

Summary
Pyroptosis is a proinflammatory mode of lytic cell death mediated by accumulation of plasma membrane (PM) macropores composed of gasdermin-family (GSDM) proteins. It facilitates two major functions in innate immunity: (i) elimination of intracellular replicative niches for pathogenic bacteria; and (ii) non-classical secretion of IL-1 family cytokines that amplify host-beneficial inflammatory responses to microbial infection or tissue damage. Physiological roles for gasdermin D (GSDMD) in pyroptosis and IL-1β release during inflammasome signaling have been extensively characterized in macrophages. This involves cleavage of GSDMD by caspase-1 to generate GSDMD macropores that mediate IL-1β efflux and progression to pyroptotic lysis. Neutrophils, which rapidly accumulate in large numbers at sites of tissue infection or damage, become the predominant local source of IL-1β in coordination with their potent microbeocidal capacity. Similar to macrophages, neutrophils express GSDMD and utilize the same spectrum of diverse inflammasome platforms for caspase-1-mediated cleavage of GSDMD. Distinct from macrophages, neutrophils possess a remarkable capacity to resist progression to GSDMD-dependent pyroptotic lysis to preserve their viability for efficient microbial killing while maintaining GSDMD-dependent mechanisms for export of bioactive IL-1β. Rather, neutrophils employ cell-specific mechanisms to conditionally engage GSDMD-mediated pyroptosis in response to bacterial pathogens that use neutrophils as replicative niches. GSDMD and pyroptosis have also been mechanistically linked to induction of NETosis, a signature neutrophil pathway that expels decondensed nuclear DNA into extracellular compartments for immobilization and killing of microbial pathogens. This review summarizes a rapidly growing number of recent studies that have produced new insights, unexpected mechanistic nuances, and some controversies regarding the regulation of, and roles for, neutrophil inflammasomes, pyroptosis, and GSDMs in diverse innate immune responses.
Keywords: gasdermin, inflammasome, interleukin-1β, NETosis, neutrophil, pyroptosis
1 |. INFLAMMASOMES, PYROPTOSIS, AND GASDERMINS: INTRODUCTION AND PERSPECTIVE
1.1 |. Initial identification of pyroptosis and gasdermin D pores as a mechanism for release of IL-1 family cytokines during inflammasome activation
Since the initial description by Jurg Tschopp in 2002,1 inflammasomes have become recognized as the critical signaling platforms for the initiation and progression of two fundamental innate immune responses.2–4 The first is production of IL-1β that, as a primary pro-inflammatory cytokine, orchestrates multiple components of the host response to microbial infection or sterile tissue damage.5–9 The second is pyroptosis, a highly regulated mode of lytic cell death first characterized by Cookson and colleagues.10–12 Pyroptosis is most often assayed by biochemically measuring release of large cytosolic proteins such as lactate dehydrogenase (LDH) to extra-cellular compartments or by morphologically tracking cell swelling and protrusion of balloon-like blebs from the surface membrane13 When triggered in macrophages infected with intracellular bacterial pathogens, pyroptosis eliminates the protected replicative compartment for those microbes by exposing them to the potent bactericidal actions of recruited neutrophils and locally activated complement.14 However, early studies revealed that pyroptosis also serves as an important mechanism for the extracellular accumulation of IL-1β (and IL-18, a related inflammatory cytokine) produced during active inflammasome signaling.15,16 IL-1β and IL-18 lack signal sequences for conventional secretion by the endoplasmic reticulum > Golgi apparatus > secretory vesicle pathway. Rather, they first accumulate as inactive precursor proteins (proIL-1β and proIL-18) in the cytosolic compartment prior to proteolytic maturation by caspase-1 in activated inflammasomes and subsequent release of the mature, biologically active cytokines into extracellular compartments.17,18 Although multiple mechanisms for the unconventional or non-classical export of mature IL-1β have been proposed,19,20 the identification in 2015 of gasdermin D (GSDMD) as the molecular effector of pyroptosis by two independent groups21,22 was rapidly followed by reports that GSDMD serves as an essential mediator of unconventional IL-1β release (reviewed in23,24).
Similar to IL-1β, GSDMD is a cytosolic substrate for proteolytic processing by caspase-1 and the resultant cleaved form of GSDMD rapidly assembles into large oligomeric pores within the plasma membrane (PM).25,26 The GDSMD pores directly mediate the efflux of cytosolic IL-1β protein27 but will also eventually collapse ionic and osmotic gradients across the PM to drive pyroptotic lysis. In the seven years since GSDMD was identified, many studies have provided compelling mechanistic support for molecular models of how GSDMD acts as a dual effector of both pyroptotic cell death and unconventional IL-1β export, the two known sequelae of inflammasome activation.25,28 However, it is important to note that Kayagaki and colleagues recently identified Ninjurin-1 (NINJ1) as an additional PM protein that integrates the ionic and osmotic perturbations elicited by GSDMD pores to coordinate the final process of PM rupture.29
1.2 |. Neutrophils as physiologically significant sources of IL-1β
Effective innate immunity involves coordination of overlapping, but non-redundant, inflammatory pathways in multiple immune effector cell types. As noted above, the functional coupling between pyroptosis and IL-1β release during inflammasome signaling was initially characterized—and remains most extensively characterized—in macrophages and other mononuclear myeloid leukocytes.23,27,30,31 However, granulocytic myeloid leukocytes, including neutrophils,31–35 mast cells,36–43 and eosinophils,44–48 are also important sources of IL-1β during multiple innate immune responses or in immune homeostasis. Given that neutrophils are the most abundant subtype of granulocyte, they are also the best characterized with respect to the assembly of diverse inflammasome platforms and modes of IL-1β release in response to diverse microbial infections, microbe-derived PAMPs (pathogen-associated molecular patterns) or host-derived DAMPs (danger-associated molecular patterns) as summarized in several recent reviews49,50 and discussed in more detail below.
Notably, several earlier studies had reported that activation of different signaling inflammasome platforms in neutrophils resulted in robust release of IL-1β in the absence of obvious pyroptotic lysis as assayed by LDH release. This included activation of NLRC4 inflammasome signaling in neutrophils infected with Salmonella typhimurium35 and activation of NLRP3 inflammasome signaling in neutrophils exposed to the pneumolysin exotoxin from Streptococcus pneumoniae51 or extracellular ATP as an agonist for ionotropic P2X7 receptors.33,52 That inflammasome-induced IL-1β release could be dissociated from pyroptosis in neutrophils ignited speculation regarding the underlying mechanisms and whether neutrophils lacked the machinery of pyroptotic induction. This speculation was soon informed by identification of GSDMD as a mediator of both IL-1β release and pyroptosis in macrophages. This motivated an initial wave of investigations regarding roles for GSDMD in neutrophil inflammasome signaling. These studies not only confirmed a key role for GSDMD in IL-1β export from inflammasome-active neutrophils but also reinforced a complex interplay between cytokine release, GSDMD subcellular trafficking, and progression to pyroptosis—or lack thereof—in these leukocytes.30,31,33,53–57 Moreover, two of those reports identified a role for GSDMD that involved coordinated permeabilization of azurophilic granules, the nuclear membrane, and the plasma membrane coincident with NETosis and release of extracellular DNA traps.54,56 Production of these extracellular traps via NETosis is per se an exceptionally active area of neutrophil biology research as summarized in recent reviews.58–60 This suggested GSDMD-dependent mechanistic links between (i) inflammasome signaling as the major pathway for regulated release of IL-1β from myeloid leukocytes; (ii) pyroptosis as a broadly expressed mode of pro-inflammatory lytic cell death; and (iii) NETosis as a signature innate immune response of neutrophils.
This review summarizes how subsequent interrogation of these links in a rapidly growing number of studies over the past three years has produced new insights, unexpected mechanistic nuances, and some controversies regarding the regulation of, and roles for, neutrophil pyroptosis in diverse innate immune responses. The reader is also referred to the excellent recent reviews by Perez-Figueroa et al.61 Rosazza et al.62 Yow et al.50 and Sollberger63 on this topic.
2 |. INFLAMMASOMES AND GASDERMINS AS CRITICAL REGULATORS OF PYROPTOSIS: EXPRESSION IN NEUTROPHILS
2.1 |. Inflammasome subtypes expressed in neutrophils
Canonical inflammasome platforms include: (i) a PYRIN interaction domain-containing initiator protein that changes conformation and self-oligomerization state upon binding particular PAMPs or sensing perturbations in cellular homeostatic parameters; (ii) the ASC adapter protein containing both PYRIN and CARD interaction motifs; and (iii) procaspase-1 as a CARD interaction domain-containing inflammatory effector protease. Details regarding the structural and functional characteristics of inflammasome signaling complexes are summarized in several recent reviews.4,64–66 In brief, oligomerized initiator protein complexes recruit ASC via PYRIN-PYRIN interactions with consequent formation of oligomerized ASC filaments. The latter recruit multiple procaspase-1 proteins via CARD-CARD interactions that facilitate proximity-induced autoproteolytic processing of the procaspase-1 molecules to generate highly active caspase-1 multimers. Seven subtypes of inflammasome platforms been identified based on the initiator protein sensors that include AIM2, CARD8, NLRC4, NLRP1, NLRP3, NLRP6, and Pyrin/MEFV. Chen et al.35 quantified mRNAs encoding these initiators in human blood neutrophils and murine bone marrow neutrophils and reported significant expression of AIM2, NLRC4, NLRP1, NLRP3, and pyrin/MEFV. Bakele et al.67 similarly described expression of mRNA transcripts for NLRP3, NLRC4, and AIM2 in highly purified preparations of human blood neutrophils.
AIM2 senses accumulation of cytosolic DNA derived from infecting pathogens or released from the host cell nucleus in response to DNA damage.68 NLRC4 acts in concert with NAIP adapters that bind PAMP ligands (flagellin, rod, and needle subunits) injected into the cytosol from bacteria that utilize type 3 secretion systems (T3SS) for delivery of their virulence factors.69 Depending on the host species, NLRP1 is conformationally activated by pathogen-derived factors that either mediate ubiquitination and limited proteosomal degradation of the NLRP1 N-terminal domain or direct cleavage of the N-terminus; these modifications liberate the NLRP1 C-terminal domain to recruit ASC.70 The NLRP3 initiator acts as a sensor of perturbed cellular homeostatic parameters that include: (i) rapid decreases in cytosolic [K+] triggered by activated host cell ion channels or microbial pore-forming exotoxins; (ii) disruption of mitochondrial or lysosomal integrity; and (iii) disorganization of the complex inter-membrane network between endosomes, the trans-Golgi apparatus and the endoplasmic reticulum.71,72 Pyrin (encoded by the MEFV/Familial Mediterranean Fever gene in humans) detects inhibition of RhoA GTPases by bacterial ADP-ribosyl transferase toxins with the resulting covalent modification disrupting the homeostatic interaction of RhoA with its downstream PKR-family kinase effectors. PKR normally phosphorylates pyrin to generate binding sites for the 14-3-3 protein that sustains pyrin in an inactive conformation; toxin modification of RhoA prevents this PKR/14-3-3 regulatory loop with consequent release of pyrin from the inhibitory complex for interaction with ASC.73,74
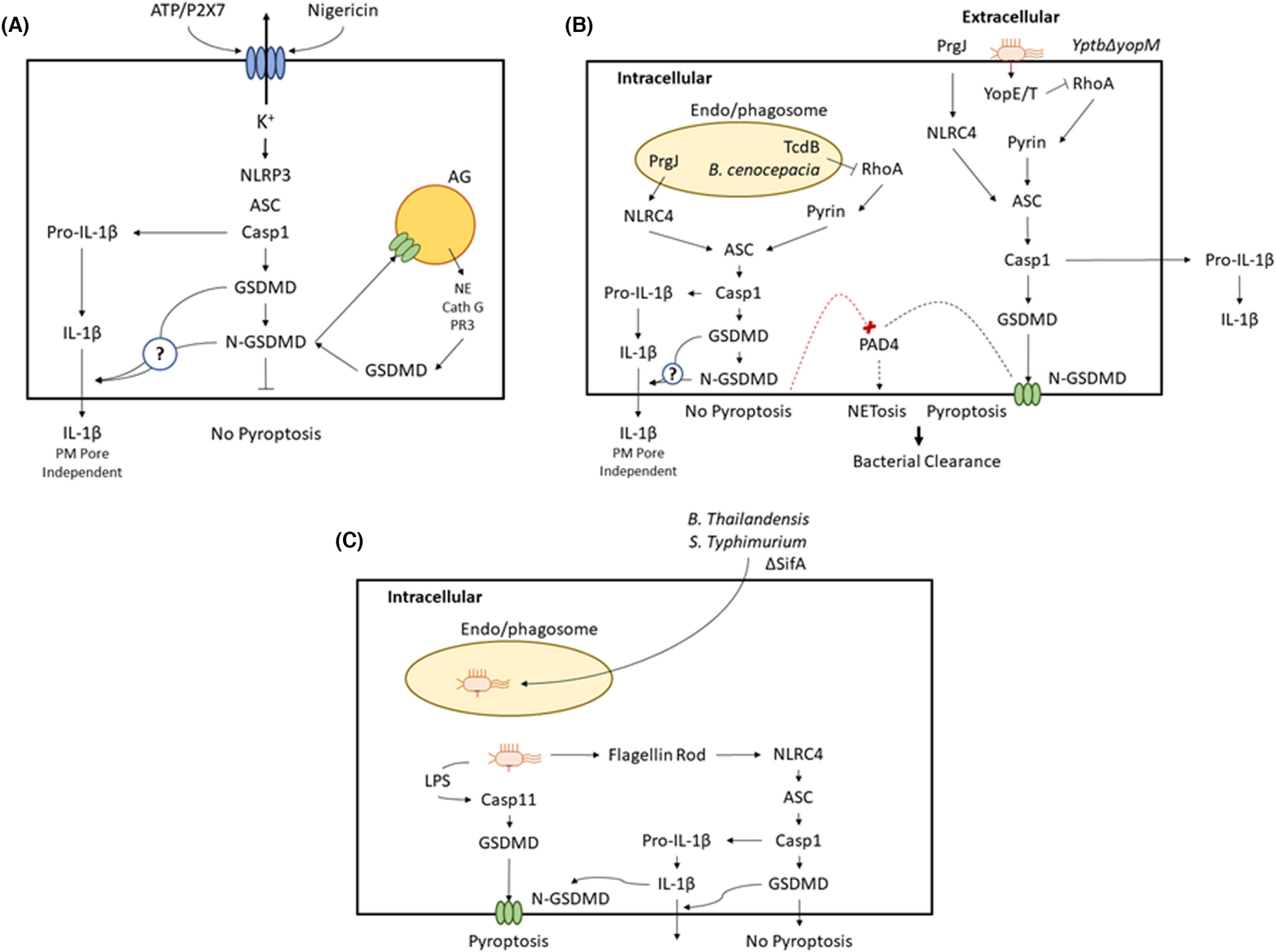
Subsequent studies have verified that neutrophils can assemble functional inflammasome platforms initiated by AIM2,75 NLRC4,35,75–79 NLRP1,75 NLRP3,32,34,51,57,75,80,81 and pyrin/MEFV.75,78 These are discussed in detail below regarding the differential induction of IL-1β release versus pyroptosis as downstream responses to inflammasome activation in neutrophils. Figure 1 illustrates the major subtypes of inflammasome platforms and their signaling pathways that have been described in neutrophils.
