

Abstract
Protein kinase R (PKR)-like endoplasmic reticulum (ER) kinase (PERK) is one of the three major sensors in the unfolded protein response (UPR). The UPR is involved in the modulation of protein synthesis as an adaptive response. Prolonged PERK activity correlates with the development of diseases and the attenuation of disease severity. Thus, the current debate focuses on the role of the PERK signaling pathway either in accelerating or preventing diseases such as neurodegenerative diseases, myelin disorders, and tumor growth and cancer. In this review, we examine the current findings on the PERK signaling pathway and whether it is beneficial or detrimental for the above-mentioned disorders.
Overview
The endoplasmic reticulum (ER) is a tubular network of membranes found within the cytoplasm of the eukaryotic cells. It is responsible for the regulation of protein, lipid and steroid biosynthesis, maintaining calcium homeostasis and calcium-dependent signaling (1–3). The ER is also the essential site of protein translation, modification and folding (4). With the help of chaperones and enzymes (5), newly formed proteins, such as integral membrane proteins and transmembrane receptors, or proteins secreted by exocytosis, are transported from the ER to the cell membrane (6). Disruption of these physiological functions leads to the accumulation of mis/unfolded proteins, causing ER stress, which further induces the unfolded protein responses (UPR) to orchestrate adaptive cellular response (7,8). Maladaptive UPR outputs trigger apoptosis (9).
In mammals, the UPR signals through three parallel ER transmembrane sensors: PKR-like endoplasmic reticulum (ER) kinase (PERK), inositol-requiring enzyme 1 (IRE1) and activating transcription factor 6α (ATF6 α). Each sensor responds to the level of mis/unfolded proteins in the luminal domain where they are in an inactivated state in a complex with an ER chaperone, immunoglobulin heavy chain-binding protein (BiP, also called glucose-regulated protein 78: GRP78) (9–12). With ER stress, the resulting BiP dissociation leads to oligomerization and trans-autophosphorylation of PERK and IRE1.
Activated PERK phosphorylates Ser51 of the α subunit of eukaryotic translation initiation factor 2 (eIF2α) (13). although phosphorylation of Ser51 of eIF2 α is associated with a temporary halt of global protein synthesis, there is a selective and heightened translation of the mRNA encoding the activating transcription factor 4 (ATF4), a key stimulator of genes that regulates autophagy, ER-associated degradation (ERAD), cell viability and apoptosis (9,14,15). To maintain a balance in global protein synthesis, dephosphorylation of eIF2α is accomplished by a growth arrest and DNA damage 34 (GADD34) and protein phosphatase 1 (PP1) complex. Upregulation of GADD34 is due to CCATT enhancer-binding protein homologous protein (CHOP), a transcription factor that is an ATF4-driven gene product (Fig. 1) (5,7,12,13,16).
Figure 1.
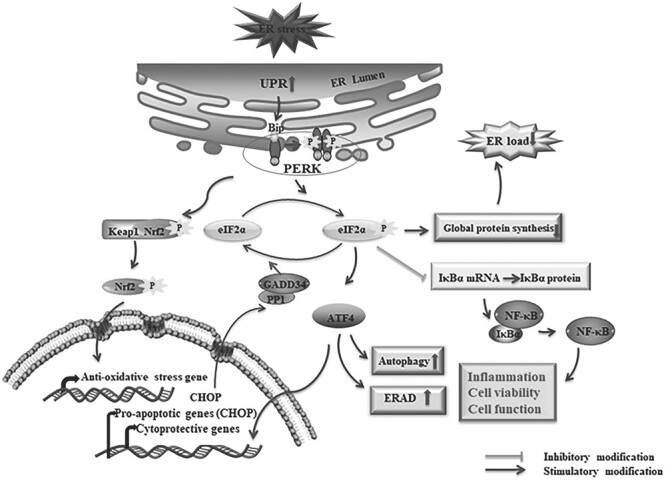
The activation of the PERK signaling pathway under ER stress. The accumulation of miss/unfolded proteins leads to activation of the PERK pathway through dissociations of BiP from the ER stress transducer. The resulting dissociations facilitate the oligomerization and autophosphorylation of PERK which further phosphorylates the eIF2α. The p-eIF2α decreases the ER load by inhibiting the global protein synthesis and induces the preferential translation of ATF4. Depending on the severity of ER stress, ATF4 induces either cytoprotective genes for cell survival or pro-apoptotic genes for apoptotic cell death as well as autophagy and ERAD-related genes. CHOP is a such kind of pro-apoptotic gene product by ATF4 induction that upregulates the GADD34 to form the complex with protein phosphatase 1 (PP1) and dephosphorylates eIF2α through the complex. In addition, the inhibition of p-eIF2α to the IκBα mRNA translation leads to dissociation of NF-κB, which mediates the transcription of several genes and induces cell functions including inflammation and cell survivability. PERK mediated phosphorylation of Nrf2, which dissociates the conjugation of Nrf2-Keap-1, resulting in accumulation and translocation of Nrf2 to the nucleus, and binds to the anti-oxidative stress gene. ‘↑’: increase; ‘↓’: decrease.
In addition, the transcription factor nuclear factor-kappa B (NF-κB) plays a crucial role in inflammatory diseases through the regulation of inflammation, cell viability and apoptotic cell death (17,18). Inactivated NF-κB remains cytoplasmic by interacting with NF-κB inhibitors (IκBs), whereas activated NF-κB goes to the nucleus upon dissociation from IκBs and stimulates transcription of target genes (19). Importantly, there is ample evidence that the activation of NF-κB is mediated by the PERK/p-eIF2α pathway through the inhibition of mRNA translation of IκBs (Fig. 1) (20–22).
Accumulation and aggregation of disease-specific mis/unfolded proteins are associated with several neurodegenerative diseases including multiple sclerosis (MS), Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS) and Huntington’s disease (HD). Several studies report the involvement of the PERK signaling pathway in these neurological diseases (6,21,23–26). There is substantial evidence that PERK activation/inhibition reduces the severity of MS (2,5,27) and reduces tumor growth, cancer progression, metastasis and angiogenesis (3,6,16,28–30).
Here, we discuss the role of the PERK signaling pathway in the pathophysiology and disease progression of the prominent neurodegenerative diseases, myelin disorder, and tumor growth and cancer, as revealed from studies using human cell lines and animal models.
PERK signaling in neurodegenerative disease
Multiple sclerosis
MS and its animal model experimental autoimmune encephalomyelitis (EAE) are chronic autoimmune inflammatory and neurodegenerative diseases of the CNS that result in demyelination, loss of oligodendrocytes and neurons, and axon degeneration (31–34). The ER stress-mediated UPR activation in MS and EAE has been well studied in these reviews and reported such activations in different cell types including oligodendrocytes (2,5,27). In MS and EAE, Lin et al. found that the activation of the PERK signaling pathway accompanied by IFN-γ ameliorated the disease’s severity and prevented demyelination, axonal damage and oligodendrocytes loss before the onset of EAE, whereas the associative functions were abrogated in PERK heterozygous deficient mice (35). To elucidate the involvement of the PERK pathway in oligodendrocytes survivability during EAE, the research team generated transgenic mice with controllable activation of the PERK pathway. Selective and moderate PERK activation in oligodendrocytes prior to EAE attack demonstrates the same effects as mentioned above (36). Moreover, consistent with the studies, increased EAE disease severity and EAE-induced oligodendrocytes loss, demyelination and axon degeneration has been noticed in oligodendrocytes-specific PERK knockout mice (37). It has also been reported that the genetic and pharmacological inactivation of GADD34 in mice and cultured hippocampus slices showed the increased phosphorylation of eIF2α in myelinating oligodendrocytes in exposure to the IFN-γ, diminished oligodendrocytes loss and hypomyelination (38). Additionally, a recent study showed that inactivation or deletion of PERK caused late-onset of oligodendrocyte dysfunction and death with impaired autophagy in young adult mice (39). The cytoprotective roles of the PERK/p-eIF2α signaling pathway is reported in oligodendrocytes in MS and EAE by mitigating the inflammation during EAE, though the internal mechanism remains elusive.
Determining which factors were really involved in such a protective mechanism requires considerable effort, as the ATF4 is the downstream and master transcription factor of the PERK/p-eIF2α pathway (34), which may have cytoprotective roles by activating pro-survival genes. Surprisingly, the inactivation of ATF4, specifically in oligodendrocytes in CNS, did not alter the EAE disease severity and did not affect oligodendrocytes loss, demyelination, axon degeneration, inflammation and neuron loss (40). Therefore, it is clear that ATF4 was not involved in the cytoprotective roles of the PERK/p- eIF2α pathway during EAE.
However, studies found that NF-κB activation in oligodendrocytes correlated with activation of the PERK pathway both in vivo and in vitro (41). In a surprising connection, data have been reported where the NF-κB activation protected oligodendrocytes against inflammation in MS and EAE (19), though the involvement of the PERK pathway was not yet studied. Additional studies should be conducted in oligodendrocytes where NF-κB activation may lead to cytoprotective effects of the PERK/p-eIF2α pathway in MS and EAE.
The regulation of the PERK pathway in neurons during MS and EAE has been poorly studied. However, a recent study revealed that neuron-specific PERK inactivation impaired disease resolution and exacerbated EAE-induced axon degeneration, neuron loss and demyelination, supporting the neuroprotective roles of the PERK/p-eIF2α pathway in MS and EAE. Interestingly, neuron-specific ATF4 inactivation did not alter such phenomena (42), while conversely, neuron-specific NF-κB ablation did not influence the neuro-axonal degeneration in EAE (43). It is undoubtedly evident that the PERK/p-eIF2α pathway is playing both cytoprotective and neuroprotective roles in MS and EAE.
