

SUMMARY
Cancers often display immune escape, but the mechanisms are incompletely understood. Herein, we identify SMYD3 as a mediator of immune escape in human papillomavirus (HPV)-negative head and neck squamous cell carcinoma (HNSCC), an aggressive disease with poor response to immunotherapy with pembrolizumab. SMYD3 depletion induces upregulation of multiple type I interferon (IFN) response and antigen presentation machinery genes in HNSCC cells. Mechanistically, SMYD3 binds to and regulates the transcription of UHRF1, encoding for a reader of H3K9me3, which binds to H3K9me3-enriched promoters of key immune-related genes, recruits DNMT1, and silences their expression. SMYD3 further maintains the repression of immune-related genes through intragenic deposition of H4K20me3. In vivo, Smyd3 depletion induces influx of CD8+ T cells and increases sensitivity to anti-programmed death 1 (PD-1) therapy. SMYD3 overexpression is associated with decreased CD8 T cell infiltration and poor response to neoadjuvant pembrolizumab. These data support combining SMYD3 depletion strategies with checkpoint blockade to overcome anti-PD-1 resistance in HPV-negative HNSCC.
Graphical Abstract
In brief
Nigam et al. show that SMYD3 depletion induces type I IFN response and APM genes in HPV-negative HNSCC cells. SMYD3 represses immune-related genes through direct transcriptional regulation of UHRF1, an H3K9me3-reader that recruits DNMT1A, and deposition of H4K20me3. SMYD3 is a rational target to overcome anti-PD-1 resistance in HPV-negative HNSCC.
INTRODUCTION
Head and neck squamous cell carcinoma (HNSCC) affects approximately 50,000 patients annually in the United States.1 While human papilloma virus (HPV)-positive patients tend to have an excellent prognosis,2 HPV-negative patients have an approximately 50% recurrence rate after treatment with chemoradiotherapy and/or surgery, and a dismal prognosis in the recurrent/metastatic (R/M) setting.3 Recently, pembrolizumab, which blocks the programmed death 1 (PD-1)/programmed death-ligand 1 (PD-L1) axis, was approved as a first-line therapy for patients with R/M HNSCC.4 However, response rates in HPV-negative HNSCC patients are as low as 19%.5-7 Thus, the elucidation of mechanisms that dictate poor CD8+ T cell infiltration in HPV-negative HNSCC is of paramount importance to improve the efficacy of immunotherapy.8,9
Epigenetic regulation mediated by histone modifications has emerged as an important cancer therapeutic avenue. The Cancer Genome Atlas (TCGA) has documented a plethora of genetic and expression alterations in chromatin modifiers in multiple cancer types, including HPV-negative HNSCC.10-13 Recent evidence supports that some of these chromatin modifiers may be involved in the regulation of antitumor immunity.14 Given the importance of immunotherapy in HPV-negative HNSCC, we previously conducted a bioinformatic interrogation of two HPV-negative HNSCC expression cohorts to identify protein methyltransferases (PMTs) and demethylases (PDMTs), a class of chromatin modifiers that are frequently altered in HPV-negative HNSCC and that are associated with the non-inflamed phenotype and could thus be biological culprits of CD8+ T cell exclusion.15 We found that the mRNA expression levels of SET and MYND-domain protein 3 (SMYD3) correlated inversely with the mRNA levels of CD8A, CD8+ T cell-attracting chemokines, such as CXCL9, CXCL10, and CXCL11, as well as several antigen presentation machinery (APM) molecules.15 Additionally, SMYD3 knockdown in HPV-negative HNSCC cell lines in vitro led to significant upregulation in the mRNA and protein levels of CXCL9, CXCL10, and CXCL11, suggesting that SMYD3 regulates the transcription of these genes.
SMYD3, a member of the SET and MYND-domain family, is a PMT that has been implicated as an oncogene in multiple cancer types, such as colorectal and hepatocellular carcinoma (HCC),16,17 pancreatic and lung adenocarcinoma,18 and breast cancer.19-21 In HCC, SMYD3 acts as a chromatin modifier in the nucleus by directly binding to DNA as a transcription factor, forming a complex with RNA polymerase II, and writing and reading H3K4me3 to activate the transcription of oncogenes involved in cell cycle and epithelial-mesenchymal transition.16,19 A few groups have also reported that SMYD3 trimethylates H4K20 (H4K20me3) to induce transcriptional repression.22,23 However, in K-ras mutant pancreatic and lung adenocarcinomas, SMYD3 directly methylates the cytoplasmic substrate MAP3K2, inhibiting its interaction with phosphatase PP2A and activating the ERK1/2 pathway.18 This suggests that the oncogenic function of SMYD3 varies by cancer type and may be mediated either through its function as a chromatin modifier/transcription factor or as a direct methylator of cytoplasmic substrates.
Given the oncogenic role of SMYD3 in many cancer types, SMYD3 inhibitors are actively in development.24-26 An alternative and promising drug platform are RNA-targeted anti-sense oligonucleotides (ASOs), which are single-stranded, chemically modified DNA oligonucleotides.27 Importantly, Kontaki et al.28 recently showed that Smyd3 ASOs decreased Smyd3 mRNA levels in vivo efficiently and halted the growth of liver tumors in a chemically induced HCC mouse model, underlining the potential of Smyd3 ASOs as a therapeutic platform.
In the present study, we identify SMYD3 as a master epigenetic regulator of antitumor immune response in HPV-negative HNSCC and provide a rationale for translational approaches combining SMYD3 ASOs with checkpoint blockade to overcome anti-PD-1 resistance in this devastating disease.
RESULTS
SMYD3 depletion is associated with upregulation of type I IFN response and APM genes in HPV-negative HNSCC cells
Based on our previously published work,15 we sought to evaluate whether SMYD3 depletion has a systemic effect on the expression of type I interferon (IFN) response and APM genes (here on referred to cumulatively as immune-related genes; see statistical analyses in section, “lists of type I IFN response and APM genes” and Tables S1 and S2).
To this end, we conducted RNA-sequencing (RNA-seq) of the HPV-negative HNSCC cell line HN-6 with endogenously high levels of SMYD3 before and after small interfering RNA (siRNA)-mediated SMYD3 knockdown for 72 h and after 24 h of IFN-γ exposure. Evaluation of the expression levels of immune-related genes revealed upregulation of multiple key type I IFN response and APM genes (Figures 1A, S1A, and S1B). A similar phenotype was reproduced with SMYD3 ASOs28 (Ionis Pharmaceuticals) (Figures 1B, S2A, and S2B) and with a SMYD3 CRISPR knockout (KO) cell line (termed 5-3) (Figures 1C, S3A, and S3B). More specifically, SMYD3 depletion using siRNAs induced upregulation of 40 out of 97 IFN-α response and 22 out of 88 APM genes (Figure S4A). SMYD3 ASO-mediated depletion upregulated 68 out of 97 IFN-α response and five out of 88 APM genes (Figure S4B). Similarly, SMYD3 depletion in the SMYD3 KO cell line induced upregulation of 73 of 97 IFN-α response genes and 24 of 88 APM genes (Figure 1D). Among the three cell systems, 28 of 97 IFN-α response genes were commonly upregulated (Figure S5). Concordantly, ingenuity pathway analysis (IPA) revealed enrichment in pathways related to inflammation in all three cell systems (Figures 1E and S6).
Figure 1. SMYD3 depletion induces upregulation of type I IFN response and APM genes in HPV-negative HNSCC cells.

(A–C) Heatmaps of type I IFN response and APM genes (RNA-seq) in HN-6 cells after SMYD3 depletion. Heatmaps of regularized log-transformed and Z score expression values. (A) HN-6 cells treated with control or a SMYD3-targeting siRNA for 72 h (three biological replicates per condition. siNC.r, control replicate; siSMYD3.r, SMYD3 siRNA replicate), and exposed to IFN-β. (B) HN-6 cells treated with PBS or SMYD3 ASOs for 72 h (two biological replicates per condition) and exposed to IFN-β. (C) Parental HN-6 and SMYD3 knockout (KO) cells (5-3 cell line) after exposure to IFN-β (three biological replicates per condition).
(D) Volcano plot showing DESeq2 results in SMYD3 KO cells (5-3 cell line) compared to parental HN-6 cells. False discovery rate (FDR), 0.1; log2FC threshold, log2 (1.3). Red triangles, IFN-α genes (upregulated, 73; downregulated, seven); blue crosses, APM genes (upregulated, 24; downregulated, 11); gray circles, other genes. Total number of genes = 19,447 (upregulated, 6,300; downregulated, 7,292).
(E) IPA reveals enrichment of pathways related to inflammation in an HPV-negative cell line (HN-6) after SMYD3 depletion (SMYD3 KO 5-3 cells).
(F) qRT-PCR (SYBR green) for immune-related genes in HN-6 (left) and HN-SCC-151 cells (right) after siRNA-mediated SMYD3 knockdown. Cells were treated with control and a SMYD3-targeting siRNA for 72 h with or without exposure to IFN-β. Technical replicates are shown. Data are represented as mean ± SEM. Student’s t test, *p < 0.05. Similar results were obtained in an independent experiment.
(G) Normalized enrichment scores (NESs) of Hallmark gene sets correlated with SMYD3 mRNA expression. Positive NES indicates pathways enriched with gene expression patterns that are positively correlated with SMYD3 mRNA. Negative NES indicates pathways enriched with gene expression patterns that are negatively correlated with SMYD3 mRNA. TCGA, Firehose Legacy, 427 HPV-negative tumor samples.
(H) Pairwise correlations between SMYD3 and type I IFN response genes in single cancer cells of a publicly available single-cell RNA-seq database.
To confirm these findings and further evaluate whether the effect of SMYD3 on the expression of immune-related genes is dependent on IFN-β exposure, we treated two HPV-negative HNSCC cell lines (HN-6, HN-SCC-151) with negative control or SMYD3-specific siRNAs for 72 h with or without exposure to IFN-β and conducted qPCR for a panel of representative immune-related genes. Results confirmed that SMYD3 depletion induced significant upregulation of multiple immune-related genes in both cell lines (Figure 1F), and, importantly, significant upregulation of these genes was observed even in the absence of IFN-β. This suggests that the effect of SMYD3 on the expression of some immune-related genes is not IFN-β dependent, and thus SMYD3 depletion could trigger an inflammatory response in cancer cells even in the setting of a “cold” tumor microenvironment.
SMYD3 mRNA expression is associated with repression of immune signatures in HPV-negative HNSCC human tumors and is negatively correlated with the expression of immune-related genes in single cancer cells
To determine whether the expression of SMYD3 is associated with enrichment of immune signatures in HPV-negative HNSCC tumor samples, we interrogated TCGA dataset of patients with primary HPV-negative HNSCC. Gene set enrichment analysis (GSEA) revealed that higher SMYD3 mRNA expression correlated with repression of immune signatures (Figure 1G).
We also analyzed a publicly available single-cell RNA-seq database of HPV-negative HNSCC tumors,29 and, consistently, SMYD3 mRNA expression correlated negatively with multiple type I IFN response and APM genes within individual cancer cells (Figures 1H and S7). These data suggest that SMYD3 is a key regulator of the expression of a broad repertoire of immune-related genes in HPV-negative HNSCC cells.
UHRF1, a reader of H3K9me3, is downregulated after transient SMYD3 depletion and regulates the transcription of immune-related genes in HPV-negative HNSCC cells
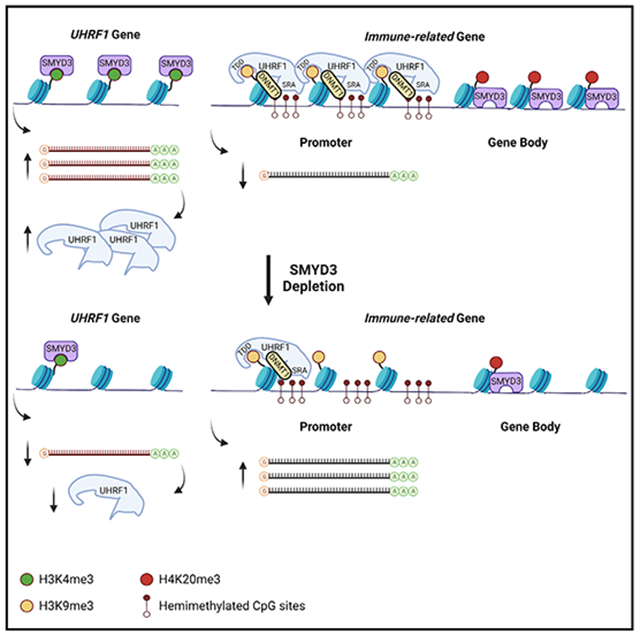
SMYD3 activates the transcription of downstream target genes by writing and reading H3K4me3,16,19 an activating histone mark; however, our data suggest a reverse phenotype, whereby SMYD3 is associated with repression of immune-related signatures. We thus reasoned that SMYD3 may regulate the expression of immune-related genes through an indirect mechanism, specifically through the upregulation of a repressive chromatin modifier or reader, which then in turn represses immune-related genes in HPV-negative HNSCC cells.
To identify candidate repressive epigenetic regulators, we curated and queried a list of 438 factors that are known to be involved in epigenetic regulation (Table S3) and sorted the genes that had significantly decreased mRNA expression upon SMYD3 depletion and encoded for chromatin factors with known repressive gene transcription functions. We found that the gene encoding the ubiquitin-like PHD and ring finger domain-containing protein 1 (UHRF1) had significantly lower mRNA expression after SMYD3 depletion (Figure S8). UHRF1 is an E3 ubiquitin ligase that recruits DNMT1 to CpG DNA methylation sites and ensures high-fidelity DNA maintenance methylation on the newly synthesized DNA strand. UHRF1 binds to and reads hemi-methylated CpG marks through its SET- and RING-associated (SRA) domain, and H3K9me3 through its tudor domain (TTD), mediating transcriptional repression of its downstream target genes.30-33 It is also associated with increased proliferation and poor prognosis in multiple cancer types.34 In order to validate UHRF1 as a downstream target of SMYD3, we performed qRT-PCR and western blotting in HN-6 and HN-SCC-151 cells after siRNA-mediated SMYD3 depletion and confirmed significant downregulation of UHRF1 both at the mRNA and protein levels in the presence or absence of IFN-β (Figures 2A, 2B, and S9).
Figure 2. UHRF1 is transcriptionally regulated by SMYD3 and silences the expression of immune-related genes in HPV-negative HNSCC cells through DNMT1 recruitment.

(A and B) SMYD3 knockdown induces downregulation of UHRF1 in HN-6 cells at the mRNA (A) and protein levels (B). Data for (A) are represented as mean ± SEM. Student’s t test, *p < 0.05.
(C) UCSC (University of California Santa-Cruz) tracks of SMYD3 and H3K4me3 in HN-6 cells. Exons and introns 1 and 2 of UHRF1 are shown.
(D) Volcano plot showing DESeq2 results of RNA-seq of HN-6 cells transfected with a UHRF1-targeting siRNA compared to control for 72 h and exposed to IFN-β. FDR < 0.1; log2FC threshold, log2 (1.3). Red triangles, IFN-α genes (upregulated, 57; downregulated, four); blue crosses, APM genes (upregulated, 12; downregulated, eight); gray circles, other genes. Total number of genes = 18,971 (upregulated, 4,438; downregulated, 4,585).
(E) Heatmaps of type I IFN response (left) and APM genes (right) in HN-6 cells after UHRF1 depletion for 3 days. HN-6 cells were treated with control (siNC) or a UHRF1-targeting siRNA (siUHRF1) for 72 h (three biological replicates per condition) and exposed to IFN-β. Cells were collected and RNA-seq was conducted. Heatmaps showing Z score of variance stabilizing transformed expression values.