FIGURE 1.
Major subtypes of inflammasome signaling platforms and their downstream sequelae in neutrophils
2.2 |. Inflammasome-independent pathways for IL-1β processing, release, and extracellular accumulation by neutrophils
Caspase-1 is the canonical processing enzyme for proteolytic cleavage of 33 kDa proIL-1β precursor protein and consequent generation of the 17 kDa mature IL-1β cytokine.82 Indeed, caspase-1 was originally designated as “interleukin-converting enzyme” or “ICE” when first isolated and molecularly characterized.83,84 However, proIL-1β can also processed by multiple serine proteases to produce biologically active IL-1β products85; these include several serine proteases (neutrophil elastase, proteinase 3, cathepsin G) stored within the abundant azurophilic granules of neutrophils. Thus, upon exocytotic release, these serine proteases can process extracellular proIL-1β that may accumulate within infected or damaged tissue loci because of pyroptosis in resident macrophages or recruited neutrophils per se86 or release of NETs.87 In addition to their capacity for extracellular processing of proIL-1β, these serine proteases can cleave cytosolic proIL-1β upon release from the permeabilized azurophilic granules that may occur in senescent neutrophils and some models of bacteria-infected neutrophils.88 Roles for serine proteases that leak from compromised azurophilic granules in the regulation of constitutive (or spontaneous) neutrophil death signaling, including the processing and activation of neutrophil gasdermins, is discussed in detail as part of Section 3. Importantly, serine protease-mediated processing of IL-1β has been characterized in both human88,89 and murine neutrophils.88 The capacity of neutrophils to generate bioactive IL-1β by both canonical inflammasome/caspase-1 pathways and alternative serine protease pathways further underscores their role as a major source of this primary proinflammatory cytokine in diverse innate immune responses.
2.3 |. Gasdermin family proteins expressed in neutrophils
The Gasdermin (GSDM) family includes six human proteins (GSDMA, GSDMB, GSDMC, GSDMD, GSDME, and Pejvakin) and ten murine proteins (GSDMA1, GSDMA2, GSDMA3, GSDMC1, GSDMC2, GSDMC3, GSDMC4, GSDMD, GSDME, and Pejvakin). with GSDMD comprising the best-characterized family member.90,91 GSDMs are expressed as ~50 kDa cytosolic pro-proteins with N-terminal effector and C-terminal regulatory domains. The C-terminal domain maintains pro-GSDMs in an auto-inhibited conformation; however, disruption of this interaction by proteolytic cleavage induces conformational rearrangement of released N-GSDM fragments (reviewed in14,15,90) to expose sites for interaction with anionic phospholipids on accessible leaflets of membrane bilayers. This facilitates N-GSDM oligomerization and insertion into the targeted bilayer to assemble macropores (10–18 nm inner diameters). Assembly of N-GSDM pores in the plasma membrane markedly increases its permeability to macromolecules (up to 20 kDa) including proteins, metabolites, ions, and major osmolytes, resulting in rapid collapse of cellular integrity to facilitate pyroptosis.25,26,92 Notably, as the biological roles for GSDM-family proteins have expanded beyond inflammasome-activated GSDMD signaling in mammalian macrophages and other immune effector cells, so has the diversity of identified GSDM expression across organisms in different phyla,93,94 the diversity or proteases—beyond the canonical inflammatory caspases—capable of cleaving the different GSDMs,95,96 and the diversity of biological roles—beyond classical pyroptosis and cytokine release—for various GDSMs.90,91,97,98
Recent functional studies have demonstrated that human and murine neutrophils express high levels of GSDMD54,56,75,78,79,99–106 and GSDME78,102 protein; analyses of the possible expression of the other GSDM proteins in neutrophils have not been reported. Although comprehensive transcriptomic profiling of GSDM family expression in neutrophils has not been reported, our RNA-Seq analysis of FACS isolated (>99%) murine neutrophil populations revealed high gene expression of GSDME and GSDMD, but no other murine GSDM family members (E. Pearlman and G.R. Dubyak, unpublished data).
2.4 |. Biology of gasdermin D versus gasdermin E in pathways for unconventional cytokine release or cell death
Given the high expression of both GSDMD and GSDME in neutrophils, it is relevant to compare the distinguishing properties of these proteins. As noted previously, GSDMD is a major target for cleavage by caspase-1 as part of multiple inflammasome signaling pathways and N-GSDMD pores that accumulate in plasma membranes function as conduits for the direct efflux of mature IL-1β. Recent cryoEM and functional analyses of N-GSDMD pores have revealed electrostatic mechanisms that favor the flux of mature IL-1β but not proIl-1β.28,107 GSDMD is also a substrate for human caspases-4 and 5 and their murine homolog, caspase-11. These latter inflammatory caspases are effector components of so-called “non-canonical” inflammasomes. Assembly of non-canonical caspase-4/11 inflammasome platforms to response to direct binding of cytosolic LPS (delivered from LPS internalized via endosomes and released from endosomes via activity of guanylate-binding proteins) has been extensively characterized in human and mouse models21,22 and recently reviewed.108,109 LPS binding induces both oligomerization and catalytic activation of caspase-4/11 with consequent cleavage of gasdermin D (GSDMD) as a major substrate. Importantly, while both caspase-1 and caspase-4/11 efficiently cleave GSDMD to generate N-GSDMD pores for pyroptosis, proIL-1β is proteolytically processed by caspase-1 but not caspase-4/11. Caspase-4/11 induced accumulation of GSDMD pores in the plasma membrane (PM) can induce sublytic PM permeabilization to drive K+ efflux sufficient for secondary assembly/activation of NLRP3 inflammasomes and caspase-1-mediated production of mature IL-1β. LPS-mediated activation of the caspase-4/11 to GSDMD signaling axis underlies the mechanism for lethal sepsis induced by infection with Gram-negative bacteria. GSDMD is also a substrate for pro-apoptotic caspases-3 and 8. GSDMD is cleaved by the pro-apoptotic initiator caspase-8 to generate pore-component N-GSDMD products similar to those produced by the inflammatory caspases,110–113 Conversely, the pro-apoptotic effector caspase-3 cleaves GSDMD within its pore-forming N-terminal and thereby suppresses accumulation of functional N-GSDMD pores.114
GSDME is the most ancestral family GSDM member with variants expressed in lower vertebrates and functional characterization in zebrafish models.90 In humans, GSDME was initially identified as DFNA5, the product of a gene associated with autosomal non-syndromic hearing loss. Human and murine GSDME are most highly expressed in epithelial tissues, whereas in other organs it is expressed at lower levels. In mammalian cells, GSDME is proteolytically activated by the apoptotic executioner caspase-3 to drive cells from apoptotic cell death to secondary necrosis (similar to pyroptosis, which is defined as cell death mediated by GSDM family members).115 Rapid efferocytotic clearance of apoptotic cells usually ensures that apoptosis is non-inflammatory. However, inefficient clearance or overwhelming induction of apoptotic cells in tissues facilitates GSDME-mediated pyroptosis to drive local inflammation. In cancer models, tumor growth is suppressed by recruited tumor cell-targeting NK cells and CTLs, and GSDME in tumor cells is activated when the serine protease granzyme B (GzmB) is delivered from NK or CTL via exocytosis of granules that store GzmB and perforin,116 which induces GSDME processing either by GzmB-mediated activation of caspase-3 that then cleaves GSDME, or by direct cleavage of GSDME by the GzmB serine protease. Importantly, in inflammasome-activated myeloid cells that lack GSDMD, the accumulation of GSDME pores can serve as an alternative pathway for efflux of mature IL-1β.117,118
It is important to emphasize that neutrophils (and macrophages) can also release IL-1β and other IL-1 family cytokines by pyroptosis-independent mechanisms that are also GSDMD-independent. Monteleone et al.55 reported that activation of NLRC4 inflammasome signaling in murine neutrophils (from wildtype C57/Bl6 or Gsdmd−/− mice) induced two distinct phases of IL-1β export that were both coordinated by electrostatic association of mature IL-1β with phosphatidylinositol-4.5-bisphosphate (PIP2) microdomains in the inner leaflet of the plasma membrane (PM); these PIP2-enriched domains protruded from the membrane surface as ruffles or blebs. At early times post-NLRC4 activation, this PM-localized IL-1β export was mediated by a GSDMD-dependent mechanism. However, with sustained NLRC4 inflammasome activation, similar amounts of IL-1β were released from control and GSDMD-deficient neutrophils. This slower GSDMD-independent phase of IL-1β export might reflect release of the PIP2-and IL-1β-enriched PM protrusions into the extracellular compartment. We have previously reported that macrophages and dendritic cells can release mature IL-1β contained within either secreted exosomes or shed PM microvesicles.119,120 In a recent study, we demonstrated that neutrophils can also release exosomes as a pathway for unconventional secretion of IL-1 family cytokines. Ratitong et al.104 used a model of elicited murine peritoneal neutrophils stimulated with particulate β-glucan (a fungal PAMP) to show that IL-1α was predominantly released within exosomes and in similar amounts from control or GSDMD-deficient neutrophils. Thus, depending on stimulus conditions, neutrophils—like other myeloid leukocytes—can release IL-1-family cytokines by unconventional vesicular mechanisms (reviewed in121,122) that are independent of GSDMD and pyroptotic lysis.
2.5 |. Similarities and differences in human versus murine neutrophil signaling pathways for inflammasome regulation, IL-1β production, and pyroptosis
Most recent studies describing neutrophil pathways for inflamma-some regulation, IL-β production, and pyroptosis at the mechanistic level have used murine neutrophil models given the ready availability of genetic models that lack expression of particular inflammasome components, GSDMD or GSDME. Mouse models also facilitate analyses of how inflammasome signaling pathways at the cellular level are integrated into particular in vivo roles for neutrophils in the host responses to microbial infection or progression of sterile inflammatory diseases. Analyses in murine neutrophils can suggest how inflammasome pathways and pyroptosis might contribute to the functions of human neutrophils in physiological or disease states. However, it is critical to consider how engagement, or lack thereof, of specific inflammasome and pyroptotic signaling pathways may reflect fundamental differences in the innate immune repertoires of murine versus human neutrophils. Indeed, as emphasized by William Nauseef in another contribution to this volume123:
Human and murine neutrophils differ with respect to representation in blood, receptors, nuclear morphology, signaling pathways, granule proteins, NADPH oxidase regulation, magnitude of oxidant and hypochlorous acid production, and their repertoire of secreted molecules. These differences often matter and can undermine extrapolations from murine studies to clinical care, as illustrated by several failed therapeutic interventions based on mouse models. Likewise, coevolution of host and pathogen undercuts fidelity of murine models of neutrophil-predominant human infections.
In this regard, it is germane to briefly summarize the current understanding and salient features of inflammasome regulation, IL-1β production and pyroptosis in human neutrophils.