Alzheimer’s disease
AD is a progressive neurodegenerative disorder pathologically characterized by the accumulation of amyloid plaques and neurofibrillary tangles, composed of amyloid-beta peptides (Aβ) and aberrantly folded microtubule-associated protein tau, respectively (44,45). It is evident that along with the gene mutations in amyloid precursor protein (APP), presenilin-1 (PS1) and presenilin-2 (PS2) (46), ER stress may also be involved in the pathology of AD. Furthermore, elevated levels of BiP/GRP78, p-PERK and p-eIF2α in the hippocampus and temporal cortex of AD neurons indicate the involvement of the UPR in the early stages of AD (47–51). Although Hamos et al. reported that increased expression of GRP78 may protect neurons from AD-specific damage (47), an involvement of the PERK pathway in the neuroprotection of AD remains unclear.
Studies show that increased levels of p-eIF2α elevated levels of BACE1 and promoted amyloidogenesis (52), as well as CREB dysfunction in the AD mouse model (53). Additionally, familial-AD-linked PS1 mutant expressed in PC12 cells and knockin (KI) mice enhanced increased levels of p-eIF2α and CHOP (54). In contrast, decreased eIF2α phosphorylation was observed in PS1 mutant expressing SK-N-SH cells (55).
Recently, the ER stress response in Aβ pathology was investigated using APP-KI, APP-single-transgenic and APP/PS1 double gene-modified AD mouse models. No ER stress response was observed in APP-KI and APP-single-transgenic mouse models, confirming that neither Aβ deposition nor APP overexpression induce detectable ER stress (56). In contrast, the APP/PS1 double gene-modified mouse, which overexpresses APP and PS1, exhibited elevated levels of p-eIF2α, and the 3xTg mouse, which expresses APP, exhibited higher levels of BiP/GRP78, CHOP and p-eIF2α, suggesting that enhanced ER stress by any modification is not always related to the AD pathology (56,57).
APP-intracellular domain (AICD) is a transcription factor that is generated from APP cleavages and stimulates transcription of CHOP. An overproduction of AICD induction of further production of CHOP (58–60) could be one of the mechanisms of neurotoxicity in AD, where downstream regulation of the PERK pathway is not directly involved (23).
More recent data show that three PERK-mediated pathways could lead to aberrant tau species (23). PERK activates (i) a tau kinase (GSK3β) that is implicated in tauopathy (61); (ii) caspases that cleave tau into cleaved-tau (cTau), an early indicator of pre-tangle pathology in AD and other tauopathies (62); and (3) phosphorylates tau (p-Tau) (63,64). This is consistent with findings in human AD brains that p-PERK immunoreactivity is observed in hippocampal neurons with p-Tau and co-localization with GSK-3β (48,65). Furthermore, reduced levels of p-Tau were observed in neuronal SK-N-SH cells (66) and rTg4510 mouse models (67) by inhibiting PERK with GSK2606414; this may support the conclusion that tau pathology may be a consequence of dysregulated PERK activity in AD. On the other hand, Hashimoto et al. did not find a relationship between the tau pathology and the ER stress markers in P301S-Tau-transgenic mice of different ages (56).
It’s important to note that the involvement of the PERK pathway in AD is complex, and the precise mechanisms and consequences of its activation in different stages of the disease are still being investigated. Research in this area aims to better understand the role of the PERK pathway and explore its potential as a therapeutic target for AD.
Parkinson’s disease
PD is a neurodegenerative movement disorder characterized by the selective loss of dopaminergic neurons along with the presence of Lewy bodies composed of α-synuclein (68). Dopaminergic neurons of the Parkinson’s brain are a prominent location for connecting the chronic activation of PERK with the molecular basis of PD. Though there is no direct link of PERK engagement with the neuropathology of PD, elevated levels of p-PERK and p-eIF2α were found in substantia nigra (SN) of the postmortem PD brains. However, the lack of colocalization of α-synuclein with p-PERK suggests that the increased levels of p-PERK or p-eIF2α are not due to the association of α-synuclein (69).
Studies also show that enhanced levels of p-eIF2α in A53T α-synuclein-induced cells of the gene mutated PD may protect cells by declining caspase (68,70). By inhibiting the GADD34, Sun et al. found that up-regulated ATF4 translation by p-eIF2α improves survival of the PD model by up-regulating parkin. Along with the elevated parkin levels, ATF4 protected the primary ventral midbrain dopaminergic neurons against cell death mediated by two PD-associated neurotoxins: 6-OHDA and MPP+ (71,72). Another study found decreased expression of parkin in the primary cortical neurons of ATF4-knockout mice (73). Therefore, ATF4 may have a neuroprotective role during parkin regulation. On the other hand, Gully et al. found that overexpression of ATF4 induces severe loss of dopamine nigral neurons in a rat model of PD (74). Interestingly, Belucci et al. found increased expression of ATF4/CREB-2 in SN of the SYN120 transgenic mouse model of PD by induction of α-synuclein accumulation (75) suggesting that the role of ATF4 may be associated with both pro-survival and pro-apoptotic functions via parkin and α-synuclein activation, respectively. Additionally, inactivation of the PERK signaling prevented neurodegeneration in a PD mice model (76) accompanied by an increase in dopamine levels and the expression of synaptic proteins (77), supporting the concept that the initial activation of the PERK pathway is related to neuroprotection, whereas the prolonged effect is supportive to neurodegeneration.
Although there is limited direct evidence specifically linking the PERK pathway to PD, it is reasonable to hypothesize that PERK activation and ER stress play a role in PD pathogenesis, given the accumulation of misfolded proteins in the disease.
Huntington’s disease
HD is an autosomal dominant, neurodegenerative disease caused by a cysteine-adenine-guanine (CAG) trinucleotide repeat expansion within the Huntington gene, leading to the pathogenic form of Huntingtin protein (Htt) (78). The PERK pathway in HD is under studied, but p-eIF2α levels are higher in neuronal PC6.3 cells expressing mutant Htt. Furthermore, pharmacological inhibition of p-eIF2α phosphatase increases phosphorylation of eIF2α, decreases mutant Htt aggregation and increases neuronal cell viability (79). However, another study demonstrated that the eIF2α phosphorylation induced autophagy and acted as a cellular defense against ER stress-mediated cell death (80). Similarly, increased levels of p-eIF2α were observed in HEK293T cells transfected with mutant Htt, suggesting that Htt overproduction induces the PERK pathway (81). Moreover, Leitman et al. reported that the eIF2α phosphorylation was increased in the striatal cells line expressing pathogenic Htt and in brains of the N171-82Q HD mouse models. Thus, the pathogenic Htt mediated eIF2α phosphorylation induced CHOP and altered protein homeostasis causing striatal cell death. Interestingly, they found an association between dephosphorylated eIF2α and cognitive decline, suggesting that the Htt pathology may be one of the sources of early cognitive impairments in HD patients by PERK activation (81).
Genz et al. recently reported that PERK activation by CCT020312 in cells and by MK-28 in mice rescued both from ER stress mediated apoptosis. Transient subcutaneous delivery of MK-28 significantly improved motor and executive functions and delayed death in R6/2 mice, showing no toxicity (82). Therefore, PERK activation by pharmacological approach can treat an aggressive HD model, suggesting a possible strategy for HD. Understanding the precise role of PERK and its interplay with other signaling pathways in HD pathology is an active area of research. Modulating the PERK pathway or targeting other components of the UPR has been investigated as a potential therapeutic approach for HD, aiming to restore ER homeostasis and alleviate the associated cellular stress. However, it is important to carefully balance the activation of PERK to avoid excessive or prolonged stress responses that may have detrimental effects on neuronal function.
Amyotrophic lateral sclerosis
ALS is a progressive neurodegenerative disease characterized by motor neuron degeneration in the spinal ventral horn, cerebral cortex, and brain stem, leading to muscular atrophy and paralysis (83,84). Most familial ALS is attributed to mutations in superoxide dismutase 1 (SOD1), TAR DNA-binding protein (TARDBP), C9orf72, and the fused in sarcoma (FUS) gene (85).
PERK activation is associated with the expressions of mutant forms of SOD1 and TDP1. In the spinal cord of ALS mouse models, the levels of p-PERK and p-eIF2α were increased in the pre-symptomatic stage, but not in the symptomatic stage (86). Moreover, along with the GPR78/BiP and CHOP, the increased expressions of PERK and p-eIF2α were found in both the stages in white gastrocnemius muscle of G93*SOD1 (ALS-Tg) mice (87), suggesting that PERK pathway activation is correlated with the pre-symptoms and not commonly with the symptoms nor disease progression. Nevertheless, neither mentioned involvement of ATF4 as a transcription factor of CHOP. However, Matus et al. reported that ATF4 deficiency attenuated the pro-apoptotic gene, leading to delayed disease onset and prolonged life span in SOD1G86R transgenic mice. This deficiency also enhanced mutant SOD1 aggregation due to alteration in the redox status in the cell, which is confirmed by in vitro studies in the motoneuron in cell line NSC34 (88). In contrast, treatment with Guanabenz, an inhibitor of GADD34 mediated dephosphorylation of eIF2α, increases phosphorylation of eIF2α, ameliorating diseases symptoms with prolonged survivability in G93A mtSOD1 transgenic mice (89).
A recent study revealed that PERK haploinsufficiency has no effect in different ALS mice models where the human SOD1 was overexpressed. This group claimed that the UPR-PERK pathway is not a therapeutic target for mutant SOD1-induced ALS (90), which contradicts previous studies. Pharmacological inhibition of PERK signaling with its downstream inhibitor ISRIB, but not with the direct PERK kinase inhibitor GSK2606414, significantly enhanced the survival of G93A SOD1-expressing neurons (91).
In addition, TDP-43 a DNA binding protein encoded by the TARDBP gene, is associated with the stress granules (92–94) and intracellular aggregates composed of mRNAs, ribosomal subunits, and various proteins. One in vitro study reported that phosphorylation of eIF2α initiated formation of stress granules (95), but did not show the exact functions of p-eIF2α in the later stage.