(F) Venn diagram showing the overlap of upregulated type I IFN response (left) and APM (right) genes in HN-6 cells treated with SMYD3 or UHRF1 siRNAs in the presence of IFN-β. FDR< 0.1; log2FC, log2(1.3).
(G) ChIP assay for HA (shades of brown), endogenous DNMT1 (shades of green), and endogenous H3K9me3 (shades of purple) followed by qRT-PCR for CXCL9, CXCL10, MX1, OAS2, and RSAD2. HN-6 cells were transfected with HA-Mock or HA-UHRF1 for 48 h and exposed to IFN-β for 24 h prior to cell collection. Data are represented as mean ± SEM. Standard error (SE) bars represent the SE of two or three technical replicates per reaction. Student’s t test, *p < 0.05, **p < 0.01, ***p < 0.001. Similar results were obtained in an independent biological replicate.
(H) ChIP assay for endogenous UHRF1 (shades of brown), DNMT1 (shades of green), and H3K9me3 (shades of purple) followed by qRT-PCR for CXCL9, CXCL10, MX1, OAS2, and RSAD2. HN-6 cells were transfected with negative control (siNC) or a SMYD3-targeting siRNA (siSMYD3) for 72 h and exposed to IFN-β for 24 h prior to cell collection. Data are represented as mean ± SEM. SE bars represent the SE of two or three technical replicates per reaction. Student’s t test, *p < 0.05, **p < 0.01, ***p < 0.001. Similar results were obtained in an independent biological replicate.
To evaluate whether SMYD3 directly binds to and regulates the transcription of UHRF1, we conducted a CUT&RUN assay for SMYD3 and H3K4me3 in HN-6 cells and found significant enrichment of SMYD3 and H3K4me3 within the gene body of UHRF1 (Figure 2C). Interestingly, Sarris et al.16 previously conducted chromatin immunoprecipitation (ChIP) for mouse Smyd3 followed by sequencing in mouse HCC cells; we analyzed the sequencing tracks from this database and consistently found enrichment of mouse Smyd3 on the promoter of Uhrf1 (Figure S10). These data support that SMYD3 binds to and transcriptionally regulates UHRF1.
To assess whether UHRF1 regulates the expression of immune-related genes, HN-6 cells were transfected with either UHRF1-targeting (siUHRF1) or control siRNAs (siNC) for 72 h and treated with or without IFN-β for 24 h, and RNA-seq was conducted. Similarly to SMYD3 depletion, UHRF1 depletion induced significant upregulation of multiple type I IFN response and APM genes, both in the presence (Figures 2D, 2E, S11A, and S11B) and absence of IFN-β exposure (Figures S12A-S12C). These results were confirmed with qPCR for a number of representative immune-related genes in HN-6 and HN-SCC-151 cells (Figures S13A and S13B). Furthermore, 47 of the type I IFN response and six of the APM genes were commonly upregulated in siSMYD3- or siUHRF1-treated HN-6 cells, suggesting that these genes are commonly affected by knockdown of either SMYD3 or UHRF1 (Figure 2F).
UHRF1 represses its downstream target genes through maintenance of CpG DNA methylation by recruiting DNTM1 and reading H3K9me3.30,35 We therefore hypothesized that UHRF1 binds to the promoters of immune-related genes enriched with H3K9me3 and recruits DNMT1 to maintain CpG DNA methylation and induce repression. To further investigate this possibility, HN-6 cells were transfected with hemagglutinin (HA)-UHRF1 or HA-Mock plasmids for 48 h and exposed to IFN-β for 24 h prior to cell collection. A ChIP assay for the HA-tag (HA-UHRF1), H3K9me3, and DNMT1 followed by qPCR showed that HA-UHRF1 and H3K9me3 were enriched in the promoters of representative immune-related genes that were upregulated with UHRF1 depletion, including CXCL9, CXCL10, MX1, OAS2, and RSAD2 (Figures 2G and S14). Concurrently, after HA-UHRF1 overexpression, DNMT1 was enriched on the promoters of some of these genes, indicating that UHRF1 recruits DNMT1 to these genes. Conversely, after 72 h of SMYD3 depletion, UHRF1 occupancy on these immune-gene promoters decreased and, on some of these genes, DNMT1 binding was decreased as well (Figures 2H and S14).
We next determined whether SMYD3 depletion was associated with a decrease in the DNA methylation levels of CpG sites in the promoters of these genes. An EPIC methylation assay was performed in HN-6 cells treated with control or SMYD3-targeting siRNAs for 72 h. One-hundred and eighteen CpG sites corresponding to the promoters of the five aforementioned immune-related genes were included in the EPIC array and over 50% of them (62 out of 118) were at least partially or close to fully methylated (beta values >0.5) at baseline, suggesting that these sites may serve as binding sites of hemi-methylated DNA for the SRA domain of UHRF1 (Table S4). While DNA methylation was detected in the promoters of these genes, we did not observe a decrease in the average CpG site DNA methylation levels after SMYD3 depletion (siNC-siSMYD3 delta beta <0.1; Table S4). This finding suggests that DNTM1, in concert with UHRF1, may mediate transcriptional repression of immune-related genes in a methyltransferase-independent manner and mainly as a scaffolding protein within the context of a repressor complex involving UHRF1.36-38
In summary, these results support that SMYD3 regulates the expression of immune-related genes through UHRF1, whereby UHRF1 reads and binds to DNA-methylated promoters enriched in H3K9me3 and recruits DNMT1 to repress these genes, likely through a methyltransferase-independent manner.
Permanent SMYD3 KO decreases the occupancy of H4K20me3 on immune-related genes in HPV-negative HNSCC cells
We then evaluated the protein levels of UHRF1 in SMYD3 KO cells. Interestingly, stable KO of SMYD3 did not perturb steady-state UHRF1 levels (Figure S15). We hypothesized that this may be related to the fact that UHRF1 is necessary for the survival of a variety of cancer cell lines39-41 and that SMYD3 KO cells compensate by upregulating UHRF1 levels back to a necessary minimum so that they can survive.
SMYD3 has been predominantly described as a transcriptional activator16,19-21; however, more recently, it has also been reported to induce transcriptional repression through deposition of H4K20me3.22,23 Based on these reports and our findings that SMYD3 depletion induced re-expression of immune-related genes, we hypothesized that SMYD3 may have a bifaceted function by acting both as a transcriptional activator of certain genes, such as UHRF1, through writing and/or reading H3K4me3, and as a transcriptional repressor of other genes, such as immune-related genes, by promoting the deposition of H4K20me3. We thus sought to assess whether SMYD3 directly silences the expression of immune-related genes through the deposition of H4K20me3. First, to evaluate whether SMYD3 affects global methylation levels of H4K20me3 in HPV-negative HNSCC cells, we conducted mass spectrometry analysis in parental HN-6 cells and two SMYD3 KO cell lines (5-2, 5-3) and found that H4K20me3 levels were significantly decreased in the SMYD3 KO cells (Figure 3A). Importantly, the protein levels of SUV420H1 and SUV420H2, which are established H4K20 methyltransferases, were stable or increased in the SMYD3 KO cell lines (Figure S16), suggesting that SMYD3 affects global levels of H4K20me3 through a mechanism independent of the protein amounts of these H4K20 methyltransferases. Although these findings do not prove that SMYD3 directly methylates H4K20, they support that SMYD3 affects the global levels of H4K20me3 in HPV-negative HNSCC cells.
Figure 3. SMYD3 depletion induces global decrease in H4K20me3 levels and decreases its occupancy on immune-related genes.

(A) Mass spectrometry (MS) quantification of H4K20me3 in HN-6 and SMYD3 KO cell lines 5-2 and 5-3. The relative percentage of H4K20me3 over all methylation states of H4K20 for HN-6 and the SMYD3 KO cell lines was generated using MS data for histone modification. Data are represented as mean ± SD. Student’st test, *p < 0.05. Error bars represent the SD generated from three biological replicates.
(B) Genomic distribution of H4K20me3 peaks using CUT&RUN assays in parental HN-6 cells compared to 5-3 cells in the presence of IFN-β.
(C) Left: genomic coordinate heatmap of differential intragenic H4K20me3 peaks with evaluable RNA-seq data annotated to 4,874 genes. At the top, distribution of peaks around the center of each peak. One representative biological replicate per condition is shown. FDR < 0.05, abs.log2FC (1.3). Right: RNA-seq heatmap of genes (n = 4,874) corresponding to differential H4K20me3 peaks, comparing HN-6 and 5-3 differential gene expression. Two biological replicates are shown. FDR < 0.1, abs.log2FC (1.3).
(D) Volcano plot of DESeq2 results of H4K20me3 intragenic peaks (total = 53,891) in HN-6 cells and 5-3 cells after 24 h of exposure to IFN-β; 49,320 H4K20me3 differential peaks were decreased and 148 H4K20me3 differential peaks were increased. FDR threshold, 0.05. Red triangles, 78 significantly decreased peaks overlapping with the promoters/TSS/gene bodies of 47 IFN-α genes; blue crosses, 17 significantly decreased peaks overlapping with the promoters/TSS/gene bodies of 14 APM genes; gray circle, other genes.
(E) Left: genomic coordinate heatmap of 84 differential intragenic H4K20me3 peaks with evaluable RNA-seq data annotated to 54 immune-related genes in HN-6 and 5-3 cells. At the top, distribution of peaks around the center of each peak. One representative biological replicate per condition is shown. FDR < 0.05, abs.log2FC (1.3). Right: RNA-seq heatmap of 54 immune-related genes corresponding to significantly decreased H4K20me3 peaks. Two biological replicates are shown. FDR <0.1, log2FC (1.3).
(F) Venn diagram showing the overlap of immune-related genes with intragenic SMYD3 (yellow circle), H4K20me3 (blue), and H3K4me3 (orange) peaks in HN-6 cells exposed to IFN-β. Absolute numbers represent genes with evaluable RNA-seq expression data. Immune-related genes with intragenic H3K4me3 peaks, 130; immune-related genes with intragenic H4K20me3 peaks, 56; immune-related genes with intragenic SMYD3 peaks, 100.
(G) Boxplot of average normalized tag counts of intragenic SMYD3 (n = 154), H4K20me3 (n = 87), and H3K4me3 (n = 152) peaks corresponding to immune-related genes with evaluable RNA-seq expression data (104 for SMYD3, 56 for H4K20me3, and 130 genes for H3K4me3 peaks) in HN-6 and 5-3 cells exposed to IFN-β. The average tag counts of three biological replicates are shown for each condition. One replicate was utilized for the 5-3 condition for the SMYD3 boxplot tag counts. Wilcoxon signed rank test was applied.
(H) UCSC browser tracks showing genome-wide mapping for SMYD3 (fuchsia), H4K20me3 (blue), and H3K4me3 (black) in HN-6 and 5-3 cells after 24 h of exposure to IFN-β. Tracks are focused on the first four introns and four exons of a representative immune-related gene, OAS1.
We then sought to evaluate the effect of SMYD3 KO on the genomic distribution of H4K20me3 and, more particularly, on immune-related genes using CUT&RUN assays in HN-6 and the SMYD3 KO cell line 5-3 after IFN-β exposure. We found 49,524 H4K20me3 peaks were called in HN-6 cells. Of these peaks, the majority (49,164 peaks, 99% of the total at baseline) were significantly decreased in the SMYD3 KO cells (FDR < 0.05), suggesting that SMYD3 has a predominant role as a positive regulator of H4K20me3 genomic deposition (Figure 3B). 37% of the differentially decreased peaks were distributed in intergenic regions and 63% in intragenic regions, including 28% in introns, 20% in exons, 7% in promoters, and the remainder in 5′ UTR and 3′ UTR (Figure 3B). We further focused our analysis on intragenic regions, given that the intragenic presence of H4K20me3 could directly contribute to the transcriptional repression of these genes. We found 9,273 genes were annotated to 30,504 H4K20me3 intragenic peaks, and, of these genes, most of them (9,272 genes) were annotated to decreased H4K20me3 peaks (Figure S17A). Among these genes, 4,874 were annotated to 15,993 differential H4K20me3 peaks and had evaluable RNA-seq data (Figure 3C, left panel, and Figure S17B). Of these 4,874 genes, 39.6% (1,929) demonstrated increased mRNA expression, signifying that SMYD3-mediated H4K20me3 is important in repressing these genes; 29.4% of these genes (1,434) had decreased mRNA expression; and 31% (1,511) showed no expression changes (FDR < 0.1, absolute log2FC >1.3) (Figure 3C, right panel).
Among the 9,272 genes with decreased H4K20me3 peaks, 60 immune-related genes were annotated to 93 decreased H4K20me3 peaks (Figures S17C and 3D; Tables S5A and S5B). Of these, 54 genes had evaluable RNA-seq data and were annotated to 84 differential H4K20me3 peaks (Figure 3E, left panel, and Figure S17D; Table S5B), with 83% (45) upregulated, 7% (four) downregulated and 9% (five) unaltered (Figure 3E, right panel). Of note, 83% of immune-related genes compared to 39.6% of all genes with decreased H4K20me3 peaks were upregulated, suggesting that H4K20me3 may be particularly important for the transcriptional repression of immune-related genes. To evaluate whether SMYD3 co-occupied the same or adjacent genomic locations with H4K20me3 within immune-related genes, we conducted co-occupancy analysis and found that SMYD3 co-occupied approximately 78% (44 out of 56) of immune-related genes with H4K20me3 in the baseline HN-6 cell state, suggesting that SMYD3 represses the expression of these genes by promoting the deposition of H4K20me3 (Figure 3F and Table S5C). Interestingly, H3K4me3, which is written and read by SMYD3, was also found to co-occupy 90% (40 out of 44) of these genes (Figure 3F and Table S5C). Furthermore, SMYD3 depletion induced a significant decrease in the average normalized tag counts corresponding to SMYD3-, H4K20me3-, as well as H3K4me3-binding sites within intragenic regions of immune-related genes (Figure 3G; Tables S5A, S5D, and S5E). Importantly, despite the decrease in the intragenic intensity of H3K4me3, which would be expected to be associated with transcriptional downregulation, the majority of immune-related genes with decreased H3K4me3 intensity (67%) were transcriptionally upregulated (Table S5F). These findings support that SMYD3 has a predominant direct repressive function on immune-related genes through H4K20me3, rather than an activating function through H3K4me3. Examples of SMYD3, H4K20me3, and H3K4me3 co-occupancy of representative immune-related genes are shown in Figures 3H and S18.
Importantly, to assess whether H4K20me3 contributed to the repression of immune-related genes together with UHRF1 after transient SMYD3 depletion, we conducted CUT&RUN assays for H4K20me3 after transient depletion of SMYD3 in HN-6 cells (siSMYD3x 3 days) and found no effect in the deposition of H4K20me3 on the promoters and gene bodies of immune-related genes (Figure S19). This finding could be explained by the longer half-life of H4K20me3, which may require a longer duration of depletion of SMYD3 to impart a global decrease in the levels of H4K20me3.
SMYD3 depletion does not increase the expression of LINE retrotransposons in HPV-negative HNSCC cells
Given multiple studies supporting the importance of re-expression of transposable elements in the stimulation of type I IFN response in cancer cells,42-44 we sought to investigate whether SMYD3 affected the expression levels of LINEs (long interspersed nuclear elements). To evaluate this possibility, we assessed the expression levels of LINEs in the RNA-seq dataset of HN-6 cells transfected with negative control or a SMYD3-targeting siRNA for 72 h with IFN-β exposure. Our analysis showed that the average mRNA expression levels of LINEs were not significantly different, suggesting a LINE-independent role of SMYD3 in regulating type I IFN responses (Figure S20). Concordantly, the Stimulator of Interferon Response CGAMP Interactor 1 (STING1) expression levels, which are expected to be upregulated with double-stranded RNA (dsRNA) intracellular stress, were not upregulated in our siSMYD3 RNA-seq dataset (Figure S21).
