of D when the Croatian Eneolitic dog is used in the test (Figure S4B), confirming the Croatian Eneolitic dog is a better source of dog ancestry.
In all the D-statistic tests we used the Andean fox as the outgroup. We considered a test to be statistically significant if the resulting z-score was smaller than -3.3 or larger than 3.3. Finally, following Admixtools’s setup, in a test of the form D(H1, H2; H3, Outgroup), positive D-statistics indicate excess shared derived alleles between H1 and H3, while negative D-statistics indicate excess shared derived alleles between H2 and H3.
TreeMix
TreeMix32 v1.13 was used to model the phylogenetic history of the Sicilian wolf and explore potential admixture events from dogs into Sicilian wolves. TreeMix was run on a subset of the samples including the four Sicilian wolves, Italian wolves, Iberian and other European wolves and ancient and modern dogs (a total of 37 samples). Starting from the pseudo-haploid SNP panel, we further restricted the analysis to sites without missing data resulting in a total of 160,233 transversion sites. We ran ten replicates for each migration event (-m), and incorporated from 0 to 4 migrations. The optional parameters -noss, -global and -k 500 were used and the tree was rooted using one of the coyote samples (Alabama coyote). The trees with the best likelihood for each number of migrations were selected using R97 and plotted using the R functions provided as part of the Treemix software. After incorporating the 1st migration (which models the Sicilian wolf as a mixture of wolf and dog) the topology of the clades involving the Sicilian wolf and Cirneco dog remains overall constant from up to 4 migration events, and none of the following inferred migrations involve the Cirneco dog or the Sicilian wolf.
Admixture graphs
To model the relationships between the Sicilian wolf, modern wolves and dogs we estimated an admixture graph using qpGraph.71 We included samples from relevant groups: Italian wolf (MW303, MW307, ItalianWolf_W050, ItalianWolf_W040), Iberian wolf (PortugueseWolf, MW122, MW127), modern dog (BoxerDog, GalgoDog, GShepDog), ancient dog (ALPO01_Croatia_Eneolithic, SOTN01_Croatia_Eneolithic, AL2397_Italy_Early), Sicilian wolf (Sic1, Sic7, Sic2) and the Andean fox. First, we identified samples that could be grouped into each of the relevant groups using the qpWave tool from AdmixTools.71 By comparing two lists of populations (left and right populations) qpWave identifies the minimum number of migrations required from the right populations to define the ancestry of the left populations. As ‘left populations’ we tested all possible pairs of Italian wolves, Iberian wolves, modern European dogs, ancient dogs and Sicilian wolves. For the ‘right populations’ we used all other samples, except the ones included among the ‘left populations’ and using the Andean fox as fixed outgroup. The qpWave tool was run using the ‘allsnps=YES’ option. Pairs of samples that were consistent with a single migration (p-value ≥ 0.05) were grouped into a population for the admixture graphs: Italian wolf (MW303, MW307 and ItalianWolf_W040), Iberian wolf (PortugueseWolf, MW122, MW127), ancient dog (ALPO01_Croatia_Eneolithic and AL2397_Italy_Early), modern European dog (GalgoDog and GShepDog) and Sicilian wolf (Sic1 and Sic7).
To efficiently explore the possible admixture graph models we used two approaches: qpBrute56,79 and a ‘base graph’ approach previously described.3 For the qpBrute approach we created a configuration file for qpBrute and performed a heuristic search of the admixture graph space. For the ‘base graph’ approach we use admixturegraph R library98 to list all possible graphs including the Italian wolf, Iberian wolf, ancient dog, and modern European dog, and fixed the Andean fox as an outgroup. We tested all possible graphs including 0, 1 and 2 migration edges. We then included the Sicilian wolf to the two graphs that fitted our data in terms of the z-score of the worst fitting f4-statistic and with a score that was not significantly different.99 The Sicilian wolf was included both as a non-admixed lineage and as an admixed lineage. In Figures S4C and S4D we show the best graph from each approach from the graphs that fitted our data.
PCAdmix
PCAdmix34 was used to infer local ancestry for the Sicilian wolves. We used 2 Cane Corso dogs, 3 Cirneco dogs, and 4 European dogs (Galgo, German shepherd, boxer, Tibetan mastiff) as representatives of the dog ancestry, and 4 Italian wolves and 6 European wolves as representatives of the wolf ancestry.
An imputed and phased dataset was prepared for PCAdmix analysis. Genotype likelihoods were called using samtools70 for 139 canid individuals in our dataset and then used as input for genotype imputation without a reference panel using Beagle v4.1.82 The imputed dataset was then phased using Beagle v5.4.83 We used the recombination map of the domestic dog genome100 for imputation and phasing.
PCAdmix was run with a window size of 0.01 cM (-wcM 0.01) and the input SNPs were pruned by minor allele frequency (-maf 0.1) and LD (-r2 0.8). We used a threshold of 0.9 for posterior probability to infer the ancestry and assigned an “unclear” ancestry for regions below this threshold. In order to evaluate the performance of the inference, we also inferred ancestry for randomly selected dogs and wolves including one wolf (wSierraMorena) with known admixed ancestry.31
Nucleotide diversity and heterozygosity in sliding windows
The genome-wide nucleotide diversity (π) was estimated for four wolf populations: 4 Sicilian wolves, 5 Italian wolves, 4 Scandinavian wolves and 6 Iberian wolves. Vcftools 0.1.1680 was run on the pseudo-haploid SNPs panel with a window of 100kbp. Rstudio81,101 and ggplot102 were used to visualise the data for each population.