In one of the earliest studies, Bakele et al.67 reported expression of mRNA transcripts for NLRP3, NLRC4, and AIM2 in highly purified preparations of human blood neutrophils, as well as release of IL-1β in response to the NLRP3 agonists, ATP/nigericin, or the AIM2 agonist, cytosolic DNA. The authors also compared the expression levels of the inflammasome regulators and magnitudes of IL-1β release in human blood neutrophils versus the peripheral blood mononuclear cells (PBMC) in the absence or presence of LPS priming; monocytes are the predominant inflammasome active leukocytes and sources of IL-1β within the PBMC fraction. Expression of mRNAs for IL-1β and NLRP3 were 5-fold lower in neutrophils versus PBMC while ASC, AIM2, and NLRC4 transcripts were 2–10-fold higher in neutrophils. Notably, the magnitudes of IL-1β release in response to ATP/nigericin were ~300-fold lower (when normalized to cell number) in neutrophils versus PBMC. Thus, the authors emphasized that even modest contamination of isolated human blood neutrophils with monocytes can distort interpretation, at the quantitative or qualitative levels, of how particular inflammasome signaling pathways are functional within human neutrophils in response to microbial infections or exposure to proinflammatory PAMPs and DAMPs. Consequently, for ex vivo analyses of inflammasome signaling in human neutrophils it is critical to include additional immunomagnetic selection steps with neutrophil preparations isolated from peripheral human blood by standard gradient centrifugation protocols. Additionally, immunofluorescence (IF) assays at the single cell level or FACS analyses can further buttress support for engagement of particular inflammasome-related signaling reactions in human neutrophils; published examples include: (i) FACS analyses of ASC speck accumulation124–127; (ii) FACS analyses of P2X7 receptor expression as signal for NLRP3 inflammasome activation33; and (iii) IF analysis of the trafficking of cleaved N-GSDMD subunits to azurophilic granules rather than the plasma membrane during NLRP3 inflammasome signaling.57
With this technical caveat in mind, most studies on inflammasome signaling and IL-1β production in human neutrophils have focused on NLRP3-based pathways in response to diverse stimuli that include: (i) Pantin-Valentine leukocidin from Staphylococcus aureus (S. aureus)128; (ii) serum amyloid A129; (iii) infection by Helicobacter pylori130; (iv) monosodium urate crystals131; (v) infection by uropathogenic E. coli132; (vi) nigericin57,127; and (vii) ATP.33,57,67 NLRC4-based inflammasome signaling in human neutrophils can be induced in response to infection with Pseudomonas aeruginosa79 or Salmonella typhimurium.35
2.6 |. Differences in human versus murine neutrophil signaling pathways for IL-1β production and regulated cell death: The example of response to Staphylococcus aureus infection
In humans, S. aureus is commensal bacteria that can cause morbidity and mortality in various contexts; homeostatic control of this commensal microbe is mediated in large part by neutrophils.123,133 Given this commensal relationship, S. aureus has evolved multiple mechanisms to resist anti-microbial defenses of human neutrophils.134 In contrast, mice are not natural targets for S. aureus infection and respond to this bacterium as an opportunistic pathogen. Differential expression of particular genes in human versus murine neutrophils, or sequence differences in the human versus murine homologs of particular gene products, underlie the distinct innate immune responses, including IL-1β production, inflammasome assembly and regulated cell death, of human versus murine neutrophils to S. aureus infection.
Cho et al.135 reported that, in mice, recruited neutrophils are the major source of IL-1beta that accumulates at dermal sites of infection and that neutrophil-derived IL-1β is critical for control and eventual clearance of the S. aureus. Moreover, the IL-1β production required ASC/NLRP3 inflammasome assembly in the neutrophils as activated by the bacterial pore-forming α-toxin that can mediate K+ efflux. Although human neutrophils can assemble NLRP3 inflammasomes in response to other S. aureus pore-forming exotoxins, including the Pantin-Valentine leukocidin,128 neutrophils that accumulate viable S. aureus within intracellular organelles utilize a distinct signaling pathway to produce bioactive IL-1β. Kremserova and Nauseef reported that this involves a highly novel signaling axis whereby the RIP3 kinase induces IL-1β processing by neutrophil serine proteases independently of inflammasome-associated caspase-1.136 Notably, although sustained signaling by this pathway will induce lytic death of the neutrophils bearing intracellular S. aureus,137 the mature IL-1β rapidly produced during the initial phase of infection is released by a non-lytic and yet-to-defined mechanism. Despite the general association of RIP3 as a critical mediator of the necroptotic pathway of lytic cell death, the role of RIP3 in inducing acute IL-1β processing/release in human neutrophils, as well as eventual neutrophil death, was independent of the MLKL (mixed lineage-kinase-like) pore-forming protein that executes necroptotic lysis.137 Thus, depending on the context of S. aureus infection (extracellular or intracellular), human neutrophils can produce IL-1β for control of this commensal pathogen by engagement of either canonical caspase-1 inflammasome signaling or a novel serine protease-based mechanism that employs the RIP3 kinase as an upstream adapter protein. Given the expanding development of antibiotic-resistant strains of S. aureus that drive human disease,138 it is relevant to consider how anti-inflammasome therapeutics, such as NLRP3 antagonists,139 may be combined with novel RIP3 or serine protease inhibitors to control this commensal pathogen.
3 |. CONSTITUTIVE NEUTROPHIL CELL DEATH: ROLES FOR THE CANONICAL APOPTOTIC MACHINERY, AZUROPHILIC GRANULE-DERIVED SERINE PROTEASES, AND GASDERMINS WITH IMPLICATIONS FOR THE ANALYSIS OF NEUTROPHIL PYROPTOSIS
3.1 |. Constitutive cell death as the driver of brief lifespan in neutrophils
Any analysis of pyroptosis as a mode of regulated cell death in neutrophils is complicated by the well-known brief lifespan (12–20 h) of these leukocytes, which reflects their continuous generation in large numbers within the bone marrow and their rapid turnover upon release into the circulation. Granulocyte precursors comprise ~60% of the human bone marrow leukocyte pool with ~1011 new neutrophils produced daily for release into the circulation under homeostatic conditions.140 The predominant experimental models for investigation of neutrophil pyroptosis are human blood neutrophils, murine bone marrow neutrophils or murine neutrophils elicited to accumulate in the peritoneum. Regardless of model, the isolated neutrophils will have a limited duration of in vitro viability that reflects the homeostatic process of progressive apoptotic induction initiated in vivo as mature neutrophils age. This homeostatic pathway of regulated death has also been termed “spontaneous” or “constitutive” neutrophil death and the underlying pathways have been extensively studied and summarized in multiple reviews.141–143 Apoptosis comprises the major mode of constitutive death but with features unique to neutrophils. Neutrophils express most of the apoptotic caspases.144–147 In vitro characterizations of apoptotic signaling with isolated neutrophils have demonstrated they express the machinery for extrinsic apoptosis induced by cell surface death receptors including the cTRAIL receptors noted above, TNFα receptors,148,149 and Fas.150,151 Neutrophils also express many proteins associated with regulation or execution of the intrinsic mitochondrial pathway including the pro-apoptotic Bax inducer of mitochondrial outer membrane permeabilization/MOMP,147,152 the cytosolic sensor of Apaf-1 adapter sensor of released cytochrome c145 and several anti-apoptotic Bcl-2 family members with Mcl-1 playing a particularly critical role.148,152–154 Studies of mice with neutrophil-specific deletion of Mcl-1 revealed a severe defect in neutrophil survival154 while mice lacking Fas or Fas ligand exhibit only mild perturbations in neutrophil survival or production.155,156 Thus, the intrinsic apoptotic pathway appears predominant in constitutive neutrophil death even though neutrophils contain only a small content of mitochondria. Indeed, some investigators have proposed that apoptotic regulation rather than oxidative metabolism and ATP synthesis is the more critical role for neutrophil mitochondria.145,146 However, as discussed below, newer studies indicate among the various intracellular organelles in neutrophils, the lysosome-like azurophilic granules play may complement or exceed the role of mitochondria in the regulation of cell death.
Notably, recent studies have also demonstrated a coordinated circadian control of neutrophil functional states, including the expression of cell death regulatory genes, granule-sequestered inflammatory mediators, and chemokine receptors, as they exit the bone marrow to initiate the timer for the constitutive aging process.157 In the absence of infection or inflammatory stress, aging neutrophils in the circulation progressively acquire increased inflammatory potential but also increased expression of CXCR4 and pro-apoptotic factors such as TRAIL receptors, as well as decreased expression of anti-apoptotic factors such as MCL-1.158,159 The upregulated CXCR4 ultimately directs trafficking of the “fully aged” neutrophils back to the SDF-1/CXCL12 producing bone marrow niche wherein their modulated expression of apoptotic regulatory factors facilitates their rapid progression to terminal apoptosis for efferocytotic clearance by bone marrow macrophages. With acquisition of infection or sterile inflammation in peripheral tissues, circulating neutrophils will execute their signature rapid and robust trafficking into those tissues, but with a hierarchy determined by relative age such that older neutrophils will redirect their migration into the inflamed tissue rather the bone marrow.160 Classically, the recruited neutrophils will be activated, execute their anti-microbial and inflammatory functions, and eventually die (via diverse cell death modes) within the peripheral tissue niche for clearance by resident macrophages.142,143
It is highly relevant to consider how the parameters that define circadian oscillations in the progression of aging and constitutive death of neutrophils in vivo might impact the design, execution and interpretation of ex vivo experiments aimed at characterization of pyroptosis in these leukocytes. Individual cells within the primary neutrophil populations isolated from human blood, mouse bone marrow, or elicited murine peritoneal leukocytes will necessarily be heterogenous regarding their relative age and progression along the spontaneous cell death pathway. As indicated previously, this progression is defined by modulated expression of various apoptotic regulatory factors that likely synergize to decrease the threshold for productive initiation of the intrinsic apoptotic signaling cascade. Those neutrophils with such decreased apoptotic thresholds may be significantly more likely to die by cell lysis when challenged by pro-pyroptotic stimuli, particularly in the absence of efferocytotic clearance by macrophages as will be the situation in ex vivo experiments with purified neutrophils. This speculation is consistent with recent studies reporting significant heterogeneity in pyroptotic progression by isolated neutrophil populations in response to diverse microbial or sterile stimuli that will be discussed in detail below.
3.2 |. Roles for azurophilic granules, neutral serine proteases, and serine protease inhibitors in the progression of constitutive neutrophil death
A defining phenotypic characteristic of neutrophils is their high content of intracellular granules that include primary azurophilic granules, secondary specific granules, gelatinase granules and secretory vesicles.161 Each granule subtype is distinguished both by its complement of resident membrane proteins and intragranular content of molecules and mediators. Azurophilic granules are lysosome-like organelles that contain multiple neutral serine proteases including neutrophil elastase (NE), proteinase 3 (PR3), and cathepsin G (CG).162 Canonically, these serine proteases play multiple physiological or pathological roles in innate immunity when delivered into extracellular compartments by exocytotic fusion of azurophilic granules with the plasma membrane, or into phagosomal compartments that may also fuse with the granules. However, these proteases can also be released into the neutrophil cytosol by conditions that compromise integrity of azurophilic granules. The activity of serine proteases within extracellular or intracellular compartments is opposed by diverse group of endogenous inhibitory proteins collectively termed as serpins (serine protease inhibitors).163 Neutrophils express high nucleo-cytosolic levels of serpinB1a and serpinB6.164 Studies by several groups have revealed that the thresholds for constitutive neutrophil death are markedly affected by perturbation of azurophilic granule integrity in combination with altered expression levels of the serpins.100,165–167 Benarafa and colleagues reported that mice with genetic ablation of serpinB1a displayed severe neutropenia reflecting a neutrophil-autonomous increase in cell death, as well as a reduced ability to clear bacteria.167 Although serpinB1a can suppress each of the three neutrophil serine proteases with varying efficacy, the increased neutrophil death and consequent neutropenia was prevented when serpinB1a deficiency in mice was combined with deletion of cathepsin G but not neutrophil elastase. Additionally, because the increased rate of death of serpinB1a-deficient neutrophils in ex vivo analyses was only modestly attenuated by a pharmacologic pan-caspase inhibitor, the study indicated that cytosolic cathepsin G may directly act as a death effector protease analogous to caspase-3.
Luo and colleagues used a model wherein isolated human blood neutrophils or elicited murine peritoneal neutrophils were incubated ex vivo for up to 48 h with tracking of various indicators of apoptosis, including binding of annexin V to phosphatidylserine flipped to the extracellular leaflet of the plasma membrane.165,166 Within 16 h, most neutrophils were positive for annexin V binding and this apoptotic progression correlated with increased accumulation of active caspase-3 effector protease and was markedly delayed by ~50% in the presence of a pan-caspase inhibitor but not inhibition of caspase-8 or caspase-9, the respective initiator caspases for extrinsic or intrinsic apoptosis. Rather, the constitutive apoptotic progression was induced by gradual leakage of PR3 serine protease from azurophilic granules into the cytosol with consequent proteolytic cleavage and activation of caspase-3 and also correlated with progressive decrease in the cytosolic content of serpinB1a. This progression was markedly delayed when the same experiments were performed with neutrophils isolated from proteinase 3-knockout mice or when control human or murine neutrophils were incubated with pharmacologic serine protease inhibitors. In contrast, the constitutive death was accelerated when the ex vivo experiments used neutrophils from serpinB1a-deficient mice Thus, constitutive neutrophil death is mediated by in large part by progressive cytosolic accumulation of the cathepsin G and proteinase 3 serine proteases in excess of counteracting inhibitory serpinB levels. These serine protease-driven pathways of neutrophil death can intersect with elements of the canonical apoptotic machinery, as with PR3 activation of caspase-3, but also drive caspase-independent death cascades.