Smyd3 depletion induces intratumoral CD8+ T cell influx and enhances the antitumor efficacy of anti-PD-1 treatment in a syngeneic mouse model of HPV-negative HNSCC
We subsequently evaluated the effect of Smyd3 depletion in the tumor immune microenvironment and its antitumor efficacy in an in vivo syngeneic mouse model of HPV-negative flank MOC1 tumors using Smyd3 ASOs.27,28 MOC1 tumors are infiltrated with low numbers of CD8+ T cells and are resistant to therapy with PD-1 inhibition, recapitulating immunotherapy-resistant HPV-negative HNSCC.45
First, Smyd3 protein levels and the feasibility of Smyd3 depletion in vitro using Smyd3 ASOs in MOC1 cells were confirmed (Figure S22A). We then treated MOC1 tumors with Smyd3 or control ASOs at two different doses, 25 mg/kg and 50 mg/kg, and mice were sacrificed on day 26 post tumor cell implantation (day 20 of ASO treatment, n = 4 or 5 per group). No significant differences in the average tumor volumes were observed, although tumors treated with the lower dose of Smyd3 ASOs were overall smaller compared to controls (Figure 4A, left top panel). Immunohistochemistry (IHC) for control or Smyd3 ASOs demonstrated successful intratumoral penetration of ASOs in the tumor microenvironment (Figure 4A, left bottom panel). Interestingly, CD8 IHC revealed significantly increased influx of CD8+ T cells in MOC1 tumors treated with 25 mg/kg of Smyd3 ASOs compared to control ASOs, while no significant difference was observed with the 50-mg/kg dose (Figure 5A, right top and bottom panels). These results support that systemic treatment with Smyd3 ASOs increased the influx of intratumoral CD8+ T cells; however, higher dosing seemed to have a deterring effect in CD8+ T cell trafficking.
Figure 4. Smyd3 depletion increases CD8+ T cell infiltration and induces significant tumor growth repression in combination with anti-PD-1 in a syngeneic mouse model of MOC1 tumors.

(A) C57BL/6 mice were injected with MOC1 cells in the right flank, and, once they reached an average tumor volume of 0.05cm3 (day 6), treatment with Smyd3 or control ASOs was initiated and continued up to day 26 post tumor implantation. Mice were sacrificed and tumors were resected and formalin-fixed. Top left: average tumor volumes, control ASO 25 mg/kg, n = 4; Smyd3 ASO 25 mg/kg, n = 5; control ASO 50 mg/kg, n = 5; Smyd3 ASO 50 mg/kg, n = 5. Top right: percentage CD8+ T cell infiltration (CD8 IHC) assessed on MOC1 sections of tumors treated with control or Smyd3 ASOs. Data are represented as mean ± SEM, Student’s t test, *p = 0.03; NS, non-significant. Bottom left, representative examples of IHC for control or Smyd3 ASOs in MOC1 tumors. Scale bar, 100 μm. Bottom right: IHC for CD8 in MOC1 tumors treated with control or Smyd3 ASOs. Scale bar, 100 μm.
(B) Left: average tumor volumes of MOC1 tumors treated with PBS (n = 10), control ASOs at 25 mg/kg (n = 10) and Smyd3 ASOs at 25 mg/kg (n = 10) and 12.5 mg/kg (n = 10). Treatment was initiated once tumors reached an average of 0.1 cm3. Differences among all groups were non-significant. Middle: average weight of mice per treatment group (day 38 post-tumor implantation). Right, qRT-PCR for Smyd3 mRNA of MOC1 tumors treated as indicated (n = 5 per group). Data are represented as mean ± SEM. Unpaired t test with Welch’s correction, *p < 0.05, ***p < 0.001.
(C) Multicolor flow cytometry of MOC1 tumors treated with PBS, control ASOs at 25 mg/kg, and Smyd3 ASOs at 25 mg/kg and at 12.5 mg/kg (day 39 post tumor implantation, n = 5 per group). Data are represented as mean ± SEM. Unpaired t test between control ASO at 25 mg/kg and Smyd3 ASO at 12.5 mg/kg, *p < 0.05.
(D) Top: average tumor volumes of flank MOC1 tumors in C57BL/6 mice treated with control ASOs plus isotype immunoglobulin (Ig) G, Smyd3 ASOs plus isotype IgG, control ASOs plus anti-PD-1, and Smyd3 ASOs plus anti-PD-1 (n = 8 mice per group). Data are represented as mean ± SEM. Unpaired t test, **p = 0.004. Bottom: hairline growth curves of flank MOC1 tumors treated with control ASOs plus anti-PD-1 and Smyd3 ASOs plus anti-PD-1. Similar results were obtained in two independent experiments.
Figure 5. SMYD3 is overexpressed in HPV-negative HNSCC tumors and is associated with CD8+ T cell infiltration and UHRF1 protein expression.

(A) SMYD3 mRNA expression levels are higher in HPV-negative HNSCC tumors compared to normal squamous epithelium (TCGA). Wilcoxon rank-sum test, ***p < 0.001.
(B) Representative images of IHC staining for SMYD3 in normal squamous epithelium, dysplastic epithelium, and HPV-negative HNSCC sections. Red arrows show examples of SMYD3-stained nuclear speckles. Scale bar, 10 μm.
(C) Distribution of IHC scores among normal squamous epithelium, dysplastic epithelium and HPV-negative HNSCC tumors. Kruskal-Wallis test, ***p < 0.001.
(D) Median IHC score for SMYD3 in normal squamous epithelium, dysplastic epithelium, and HPV-negative HNSCC tumors. Dunn test for multiple comparisons, *p < 0.05, **p < 0.01.
(E) Scatterplots of protein abundance showing correlations for SMYD3, UHRF1, and CD8A. Correlations were conducted in 108 HPV-negative HNSCC tumor samples of the CPTAC. R, Pearson correlation coefficient.
(F) Representative images of IHC in HPV-negative HNSCC tumor sections with high SMYD3/low CD8A or low SMYD3/high CD8A staining (left, scale bar, 100 μm) and high SMYD3/high UHRF1 or low SMYD3/low UHRF1 staining (right, scale bar, 50 μm).
To validate these observations and to further explore the effect of Smyd3 ASOs in the tumor microenvironment, we conducted multicolor flow cytometry of MOC1 tumors treated with PBS, control ASOs at 25 mg/kg, Smyd3 ASOs at 25 mg/kg, and Smyd3 ASOs at 12.5 mg/kg (n = 10 per group). We opted to treat mice at an even lower dose of Smyd3 ASOs (12.5 mg/kg), considering that 25 mg/kg induced greater influx of CD8+ T cells compared to 50 mg/kg and that an even lower dose could be associated with an even greater influx of CD8+ T cells. As per the aforementioned experiment, no significant differences were observed in the tumor volumes between the groups, although MOC1 tumors treated with Smyd3 ASOs at 12.5 mg/kg tended to have smaller average tumor volumes compared to the control group (Figure 4B, left panel). The treatment with ASOs was tolerated well at all doses (Figure 4B, middle panel), and mice were sacrificed on day 39 post tumor cell implantation (day 24 of treatment). MOC1 tumors were surgically resected, and tumor RNA extraction and multicolor flow were conducted (n = 5 tumors per group). Smyd3 mRNA levels from tumor extracts were decreased in a dose-dependent manner, with approximately a 50% decrease and a 75% decrease observed in tumors treated with Smyd3 ASOs at 12.5 mg/kg and 25 mg/kg respectively (Figure 4B, right panel). CD8+, CD4+ T cells and macrophages were significantly increased in the MOC1 tumors treated with Smyd3 ASOs at 12.5 mg/kg compared to all other groups (Figures 4C and S22B). Regulatory T cells (Tregs) and granulocytic MDSCs were not increased; however, an influx of monocytic MDSCs was observed in the tumors treated with the Smyd3 ASOs at 12.5 mg/kg. Major histocompatibility complex (MHC) class I H-2Kb and PD-L1 were also significantly upregulated on MOC1 tumor cells. Furthermore, PD-1 expression was significantly upregulated on CD8+ T cells, suggesting a state of exhaustion.
We then sought to evaluate whether Smyd3 ASOs could synergize with anti-PD-1 therapy. Mice with flank MOC1 tumors were treated with control ASOs at 12.5 mg/kg plus anti-PD-1 or Smyd3 ASOs at 12.5 mg/kg plus anti-PD-1. The group treated with Smyd3 ASOs plus anti-PD-1 demonstrated a statistically significant decrease in the average MOC1 tumor volumes compared to control ASOs plus anti-PD-1 (Figure 4D). Four out of eight mice treated with Smyd3 ASOs plus anti-PD-1 were cured, while two out of eight mice had significant tumor regressions (0.06cm3 and 0.007 cm3 at the time of sacrifice). On the other hand, the MOC1 tumors in two out of eight mice of the Smyd3 ASO combination group “escaped” the treatment effect and grew similarly to the control ASO plus anti-PD-1-treated group.
The above data support that Smyd3 depletion induces favorable changes in the MOC1 tumor microenvironment with influx of CD8+, CD4+ T cells and macrophages, while also upregulating the expression of H-2Kb and PD-L1 on the cell surface of MOC1 tumor cells. These changes are associated with sensitization of MOC1 tumors to anti-PD-1 therapy, with deep responses and complete tumor regressions in mice treated with systemic Smyd3 ASOs plus anti-PD-1 therapy.
SMYD3 is overexpressed and is associated with CD8+ T cell infiltration and UHRF1 protein expression in human HPV-negative HNSCC tumors
To assess the expression levels of SMYD3 in HPV-negative HNSCC tumors, the HPV-negative HNSCC database of TCGA was interrogated and results showed that SMYD3 mRNA levels were significantly higher in HPV-negative HNSCC tumors compared to normal squamous epithelium (Figure 5A). To assess the pattern of expression and validate the aforementioned results at the protein level, we conducted IHC for SMYD3 in 64 HPV-negative HNSCC tumors as well as in 10 available normal and seven dysplastic buccal squamous epithelium samples (Figure 5B and Table S6). The pattern of SMYD3 staining was predominantly cytoplasmic; however, SMYD3-stained nuclear speckles were also observed, indicating the presence of SMYD3 in the nucleus of oral dysplastic and HPV-negative HNSCC tumor cells (Figure 5B). Analysis of the staining results using a semiquantitative scoring scale (0, +1, +2, +3) revealed that the percentage of samples with IHC score of +3 increased significantly from normal, to dysplastic epithelium, and then to squamous cell carcinoma samples (Figure 5C). Furthermore, approximately 80% of tumors showed strong staining (+2, +3). Concordantly, the median IHC score was significantly higher in dysplastic and HNSCC samples compared to normal squamous samples (Dunn test for multiple comparisons, p < 0.05) (Figure 5D). These data support a potential role of SMYD3 in the oncogenesis of HPV-negative HNSCC.
Furthermore, we sought to evaluate the relative protein levels of SMYD3 in the cancer cell compartment compared to the stroma of HPV-negative HNSCC tumors. The average H score of the cancer to the stroma compartment HPV-negative HNSCC sections was significantly higher (Wilcoxon rank-sum test, p = 3.6 × 10−6) (Figure S23A). This finding was further corroborated by the significantly higher mRNA expression of SMYD3 in HPV-negative HNSCC cancer cells compared to other cell subtypes, such as T cells, B cells, and fibroblasts, of a publicly available single-cell RNA-seq database29 (Figure S23B). These data support a more prominent role of SMYD3 in regulating biological functions in cancer cells and less in stroma/immune cells; however, this needs to be more definitively answered through experimental interrogation.
We then evaluated the association between SMYD3 protein levels and CD8 T cell infiltration in HPV-negative HNSCC tumors. To this end, we utilized the Clinical Proteomic Tumor Analysis Consortium (CPTAC) database26 and conducted correlations between SMYD3 and CD8A in the respective HPV-negative cohort of tumor samples (n = 108). As expected, we found that tumors with higher SMYD3 protein levels had significantly lower CD8A protein expression, suggesting lower CD8+ T cell infiltration (Pearson R = −0.2, p = 0.038) (Figure 5E). Given that UHRF1 was found to be a downstream target of SMYD3 and a mediator of repression of immune-related genes, we also evaluated the correlation between SMYD3 and UHRF1, as well as UHRF1 and CD8A protein levels using the same database. UHRF1 protein levels were found positively correlated with SMYD3 (Pearson R = 0.23, p = 0.018), and negatively correlated with CD8A (Pearson R = −0.37, p < 0.001) (Figure 5E). We also validated these correlations between SMYD3 and CD8A, as well as SMYD3 and UHRF1 in our aforementioned cohort of HPV-negative HNSCC tumor samples by conducting IHC for CD8A and UHRF1; however, the correlation between UHRF1 and CD8A was not observed, likely due to the lower sample size of our validation cohort (Figure S24). Representative examples of IHC staining for SMYD3, CD8A, and UHRF1 are shown in Figure 5F.
SMYD3 protein levels predict pathologic response to neoadjuvant pembrolizumab
We then sought to evaluate whether SMYD3 expression levels predict response to treatment with pembrolizumab. To this end, we used a published RNA-seq database of 20 evaluable patients with newly diagnosed HPV-negative oral cavity HNSCC who were treated with one dose of neoadjuvant pembrolizumab and then were surgically resected 2–3 weeks after the treatment to evaluate pathologic tumor response (PTR) and clinical to pathological downstaging.46 Higher baseline SMYD3 mRNA levels were significantly associated with no clinical to pathological downstaging (Wilcoxon rank-sum test, p = 0.02; Figure 6A, left graph). While higher baseline UHRF1 mRNA levels were also observed in patients without clinical to pathological downstaging, this difference did not reach statistical significance (Wilcoxon rank-sum test, p = 0.49; Figure 6A, middle graph), similarly to lower baseline CD8A mRNA levels (Wilcoxon rank-sum test, p = 0.09; Figure 6A, right graph).
Figure 6. SMYD3 expression predicts response to neoadjuvant pembrolizumab and determines survival when combined with UHRF1 expression.

(A) Boxplot showing the correlation between baseline SMYD3 (left), UHRF1 (middle), and CD8A (right) mRNA levels and clinical to pathological downstaging after one dose of neoadjuvant pembrolizumab in 20 oral cavity, treatment-naive, HPV-negative HNSCC patients. Sixteen patients without and four patients with clinical to pathological downstaging. Wilcoxon test for SMYD3, p = 0.022; for UHRF1, p = 0.49; for CD8A, p = 0.09.
(B and C) Kaplan-Meier OS (n = 61 patients) (B) and PFS curve (n = 54) (C) in HPV-negative HNSCC patients of the CPTAC. Patients with higher SMYD3/UHRF1 protein levels at baseline had significantly worse OS and PFS.
Combined SMYD3 and UHRF1 protein levels predict overall and progression-free survival in patients with primary HPV-negative HNSCC
We then assessed whether baseline SMYD3 and UHRF1 protein levels correlate with overall survival (OS) and progression-free survival (PFS). Using the CPTAC patient database, patients with higher combined baseline SMYD3 and UHRF1 protein levels had significantly worse OS (Figure 6B) and PFS (Figure 6C) compared to patients with lower combined protein levels. A similar trend was observed in our smaller University of Chicago patient database; however, this did not reach statistical significance (Figures S25A-S25D). Interestingly, though, combined protein expression of SMYD3, UHRF1, and CD8A at baseline predicted a significant impact on both PFS and OS. More specifically, patients with high baseline SMYD3 and UHRF1 and low CD8A protein levels had significantly worse PFS and OS compared to other patient subgroups (Figures S25E and S25F).