Wang and colleagues later found the upregulation of the p-eIF2a and CHOP after overexpressing TDP-43-WT and mutant A315T in neural SH-SY5Y cells (96–98). Inhibition of PERK using GSK2606414 reduced eIF2α phosphorylation and rescued TDP-43-mediated neurotoxicity in Drosophila and mammalian neurons. Inhibition of GADD34 (eIF2α phosphatase) also enhances eIF2α phosphorylation and accelerates TDP-43-induced neurotoxicity (99,100).
The expansion of the GGGGCC (G4C2) hexanucleotide in the chromosome 9 open reading frame 72 (C9orf72) gene accounts for 10% of ALS patients (99). Transcriptome analysis in the cerebellum and frontal cortex of C9orf72-ALS patients revealed altered UPR-related gene expression such as ATF4 and CHOP (101). Moreover, the mRNA levels of ATF4, CHOP and GADD34 were also upregulated by poly-PR expression in K562 cell lines (102) and in SH-SY5Y cells (103). All these data indicated that ER stress is related to C9ORF72-mediated neurodegeneration (99).
To conclude, PERK signaling may have opposite functions in ALS pathology. The ATF4 production may lead to neuronal loss, whereas eIF2α phosphorylation was neuroprotective. However, the effect of PERK pathway in ALS is remained inconclusive.
Tumor growth and cancer
ER stress, especially PERK, plays a role in tumor growth and cancers. Hypoxia is one of the dynamic features of the tumor microenvironment that is associated with the rapid cancer progression and induction of metastasis (104). However, the PERK pathway is correlated with tumor growth (104–106) as well as cancer invasion and metastasis on the molecular level (28,107). Studies show that the PERK pathway, including p-eIF2α, confers the survival advantages for the tumor cells under hypoxia (108), although apoptotic cell death also occurred by ATF4-CHOP activation (106,109) indicating the paradoxical phenomena of the PERK pathway.
Several studies found increased levels of p-eIF2α in the cancer cells, including bronchioloalveolar carcinoma (110), Hodgkin’s lymphoma (111), benign and malignant melanocyte, and colonic epithelial neoplasms (112) as well as gastrointestinal carcinoma (113). Consistent with these studies, Guo et al. found elevated levels of p-eIF2α in breast cancer cells, suggesting that ER stress, more specifically the PERK pathway, plays an important role in the initiation of tumor formation (114). In contrast, a decreased level of p-eIF2α has been reported in human osteosarcoma, a common bone tumor of children and young adults (115).
Despite these findings, the role of the PERK pathway in tumor growth and cancer initiation and progression is not fully understood. According to some studies, the PERK pathway is involved in promoting tumor growth, yet also involved in tumor cell death (3). For example, apoptotic cell death in osteosarcoma cells and the resulting decreased tumor growth have been reported in vitro and in vivo studies where the CYT997-mediated PERK/p-eIF2α/CHOP signaling pathway was up-regulated (116). Similarly, Wang et al. have revealed the involvement of the PERK/p-eIF2α pathway with increased levels of BiP, p-PERK, p-eIF2α in paraquat-induced human lung epithelial-like-A549 cell apoptosis (117) and with increased levels of BiP, PERK, p-eIF2α, ATF4, CHOP in pterostilbene-induced autophagy-dependent cell death in human hepatocellular carcinoma cells (118). On the other hand, reduced tumor growth, impaired angiogenesis, reduced vascularity and viability were observed in PERK-deficient mice (119,120).
Glioblastoma multiforme (GBM) is an aggressive brain tumor in which the involvement of the PERK pathway is not extensively studied. Moreover, PERK was found to stimulate GBM growth (121), though the mechanism was not fully described. Dadey et al. reported that the PERK/p-eIF2α/ATF4 pathway modulated the cell viability by pro-survival activity in irradiated GBM (122). A recent in vitro study showed that the PERK promoted the cell proliferation and migration in glioblastoma stem cells (123) and angiogenesis by interacting with peptidyl glycine α-amidating monooxygenase (PAM) in glioblastoma cell lines (124). Similarly, PERK activation inhibited the growth and invasion of pancreatic cancer cells in vitro and in vivo, while PERK inhibition had the opposite effects (125).
Accordingly, PERK-dependent signaling facilitated the formation of mammary tumors in aged PERK-null mice but not in the normal mammary tissue (126), and the PERK-deficient signaling led to the formation of the same tumor types (127). Studies found that inhibition of the PERK pathway can suppress breast cancer growth and metastasis (128,129). Moreover, PERK, ATF4 and lysosomal-associated membrane protein 3 (LAMP3) mediated cell migration was reported in hypoxia-induced breast cancer (130) that supports the increased expression of ATF4 in several solid tumor types (131) and in resulting cancer cell survival (132). It was also identified that ATF4 overexpression facilitates progression and fosters the malignancy of tumors via increasing their proliferation, vessel growth, cell migration and metastasis (3,133–136). Thus, the activation (137) and inactivation of the PERK (128) pathway may have dual effects on the different steps of tumorigenesis and carcinogenesis, though the definite role remains controversial.
Closing Comments
Accumulating evidence data suggests that the PERK pathway is a potent candidate for studying the cellular and molecular mechanisms of various disease progressions as well as their remediation. It is evident that the PERK pathway and its downstream molecules are implicated in many neurodegenerative diseases, as well as tumor and cancer growth. Nonetheless, they are also involved in impeding the progress of these disorders through their controlled mechanisms (Fig. 2 and Table 1).
Figure 2.
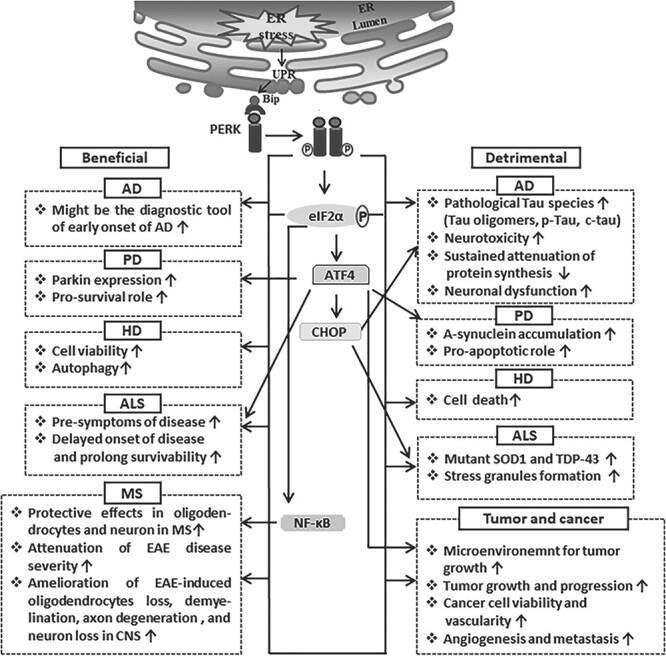
The beneficial and detrimental roles of the PERK signaling pathway in AD, PD, HD, ALS, MS and cancer.
Table 1.