We used ANGSD to estimate the heterozygosity of each sample, by calculating the folded site frequency spectrum (SFS) on the autosomal chromosomes. We estimated genotype likelihoods for each of the samples independently using ANGSD’s GATK (-GL 2) model (doSaf 1 -fold 1), removing transitions (-rmtrans 1), bases with quality score lower than 20 (-minQ 20) and reads with mapping quality lower than 20 (-minmapq 20). The dog reference genome was used both as reference and as ancestral (- ref and -anc options) and the repeat regions were masked using a repeat mask file (-sites). The chromosomes were partitioned into overlapping windows of size 1 Mb with a step size of 500 kb using the BEDTools76 windows tool. Windows shorter than 1Mb at the end of the chromosomes were discarded. The SFS for each window was estimated using the realSFS utility tool provided in ANGSD and subsequently the final heterozygosity per window was calculated as the ratio of heterozygous sites/total sites. Rstudio, ggplot2 and dplyr103 were used to visualise the heterozygosity level at each window in the form of a violin plot for each individual.
ROHan
To assess the extent of recent inbreeding, we estimated the length and abundance of segments of runs of homozygosity (ROH) using ROHan. We estimated ROHs on two genomes of Sicilian wolf (Sic1 and Sic2) with ∼5x (the acceptable minimum coverage for ancient genomes when post-mortem deamination is <=0.0184), 2 Pleistocene wolves with minimum coverage of 7x, and 11 modern wolves with minimum coverage of 5x. Before running ROHan, we created a bed file containing mappable regions in the reference dog genome (CanFam3.1) to account for divergence between the genomes of the wolf and the dog. Then, a file listing only the autosomal chromosomes was created to restrict our analysis to the autosomal regions. To obtain an estimate of the expected number of segregating sites in the ROH segments (--rohmu) required by ROHan, we estimated background heterozygosity using the Watterson’s theta formula implemented in ROHan. In this formula, the number of segregating sites is calculated as four times the mutation rate multiplied by the effective population size.85 We consider that the effective population size of contemporary grey wolf populations is ∼1,000 following (Pacheco et al., 2022)104 and a mutation rate of 1x10-8, resulting in an expected number of 4x10-4 segregating sites, which means that in a ROH segment it will be lower at 2x10-4. We also observed the deamination profile of the ancient genomes using bam2prof (included in ROHan) and determined that for most of the ancient genomes the damage levels were off at -q 20. The bam2prof results were then used on the ROHan call for the Sicilian wolf genomes (--deam5p and --deam3p).
We calculated the number of segments (NROH), mean length (LROH), and total sum of runs of homozygosity (SROH) in the autosomal region of each sample from the .hmmrohl output of ROHan using an R script. The genomic coefficient of inbreeding was calculated in the same script by dividing the SROH by the total number of validated sites by ROHan available on the .hEst file. The plots in Figures 3C and 3D show the middle mid-point estimates from the three mid-point estimates provided by ROHan (min, mid, and max).
Transversion load
To assess the mutational load in the two Sicilian and 13 reference wolf genomes where we calculated ROH, we used the SIFT scores as measure of mutation’s deleteriousness based on the change it causes in the protein,105 which is available for the domestic dog, Canis familiaris (CanFam3.1), through Variant Effect Predictor (VEP).106 We first extracted positions of coding sequences (CDs) from the domestic dog genome available from the GFF file from Ensembl (https://www.ensembl.org/info/genome/variation/prediction/predicted_data.html#consequences) (release 104; accessed: 15 July 2022). The VEP was used to get SIFT scores at the positions at the CDs, limiting to sites marked as canonical (--pick 1) with scores smaller than 0.05 to get the most deleterious positions. As SIFT scores differ according to base, VEP was run for all four possible variants (A, C, G, or T). This SIFT reference file was then run with a custom script that weights the scores based on the genotype probability in that variant’s position (see below for how genotype probabilities were estimated). The mutational load was calculated in this set of predetermined sites that were attainable from all compared samples (a total of 11,021,945 sites that were present in all 15 samples compared in Figure 3).
As SIFT scores are the normalised probability that an amino acid change is tolerated, with scores lower than 0.05 predicted as deleterious,105 the magnitude of SIFT scores is negatively correlated with the deleteriousness of a mutation (i.e. the smaller the score, the more deleterious the mutation is). To generate a more intuitive metric that can use used to compare the deleteriousness across samples, we estimated the complement score () by subtracting 1 from the score for a each derived allele :
We then estimate the mutational load at site , by multiplying by the genotype probability of allele at site . For each site () where the ancestral allele () is known, three different derived alleles () are possible given that ≠ and ,a ∈ . Each of these derived alleles have their own complement score (, and ). The homozygous derived load for each site is thus:
| (Equation 1) |
where is the genotype probability of genotype that is homozygous for allele in position . Thus, for a genome with a total of n positions, the total sum of conservation scores in homozygous positions () is simply the sum of homozygous derived score (Equation 1) across sites:
| (Equation 2) |
The entire homozygous load () for a genome with sites is thus:
| (Equation 3) |
When dealing with ancient genomes, removing transitions is desirable to exclude errors derived from post-mortem DNA damage. In other words, homozygous transversion load is calculated using Equation 1 for ∈ {C,T} only when ∈ {A,G} and for ∈ {A,G} only when ∈ {C,T}. As SIFT scores are different for different alleles, the homozygous transversion load () for a genome with sites is:
| (Equation 4) |
where is the genotype probability for homozygous transversion site 1 and is its respective conservation score, is the genotype probability for homozygous transversion site 2 and is its respective conservation score, and is the probability of homozygous transversion genotype.
