

Abstract
Objectives
To evaluate the safety, efficacy and response duration of four different dosing regimens of dazodalibep (DAZ), a non-antibody biological antagonist of CD40L, in patients with rheumatoid arthritis (RA).
Methods
This double-blind study included adult patients with moderate-to-severe active RA with a positive test for serum rheumatoid factor and/or anticitrullinated protein antibodies, an inadequate response to methotrexate, other conventional disease-modifying antirheumatic drugs or tumour necrosis factor-α inhibitors, and no prior treatment with B-cell depleting agents. Eligible participants were randomised equally to five groups receiving intravenous infusions of DAZ or placebo. The primary endpoint was the change from baseline in the Disease Activity Score-28 with C reactive protein (DAS28-CRP) at day 113. Participants were followed through day 309.
Results
The study randomised 78 eligible participants. The change from baseline in DAS28-CRP (least squares means±SE) at day 113 was significantly greater for all DAZ groups (−1.83±0.28 to −1.90±0.27; p<0.05) relative to PBO (−1.06±0.26); significant reductions in DAS28-CRP were also observed for all DAZ groups at day 309. The distribution of adverse events was generally balanced among DAZ and PBO groups (74% and 63%, respectively). There were four serious adverse events deemed by investigators to be unrelated to study medication.
Conclusions
DAZ treatment for all dosage regimens significantly reduced DAS28-CRP at day 113 relative to PBO. The safety data suggest an acceptable safety and tolerability profile. Treatment effects at day 113 and the prolonged duration of responses after DAZ cessation support the use of longer dosing intervals.
Trial registration number
Keywords: therapeutics; pharmacokinetics; arthritis, rheumatoid
WHAT IS ALREADY KNOWN ON THIS TOPIC
Dazodalibep (DAZ) is a novel non-antibody biological antagonist of CD40L under investigation for the treatment of autoimmune disorders.
The CD40/CD40L pathway plays a critical role in mounting cellular and humoral immune responses and has been implicated in the pathophysiology of rheumatoid arthritis and other autoimmune disorders.
Prior studies of anti-CD40L monoclonal antibodies (mAb) targeting the CD40/CD40L pathway were associated with thromboembolic complications related to the Fc portion of their mAb structure.
Treatment with DAZ led to higher, durable response rates compared with placebo in a double-blind, phase Ib study in patients with moderate-to-severe rheumatoid arthritis and was not associated with thrombotic complications or other major safety concerns.
WHAT THIS STUDY ADDS
In MIDORA (Mechanistic Insight and Dose Optimization in Rheumatoid Arthritis), a randomised, phase II trial of different dosage regimens of DAZ therapy for moderate-to-severe rheumatoid arthritis, the primary efficacy endpoint was achieved with statistical significance at all DAZ dosage regimens relative to placebo.
DAZ was associated with an acceptable safety and tolerability profile with no reported thromboembolic events; however, there was an imbalance in COVID-19 infections.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
The results provide further evidence of the potential therapeutic benefits of targeting the CD40/CD40L pathway in rheumatoid arthritis and other autoimmune disorders.
The safety data from this study are reassuring and suggest that DAZ therapy has an acceptable balance of potential risks and benefits.
Introduction
The CD40/CD40L pathway has been implicated as a key driver in mounting cellular and humoral immune responses and likely plays a role in the pathophysiology of several autoimmune diseases.1–3 The CD40 receptor is a member of the tumour necrosis factor (TNF) family and is expressed on the plasma membrane of antigen-presenting cells, including macrophages, dendritic cells and B cells.4 5 The cognate ligand for CD40 is CD40L (also known as CD154), which is expressed on the plasma membrane of T cells and other cell types, including platelets.6 7 The CD40/CD40L pathway plays a critical role in B cell and T cell activation.1 8 Activation of CD40 is critical for germinal centre (GC) formation, immunoglobulin-class switching and expression of cytokines such as interferon-α, TNFα and interleukin (IL)-6.1 9 10 While the potential therapeutic utility of inhibiting the CD40/CD40L pathway was suggested in previous studies of anti-CD40L monoclonal antibodies (mAbs) in patients with lupus and other autoimmune disorders, the development of these biologics was halted more than two decades ago due to thromboembolic complications found to be secondary to platelet activation mediated by the Fc portion of the mAb.11–16
Dazodalibep (DAZ), a novel non-antibody biological antagonist of CD40L, was designed to circumvent Fc-dependent platelet activation. It comprises two Tn3 proteins fused to human serum albumin.17 18 Tn3, derived from the third fibronectin type III domain of human tenascin-C, is structurally similar to immunoglobulins and exhibits protein folds analogous to the complementarity-determining regions of an antibody. Notably, DAZ was designed to lack an Fc domain; instead, human serum albumin was covalently linked to the bivalent CD40L binding domain to avoid any Fc-mediated effects and extend its half-life.18 DAZ did not induce platelet aggregation in non-clinical studies,19 and no platelet-related safety concerns have been observed thus far in treated subjects.18
In a phase Ib trial of DAZ in 53 patients with moderate-to-severe rheumatoid arthritis (RA), intravenous DAZ 1000 mg and 1500 mg administered every other week for 12 weeks rapidly improved RA disease activity compared with placebo (PBO), with maintenance of clinical responses during the 3-month follow-up period.20 Here, we report the results of a phase II trial (MIDORA; NCT04163991) of DAZ in adult patients with moderate-to-severe RA assessing the safety, efficacy and duration of clinical response after treatment with four different dosage regimens of DAZ. Additionally, we assessed the impact of DAZ treatment on exploratory biomarkers of RA disease activity and biomarkers related to its presumed mechanism of action.
Methods
Study design
This was a multicentre, randomised, double-blind, PBO-controlled, parallel-cohort clinical trial to evaluate the safety and efficacy of DAZ in adult patients with active, moderate-to-severe, adult-onset RA who tested positive for serum rheumatoid factor (RF) and/or anticitrullinated protein antibodies (ACPA), had an inadequate response to methotrexate (MTX), other conventional disease-modifying antirheumatic drugs (cDMARDs) or an anti-TNFα agent, and had not previously received a B-cell depleting agent.
Eligible participants were randomly allocated to five groups in a 1:1:1:1:1 ratio to receive one of four intravenous dosage regimens of DAZ or PBO (online supplemental figure S1) in addition to their ongoing baseline treatment. An interactive web response system was used for block randomisation (block size=5). A participant was considered randomised when the investigator had notified the system that they had met eligibility criteria and the system provided the assignment of treatment group. Participants, investigators, site staff, sponsor and evaluating staff were blinded to treatment assignment.
rmdopen-2023-003317supp001.pdf (800.6KB, pdf)
No change in background treatment was allowed for 12 weeks (day 85), at which time rescue therapy could be instituted if they had persistently active disease. Rescue therapy was considered as any new or intensified cDMARD therapy or initiation of biological DMARD (bDMARD) treatment for RA, including initiation of any new cDMARD or increase in dose of a concomitant cDMARD therapy, initiation of a bDMARD or JAK inhibitor, increase in baseline corticosteroid dose, an intra-articular steroid injection >40 mg methylprednisolone (or its equivalent) or more than one intra-articular steroid injection of any dose. One intra-articular steroid injection ≤40 mg methylprednisolone in one joint was permitted and not considered rescue therapy. All participants were followed at least through the primary analysis (day 113), and those who had not instituted rescue therapy, were followed through day 309 to determine the duration of clinical response. Study visits occurred on days 1, 15, 29, 57, 85, 113, 141, 169, 197, 225, 253, 281 and 309.