3.3 |. Roles for gasdermin D in constitutive neutrophil cell death progression
Subsequent studies by the two groups who characterized roles for azurophilic granule-derived serine proteases in constitutive neutro-phil cell death identified gasdermin D as an additional substrate for proteolytic activation by these serine proteases. Kambara et al.99 initially used a mouse model of acute bacterial peritonitis (intraperitoneal injection with Gram-negative E. coli) to compare the ability of wildtype (WT) versus GSDMD-deficient (Gsdmd−/−) mice to control and clear the infection. WT mice utilize multiple innate immune responses to first suppress E. coli proliferation and then clear residual bacteria to minimize dissemination to peripheral tissues. Unexpectedly, Gsdmd−/− mice were significantly more efficient in reducing the numbers of viable E. coli and suppressing dissemination. This effect of gasdermin D deficiency to enhance in vivo host bactericidal activity was linked to enhanced accumulation of viable neutrophils within the infected peritoneum due to delay in the death of neutrophils that typically occurs in the course of executing their microbe killing roles.142,143 Experiments using isolated murine neutrophils incubated in vitro for increasing times verified that cultures of GSDMD-deficient neutrophils contained 40% more viable cells at 24 h than cultures of WT neutrophils. The accelerated decrease in viability of the cultured WT neutrophils reflected progressive cleavage of GSDMD to produce accumulation of the membrane pore-forming N-terminal domain fragments as the azurophilic granules gradually lost integrity and released serine proteases into the cytosol. Biochemical experiments indicated that GSDMD cleavage was predominantly catalyzed by released neutrophil elastase at a site seven amino acids upstream of the canonical Asp-275 (human)/Asp-276 (murine) residue targeted by caspase-1/4/5/11. Ectopic expression in HEK293 cells of the N-GSDMD product produced by elastase induced robust LDH release indicative of pyroptotic lysis. Thus, the gradual loss of azurophilic granule integrity that defines aging neutrophils can facilitate their constitutive death by two parallel pathways involving the proteinase 3-mediated accumulation of active caspase-3 and elastase mediated accumulation of active N-GSDMD pores.
In a follow-up to the Benarafa group’s identification of azurophilic granule-released cathepsin G in the cytosol as a major driver of constitutive neutrophil death, Burgener et al.100 observed that this serine protease also efficiently cleaves GSDMD in murine neutrophils at a Leu-274 residue upstream of the Asp-276 site for caspase-1//4/5/11 proteolysis. However, in contrast to the findings of Kambara et al.99 with elastase-cleaved GSDMD, this cleavage of neutrophil GSDMD by cathepsin G did not significantly accelerate constitutive death of isolated neutrophils cultured ex vivo. Moreover, co-deletion of GSDMD with serpinB1a did not reduce the severe neutropenia that characterize serpinB1a knockout mice. Proteinase 3 and elastase (also released from leaky azurophilic granules) also cleaved GSDMD but into multiple fragments that were rapidly degraded. The underlying reasons for the differential roles of various serine proteases in GSDMD processing as reported by Kambara et al.99 versus Burgener et al.100 remain unclear. Regardless, the ability of these non-caspase proteases to mediate GSDMD processing in neutrophils provides additional perspective for appreciating how pyroptotic regulation in these cells can significantly diverge from the canonical mechanisms established in macrophages.
4 |. DISSOCIATION OF IL-1b RELEASE FROM PYROPTOSIS IN NEUTROPHILS: PERSPECTIVE FROM RECENT INSIGHTS USING CANONICAL MACROPHAGE MODELS AND CONSIDERATION OF NEUTROPHIL-SPECIFIC BIOLOGY
4.1 |. New mechanistic insights regarding the complex regulation of inflammasome signaling and pyroptosis in myeloid leukocytes
The paradigm for canonical inflammasome signaling, as mainly defined in macrophages and dendritic cells, is that IL-1β release and pyroptosis represent parallel functions of the caspase-1-generated N-GSDMD pores that form in plasma membranes (PM).23,27,31 However, this mode has been refined by recent mechanistic nuances that include: (i) suppression of pyroptosis by membrane repair mechanisms involving removal of PM N-GSDMD pores168; (ii) the ability of pro-apoptotic caspase-8 to cleave both proIL-1β and GSDMD110,113,169; (iii) the ability of N-GSDMD pores to permeabilize mitochondria and drive apoptotic signaling170–172; (iv) the ability of caspase-1 to drive apoptotic signaling cascades leading to gasdermin E (GSDME)-mediated IL-1β release117,118,173 and secondary pyroptosis53 in GSDMD-deficient macrophages; and (v) modulation of functional N-GSDMD pore assembly in the PM by the Ragulator/Rag small GTPase module canonically associated with mTOR signaling.174 It is particularly noteworthy that GSDMD-mediated IL-1β export can be uncoupled from pyroptosis in murine dendritic cells and macrophages in which atypical inflammasome signaling is activated by oxidized phosphocholine lipid metabolites.175,176 Sustained IL-1β release from these so-called hyperactive but viable dendritic cells that have trafficked to the tumor-draining lymph nodes in implanted mouse cancer models is sufficient to facilitate polarization of anti-tumor T cells for durable suppression of tumor growth in vivo.177,178
4.2 |. Roles for neutrophil-specific phenotypic characteristics in the regulation of inflammasome signaling and pyroptosis
It is important to consider how these recently refined mechanisms for how GSDMD or GSDME can differentially regulate IL-1β release and pyroptosis in macrophages, might be utilized and further adapted by neutrophils to provide alternative pathways for secretion of bioactive IL-1β coupled with, or dissociated from, lytic cell death responses. Neutrophils are defined by their ability to execute non-redundant innate immune functions distinct from those executed by macrophages or dendritic cells. While tissue resident macrophages are the earliest myeloid first responders to bacterial (and other microbial) pathogens, the rapid, massive recruitment of neutrophils to the sites of infection makes them the predominant myeloid cell type and major source of IL-1β in the secondary phase of an innate immune response. As discussed previously, neutrophils can process and release mature IL-1β via the standard intracellular inflammasome/caspase-1 cascades. However, this can be reinforced by their capacity for exocytosis of azurophilic granule serine proteases, including elastase, cathepsin G and proteinase 3, that can process extracellular proIL-1β, released from dying neutrophils or macrophages, into bioactive IL-1β.85,86 Additionally, these serine proteases, when released into the cytosol from leaky granules, can cleave intracellular proIL-1β and, as noted previously, GSDMD.54,99,100 The expression of specialized machinery that facilitates NETosis provides neutrophils and other granulocytes with an additional pathway to employ externalized DNA and granule proteins as extracellular traps for microbial immobilization and killing.59,179,180
Neutrophils play important roles in the control and clearance of bacteria that utilize macrophages as a protected replicative niche. GSDMD-mediated pyroptosis of the infected macrophages serves both to release IL-1β (and other mediators) as an efficacious neutrophil attractant and activator and to trap bacteria within the macrophage corpses.181,182 This facilitates efficient efferocytosis of the dead macrophages and entrapped bacteria by the recruited neutrophils.183 Because the overall efficacy of this bacterial clearance mechanism would be greatly reduced by pyroptosis of the recruited neutrophils, it is teleologically plausible to consider evolutionary selection of mechanisms that favored attenuation of pyroptotic progression in neutrophils while preserving their ability to produce and release IL-1β for feed-forward amplification of the neutrophil recruitment process. Conversely, control of bacterial pathogens that utilize neutrophils as an abundant replicative niche will enhanced by signaling pathways that license neutrophil pyroptosis to release the microbes for killing by alternative innate immune mechanisms. Indeed, new investigations that are reviewed below provide strong support for stimulus-specific mechanisms that allow neutrophils to release bioactive IL-1β while either resisting or engaging pyroptotic progression. These mechanisms involve a remarkable interplay between some of the defining features of neutrophil cell biology and the neutrophil responses to virulence factors employed by bacterial pathogens to subvert the diverse neutrophil-mediated anti-microbial pathways.
5 |. NEUTROPHIL INFLAMMASOME SIGNALING PATHWAYS THAT FACILITATE RELEASE OF IL-1b WITH HIGH RESISTANCE TO PYROPTOSIS
5.1 |. NLRP3/caspase-1 inflammasome signaling in neutrophils
In contrast to most inflammasome initiator/sensors that are stimulated in response to direct binding of PAMPs or the activity of virulence factor enzymes, NLRP3 undergoes conformational activation in response to perturbation of fundamental host cell homeostatic parameters with decreased cytosolic [K+] being the best-characterized.71 Although the obligatory K+ efflux can be rapidly induced by the K+/H+ exchanger ionophore nigericin as a convenient investigational tool, this stimulus lacks physiological relevance. Physiological stimuli include endogenous agonists that gate the opening of host cell K+-permeable channels or pore-forming exotoxins produced by certain extracellular bacteria. Extracellular ATP-gated P2X7 receptor channels are the best-characterized endogenous activators of NLRP3 inflammasome signaling,121,184,185 while pneumolysin from Streptococcus pneumoniae is a validated pore-forming toxin that drives K+ efflux-mediated NLRP3 activation.51,185,186 We reported that P2X7 receptor stimulation33,57 and pneumolysin,51 as well as nigericin33,57 induce NLRP3 inflammasome activation in murine and human neutrophils to drive robust release of IL-1β but not LDH as an index of pyroptosis. Heilig et al.31 similarly observed significant IL-1β secretion from nigericin-stimulated neutrophils that was dependent on GSDMD but with minimal loss of viability. We also found that the ability of ATP, nigericin or pneumolysin to induce IL-1β release was correlated with cleavage of GSDMD in wildtype neutrophils and was suppressed in neutrophils from GSDMD-knockout mice.57
Other studies have reported that limited accumulation of N-GSDMD pores in the plasma membrane of dendritic cells can be sufficient to mediate significant IL-1β efflux but insufficient to drive pyroptotic lysis.27,31 These findings were predicated on the observation that sublytic numbers of plasma membrane GSDMD pores in these dendritic cells were sufficient to mediate uptake of propidium, a normally impermeant small organic divalent cation, that forms a fluorescent adduct upon binding to intracellular DNA. Thus, the GSDMD pores functioned as conduits for efflux of intra-cellular IL-1β and influx of extracellular propidium, but not sufficient drivers of pyroptotic lysis. In contrast, we found that caspase-1-derived N-GSDMD products in NLRP3-inflammasome activated neutrophils did not significantly localize to the plasma membranes to form propidium-permeable pores or drive pyroptosis but were required for IL-1β secretion.57 This absence of neutrophil plasma membrane pores correlated with alternative subcellular trafficking whereby N-GSDMD predominantly associated with the very abundant azurophilic secretory granules and also with LC3+ autophagosomes. N-GSDMD trafficking to the azurophilic granules caused leakage of neutrophil elastase into the cytosol, resulting in secondary cleavage of GSDMD to an alternative N-GSDMD product. Analyses using ATG7-deficient neutrophils indicated that IL-1β is secreted via an autophagy machinery-assisted mechanism. These findings revealed fundamental differences in N-GSDMD trafficking between neutrophils and macrophages/dendritic cells that may explain the neutrophil-specific resistance to pyroptotic progression during canonical inflammasome activation. In this model proposed by Karmakar et al.57 abundant intracellular organelles within neutrophils act as sinks for the predominant binding of N-GSDMD cleavage products as they generated by the active caspase-1 associated with the oligomerized NLRP3 inflammasome platforms. The high number and tight packing of the neutrophil granule subtypes may additionally act as a diffusional barrier that retards trafficking of the N-GSDMD products to the inner leaflet of the plasma membrane.
Notably, a very recent study by Chauhan et al.75 reported that ATP-or nigericin-induced activation of NLRP3 inflammasomes in murine neutrophils resulted in a GSDMD-dependent secretion of IL-1β but also GSDMD-dependent uptake of SytoxGreen (a DNA-intercalating dye similar to propidium) indicative of accumulated N-GSDMD pores in the plasma membrane. However, the number of SytoxGreen+ neutrophils in the stimulated conditions was quite modest (~15% vs. 5–7% in unstimulated neutrophils) even after 100 minutes. Nigericin stimulation for 100 minutes also resulted in extracellular accumulation of GAPDH (a 36 kDa cytosolic protein) indicative of pyroptotic lysis. Chauhan et al.75 emphasized that “although these inflammasome stimuli induced consistent and time-dependent SytoxGreen incorporation in neutrophils, the observed signal appeared less prominent than usually observed in macrophages.” Thus, relative to the “N-GSDMD sink” model proposed by Karmakar et al.57 the observations of Chauhan et al.75 would support a scenario wherein some N-GSDMD fragments can diffuse through the packed network of intracellular granules to drive accumulation of plasma membrane N-GSDMD pores sufficient to support pyroptotic progression in a limited number (~10%) of the neutrophil population. As noted in Section 3.1, individual cells within the primary neutrophil populations isolated from mouse bone marrow will be heterogenous regarding their relative age and progression along the spontaneous apoptotic cell death pathway. Older neutrophils with a lower apoptotic threshold may progress to cell lysis when challenged by pro-pyroptotic stimuli for sustained durations. Moreover, the observed ability of NLRP3 inflammasome-generated N-GSDMD to induce permeabilization of azurophilic granules and release of serine proteases into the cytosol raises the possibility that these proteases can drive feed-forward acceleration of the processing of both GSDMD and GSDME (the latter via a proteinase 3 > caspase-3 > GSDME cascade). As discussed below, studies with other inflammasome platforms in neutrophils indicate that GSDMD and GSMDE can be differentially engaged by parallel signaling pathways to additively drive pyroptosis.