Estimating genotype probabilities used in the estimation of the homozygous transversion load
The genotype probability was obtained from the genotype likelihood values obtained from ANGSD69 with minimum base quality 20 (-minQ 20), minimum mapping quality 20 (-minMapQ 20), 5 edge bases trimmed (-trim 5), genotype likelihood estimation following GATK model (-GL 2) to have an output the log likelihoods of all 10 genotypes (-doGlf 4). A framework employing a Bayesian framework described in Plassais et al. (2022)107 was used to convert the genotype likelihood to a genotype probability using a custom script made available in a GitHub repository (Greer, 2020,51 all_genotype_probability_v3.py).
Estimating the ancestral allele
As the above equations require the ancestral state of each position, we inferred the ancestral allele by mapping five species representing taxa that are ancestral to the genus Canidae to enable the comparison of all taxa within the Canidae family. These ancestral taxa were used to polarise the alleles and included: domestic cat (Felis catus), polar bear (Ursus maritimus), spotted hyena (Crocuta crocuta), red panda (Ailurops fulgens), and walrus (Odobenus rosmarus). These taxa were inferred to be ancestral to the Canidae group using the mammal tree of life.108 The domestic cat (Felis_catus_9.0, GCA_000181335.4) and polar bear (UrsMar_1.0, GCA_000687225.1) fasta files were downloaded from Ensembl, while the hyena, red panda, and walrus reference genomes were generated by the DNA Zoo consortium following the Juice Box protocol.109,110 The walrus and red panda assemblies were generated based on available draft genomes.111,112 The reference-quality genomes of each of the taxa, available as a fasta file, were split into 500bp fragments and aligned to the CanFam3.1 reference genome using bwa-mem.113 The resulting BAM files were merged and a consensus sequence was called using ANGSD -doFasta 2.
Acknowledgments
This work was supported by ERC Consolidator Award 681396 Extinction Genomics, DNRF143 Center for Evolutionary Hologenomics, and the Norwegian Environment Agency (project 18088069). The National Science Centre, Poland supported S. Nowak (grant No. 2020/39/B/NZ9/01829) and R.W. Mysłajek (grant No. 2019/35/O/NZ8/01550). G.H-A. is supported by the Consejo Nacional de Ciencia y Tecnología from Mexico (CONACyT 576734). R.Godinho is supported by the Portuguese Foundation for science and Technology (DL57/2016/CP1440). U. Saarma and M. Hindrikson were supported by funding from the Estonian Ministry of Education and Research (grants PRG1209 and PSG715). We would like to thank Luca Sineo for helping collect the Sicilian wolf samples and Domenico Tricomi, together with the owners of the Cirneco dell’Etna dogs for their collaboration in this project. We are deeply grateful to the curators of the museums who allowed us to sample the historical Sicilian wolves: Paolo Agnelli of the Museum of Natural History of the University of Florence, zoology section "La Specola", Fabio Lo Bono of the Civic Museum "Baldassare Romano" in Termini Imerese (PA), Enrico Bellia of the Museum of Zoology "P. Doderlein", SIMUA, Palermo (PA) and Ferdinando Maurici and Fabio Lo Valvo from Regional Museum of Terrasini (PA). We also acknowledge Davide Palumbo for his valuable inputs and support at the beginning of this collaboration. We also thank the staff of the Danish National High-throughput Sequencing Center and BGI Denmark for their support in data generation and the Danish National Supercomputer for Life Sciences at the DTU National Lifescience Center at Technical University of Denmark (DTU) for facilitating the data analysis process.
Author contributions
This genome scale study of the Sicilian wolf was initiated by M.M.C., E.C., and M.T.P.G. to build upon the findings of a prior mtDNA analyses of the Sicilian wolf led by F.M.A. and L.R. M.M.C., J.R.-M., M.H-S.S., E.C, and S.G. designed the research. F.M.A., L.R., E.C., S.L.B., and M.M.C. contributed with material, information, and logistics to the historical Sicilian samples. J.A., I.K., La.B., Li.B., J.M., M.A., U.S., M.H., P.H., B.C.B., C.N., R.Q., S.S., L.P., S.N., R.W.M., S.L.B., P.C., Lu.B., C.V., H.K.S., and O.S. contributed material information, logistic and/or contributed to modern samples. M.M.C. did historical DNA molecular work, supervised by O.S., M.H.S.S., and M.T.P.G. C.H.S.-O., L.T.L., I.F., and C.G.C. coordinated logistics and/or did modern DNA molecular work, supervised by M.M.C., M.H.S.S., and M.T.P.G. M.M.C. and G.H.-A. did initial processing of the modern raw sequencing data, supervised by S.G. and M.T.P.G. M.M.C. performed the population genomic, nucleotide diversity, and heterozygosity analyses with input from J.R.-M. A.C. and J.R.-M did admixture graph analyses. S.G.A. performed the runs of homozygosity and genomic load analyses, with input from L.F. X.S. performed the local ancestry inference analysis. M.M.C. wrote the paper with input from J.R.-M., S.G., L.R., F.M.A, M.-H.S.S. and all other authors.
Declaration of interests
The authors declare no competing interests.
Published: July 10, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107307.
Contributor Information
Marta Maria Ciucani, Email: ciucani@palaeome.org.
Shyam Gopalakrishnan, Email: shyam.gopalakrishnan@sund.ku.dk.
Supplemental information
Data and code availability
-
•
Sequencing data generated in this study have been deposited at the European Nucleotide Archive (ENA) under accession number PRJEB57290 and are publicly available as of the date of publication.
-
•
This paper does not report any original code.
-
•
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.