Participants randomised to PBO or DAZ 1500 mg×4 received four infusions of saline or DAZ, respectively, on days 1, 15, 29 and 57. DAZ infusions were administered on day 1 for the DAZ 3000 mg×1 group and on days 1 and 57 for the DAZ 1500 mg×2 and DAZ 3000 mg×2 groups. PBO was given on dosing days when DAZ was not administered.
Patients
Key inclusion criteria
The study population included adult participants with a confirmed RA diagnosis at least 6 months prior to screening according to the EULAR/American College of Rheumatology 2010 criteria.21 Participants were required at screening to have moderate-to-severe disease activity as defined by a Disease Activity Score-28 with C reactive protein (DAS28-CRP) score >3.2, tender joint count (TJC) ≥4 and swollen joint count (SJC) ≥4, and to be positive for RF and/or ACPA. Participants were also required to have been treated with MTX (7.5–25.0 mg/week), with or without a concomitant cDMARD via the same route of administration for at least 12 weeks and without a change in dose for at least 6 weeks prior to screening. If not receiving MTX therapy due to intolerance or contraindication, participants must have been treated with one or more cDMARD for at least 12 weeks prior to screening without a change in dose during the past 6 weeks.
Key exclusion criteria
Participants were excluded if they had a prior or current inflammatory joint disease other than RA, severe interstitial lung disease or prior use of B-cell depleting or anti-CD40L agent. Participants were also excluded if they had received any anti-TNFα biological agent within 8 weeks prior to screening (discontinuation could have been for any reason: lack of efficacy, safety/tolerability issues or lack of access to drug). In addition, participants were excluded if they had received any bDMARD with a mechanism of action other than direct TNF blockade, including, abatacept, anti-IL-6R (tocilizumab, sarilumab), rituximab or any JAK inhibitor, within 12 weeks or within five half-lives of the drug (whichever is longer) prior to screening. Additionally, participants were excluded if they had received any experimental therapy within 12 weeks or within five half-lives of the drug (whichever is longer) prior to screening.
Other exclusion criteria included a history of confirmed deep venous thrombosis or arterial thromboembolism within 2 years of enrolment, history of recurrent deep venous thrombosis or arterial thromboembolism and participants with risk factors for venous thromboembolism or arterial thrombosis (eg, immobilisation or major surgery within 12 weeks before screening), prothrombotic status (including, but not limited to, known congenital or inherited deficiency of antithrombin III, protein C, protein S or confirmed diagnosis of catastrophic antiphospholipid syndrome).
Assessments
Efficacy
The primary endpoint was the change from baseline to day 113 in DAS28-CRP. DAS28-CRP was assessed at each study visit through day 309. The DAS28-CRP is a composite score that includes an assessment of 28 specified joints for TJC and SJC, a patient global assessment of disease activity (PGA) and CRP level (mg/L).
Secondary endpoints included the proportion of participants at day 113 with DAS28-CRP-defined clinical remission (DAS28-CRP <2.6) and time to institution of rescue medication. Established definitions of remission and high disease activity are DAS28-CRP <2.6 and DAS28-CRP >5.1, respectively.22 23 Exploratory efficacy assessments through day 309 included the change in Clinical Disease Activity Index (CDAI), Simplified Disease Activity Index (SDAI), TJC, SJC, physician global assessment of disease activity (MDGA), PGA and Health Assessment Questionnaire (including pain) score.
See online supplemental information for pharmacokinetic (PK), immunogenicity and biomarker methodology.
Safety
Safety was monitored through day 309 and included the incidence of treatment-emergent adverse events (AEs), serious adverse events (SAEs) and adverse events of special interest (AESI).
Statistical analysis
The planned sample size of 75 participants (15 participants per treatment group) provided approximately 80% power to detect a difference of 1.2 in the primary efficacy endpoint between the DAZ and PBO treatment groups assuming an SD of 1.25, at a two-sided statistical significance level of 0.10 using a two-sample t-test.
The primary efficacy endpoint and other continuous endpoints were analysed using a mixed model for repeated measures (MMRM) analysis with treatment, visit, visit by treatment interaction and corresponding baseline value included in the model. Data collected after administration of rescue medication were not included in the analysis. Data were included if they were collected after treatment discontinuation for reasons other than rescue. Missing data were handled by the MMRM approach.
For the primary efficacy endpoint analysis, the type I error rate was controlled at the 0.1 level (two-sided) using the following sequential testing strategy: step 1—the primary endpoint was tested for the DAZ 1500 mg×4 groups compared with PBO; step 2—if the p value was ≤0.1 in step 1, the primary endpoint was tested for the DAZ 3000 mg×2 groups compared with PBO; step 3—if the p value was ≤0.1 in both step 1 and step 2, the primary endpoint was tested for the DAZ 1500 mg×2 and DAZ 3000 mg×1 group compared with PBO using the Hochberg’s method.24
The proportion of participants achieving clinical remission at day 113 was analysed using a logistic regression model, with treatment and baseline value included in the model. Participants who received rescue medication before day 113 or had prematurely discontinued before day 113 were considered non-responders. Time to start of rescue medication for RA was analysed using the Cox proportional hazards model with treatment group included in the model; participants not receiving rescue medication were censored at their end of study date.
Safety data were summarised descriptively and by treatment group.
Results
Baseline demographics and disease characteristics and disposition of participants
MIDORA was conducted at 19 sites in the USA and Poland from December 2019 to December 2021. Of 175 patients screened, 78 were randomised and dosed. The mean (SD) age of participants was 56.3 (12.7) years, and the majority were female (79.5%), white (93.6%) and not Hispanic or Latino (96.2%). Demographics and baseline disease characteristics were generally similar across arms, except for RF+ proportion and mean CRP levels (table 1). The average (SD) disease duration was 12.5 (9.8) years.
Table 1.