Involvement of PERK pathway in neuropathology and oncology
| Neuro-degenerative diseases | Gene modification | Lines | Regulation | Techniques, brain area | Conclusive remarks | References |
|---|---|---|---|---|---|---|
| AD | – | Human postmortem brain tissue of AD | p-PERK ↑ | IHC; temporal cortex (mid), hippocampus | Neuroprotective at the initial stage but neurodegenerative in sustained activation | (48,49) |
| – | Tg2576, 5XFAD Tg |
p-eIF2α ↑ | WB, IHC; whole brain | Aβ overproduction | (52) | |
| PS1 mutant | C57BL/6 · 129/Sv | p-eIF2α ↑ CHOP ↑ |
WB; cortex, hippocampus | Increased cell death in familial AD | (54) | |
| APP | AppNL-G-F | p-eIF2α → CHOP → |
WB; cortex, hippocampus | ER stress not related to AD pathology | (57) | |
| Tg2576 | ||||||
| APP23 | ||||||
| APP/PS1 | APP(Swe)-Tg, PS(∆E9)-Tg | p-eIF2α ↑ CHOP ↑ |
||||
| APP/PS1/Mapt | 3XTg | |||||
| Transfection | APP-FLAG, C83-FLAG and AICD-FLAG | CHOP ↑ | WB, ELISA, MTT | Increased cell death | (60) | |
| Tau mutant | rTg4510 | p-PERK ↓ p-eIF2α ↓ |
WB, IHC; hippocampus | Prevented Tau mediated neurodegeneration | (67) | |
| APP/PS1 | P301S-Tau-Tg | p-eIF2α → CHOP → |
WB; hippocampus | No relation with AD pathogenesis | (56) | |
| APP/PS1 | C57BL/6 | p-PERK ↑ p-eIF2α ↑ |
WB: cerebral cortex | – | (51) | |
| PD | Dopaminergic neuron | Human autopsy brain | p-PERK ↑ p-eIF2α ↑ |
IHC; substantia nigra | No colocalization with α- synuclein | (69) |
| Co-transfection of A53T α-synuclein | PC12 cell | p-PERK ↑ | WB | Protected cell death | (68) | |
| Transfection | PC12 cell | ATF4 ↑ | WB; ventral midbrain | Promoted neuronal survival | (71,72) | |
| rAAV- mediated gene transfer | Sprague–Dawley rat | ATF4 ↑ | WB, IHC; substantia nigra | Neuron loss | (74) | |
| TH neuron | C57BL/6 | p-PERK ↑ p-eIF2α ↑ ATF4 ↑ |
WB; brain | Stress-mediated neuronal apoptosis | (76) | |
| HD | Transfection | PC6.3 cells | p-eIF2α ↑ | WB, ICC | Increased cell viability | (79) |
| Transfection | HEK293T | p-eIF2α ↑ | WB, ICC | Increased Htt expression | (81) | |
| Transfection | Striatal cells | p-eIF2α ↑ | WB, ICC | Decreased cell viability | ||
| Human htt | N171-82Q | p-eIF2α ↑ | IHC; striatum | Cell death | ||
| Human htt | STHdhQ111/111 | p-PERK ↑ p-eIF2α ↑ ATF4 ↑ CHOP ↑ GADD34↑ |
WB, qPCR | Reduced cytotoxicity | (82) | |
| R6/2 TG mice | p-PERK ↑ p-eIF2α ↑ |
IHC; striatum | Improved motor and executive functions | |||
| ALS | Human SOD1 | SOD1-G93A Tg | p-PERK ↑ p-eIF2α ↑ |
WB, IHC; spinal cord | Motor neuron degeneration | (86) |
| Mutant SOD1 | G93A*SOD1 Tg | PERK ↑ p-eIF2α ↑ CHOP ↑ |
WB; muscle | Muscle atrophy and weakness | (87) | |
| Mutant SOD1 | ATF4−/− -SOD1G86R Tg |
ATF4 ↓ | WB, IHC; spinal cord | Delayed disease onset and prolonged the life span | (88) | |
| Mutant SOD1 | SOD1-G93A Tg | p-eIF2α ↑ | WB; spinal cord | Delayed disease onset and prolonged survival | (89) | |
| Human SOD1 | SOD1-G93A and others | PERK ↓↑ GADD34↓↑ CHOP ↓↑ |
WB, IHC; spinal cord | No effect | (90) | |
| HA-TDP-43 and HA-TDP-43A315T | SH-SY5Y cells | p-eIF2α ↑ CHOP ↑ |
WB | Neuronal toxicity | (97) | |
| Mutant TDP-43 | Flies and rat cortical neuron | p-eIF2α ↑ | WB, IHC | TDP-43 toxicity | (100) | |
| C9orf72 gene | SH-SY5Y cells | CHOP ↑ | WB | Neuronal death | (103) | |
| MS | PERK mutation | C57BL/6 mice with | PERK ↑ p-eIF2α ↑ |
IHC; lumbar spinal cord | Attenuation of disease severity, amelioration of oligodendrocytes loss, demyelination and axon degeneration | (35) |
| IFN-γCNS+; Perk+/+ | ||||||
| PERK transgene | PLP/Fv2E-PERK | (36) | ||||
| PERK knockout | C57BL/6 mice with OL-PERK ko/ko | – | WB, IHC; lumbar spinal cord | Increased oligodendrocytes loss, demyelination and axon degeneration | (37) | |
| Mutant GADD34 | GFAP/tTA; TRE/IFN-γ; GADD34 mice | p-eIF2α ↑ | WB, IHC, EM; brain, lumbar spinal cord | Amelioration of oligodendrocytes loss and hypomyelination | (38) | |
| ATF4 knockout | ATF4loxP/loxP; CNP/Cre mice | WB, IHC; brain, lumbar spinal cord | No effects in oligodendrocytes loss, demyelination, axon degeneration, inflammation and neuron loss | (118) | ||
| PERK inactivation | PERKloxP/loxP; Thy1/CreERT2 mice | – | WB, IHC; brain, lumbar spinal cord | impaired disease resolution and exacerbated EAE-induced axon degeneration, neuron loss and demyelination | (42) | |
| Tumor growth and cancer | Transfection | MCF7, T47D, BT474, BT549, ZR-75-30, Hs578T, MDA-MB-157 and MDA.MB.231 | PERK → | WB, IHC | Cancer invasion and metastasis | (107) |
| – | Melanocytic navi and melanoma | p-eIF2α ↑ | IHC | Cancer initiation and progression | (112) | |
| – | Tumor tissue of breast cancer | p-eIF2α ↑ | IHC | Prognostic tool for breast cancer | (114) | |
| Human osteosarcoma cell | MG63, 143B, KHOS and HOS | p-eIF2α ↓ | WB | Anti-proliferation | (115) | |
| BALB/c-nu mice | PERK ↑ p-eIF2α ↑ CHOP ↑ |
WB, IHC | Decreased tumor growth | (116) | ||
| Human osteosarcoma cell | 143B, SJSA, MG63 and U2OS | |||||
| PERK-deficient | PKO-βTag | – | IHC; pancreata | Reduced proliferation, vascularity, the viability in insulinomas | (119) | |
| Human glioblastoma cell | D54, LN827 | PERK ↑ p-eIF2α ↑ ATF4 ↑ |
WB | Increased cell viability | (122) | |
| glioblastoma cell | LN308, LN229T, NCH82 | PERK ↑ p-eIF2α ↑ |
WB, IF | Regulation of angiogenesis | (124) | |
| Human glioblastoma cell | U87, U251 | ATF4 ↑ | WB, IHC | Promoted tumor angiogenesis | (133) | |
| Transfection | MDA-MB-231 cells | Knockdown of PERK and ATF4 | Transwell and gap closure assay | Reduced migration of breast cancer cell | (130) | |
| Breast cancer | MDA-MB-231 and 468 cells | PERK ↑ p-eIF2α ↑ ATF4 ↑ CHOP↑ |
WB | Apoptotic cell death | (128,137) | |
| Breast cancer | MDA-MB-453, CAL-148, HCC2185 and MFM-223 | PERK ↑ p-eIF2α ↑ ATF4 ↑ |
WB, qRT-PCR | Inhibit androgen receptors activity | (138) | |
| Prostate cancer | LNCap, C4-2 and 22RV1 | |||||
| Human prostate cancer cells | LNCaP, VCaP, 22Rv1 | ATF4 ↑ | WB, IHC | Increased growth and survivability | (135) |
‘↑’ indicates ‘increase"‘↓’ indicates ‘decrease"‘→’ inicates ‘no changes’
In addition, neuronal dysfunction with learning and memory deficits in neurodegenerative disorders might be the resulting inhibitions of protein synthesis, which are necessary for synapse formation. Therefore, to reduce a load of mis/unfolded proteins by ER stress response, it is more important to inhibit the global proteins translation.
It is hard to designate a single role for the PERK activation pathways as there are several downstream pathways that seem to determine if activation is as beneficial or detrimental. Instead, it would be better to judge the activity of the PERK pathway depending on its activation or inhibition (Table 2). According to the data of various studies, the activation of the PERK pathway is sometimes detrimental, but pharmacological inhibition of the pathway becomes beneficial for the same pathological conditions. For example, the pharmacological activation of PERK pathway is protective in models of neurodegenerative, such as MS and HD, whereas the inhibition of PERK pathway is protective in models in AD, PD and ALS. Taken as a whole, the PERK pathway might be considered a scientific blessing as the therapeutic target (Table 3) in different diseases regarding their switch-on or -off systems. Future studies aiming to identify the time points in which the PERK pathway is active will help identify an early therapeutic window.
Table 2.
Summarized roles of the PERK signaling pathway
| Diseases | Switch of the PERK pathway | Neurological role | Summery | References |
|---|---|---|---|---|
| AD | PERK activation | Neurodegenerative | Detrimental | (48,49) |
| PERK inhibition | Reduced tauopathy | Beneficial | (67) | |
| p-eIF2α activation | Cell death | Detrimental | (54,60) | |
| PD | PERK inhibition | Neuroprotection | Beneficial | (75,139) |
| p-eIF2α activation | Neuroprotection | Beneficial | (71) | |
| HD | PERK inhibition | Neuroprotection | Beneficial | (81) |
| p-eIF2α activation | Neuronal death | Detrimental | ||
| Neuroprotection | Beneficial | (79) | ||
| PERK activation | Neuroprotection | Beneficial | (82) | |
| ALS | p-PERK activation | Motorneuron degeneration, muscle atrophy | Detrimental | (86,87) |
| p-eIF2α activation | ||||
| p-eIF2α activation | Prolong survivability | Beneficial | (89) | |
| PERK inhibition | Neuroprotection | Beneficial | (99,100) | |
| MS | PERK activation | cytoprotection | Beneficial | (35,36,38) |
| PERK inhibition | Loss of cyto/neuroprotective effects | Detrimental | (39,42) | |
| Tumor growth and cancer | p-eIF2α activation | Cancer initiation and progression | Detrimental | (105,112) |
| PERK activation | Decreased tumor growth/cell death | Beneficial | (116,120) | |
| Increased cell viability | Detrimental | (122) | ||
| Angiogenesis | Detrimental | (124) | ||
| PERK inhibition | Reduced metastasis | Beneficial | (130) |
Table 3.
| Name of the drugs | Therapeutic target | Functions | p-eIF2α | Disease effects | Disease |
|---|---|---|---|---|---|
| CCT020312 | PERK | Activation | ↑ | Beneficial | MS, cancer |
| MK-28 | PERK | Activation | ↑ | Beneficial | HD |
| CCT020312 | PERK | Activation | ↑ | Beneficial | HD |
| GSK2606414 | PERK | Inhibition | ↓ | Beneficial | AD, PD, ALS |
| GSK2656157 | PERK | Inhibition | ↓ | Beneficial | Cancer |
| Salubrinal | GADD34 | Inhibition | ↑ | Beneficial | MS, ALS |
| Guanabenz | GADD34 | Inhibition | ↑ | Beneficial | MS, ALS |
| Sephin1 | GADD34 | Inhibition | ↑ | Beneficial | MS, ALS |
‘↑’ indicates ‘increase"‘↓’ indicates ‘decrease"‘→’ inicates ‘no changes’
Acknowledgements
The authors would like to thank Dr Ekhtear Hossain, Assistant Professor, Department of Biological Sciences and Chemistry, Southern University and A&M College, for his meaningful discussion. The authors also acknowledge Laura Berg and Lisa Duvick, Department of Laboratory Medicine and Pathology, Institute for Translational Neuroscience, University of Minnesota, for editing this manuscript.
Conflict of Interest statement. None declared.
Contributor Information
Gourango Talukdar, Institute for Translational Neuroscience and Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, MN 55455, USA; Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, MN 55455, USA; Department of Neuroscience, University of Minnesota, Minneapolis, MN 55455, USA.