5.2 |. NLRC4/caspase-1 inflammasome signaling in neutrophils
Chen et al.35 first reported that infection of neutrophils with Salmonella enterica serovar Typhimurium triggered NLRC4 inflammasome assembly to drive caspase-1 mediated IL-1β release in the absence of LDH release indicative of pyroptosis. Several subsequent studies have investigated the underlying mechanisms with particular emphasis on the role of GSDMD as a target for NLRC4-triggered caspase-1 and as a mediator of IL-1β export.56,77 Chen et al.56 confirmed that infection of neutrophils with Salmonella induced caspase-1 activation and IL-1β release in the absence of pyroptosis, but observed that active caspase-1 only weakly processed GSDMD in comparison with caspase-11; similar results were obtained when neutrophils were transfected with purified flagellin as a direct activator of NLRC4. Kovacs et al.77 employed a modified approach to deliver a different PAMP activator of NLRC4 into neutrophils; this was a chimeric protein (PrgJ-Tox) comprising PrgJ, a component of the type 3 secretion system needle, and Anthrax lethal toxin; the lethal toxin component facilitates very efficient cytosolic delivery of the chimera. This resulted in robust activation of caspase-1 mediated processing of GSDMD and GSDMD-dependent release of IL-1β but not LDH. In contrast, treatment of macrophages with PrgJ-Tox elicited a coordinated caspase-1- and GSDMD-dependent release of both IL-1β and LDH. Together, these findings indicate that, despite the ability of the NLRC4-caspase-1 axis to drive robust GSDMD processing and GSDMD-dependent IL-1β release in neutrophils, GSDMD does not induce pyroptotic cell lysis. Importantly, while defining this resistance of neutrophils to GSDMD-dependent pyroptosis downstream of canonical inflammasomes that employ caspase-1 as the effector protease, both Chen et al.56 and Kovacs et al.77 subjected neutrophils to diverse stimuli that result in accumulation of cytosolic LPS, the driver of non-canonical inflammasome platforms that employ caspase-4 (human) or caspase-11 (murine) as the effector proteases. As described below, these stimulus conditions resulted in caspase-11-mediated processing of GSDMD that induced lytic death of the neutrophils via pyroptosis or NETosis.
6 |. NEUTROPHIL INFLAMMASOME SIGNALING PATHWAYS THAT FACILITATE RELEASE OF IL-1b WITH LOW OR CONDITIONAL RESISTANCE TO PYROPTOSIS
6.1 |. Non-canonical caspase-11 inflammasome signaling in neutrophils
The studies of Kovacs et al.77 were framed within the larger question of how mice control infection by Burkholderia thailandensis as a model intracellular bacterium that uses its type 3 secretion system to escape from phagosomes into the cytosol. Earlier investigations had established that wildtype mice effectively control this pathogen by a caspase-11-dependent mechanism, but that the bacteria also engage NLRC4-caspase-1 signaling platforms.187,188 After establishing that neutrophils are critical immune effector cells for control of B. thailandensis, Kovacs et al.77 compared infection with B. thailandensis in neutrophils isolated from wildtype, caspase-1-deficient, or caspase-11-deficient mice. Similar GSDMD processing was observed in all three genotypes indicating that GSDMD was effectively targeted by both caspase-1 and caspase-11. As expected, IL-1β release was strictly dependent on NLRC4-regulated caspase-1; however, caspase-11, but not caspase-1, was required for pyroptotic lysis. The absence of pyroptosis in the caspase-11-knockout neutrophils correlated with robust replication of the intracellular bacteria. When LPS was directly transfected into the three neutrophil genotypes, IL-1β release was detected only in wildtype cells because intracellular LPS cannot activate the NLRC4-caspase-1 axis while caspase-11 cannot process proIL-1β. Thus, engagement of the NLRC4-caspase-1 pathway was sufficient to drive GSDMD processing for robust release of IL-1β but not pyroptotic lysis. Parallel processing of GSDMD by LPS-activated caspase-11 was obligatory for effective neutrophil pyroptosis. Together, these findings demonstrate coordinate activity of these neutrophil inflammatory caspases in a physiologically relevant model of infection.
Chen et al.56 also infected neutrophils with either of two Gram-negative bacteria species that escape the vacuole compartment to establish cytosolic residence, Citrobacter rodentium and the Salmonella ΔsifA strain, as well as transfection with LPS, to interrogate roles for the caspase-11/GSDMD signaling axis in cell death and control of bacterial growth. Intracellular LPS induced robust release of IL-1β and LDH that correlated with cleavage of GSDMD. These effects were markedly attenuated in neutrophils from mice with genetic deletion of capase-11 or GSDMD. The intracellular LPS-induced stimulation of IL-1β release reflected secondary activation of NLRP3 inflammasomes via K+ efflux mediated by caspase-11-induced accumulation of plasma membrane GSDMD pores. Notably, responses to the LPS/caspase-11/GSDMD signaling cascade correlated with induction of late-stage NETosis as indicated by release of extracellular DNA, citrullinated histone H3, and myeloperoxidase. These indices of NETosis were absent in Casp11−/− and Gsdmd−/− neutrophils. In addition to driving late-stage NETosis, caspase-11, and GSDMD were required for key early signaling events that initiate the NETosis cascade including disruption of nuclear membrane integrity, cleavage of intranuclear histone H3, chromatin decondensation, and expansion of nuclear volume. Together, these observations supported a model wherein caspase-11-mediated production of N-GSDMD facilitates coordinated accumulation of N-GSDMD pores in the nuclear membrane, granule membranes, and plasma membrane. The nuclear membrane N-GSDMD pores mediate influx of active caspase-11 into the intranuclear compartment to cleave histone H3 and thereby facilitate chromatin decondensation and mixing of the nuclear, azurophilic granule, and cytosolic compartments prior to expulsion of chromatin, granule proteins, and cytosolic proteins (e.g., LDH) via the compromised plasma membrane barrier driven by accumulating N-GSDMD pores. Human neutrophils infected with C. rodentium and murine neutrophils infected with Salmonella ΔsifA displayed a similar NETotic phenotype which was absent in Casp11−/− or Gsdmd−/− neutrophils; these deficient cells were also characterized by increased numbers of intracellular bacteria. Thus, activation of non-canonical caspase-11 inflammasomes in neutrophils elicits a lytic cell death phenotype with mixed features of pyroptosis and NETosis. In a separate study, Oh et al.103 reported that neutrophils infected with a mutant Shigella flexneri strain that lacks OspC3, a type 3 secretion system (T3SS) virulence factor, engaged a similar caspase-11 > GSDMD pathway to drive pyroptosis and loss of the replicative niche in neutrophils. However, wildtype S. flexneri that express OspC3 suppressed this signaling axis via OspC3 binding to, and inhibition of, caspase-11. This suppression underlies, in part, the ability of S. flexneri to evade host defenses for systemic dissemination.
6.2 |. Canonical NLRC4/caspase-1 or Pyrin/caspase-1 inflammasome signaling in neutrophils infected with extracellular pathogenic bacteria
Recent studies have investigated how infection of neutrophils with extracellular bacterial pathogens can induce assembly of canonical caspase-1-based inflammasomes that facilitate robust IL-1β release but with conditional resistance to pyroptosis. Remarkably, this conditional resistance to caspase-1-mediated pyroptosis predominantly reflects the cellular route by which bacterial PAMPs and/or virulence factor exotoxins are delivered into the neutrophil cytosol.
Santoni et al.79 infected murine neutrophils with Pseudomonas aeruginosa, an extracellular pathogen that uses a T3SS to deliver NLCR4-activating PAMPs (flagellin and T3SS injection needle components) into the cytosol. The delivery routes include direct injection through the plasma membrane by extracellular bacteria (which is predominant) or injection across the phagosomal membrane by internalized bacteria. Multiple P. aeruginosa strains induced release of both IL-1β and LDH; these responses were greatly attenuated in neutrophils that lacked expression of NLRC4, caspase-1 or GSDMD. Strains lacking the ExoS ADP-ribosyltransferase triggered more robust responses; this indicates that ExoS somehow limits assembly of functional NLRC4 inflammasomes. Thus, P. aeruginosa, in contrast to Salmonella enterica serovar Typhimurium,35,56 can direct NLRC4 inflammasomes to utilize caspase-1 for both IL-1β processing/release and GSDMD-mediated pyroptosis. Given the apparent stimulus type-dependent involvement of GSDMD in regulating NETosis,56 Santoni et al.79 tested the ability of P. aeruginosa to engage this signaling axis. They found bacterial infection induced citrullination of histone H3 and decondensation of nuclear DNA and these responses required NLRC4, caspase-1 and GSDMD. Furthermore, the H3 histone citrullination and DNA decondensation required Ca2+ influx through the plasma membrane GSDMD pores to activate PAD4 (protein arginine deaminase 4) which catalyzes citrullination. However, despite the ability of this signaling axis to coordinate pyroptosis and DNA decondensation, the pyroptotic neutrophils retained the expanded chromatin network and did not expel it into the extracellular compartment as NETs.
We recently demonstrated that P. aeruginosa activates the NLRP3 rather than the NLRC4 inflammasome in murine neutrophils (though not macrophages) when infected with Pseudomonas aeruginosa expressing the ExoS-ADPRT virulence factor delivered via the T3SS.189 A critical factor in determining this skewing to NLRP3-versus NLRC4-inflammasomes (as described by Santoni et al.79) is priming the neutrophils with LPS before infection, or using elicited neutrophils from the inflamed peritoneal cavity, both of which conditions upregulate NLRP3 expression. We also observed that GSDMD is required for IL-1β release and pyroptosis in the P. aeruginosa-infected neutrophils but is entirely dispensable for NETosis.
Yersinia species predominantly function as extracellular bacteria that also use their contact-dependent T3SS to inject a wide variety of Yersinia outer protein (Yop) virulence effectors into host cells.190 Different Yop effectors will target particular host cell innate immune signaling components including those which regulate inflammasome assembly or activity.74 For example, YopJ inhibits the kinase components that couple Toll-like receptors to the NFκB pathway required for transcriptional induction of anti-apoptotic factors and inflammatory cytokines including proIL-1β. YopE and YopT suppress activity of the RhoA small GTPase required for phagocyotic sequestration/killing of the bacteria. However, as noted previously, RhoA is also a regulator of the pyrin inflammasome by acting as negative regulator of PKR-family kinases that phosphorylate pyrin to generate interaction sites for 14-3-3 protein that sustains pyrin in an inactive conformation. Thus, myeloid cells can counteract the suppressed phagocytosis caused by YopE/YopT inhibition of RhoA by depressing 14-3-3/pyrin complexes to liberate pyrin for auto-oligomerization and recruitment of ASC and caspase-1 and consequent cleavage of proIL-1β and GSDMD; in macrophages this signaling axis is sufficient to drive pyroptosis.191 However, in the “arms race” with the host, Yersinia utilize YopM to sustain PKR-mediated phosphorylation of pyrin in its inactive complex with 14-3-3 and thereby attenuate caspase-1 mediated IL-1β release and GSDMD-dependent pyroptosis.
Oh et al.78 recently adapted this model to investigate how neutrophils engage Yersinia infection via inflammasome and pyroptotic signaling pathways. This is germane because neutrophils comprise a highly abundant cell type recruited to sites of Yersinia infection.192,193 The inflammasome and pyroptotic responses of LPS + interferon-γ-primed murine neutrophils to infection with a control Yersinia pseudotuberculosis strain (Yptb-WT) versus a strain lacking YopM (Yptb-ΔyopM) were compared. As expected, neutrophils infected with Yptb-ΔyopM produced and released ~10-fold greater amounts of mature IL-1β than Yptb-WT-infected cells. Although a modest caspase-1-independent LDH release indicative of pyroptosis was observed in neutrophils infected with either strain, the lytic response to Yptb-ΔyopM was 50% greater and the additional death was mediated by caspase-1. The caspase-1-independent neutrophil lysis observed with both strains reflected engagement of a caspase-8 > caspase-3 > GSDME pyroptotic cascade previously described in Yersinia-infected neutrophils by Chen et al.102 Notably, Oh et al. observed that the modest neutrophil lysis induced by Yptb-WT was not reduced in GSDMD-deficient cells, but was completely suppressed in neutrophils deficient in GSDME or doubly deficient in GSDME and GSDMD. The additional cell lysis induced by Yptb-ΔyopM was reversed in GSDMD knockout neutrophils but not GSDME-deficient cells. In contrast, the amplified release of IL-1β stimulated by Yptb-ΔyopM was not inhibited in either GSDMD-deficient neutrophils or GSDME-deficient cells but was markedly suppressed in neutrophils doubly deficient in GSDME and GSDMD. Together, these findings indicate that pyrin-dependent caspase-1 drives neutrophil pyroptosis via engagement of parallel signaling pathways that result in accumulation of both N-GSDMD and N-GSDME membrane pores; each complement of pores is sufficient for maximal IL-1β release such that ablation of both complements is required to significantly attenuate export of the mature cytokine.