Demographic and baseline disease characteristics
| Parameter | PBO N=16 |
DAZ 3000 mg×1 N=16 |
DAZ 1500 mg×2 N=16 |
DAZ 3000 mg×2 N=15 |
DAZ 1500 mg×4 N=15 |
Total N=78 |
| Age, mean (SD) | 56.3 (14.0) | 53.2 (10.7) | 59.0 (12.2) | 56.5 (12.2) | 56.4 (15.2) | 56.3 (12.7) |
| Female, n (%) | 12 (75.0) | 14 (87.5) | 11 (68.8) | 13 (86.7) | 12 (80.0) | 62 (79.5) |
| Race, n (%) | ||||||
| Black/African-American | 2 (12.5) | 0 | 0 | 1 (6.7) | 1 (6.7) | 4 (5.1) |
| White | 14 (87.5) | 16 (100) | 15 (93.8) | 14 (93.3) | 14 (93.3) | 73 (93.6) |
| Other | 0 | 0 | 1 (6.3) | 0 | 0 | 1 (1.3) |
| Ethnicity, n (%) | ||||||
| Hispanic or Latino | 1 (6.3) | 1 (6.3) | 1 (6.3) | 0 | 0 | 3 (3.8) |
| Non-Hispanic or Latino | 15 (93.8) | 15 (93.8) | 15 (93.8) | 15 (100) | 15 (100) | 75 (96.2) |
| BMI, mean (SD), kg/m2 | 27.6 (5.5) | 27.6 (3.8) | 28.0 (4.8) | 30.0 (6.0) | 30.1 (7.6) | 28.6 (5.6) |
| DAS28-CRP, mean (SD) | 5.4 (1.1) | 5.5 (0.9) | 5.9 (0.7) | 5.5 (0.6) | 5.8 (0.7) | 5.6 (0.8) |
| CRP, mean (SD), mg/mL | 17.6 (23.7) | 7.2 (12.4) | 14.9 (16.1) | 8.8 (9.2) | 14.1 (13.9) | 12.6 (16.0) |
| RF+, n (%) | 14 (87.5) | 12 (75.0) | 15 (93.8) | 12 (80.0) | 8 (53.3) | 61 (78.2) |
| CDAI, mean (SD) | 36.5 (10.5) | 39.0 (10.2) | 43.5 (10.7) | 35.7 (8.8) | 39.3 (8.8) | 38.9 (10.0) |
| SDAI, mean (SD) | 38.3 (11.6) | 39.7 (10.7) | 45.0 (10.8) | 36.6 (8.9) | 40.7 (9.3) | 40.1 (10.4) |
| TJC, mean (SD) | 13.3 (5.0) | 14.9 (5.1) | 17.5 (5.3) | 13.9 (4.1) | 15.3 (4.8) | 15.0 (5.0) |
| SJC, mean (SD) | 10.5 (4.0) | 9.8 (4.5) | 11.9 (5.1) | 9.3 (3.8) | 9.9 (4.1) | 10.3 (4.3) |
| PGA, mean (SD), mm VAS | 56.6 (21.2) | 71.3 (13.0) | 65.9 (18.9) | 64.6 (16.9) | 69.0 (16.4) | 65.4 (17.8) |
| MDGA, mean (SD), mm VAS | 71.0 (15.3) | 71.9 (12.6) | 75.3 (14.7) | 59.9 (18.3) | 72.4 (12.0) | 70.2 (15.3) |
BMI, body mass index; CDAI, Clinical Disease Activity Index; CRP, C reactive protein; DAS28-CRP, Disease Activity Score in 28 joints using C reactive protein; DAZ, dazodalibep; MDGA, physician global assessment of disease activity; PBO, placebo; PGA, patient global assessment of disease activity; RF, rheumatoid factor; SDAI, Simplified Disease Activity Index; SJC, swollen joint count; TJC, tender joint count; VAS, visual analogue scale.
At baseline, all participants were receiving at least one cDMARD with almost 90% (70/78) receiving MTX. The mean (SD) MTX dose was 17.46 (4.49) mg/week. Approximately 42% (33/78) were receiving a glucocorticoid for the treatment of RA at a mean (SD) prednisone-equivalent dose of 6.09 (2.43) mg/day. Baseline use of any cDMARD was balanced between the treatment groups; however, there was an imbalance in glucocorticoid use (PBO: 50.0%; DAZ groups: 33.3%–43.8%). Approximately 21% (16/78) of participants had received prior anti-TNFα agents. A total of 73 (94%) participants completed treatment, and 65 (83%) completed the study follow-up to day 309 (figure 1). Thirteen participants discontinued the study (AE: 1 (1.3%); death: 1 (1.3%); lost to follow-up: 1 (1.3%); withdrawal: 7 (9.0%); other: 3 (3.8%)).
Figure 1.

Disposition of MIDORA participants. Data reported as n (%). DAZ, dazodalibep.
Efficacy
A statistically significant improvement in DAS28-CRP score at day 113, the primary endpoint, was observed in all DAZ groups relative to PBO (LS mean±SE); DAZ 3000 mg×1: −1.90±0.27 (p=0.0296); DAZ 1500 mg×2: −1.87±0.27 (p=0.0355); DAZ 3000 mg×2: −1.87±0.27 (p=0.0364); DAZ 1500 mg×4: −1.83±0.28 (p=0.0478); PBO: −1.06±0.26 (figure 2, table 2). The DAS28-CRP component scores are presented in online supplemental figure S2. Reductions from baseline in DAS28-CRP were also found in all DAZ groups at day 309. In the DAZ 3000 mg×2 groups, separation from PBO was achieved at all timepoints after day 113.
Figure 2.
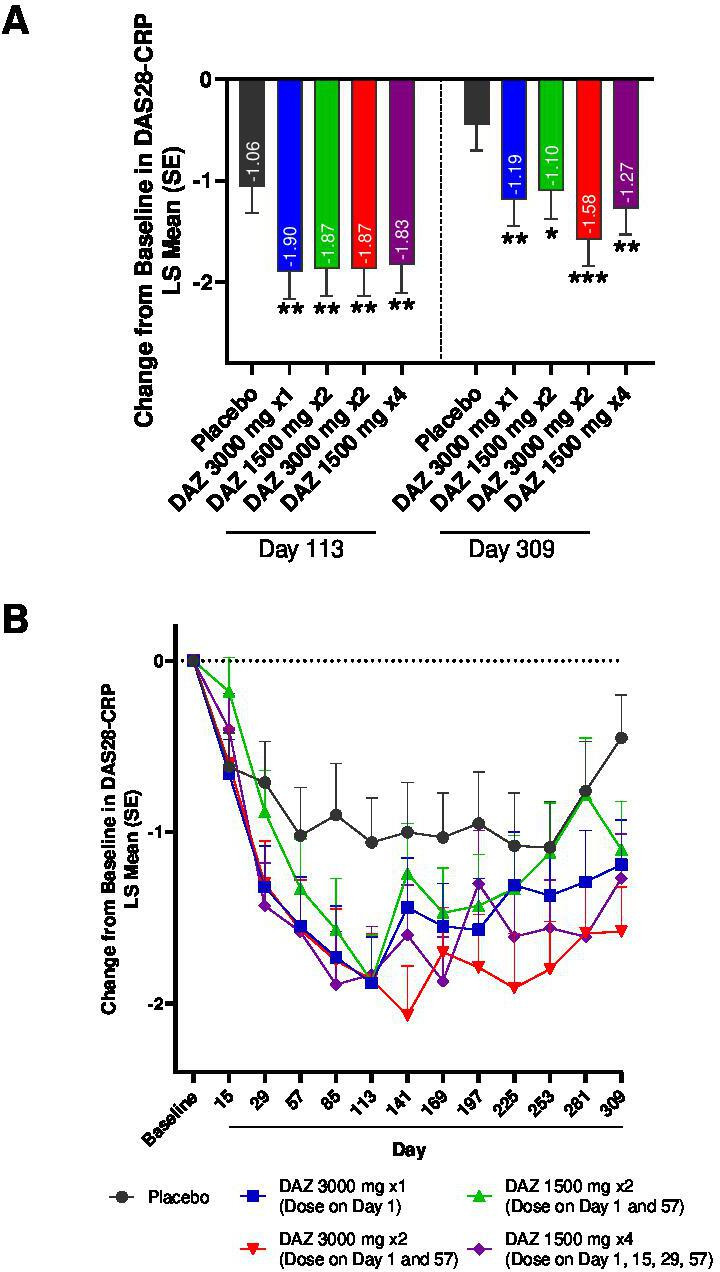
Change from baseline in DAS28-CRP. (A) MMRM results for DAS28-CRP at day 113 and day 309; *p<0.1; **p<0.05; ***p<0.01. (B) Change from baseline in DAS28-CRP plotted by study visit. DAS28-CRP, Disease Activity Score in 28 joints using C reactive Protein; DAZ, dazodalibep; LS, least squares; MMRM, mixed model for repeated measures.