Harry T Orr, Institute for Translational Neuroscience and Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, MN 55455, USA; Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, MN 55455, USA.
Zhixin Lei, Institute for Translational Neuroscience and Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, MN 55455, USA; Department of Neuroscience, University of Minnesota, Minneapolis, MN 55455, USA.
Funding
National Institute of Health (NINDS/NS127248 to G.T.).
Authors’ contributions
G.T., H.T.O. and Z.L. wrote and revised this manuscript. All the authors read and approved the final version.
References
- 1. Kaufman, R.J. (1999) Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev., 13, 1211–1233. [DOI] [PubMed] [Google Scholar]
- 2. Lin, W. and Popko, B. (2009) Endoplasmic reticulum stress in disorders of myelinating cells. Nat. Neurosci., 12, 379–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Siwecka, N., Rozpędek, W., Pytel, D., Wawrzynkiewicz, A., Dziki, A., Dziki, Ł., Diehl, J.A. and Majsterek, I. (2019) Dual role of endoplasmic reticulum stress-mediated unfolded protein response signaling pathway in carcinogenesis. Int. J. Mol. Sci., 20, 4354. 10.3390/ijms20184354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Harding, H.P., Zhang, Y. and Ron, D. (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature, 397, 271–274. [DOI] [PubMed] [Google Scholar]
- 5. Lin, W. and Stone, S. (2020) Unfolded protein response in myelin disorders. Neural Regen. Res., 15, 636. 10.4103/1673-5374.266903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Scheper, W. and Hoozemans, J.J.M. (2015) The unfolded protein response in neurodegenerative diseases: a neuropathological perspective. Acta Neuropathol. (Berl.), 130, 315–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ron, D. and Walter, P. (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol., 8, 519–529. [DOI] [PubMed] [Google Scholar]
- 8. Wu, J. and Kaufman, R.J. (2006) From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ., 13, 374–384. [DOI] [PubMed] [Google Scholar]
- 9. Hetz, C. and Papa, F.R. (2018) The unfolded protein response and cell fate control. Mol. Cell, 69, 169–181. [DOI] [PubMed] [Google Scholar]
- 10. Cao, S.S. and Kaufman, R.J. (2012) Unfolded protein response. Curr. Biol., 22, R622–R626.. [DOI] [PubMed] [Google Scholar]
- 11. Hetz, C., Chevet, E. and Oakes, S.A. (2015) Proteostasis control by the unfolded protein response. Nat. Cell Biol., 17, 829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang, M. and Kaufman, R.J. (2016) Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature, 529, 326–335. [DOI] [PubMed] [Google Scholar]
- 13. Walter, P. and Ron, D. (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science, 334, 1081–1086. [DOI] [PubMed] [Google Scholar]
- 14. Almeida, L.M., Pinho, B.R., Duchen, M.R. and Oliveira, J.M.A. (2022) The PERKs of mitochondria protection during stress: insights for PERK modulation in neurodegenerative and metabolic diseases. Biol. Rev., 97, 1737–1748. [DOI] [PubMed] [Google Scholar]
- 15. Tabas, I. and Ron, D. (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol., 13, 184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xiang, C., Wang, Y., Zhang, H. and Han, F. (2017) The role of endoplasmic reticulum stress in neurodegenerative disease. Apoptosis, 22, 1–26. [DOI] [PubMed] [Google Scholar]
- 17. Nakajima, S. and Kitamura, M. (2013) Bidirectional regulation of NF-κB by reactive oxygen species: a role of unfolded protein response. Free Radic. Biol. Med., 65, 162–174. [DOI] [PubMed] [Google Scholar]
- 18. Yue, Y., Stone, S. and Lin, W. (2018) Role of nuclear factor κB in multiple sclerosis and experimental autoimmune encephalomyelitis. Neural Regen. Res., 13, 1507. 10.4103/1673-5374.237109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stone, S., Jamison, S., Yue, Y., Durose, W., Schmidt-Ullrich, R. and Lin, W. (2017) NF-κB activation protects oligodendrocytes against inflammation. J. Neurosci., 37, 9332–9344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Deng, J., Lu, P.D., Zhang, Y., Scheuner, D., Kaufman, R.J., Sonenberg, N., Harding, H.P. and Ron, D. (2004) Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol. Cell. Biol., 24, 10161–10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jiang, H.-Y., Wek, S.A., McGrath, B.C., Scheuner, D., Kaufman, R.J., Cavener, D.R. and Wek, R.C. (2003) Phosphorylation of the subunit of eukaryotic initiation factor 2 is required for activation of NF- B in response to diverse cellular stresses. Mol. Cell. Biol., 23, 5651–5663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lei, Z., Yue, Y., Stone, S., Wu, S. and Lin, W. (2020) NF-κB activation accounts for the cytoprotective effects of PERK activation on oligodendrocytes during EAE. J. Neurosci., 40, 6444–6456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bell, M.C., Meier, S.E., Ingram, A.L. and Abisambra, J.F. (2016) PERK-opathies: an endoplasmic reticulum stress mechanism underlying neurodegeneration. CAR, 13, 150–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Halliday, M., Hughes, D. and Mallucci, G.R. (2017) Fine-tuning PERK signaling for neuroprotection. J. Neurochem., 142, 812–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ohno, M. (2018) PERK as a hub of multiple pathogenic pathways leading to memory deficits and neurodegeneration in Alzheimer’s disease. Brain Res. Bull., 141, 72–78. [DOI] [PubMed] [Google Scholar]
- 26. Shacham, T., Patel, C. and Lederkremer, G.Z. (2021) PERK pathway and neurodegenerative disease: to inhibit or to activate? Biomol. Ther., 11, 354. 10.3390/biom11030354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stone, S. and Lin, W. (2015) The unfolded protein response in multiple sclerosis. Front. Neurosci., 9, 264. 10.3389/fnins.2015.00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cubillos-Ruiz, J.R., Bettigole, S.E. and Glimcher, L.H. (2017) Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell, 168, 692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hsu, S.-K., Chiu, C.-C., Dahms, H.-U., Chou, C.-K., Cheng, C.-M., Chang, W.-T., Cheng, K.-C., Wang, H.-M.D. and Lin, I.-L. (2019) Unfolded protein response (UPR) in survival, dormancy, immunosuppression, metastasis, and treatments of cancer cells. Int. J. Mol. Sci., 20, 2518. 10.3390/ijms20102518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yadav, R.K., Chae, S.-W., Kim, H.-R. and Chae, H.J. (2014) Endoplasmic reticulum stress and cancer. J. Cancer Prev., 19, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Frohman, E.M., Racke, M.K. and Raine, C.S. (2006) Multiple sclerosis—the plaque and its pathogenesis. N. Engl. J. Med., 354, 942–955. [DOI] [PubMed] [Google Scholar]
- 32. Lassmann, H. (2007) Experimental models of multiple sclerosis. Rev. Neurol. (Paris), 163, 651–655. [DOI] [PubMed] [Google Scholar]
- 33. Lassmann, H. and Bradl, M. (2017) Multiple sclerosis: experimental models and reality. Acta Neuropathol. (Berl.), 133, 223–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reich, D.S., Lucchinetti, C.F. and Calabresi, P.A. (2018) Multiple sclerosis. N. Engl. J. Med., 378, 169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lin, W., Bailey, S.L., Ho, H., Harding, H.P., Ron, D., Miller, S.D. and Popko, B. (2007) The integrated stress response prevents demyelination by protecting oligodendrocytes against immune-mediated damage. J. Clin. Invest., 117, 448–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lin, W., Lin, Y., Li, J., Fenstermaker, A.G., Way, S.W., Clayton, B., Jamison, S., Harding, H.P., Ron, D. and Popko, B. (2013) Oligodendrocyte-specific activation of PERK signaling protects mice against experimental autoimmune encephalomyelitis. J. Neurosci., 33, 5980–5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hussien, Y., Cavener, D.R. and Popko, B. (2014) Genetic inactivation of PERK signaling in mouse oligodendrocytes: normal developmental myelination with increased susceptibility to inflammatory demyelination: oligodendrocyte-specific PERK inactivation. Glia, 62, 680–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lin, W., Kunkler, P.E., Harding, H.P., Ron, D., Kraig, R.P. and Popko, B. (2008) Enhanced integrated stress response promotes myelinating oligodendrocyte survival in response to interferon-γ. Am. J. Pathol., 173, 1508–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stone, S., Wu, S., Nave, K.-A. and Lin, W. (2020) The UPR preserves mature oligodendrocyte viability and function in adults by regulating autophagy of PLP. JCI Insight, 5, e132364. 10.1172/jci.insight.132364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yue, Y., Stanojlovic, M., Lin, Y., Karsenty, G. and Lin, W. (2019) Oligodendrocyte-specific ATF4 inactivation does not influence the development of EAE. J. Neuroinflammation, 16, 23. 10.1186/s12974-019-1415-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lin, Y., Jamison, S. and Lin, W. (2012) Interferon-γ activates nuclear factor-κ B in oligodendrocytes through a process mediated by the unfolded protein response. PLoS One, 7, e36408. 10.1371/journal.pone.0036408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stone, S., Yue, Y., Stanojlovic, M., Wu, S., Karsenty, G. and Lin, W. (2019) Neuron-specific PERK inactivation exacerbates neurodegeneration during experimental autoimmune encephalomyelitis. JCI Insight, 4, e124232. 10.1172/jci.insight.124232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee, D.-H., Kubera, K., Rosenthal, B., Kaltschmidt, B., Kaltschmidt, C., Gold, R. and Linker, R.A. (2012) Neuronal NF-κB ablation does not influence neuro-axonal degeneration in experimental autoimmune demyelination. J. Neuroimmunol., 246, 38–42. [DOI] [PubMed] [Google Scholar]