References
- 1.Fan Z., Silva P., Gronau I., Wang S., Armero A.S., Schweizer R.M., Ramirez O., Pollinger J., Galaverni M., Ortega Del-Vecchyo D., et al. Worldwide patterns of genomic variation and admixture in gray wolves. Genome Res. 2016;26:163–173. doi: 10.1101/gr.197517.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loog L., Thalmann O., Sinding M.-H.S., Schuenemann V.J., Perri A., Germonpré M., Bocherens H., Witt K.E., Samaniego Castruita J.A., Velasco M.S., et al. Ancient DNA suggests modern wolves trace their origin to a Late Pleistocene expansion from Beringia. Mol. Ecol. 2020;29:1596–1610. doi: 10.1111/mec.15329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramos-Madrigal J., Sinding M.-H.S., Carøe C., Mak S.S.T., Niemann J., Samaniego Castruita J.A., Fedorov S., Kandyba A., Germonpré M., Bocherens H., et al. Genomes of Pleistocene Siberian wolves uncover multiple extinct wolf lineages. Curr. Biol. 2021;31:198–206.e8. doi: 10.1016/j.cub.2020.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergström A., Stanton D.W.G., Taron U.H., Frantz L., Sinding M.-H.S., Ersmark E., Pfrengle S., Cassatt-Johnstone M., Lebrasseur O., Girdland-Flink L., et al. Grey wolf genomic history reveals a dual ancestry of dogs. Nature. 2022;607:313–320. doi: 10.1038/s41586-022-04824-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gopalakrishnan S., Sinding M.-H.S., Ramos-Madrigal J., Niemann J., Samaniego Castruita J.A., Vieira F.G., Carøe C., de Manuel Montero M., Kuderna L., Serres A., et al. Interspecific gene flow shaped the evolution of the genus Canis. Curr. Biol. 2019;29:4152. doi: 10.1016/j.cub.2019.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hindrikson M., Remm J., Pilot M., Godinho R., Stronen A.V., Baltrūnaité L., Czarnomska S.D., Leonard J.A., Randi E., Nowak C., et al. Wolf population genetics in Europe: a systematic review, meta-analysis and suggestions for conservation and management. Biol. Rev. Camb. Philos. Soc. 2017;92:1601–1629. doi: 10.1111/brv.12298. [DOI] [PubMed] [Google Scholar]
- 7.Randi E. Genetics and conservation of wolves Canis lupus in Europe. Mamm Rev. 2011;41:99–111. [Google Scholar]
- 8.Leonard J.A. Ecology drives evolution in grey wolves. Evol. Ecol. Res. 2014;16:461–473. [Google Scholar]
- 9.Leonard J.A., Echegaray J., Randi E., Vilà C., Gompper M.E. Free-ranging dogs and wildlife conservation. 2013. Impact of hybridization with domestic dogs on the conservation of wild canids; pp. 170–184. [Google Scholar]
- 10.Randi E., Lucchini V., Christensen M.F., Mucci N., Funk S.M., Dolf G., Loeschcke V. Mitochondrial DNA variability in Italian and east European wolves: Detecting the consequences of small population size and hybridization. Conserv. Biol. 2000;14:464–473. [Google Scholar]
- 11.Boggiano F., Ciofi C., Boitani L., Formia A., Grottoli L., Natali C., Ciucci P. Detection of an East European wolf haplotype puzzles mitochondrial DNA monomorphism of the Italian wolf population. Mamm. Biol. 2013;78:374–378. [Google Scholar]
- 12.VonHoldt B.M., Pollinger J.P., Earl D.A., Knowles J.C., Boyko A.R., Parker H., Geffen E., Pilot M., Jedrzejewski W., Jedrzejewska B., et al. A genome-wide perspective on the evolutionary history of enigmatic wolf-like canids. Genome Res. 2011;21:1294–1305. doi: 10.1101/gr.116301.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Montana L., Caniglia R., Galaverni M., Fabbri E., Randi E. A new mitochondrial haplotype confirms the distinctiveness of the Italian wolf (Canis lupus) population. Mamm. Biol. 2017;84:30–34. [Google Scholar]
- 14.Angelici F.M., Rossi L. A new subspecies of grey wolf (Carnivora Canidae), recently extinct, from Sicily, Italy. bioRxiv. 2018 doi: 10.1101/320655. Preprint at. [DOI] [Google Scholar]
- 15.Angelici F.M., Ciucani M.M., Angelini S., Annesi F., Caniglia R., Castiglia R., Fabbri E., Galaverni M., Palumbo D., Ravegnini G., et al. The Sicilian wolf: genetic identity of a recently extinct insular population. Zool. Sci. (Tokyo) 2019;36:189–197. doi: 10.2108/zs180180. [DOI] [PubMed] [Google Scholar]
- 16.Reale S.C., Farber M.K., Cumbo V., Sammarco I., Bonanno F., Spinnato A., Seminara S. Biodiversity lost: the phylogenetic relationships of a complete mitochondrial DNA genome sequenced from the extinct wolf population of Sicily. Mamm. Biol. 2019;38:1–3. [Google Scholar]
- 17.David Mech L., Boitani L. University of Chicago Press; 2007. Wolves: Behavior, Ecology, and Conservation. [Google Scholar]
- 18.Salvatori V. Council of Europe; 2005. Report on the Conservation Status and Threats for Wolf (Canis Lupus) in Europe. [Google Scholar]
- 19.Sastre N., Vilà C., Salinas M., Bologov V.V., Urios V., Sánchez A., Francino O., Ramírez O. Signatures of demographic bottlenecks in European wolf populations. Conserv. Genet. 2011;12:701–712. [Google Scholar]
- 20.Sarà M. Soc. messinese di Storia Patria, Messina; 1999. Il “Catalogo dei Mammiferi della Sicilia” rivisitato. [Google Scholar]
- 21.Palumbo F.M. Catalogo dei mammiferi della Sicilia. Annali di Agricoltura Siciliana. 1868;12:3–123. [Google Scholar]