Table 2.
Primary and secondary efficacy endpoints
| PBO N=16 |
DAZ 3000 mg×1 N=16 |
DAZ 1500 mg×2 N=16 |
DAZ 3000 mg×2 N=15 |
DAZ 1500 mg×4 N=15 |
|
| DAS28-CRP, n | 15 | 14 | 15 | 14 | 14 |
| LS mean CFB (SE) | −1.06 (0.26) | −1.90 (0.27) | −1.87 (0.27) | −1.87 (0.27) | −1.83 (0.28) |
| LS mean difference (SE) | −0.84 (0.38) | −0.81 (0.38) | −0.81 (0.38) | −0.77 (0.38) | |
| P value | 0.0296 | 0.0355 | 0.0364 | 0.0478 | |
| DAS28-CRP <2.6, n | 16 | 16 | 16 | 15 | 15 |
| Responder, n (%) | 3 (18.8) | 3 (18.8) | 1 (6.3) | 2 (13.3) | 2 (13.3) |
| OR (90% CI) | 1.4 (0.2 to 8.0) | 0.6 (<0.1 to 5.5) | 0.9 (0.1 to 5.7) | 0.9 (0.2 to 5.1) | |
| P value | 0.7750 | 0.6974 | 0.9318 | 0.9108 | |
| Time to start rescue medication | |||||
| Rescued, n (%) | 1 (6.3) | 1 (6.3) | 1 (6.3) | 3 (20.0) | 1 (6.7) |
| Censored, n (%) | 15 (93.8) | 15 (93.8) | 15 (93.8) | 12 (80.0) | 14 (93.3) |
| HR (90% CI) | 1.04 (0.10 to 10.60) | 1.04 (0.10 to 10.60) | 3.01 (0.45 to 20.09) | 1.04 (0.10 to 10.60) | |
| P value | 0.9805 | 0.9805 | 0.3407 | 0.9805 | |
DAS28-CRP score (primary endpoint) and DAS28-CRP-defined clinical remission (DAS28-CRP <2.6; secondary endpoint) at day 113; time to start rescue medication (secondary endpoint) through day 309.
CFB, change from baseline; DAS28-CRP, Disease Activity Score in 28 joints using C reactive protein; LS, least-squares; PBO, placebo.
The secondary efficacy endpoints are presented in table 2. DAS28-CRP clinical remission rates were low and similar in all groups (PBO: 18.8%; DAZ groups: 6.3%–18.8%), but fewer DAZ participants had high disease activity at day 113 (PBO group: 40.0%; DAZ groups: 7.1%–14.3%). Few participants (PBO: n=1/16; DAZ: n=6/62) received rescue medication (none before day 113), and time to rescue medication did not differ between DAZ and PBO.
The change from baseline (LS mean±SE) scores for exploratory efficacy endpoints are presented in table 3. A greater reduction in SDAI score at day 113 was achieved in all DAZ groups and ranged from −21.20 to −23.99 (p=0.0944 for DAZ 1500 mg×4; p<0.05 for all other DAZ groups) compared with PBO (−14.17). The ranges were similar for CDAI scores. The magnitude of improvement in SJC at day 113 was greater in the four DAZ groups (−6.5 to −8.2) compared with PBO (−4.4) with separation from PBO occurring in the DAZ 3000 mg×1 (p=0.0895), DAZ 1500 mg×2 (p=0.0081) and DAZ 3000 mg×2 (p=0.0317) groups. Improvement in TJC at day 113 in the DAZ groups ranged from −8.0 to −9.8 (PBO: −5.8); separation from PBO was achieved for the DAZ 3000 mg×1 (p=0.0542) and DAZ 3000 mg×2 (p=0.0712) groups.
Table 3.
Exploratory efficacy endpoints
| PBO N=16 |
DAZ 3000 mg×1 N=16 |
DAZ 1500 mg×2 N=16 |
DAZ 3000 mg×2 N=15 |
DAZ 1500 mg×4 N=15 |
|
| CDAI, n | 15 | 14 | 15 | 14 | 14 |
| LS mean CFB (SE) | −13.47 (2.87) | −22.22 (2.89) | −23.28 (2.93) | −22.98 (2.94) | −20.84 (2.98) |
| LS mean difference (SE) | −8.74 (4.07) | −9.81 (4.13) | −9.50 (4.09) | −7.36 (4.14) | |
| P value | 0.0356 | 0.0204 | 0.0233 | 0.0799 | |
| SDAI, n | 15 | 14 | 15 | 14 | 14 |
| LS mean CFB (SE) | −14.17 (2.87) | −22.61 (2.90) | −23.99 (2.93) | −22.95 (2.94) | −21.20 (2.98) |
| LS mean difference (SE) | −8.44 (4.07) | −9.82 (4.12) | −8.78 (4.10) | −7.03 (4.14) | |
| P value | 0.0424 | 0.0202 | 0.0360 | 0.0944 | |
| TJC, n | 15 | 14 | 15 | 14 | 14 |
| LS mean CFB (SE) | −5.8 (1.5) | −9.8 (1.5) | −9.1 (1.5) | −9.6 (1.5) | −8.0 (1.5) |
| LS mean difference (SE) | −4.1 (2.1) | −3.4 (2.1) | −3.8 (2.1) | −2.3 (2.1) | |
| P value | 0.0542 | 0.1168 | 0.0712 | 0.2849 | |
| SJC, n | 15 | 14 | 15 | 14 | 14 |
| LS mean CFB (SE) | −4.4 (1.0) | −6.8 (1.0) | −8.2 (1.0) | −7.5 (1.0) | −6.5 (1.0) |
| LS mean difference (SE) | −2.4 (1.4) | −3.8 (1.4) | −3.1 (1.4) | −2.1 (1.4) | |
| P value | 0.0895 | 0.0081 | 0.0317 | 0.1429 | |
| PGA, n | 15 | 14 | 15 | 14 | 14 |
| LS mean CFB (SE) | −7.1 (5.3) | −19.8 (5.5) | −15.4 (5.3) | −21.6 (5.5) | −21.9 (5.5) |
| LS mean difference (SE) | −12.7 (7.7) | −8.4 (7.6) | −14.5 (7.6) | −14.9 (7.7) | |
| P value | 0.1030 | 0.2724 | 0.0612 | 0.0588 | |
| MDGA, n | 15 | 14 | 15 | 14 | 14 |
| LS mean CFB (SE) | −26.1 (5.3) | −38.0 (5.4) | −40.4 (5.4) | −38.2 (5.6) | −42.1 (5.6) |
| LS mean difference (SE) | −12.0 (7.6) | −14.3 (7.6) | −12.1 (7.7) | −16.0 (7.7) | |
| P value | 0.1215 | 0.0642 | 0.1213 | 0.0425 | |
| HAQ, n | 15 | 14 | 15 | 14 | 14 |
| LS mean CFB (SE) | −0.27 (0.10) | −0.31 (0.11) | −0.16 (0.10) | −0.27 (0.11) | −0.26 (0.11) |
| LS mean difference (SE) | −0.04 (0.15) | 0.11 (0.15) | −0.01 (0.15) | 0.00 (0.15) | |
| P value | 0.7692 | 0.4784 | 0.9641 | 0.9749 | |
| Pain, n | 15 | 14 | 15 | 14 | 14 |
| LS mean CFB (SE) | −8.8 (5.0) | −14.1 (5.0) | −9.9 (4.9) | −19.7 (5.1) | −21.8 (5.1) |
| LS mean difference (SE) | −5.3 (7.1) | −1.1 (7.0) | −10.9 (7.1) | −13.0 (7.2) | |
| P value | 0.4598 | 0.8739 | 0.1284 | 0.0754 |
Exploratory efficacy endpoints at day 113.