- 44. Alzheimer’s Association, Thies, W. and Bleiler, L. (2013) 2013 Alzheimer’s disease facts and figures. Alzheimers Dement., 9, 208–245. [DOI] [PubMed] [Google Scholar]
- 45. Jiang, T., Yu, J.-T. and Tan, L. (2012) Novel disease-modifying therapies for Alzheimer’s disease. J. Alzheimers Dis. JAD, 31, 475–492. [DOI] [PubMed] [Google Scholar]
- 46. Cruchaga, C., Chakraverty, S., Mayo, K., Vallania, F.L.M., Mitra, R.D., Faber, K., Williamson, J., Bird, T., Diaz-Arrastia, R., Foroud, T.M.et al. (2012) Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer’s disease families. PLoS One, 7, e31039. 10.1371/journal.pone.0031039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hamos, J.E., Oblas, B., Pulaski-Salo, D., Welch, W.J., Bole, D.G. and Drachman, D.A. (1991) Expression of heat shock proteins in Alzheimer’s disease. Neurology, 41, 345–350. [DOI] [PubMed] [Google Scholar]
- 48. Hoozemans, J.J.M., vanHaastert, E.S., Nijholt, D.A.T., Rozemuller, A.J.M., Eikelenboom, P. and Scheper, W. (2009) The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am. J. Pathol., 174, 1241–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hoozemans, J.J.M., Veerhuis, R., Van Haastert, E.S., Rozemuller, J.M., Baas, F., Eikelenboom, P. and Scheper, W. (2005) The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. (Berl.), 110, 165–172. [DOI] [PubMed] [Google Scholar]
- 50. Lanzillotta, C., Zuliani, I., Tramutola, A., Barone, E., Blarzino, C., Folgiero, V., Caforio, M., Valentini, D., Villani, A., Locatelli, F.et al. (2021) Chronic PERK induction promotes Alzheimer-like neuropathology in Down syndrome: insights for therapeutic intervention. Prog. Neurobiol., 196, 101892. 10.1016/j.pneurobio.2020.101892. [DOI] [PubMed] [Google Scholar]
- 51. Wang, F., Gu, Y., Xu, C., Du, K., Zhao, C., Zhao, Y. and Liu, X. (2022) Transplantation of fecal microbiota from APP/PS1 mice and Alzheimer’s disease patients enhanced endoplasmic reticulum stress in the cerebral cortex of wild-type mice. Front. Aging Neurosci., 14, 858130. 10.3389/fnagi.2022.858130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. O’Connor, T., Sadleir, K.R., Maus, E., Velliquette, R.A., Zhao, J., Cole, S.L., Eimer, W.A., Hitt, B., Bembinster, L.A., Lammich, S.et al. (2008) Phosphorylation of the translation initiation factor eIF2α increases BACE1 levels and promotes amyloidogenesis. Neuron, 60, 988–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Devi, L. and Ohno, M. (2014) PERK mediates eIF2α phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer’s disease. Neurobiol. Aging, 35, 2272–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Milhavet, O., Martindale, J.L., Camandola, S., Chan, S.L., Gary, D.S., Cheng, A., Holbrook, N.J. and Mattson, M.P. (2002) Involvement of Gadd153 in the pathogenic action of presenilin-1 mutations: Gadd153 and presenilin-1 mutations. J. Neurochem., 83, 673–681. [DOI] [PubMed] [Google Scholar]
- 55. Katayama, T., Imaizumi, K., Honda, A., Yoneda, T., Kudo, T., Takeda, M., Mori, K., Rozmahel, R., Fraser, P., George-Hyslop, P.S. and Tohyama, M. (2001) Disturbed activation of endoplasmic reticulum stress transducers by familial Alzheimer’s disease-linked Presenilin-1 mutations. J. Biol. Chem., 276, 43446–43454. [DOI] [PubMed] [Google Scholar]
- 56. Hashimoto, S. and Saido, T.C. (2018) Critical review: involvement of endoplasmic reticulum stress in the aetiology of Alzheimer’s disease. Open Biol., 8, 180024. 10.1098/rsob.180024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hashimoto, S., Ishii, A., Kamano, N., Watamura, N., Saito, T., Ohshima, T., Yokosuka, M. and Saido, T.C. (2018) Endoplasmic reticulum stress responses in mouse models of Alzheimer’s disease: overexpression paradigm versus knockin paradigm. J. Biol. Chem., 293, 3118–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Copanaki, E., Schürmann, T., Eckert, A., Leuner, K., Müller, W.E., Prehn, J.H.M. and Kögel, D. (2007) The amyloid precursor protein potentiates CHOP induction and cell death in response to ER Ca2+ depletion. Biochim. Biophys. Acta BBA - Mol. Cell Res., 1773, 157–165. [DOI] [PubMed] [Google Scholar]
- 59. Kögel, D., Concannon, C.G., Müller, T., König, H., Bonner, C., Poeschel, S., Chang, S., Egensperger, R. and Prehn, J.H.M. (2012) The APP intracellular domain (AICD) potentiates ER stress-induced apoptosis. Neurobiol. Aging, 33, 2200–2209. [DOI] [PubMed] [Google Scholar]
- 60. Takahashi, K., Niidome, T., Akaike, A., Kihara, T. and Sugimoto, H. (2009) Amyloid precursor protein promotes endoplasmic reticulum stress-induced cell death via C/EBP homologous protein-mediated pathway. J. Neurochem., 109, 1324–1337. [DOI] [PubMed] [Google Scholar]
- 61. Hanger, D.P., Hughes, K., Woodgett, J.R., Brion, J.P. and Anderton, B.H. (1992) Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett., 147, 58–62. [DOI] [PubMed] [Google Scholar]
- 62. Jiang, H.-Y. and Wek, R.C. (2005) Phosphorylation of the α-subunit of the eukaryotic initiation factor-2 (eIF2α) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J. Biol. Chem., 280, 14189–14202. [DOI] [PubMed] [Google Scholar]
- 63. Newman, J., Rissman, R.A., Sarsoza, F., Kim, R.C., Dick, M., Bennett, D.A., Cotman, C.W., Rohn, T.T. and Head, E. (2005) Caspase-cleaved tau accumulation in neurodegenerative diseases associated with tau and alpha-synuclein pathology. Acta Neuropathol. (Berl.), 110, 135–144. [DOI] [PubMed] [Google Scholar]
- 64. Rohn, T.T., Rissman, R.A., Davis, M.C., Kim, Y.E., Cotman, C.W. and Head, E. (2002) Caspase-9 activation and caspase cleavage of tau in the Alzheimer’s disease brain. Neurobiol. Dis., 11, 341–354. [DOI] [PubMed] [Google Scholar]
- 65. Nijholt, D.A., vanHaastert, E.S., Rozemuller, A.J., Scheper, W. and Hoozemans, J.J. (2012) The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J. Pathol., 226, 693–702. [DOI] [PubMed] [Google Scholar]
- 66. van der Harg, J.M., Nölle, A., Zwart, R., Boerema, A.S., vanHaastert, E.S., Strijkstra, A.M., Hoozemans, J.J. and Scheper, W. (2014) The unfolded protein response mediates reversible tau phosphorylation induced by metabolic stress. Cell Death Dis., 5, e1393–e1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Radford, H., Moreno, J.A., Verity, N., Halliday, M. and Mallucci, G.R. (2015) PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. (Berl.), 130, 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Smith, W.W., Jiang, H., Pei, Z., Tanaka, Y., Morita, H., Sawa, A., Dawson, V.L., Dawson, T.M. and Ross, C.A. (2005) Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum. Mol. Genet., 14, 3801–3811. [DOI] [PubMed] [Google Scholar]
- 69. Hoozemans, J.J.M., vanHaastert, E.S., Eikelenboom, P., deVos, R.A.I., Rozemuller, J.M. and Scheper, W. (2007) Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun., 354, 707–711. [DOI] [PubMed] [Google Scholar]
- 70. Colla, E., Coune, P., Liu, Y., Pletnikova, O., Troncoso, J.C., Iwatsubo, T., Schneider, B.L. and Lee, M.K. (2012) Endoplasmic reticulum stress is important for the manifestations of—synucleinopathy. Vivo J. Neurosci., 32, 3306–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sun, X., Aimé, P., Dai, D., Ramalingam, N., Crary, J.F., Burke, R.E., Greene, L.A. and Levy, O.A. (2018) Guanabenz promotes neuronal survival via enhancement of ATF4 and parkin expression in models of Parkinson disease. Exp. Neurol., 303, 95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sun, X., Liu, J., Crary, J.F., Malagelada, C., Sulzer, D., Greene, L.A. and Levy, O.A. (2013) ATF4 protects against neuronal death in cellular Parkinson’s disease models by maintaining levels of parkin. J. Neurosci., 33, 2398–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bouman, L., Schlierf, A., Lutz, A.K., Shan, J., Deinlein, A., Kast, J., Galehdar, Z., Palmisano, V., Patenge, N., Berg, D.et al. (2011) Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ., 18, 769–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gully, J.C., Sergeyev, V.G., Bhootada, Y., Mendez-Gomez, H., Meyers, C.A., Zolotukhin, S., Gorbatyuk, M.S. and Gorbatyuk, O.S. (2016) Up-regulation of activating transcription factor 4 induces severe loss of dopamine nigral neurons in a rat model of Parkinson’s disease. Neurosci. Lett., 627, 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bellucci, A., Navarria, L., Zaltieri, M., Falarti, E., Bodei, S., Sigala, S., Battistin, L., Spillantini, M., Missale, C. and Spano, P. (2011) Induction of the unfolded protein response by α-synuclein in experimental models of Parkinson’s disease: α-Synuclein accumulation induces the UPR. J. Neurochem., 116, 588–605. [DOI] [PubMed] [Google Scholar]