DISCUSSION
Deciphering mechanisms that drive checkpoint inhibitor resistance in HPV-negative HNSCC is of paramount importance to increase the percentage of patients that benefit from immunotherapy. Our work reveals that SMYD3 depletion by siRNAs, ASOs, or CRISPR increases cancer cell sensitivity to IFN-β and derepresses type I IFN response and APM genes in HPV-negative HNSCC cell lines through two temporally distinct, chromatin-based mechanisms that support a bifaceted function of SMYD3 both as a transcriptional activator and a transcriptional repressor: (1) SMYD3 binds to and activates the expression of the H3K9me3-reader UHRF1 through H3K4me3, which then mediates repression of immune-related genes through recruitment of DNMT1 on their promoters; (2) SMYD3 binds to and mediates the deposition of H4K20me3 on immune-related genes (Figure 7).
Figure 7. Schematic of the mechanism of SMYD3-mediated repression of immune-related genes.

We found that SMYD3 and H3K4me3 co-occupy UHRF1 and that transient SMYD3 knockdown induces downregulation of UHRF1. In turn, UHRF1 and H3K9me3 were found enriched in the promoters of type I IFN response genes CXCL9, CXCL10, MX1, OAS2, and RSAD2. At least 50% of the CpG sites within the promoters of these genes were found to be partially or close to fully DNA methylated, suggesting that these sites may serve as binding sites of hemi-methylated DNA for UHRF1. Furthermore, UHRF1 overexpression was associated with enrichment of DNMT1 in the promoters of some of these genes, implying that UHRF1 silences their expression by binding on their promoters and recruiting DNMT1. Despite the fact that DNMT1 occupancy was decreased in the promoters of these genes after SMYD3 depletion, we did not observe a decrease in the DNA methylation levels in the promoters of these genes. This finding suggests that DNMT1 represses immune-related genes in a methyltransferase-independent manner and as a scaffolding protein, forming a repressor complex with UHRF1 that recognizes and reads methylated CpG sites and H3K9me3 on immune-related genes. A methyltransferase-independent function of DNMT1 has been reported by various groups.36-38 Identifying interacting proteins that constitute a repressor complex with UHRF1 and DNMT1 would be important to further understand the mechanism through which UHRF1 silences immune-related genes. Furthermore, deciphering the mechanisms of enrichment of H3K9me3 on immune-related genes merits further investigation, as reversal of this repressive mark may impart synergistic or additive effects to SMYD3 depletion toward derepression of type I IFN response and APM genes.
While we expected that the UHRF1-mediated mechanism of immune-related gene repression would also apply in the SMYD3 CRISPR KO cells, we found that UHRF1 levels were preserved, most likely because its expression may be necessary for the survival of HPV-negative HNSCC cells. The fact that the UHRF1-mediated mechanism could not be validated in our CRISPR SMYD3 KO cells may be explained by the fact that cells with complete genetic KO of a target gene may evolve different biological mechanisms to survive and maintain certain phenotypes. In these SMYD3 KO cells, our data support that immune-gene derepression was mediated through depletion of H4K20me3, a repressive mark, from intragenic regions, which may be the predominant mechanism of immunomodulation in permanently SMYD3-depleted cells. Conversely, while transient SMYD3 depletion did not induce a decrease in the deposition of H4K20me3 on immune-related genes (Figure S19), it could be speculated that this mechanism may start predominating with longer and deeper depletion of SMYD3. From a translational standpoint, this may be recapitulating cancer cells within the tumor microenvironment that have evolved to survive through sustained SMYD3 depletion, albeit they still maintain the phenotype of enhanced type I IFN responsiveness.
The proposed function of SMYD3 as an activator and repressor of specific gene sets has been previously reported as a paradigm by Kim et al.,47 whereby enhancer of zeste homolog 2 (EZH2), a writer of the repressive H3K27me3 mark, was found to function as a transcriptional activator, binding to the promoter of the androgen receptor gene and inducing its transcription in a methylation- and polycomb-independent manner. Our data show that SMYD3 binds to and co-occupies the gene body of UHRF1 with H3K4me3, inducing its transcriptional activation, while, concurrently, it co-occupies the promoters and/or gene bodies of immune-related genes with H4K20me3, inducing their transcriptional repression. Interestingly, SMYD3 and H4K20me3 were found to co-occupy the promoters or gene bodies of CXCL9, OAS2, MX1, and RSAD2, where UHRF1 was also found to bind, but not the promoter of CXCL10, suggesting that UHRF1 may act synergistically and/or be complementary to H4K20me3 to repress immune-related genes in the basal state of HPV-negative HNSCC cells.
Previously reported epigenetic mechanisms of immune-related gene silencing pertain to the repression of endogenous retroviral elements (ERVs) or dsRNAs, type I IFN response and APM genes through DNA methylation, EZH2-mediated H3K27 trimethylation, and SETDB1-mediated H3K9 trimethylation in melanoma or non-small cell lung cancer cell lines.14,48-51 More specifically, inhibition of DNA methylation alone or together with histone deacetylase inhibitors induces re-expression of ERVs and activates type I IFN responses in mouse melanoma models.42,50 SETDB1 amplification has been associated with resistance to PD-1 inhibition, while SETDB1 loss derepresses transposable elements (TEs), immunostimulatory genes, and TE-encoded retroviral antigens through erasure of H3K9me3, inducing TE-specific CD8+ T cell antitumor immune responses in melanoma and Lewis lung carcinoma mouse models.50 Additionally, lysine-specific histone demethylase 1 (LSD1) represses the expression of ERVs and increases the stability of certain RNA-induced silencing complex components, attenuating type I IFN responses in breast cancer cells.43 Accordingly, LSD1 depletion increases the expression of ERVs and activates type I IFN responses, inducing T cell infiltration and antitumor immunity in mouse melanoma models.43
Other studies have demonstrated the importance of EZH2-mediated deposition of H3K27me3 directly on key Th1-type chemokine genes CXCL9 and CXCL10. More specifically, EZH2 and DNA-methyltransferase 1 (DNMT-1) transcriptionally repress CXCL9 and CXCL10, and inhibition of both EZH2 and DNMT-1 increases CD8+ T cell trafficking, reduces tumor growth, and improves the efficacy of PD-1 blockade in mouse ovarian cancer models.47 Similarly, EZH2-mediated transcriptional repression of CXCL9 and CXCL10 through H3K27 trimethylation has been reported in colon cancer cells.48 EZH2 silences the B2m promoter through H3K27me3 in MOC1 cells, and EZH2 inhibition enhances antigen presentation and responses to anti-PD-1 immunotherapy in the anti-PD-1 MOC1 resistant mouse model of HPV-negative HNSCC.52 An important question to decipher is whether different repressive histone marks, such as H3K27me3 or H4K20me3, have mutually exclusive, complementary, or redundant functions in the repression of immune-related genes, thus allowing for synergistic roles or the development of resistance mechanisms through the respective epigenetic regulators governing their deposition or erasure.
Smyd3 ASOs induced marked influx of CD8+ T cells, CD4+ T cells, and macrophages and markedly upregulated the expression of MHC class H-2Kb in MOC1 tumors, inducing sensitization of MOC1 flank tumors to anti-PD-1 therapy. Tregs and granulocytic MDSCs, which have also been shown to play a significant role as immunosuppressive immune cell subsets in HPV-negative HNSCC,53 were not increased with Smyd3 ASO treatment. However, Smyd3 ASOs induced upregulation of PD-1 and PD-L1, indicating an exhaustive state of the CD8+ T cells, which may explain why monotherapy with Smyd3 ASOs did not induce significant tumor growth restraint despite the influx of CD8+ T cells. In this setting, combination treatment of MOC1 tumors with Smyd3 ASOs and anti-PD-1 led to cures and marked tumor growth restraint (Figure 4D). Smyd3 ASO treatment also induced influx of monocytic MDSCs, which may explain why two out of eight treated mice escaped the treatment effect of the Smyd3 ASOs and anti-PD-1 combination. Further interrogation of possible mechanisms of escape is warranted and ongoing, with a focus on potential competing and redundant functions of other chromatin modifiers that could orchestrate the deposition of repressive histone marks on immune-related genes. Another important observation is that a higher dose of Smyd3 ASOs did not induce influx of CD8 and CD4+ T cells, suggesting a deleterious effect of higher Smyd3 ASO dosage on the intratumoral trafficking of T cells. As such, the elucidation of the differential effect of Smyd3 depletion on cancer versus immune cells would be of paramount importance.
Approximately 80% of HPV-negative HNSCC tumor sections in our patient cohort overexpressed SMYD3, and increased baseline SMYD3 mRNA expression in HPV-negative HNSCC tumor samples predicted poor pathologic response to neoadjuvant pembrolizumab, supporting that SMYD3 may abrogate therapeutic responses to pembrolizumab. Furthermore, high baseline SMYD3 protein levels in combination with high UHRF1, as well as low CD8A levels, predicted poor PFS and OS in newly diagnosed HPV-negative HNSCC patients. The fact that chemoradiotherapy is an integral part of the treatment of HNSCC and that effective antitumor immunity is necessary for its efficacy in HNSCC54,55 could explain this finding.
Interestingly, a number of studies have suggested that certain highly inducible inflammatory genes are maintained in transcriptional repression by nuclear repressor complexes, such as the nuclear repressor coreceptor 1 (NCoR), the corepressor of REST (CoREST), or the glucocorticoid receptor (GR).56-63 Upon appropriate pro-inflammatory stimuli, these transcriptional repressor complexes are removed and the inflammatory genes transition from their basal, silenced state to an actively transcribed state. Along these lines, Stender et al.64 previously reported the importance of H4K20 methylation/demethylation in the transcriptional regulation of certain TLR4-responsive inflammatory genes through SMYD5, which trimethylates H4K20, and PHF2, a H4K20 demethylase that is activated upon lipopolysaccharide (LPS)-induced TLR4 stimulation. SMYD5 was found to be a component of the repressor NCoR complex, and, importantly, knockdown of the other four members of the SMYD family in mouse macrophages did not induce upregulation of TLR4-responsive inflammatory genes, indicating distinct biological roles for each SMYD family member. Furthermore, SMYD5 knockdown did not alter the inhibitory effect of the GR receptor on inflammatory genes, signifying a pathway-specific mechanism of repression of these genes. In another study,65 Smyd2 was found to repress the expression of Il-6 and Tnf through H3K36 dimethylation of their promoters, inhibiting macrophage activation and M1 polarization, and to increase the expression of Tgf-β, which promoted Treg differentiation. Nagata et al.66 also reported that Smyd3 induces the expression of Foxp3 through a Tgf-β/Smad3-dependent mechanism and promotes Treg differentiation, and that Smyd3 KO mice demonstrated excessive RSV-induced pulmonary inflammation secondary to uncontrolled inflammatory responses. These data suggest that the SMYD family members may have distinct biological functions in the regulation and optimization of anti-viral and anti-bacterial immunity.
Our study provides important insights into the function of SMYD3 as a repressor of antitumor immunity and driver of resistance to anti-PD-1 therapy in HPV-negative HNSCC. As a therapeutic targeting strategy for SMYD3, ASOs provide a promising platform and could overcome the significant problem of specificity when targeting methyltransferase enzymes. Furthermore, ASOs have the potential to abolish oncogenic mechanisms induced not only by enzymatic hyperactivity due to mutations but also through overexpression of the target gene. The latter is relevant for SMYD3, which is overexpressed but rarely mutated (<2%) in HPV-negative HNSCC (Figure S26). Two ASOs have been US Food and Drug Administration (FDA) approved for the treatment of hypercholesterolemia and spinal muscular atrophy,27 underscoring the translational feasibility of this drug platform. These data support a rational translational strategy, whereby HPV-negative HNSCC patients could be stratified by baseline SMYD3 protein levels and treated with combinatorial SMYD3 ASOs with PD-1 checkpoint inhibition to increase efficacy of checkpoint immunotherapy.
Limitations of the study
Although our experiments support that UHRF1 binds to and represses a panel of immune-related genes by recruiting DNMT1, genome-wide mapping for UHRF1 and DNMT1 would be needed to further validate this mechanism across multiple immune-related genes. Furthermore, additional functional experiments would be necessary to assess whether DNMT1 functions in a methyltransferase-independent manner in concert with UHRF1 to repress immune-related genes. While our findings seem to support a bifaceted function of SMYD3 as an activator and repressor of different gene sets within the same cell context, these data need to be interpreted with caution, and further investigation is needed to delineate the specific mechanisms through which SMYD3 functions as a transcriptional activator in certain gene sets versus a repressor in other gene sets. Additionally, whether SMYD3 directly methylates H4K20 still remains unclear, and while we showed that the protein levels of established H4K20 methyltransferases SUV420H1/H2 remain stable or are increased after SMYD3 KO, it cannot be excluded that SMYD3 may cooperate through methylation-dependent or -independent mechanisms with SUV420H1/H2 to promote the genomic deposition of H4K20me3. Further, 37% of the H4K20me3 peaks affected by SMYD3 KO were intergenic, suggesting that SMYD3 may also affect the function of enhancers of immune-related genes; genome-wide mapping for activating enhancer marks, such as H3K4me1 and H3K27Ac, could elucidate this possibility and is ongoing by our group. Finally, although SMYD3 was present in the nucleus of HPV-negative HNSCC cells, it was predominantly expressed in the cytoplasm of HPV-negative HNSCC tumor cells. Given that non-histone substrates of SMYD3 have been reported, such as MAP3K2,18 it cannot be excluded that the immune-repressive effect of SMYD3 may also be partially mediated through a non-histone cytoplasmic substrate.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Vassiliki Saloura (vassiliki.saloura@nih.gov).
Materials availability
Plasmids generated in this study have been deposited to Addgene [pCE0482, Addgene plasmid ID#202621]. CRISPR KO cell lines generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
RNA-seqand CUT&RUN DNA-seq data have been deposited at GEO with accession ID# GSE233495 and are publicly available as of the date of publication.
This paper does not report original code.
All raw data reported in this paper will be shared by the lead contact upon request. Any additional information required to re-analyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT PARTICIPANT DETAILS
In vivo mouse experiments
All mouse experiments and procedures performed were conducted in a fully accredited animal housing facility at the National Institutes of Health, Bethesda, MD. All animal experimental protocols were approved by the NCI-Bethesda Animal Care and Use Committee. 4-6 week-old female C57BL/6 mice were purchased from Taconic and used for the described experiments. The study designs and animal usage were conducted accordingly to all applicable guidelines by the NCI-Bethesda Animal Care and Use Committee. Mice were randomly assigned to experimental groups. An established, DMBA (7,12-dimethylbenz(a)anthracene)-induced, mouse oral carcinoma 1 (MOC1) cell line in a syngeneic C57BL/6 mouse model of MOC1 flank tumors was utilized.44 MOC1 cells were grown in vitro and were inoculated by subcutaneous injections of 5 million MOC1 cells in suspension using Matrigel (Corning), in the right flanks of C57BL/6 mice. Once flank tumors reached an average volume of 0.1cm3, mice were randomized into treatment groups and treatment was initiated according to each experiment. Mice were treated with control ASOs or Smyd3 ASOs at concentrations ranging from 12.5 m/kg to 50 mg/kg or PBS, as described in each experiment, for 5 days per week with subcutaneous injections. Intraperitoneal injections of isotype IgG (InVivoPlus rat IgG2a, 2A3, BioXCell) or anti-PD-1 (InVivoPlus anti-mouse PD-1, RMP1-14, BioXCell) were conducted at 200ug/injection, twice weekly. Tumor length (L) and width (W) were measured twice weekly with calipers and tumor volumes were calculated using the formula LxW^2/2. Weights were measured twice weekly.