CDAI, Clinical Disease Activity Index; CFB, change from baseline; DAZ, dazodalibep; HAQ, Health Assessment Questionnaire; LS, least-squares; MDGA, physician global assessment of disease activity; PBO, placebo; PGA, patient global assessment of disease activity; SDAI, Simplified Disease Activity Index; SJC, swollen joint count; TJC, tender joint count.
At day 113, there was a greater magnitude of improvement in PGA score in each of the DAZ groups (−15.4 to −21.9) compared with PBO (−7.1); separation from PBO was achieved for the DAZ 3000 mg×2 and DAZ 1500 mg×4 groups (p=0.0612 and p=0.0588, respectively). Similarly, there was numerically greater improvement from baseline in MDGA score at day 113 in DAZ groups (−38.0 to −42.1) relative to PBO (−26.1), with separation from PBO in the DAZ 1500 mg×2 groups (p=0.0642) and DAZ 1500 mg×4 groups (p=0.0425). A substantially greater change from baseline in HAQ-Pain score at day 113 was observed in the DAZ 1500 mg×4 groups relative to PBO (−21.8 vs −8.8; p=0.0754). The magnitude of change from baseline in HAQ-Pain score at day 113 was −14.1, –9.9 and −19.7 in the DAZ 3000 mg×1, DAZ 1500 mg×2 and DAZ 3000 mg×2 groups, respectively.
Pharmacokinetics and immunogenicity
Mean plasma concentration-time profiles for DAZ are shown in figure 3, and a summary of the PK parameters is shown in online supplemental table S1. DAZ plasma concentrations were below the limit of quantification for all participants receiving PBO. Following infusion of DAZ (1500 and 3000 mg), the mean concentration-time profiles showed parallel terminal phase and DAZ area under the concentration-time curve (AUC) and maximal concentration (Cmax) that increased in an approximately dose-proportional manner. After the first dose, the mean Cmax values ranged from 421 to 476 µg/mL and 860–877 µg/mL in DAZ 1500 mg and 3000 mg groups, respectively; the mean AUC from time 0 to day 56 was 4280 hours×μg/mL and 7280–7870 hours×μg/mL. The terminal elimination half-life (t1/2) was comparable across all DAZ groups (9–10 days).
Figure 3.
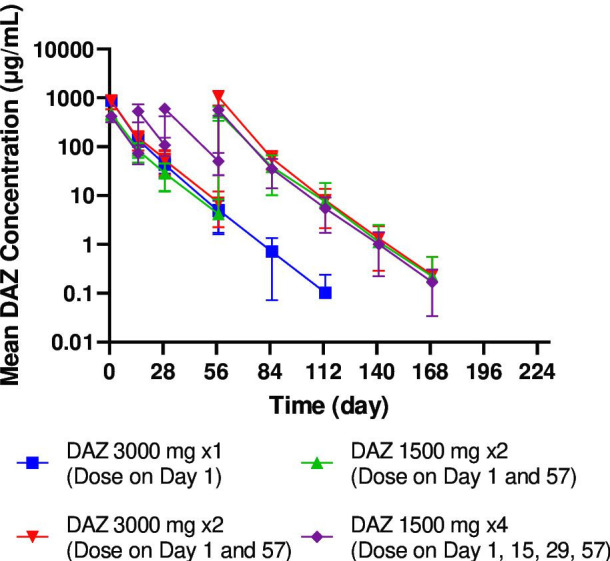
Concentration-time profile of DAZ. The mean (±SD) concentration-time profiles of DAZ following intravenous infusion. DAZ, dazodalibep.
None of the 62 participants treated with DAZ had ADA detected at baseline, and no participants in the PBO group tested ADA positive. A total of 19 participants (30.6%) tested ADA positive postbaseline (online supplemental table S2), with the earliest detectable ADA observed at day 85. Titres ranged from <60 to 480. Participants with positive ADA results had slightly lower exposure and slightly higher total body clearance (CL) when compared with participants with negative ADA results (online supplemental table S3 and online supplemental figure S3), however, there was no clear impact of ADAs on the PK profile based on a comparison of CL between the first and second dose.
Biomarkers
The impact of DAZ treatment on RF and ACPA levels (adjusted geometric mean ratio to baseline (90% CI)) is shown in figure 4A–B. At day 113, RF levels dropped and separated from PBO for all DAZ groups (p≤0.0007) with values ranging from 0.57 (0.49 to 0.66) to 0.77 (0.66 to 0.89) IU/mL for DAZ groups relative to 1.20 (1.04 to 1.39) IU/mL for PBO. Separation was observed starting at day 57 and sustained through day 113 (p≤0.0035) for all DAZ dosage regimens. The changes in RF isotype levels are presented in online supplemental figure S4. ACPA levels were reduced for the DAZ 3000 mg×1 group (0.69 (0.52 to 0.91) U/mL; p=0.0584) and DAZ 1500 mg×4 groups (0.62 (0.47 to 0.82) U/mL; p=0.0199) compared with PBO (1.08 (0.83 to 1.42)) at day 113. The groups receiving DAZ 1500 mg×2 and DAZ 3000 mg×2 showed a reduction in ACPA levels relative to PBO at day 113 (0.82 (0.62 to 1.07) and 0.84 (0.63 to 1.11) U/mL, respectively), but these differences were modest and not statistically significant. Serum total IgG, IgA and IgM is presented in online supplemental figure S5.
Figure 4.

DAZ biomarkers. Line plots of DAZ biomarkers by study visit for (A) RF, (B) ACPA, (C) total sCD40L and (D) CXCL13. ACPA, anticitrullinated protein antibody; CXCL13, C-X-C motif chemokine ligand 13; DAZ, dazodalibep; MAD, median absolute deviation; RF, rheumatoid factor; sCD40L, soluble CD40L.