- 76. Li, Y., Fan, H., Sun, R., Jia, L., Yang, L., Zhang, H., Jin, X., Xiao, B., Ma, C. and Chai, Z. (2023) Wuzi Yanzong pill plays a neuroprotective role in Parkinson’s disease mice via regulating unfolded protein response mediated by endoplasmic reticulum stress. Chin. J. Integr. Med., 29, 19–27. [DOI] [PubMed] [Google Scholar]
- 77. Mercado, G., Castillo, V., Soto, P., López, N., Axten, J.M., Sardi, S.P., Hoozemans, J.J.M. and Hetz, C. (2018) Targeting PERK signaling with the small molecule GSK2606414 prevents neurodegeneration in a model of Parkinson’s disease. Neurobiol. Dis., 112, 136–148. [DOI] [PubMed] [Google Scholar]
- 78. Macdonald, M. (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell, 72, 971–983. [DOI] [PubMed] [Google Scholar]
- 79. Reijonen, S., Putkonen, N., Nørremølle, A., Lindholm, D. and Korhonen, L. (2008) Inhibition of endoplasmic reticulum stress counteracts neuronal cell death and protein aggregation caused by N-terminal mutant huntingtin proteins. Exp. Cell Res., 314, 950–960. [DOI] [PubMed] [Google Scholar]
- 80. Kouroku, Y., Fujita, E., Tanida, I., Ueno, T., Isoai, A., Kumagai, H., Ogawa, S., Kaufman, R.J., Kominami, E. and Momoi, T. (2007) ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ., 14, 230–239. [DOI] [PubMed] [Google Scholar]
- 81. Leitman, J., Barak, B., Benyair, R., Shenkman, M., Ashery, U., Hartl, F.U. and Lederkremer, G.Z. (2014) ER stress-induced eIF2-alpha phosphorylation underlies sensitivity of striatal neurons to pathogenic huntingtin. PLoS One, 9, e90803. 10.1371/journal.pone.0090803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ganz, J., Shacham, T., Kramer, M., Shenkman, M., Eiger, H., Weinberg, N., Iancovici, O., Roy, S., Simhaev, L., Da’adoosh, B.et al. (2020) A novel specific PERK activator reduces toxicity and extends survival in Huntington’s disease models. Sci. Rep., 10, 6875. 10.1038/s41598-020-63899-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Cleveland, D.W. and Rothstein, J.D. (2001) From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat. Rev. Neurosci., 2, 806–819. [DOI] [PubMed] [Google Scholar]
- 84. Gordon, P. (2013) Amyotrophic lateral sclerosis: an update for 2013 clinical features, pathophysiology, management and therapeutic trials. Aging Dis., 04, 295–310 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Swinnen, B. and Robberecht, W. (2014) The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol., 10, 661–670. [DOI] [PubMed] [Google Scholar]
- 86. Nagata, T., Ilieva, H., Murakami, T., Shiote, M., Narai, H., Ohta, Y., Hayashi, T., Shoji, M. and Abe, K. (2007) Increased ER stress during motor neuron degeneration in a transgenic mouse model of amyotrophic lateral sclerosis. Neurol. Res., 29, 767–771. [DOI] [PubMed] [Google Scholar]
- 87. Chen, D., Wang, Y. and Chin, E.R. (2015) Activation of the endoplasmic reticulum stress response in skeletal muscle of G93A*SOD1 amyotrophic lateral sclerosis mice. Front. Cell. Neurosci., 9, 170. 10.3389/fncel.2015.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Matus, S., Lopez, E., Valenzuela, V., Nassif, M. and Hetz, C. (2013) Functional contribution of the transcription factor ATF4 to the pathogenesis of amyotrophic lateral sclerosis. PLoS One, 8, e66672. 10.1371/journal.pone.0066672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wang, L., Popko, B., Tixier, E. and Roos, R.P. (2014) Guanabenz, which enhances the unfolded protein response, ameliorates mutant SOD1-induced amyotrophic lateral sclerosis. Neurobiol. Dis., 71, 317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Dzhashiashvili, Y., Monckton, C.P., Shah, H.S., Kunjamma, R.B. and Popko, B. (2019) The UPR-PERK pathway is not a promising therapeutic target for mutant SOD1-induced ALS. Neurobiol. Dis., 127, 527–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bugallo, R., Marlin, E., Baltanás, A., Toledo, E., Ferrero, R., Vinueza-Gavilanes, R., Larrea, L., Arrasate, M. and Aragón, T. (2020) Fine tuning of the unfolded protein response by ISRIB improves neuronal survival in a model of amyotrophic lateral sclerosis. Cell Death Dis., 11, 397. 10.1038/s41419-020-2601-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Dewey, C.M., Cenik, B., Sephton, C.F., Dries, D.R., Mayer, P., Good, S.K., Johnson, B.A., Herz, J. and Yu, G. (2011) TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol. Cell. Biol., 31, 1098–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Liu-Yesucevitz, L., Bilgutay, A., Zhang, Y.-J., Vanderwyde, T., Citro, A., Mehta, T., Zaarur, N., McKee, A., Bowser, R., Sherman, M., Petrucelli, L. and Wolozin, B. (2010) Tar DNA binding Protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One, 5, e13250. 10.1371/journal.pone.0013250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. McDonald, K.K., Aulas, A., Destroismaisons, L., Pickles, S., Beleac, E., Camu, W., Rouleau, G.A. and Vande Velde, C. (2011) TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum. Mol. Genet., 20, 1400–1410. [DOI] [PubMed] [Google Scholar]
- 95. Kedersha, N.L., Gupta, M., Li, W., Miller, I. and Anderson, P. (1999) RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J. Cell Biol., 147, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. de Mena, L., Lopez-Scarim, J. and Rincon-Limas, D.E. (2021) TDP-43 and ER stress in neurodegeneration: friends or foes? Front. Mol. Neurosci., 14, 772226. 10.3389/fnmol.2021.772226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wang, X., Zhou, S., Ding, X., Ma, M., Zhang, J., Zhou, Y., Wu, E. and Teng, J. (2015) Activation of ER stress and autophagy induced by TDP-43 A315T as pathogenic mechanism and the corresponding histological changes in skin as potential biomarker for ALS with the mutation. Int. J. Biol. Sci., 11, 1140–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zhao, C., Liao, Y., Rahaman, A. and Kumar, V. (2022) Towards understanding the relationship between ER stress and unfolded protein response in amyotrophic lateral sclerosis. Front. Aging Neurosci., 14, 892518. 10.3389/fnagi.2022.892518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Jeon, Y.-M., Kwon, Y., Lee, S. and Kim, H.-J. (2023) Potential roles of the endoplasmic reticulum stress pathway in amyotrophic lateral sclerosis. Front. Aging Neurosci., 15, 1047897. 10.3389/fnagi.2023.1047897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kim, H.-J., Raphael, A.R., LaDow, E.S., McGurk, L., Weber, R.A., Trojanowski, J.Q., Lee, V.M.-Y., Finkbeiner, S., Gitler, A.D. and Bonini, N.M. (2014) Therapeutic modulation of eIF2α phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat. Genet., 46, 152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Freibaum, B.D. and Taylor, J.P. (2017) The role of dipeptide repeats in C9ORF72-related ALS-FTD. Front. Mol. Neurosci., 10, 35. 10.3389/fnmol.2017.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kramer, N.J., Haney, M.S., Morgens, D.W., Jovičić, A., Couthouis, J., Li, A., Ousey, J., Ma, R., Bieri, G., Tsui, C.K.et al. (2018) CRISPR–Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity. Nat. Genet., 50, 603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Wang, R., Xu, X., Hao, Z., Zhang, S., Wu, D., Sun, H., Mu, C., Ren, H. and Wang, G. (2019) Poly-PR in C9ORF72-related amyotrophic lateral sclerosis/frontotemporal dementia causes neurotoxicity by clathrin-dependent endocytosis. Neurosci. Bull., 35, 889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Fels, D.R. and Koumenis, C. (2006) The PERK/eIF2α/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer Biol. Ther., 5, 723–728. [DOI] [PubMed] [Google Scholar]
- 105. Li, Z., Ge, Y., Dong, J., Wang, H., Zhao, T., Wang, X., Liu, J., Gao, S., Shi, L., Yang, S., Huang, C. and Hao, J. (2022b) BZW1 facilitates glycolysis and promotes tumor growth in pancreatic ductal adenocarcinoma through potentiating eIF2α phosphorylation. Gastroenterology, 162, 1256–1271.e14. 10.1053/j.gastro.2021.12.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Rozpedek, W., Pytel, D., Mucha, B., Leszczynska, H., Diehl, J.A. and Majsterek, I. (2016) The role of the PERK/eIF2α/ATF4/CHOP signaling pathway in tumor progression during endoplasmic reticulum stress. Curr. Mol. Med., 16, 533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Feng, Y.-X., Jin, D.X., Sokol, E.S., Reinhardt, F., Miller, D.H. and Gupta, P.B. (2017) Cancer-specific PERK signaling drives invasion and metastasis through CREB3L1. Nat. Commun., 8, 1079. 10.1038/s41467-017-01052-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Liu, L., Wise, D.R., Diehl, J.A. and Simon, M.C. (2008) Hypoxic reactive oxygen species regulate the integrated stress response and cell survival. J. Biol. Chem., 283, 31153–31162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Salvagno, C., Mandula, J.K., Rodriguez, P.C. and Cubillos-Ruiz, J.R. (2022) Decoding endoplasmic reticulum stress signals in cancer cells and antitumor immunity. Trends Cancer, 8, 930–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Rosenwald, I.B., Hutzler, M.J., Wang, S., Savas, L. and Fraire, A.E. (2001) Expression of eukaryotic translation initiation factors 4E and 2alpha is increased frequently in bronchioloalveolar but not in squamous cell carcinomas of the lung. Cancer, 92, 2164–2171. [DOI] [PubMed] [Google Scholar]