Human participants
Informed consent was obtained from all participants and the study was approved by the Institutional Review Board of the University of Chicago (IRB#8980). Tissue microarrays containing clinically annotated, de-identified patient HNSCC tumor samples were obtained from the Human Tissue Research Center of the University of Chicago Pathology Department. Tumor site, tissue of origin, TNM stage, age, gender, smoking history and survival data are available through Table S6. Ancestry, race, ethnicity and socioeconomic status of the study participants can be provided by the lead contact upon request.
Cell lines
HPV-negative squamous cell carcinoma cell lines HN-6 (oral cavity, male sex) and HN-SCC-151 (tongue) were derived from patients with locoregionally advanced HNSCC and were kindly provided by Dr. Tanguy Seiwert (University of Chicago). The sex of HN-SCC-151 cells is unavailable due to lack of whole-exome sequencing data comparing this cell line with a normal control cell line. HN-6 cells were maintained in DMEM medium (Thermo Fisher Scientific) and HN-SCC-151 cells in DMEM/F12 medium (Thermo Fisher Scientific). Media were supplemented with 10% fetal bovine serum (Cytiva), 1% penicillin/streptomycin (Vita Scientific), and 2 nM L-glutamine (Thermo Fisher Scientific). Cell lines were cultured at 37°C.
Generation of SMYD3 knockout cell lines using CRISPR
SMYD3 CRISPR knockout cell lines (SMYD3 KO 5-2, 5-3) were generated from parental HN-6 cells using clustered regularly inter-spaced short palindromic repeats (CRISPR/Cas9) technology. Briefly, four candidate guide RNAs targeting SMYD3 were designed using sgRNA Scorer 2.0 and tested for activity in 293T cells. Of the four guide RNAs, candidate 785 was determined to be most effective. Oligonucleotides corresponding to this guide RNA were subsequently annealed and ligated into BbsI digested pX458 back-bone68 to generate vector pCE0482. Plasmids were then miniprepped and verified by Sanger sequencing. pCE0482 has been deposited in Addgene under plasmid ID: 202621. pSpCas9(BB)-2A-GFP (PX458) was a gift from Feng Zhang (Addgene plasmid # 48138; http://n2t.net/addgene:48138; RRID:Addgene_48138). Cells were then transfected with pCE0482 and single GFP-expressing HN-6 cells were sorted using flow cytometry to generate clonal populations. Subsequently, SMYD3 expression levels were assessed by Western blotting to confirm efficiency of knockout (Figure S15).
METHOD DETAILS
siRNA transfections
siRNA oligonucleotides were purchased from Sigma-Aldrich to target the human SMYD3 mRNA (SASI_Hs02_0035–5988) and UHRF1 mRNA (SASI_Hs02_00311672). For convention, SMYD3-targeting siRNA is referred to as siSMYD3 and UHRF1-targeting siRNA is referred to as siUHRF1. The negative control siRNA was purchased from Dharmacon (siRNA negative control Dharmacon ON-TARGET plus control pool, #D-001810-10-20). HNSCC cells were plated overnight in 10cm dishes and were transfected with siRNA duplexes (50 nM final concentration) using Lipofectamine RNAimax (Thermo Fisher Scientific) for 72h (3 days) or 144 h (6 days). For 6 days of siRNA transfection, re-transfections were performed on day 3. Experiments were performed in biological triplicates.
Interferon-β treatment
For all experiments, interferon-β (R&D systems Inc) treatments were performed at a concentration of 1000U/mL for 24 h. When siRNA transfections were performed for a 72 h total duration, interferon-β treatment was initiated at the 48 h time point and lasted for 24 h prior to cell collection.
Western blotting
Nuclear extracts were prepared using the Nuclear Complex Co-IP kit (Active Motif) and 10–20 μg of each extract was loaded to examine protein levels of SMYD3, UHRF1, SUV420H1, SUV420H2 and histone H3. Primary antibodies used were anti-SMYD3 (Abcam, 1:1000), anti-UHRF1 (Cell Signaling Technologies, dilution 1:1000), anti-SUV420H1 (ThermoFisher, dilution 1:1000), anti-SUV420H2 (ThermoFisher, dilution 1:1000), and anti-histone H3 (Abcam, dilution 1:20000). Densitometry of all the western blots was performed using ImageJ software. Similar results were obtained in two separate biological replicates.
Quantitative real-time PCR
Primers for human GAPDH (housekeeping gene), SMYD3, UHRF1, TRIM21, STAT1, OASL, IFNGR1, OAS3, CD274, OAS2, DHX58, NCOA7, MX1, MX2, GBP2, RSAD2, TAP2, CANX, CXCL9, and CXCL10 were purchased from Sigma-Aldrich. RNA extraction was performed using the Zymo Research Direct-zol RNA miniprep kit (Zymo Research). cDNA conversion was performed using the Invitrogen SuperScript III First-Strand Synthesis System (Invitrogen). PCR was conducted in technical triplicates using either SYBR Select Master Mix (Applied Biosystems) or GreenLink qPCR Master-Mix (BioLink Laboratories). PCR reactions were performed using CFX96 Touch Real-Time PCR Detection System. Results were reproduced in two separate biological replicates.
ChIP-qPCR
ChIP-qPCR was performed in HN-6 cells for HA-UHRF1, UHRF1, DNMT1 and H3K9me3. Briefly, the cells were seeded in 15cm dishes, and were transfected at 60% confluence. For the UHRF1 overexpression ChIP, cells were transiently transfected with the either HA-Mock or HA-UHRF1 plasmids (SinoBiological) for 48h, and for SMYD3 knockdown ChIP, the cells were transfected with negative control or SMYD3-targeting siRNAs for 72h (Sigma-Aldrich). Cells were treated with 1000U/mL of human interferon-β (R&D systems Inc.) for 24h prior to cell collection. Cells were trypsinized and cross-linked with 1% formaldehyde for 10 min. Cross-linking was stopped by 125mM Glycine. The cells were washed twice with 1X PBS and lysed using ChIP Lysis Buffer for 1 h. Samples were sonicated using Bioruptor (Diagenode) at 15s on and 30s off for a total of 23 cycles. The sonicated samples were centrifuged, and the supernatant was diluted 5-fold using ChIP dilution buffer. 50ul of the diluted samples was kept as input and the remaining was used for immunoprecipitation. The diluted samples were precleared with Dynabeads Protein G (Invitrogen) for 1h at 4C and incubated overnight with 5-10μg of the respective antibody (10μg anti-HA: Cell Signaling Technologies, 10ug human anti-UHRF1: ThermoFisher, 5ug human anti-DNMT1: ThermoFisher, 10ug anti-H3K9me3: Diagenode INC). Next day, the Dynabeads Protein G beads were washed and incubated with the sample-antibody for 2h at 4C. The bead-antibody-chromatin complex was washed with the ChIP buffers and finally the DNA was eluted. The Input DNA and the IP eluted DNA were treated with RNAse for 1 h at 37C, and then reverse cross-linked overnight at 65C in proteinase K. The samples were purified using phenol-chloroform precipitation and resuspended in 100μL H2O. Equal volumes of IP and Input DNA were tested for human ChIP primers specific to the promoter regions of different genes listed in the primers. The PCR was performed in multiple technical duplicates with SYBR Select Master-Mix using the ViiA 7 AB applied biosystems PCR Detection System. The IP/Input for each target was calculated using the formula: [Quantity IP/(Quantity Input *50) (50 represents the input dilution factor). For the HA-UHRF1 overexpression system, the enrichment as Relative IP/Input was calculated as follows: [(IP-UHRF1/Input)/(IP-Mock/Input)]. For the knockdown system, the enrichment as Relative IP/Input was calculated as follows: [(IP-siSMYD3/Input)/(IP-siNC/Input)]. Quantities of 2 or 3 technical replicates and at 1, 2 or 3 biological replicates were used for the analysis.
RNA-seq
RNA-seq was performed in HN-6 cells treated with SMYD3 targeting siRNAs, SMYD3 ASOs and in the CRISPR SMYD3 knockout cell line 5-3. For the siRNA mediated SMYD3 knockdown, HN-6 cells were plated overnight at 40% confluence and transfected with siNC or siSMYD3 using Lipofectamine RNAimax for 72h, with exposure to human IFN-β for 24h. For the SMYD3 ASO treatment, 40% confluent HN-6 cells were treated with SMYD3 ASOs or PBS for 72h, with exposure to human IFN-β for 24h. For the CRISPR SMYD3 knockout cell lines, parental HN-6 cells and the CRISPR SMYD3 knockout cell line 5-3 were plated at 60% confluence, and collected after exposure to human IFN-β for 24h. RNA-seq was also performed in HN-6 cells treated with UHRF1 targeting siRNAs; briefly, HN-6 cells were plated overnight at 40% confluence and transfected with siNC or siUHRF1 using Lipofectamine RNAimax for 72h, with or without exposure to human IFN-β for 24h.
Following completion of incubation, cells were trypsinized, washed twice with PBS, centrifuged and processed for RNA extraction (Direct-zol RNA miniprep kit, Zymo Research). Three biological replicates for each sample were processed to extract RNA, quantified using Qubit and sequenced. Samples were pooled and sequenced on NextSeq or NovaSeq using the Illumina TruSeq Stranded mRNA Library Prep kit (Illumina) and paired-end sequencing. The samples have 68 to 130 million pass filter reads. Reads of the samples were trimmed for adapters and low-quality bases using Cutadapt before alignment with the reference genome (hg38) and the annotated transcripts using STAR. The samples had 62-75% non-duplicate reads. In addition, the gene expression quantification analysis was performed for all samples using STAR/RSEM tools. The raw counts are provided as part of the data delivery.
CUT&RUN assays and DNA-sequencing
HN-6 and SMYD3 KO 5-3 cells or HN-6 cells transfected with control or SMYD3 targeting siRNAs for 72h were grown in 10cm dishes in DMEM medium supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin and 2nM L-glutamine, and were treated with human IFN-β at 1000U/mL for 24h prior to cell collection. For CUT&RUN assays, the 14–1048 CUT&RUN kit by EpiCypher was utilized according to EpiCypher’s protocol. Briefly, CUTANA spike-in dNuc controls (H3K4me0, 1, 2, 3) were mixed together with washed streptavidin (SA) beads in 4 separate 1.5mL tubes, and incubated for 30 min at RT on nutator. Concanavalin (ConA) beads were activated using cold bead activation buffer, washed twice using a magnet, resuspended in cold activation buffer, added at 10uL/sample in separate strip tubes (1 tube per experimental sample) and kept on ice. 500,000 cells per experimental condition were obtained after trypsinization from respective cell culture dishes (1 10cm dish per biological replicate) and were washed with PBS x 3 to remove excess trypsin. Cells that were designated for the SMYD3 CUT&RUN assay were resuspended in cell culture medium and lightly cross-linked with 0.25% HCHO for 2min, and the reaction was stopped with 125mM Glycine. Cells were then resuspended in 100uL/sample of RT wash buffer and washed twice at 600xg for 3min. After the final wash, cell pellets were resuspended in 105uL of RT wash buffer, and 100uL per sample were aliquoted into each 8-strip tube containing 10uL of activated beads. The cell-bead slurries were incubated on the benchtop for 10 min at RT to allow for adsorption of the cells to the beads. After the incubation, the slurries were placed on a magnet and a small aliquot of the supernatant was obtained to confirm adsorption of cells to the beads (binding efficacy >93% of cell input). The supernatants were completed removed and the cell-bead slurries were then exposed to cold antibody buffer and vortexed. The CUTANA H3K4MetSTat spike-in control dNucs were added to designated positive (H3K4me3) and negative (IgG) control tubes. Then, 0.5ug of antibodies to H4K20me3 (ThermoFisher), H3K4me3 (EpiCypher) or SMYD3 (Abcam) were added to each designated experimental tube. Biological triplicates were used for each experimental condition. The samples were incubated overnight on a nutator at 4°C. Next day, the 8-strip tubes containing the samples were placed on a magnet until the slurries cleared, supernatants were removed and the cell-beads were washed twice with cold cell permealization buffer. After the final wash, 50uL of the cold cell permealization buffer was added to the cell-bead slurries, and then 2.5uL of pAG-MNase was added to each sample. Samples were incubated for 10 min at RT and the 8-strip tubes were placed back on a magnet. Supernatants were removed and cell-beads complexes were washed twice with cold cell permealization buffer. After the final wash, 50uL of cell cell permealization buffer was added in each sample, and targeted chromatin digestion followed by adding 1uL of chromatin digest additive to each sample. Strips were incubated for 2h at 4°C on a nutator and the reaction was stopped using Stop buffer. 0.5ng of spike-in Ecoli DNA was added to each sample and samples were incubated for 10 min at 37°C in a thermocycler. The strips were then placed on a magnet and the supernatants containing the CUT&RUN enriched DNA were transferred to new tubes. DNA was purified per EpiCypher’s protocol, and library construction was conducted using the TruSeq ChIP (Illumina) (H4K20me3 for siNC/siSMYD3 treated cells) or IDT 2S Plus (IDT) (H4K20me3, H3K4me3, SMYD3 for SMYD3 KO cells) DNA library prep kits. Nucleic acid size selection to enrich for fragment sizes between 200 and 500bp was conducted using SPRIselect (Beckman Coulter Life Sciences). Samples were pooled and sequenced on NextSeq2000 with paired-end sequencing. All the samples had yields between 53 and 80 million pass filter reads. Samples were trimmed for adapters using Cutadapt before the alignment. The trimmed reads were aligned with hg38 reference using Bowtie2 alignment. All the samples had library complexity with percent non-duplicated reads ranging from 75 to 88%.
EPIC DNA methylation arrays
HN-6 cells were cultured in vitro in biological triplicates (one 10cm dish per biological replicate) and treated with control or a SMYD3-targeting siRNA (siSMYD3#1) for 72h, with exposure to IFN-β for 24h prior to collection. Cells were trypsinized and pelleted, and genomic DNA extraction was conducted using the DNeasy Blood&Tissue kit (Qiagen). The Infinium MethylationEPIC BeadChip Kit (Qiagen) was utilized per manufacturer’s protocol to quantify DNA methylation differences in HN-6 cells before and after SMYD3 depletion.
Immunohistochemistry
Formalin-fixed, paraffin embedded tissue microarrays containing clinically annotated, de-identified patient tumor samples were obtained from the Human Tissue Research Center of the University of Chicago Pathology Department (IRB#8980). The IHC staining was approved by the Institutional Review Board of the University of Chicago (IRB#18-0468-AM002). 64 HPV-negative HSNCC tumors, 10 dysplastic lesions and 10 samples from normal buccal epithelium which stained for SMYD3 (Abcam), CD8A (DAKO)) and UHRF1 (Abcam) using immunohistochemistry (IHC). For the SMYD3 IHC, the staining was performed on Leica Bond RX automated stainer. After deparaffinization and rehydration, tissue sections were treated with antigen retrieval solution (Leica Microsystems) with heat near 100°C for 20 min. The anti-SMYD3 antibody (1:400) was applied on tissue sections for 1 h incubation at room temperature. The antigen-antibody binding was detected with Leica Bond Polymer Refine Detection system (Leica Microsystems) and the slides were covered with cover glasses. For the CD8A IHC, the slides were stained on Leica Bond RX automatic stainer using the protocol “HTRC Bond DAB Refine”. Epitope retrieval solution I (Leica Biosystems) was used for 20 min. The anti-CD8 antibody (1:400) was applied on tissue sections for 25 min incubation and the antigen-antibody binding was detected with Bond polymer refine detection (Leica Biosystems). The tissue sections were covered with cover glasses. For the UHRF1 IHC, after deparaffinization and rehydration, tissue sections were treated with antigen retrieval buffer (DAKO) in a steamer for 20 min. The anti-UHRF1 antibody (1:200) was applied on tissue sections for 1 h incubation at room temperature in a humidity chamber. Following TBS wash, the antigen-antibody binding was detected by Bond Polymer Refine Detection (Leica Microsystems) and DAB+ chromogen (DAKO). Tissue sections were briefly immersed in hematoxylin for counterstaining and were covered with cover glasses.