The effect of DAZ treatment on total soluble CD40L (sCD40L) and C-X-C motif chemokine ligand 13 (CXCL13) levels is presented in figure 4C–D. Increases from baseline in total sCD40L (median±median absolute deviation) were observed in all DAZ dosage groups at day 15, the earliest sampling timepoint. Increases peaked about day 85 in the groups receiving multiple doses of DAZ and returned to baseline values by day 197. In the 3000 mg×1 group, sCD40L levels peaked at day 57 and returned to baseline earlier, at day 141. Maximum reduction in CXCL13 levels was observed in all DAZ groups at day 29 (p≤0.0013 for all DAZ groups) with DAZ 1500 mg×4 maintaining reduction at day 57. An additional reduction in CXCL13 levels was observed relative to PBO at day 85 in cohorts receiving DAZ at day 57 (DAZ 1500 mg×2: p=0.0017, DAZ 3000 mg×2: p=0.0006 and DAZ 1500 mg×4; p=0.0002). CXCL13 levels in these groups began to recover after day 85 and returned to baseline levels by day 141, while in the DAZ 3000 mg×1 group, CXCL13 levels returned by baseline by approximately day 85.
Safety
Treatment-emergent AEs are summarised in table 4. The majority (91.0% (71/78)) of participants received all four doses of study medication (DAZ groups: 80.0%–93.8%; PBO: 93.8%). The proportion of participants reporting at least one AE was relatively balanced among treatment groups (DAZ groups: 74.2% (46/62); PBO: 62.5% (10/16)). Similarly, the proportion of participants with AEs deemed by investigators to be related to study medication was similar between DAZ groups (14.3%–23.5%) and PBO (25.0%). There were four SAEs in three participants randomised to a DAZ group, all considered by the investigators to be unrelated to study drug: one nephrolithiasis, one COVID-19 infection that occurred the evening of dose 1 (also an AESI) and one participant hospitalised for COVID-19 pneumonia (also an AESI) who died from unknown cause 3 days after discharge (233 days after last dose). Full safety narratives pertaining to the SAEs observed on study are presented in the online supplemental information. There were no SAEs in the PBO group.
Table 4.
Treatment-emergent AEs
| Parameter, n (%) preferred term |
PBO N=16 |
DAZ 3000 mg×1 N=18 |
DAZ 1500 mg×2 N=17 |
DAZ 3000 mg×2 N=13 |
DAZ 1500 mg×4 N=14 |
DAZ total N=62 |
| ≥1 AE | 10 (62.5) | 10 (55.6) | 14 (82.4) | 11 (84.6) | 11 (78.6) | 46 (74.2) |
| ≥1 AE related to study drug | 4 (25.0) | 3 (16.7) | 4 (23.5) | 2 (15.4) | 2 (14.3) | 11 (17.7) |
| ≥1 AE leading to death | 0 | 0 | 1 (5.9) | 0 | 0 | 1 (1.6) |
| ≥1 AE leading to discontinuation | 0 | 1 (5.6) | 0 | 0 | 0 | 1 (1.6) |
| ≥1 SAE | 0 | 1 (5.6) | 1 (5.9) | 0 | 1 (7.1) | 3 (4.8) |
| ≥1 SAE related to study drug | 0 | 0 | 0 | 0 | 0 | 0 |
| ≥1 AESI | 0 | 1 (5.6) | 1 (5.9) | 0 | 0 | 2 (3.2) |
| Most common AEs* | ||||||
| COVID-19 | 1 (6.3) | 2 (11.1) | 3 (17.6) | 0 | 3 (21.4) | 8 (12.9) |
| Rheumatoid arthritis | 1 (6.3) | 0 | 3 (17.6) | 2 (15.4) | 1 (7.1) | 6 (9.7) |
| Alanine aminotransferase increased |
0 | 1 (5.6) | 2 (11.8) | 1 (7.7) | 0 | 4 (6.5) |
| Spinal osteoarthritis | 0 | 2 (11.1) | 2 (11.8) | 0 | 0 | 4 (6.5) |
| Upper respiratory tract infection |
1 (6.3) | 1 (5.6) | 0 | 3 (23.1) | 0 | 4 (6.5) |
| Hypertension | 0 | 1 (5.6) | 0 | 1 (7.7) | 1 (7.1) | 3 (4.8) |
| Nasopharyngitis | 0 | 0 | 1 (5.9) | 1 (7.7) | 1 (7.1) | 3 (4.8) |
| Osteoarthritis | 0 | 0 | 0 | 3 (23.1) | 0 | 3 (4.8) |
| Pharyngitis | 0 | 0 | 0 | 2 (15.4) | 1 (7.1) | 3 (4.8) |
| Anaemia | 1 (6.3) | 2 (11.1) | 0 | 0 | 0 | 2 (3.2) |
| Arthralgia | 0 | 0 | 1 (5.9) | 0 | 1 (7.1) | 2 (3.2) |
| Aspartate aminotransferase increased |
0 | 0 | 2 (11.8) | 0 | 0 | 2 (3.2) |
| Back pain | 0 | 1 (5.6) | 1 (5.9) | 0 | 0 | 2 (3.2) |
| Hypercholesterolaemia | 0 | 1 (5.6) | 0 | 1 (7.7) | 0 | 2 (3.2) |
| Lymphopenia | 0 | 0 | 2 (11.8) | 0 | 0 | 2 (3.2) |
| Pyrexia | 0 | 0 | 1 (5.9) | 0 | 1 (7.1) | 2 (3.2) |
| Cystitis | 2 (12.5) | 0 | 0 | 0 | 1 (7.1) | 1 (1.6) |
| Urinary tract infection | 2 (12.5) | 0 | 1 (5.9) | 0 | 0 | 1 (1.6) |
*Reported in ≥2 participants in PBO group or ≥2 participants in combined DAZ groups.
AE, adverse event; AESI, adverse event of special interest; PBO, placebo; SAE, serious adverse event.
There was no imbalance of events in the infection and infestation system organ class between DAZ and PBO (32.3% and 31.3%, respectively); however, an imbalance in COVID-19 infections was observed, with COVID-19 infection reported by 12.9% (8/62) of DAZ recipients compared with 6.3% (1/16) who received PBO. In DAZ groups, one COVID-19 event occurred on the evening of dose 1, the others 51–279 days after dose 1 (16–223 days from most recent dose). Except for the two SAEs related to COVID-19 (COVID-19 and COVID-19 pneumonia), which were considered to be severe, other COVID-19 AEs were mild-to-moderate in severity. No participant with a COVID-19 infection during the trial was intubated or treated in an intensive care unit.