- 22.Migneco M. Catania: Stabilimento tipografico M. Galati; 1897. Considerazioni ed appunti sul cane cirneco. 17. [Google Scholar]
- 23.Ghigi A. Ricerche faunistiche e sistematiche sui Mammiferi d’Italia che formano oggetto di caccia. Natura. 1911:1–51. [Google Scholar]
- 24.Zimen E., Boitani L. Number and distribution of wolves in Italy. Z. Säugetierkunde. 1975;40:102–112. [Google Scholar]
- 25.Lucchini V., Galov A., Randi E. Evidence of genetic distinction and long-term population decline in wolves (Canis lupus) in the Italian Apennines. Mol. Ecol. 2004;13:523–536. doi: 10.1046/j.1365-294x.2004.02077.x. [DOI] [PubMed] [Google Scholar]
- 26.Altobello G. Fauna dell’Abruzzo e del Molise - Mammiferi - Carnivori. 1921. http://www.storiadellafauna.it/scaffale/testi/alto/Carnivo.htm
- 27.Ciucani M.M., Palumbo D., Galaverni M., Serventi P., Fabbri E., Ravegnini G., Angelini S., Maini E., Persico D., Caniglia R., et al. Old wild wolves: ancient DNA survey unveils population dynamics in Late Pleistocene and Holocene Italian remains. PeerJ. 2019;7:e6424. doi: 10.7717/peerj.6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koupadi K., Fontani F., Ciucani M.M., Maini E., De Fanti S., Cattani M., Curci A., Nenzioni G., Reggiani P., Andrews A.J., et al. Population dynamics in Italian Canids between the Late Pleistocene and Bronze Age. Genes. 2020;11:1409. doi: 10.3390/genes11121409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Freedman A.H., Gronau I., Schweizer R.M., Ortega-Del Vecchyo D., Han E., Silva P.M., Galaverni M., Fan Z., Marx P., Lorente-Galdos B., et al. Genome sequencing highlights the dynamic early history of dogs. PLoS Genet. 2014;10:e1004016. doi: 10.1371/journal.pgen.1004016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alexander D.H., Novembre J., Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gómez-Sánchez D., Olalde I., Sastre N., Enseñat C., Carrasco R., Marques-Bonet T., Lalueza-Fox C., Leonard J.A., Vilà C., Ramírez O. On the path to extinction: Inbreeding and admixture in a declining grey wolf population. Mol. Ecol. 2018;27:3599–3612. doi: 10.1111/mec.14824. [DOI] [PubMed] [Google Scholar]
- 32.Pickrell J.K., Pritchard J.K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012;8:e1002967. doi: 10.1371/journal.pgen.1002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kozlov A.M., Darriba D., Flouri T., Morel B., Stamatakis A. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics. 2019;35:4453–4455. doi: 10.1093/bioinformatics/btz305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brisbin A., Bryc K., Byrnes J., Zakharia F., Omberg L., Degenhardt J., Reynolds A., Ostrer H., Mezey J.G., Bustamante C.D. PCAdmix: principal components-based assignment of ancestry along each chromosome in individuals with admixed ancestry from two or more populations. Hum. Biol. 2012;84:343–364. doi: 10.3378/027.084.0401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niemann J., Gopalakrishnan S., Yamaguchi N., Ramos-Madrigal J., Wales N., Gilbert M.T.P., Sinding M.-H.S. Extended survival of Pleistocene Siberian wolves into the early 20th century on the island of Honshū. iScience. 2021;24:101904. doi: 10.1016/j.isci.2020.101904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Segawa T., Yonezawa T., Mori H., Kohno A., Kudo Y., Akiyoshi A., Wu J., Tokanai F., Sakamoto M., Kohno N., et al. Paleogenomics reveals independent and hybrid origins of two morphologically distinct wolf lineages endemic to Japan. Curr. Biol. 2022;32:2494–2504.e5. doi: 10.1016/j.cub.2022.04.034. [DOI] [PubMed] [Google Scholar]
- 37.Kardos M., Åkesson M., Fountain T., Flagstad Ø., Liberg O., Olason P., Sand H., Wabakken P., Wikenros C., Ellegren H. Genomic consequences of intensive inbreeding in an isolated wolf population. Nat. Ecol. Evol. 2018;2:124–131. doi: 10.1038/s41559-017-0375-4. [DOI] [PubMed] [Google Scholar]
- 38.Robinson J.A., Räikkönen J., Vucetich L.M., Vucetich J.A., Peterson R.O., Lohmueller K.E., Wayne R.K. Genomic signatures of extensive inbreeding in Isle Royale wolves, a population on the threshold of extinction. Sci. Adv. 2019;5:eaau0757. doi: 10.1126/sciadv.aau0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dussex N., van der Valk T., Morales H.E., Wheat C.W., Díez-Del-Molino D., von Seth J., Foster Y., Kutschera V.E., Guschanski K., Rhie A., et al. Population genomics of the critically endangered kākāpō. Cell Genom. 2021;1:100002. doi: 10.1016/j.xgen.2021.100002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ciucani M.M., Jensen J.K., Sinding M.-H.S., Smith O., Lucenti S.B., Rosengren E., Rook L., Tuveri C., Arca M., Cappellini E., et al. Evolutionary history of the extinct Sardinian dhole. Curr. Biol. 2021;31:5571–5579.e6. doi: 10.1016/j.cub.2021.09.059. [DOI] [PubMed] [Google Scholar]
- 41.Pemberton T.J., Absher D., Feldman M.W., Myers R.M., Rosenberg N.A., Li J.Z. Genomic patterns of homozygosity in worldwide human populations. Am. J. Hum. Genet. 2012;91:275–292. doi: 10.1016/j.ajhg.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Greer, D. (2020). Extinction Genomics: The Case of Grey Wolves Year.