The stained tissue microarray slides were then scanned and digital image analysis algorithms were developed in QuPath v.0.3.0 (Belfast, UK). Digital slides were reviewed for 64 head and neck cancer tumors and included H&E stained sections along with IHC on serial sections for SMYD3, UHRF1 and CD8A. All tissue sections had minimal artifact and staining pattern were consistent with specific staining. For each case, tumor was annotated to exclude normal tissue, artifact and necrotic regions. For SMYD3 and UHRF1 assessment, only tumor compartments were annotated from each core, while for the CD8A assessment, both the tumor and stroma compartments were evaluated together in each core. Parameters for cell detection were optimized and validated. SMYD3 immunolabeling was commonly positive in cancer cells, most commonly with a cytoplasmic pattern but with a detectable nuclear pattern too, while UHRF1 immunolabeling was predominantly nuclear. SMYD3 and UHRF1 immunolabeling were reported with an H-score. SMYD3 H-scores ranged from 84.14 to 295.65, while UHRF1 H-scores ranged from 0.3 to 213.53. CD8A scoring was reported as CD8% in each tumor core and ranged from 0.03 to 20.66%. Semiquantitative assessment for SMYD3 (scale 0, +1, +2, +3) was also conducted.
For the IHC staining of ASOs and mouse CD8A of the MOC1 tumor sections, slides were stained with rabbit polyclonal ASO antibody (Ionis) or anti-CD8A (SinoBiological) on a Ventana Ultra staining system. ASO slides were treated enzymatically with trypsin (Sigma-Aldrich). The slides were then blocked with endogenous biotin blocking kit (Ventana) and normal goat serum (Jackson Immuno Labs). The primary antibodies were diluted with discovery antibody diluent (Ventana) (1:2500 for anti-CD8A) and incubated for 1 h at 37°c. The antibodies were detected with biotin labeled goat anti-rabbit secondary antibody (Jackson Immuno Labs). The secondary antibody was labeled with DABMap Kit (Ventana). Images were scanned on a Hamamatsu S360 scanner at 20X resolution.
Multicolor flow cytometry
For the multicolor flow of MOC1 tumors, mice were euthanized and flank MOC1 tumors were surgically resected, and mechanically and chemically digested into single-cell suspensions using the gentleMACS Dissociator and the mouse tumor dissociation kit (Miltenyi Biotec), per manufacturer’s protocol. Single-cell suspensions were filtered through 70uμ filters and washed with 1% BSA in PBS. Samples were incubated with anti-CD16/32 (Biolegend) antibody to block nonspecific staining. Subsequently, the primary antibodies were added and incubation for 30min was carried out in the dark. Cell surface staining was performed using fluorophore-conjugated anti-mouse CD45.2 (clone 104), CD3 (145-2C11), CD4 (GK1.5), CD8 (53–6.7), CD31 (390), PDGFR (APA5), PD-L1 (10F.9G2), H2-Kb (AF6-88.5), PD-1 (RMP1-30), CD11b (M1/70), Ly6G (1A8), Ly6C (HK1.4), CD11c (N418) and F4/80 (BM8) from Biolegend. FoxP3+ regulatory T cell staining was performed with the mouse regulatory T cell Staining Kit (eBioscience) as per manufacturer’s protocol. Cell viability was assessed with Sytox (Thermo Fisher Scientific) or Zombie (Biolegend) cell viability dyes. All analyses were performed on a BD Fortessa analyzer running FACSDiva software and interpreted using FlowJo V.X10.0.7r2.
Mass spectrometry
Histone purification and chemical labeling of histone peptides
Core Histones were purified from cultured cells using the Histone Purification Mini Kit (Active Motif) according to the manufacturer’s instructions. The purified histones were desalted using the Zeba spin columns (Thermo Fisher Scientific) prior to downstream procedures. The purity of the core histones was checked by running an SDS PAGE gel. Peptides were desalted using C18 spin columns (Thermo Fisher Scientific) according to manufacturer’s protocol. The samples were vacuum centrifuged to dryness and stored at −80°C until analysis by mass spectrometry.
Mass spectrometry acquisition
The dried peptide fractions were reconstituted in 0.1%TFA and subjected to nanoflow liquid chromatography (Thermo Ultimate 3000RSLC nano LC system, Thermo Fisher Scientific) coupled to an Orbitrap Eclipse mass spectrometer (Thermo Fisher Scientific). Peptides were separated using a low pH gradient using a 0–50% ACN over 60 min in mobile phase containing 0.1% formic acid at 300 nL/min flow rate. MS scans were performed in the Orbitrap analyser at a resolution of 120,000 with an ion accumulation target set at 4e5 and max IT set at 50ms over a mass range of 200–1400 m/z.
Histone modification quantitation
Acquired MS/MS spectra were searched against a human uniprot protein database along with a contaminant protein database, using a SEQUEST and percolator validator algorithms in the Proteome Discoverer 2.4 software (Thermo Fisher Scientific). For the identification of histone post-translational modifications (PTM), the following dynamic modifications at lysine residues were used: unmodified (56.026 Da), monomethylation (70.042 Da), demethylation (28.031 Da), trimethylation (42.047 Da) and acetylation (42.011 Da). Trypsin was specified as the proteolytic enzyme, with up to 2 missed cleavage sites allowed. The sum of the MS1 intensity from all the identified PTM modified peptide represents the total amount of peptide present in the sample. This sum of MS1 intensities was normalized against all the samples and proportion so the different methylation states were calculated and displayed as percentages of the overall peptide.
QUANTIFICATION AND STATISTICAL ANALYSIS
Lists of type I IFN response and APM genes
Based on previous publications,42,43 we comprised a list of type I IFN response and APM genes (Table S1). Bioinformatic interrogation of this gene list is shown in Figure 1. Genes that were not found to be expressed at the mRNA level in the RNA-seq databases of each cell system presented in Figures 1A-1C were omitted. We also interrogated the gene set from the HALLMARK_INTERFERON_ALPHA_RESPONSE (IFNa response genes) and the KEGG_ANTIGEN_PROCESSING_AND_PRESENTATION (APM genes), which are available in the Molecular Signature Database (MSigDB) gene sets (Table S2, https://www.gsea-msigdb.org/gsea/msigdb/index.jsp). Bioinformatic interrogation of these gene lists are shown in Figures S1, S2 and S3.
RNA-seq heatmaps for type I IFN response and APM genes
RNA-Seq data were quantitated to obtain raw tag counts at the gene level using either HTSeq or featureCount. The raw tag count data was variance stabilizing transformed using VST function in DESeq2 R library, and Z score of the transformed data was obtained to color code for heatmap. For clustered heatmaps, pheatmap R library was used with Euclidean distance and ward.D2 clustering options. Significance of gene expression changes was evaluated using DESeq2 R library and determined based on Wald-statistics (FDR<0.1) and shrunken log2 fold-change (>log2(1.3), <−log2(1.3)) using ahsr method available from DESeq2 library.
IPA analysis of DEGs
Ingenuity Pathway Analysis (IPA) was used to identify significantly enriched canonical pathways based on DEGs. To obtain manage-able number of DEGs for IPA core analysis, different FDR and LFC were applied; siSMYD3 vs. HN6 (991 DEGs, FDR<0.05, LFC < −1 or LFC >1), SMYD3 ASOs vs. HN6 (1401 DEGs, FDR<0.05), CRISPR KO vs. HN6 (3714 DEGs, FDR<0.01). Core analysis was performed using default options.
GSEA analysis of TCGA data
For the gene set enrichment analysis (GSEA), 422 TCGA HPV-negative tumor samples (Firehose Legacy) with mRNA expression data (data_RNA_Seq_v2_mRNA_median_Zscores.txt) were analyzed and a ranked gene list was obtained based on Pearson’s correlation of each gene with SMYD3 expression. This list was used as input to the pre-ranked GSEA against the MsigDB’s Hallmark gene sets (h.all.v7.4.symbols.gmt). The analysis was performed using fgsea R library.
Expression of retrotransposons in mRNA-Seq data
For the evaluation of differences in the expression levels of retransposons and other repetitive elements, RNA-seq retrotransposon quantification and analysis was performed using homer and bedtools. Briefly, genomic coordinates (hg38) of LINE and other repetitive elements were obtained from the UCSC (University of California Santa-Cruz) genome browser consortium and quantified using the annotatePeaks.pl script, by homer.
CUT&RUN analysis
Raw fastq files were trimmed with Cutadapt 3.4 and aligned using bowtie2-2.4.4 against hg38. For spike-in controls, the trimmed FASTQ files were aligned against the Escherichia coli MG1655 genome. Duplicated reads were removed using Picard tools 2.25.0, and normalization factors were derived based on the uniquely mapped fragments in the corresponding spike-in control data. The enriched regions with H4K20me3, SMYD3 and H3K4me3 signals were identified using GoPeaks spike-in normalization, with default “broad” (H4K20me3, SMYD3) or “narrow” (H3K4me3, SMYD3) parameters applied. Differential analysis was performed using DESeq2 to obtain a final list of regions for downstream analysis (FDR<0.05, abs(log2FC) > 1.3).
Annotation of peaks (CUT&RUN)
Each peak/enriched region in CUT&RUN assays was annotated with the nearby gene displaying the shortest distance between TSS and the center of each peak using the ChipSeeker. Pre-identified immune-related genes were used to annotate heatmap. Genome browser track and genomic coordinate heatmaps were obtained using deepTools. To identify CUT&RUN peaks (H4K20me3, H3K4me3, SMYD3) overlapping with immune response genes either at their gene body or promoter region (<2kb from TSS), genomic coordinates of gene boundaries of the immune response related genes (Table S5) were extracted using BioMart (v. 2.54.0) R library Ensembl GRCh38.p13 database. To account for the promoter region, we extended the coordinate of each gene boundary by 2kb upstream. Then GenomicRanges (v.1.50.2) R library was used to find overlapping CUT&RUN peaks and genes based on their extended gene boundary.
Volcano plots
For all volcano plots, EnhancedVolcano R library was used.
Single-cell RNA-sequencing analysis
Published single-cell RNA-seq data were obtained from and processed as described by Puram et al.29 Processed expression data were downloaded from Gene Expression Omnibus (GSE103322) and subjected to log2 transformation after adding one to each value. Analysis was performed using R V.4.1.2; heatmaps were generated using pheatmap. Pearson correlation was used for correlation-based analysis.
Statistical analyses for in vitro and in vivo experiments
The Student t-test was used to compare groups of biological or technical replicates for qPCR and MS experiments, as well as to compare the CD8 T cell infiltration assessed by IHC in control or Smyd3 ASO treated MOC1 tumors. Unpaired t test was used to compare tumor volumes, cell subsets, and immune and cancer cell markers of MOC1 tumors treated with control ASOs, PBS or Smyd3 ASOs.
Statistical analyses for clinical datasets
To compare IHC scores among normal, dysplastic buccal squamous epithelium and HNSCC, the Kruskal-Wallis test followed by Dunn’s post hoc test with Benjamini-Hochberg (BH) procedure for multiple comparisons was performed using stats and FSA R libraries. Pearson correlations between SMYD3 and CD8A, SMYD3 and UHRF1, and UHRF1 and CD8A protein levels (CPTAC protein abundance levels and QuPath scores) were obtained using stats R library.
Associations between clinical to pathological downstaging and pretreatment mRNA expression levels of SMYD3, UHRF1, and CD8A were evaluated using published mRNA data (dbGaP, phs001623). The TPM values of SMYD3, UHRF1, and CD8A mRNA expression were used to perform the Wilcoxon rank-sum test between groups with (n = 4) and without (n = 16) clinical to pathological downstaging.
For the survival analysis using the CPTAC HNSCC database (n = 108 patients), protein abundance and clinical data were extracted from the CPTAC data portal (https://proteomics.cancer.gov/data-portal, http://linkedomics.org/data_download/CPTAC-HNSCC). The R function “surv_cutpoint” (package survminer) was applied to identify optimal cutpoints that correspond to the most significant relation with OS and PFS outcomes for proteins SMYD3 and UHRF1. Using 60% and 52% percentiles as cutpoints for SMYD3 and UHRF1 respectively in the OS analysis, and 37% and 49% percentiles in the PFS analysis, the data were dichotomized to high and low protein expression groups. The intersection of the two high-expression groups were defined as High SMYD3_High UHRF1, and the intersection of the two low-expression groups were defined as Low SMYD3_Low UHRF1. The progression-free (PFS) and overall survival (OS) curves were obtained using the Kaplan-Meier (KM) method and were compared using the log rank test. The Cox proportional hazards model was used to estimate hazard ratios (HRs) with 95% confidence intervals (CIs). Similarly, for the survival analysis using the University of Chicago HPV-negative HNSCC database (n = 35 patients), patients were partitioned into four groups based on dichotomized values of SMYD3, UHRF1 and CD8A QuPath scores derived from the FFPE clinically annotated tissue microarrays. The top 15% of SMYD3, 30% of UHRF1 and 35% of CD8A QuPath scores were designated as “high”, while the rest were designated as “low” for both the OS and PFS analysis.