Given that other active caspase-1 inflammasomes in neutrophils drive GSDMD-dependent IL-1β export in the absence of plasma membrane N-GSDMD pores and consequent pyroptosis (see Section 5), Oh et al.78 designed experiments to test whether or not activation of pyrin inflammasome by stimuli other than Yptb-ΔyopM infection will facilitate caspase-1-dependent pyroptosis in neutrophils. They used Clostridium difficile toxin B (TcdB), which enters cells via endosomal uptake followed by release into the cytosol upon endosomal acidification; cytosolic TcdB catalyzes ADP-ribosylation and inactivation of RhoA, which is sensed by the PKR/pyrin/14-3-3 cassette to liberate active pyrin. While TcdB treatment triggered robust caspase-1-dependent IL-1β release and GSDMD cleavage—equivalent in magnitude to that in Yptb-ΔyopM-infected neutrophils, it did not induce pyroptosis. Conversely, the same treatment protocols in murine macrophages produced robust IL-1β release and pyroptosis. The investigators next used electroporation to directly deliver TcdB into the cytosol, thereby bypassing the endosomal delivery route. Electroporation of TcdB into neutrophils induced caspase-1-dependent and N-GSDMD-dependent release of both IL-1β and LDH indicative of pyroptosis. Thus, the active caspase-1 within assembled pyrin inflammasomes differentially drives, or does not drive, pyroptosis based on the delivery route for the stimuli that initiates conformational activation of pryrin: direct delivery via influx across the plasma membrane facilitates pyroptosis while indirect delivery from the endosomal compartment does not.
Oh et al.78 tested the general applicability of this apparent delivery route-specific induction of neutrophil pyroptosis by caspase-1 based inflammasome by showing that activation of NLRC4 inflammasomes is coupled to pyroptosis in response to P. aeruginosa, which utilizes contact-mediated T3SS assembly in the plasma membrane to inject NLRC4-inducing PAMPs. Conversely, neutrophils resisted pyroptosis when NLRC4 PAMPS are injected via T3SS assembled within phagosomal membranes by internalized Salmonella typhimurium. These findings reinforced the similar observations of differential pyroptotic progression in P. aeruginosa-infected neutrophils versus S. typhimurium-infected neutrophils as respectively reported by Santoni et al.79 and Chen et al.56
Finally, Oh et al.78 described the ability of Yptb-ΔyopM infection to facilitate NETosis downstream of active pyrin inflammasome assembly and accumulation of pro-pyroptotic N-GSDMD pores in the plasma membrane. Similar to the findings of Santoni et al.79 with P. aeruginosa-infected neutrophils, PAD4-mediated H3 histone citrullination was triggered by Ca2+ influx through the plasma membrane N-GSDMD pores that accumulated in Yptb-ΔyopM infected neutrophils. Dissimilar from the observations of Santoni et al.79 the PAD4-mediated H3 histone citrullination induced in Yptb-ΔyopM infected cells correlated with expulsion of the decondensed chromatin into the extracellular space as NETs surrounding the lysed neutrophils.
7 |. INTEGRATED MODEL FOR HOW DIFFERENT INFLAMMASOME SIGNALING PLATFORMS FACILITATE CONDITIONAL RESISTANCE OR PROGRESSION TO PYROPTOSIS IN NEUTROPHILS
7.1 |. Roles for subcellular localization of GSDMD cleavage reactions that generate pore-forming N-GSDMD
The findings of Oh et al.78 provided significant new insights regarding the mechanisms by which neutrophils conditionally engage GSDMD-dependent pyroptosis based on the presumed subcellular location of particular inflammasome caspase-1 complexes that cleave GSDMD to generate the pore-forming N-GSDMD products. When either pyrin-or NLRC4-inflammasome activation is initiated by PAMP stimuli delivered from intracellular endosomal or phagosomal compartments, caspase-1-mediated generation of N-GSDMD subunits does not result in pyroptosis, which requires assembly and accumulation of oligomerized N-GSDMD macropores in the plasma membrane. Conversely, when PAMPs for conformational activation of pyrin or NLRC4 are directly delivered through the plasma membrane barrier by T3SS injection needles or electroporation, N-GSDMD subunits are apparently generated within cytosolic locations that facilitate efficient association with the inner leaflet of the plasma membrane to generate the macropores required for pyroptosis. It is important to emphasize that macrophages execute efficient pyroptotic progression regardless of which PAMP delivery route is used to trigger activation of pyrin or NLRC4 inflammasomes. This suggests important roles for unique cell biological features that differentiate neutrophils from macrophages; these include their polymorphonuclear organization and high content of specialized granules and secretory vesicles distributed as densely packed clusters throughout the neutrophil cytoplasm. Several defining aspects of basic inflammasome and gasdermin biology need to be considered in assessing such potential roles for neutrophil subcellular organization.
First, upon activation, canonical ASC-containing inflammasomes progressively assemble into the large macromolecular complexes morphologically identified as the so-called “ASC specks.” In macrophages and other mononuclear cells, ASC-containing inflammasomes typically coalesce into a single centrally located ASC speck adjacent to the nucleus.4,72 Although few studies have directly imaged ASC specks in neutrophils, Karmakar et al.51 reported that neutrophils accumulate multiple and smaller ASC specks in response to NLRP3 inflammasome activation by pneumolysin. This likely reflects effects of polymorphonuclear architecture to favor formation of multiple specks adjacent to individual lobes of the atypical nucleus. Recent studies in macrophages have also demonstrated that the initial formation of NLRP3 oligomers is seeded at discrete endosomal loci.194,195 As discussed in Section 5.1, neutrophils containing activated NLRP3 inflammasomes are particularly resistant to pyroptosis31,33,51,57 with delayed lysis in only a small percentage of cells.75 It is plausible that the unique cell biology of neutrophils enforces highly compartmentalized foci of NLRP3 inflammasome activity within organelle networks comprising endosomes and the multiple interlobular specks that are further constrained by proximity to the densely packed granules and secretory vesicles. Consequently, the generation of pore-forming N-GSDMD products would also be compartmentalized at these focal and constrained intracellular sites. When combined with its high avidity for anionic phospholipids at membrane surfaces, N-GSDMD products generated at these intracellular foci will encounter significant biophysical barriers for efficacious trafficking to the neutrophil plasma membrane. This scenario is supported by the observations of Karmakar et al.57 that N-GDSMD accumulates within discrete intracellular puncta in NLRP3 inflammasome-activated neutrophils in contrast to plasma membrane localization in NLRP3 inflammasome-activated macrophages.
At the opposite end of the spectrum, activation of non-canonical caspase-11 inflammasomes in neutrophils generates N-GSDMD products that readily assemble into functional plasma membrane macropores to facilitate effective pyroptotic progression.56,77,103 This is consistent with the biochemically distinct mechanism for assembling active caspase-11 inflammasomes versus those for assembling canonical caspase-1 containing inflammasomes. Caspase-11 activation requires only auto-dimerization induced by LPS binding108,109 and is independent of ASC and localization in discrete specks at particular subcellular sites. Consequently, active caspase-11 dimers and their generated N-GSDMD products will be distributed throughout the neutrophil cytoplasm to readily facilitate assembly of functional N-GSDMD macropores in the neutrophil plasma membrane and also different intracellular organelles. This scenario is supported by the observations of Chen et al.56 that N-GDSMD generated by active caspase-11 in neutrophils could coordinately target the plasma membrane, nuclear membrane, and granule membranes to change their permeability properties.
Pyrin and NLRC4, like NLRP3, also recruit ASC to generate active caspase-1-based inflammasomes for cleavage of GSDMD. Thus, these inflammasomes should also be assembled within a discrete ASC speck or specks at particular subcellular locations in neutrophils. The findings of Oh et al.78 clearly suggest that pyrin-or NLRC4 inflammasomes can differentially target or “channel” their N-GSDMD products to the plasma membrane depending on whether their activating PAMPs are delivered through the plasma membrane barrier or via release from intracellular endosomes/phagosomes. However, the structural basis for this conditional trafficking of N-GSDMD products pyrin or NLRC4 inflammasomes remains to be determined. Might PAMP delivery via the plasma membrane route induce assembly of “micro” pyrin/ASC or NLRC4/ASC specks adjacent to the plasma membrane to favor assembly of N-GSDMD pores in that neutrophil compartment? Might the trafficking of N-GSDMD, generated at conventional, centrally located pyrin/ASC or NLRC4/ASC specks, be modulated by other signals or changes in neutrophil subcellular organization induced by PAMP delivery via the plasma membrane route? Given the roles for these neutrophil inflammasomes in the control of several bacterial pathogens, these are important areas for future investigations.
7.2 |. Roles for azurophilic granules and granule-derived serine proteases released into the cytosol
The highly abundant azurophilic granules may modulate the ability of canonical inflammasomes to engage or resist GSDMD-dependent pyroptosis in two ways. First, as previously described, they comprise a quantitively significant membrane sink for binding N-GSDMD products produced by canonical caspase-1-containing inflammasomes.57 By mass action effects, this reduces the pool of N-GSDMD products available for assembly of N-GSDMD macropores in the plasma membrane which is required for effective pyroptosis. Second, the assembly of functional N-GSDMD macropores in the azurophilic granule membranes will per se compromise the permeability properties of those membranes such that neutral serine proteases and other granule contents can be released in the neutrophil cytosol. As discussed in Section 3, all of the most abundant granule proteases (cathepsin G, elastase, proteinase 3) can cleave GSDMD at multiple sites to produce a suite of N-GSDMD products, some of which are pore-competent. The studies of Kambara et al.99 established a role for elastase-cleaved GSDMD in modulating the progression of constitutive neutrophil death and the numbers of viable neutrophils available for control of bacterial pathogens in densely infected tissue compartments. Burgener et al.100 established that cathepsin G-cleaved GSDMD could similarly accelerate neutrophil cell death. Within the context of canonical inflammasome signaling, the secondary generation of serine protease-processed and pore-competent N-GSDMD products may add to the pool of caspase-1 processed N-GSDMD products available for assembly of plasma membrane macropores to accelerate pyroptotic progression. Alternatively, cytosolic accumulation of granule serine proteases may drive proteolytic cascades that inactivate the pore-forming ability of caspase-1 cleaved N-GSDMD products and thus facilitate resistance to pyroptotic progression. For example, pro-apoptotic caspase-3 cleaves GSDMD within its pore-forming N-terminal domain to suppress assembly of functional N-GSDMD macropores.114 Within the context of constitutive neutrophil cell death, proteinase 3 (PR3) released into the neutrophil activates caspase-3 to drive apoptotic progression for clearance of senescent neutrophils.165,166 However, within the context of inflammasome signaling in neutrophils, this PR3 > caspase-3 cascade would inactivate GSDMD to promote resistance to pyroptotic progression. Assessing the contribution of azurophilic granule serine proteases to pyroptotic resistance or progression in different models of neutrophil inflammasome signaling is another important area for future investigation.
7.3 |. Roles for activated GSMDE
GSDME is highly expressed in neutrophils and has the capacity to generate plasma membrane macropores upon cleavage by the apoptotic effector caspase-3. Chen et al.102 observed that when isolated neutrophils from wildtype versus GSDME-deficient mice were incubated in vitro, the GSDME-deficient cells retained viability for longer durations. This indicated that GSDME can drive spontaneous neutrophil death likely via caspase-3 activated by proteinase 3 released from leaky azurophilic granules. Thus, it is relevant to consider how production of pore-competent N-GSDME products during activation of certain inflammasome signaling cascades in neutrophils may modulate pyroptotic progression. Analysis of macrophages deficient in GDSDM have indicated that GSDME can mediate both IL-1β release and lytic cell death in response to activators of several canonical inflammasome platforms.117,118,170,173 However, when Chen et al.102 compared the responses of macrophages versus neutrophils to infection with a YopJ-expressing Yersinia strain, they observed that macrophages engaged RIPK1 to activate caspase-8, which directly cleaved GSDMD to drive macrophage pyroptosis. In contrast, neutrophils engaged RIPK1 to activate a caspase-8 > caspase-3 protease cascade, which bypassed GSDMD and cleaved GSDME to drive neutrophil pyroptosis. Studies with GSDMD-knockout or GSDME-knockout myeloid cells confirmed that GSDMD, but not GSDME, was required for macrophage death, while the opposite was true for neutrophils. Furthermore, as described in Section 6.2, Oh et al.78 used a Yersinia strain deficient in the YopM virulence factor to drive pyrin-dependent caspase-1 signaling responses in wildtype neutrophils versus neutrophils lacking GSDMD, GSDME or both GSDMs. They determined that pyrin-dependent caspase-1 induced pyroptosis via two parallel signaling pathways involving accumulation of both N-GSDMD and N-GSDME membrane pores and that ablation of both GSDMs was required to suppress release of IL-1β. Thus, neutrophil inflammasome signaling networks are distinctively wired to use GSDME for important innate immune responses, including pyroptosis, to control particular pathogens. Future studies are needed to determine whether engagement of GSDME cleavage and pore formation modulates pyroptotic resistance or progression regulated by other inflammasome platforms (NLRP3, NLRC4, non-canonical caspase-4/11) in neutrophils.