Discussion
In this phase II trial of adults with moderate-to-severe RA, DAZ treatment significantly improved RA disease activity in all dosage groups as assessed by the change from baseline in DAS28-CRP at day 113 (primary endpoint). The observed improvement with DAZ treatment in DAS28-CRP scores was supported by the favourable effects of this agent on multiple components of the composite score. Although with some fluctuation, much of the improvement in disease activity appeared to be sustained in the DAZ treatment groups relative to PBO through day 197, despite receipt of the last dose at day 57 by participants in the 1500 mg×2, 3000 mg×2 and 1500 mg×4 groups and administration of a single dose at day 1 in the 3000 mg×1 group. A dose-response relationship was previously demonstrated in the phase Ib trial of DAZ in patients with RA that investigated doses ranging from 75 to 1500 mg administered every other week.18 The current study was not designed to formally test dose-response but to expand on the findings of the phase Ib study and explore the potential efficacy of higher doses of DAZ administered at intervals substantially longer than the every 2-week dosing interval tested in the phase Ib study. The efficacy observed across all dosage regimens strongly supports further investigation of dosing intervals of 4 weeks and longer, particularly at the 3000 mg dose. Notably, the improvement in RA disease activity was achieved without evidence of thromboembolic complications, as expected based on the lack of an Fc domain in DAZ and consistent with previous non-clinical/clinical studies.18 19
During the study, we observed an imbalance in COVID-19 infections between the PBO and DAZ groups. It is possible that DAZ treatment may increase the risk for COVID-19 infection, which would comport with its known mechanisms of action. However, given the small sample size, particularly in the PBO group, the potential differences in epidemiological risk and other possible confounding factors (study began in the pre-COVID-19 era, and COVID-19 disease activity varied widely by region), we are uncertain about the extent to which DAZ therapy predisposes to COVID-19-related illness and its severity; we will closely monitor for COVID-19 infection in future studies with appropriate safety guidelines in place. SARS-CoV2 vaccination became available during the study period (later in Poland than in the USA) and as a result, only 44.9% of participants randomised to DAZ treatment were vaccinated before or during the study; only one of the participants who developed a COVID-19 infection during the trial reported having been vaccinated prior to developing this infection. Despite this lack of protective immunity to the SARS-CoV2 virus, no participant was severely ill due to COVID-19. An autopsy was not performed on the participant experiencing the SAE of death, and the cause of death could not be determined.
DAZ PK exposures (AUC and Cmax) were dose-proportional over the twofold dose range tested and the t1/2 was comparable across groups. Approximately 31% of participants receiving DAZ had detectable ADAs, which were, at most, only four-fold greater than the lowest minimum detectable titre level (60); there was no clear impact of ADAs on the PK profile based on a comparison of CL between the first and second dose. Moreover, ADA-positive participants exhibited lower exposures after the first dose on day 15, which was much earlier than when ADAs were first detected (the earliest positive ADA timepoint was day 85), suggesting many ADA-positive participants exhibited overall high CL initially, and that the occurrence of ADA is unlikely to impact efficacy. Interestingly, the clinical effect on disease activity seemed to be maintained beyond the last detectable level of DAZ in the circulation, particularly in the DAZ 3000 mg×2 groups, where most of the clinical response was maintained to at least day 253.
Total sCD40L and CXCL13 levels were evaluated as biomarkers of proximal and distal target engagement relevant to the CD40L/CD40 pathway. CXCL13, a biomarker of GC activity, is secreted by T follicular helper cells and is a key factor in the regulation of lymphoid organ development, ectopic GC development and plasma cell differentiation.25 Elevated plasma levels are found in patients with active RA and other autoimmune diseases.26 27 DAZ, in binding to CD40L, prevents the interaction between B and T cells through this pathway and has been shown to dissolve established GCs.19 Total sCD40L levels increased from baseline through day 85, indicative of DAZ binding sCD40L. The increase in total sCD40L levels was independent of DAZ dose consistent with maximum accumulation being reached at doses >300 mg.18 The reduction in serum CXCL13 levels likely reflects at least in part the inhibitory effect of DAZ on GCs and may be a leading indicator of clinical and serological responses. CXCL13 is also secreted by synovial plasmacytoid dendritic cells and T peripheral helper cells28; however, the impact of DAZ on these signalling pathways remains to be fully elucidated. Significant reductions in RF were seen in all dosage cohorts by day 113, with modest reductions in ACPA levels by day 113 to day 169. These reductions reached their nadir and were sustained after CXCL13 had returned to baseline. Taken together, the clinical efficacy and biomarker responses, the results suggest that the interval between DAZ doses could be greater than every 2 weeks and yet produce a significant treatment benefit in RA.
The study population assessed was predominantly white, and not Hispanic or Latino, and therefore, the results reported here may not be generalisable to other racial or ethnic groups. Moreover, this study was designed to explore the effect of a short (8 weeks or less) treatment period on the duration of clinical responses. Longer studies with repeated treatments at extended intervals are needed to establish the efficacy of DAZ for the chronic treatment of RA. Notably, the small sample sizes in each of the dosage groups prevent us from making any definitive interpretations about the effect of treatment on the secondary and exploratory endpoints, although positive trends (greater numerical improvement in DAZ groups relative to PBO) were observed for many of the exploratory efficacy endpoints. Additionally, a total of 13 (16.7%) participants discontinued the study (DAZ groups: 12; PBO: 1), which may have impacted our interpretation of the results during the follow-up period at timepoints after day 113.
In summary, the initially observed efficacy of DAZ therapy for RA shown in a phase Ib study was confirmed by the results here and provides additional information regarding its dosing, PK, duration of clinical response and impact on biomarkers related to its mechanism of action. These study results support the continued evaluation of DAZ for the treatment of RA and other autoimmune disorders. DAZ dosing with more widely spaced intervals should be investigated in future clinical trials as they may be sufficient to provide sustained clinical benefits and allow for a greater margin of safety.
Acknowledgments
Medical writing support was provided by Brendan Lujan, PhD, of Horizon Therapeutics. The authors thank Tiffany Ward and Nanette Mittereder of Horizon Therapeutics for sample testing efforts, and Jörn Drappa and Bill Rees for providing critical review of the manuscript.
Footnotes
Correction notice: This article has been corrected since it was first published online. Figure 4 has been updated.
Contributors: Provided substantial contributions to the conception or design of the work; or acquisition, analysis or interpretation of data for the work: AK, LW, IA, MG, JF, GI, EWS. Drafting the work or revising it critically for important intellectual content: AK, LW, IA, MG, JF, GI, EWS. Final approval of the version to be published: AK, LW, IA, MG, JF, GI, EWS. Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved: AK, LW, IA, MG, JF, GI, EWS.
Funding: This study was funded by Horizon Therapeutics.
Competing interests: AK has received study funding, medical writing support, and article processing charges from Amgen. He has received consulting fees from AXDEV Group, Pfizer, Janssen, Boehringer Ingelheim, AbbVie, Flexion, Gilead, Grunenthal, Orion, Regeneron, Sun Pharma Advance Research, and ECOR1. He has received payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events from Merck & Co, Eli Lilly, Novartis, Pfizer, Flexion, AbbVie, Amgen, Genentech, Regeneron, UCB, Horizon, and GSK. He has participated in a data safety monitoring board for AbbVie and Amgen. He has been part of a board or advisory board for AbbVie, Bendcare, Boehringer Ingelheim, ChemoCentryx, Flexion, Gilead, Grunenthal, Horizon, Eli Lilly, Janssen, Pfizer, Regeneron, UCB, and Novartis. He has stock or stock options in Pfizer, GSK, Gilead, Novartis, and Amgen. LW, IA are current employees of Horizon Therapeutics and own stock. MG, JF and GI are former employees of Horizon Therapeutics and own stock. EWS has consulted for Horizon Therapeutics, Bristol Myers Squibb, CSL Behring, Resolve Therapeutics, and Sonoma Biotherapeutics, and receives royalties from UpToDate.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data are available on reasonable request. These data are not available in a repository but available on reasonable request.
Ethics statements
Patient consent for publication
Not applicable.
Ethics approval
This study was approved by Copernicus Group Independent Review Board (Tracking Number: 20192633). Participants gave informed consent to participate in the study before taking part.