CPTAC analysis
The specimens collection and sample processing of 108 HPV(−) head and neck squamous cell carcinomas (HNSCCs) can be found in Huang et al.69 and the CPTAC data portal (PDC000221, https://pdc.cancer.gov/pdc/study/PDC000221). Processed proteomics data files were downloaded via LinkedOmics: http://www.linkedomics.org. The gene-level proteomics data were represented as normalized mass spectrometric intensity and log2 transformed.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Human anti-SMYD3 | Abcam | Cat# ab187149 |
| Human anti-UHRF1 | Cell Signaling Technologies | Cat# D6G8E |
| Human anti-UHRF1 | Abcam | Cat# ab194236 |
| Human anti-HA | Cell Signaling Technologies | Cat# 3724S; RRID:AB_1549585 |
| Human anti-H4K20me3 | ThermoFisher | Cat# MA5-36090; RRID:AB_2884049 |
| Human anti-SUV420H1 | ThermoFisher | Cat# PA5-69767; RRID:AB_2689479 |
| Human anti-SUV420H2 | ThermoFisher | Cat# PA5-109891; RRID:AB_2855302 |
| Human anti-H3K4me3 | EpiCypher | Cat# 13-0041 |
| Human anti-H3K9me3 | Abcam | Cat# ab176916; RRID:AB_2797591 |
| Human anti-H3K9me3 | Diagenode INC | Cat# C15410056 |
| Human anti-H3 | Abcam | Cat# ab1791; RRID:AB_302613 |
| Human anti-DNMT1 | ThermoFisher | Cat# PA5-30581; RRID:AB_2548055 |
| Human anti-CD8A | DAKO | Cat# M7103; RRID:AB_2075537 |
| Human anti-UHRF1 | ThermoFisher | Cat# PA5-29884; RRID:AB_2547358 |
| Goat anti-rabbit secondary | Ventana | Cat# 760-124 |
| Goat anti-rabbit secondary | Jackson Immuno Labs | Cat# 111-005-003; RRID:AB_2337913 |
| Mouse anti-CD8A | SinoBiological | Cat# 50389-T26 |
| Rat anti-IgG2a | BioXCell | Cat# BE0089; RRID:AB_1107769 |
| Mouse anti-PD-1 (CD279) | BioXCell | Cat# BE0146; RRID:AB_10949053 |
| Mouse anti-CD16/32 | Biolegend | Cat# 156619; RRID:AB_2922485 |
| Mouse anti-CD45.2 (104) | Biolegend | Cat# 109815; RRID:AB_492869 |
| Mouse anti-CD3 (145-2C11) | Biolegend | Cat# 317317; RRID:AB_1937213 |
| Mouse anti-CD4 (GK1.5) | Biolegend | Cat# 100437; RRID:AB_10900241 |
| Mouse anti-CD8 (53–6.7) | Biolegend | Cat# 344737 |
| Mouse anti-CD31 (390) | Biolegend | Cat# 102417; RRID:AB_830756 |
| Mouse anti-PDGFR (APA5) | Biolegend | Cat# 135907; RRID:AB_2043969 |
| Mouse anti-PD-LI (10F.9G2) | Biolegend | Cat# 124307; RRID:AB_2073557 |
| Mouse anti-H2-Kb (AF6-88.5) | Biolegend | Cat# 116503; RRID:AB_313730 |
| Mouse anti-PD-1 (RMP1-30) | Biolegend | Cat# 109119; RRID:AB_2566640 |
| Mouse anti-CD11b (M1/70) | Biolegend | Cat# 101211; RRID:AB_312794 |
| Mouse anti-Ly6G (1A8) | Biolegend | Cat# 127614; RRID:AB_2227348 |
| Mouse anti-Ly6C (HK1.4) | Biolegend | Cat# 128001; RRID:AB_1134213 |
| Mouse anti-CD11c (N418) | Biolegend | Cat# 117307; RRID:AB_313776 |
| Mouse anti-F4/80 (BM8) | Biolegend | Cat# 123141; RRID:AB_2563667 |
| Biological samples | ||
| Human HNSCC Tumor Tissue Microarrays | Human Tissue Research Center of the University of Chicago Pathology Department | IRB#8980 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant human interferon-β | R & D systems Inc. | Cat# 8499-IF-010/CF |
| Critical commercial assays | ||
| CUT&RUN Kit | Epicypher | Cat# 14-1048 |
| Nuclear complex Co-IP kit | Active Motif | Cat# 54001 |
| Direct-zol RNA miniprep kit | Zymo Research | Cat# R2072 |
| TruSeq ChIP Library Prep Kit | Illumina | Cat# IP-202-1012 |
| IDT 2S Plus DNA Library Prep Kit | IDT | Cat# 10009878 |
| TruSeq Stranded mRNA Library Prep kit | Illumina | Cat# 20020594 |
| Dneasy Blood&Tissue Kit | Qiagen | Cat# 69504 |
| Infinium MethylationEPIC BeadChip Kit | Qiagen | Cat# WG-317-1001 |
| Mouse tumor dissociation kit | Miltenyi Biotec | Cat# 130-096-730 |
| Mouse regulatory T cell staining kit | eBioscience | Cat# 88-8111-40 |
| Histone Purification Mini Kit | Active Motif | Cat# 40026 |
| Deposited data | ||
| RNA-seq | GEO | Accession ID: GSE233495 |
| CUT&RUN DNA-seq | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE233495 | |
| Experimental models: Cell lines | ||
| HN-6 | Dr. Tanguy Seiwert, University of Chicago | CVCL_8129 |
| HN-SCC-151 | Dr. Tanguy Seiwert, University of Chicago | CVCL_RK55 |
| MOC1 | Dr. Clint Allen, NIDCD | CVCL_ZD32 |
| Experimental models: Organisms/strains | ||
| C57BL/6 mice | Taconic | https://www.taconic.com/mouse-model/black-6-b6ntac |
| Oligonucleotides | ||
| siRNA for SMYD3 | Millipore-Sigma | SASI_Hs02_0035-5988 |
| siRNA for UHRF1 | Millipore-Sigma | SASI_Hs02_00311672 |
| non-targeting negative control siRNA | Dharmacon | Cat# D-001810-10-20 |
| qRT-PCR primers for GAPDH | Sigma-Aldrich | https://www.sigmaaldrich.com |
| qRT-PCR primers for SMYD3 | Sigma-Aldrich | https://www.sigmaaldrich.com |
| qRT-PCR primers for UHRF1 | Sigma-Aldrich | https://www.sigmaaldrich.com |
| qRT-PCR primers for TRIM21 | Sigma-Aldrich | https://www.sigmaaldrich.com |
| qRT-PCR primers for STAT1 | Sigma-Aldrich | https://www.sigmaaldrich.com |
| qRT-PCR primers for OASL | Sigma-Aldrich | https://www.sigmaaldrich.com |
| qRT-PCR primers for IFNGR1 | Sigma-Aldrich | https://www.sigmaaldrich.com |
| qRT-PCR primers for OAS3 | Sigma-Aldrich | https://www.sigmaaldrich.com |
| qRT-PCR primers for CD274 | Sigma-Aldrich | https://www.sigmaaldrich.com |
| qRT-PCR primers for OAS2 | Sigma-Aldrich | https://www.sigmaaldrich.com |
| qRT-PCR primers for DHX58 | Sigma-Aldrich | https://www.sigmaaldrich.com |
| qRT-PCR primers for NCOA7 | Sigma-Aldrich | https://www.sigmaaldrich.com |
| qRT-PCR primers for MX1 | Sigma-Aldrich | https://www.sigmaaldrich.com |
| qRT-PCR primers for MX2 | Sigma-Aldrich | https://www.sigmaaldrich.com |
| qRT-PCR primers for GBP2 | Sigma-Aldrich | https://www.sigmaaldrich.com |
| qRT-PCR primers for RSAD2 | Sigma-Aldrich | https://www.sigmaaldrich.com |
| qRT-PCR primers for TAP2 | Sigma-Aldrich | https://www.sigmaaldrich.com |
| qRT-PCR primers for CANX | Sigma-Aldrich | https://www.sigmaaldrich.com |
| qRT-PCR primers for CXCL9 | Sigma-Aldrich | https://www.sigmaaldrich.com |
| qRT-PCR primers for CXCL10 | Sigma-Aldrich | https://www.sigmaaldrich.com |
| Smyd3 ASOs | Ionis Pharmaceuticals | N/A |
| ChIP Primers for CXCL9 | Sigma-Aldrich | CXCL9-F: 5′-TGCACTCCAATCAGAACCAG-3′ CXCL9-R: 5′-CCAATACAGGAGTGACTTGGAAC-3′ |
| ChIP Primers for CXCL10 | Sigma-Aldrich | CXCL10-F: 5′-TCCCTCCCTAATTCTGATTGG-3′ CXCL10-R: 5′-AGCAGAGGGAAATTCCGTAAC-3′ |
| ChIP Primers for MX1 | Sigma-Aldrich | MX1-F: 5′ GGGACAGGCATCAACAAAGCC 3′ MX1-R: 5′ GCCCTCTCTTCTTCCAGGCAAC 3′ |
| ChIP Primers for OAS2 | Sigma-Aldrich | OAS2-F: 5′ CGCTGCAGTGGGTGGAGAGA3′ OAS2-R: 5′ GCCGGCAAGACAGTGAATGG 3′ |
| ChIP Primers for RSAD2 | Sigma-Aldrich | RSAD2-F: 5′ CCAATGACAGGTTGCTCAGA 3′ RSAD2-R: 5′ CAGCTGCTGCTTTCTCCTCT 3′ |
| Recombinant DNA | ||
| pCE0482 (SMYD3 KO CRISPR plasmid) | This paper | Addgene plasmid ID#202621 |
| HA-MOCK plasmid | SinoBiological | Cat# CV013 |
| HA-UHRF1 plasmid | SinoBiological | Cat# HG17896-CY |
| Software and algorithms | ||
| ImageJ v1.53c | NIH | https://imagej.nih.gov/ij |
| GraphPad Prism v9.5.1 | GraphPad software | https://www.graphpad.com |
| QuPath v.0.3.0 | Bankhead et al.67 | https://qupath.github.io/ |
| FACSDiva | BD biosciences | https://www.bdbiosciences.com/en-us/products/software/ |
| FlowJo V.X10.0.7r2 | BD biosciences | https://www.flowjo.com |
| BioRender | BioRender | https://www.biorender.com/ |
| Proteome Discoverer 2.4 software | Thermo Scientific | https://www.thermofisher.com/us/en/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/multi-omics-data-analysis/proteome-discoverer-software.html |
| Ingenuity pathway, 2021-2023 | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/ | |
| htseq 0.11.4 | https://htseq.readthedocs.io/en/master/index.html | |
| subread 2.0.1 | https://github.com/ShiLab-Bioinformatics/subread | |
| deeptools 3.5.1 | https://deeptools.readthedocs.io/en/develop/index.html | |
| samtools 1.11 | https://sourceforge.net/projects/samtools/files/samtools/1.11/ | |
| bedtools 2.30.0 | https://bedtools.readthedocs.io/en/latest/index.html | |
| pheatmap_1.0.12 | https://cran.r-project.org/web/packages/pheatmap/index.html | |
| ChIPseeker_1.34.1 | https://bioconductor.org/packages/release/bioc/html/ChIPseeker.html | |
| TxDb.Hsapiens.UCSC.hg38.knownGene_3.16.0 | https://bioconductor.org/packages/release/data/annotation/html/TxDb.Hsapiens.UCSC.hg38.knownGene.html | |
| GenomicRanges_1.50.2 | https://bioconductor.org/packages/release/bioc/html/GenomicRanges.html | |
| DESeq2_1.38.3 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html | |
| fgsea_1.24.0 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html | |
| EnhancedVolcano_1.16.0 | https://bioconductor.org/packages/release/bioc/html/EnhancedVolcano.html | |
| eulerr_6.1.1 | https://cran.r-project.org/web/packages/eulerr/index.html | |
| survminer_0.4.9 | https://cran.r-project.org/web/packages/survminer/index.html | |
| survival_3.4–0 | https://cran.r-project.org/web/packages/survival/index.html | |
| Other | ||
| DMEM medium | Thermo Fisher Scientific | Cat# 11965092 |
| DMEM/F12 | Thermo Fisher Scientific | Cat# 11320033 |
| Fetal bovine serum | Cytiva | Cat# SH30071.03 |
| Penicillin/streptomycin | Vita Scientific | Cat# 120-095-721 |
| L-glutamine | Thermo Fisher Scientific | Cat# 17605E |
| Lipofectamine RNAimax | Thermo Fisher Scientific | Cat# 13778150 |
| Trypsin EDTA 0.25% | Sigma | Cat# T8003 |
| Fugene HD | Active Motif | Cat# 32042 |
| SuperScript III First-Strand Synthesis System | Invitrogen | Cat# 18080051 |
| SYBR Select Master Mix | Applied Biosystems | Cat# 4472908 |
| GreenLink qRT-PCR Master Mix | BioLink Laboratories | Cat# 16-2000 |
| Dynabeads Protein G | Invitrogen | Cat# 10004D |
| SPRIselect | Beckman Coulter Life Sciences | Cat# B23318 |
| Antigen retrieval solution | Leica Microsystems | Cat# AR9640 |
| Bond Polymer Refine Detection | Leica Microsystems | Cat# DS9800 |
| Epitope retrieval solution | Leica Microsystems | Cat# AR9961 |
| Antigen retrieval buffer | DAKO | Cat# S2367 |
| DAB+ chromogen | DAKO | Cat# K3468 |
| Endogenous Biotin Blocking Kit | Ventana | Cat# 760-050 |
| Normal Goat Serum | Jackson Immuno Labs | Cat# 005-000-121 |
| Discovery Antibody Diluent | Ventana | Cat# 760-108 |
| DABMap Kit | Ventana | Cat# 760-124 |
| Matrigel | Corning | Cat# 354277 |
| Sytox cell viability dye | Thermo Fisher Scientific | Cat# S34857 |
| Zombie cell viability dye | Biolegend | Cat# 423117 |
Highlights.
SMYD3 depletion derepresses type I IFN response and APM genes in HPV-negative HNSCC cells
SMYD3 represses immune genes through UHRF1, an H3K9me3-reader, and deposition of H4K20me3
Smyd3 ASOs increase CD8+ T cell influx and sensitize MOC1 tumors to anti-PD-1 therapy
SMYD3 mRNA levels predict response to neoadjuvant pembrolizumab in HPV-negative HNSCC
ACKNOWLEDGMENTS
This work was funded by the Intramural Research Program of the National Cancer Institute, National Institutes of Health (contract no. HHSN261201500003I).
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112823.
DECLARATION OF INTERESTS
L.R. is currently an employee of Delfi Diagnostics in Baltimore, MD, USA. X.L. was an employee and shareholder of Ionis Pharmaceuticals in Carlsbad, CA, USA during the conduct of this study. Y.N. is employed as the president of the National Institutes of Biomedical Innovation, Health and Nutrition, and has 6% of stocks of OncoTherapy Science. J.L. has research support by Epizyme.