8 |. ROLES FOR INFLAMMASOME AND GASDERMIN D SIGNALING IN THE REGULATION OF NETosis
In 1996, Takei et al.196 reported that phorbol esters induced the rapid killing of neutrophils with morphological features distinct from apoptotic or necrotic death. In 2004, Zychlinsky et al.197 observed that this atypical neutrophil death correlated with the release of nuclear chromatin complexes that were termed neutrophil extracellular traps (NETs) and proposed to function as physiological components of the neutrophil innate immune response repertoire. NETosis, the death response associated with release of NETs, has developed into an exceptionally active area of neutrophil biology research as summarized in recent reviews,58–60 but also comprises a somewhat contentious topic.198 In 2018, independent studies by Chen et al.56 and Sollberger et al.54 described roles for GSDMD in the coordinated permeabilization of azurophilic granules, the nuclear membrane, and the plasma membrane coincident with NETosis and release of extracellular DNA traps.54,56 Those reports have sparked subsequent investigations at two different levels: (i) reductionist experiments to further define mechanistic relationships between inflammasome signaling pathways that target GSDMD cleavage for pyroptotic progression and the various stimuli and signaling pathways that regulate different modes of NETosis; and (ii) in vivo experiments that use mice with global GSDMD deficiency or neutrophil-specific GSDMD depletion, in concert with pharmacological GSDMD inhibition, to interrogate roles for GSDMD in disease models of organ dysfunction previously linked to pathogenic neutrophil recruitment and NETosis. With regard to pharmacological agents, disulfiram, an FDA-approved therapeutic for alcoholism, has been shown to covalently modify a critical cysteine in N-GSDMD required for the conformational changes that mediate membrane insertion and pore formation.199
As described in Section 6.1, Chen et al.56 demonstrated that LPS activation of non-canonical caspase-11 inflammasomes in neutrophils induced GSDMD-dependent pyroptosis that correlated with induction of late-stage NETosis markers including accumulation of extracellular DNA, citrullinated histone H3 and myeloperoxidase. Induction of these NETosis markers was prevented in Casp11−/− or Gsdmd−/− neutrophils. Oh et al.78 used Yersinia-infected neutrophils to show that activation of pyrin inflammasomes induced assembly of pro-pyroptotic N-GSDMD pores in the plasma membrane that preceded the development of NETosis markers including PAD4-mediated H3 histone citrullination, chromatin decondensation, and expulsion of the decondensed chromatin into the extracellular space surrounding the lysed pyroptotic neutrophils. Ca2+ influx through plasma membrane N-GSDMD pores was the critical upstream signal for PAD4 activation and development of downstream indices of NETosis; PAD4-dependent H3 histone citrullination was absent in neutrophils lacking expression of caspase-1 or GSDMD. As previously described in Section 6.1, Santoni et al.79 demonstrated that P. aeruginosa infection induced NLRC4-caspase-1 inflammasomes in neutrophils, which licensed assembly of plasma membrane N-GSDMD pores for efficient pyroptotic progression that correlated with citrullination of histone H3 and decondensation of nuclear DNA. Similar to the findings of Oh et al.78 with Yersinia-infected neutrophils, the ability of P. aeruginosa neutrophils to accumulate the citrullinated H3 histone decondense DNA was mediated by Ca2+ influx through the N-GSDMD macropores However, the pyroptotic neutrophils did not release the expanded chromatin into the extracellular compartment as NETs. A plausible reason for this disparity from the NETosis observed in Yersinia-infected neutrophils is that the latter were primed with LPS plus IFNγ before infection, while Santoni et al.79 directly infected unprimed neutrophils with P. aeruginosa. LPS/IFNγ priming induces expression of multiple proinflammatory proteins that may modulate thresholds for efficient progression of pyroptosis and/or NETosis. Thus, activation of non-canonical caspase-11 inflammasomes, canonical pyrin/caspase-1 inflammasomes and canonical NLRC4/ caspase-1 inflammasomes in neutrophils can drive GSDMD-dependent pyroptosis as an upstream inducer of NETosis. Conversely, Chauhan et al.75 used GSDMD-deficient neutrophils to demonstrate GSDMD was completely dispensable for the activation of NETosis by phorbol ester, the well-established inducer of NADPH oxidase-dependent NETosis. We have also observed identical rates and extents of phorbol ester-induced NETosis in neutrophils isolated from wildtype C57/Bl6 mice, Gsdmd−/− mice, or Gsdme−/− mice.189 Together, these recent reports suggest a contingent, rather than obligatory, role for GSDMD in regulating NETosis. Clearly, more investigation is required to further clarify the signaling pathways that underlie this contingent role for GSDMD in an important component of neutrophil-mediated innate immunity.
Several recent studies have described how GSDMD-dependent proinflammatory neutrophil death pathways (pyroptosis or NETosis) can contribute to organ dysfunction in different disease models. Jiang et al.200 examined pathogenic roles for neutrophil GSDMD signaling in a mouse model of myocardial ischemic injury. Earlier studies had established roles for NLRP3 signaling by recruited myeloid leukocytes in the exacerbation of acute ischemic injury.201 Jiang et al.200 found that neutrophils were the major recruited source of active NLRP3 inflammasomes within the damaged myocardium in the initial 24 hours post-ischemia. Mice with global knockout of GSDMD or wildtype mice treated with disulfiram had reduced myocardial damage, improved cardiac output, and enhanced survival. Notably, using bone marrow chimera models, Jiang et al.200 determined that neutrophil-selective knockout of GSDMD provided most of the improved cardiac function and enhanced survival post-ischemia. Xie et al.202 employed a mouse model of intratracheal LPS-induced acute respiratory distress syndrome (ARDS) to demonstrate significant correlation between the development of ARDS-related lung injury and the accumulation of released NETs in the lungs. ARDS injury and pulmonary NET accumulation were markedly attenuated in mice with conditional knockout of GSDMD in neutrophils or in wildtype mice treated with disulfiram.
Silva et al.105 used the mouse model of polymicrobial systemic sepsis induced by cecal ligation and puncture (CLP) to verify previous observations that mice with global GSDMD knockout were protected from the lethal sepsis and dysfunction of multiple organs that defines the integrated response to CLP. Wildtype mice treated with disulfiram were similarly protected. Sera and tissue from the dysfunctional organs (e.g., lung) of CLP-challenged wildtype mice were characterized by increased levels of NETosis markers (DNA-myeloperoxidase complexes). Transfer of wildtype neutrophils expressing GSDMD into the mice with global GSDMD deficiency reversed the protection of their organs from CLP-induced sepsis, and also increased their serum levels of NETosis markers. Based on these observations, Silva et al.105 concluded that GSDMD signaling in the neutrophil compartment and GSDMD-dependent NETosis, in particular, are major drivers of organ dysfunction during polymicrobial systemic sepsis. Consequently, they proposed that indirect inhibition of NETosis by pharmacologically targeting the upstream GSDMD driver may be an efficacious anti-sepsis therapy.
Conversely, Liu et al.203 demonstrated that induction of CLP sepsis in mice with conditional knockout of GSDMD in neutrophils surprisingly increased septic lethality and extent of tissue damage and multi-organ dysfunction. Thus, deletion of neutrophil GSDMD did not protect the mice from polymicrobial sepsis but rather elicited more severe sepsis. This is an opposite outcome to the mechanistic conclusions and model described by Silva et al.105 Liu et al.203 confirmed that global deletion of GSDMD did protect the mice from lethal sepsis and correlated with reduced levels of inflammatory cytokines in these mice. In contrast, mice with neutrophil-specific GSDMD deficiency had higher levels of inflammatory cytokines and also greater bacterial loads. This suggested that neutrophils lacking GSDMD provide a replicative niche for intracellular bacterial pathogens that would be included in the poly-microbial mix released from the gut into the peritoneum by CLP. In neutrophils harboring such intracellular bacterial pathogens, GSDMD-dependent pyroptosis is host-beneficial because it both removes a protected replicative niche and exposes the released bacteria to killing by alternative anti-microbial mechanisms such as complement or NETs. In the absence of GSDMD signaling in those neutrophils, the overall bacterial killing capacity of the host may be compromised leading to greater and more prolonged infection and hyperinflammatory cytokine production to the detriment of the host.
Description of this last experimental scenario provides an apt and relevant conclusion to this review as it starkly illustrates the review’s two major themes (i) that neutrophils conditionally engage GSDMD-mediated pyroptosis as a defensive response against bacterial pathogens that use neutrophils as replicative niches; (ii) that neutrophils possess a remarkable capacity to resist progression to GSDMD-dependent pyroptotic lysis to preserve their viability for efficient microbial killing while maintaining GSDMD-dependent mechanisms for export of bioactive IL-1β to the overall benefit of the host.
Funding information
National Institutes of Health, Grant/Award Number: P01-AI141350, R01-EY014362 and R21-EY032662
Footnotes
CONFLICT OF INTEREST
There is no conflict of interest to declare.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–426. [DOI] [PubMed] [Google Scholar]
- 2.Place DE, Kanneganti TD. Recent advances in inflammasome biology. Curr Opin Immunol. 2017;50:32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lamkanfi M, Dixit VM. In Retrospect: The inflammasome turns 15. Nature. 2017;548(7669):534–535. [DOI] [PubMed] [Google Scholar]
- 4.Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16(7):407–420. [DOI] [PubMed] [Google Scholar]
- 5.Shouval DS, Biswas A, Kang YH, et al. Interleukin 1beta mediates intestinal inflammation in mice and patients with interleukin 10 receptor deficiency. Gastroenterology. 2016;151(6):1100–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rathinam VA, Fitzgerald KA. Inflammasome complexes: emerging mechanisms and effector functions. Cell. 2016;165(4):792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bersudsky M, Luski L, Fishman D, et al. Non-redundant properties of IL-1alpha and IL-1beta during acute colon inflammation in mice. Gut. 2014;63(4):598–609. [DOI] [PubMed] [Google Scholar]
- 8.Coccia M, Harrison OJ, Schiering C, et al. IL-1beta mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J Exp Med. 2012;209(9):1595–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franchi L, Munoz-Planillo R, Nunez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. 2012;13(4):325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7(2):99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fink SL, Bergsbaken T, Cookson BT. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc Natl Acad Sci USA. 2008;105(11):4312–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006;8(11):1812–1825. [DOI] [PubMed] [Google Scholar]
- 13.Feng Y, Huang X. Methodology for comprehensive detection of pyroptosis. Methods Mol Biol. 2021;2255:149–157. [DOI] [PubMed] [Google Scholar]
- 14.Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42(4):245–254. [DOI] [PubMed] [Google Scholar]
- 15.Kovacs SB, Miao EA. Gasdermins: effectors of pyroptosis. Trends Cell Biol. 2017;27(9):673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev. 2017;277(1):61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev. 2018;281(1):8–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity. 2013;39(6):1003–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kimura T, Jia J, Claude-Taupin A, et al. Cellular and molecular mechanism for secretory autophagy. Autophagy. 2017;13(6):1084–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ponpuak M, Mandell MA, Kimura T, Chauhan S, Cleyrat C, Deretic V. Secretory autophagy. Curr Opin Cell Biol. 2015;35:106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kayagaki N, Stowe IB, Lee BL, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526(7575):666–671. [DOI] [PubMed] [Google Scholar]
- 22.Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–665. [DOI] [PubMed] [Google Scholar]
- 23.Lieberman J, Wu H, Kagan JC. Gasdermin D activity in inflammation and host defense. Sci Immunol. 2019;4(39):eaar1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evavold CL, Kagan JC. Diverse control mechanisms of the interleukin-1 cytokine family. Front Cell Dev Biol. 2022;10:910983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu X, Zhang Z, Ruan J, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535(7610):153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding J, Wang K, Liu W, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535(7610):111–116. [DOI] [PubMed] [Google Scholar]
- 27.Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity. 2018;48(1):35–44.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xia S, Zhang Z, Magupalli VG, et al. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature. 2021;593(7860):607–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kayagaki N, Kornfeld OS, Lee BL, et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature. 2021;591(7848):131–136. [DOI] [PubMed] [Google Scholar]