References
- 1.Karnell JL, Rieder SA, Ettinger R, et al. Targeting the CD40-CD40L pathway in autoimmune diseases: humoral immunity and beyond. Adv Drug Deliv Rev 2019;141:92–103. 10.1016/j.addr.2018.12.005 [DOI] [PubMed] [Google Scholar]
- 2.Daoussis D, Antonopoulos I, Andonopoulos AP, et al. Increased expression of CD154 (CD40L) on stimulated T-cells from patients with psoriatic arthritis. Rheumatology (Oxford) 2007;46:227–31. 10.1093/rheumatology/kel229 [DOI] [PubMed] [Google Scholar]
- 3.Desai-Mehta A, Lu L, Ramsey-Goldman R, et al. Hyperexpression of CD40 ligand by B and T cells in human lupus and its role in pathogenic autoantibody production. J Clin Invest 1996;97:2063–73. 10.1172/JCI118643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Croft M, Benedict CA, Ware CF. Clinical targeting of the TNF and TNFR Superfamilies. Nat Rev Drug Discov 2013;12:147–68. 10.1038/nrd3930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banchereau J, Dubois B, Fayette J, et al. Functional Cd40 antigen on B cells, Dendritic cells and fibroblasts. Adv Exp Med Biol 1995;378:79–83. [DOI] [PubMed] [Google Scholar]
- 6.Grewal IS, Flavell RA. A central role of CD40 ligand in the regulation of CD4+ T-cell responses. Immunol Today 1996;17:410–4. 10.1016/0167-5699(96)10030-x [DOI] [PubMed] [Google Scholar]
- 7.van Kooten C, Banchereau J. CD40-CD40 ligand. J Leukoc Biol 2000;67:2–17. 10.1002/jlb.67.1.2 [DOI] [PubMed] [Google Scholar]
- 8.Ford ML, Adams AB, Pearson TC. Targeting co-stimulatory pathways: transplantation and autoimmunity. Nat Rev Nephrol 2014;10:14–24. 10.1038/nrneph.2013.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawabe T, Naka T, Yoshida K, et al. The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity 1994;1:167–78. 10.1016/1074-7613(94)90095-7 [DOI] [PubMed] [Google Scholar]
- 10.Stout RD, Suttles J. The many roles of CD40 in cell-mediated inflammatory responses. Immunol Today 1996;17:487–92. 10.1016/0167-5699(96)10060-i [DOI] [PubMed] [Google Scholar]
- 11.Boumpas DT, Furie R, Manzi S, et al. A short course of Bg9588 (anti-CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum 2003;48:719–27. 10.1002/art.10856 [DOI] [PubMed] [Google Scholar]
- 12.Vazquez-Lombardi R, Phan TG, Zimmermann C, et al. Challenges and opportunities for non-antibody scaffold drugs. Drug Discov Today 2015;20:1271–83. 10.1016/j.drudis.2015.09.004 [DOI] [PubMed] [Google Scholar]
- 13.Dumont FJ. IDEC-131. IDEC/Eisai. Curr Opin Investig Drugs 2002;3:725–34. [PubMed] [Google Scholar]
- 14.Kawai T, Andrews D, Colvin RB, et al. Thromboembolic complications after treatment with Monoclonal antibody against CD40 ligand. Nat Med 2000;6:114. 10.1038/72162 [DOI] [PubMed] [Google Scholar]
- 15.Langer F, Ingersoll SB, Amirkhosravi A, et al. The role of CD40 in CD40L- and antibody-mediated platelet activation. Thromb Haemost 2005;93:1137–46. 10.1160/TH04-12-0774 [DOI] [PubMed] [Google Scholar]
- 16.Robles-Carrillo L, Meyer T, Hatfield M, et al. Anti-Cd40L immune complexes potently activate platelets in vitro and cause thrombosis in FCGR2A transgenic mice. J Immunol 2010;185:1577–83. 10.4049/jimmunol.0903888 [DOI] [PubMed] [Google Scholar]
- 17.Oganesyan V, Ferguson A, Grinberg L, et al. Fibronectin type III domains engineered to bind Cd40L: cloning, expression, purification, crystallization and preliminary X-ray diffraction analysis of two complexes. Acta Crystallogr Sect F Struct Biol Cryst Commun 2013;69(Pt 9):1045–8. 10.1107/S1744309113022847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karnell JL, Albulescu M, Drabic S, et al. A CD40L-targeting protein reduces autoantibodies and improves disease activity in patients with autoimmunity. Sci Transl Med 2019;11. 10.1126/scitranslmed.aar6584 [DOI] [PubMed] [Google Scholar]
- 19.Nicholson SM, Casey KA, Gunsior M, et al. The enhanced Immunopharmacology of VIB4920, a novel Tn3 fusion protein and CD40L antagonist, and assessment of its safety profile in cynomolgus monkeys. Br J Pharmacol 2020;177:1061–76. 10.1111/bph.14897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ali O, Wang L, Xu W, et al. OP0120 duration of clinical efficacy following treatment with VIB4920 in subjects with moderate to severe rheumatoid arthritis. Ann Rheum Dis 2021;80:67. 10.1136/annrheumdis-2021-eular.2544 [DOI] [Google Scholar]
- 21.Aletaha D, Neogi T, Silman AJ, et al. 2010 rheumatoid arthritis classification criteria: an American College of rheumatology/European League against rheumatism collaborative initiative. Arthritis Rheum 2010;62:2569–81. 10.1002/art.27584 [DOI] [PubMed] [Google Scholar]
- 22.Anderson J, Caplan L, Yazdany J, et al. Rheumatoid arthritis disease activity measures: American college of rheumatology recommendations for use in clinical practice. Arthritis Care Res (Hoboken) 2012;64:640–7. 10.1002/acr.21649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wells G, Becker J-C, Teng J, et al. Validation of the 28-joint disease activity score (DAS28) and European League against rheumatism response criteria based on C-reactive protein against disease progression in patients with rheumatoid arthritis, and comparison with the DAS28 based on Erythrocyte sedimentation rate. Ann Rheum Dis 2009;68:954–60. 10.1136/ard.2007.084459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hochberg Y. A sharper bonferroni procedure for multiple tests of significance. Biometrika 1988;75:800–2. 10.1093/biomet/75.4.800 [DOI] [Google Scholar]
- 25.Yoshitomi H. Peripheral helper T cell responses in human diseases. Front Immunol 2022;13:946786. 10.3389/fimmu.2022.946786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu T, Pan Z, Zhang N. Elevated Cxcl13 in primary Sjögren’s syndrome and its correlation with disease activity: a systematic review and meta-analysis. Clin Rheumatol 2022;41:2791–802. 10.1007/s10067-022-06210-2 [DOI] [PubMed] [Google Scholar]
- 27.Jones JD, Hamilton BJ, Challener GJ, et al. Serum C-X-C motif chemokine 13 is elevated in early and established rheumatoid arthritis and correlates with rheumatoid factor levels. Arthritis Res Ther 2014;16:R103. 10.1186/ar4552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshitomi H, Ueno H. Shared and distinct roles of T peripheral helper and T follicular helper cells in human diseases. Cell Mol Immunol 2021;18:523–7. 10.1038/s41423-020-00529-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
rmdopen-2023-003317supp001.pdf (800.6KB, pdf)
Data Availability Statement
Data are available on reasonable request. These data are not available in a repository but available on reasonable request.