- 43.Bergström A., Frantz L., Schmidt R., Ersmark E., Lebrasseur O., Girdland-Flink L., Lin A.T., Storå J., Sjögren K.G., Anthony D., et al. Origins and genetic legacy of prehistoric dogs. Science. 2020;370:557–564. doi: 10.1126/science.aba9572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Montana L., Caniglia R., Galaverni M., Fabbri E., Ahmed A., Bolfíková B.Č., Czarnomska S.D., Galov A., Godinho R., Hindrikson M., et al. Combining phylogenetic and demographic inferences to assess the origin of the genetic diversity in an isolated wolf population. PLoS One. 2017;12:e0176560. doi: 10.1371/journal.pone.0176560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silva P., Galaverni M., Ortega-Del Vecchyo D., Fan Z., Caniglia R., Fabbri E., Randi E., Wayne R., Godinho R. Genomic evidence for the Old divergence of Southern European wolf populations. Proc. Biol. Sci. 2020;287:20201206. doi: 10.1098/rspb.2020.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Auton A., Rui Li Y., Kidd J., Oliveira K., Nadel J., Holloway J.K., Hayward J.J., Cohen P.E., Greally J.M., Wang J., et al. Genetic recombination is targeted towards gene promoter regions in dogs. PLoS Genet. 2013;9:e1003984. doi: 10.1371/journal.pgen.1003984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.vonHoldt B.M., Cahill J.A., Fan Z., Gronau I., Robinson J., Pollinger J.P., Shapiro B., Wall J., Wayne R.K. Whole-genome sequence analysis shows that two endemic species of North American wolf are admixtures of the coyote and gray wolf. Sci. Adv. 2016;2:e1501714. doi: 10.1126/sciadv.1501714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gopalakrishnan S., Sinding M.S., Ramos-Madrigal J., Niemann J., Samaniego Castruita J.A., Vieira F.G., Carøe C., Montero M.M., Kuderna L., et al. Interspecific gene flow shaped the evolution of the Genus Canis. Curr. Biol. 2018;28:3441–3449.e5. doi: 10.1016/j.cub.2018.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang W., Fan Z., Han E., Hou R., Zhang L., Galaverni M., Huang J., Liu H., Silva P., Li P., et al. Hypoxia adaptations in the grey wolf (Canis lupus chanco) from Qinghai-Tibet Plateau. PLoS Genet. 2014;10:e1004466. doi: 10.1371/journal.pgen.1004466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang G.-D., Zhai W., Yang H.-C., Wang L., Zhong L., Liu Y.-H., Fan R.-X., Yin T.-T., Zhu C.-L., Poyarkov A.D., et al. Out of southern East Asia: the natural history of domestic dogs across the world. Cell Res. 2016;26:21–33. doi: 10.1038/cr.2015.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sinding M.-H.S., Gopalakrishan S., Vieira F.G., Samaniego Castruita J.A., Raundrup K., Heide Jørgensen M.P., Meldgaard M., Petersen B., Sicheritz-Ponten T., Mikkelsen J.B., et al. Population genomics of grey wolves and wolf-like canids in North America. PLoS Genet. 2018;14:e1007745. doi: 10.1371/journal.pgen.1007745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang G.-D., Zhai W., Yang H.-C., Fan R.-X., Cao X., Zhong L., Wang L., Liu F., Wu H., Cheng L.-G., et al. The genomics of selection in dogs and the parallel evolution between dogs and humans. Nat. Commun. 2013;4:1860. doi: 10.1038/ncomms2814. [DOI] [PubMed] [Google Scholar]
- 53.Sinding M.-H.S., Gopalakrishnan S., Ramos-Madrigal J., de Manuel M., Pitulko V.V., Kuderna L., Feuerborn T.R., Frantz L.A.F., Vieira F.G., Niemann J., et al. Arctic-adapted dogs emerged at the Pleistocene-Holocene transition. Science. 2020;368:1495–1499. doi: 10.1126/science.aaz8599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Frantz L.A.F., Mullin V.E., Pionnier-Capitan M., Lebrasseur O., Ollivier M., Perri A., Linderholm A., Mattiangeli V., Teasdale M.D., Dimopoulos E.A., et al. Genomic and archaeological evidence suggest a dual origin of domestic dogs. Science. 2016;352:1228–1231. doi: 10.1126/science.aaf3161. [DOI] [PubMed] [Google Scholar]
- 55.Botigué L.R., Song S., Scheu A., Gopalan S., Pendleton A.L., Oetjens M., Taravella A.M., Seregély T., Zeeb-Lanz A., Arbogast R.M., et al. Ancient European dog genomes reveal continuity since the Early Neolithic. Nat. Commun. 2017;8:16082. doi: 10.1038/ncomms16082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ní Leathlobhair M., Perri A.R., Irving-Pease E.K., Witt K.E., Linderholm A., Haile J., Lebrasseur O., Ameen C., Blick J., Boyko A.R., et al. The evolutionary history of dogs in the Americas. Science. 2018;361:81–85. doi: 10.1126/science.aao4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Feuerborn T.R., Carmagnini A., Losey R.J., Nomokonova T., Askeyev A., Askeyev I., Askeyev O., Antipina E.E., Appelt M., Bachura O.P., et al. Modern Siberian dog ancestry was shaped by several thousand years of Eurasian-wide trade and human dispersal. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2100338118. e2100338118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wiedmer M., Oevermann A., Borer-Germann S.E., Gorgas D., Shelton G.D., Drögemüller M., Jagannathan V., Henke D., Leeb T. A RAB3GAP1 SINE insertion in Alaskan huskies with polyneuropathy, ocular abnormalities, and neuronal vacuolation (POANV) resembling human warburg micro syndrome 1 (WARBM1) G3 (Bethesda) 2016;6:255–262. doi: 10.1534/g3.115.022707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Decker B., Davis B.W., Rimbault M., Long A.H., Karlins E., Jagannathan V., Reiman R., Parker H.G., Drögemüller C., Corneveaux J.J., et al. Comparison against 186 canid whole-genome sequences reveals survival strategies of an ancient clonally transmissible canine tumor. Genome Res. 2015;25:1646–1655. doi: 10.1101/gr.190314.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Metzger J., Nolte A., Uhde A.-K., Hewicker-Trautwein M., Distl O. Whole genome sequencing identifies missense mutation in MTBP in Shar-Pei affected with Autoinflammatory Disease (SPAID) BMC Genom. 2017;18:348. doi: 10.1186/s12864-017-3737-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marsden C.D., Ortega-Del Vecchyo D., O’Brien D.P., Taylor J.F., Ramirez O., Vilà C., Marques-Bonet T., Schnabel R.D., Wayne R.K., Lohmueller K.E. Bottlenecks and selective sweeps during domestication have increased deleterious genetic variation in dogs. Proc. Natl. Acad. Sci. USA. 2016;113:152–157. doi: 10.1073/pnas.1512501113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lindblad-Toh K., Wade C.M., Mikkelsen T.S., Karlsson E.K., Jaffe D.B., Kamal M., Clamp M., Chang J.L., Kulbokas E.J., 3rd, Zody M.C., et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438:803–819. doi: 10.1038/nature04338. [DOI] [PubMed] [Google Scholar]
- 63.Meyer M., Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010;2010 doi: 10.1101/pdb.prot5448. https://pubmed.ncbi.nlm.nih.gov/20516186/ [DOI] [PubMed] [Google Scholar]
- 64.Mak S.S.T., Gopalakrishnan S., Carøe C., Geng C., Liu S., Sinding M.-H.S., Kuderna L.F.K., Zhang W., Fu S., Vieira F.G., et al. Comparative performance of the BGISEQ-500 vs Illumina HiSeq2500 sequencing platforms for palaeogenomic sequencing. Gigascience. 2017;6:1–13. doi: 10.1093/gigascience/gix049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schubert M., Ermini L., Der Sarkissian C., Jónsson H., Ginolhac A., Schaefer R., Martin M.D., Fernández R., Kircher M., McCue M., et al. Characterization of ancient and modern genomes by SNP detection and phylogenomic and metagenomic analysis using PALEOMIX. Nat. Protoc. 2014;9:1056–1082. doi: 10.1038/nprot.2014.063. [DOI] [PubMed] [Google Scholar]
- 66.Schubert M., Lindgreen S., Orlando L. AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes. 2016;9:88. doi: 10.1186/s13104-016-1900-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Korneliussen T.S., Albrechtsen A., Nielsen R. ANGSD: analysis of next generation sequencing data. BMC Bioinformatics. 2014;15:356. doi: 10.1186/s12859-014-0356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Patterson N., Moorjani P., Luo Y., Mallick S., Rohland N., Zhan Y., Genschoreck T., Webster T., Reich D. Ancient admixture in human history. Genetics. 2012;192:1065–1093. doi: 10.1534/genetics.112.145037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jónsson H., Ginolhac A., Schubert M., Johnson P.L.F., Orlando L. mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics. 2013;29:1682–1684. doi: 10.1093/bioinformatics/btt193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rambaut A. FigTree V. 1.4.0. 2012. http://tree.bio.ed.ac.uk/software/figtree/
- 76.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Minh B.Q., Schmidt H.A., Chernomor O., Schrempf D., Woodhams M.D., von Haeseler A., Lanfear R. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic Era. Mol. Biol. Evol. 2020;37:1530–1534. doi: 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang C., Rabiee M., Sayyari E., Mirarab S. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics. 2018;19:153. doi: 10.1186/s12859-018-2129-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu L., Bosse M., Megens H.-J., Frantz L.A.F., Lee Y.-L., Irving-Pease E.K., Narayan G., Groenen M.A.M., Madsen O. Genomic analysis on pygmy hog reveals extensive interbreeding during wild boar expansion. Nat. Commun. 2019;10:1992. doi: 10.1038/s41467-019-10017-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Danecek P., Auton A., Abecasis G., Albers C.A., Banks E., DePristo M.A., Handsaker R.E., Lunter G., Marth G.T., Sherry S.T., et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Allaire, J. (2012). RStudio: Integrated Development Environment for R. Boston, MA.