REFERENCES
- 1.Siegel RL, Miller KD, Wagle NS, and Jemal A (2020). Cancer Statistics, 2020. CA Cancer J. Clin 70, 7–30. 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Ang KK, and Sturgis EM (2012). Human papillomavirus as a marker of the natural history and response to therapy of head and neck squamous cell carcinoma. Semin. Radiat. Oncol 22, 128–142. 10.1016/j.semradonc.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 3.Chaturvedi AK, Engels EA, Pfeiffer RM, Hernandez BY, Xiao W, Kim E, Jiang B, Goodman MT, Sibug-Saber M, Cozen W, et al. (2011). Human Papillomavirus and rising oropharyngeal cancer incidence in the United States. J. Clin. Oncol 29, 4294–4301. 10.1200/JCO.2011.36.4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burtness B, Harrington KJ, Greil R, Soulières D, Tahara M, de Castro G Jr., Psyrri A, Basté N, Neupane P, Bratland Å, et al. (2019). Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet 394, 1915–1928. 10.1016/S0140-6736(19)32591-7. [DOI] [PubMed] [Google Scholar]
- 5.Ferris RL (2015). Immunology and immunotherapy of head and neck cancer. J. Clin. Oncol 33, 3293–3304. 10.1200/JCO.2015.61.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saloura V, Izumchenko E, Zuo Z, Bao R, Korzinkin M, Ozerov I, Zhavoronkov A, Sidransky D, Bedi A, Hoque MO, et al. (2019). Immune profiles in primary squamous cell carcinoma of the head and neck. Oral Oncol. 96, 77–88. 10.1016/j.oraloncology.2019.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanna GJ, Lizotte P, Cavanaugh M, Kuo FC, Shivdasani P, Frieden A, Chau NG, Schoenfeld JD, Lorch JH, Uppaluri R, et al. (2018). Frameshift events predict anti–PD-1/L1 response in head and neck cancer. JCI Insight 3, 98811. 10.1172/jci.insight.98811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, Caux C, and Depil S (2019). Cold tumors: a therapeutic challenge for immunotherapy. Front. Immunol 10, 168. 10.3389/fimmu.2019.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duan Q, Zhang H, Zheng J, and Zhang L (2020).Turning cold into hot: firing up the tumor microenvironment. Trends Cancer 6, 605–618. 10.1016/j.trecan.2020.02.022. [DOI] [PubMed] [Google Scholar]
- 10.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leemans CR, Snijders PJF, and Brakenhoff RH (2018). The molecular landscape of head and neck cancer. Nat. Rev. Cancer 18, 269–282. 10.1038/nrc.2018.11. [DOI] [PubMed] [Google Scholar]
- 13.Hamamoto R, Saloura V, and Nakamura Y (2015). Critical roles of non-histone protein lysine methylation in human tumorigenesis. Nat. Rev. Cancer 15, 110–124. 10.1038/nrc3884. [DOI] [PubMed] [Google Scholar]
- 14.Cao J, and Yan Q (2020). Cancer epigenetics, tumor immunity, and immunotherapy. Trends Cancer 6, 580–592. 10.1016/j.trecan.2020.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vougiouklakis T, Bao R, Nakamura Y, and Saloura V (2017). Protein methyltransferases and demethylases dictate CD8+ T-cell exclusion in squamous cell carcinoma of the head and neck. Oncotarget 8, 112797–112808. 10.18632/oncotarget.22627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sarris ME, Moulos P, Haroniti A, Giakountis A, and Talianidis I (2016). Smyd3 is a transcriptional potentiator of multiple cancer-promoting genes and required for liver and colon cancer development. Cancer Cell 29, 354–366. 10.1016/j.ccell.2016.01.013. [DOI] [PubMed] [Google Scholar]
- 17.Bernard BJ, Nigam N, Burkitt K, and Saloura V (2021). SMYD3: a regulator of epigenetic and signaling pathways in cancer. Clin. Epigenet 13, 45. 10.1186/s13148-021-01021-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mazur PK, Reynoird N, Khatri P, Jansen PWTC, Wilkinson AW, Liu S, Barbash O, Van Aller GS, Huddleston M, Dhanak D, et al. (2014). SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature 510, 283–287. 10.1038/nature13320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamamoto R, Furukawa Y, Morita M, limura Y, Silva FP, Li M, Yagyu R, and Nakamura Y (2004). SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat. Cell Biol 6, 731–740. 10.1038/ncb1151. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Xie BH, Lin WH, Huang YH, Ni JY, Hu J, Cui W, Zhou J, Shen L, Xu LF, et al. (2019). Amplification of SMYD3 promotes tumorigenicity and intrahepatic metastasis of hepatocellular carcinoma via upregulation of CDK2 and MMP2. Oncogene 38, 4948–4961. 10.1038/s41388-019-0766-x. [DOI] [PubMed] [Google Scholar]
- 21.Fenizia C, Bottino C, Corbetta S, Fittipaldi R, Floris P, Gaudenzi G, Carra S, Cotelli F, Vitale G, and Caretti G (2019). SMYD3 promotes the epithelial-mesenchymal transition in breast cancer. Nucleic Acids Res. 47, 1278–1293. 10.1093/nar/gky1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Foreman KW, Brown M, Park F, Emtage S, Harriss J, Das C, Zhu L, Crew A, Arnold L, Shaaban S, and Tucker P (2011). Structural and functional profiling of the human histone methyltransferase SMYD3. PLoS One 6, e22290. 10.1371/journal.pone.0022290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang Y, Lyu T, Che X, Jia N, Li Q, and Feng W (2019). Overexpression of SMYD3 in ovarian cancer is associated with ovarian cancer proliferation and apoptosis via methylating H3K4 and H4K20. J. Cancer 10, 4072–4084. 10.7150/jca.29861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peserico A, Germani A, Sanese P, Barbosa AJ, Di Virgilio V, Fittipaldi R, Fabini E, Bertucci C, Varchi G, Moyer MP, et al. (2015). A SMYD3 small-molecule inhibitor impairing cancer cell growth. J. Cell. Physiol 230, 2447–2460. 10.1002/jcp.24975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mitchell LH, Boriack-Sjodin PA, Smith S, Thomenius M, Rioux N, Munchhof M, Mills JE, Klaus C, Totman J, Riera TV, et al. (2016). Novel oxindole sulfonamides and sulfamides: EPZ031686, the first orally bioavailable small molecule SMYD3 inhibitor. ACS Med. Chem. Lett 7, 134–138. 10.1021/acsmedchemlett.5b00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang C, Liew SS, Lin GR, Poulsen A, Ang MJY, Chia BCS, Chew SY, Kwek ZP, Wee JLK, Ong EH, et al. (2019). Discovery of irreversible inhibitors targeting histone methyltransferase, SMYD3. ACS Med. Chem. Lett 10, 978–984. 10.1021/acsmed-chemlett.9b00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crooke ST, Witztum JL, Bennett CF, and Baker BF (2018). RNA-targeted therapeutics. Cell Metab. 27, 714–739. 10.1016/j.cmet.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 28.Kontaki H, Koukaki M, Vasilarou M, Giakountis A, Deligianni E, Luo X, Kim Y, and Talianidis I (2021). Targeting Smyd3 by next-generation antisense oligonucleotides suppresses liver tumor growth. iScience 24, 102473. 10.1016/j.isci.2021.102473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, Rodman C, Luo CL, Mroz EA, Emerick KS, et al. (2017). Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 171, 1611–1624.e24. 10.1016/j.cell.2017.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rothbart SB, Krajewski K, Nady N, Tempel W, Xue S, Badeaux AI, Barsyte-Lovejoy D, Martinez JY, Bedford MT, Fuchs SM, et al. (2012). Association of UHRF1 with methylated H3K9 directs the maintenance of DNA methylation. Nat. Struct. Mol. Biol 19, 1155–1160. 10.1038/nsmb.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng J, Yang Y, and Fang J (2013). Structural insight into coordinated recognition of trimethylated histone H3 lysine 9 (H3K9me3) by the plant homeodomain (PHD) and tandem tudor domain (TTD) of UHRF1 (Ubiquitin-like, containing PHD and RING finger domains, 1) Protein. J. Biol. Chem 288, 1329–1339. 10.1074/jbc.M112.415398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karagianni P, Amazit L, and Qin J (2008). ICBP90, a Novel Methyl K9 H3 Binding Protein Linking Protein Ubiquitination with Heterochromatin Formation. Mol. Cell. Biol, 705–717. 10.1128/MCB.01598-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie S, Jakoncic J, and Qian C (2012). UHRF1 double tudor domain and the adjacent PHD finger act together to recognize K9me3-containing histone H3 tail. J. Mol. Biol 415, 318–328. 10.1016/j.jmb.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 34.Kong X, Chen J, Xie W, Brown SM, Cai Y, Wu K, Fan D, Nie Y, Yegnasubramanian S, Tiedemann RL, et al. (2019). Defining UHRF1 domains that support maintenance of human colon cancer DNA methylation and oncogenic properties. Cancer Cell 35, 633–648.e7. 10.1016/j.ccell.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu X, Gao Q, Li P, Zhao Q, Zhang J, Li J, Koseki H, and Wong J (2013). UHRF1 targets DNMT1 for DNA methylation through cooperative binding of hemi-methylated DNA and methylated H3K9. Nat. Commun 4, 1563. 10.1038/ncomms2562. [DOI] [PubMed] [Google Scholar]
- 36.Clements EG, Mohammad HP, Leadem BR, Easwaran H, Cai Y, Van Neste L, and Baylin SB (2012). DNMT1 modulates gene expression without its catalytic activity partially through its interactions with histone-modifying enzymes. Nucleic Acids Res. 40, 4334–4346. 10.1093/nar/gks031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mohan KN (2022). DNMT1: catalytic and non-catalytic roles in different biological processes. Epigenomics 14, 629–643. 10.2217/epi-2022-0035. [DOI] [PubMed] [Google Scholar]
- 38.Rountree MR, Bachman KE, and Baylin SB (2000). DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat. Genet 25, 269–277. 10.1038/77023. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Q, Qiao L, Wang X, Ding C, and Chen JJ (2018). UHRF1 epigenetically down-regulates UbcH8 to inhibit apoptosis in cervical cancer cells. Cell Cycle 17, 300–308. 10.1080/15384101.2017.1403686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J, Wang R, Hu X, Gao Y, Wang Z, Li J, and Wong J (2019). Activated MEK/ERK pathway drives widespread and coordinated overexpression of UHRF1 and DNMT1 in cancer cells. Sci. Rep 9, 907. 10.1080/15384101.2017.1403686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mudbhary R, Hoshida Y, Chernyavskaya Y, Jacob V, Villanueva A, Fiel MI, Chen X, Kojima K, Thung S, Bronson RT, et al. (2014). UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell 25, 196–209. 10.1016/j.ccr.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et al. (2015). Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous Retroviruses. Cell 162, 974–986. 10.1016/j.cell.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, Li Y, Chen H, Yang H, Hsu PH, et al. (2018). LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell 174, 549–563.e19. 10.1016/j.cell.2018.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kong Y, Rose CM, Cass AA, Williams AG, Darwish M, Lianoglou S, Haverty PM, Tong AJ, Blanchette C, Albert ML, et al. (2019). Transposable element expression in tumors is associated with immune infiltration and increased antigenicity. Nat. Commun 10, 5228. 10.1038/s41467-019-13035-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Judd NP, Allen CT, Winkler AE, and Uppaluri R (2012). Comparative analysis of tumor-infiltrating lymphocytes in a syngeneic mouse model of oral cancer. Otolaryngol. Head Neck Surg 147, 493–500. 10.1177/0194599812442037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Uppaluri R, Campbell KM, Egloff AM, Zolkind P, Skidmore ZL, Nussenbaum B, Paniello RC, Rich JT, Jackson R, Pipkorn P, et al. (2020). Neoadjuvant and adjuvant pembrolizumab in resectable locally advanced, human Papillomavirus-unrelated head and neck cancer: a multicenter, phase II trial. Clin. Cancer Res 26, 5140–5152. 10.1158/1078-0432.CCR-20-1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim J, Lee Y, Lu X, Song B, Fong KW, Cao Q, Licht JD, Zhao JC, and Yu J (2018). Polycomb- and methylation-independent roles of EZH2 as a transcription activator. Cell Rep. 25, 2808–2820.e4. 10.1016/j.celrep.2018.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, Sun Y, Zhao E, Vatan L, Szeliga W, et al. (2015). Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253. 10.1038/nature15520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nagarsheth N, Peng D, Kryczek I, Wu K, Li W, Zhao E, Zhao L, Wei S, Frankel T, Vatan L, et al. (2016). PRC2 epigenetically silences Th1-type chemokines to suppress effector T- cell trafficking in colon cancer. Cancer Res. 76, 275–282. 10.1158/0008-5472.CAN-15-1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Topper MJ, Vaz M, Chiappinelli KB, DeStefano Shields CE, Niknafs N, Yen RWC, Wenzel A, Hicks J, Ballew M, Stone M, et al. (2017). Epigenetic therapy ties MYC depletion to reversing immune evasion and treating lung cancer. Cell 171, 1284–1300.e21. 10.1016/j.cell.2017.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Griffin GK, Wu J, Iracheta-Vellve A, Patti JC, Hsu J, Davis T, Dele-Oni D, Du PP, Halawi AG, Ishizuka JJ, et al. (2021). Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature 595, 309–314. 10.1038/s41586-021-03520-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou L, Mudianto T, Ma X, Riley R, and Uppaluri R (2020). Targeting EZH2 enhances antigen presentation, antitumor immunity, and circumvents Anti-PD-1 resistance in head and neck cancer. Clin. Cancer Res 26, 290–300. 10.1158/1078-0432.CCR-19-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Greene S, Robbins Y, Mydlarz WK, Huynh AP, Schmitt NC, Friedman J, Horn LA, Palena C, Schlom J, Maeda DY, et al. (2020). Inhibition of MDSC trafficking with SX-682, a CXCR1/2 inhibitor, enhances NK-cell immunotherapy in head and neck cancer models. Clin. Cancer Res 26, 1420–1431. 10.1158/1078-0432.CCR-19-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ye W, Gunti S, Allen CT, Hong Y, Clavijo PE, Van Waes C, and Schmitt NC (2020). ASTX660, an antagonist of cIAP1/2 and XIAP, increases antigen processing machinery and can enhance radiation-induced immunogenic cell death in preclinical models of head and neck cancer. Oncoimmunology 9, 1710398. 10.1080/2162402X.2019.1710398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xiao R, An Y, Ye W, Derakhshan A, Cheng H, Yang X, Allen C, Chen Z, Schmitt NC, and Van Waes C (2019). Dual antagonist of cIAP/XIAP ASTX660 sensitizes HPV− and HPV+ head and neck cancers to TNFα, TRAIL, and radiation therapy. Clin. Cancer Res 25, 6463–6474. 10.1158/1078-0432.CCR-18-3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ghisletti S, Huang W, Jepsen K, Benner C, Hardiman G, Rosenfeld MG, and Glass CK (2009). Cooperative NCoR/SMRT interactions establish a corepressor-based strategy for integration of inflammatory and antiinflammatory signaling pathways. Genes Dev. 23, 681–693. 10.1101/gad.1773109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hargreaves DC, Horng T, and Medzhitov R (2009). Control of inducible gene expression by signal-dependent transcriptional elongation. Cell 138, 129–145. 10.1016/j.cell.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hoberg JE, Popko AE, Ramsey CS, and Mayo MW (2006). IkappaB kinase alpha-mediated derepression of SMRT potentiates acetylation of RelA/p65 by p300. Mol. Cell Biol 26, 457–471. 10.1128/MCB.26.2.457-471.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang W, Ghisletti S, Perissi V, Rosenfeld MG, and Glass CK (2009). Transcriptional integration of TLR2 and TLR4 signaling at the NCoR derepression checkpoint. Mol. Cell 35, 48–57. 10.1016/j.molcel.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ogawa S, Lozach J, Jepsen K, Sawka-Verhelle D, Perissi V, Sasik R, Rose DW, Johnson RS, Rosenfeld MG, and Glass CK (2004). A nuclear receptor corepressor transcriptional checkpoint controlling activator protein 1-dependent gene networks required for macrophage activation. Proc. Natl. Acad. Sci. USA 101, 14461–14466. 10.1073/pnas.0405786101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, and Glass CK (2005). A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature 437, 759–763. 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, Gage FH, and Glass CK (2009). A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 137, 47–59. 10.1016/j.cell.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Flammer JR, and Rogatsky I (2011). Minireview: Glucocorticoids in autoimmunity: unexpected targets and mechanisms. Mol. Endocrinol 25, 1075–1086. 10.1210/me.2011-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stender JD, Pascual G, Liu W, Kaikkonen MU, Do K, Spann NJ, Boutros M, Perrimon N, Rosenfeld MG, and Glass CK (2012). Control of proinflammatory gene programs by regulated trimethylation and demethylation of histone H4K20. Mol. Cell 48, 28–38. 10.1016/j.molcel.2012.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu G, Liu G, Xiong S, Liu H, Chen X, and Zheng B (2015). The histone methyltransferase Smyd2 is a negative regulator of macrophage activation by suppressing interleukin 6 (IL-6) and tumor necrosis factor alpha (TNF-alpha) production. J. Biol. Chem 290, 5414–5423. 10.1074/jbc.M114.610345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nagata D.E.d.A., Ting H-A, Cavassani KA, Schaller MA, Mukherjee S, Ptaschinski C, Kunkel SL, and Lukacs NW (2015). Epigenetic control of Foxp3 by SMYD3 H3K4 histone methyltransferase controls iTreg development and regulates pathogenic T-cell responses during pulmonary viral infection. Mucosal Immunol. 8, 1131–1143. 10.1038/mi.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG,et al. (2017). QuPath: Open source software for digital pathology image analysis. Scientific Reports 7, 16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang C, Chen L, Savage SR, Eguez RV, Dou Y, Li Y, da Veiga Leprevost F, Jaehnig EJ, Lei JT, Wen B, et al. (2021). Proteogenomic insights into the biology and treatment of HPV-negative head and neck squamous cell carcinoma. Cancer Cell 39, 361–379.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seqand CUT&RUN DNA-seq data have been deposited at GEO with accession ID# GSE233495 and are publicly available as of the date of publication.
This paper does not report original code.
All raw data reported in this paper will be shared by the lead contact upon request. Any additional information required to re-analyze the data reported in this paper is available from the lead contact upon request.