- 111. Rosenwald, I.B., Koifman, L., Savas, L., Chen, J.-J., Woda, B.A. and Kadin, M.E. (2008) Expression of the translation initiation factors eIF-4E and eIF-2α is frequently increased in neoplastic cells of Hodgkin lymphoma. Hum. Pathol., 39, 910–916. [DOI] [PubMed] [Google Scholar]
- 112. Rosenwald, I.B., Wang, S., Savas, L., Woda, B. and Pullman, J. (2003) Expression of translation initiation factor eIF-2 alpha is increased in benign and malignant melanocytic and colonic epithelial neoplasms. Cancer, 98, 1080–1088. [DOI] [PubMed] [Google Scholar]
- 113. Lobo, M.V., Martín, M.E., Pérez, M.I., Alonso, F.J., Redondo, C., Alvarez, M.I. and Salinas, M. (2000) Levels, phosphorylation status and cellular localization of translational factor eIF2 in gastrointestinal carcinomas. Histochem. J., 32, 139–150. [DOI] [PubMed] [Google Scholar]
- 114. Guo, L., Chi, Y., Xue, J., Ma, L., Shao, Z. and Wu, J. (2017) Phosphorylated eIF2α predicts disease-free survival in triple-negative breast cancer patients. Sci. Rep., 7, 44674. 10.1038/srep44674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Wimbauer, F., Yang, C., Shogren, K.L., Zhang, M., Goyal, R., Riester, S.M., Yaszemski, M.J. and Maran, A. (2012) Regulation of interferon pathway in 2-methoxyestradiol-treated osteosarcoma cells. BMC Cancer, 12. 10.1186/1471-2407-12-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wang, Z., Yin, F., Xu, J., Zhang, T., Wang, G., Mao, M., Wang, Z., Sun, W., Han, J., Yang, M.et al. (2019b) CYT997(Lexibulin) induces apoptosis and autophagy through the activation of mutually reinforced ER stress and ROS in osteosarcoma. J. Exp. Clin. Cancer Res., 38, 44. 10.1186/s13046-019-1047-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Wang, R., Sun, D.-Z., Song, C.-Q., Xu, Y.-M., Liu, W., Liu, Z. and Dong, X.-S. (2018) Eukaryotic translation initiation factor 2 subunit α (eIF2α) inhibitor salubrinal attenuates paraquat-induced human lung epithelial-like A549 cell apoptosis by regulating the PERK-eIF2α signaling pathway. Toxicol. In Vitro, 46, 58–65. [DOI] [PubMed] [Google Scholar]
- 118. Yu, C.-L., Yang, S.-F., Hung, T.-W., Lin, C.-L., Hsieh, Y.-H. and Chiou, H.-L. (2019) Inhibition of eIF2α dephosphorylation accelerates pterostilbene-induced cell death in human hepatocellular carcinoma cells in an ER stress and autophagy-dependent manner. Cell Death Dis., 10, 418. 10.1038/s41419-019-1639-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Gupta, S., McGrath, B. and Cavener, D.R. (2009) PERK regulates the proliferation and development of insulin-secreting beta-cell tumors in the endocrine pancreas of mice. PLoS One, 4, e8008. 10.1371/journal.pone.0008008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Mandula, J.K., Chang, S., Mohamed, E., Jimenez, R., Sierra-Mondragon, R.A., Chang, D.C., Obermayer, A.N., Moran-Segura, C.M., Das, S., Vazquez-Martinez, J.A.et al. (2022) Ablation of the endoplasmic reticulum stress kinase PERK induces paraptosis and type I interferon to promote anti-tumor T cell responses. Cancer Cell, 40, 1145–1160.e9. 10.1016/j.ccell.2022.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Peñaranda Fajardo, N.M., Meijer, C. and Kruyt, F.A.E. (2016) The endoplasmic reticulum stress/unfolded protein response in gliomagenesis, tumor progression and as a therapeutic target in glioblastoma. Biochem. Pharmacol., 118, 1–8. [DOI] [PubMed] [Google Scholar]
- 122. Dadey, D.Y.A., Kapoor, V., Khudanyan, A., Thotala, D. and Hallahan, D.E. (2018) PERK regulates glioblastoma sensitivity to ER stress although promoting radiation resistance. Mol. Cancer Res., 16, 1447–1453. [DOI] [PubMed] [Google Scholar]
- 123. Khoonkari, M., Liang, D., Lima, M.T., van derLand, T., Liang, Y., Sun, J., Dolga, A., Kamperman, M., vanRijn, P. and Kruyt, F.A.E. (2022) The unfolded protein response sensor PERK mediates stiffness-dependent adaptation in glioblastoma cells. Int. J. Mol. Sci., 23, 6520. 10.3390/ijms23126520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Soni, H., Bode, J., Nguyen, C.D.L., Puccio, L., Neßling, M., Piro, R.M., Bub, J., Phillips, E., Ahrends, R., Eipper, B.A., Tews, B. and Goidts, V. (2020) PERK-mediated expression of peptidylglycine α-amidating monooxygenase supports angiogenesis in glioblastoma. Oncogene, 9, 18. 10.1038/s41389-020-0201-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Dai, H., Shen, K., Yang, Y., Su, X., Luo, Y., Jiang, Y., Shuai, L., Zheng, P., Chen, Z. and Bie, P. (2019) PUM1 knockdown prevents tumor progression by activating the PERK/eIF2/ATF4 signaling pathway in pancreatic adenocarcinoma cells. Cell Death Dis., 10, 595. 10.1038/s41419-019-1839-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Bobrovnikova-Marjon, E., Grigoriadou, C., Pytel, D., Zhang, F., Ye, J., Koumenis, C., Cavener, D. and Diehl, J.A. (2010) PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene, 29, 3881–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Sequeira, S.J., Ranganathan, A.C., Adam, A.P., Iglesias, B.V., Farias, E.F. and Aguirre-Ghiso, J.A. (2007) Inhibition of proliferation by PERK regulates mammary acinar morphogenesis and tumor formation. PLoS One, 2, e615. 10.1371/journal.pone.0000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Li, X., Zheng, J., Chen, S., Meng, F., Ning, J. and Sun, S. (2021) Oleandrin, a cardiac glycoside, induces immunogenic cell death via the PERK/elF2α/ATF4/CHOP pathway in breast cancer. Cell Death Dis., 12, 314. 10.1038/s41419-021-03605-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zheng, Y., Liu, P., Wang, N., Wang, S., Yang, B., Li, M., Chen, J., Situ, H., Xie, M., Lin, Y. and Wang, Z. (2019) Betulinic acid suppresses breast cancer metastasis by targeting GRP78-mediated glycolysis and ER stress apoptotic pathway. Oxidative Med. Cell. Longev., 2019, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Nagelkerke, A., Bussink, J., Mujcic, H., Wouters, B.G., Lehmann, S., Sweep, F.C. and Span, P.N. (2013) Hypoxia stimulates migration of breast cancer cells via the PERK/ATF4/LAMP3-arm of the unfolded protein response. Breast Cancer Res., 15, R2. 10.1186/bcr3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Wortel, I.M.N., van derMeer, L.T., Kilberg, M.S. and vanLeeuwen, F.N. (2017) Surviving stress: modulation of ATF4-mediated stress responses in normal and malignant cells. Trends Endocrinol. Metab., 28, 794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Pike, L.R.G., Singleton, D.C., Buffa, F., Abramczyk, O., Phadwal, K., Li, J.-L., Simon, A.K., Murray, J.T. and Harris, A.L. (2013) Transcriptional up-regulation of ULK1 by ATF4 contributes to cancer cell survival. Biochem. J., 449, 389–400. [DOI] [PubMed] [Google Scholar]
- 133. Chen, D., Fan, Z., Rauh, M., Buchfelder, M., Eyupoglu, I.Y. and Savaskan, N. (2017) ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene, 36, 5593–5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Dey, S., Sayers, C.M., Verginadis, I.I., Lehman, S.L., Cheng, Y., Cerniglia, G.J., Tuttle, S.W., Feldman, M.D., Zhang, P.J.L., Fuchs, S.Y., Diehl, J.A. and Koumenis, C. (2015) ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J. Clin. Invest., 125, 2592–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Pällmann, N., Livgård, M., Tesikova, M., Zeynep Nenseth, H., Akkus, E., Sikkeland, J., Jin, Y., Koc, D., Kuzu, O.F., Pradhan, M.et al. (2019) Regulation of the unfolded protein response through ATF4 and FAM129A in prostate cancer. Oncogene, 38, 6301–6318. [DOI] [PubMed] [Google Scholar]
- 136. Zeng, H., Zhang, J., Du, Y., Wang, J., Ren, Y., Li, M., Li, H., Cai, Z., Chu, Q. and Yang, C. (2016) Crosstalk between ATF4 and MTA1/HDAC1 promotes osteosarcoma progression. Oncotarget, 7, 7329–7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Coker-Gurkan, A., Can, E., Sahin, S., Obakan-Yerlikaya, P. and Arisan, E.-D. (2021) Atiprimod triggered apoptotic cell death via acting on PERK/eIF2α/ATF4/CHOP and STAT3/NF-ΚB axis in MDA-MB-231 and MDA-MB-468 breast cancer cells. Mol. Biol. Rep., 48, 5233–5247. [DOI] [PubMed] [Google Scholar]
- 138. Li, X., Zhou, D., Cai, Y., Yu, X., Zheng, X., Chen, B., Li, W., Zeng, H., Hassan, M., Zhao, Y. and Zhou, W. (2022a) Endoplasmic reticulum stress inhibits AR expression via the PERK/eIF2α/ATF4 pathway in luminal androgen receptor triple-negative breast cancer and prostate cancer. NPJ Breast Cancer, 8, 2. 10.1038/s41523-021-00370-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Celardo, I., Costa, A.C., Lehmann, S., Jones, C., Wood, N., Mencacci, N.E., Mallucci, G.R., Loh, S.H.Y. and Martins, L.M. (2016) Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease. Cell Death Dis., 7, e2271–e2271. [DOI] [PMC free article] [PubMed] [Google Scholar]