- 82.Browning B.L., Browning S.R. Genotype Imputation with Millions of Reference Samples. Am. J. Hum. Genet. 2016;98:116–126. doi: 10.1016/j.ajhg.2015.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Browning B.L., Tian X., Zhou Y., Browning S.R. Fast two-stage phasing of large-scale sequence data. Am. J. Hum. Genet. 2021;108:1880–1890. doi: 10.1016/j.ajhg.2021.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Renaud G., Hanghøj K., Korneliussen T.S., Willerslev E., Orlando L. Joint estimates of heterozygosity and runs of homozygosity for modern and ancient samples. Genetics. 2019;212:587–614. doi: 10.1534/genetics.119.302057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Watterson G.A. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- 86.Dabney J., Meyer M., Paabo S. Ancient DNA Damage. Cold Spring Harb. Perspect. Biol. 2013;5:a012567. doi: 10.1101/cshperspect.a012567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Campos P.F., Gilbert M.T.P. DNA Extraction from Keratin and Chitin. Methods Mol. Biol. 2019;1963:57–63. doi: 10.1007/978-1-4939-9176-1_7. [DOI] [PubMed] [Google Scholar]
- 88.Carøe C., Gopalakrishnan S., Vinner L., Mak S.S.T., Sinding M.H.S., Samaniego J.A., Wales N., Sicheritz-Pontén T., Gilbert M.T.P. Single-tube library preparation for degraded DNA. Methods Ecol. Evol. 2018;9:410–419. [Google Scholar]
- 89.Sirén K., Mak S.S.T., Melkonian C., Carøe C., Swiegers J.H., Molenaar D., Fischer U., Gilbert M.T.P. Taxonomic and functional characterization of the microbial community during spontaneous in vitro fermentation of riesling must. Front. Microbiol. 2019;10:697. doi: 10.3389/fmicb.2019.00697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Toolkit P. Broad Institute; 2018. Picard Toolkit. [Google Scholar]
- 91.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M., et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Becker D., Minor K.M., Letko A., Ekenstedt K.J., Jagannathan V., Leeb T., Shelton G.D., Mickelson J.R., Drögemüller C. A GJA9 frameshift variant is associated with polyneuropathy in Leonberger dogs. BMC Genom. 2017;18:662. doi: 10.1186/s12864-017-4081-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Plassais J., Kim J., Davis B.W., Karyadi D.M., Hogan A.N., Harris A.C., Decker B., Parker H.G., Ostrander E.A. Whole genome sequencing of canids reveals genomic regions under selection and variants influencing morphology. Nat. Commun. 2019;10:1489. doi: 10.1038/s41467-019-09373-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Behr A.A., Liu K.Z., Liu-Fang G., Nakka P., Ramachandran S. pong: fast analysis and visualization of latent clusters in population genetic data. Bioinformatics. 2016;32:2817–2823. doi: 10.1093/bioinformatics/btw327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hoang D.T., Chernomor O., von Haeseler A., Minh B.Q., Vinh L.S. UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018;35:518–522. doi: 10.1093/molbev/msx281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kalyaanamoorthy S., Minh B.Q., Wong T.K.F., von Haeseler A., Jermiin L.S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017;14:587–589. doi: 10.1038/nmeth.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Computing R., et al. R Core Team; 2013. R: A Language and Environment for Statistical Computing. [Google Scholar]
- 98.Leppälä K., Nielsen S.V., Mailund T. admixturegraph: an R package for admixture graph manipulation and fitting. Bioinformatics. 2017;33:1738–1740. doi: 10.1093/bioinformatics/btx048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lipson M., Reich D. A working model of the deep relationships of diverse modern human genetic lineages outside of Africa. Mol. Biol. Evol. 2017;34:889–902. doi: 10.1093/molbev/msw293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Campbell C.L., Bhérer C., Morrow B.E., Boyko A.R., Auton A. A pedigree-based map of recombination in the domestic dog genome. G3. 2016;6:3517–3524. doi: 10.1534/g3.116.034678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.RStudio Team (2020). RStudio: Integrated Development Environment for R.
- 102.Wickham H. Springer-Verlag; 2016. ggplot2: Elegant Graphics for Data Analysis. [Google Scholar]
- 103.Hadley Wickham, R.F., Henry, L., Müller, K., et al. (2018). Dplyr: A Grammar of Data Manipulation. Version 0. 7 6.
- 104.Pacheco C., Stronen A.V., Jędrzejewska B., Plis K., Okhlopkov I.M., Mamaev N.V., Drovetski S.V., Godinho R. Demography and evolutionary history of grey wolf populations around the Bering Strait. Mol. Ecol. 2022;31:4851–4865. doi: 10.1111/mec.16613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R.S., Thormann A., Flicek P., Cunningham F. The ensembl variant effect predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Plassais J., vonHoldt B.M., Parker H.G., Carmagnini A., Dubos N., Papa I., Bevant K., Derrien T., Hennelly L.M., Whitaker D.T., et al. Natural and human-driven selection of a single non-coding body size variant in ancient and modern canids. Curr. Biol. 2022;32:889–897.e9. doi: 10.1016/j.cub.2021.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Upham N.S., Esselstyn J.A., Jetz W. Inferring the mammal tree: Species-level sets of phylogenies for questions in ecology, evolution, and conservation. PLoS Biol. 2019;17:e3000494. doi: 10.1371/journal.pbio.3000494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dudchenko O., Shamim M.S., Batra S.S., Durand N.C., Musial N.T., Mostofa R., Pham M., Glenn St Hilaire B., Yao W., Stamenova E., et al. The Juicebox Assembly Tools module facilitates de novo assembly of mammalian genomes with chromosome-length scaffolds for under $1000. bioRxiv. 2018 doi: 10.1101/254797. Preprint at. [DOI] [Google Scholar]
- 110.Dudchenko O., Batra S.S., Omer A.D., Nyquist S.K., Hoeger M., Durand N.C., Shamim M.S., Machol I., Lander E.S., Aiden A.P., et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 2017;356:92–95. doi: 10.1126/science.aal3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Foote A.D., Liu Y., Thomas G.W.C., Vinař T., Alföldi J., Deng J., Dugan S., van Elk C.E., Hunter M.E., Joshi V., et al. Convergent evolution of the genomes of marine mammals. Nat. Genet. 2015;47:272–275. doi: 10.1038/ng.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hu Y., Wu Q., Ma S., Ma T., Shan L., Wang X., Nie Y., Ning Z., Yan L., Xiu Y., et al. Comparative genomics reveals convergent evolution between the bamboo-eating giant and red pandas. Proc. Natl. Acad. Sci. USA. 2017;114:1081–1086. doi: 10.1073/pnas.1613870114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013 doi: 10.48550/arXiv.1303.3997. Preprint at. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Sequencing data generated in this study have been deposited at the European Nucleotide Archive (ENA) under accession number PRJEB57290 and are publicly available as of the date of publication.
-
•
This paper does not report any original code.
-
•
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.