- 30.Kanneganti A, Malireddi RKS, Saavedra PHV, et al. GSDMD is critical for autoinflammatory pathology in a mouse model of Familial Mediterranean Fever. J Exp Med. 2018;215(6):1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heilig R, Dick MS, Sborgi L, Meunier E, Hiller S, Broz P. The Gasdermin-D pore acts as a conduit for IL-1beta secretion in mice. Eur J Immunol. 2018;48(4):584–592. [DOI] [PubMed] [Google Scholar]
- 32.Goldberg EL, Asher JL, Molony RD, et al. beta-hydroxybutyrate deactivates neutrophil NLRP3 inflammasome to relieve gout flares. Cell Rep. 2017;18(9):2077–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karmakar M, Katsnelson MA, Dubyak GR, Pearlman E. Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1beta secretion in response to ATP. Nat Commun. 2016;7:10555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen KW, Bezbradica JS, Gross CJ, et al. The murine neutrophil NLRP3 inflammasome is activated by soluble but not particulate or crystalline agonists. Eur J Immunol. 2016;46(4):1004–1010. [DOI] [PubMed] [Google Scholar]
- 35.Chen KW, Gross CJ, Sotomayor FV, et al. The neutrophil NLRC4 inflammasome selectively promotes IL-1beta maturation without pyroptosis during acute Salmonella challenge. Cell Rep. 2014;8(2):570–582. [DOI] [PubMed] [Google Scholar]
- 36.Suurmond J, Habets KL, Dorjee AL, Huizinga TW, Toes RE. Expansion of Th17 cells by human mast cells is driven by inflammasome-independent IL-1beta. J Immunol. 2016;197(11):4473–4481. [DOI] [PubMed] [Google Scholar]
- 37.Kuhny M, Hochdorfer T, Ayata CK, Idzko M, Huber M. CD39 is a negative regulator of P2X7-mediated inflammatory cell death in mast cells. Cell Commun Signal. 2014;12:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakamura Y, Franchi L, Kambe N, Meng G, Strober W, Nunez G. Critical role for mast cells in interleukin-1beta-driven skin inflammation associated with an activating mutation in the nlrp3 protein. Immunity. 2012;37(1):85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kono H, Orlowski GM, Patel Z, Rock KL. The IL-1-dependent sterile inflammatory response has a substantial caspase-1-independent component that requires cathepsin C. J Immunol. 2012;189(7):3734–3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kambe N, Nakamura Y, Saito M, Nishikomori R. The inflammasome, an innate immunity guardian, participates in skin urticarial reactions and contact hypersensitivity. Allergol Int. 2010;59(2):105–113. [DOI] [PubMed] [Google Scholar]
- 41.Guma M, Kashiwakura J, Crain B, et al. JNK1 controls mast cell degranulation and IL-1{beta} production in inflammatory arthritis. Proc Natl Acad Sci USA. 2010;107(51):22122–22127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakamura Y, Kambe N, Saito M, et al. Mast cells mediate neutrophil recruitment and vascular leakage through the NLRP3 inflammasome in histamine-independent urticaria. J Exp Med. 2009;206(5):1037–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guma M, Ronacher L, Liu-Bryan R, Takai S, Karin M, Corr M. Caspase 1-independent activation of interleukin-1beta in neutrophil-predominant inflammation. Arthritis Rheum. 2009;60(12):3642–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Esnault S, Kelly EA, Johnson SH, et al. Matrix metalloproteinase-9-dependent release of IL-1beta by human eosinophils. Mediat Inflamm. 2019;2019:7479107–7479111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Palacios-Macapagal D, Connor J, Mustelin T, Ramalingam TR, Wynn TA, Davidson TS. Cutting edge: eosinophils undergo caspase-1-mediated pyroptosis in response to necrotic liver cells. J Immunol. 2017;199(3):847–853. [DOI] [PubMed] [Google Scholar]
- 46.Jung Y, Wen T, Mingler MK, et al. IL-1beta in eosinophil-mediated small intestinal homeostasis and IgA production. Mucosal Immunol. 2015;8(4):930–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tran HB, Lewis MD, Tan LW, et al. Immunolocalization of NLRP3 inflammasome in normal murine airway epithelium and changes following induction of ovalbumin-induced airway inflammation. J Allergy. 2012;2012:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheung PF, Wong CK, Lam CW. Molecular mechanisms of cytokine and chemokine release from eosinophils activated by IL-17A, IL-17F, and IL-23: implication for Th17 lymphocytes-mediated allergic inflammation. J Immunol. 2008;180(8):5625–5635. [DOI] [PubMed] [Google Scholar]
- 49.Tyrkalska SD, Candel S, Mulero V. The neutrophil inflammasome. Dev Comp Immunol. 2021;115:103874. [DOI] [PubMed] [Google Scholar]
- 50.Yow SJ, Yeap HW, Chen KW. Inflammasome and gasdermin signaling in neutrophils. Mol Microbiol. 2022;117(5):961–972. [DOI] [PubMed] [Google Scholar]
- 51.Karmakar M, Katsnelson M, Malak HA, et al. Neutrophil IL-1β processing induced by pneumolysin is mediated by the NLRP3/ASC inflammasome and caspase-1 activation and is dependent on K+ efflux. J Immunol. 2015;194(4):1763–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mankan AK, Dau T, Jenne D, Hornung V. The NLRP3/ASC/Caspase-1 axis regulates IL-1beta processing in neutrophils. Eur J Immunol. 2012;42(3):710–715. [DOI] [PubMed] [Google Scholar]
- 53.Tsuchiya K, Nakajima S, Hosojima S, et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat Commun. 2019;10(1):2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sollberger G, Choidas A, Burn GL, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. 2018;3(26):eaar6689. [DOI] [PubMed] [Google Scholar]
- 55.Monteleone M, Stanley AC, Chen KW, et al. Interleukin-1beta maturation triggers its relocation to the plasma membrane for gasdermin-D-dependent and -independent secretion. Cell Rep. 2018;24(6):1425–1433. [DOI] [PubMed] [Google Scholar]
- 56.Chen KW, Monteleone M, Boucher D, et al. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci Immunol. 2018;3(26):eaar6676. [DOI] [PubMed] [Google Scholar]
- 57.Karmakar M, Minns M, Greenberg EN, et al. N-GSDMD trafficking to neutrophil organelles facilitates IL-1β release independently of plasma membrane pores and pyroptosis. Nat Commun. 2020;11(1):2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Papayannopoulos V Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18(2):134–147. [DOI] [PubMed] [Google Scholar]
- 59.Sollberger G, Tilley DO, Zychlinsky A. Neutrophil extracellular traps: the biology of chromatin externalization. Dev Cell. 2018;44(5):542–553. [DOI] [PubMed] [Google Scholar]
- 60.Thiam HR, Wong SL, Wagner DD, Waterman CM. Cellular mechanisms of NETosis. Annu Rev Cell Dev Biol. 2020;36:191–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pérez-Figueroa E, Álvarez-Carrasco P, Ortega E, Maldonado-Bernal C. Neutrophils: many ways to die. Front Immunol. 2021;12:631821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rosazza T, Warner J, Sollberger G. NET formation – mechanisms and how they relate to other cell death pathways. FEBS J. 2021;288(11):3334–3350. [DOI] [PubMed] [Google Scholar]
- 63.Sollberger G Approaching neutrophil pyroptosis. J Mol Biol. 2022;434(4):167335. [DOI] [PubMed] [Google Scholar]
- 64.Christgen S, Place DE, Kanneganti TD. Toward targeting inflammasomes: insights into their regulation and activation. Cell Res. 2020;30(4):315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paerewijck O, Lamkanfi M. The human inflammasomes. Mol Asp Med. 2022;88:101100. [DOI] [PubMed] [Google Scholar]
- 66.Zheng D, Liwinski T, Elinav E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discov. 2020;6:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bakele M, Joos M, Burdi S, et al. Localization and functionality of the inflammasome in neutrophils. J Biol Chem. 2014;289(8):5320–5329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lugrin J, Martinon F. The AIM2 inflammasome: sensor of pathogens and cellular perturbations. Immunol Rev. 2018;281(1):99–114. [DOI] [PubMed] [Google Scholar]
- 69.Kay C, Wang R, Kirkby M, Man SM. Molecular mechanisms activating the NAIP-NLRC4 inflammasome: implications in infectious disease, autoinflammation, and cancer. Immunol Rev. 2020;297(1):67–82. [DOI] [PubMed] [Google Scholar]
- 70.Taabazuing CY, Griswold AR, Bachovchin DA. The NLRP1 and CARD8 inflammasomes. Immunol Rev. 2020;297(1):13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu J, Núñez G. The NLRP3 inflammasome: activation and regulation. Trends Biochem Sci. 2022. doi: 10.1016/j.tibs.2022.10.01. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Akbal A, Dernst A, Lovotti M, Mangan MSJ, McManus RM, Latz E. How location and cellular signaling combine to activate the NLRP3 inflammasome. Cell Mol Immunol. 2022;19(11):1201–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Loeven NA, Medici NP, Bliska JB. The pyrin inflammasome in host-microbe interactions. Curr Opin Microbiol. 2020;54:77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Malik HS, Bliska JB. The pyrin inflammasome and the Yersinia effector interaction. Immunol Rev. 2020;297(1):96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chauhan D, Demon D, Vande Walle L, et al. GSDMD drives canonical inflammasome-induced neutrophil pyroptosis and is dispensable for NETosis. EMBO Rep. 2022;23(10):e54277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ryu JC, Kim MJ, Kwon Y, et al. Neutrophil pyroptosis mediates pathology of P. aeruginosa lung infection in the absence of the NADPH oxidase NOX2. Mucosal Immunol. 2017;10(3):757–774. [DOI] [PubMed] [Google Scholar]
- 77.Kovacs SB, Oh C, Maltez VI, et al. Neutrophil Caspase-11 is essential to defend against a cytosol-invasive bacterium. Cell Rep. 2020;32(4):107967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Oh C, Li L, Verma A, et al. Neutrophil inflammasomes sense the subcellular delivery route of translocated bacterial effectors and toxins. Cell Rep. 2022;41(8):111688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Santoni K, Pericat D, Gorse L, et al. Caspase-1-driven neutrophil pyroptosis and its role in host susceptibility to Pseudomonas aeruginosa. PLoS Pathog. 2022;18(7):e1010305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Paget C, Doz-Deblauwe E, Winter N, Briard B. Specific NLRP3 inflammasome assembling and regulation in neutrophils: relevance in inflammatory and infectious diseases. Cells. 2022;11(7):1188. doi: 10.3390/cells11071188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Son S, Yoon SH, Chae BJ, et al. Neutrophils facilitate prolonged inflammasome response in the DAMP-rich inflammatory milieu. Front Immunol. 2021;12:746032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Afonina IS, Muller C, Martin SJ, Beyaert R. Proteolytic processing of interleukin-1 family cytokines: variations on a common theme. Immunity. 2015;42(6):991–1004. [DOI] [PubMed] [Google Scholar]
- 83.Thornberry NA. Interleukin-1 beta converting enzyme. Methods Enzymol. 1994;244:615–631. [DOI] [PubMed] [Google Scholar]
- 84.Thornberry NA, Bull HG, Calaycay JR, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. 1992;356(6372):768–774. [DOI] [PubMed] [Google Scholar]
- 85.Netea MG, van de Veerdonk FL, van der Meer JW, Dinarello CA, Joosten LA. Inflammasome-independent regulation of IL-1-family cytokines. Annu Rev Immunol. 2015;33:49–77. [DOI] [PubMed] [Google Scholar]
- 86.Clancy DM, Sullivan GP, Moran HBT, et al. Extracellular neutro-phil proteases are efficient regulators of IL-1, IL-33, and IL-36 cytokine activity but poor effectors of microbial killing. Cell Rep. 2018;22(11):2937–2950. [DOI] [PubMed] [Google Scholar]
- 87.Clancy DM, Henry CM, Sullivan GP, Martin SJ. Neutrophil extra-cellular traps can serve as platforms for processing and activation of IL-1 family cytokines. FEBS J. 2017;284(11):1712–1725. [DOI] [PubMed] [Google Scholar]
- 88.Karmakar M, Sun Y, Hise AG, Rietsch A, Pearlman E. Cutting edge: IL-1beta processing during Pseudomonas aeruginosa infection is mediated by neutrophil serine proteases and is independent of NLRC4 and caspase-1. J Immunol. 2012;189(9):4231–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Keitelman IA, Shiromizu CM, Zgajnar NR, et al. The interplay between serine proteases and caspase-1 regulates the autophagy-mediated secretion of Interleukin-1 beta in human neutrophils. Front Immunol. 2022;13:832306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Broz P, Pelegrin P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol. 2019;20:143–157. doi: 10.1038/s41577-019-0228-2 [DOI] [PubMed] [Google Scholar]
- 91.Orning P, Lien E, Fitzgerald KA. Gasdermins and their role in immunity and inflammation. J Exp Med. 2019;216(11):2453–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sborgi L, Rühl S, Mulvihill E, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016;35(16):1766–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Angosto-Bazarra D, Alarcón-Vila C, Hurtado-Navarro L, Baños MC, Rivers-Auty J, Pelegrín P. Evolutionary analyses of the gasdermin family suggest conserved roles in infection response despite loss of pore-forming functionality. BMC Biol. 2022;20(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Daskalov A, Glass NL. Gasdermin and gasdermin-like pore-forming proteins in invertebrates, fungi and bacteria. J Mol Biol. 2022;434(4):167273. [DOI] [PubMed] [Google Scholar]
- 95.Liu Z, Busscher BM, Storl-Desmond M, Xiao TS. Mechanisms of gasdermin recognition by proteases. J Mol Biol. 2022;434(4):167274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu X, Lieberman J. Knocking ‘em dead: pore-forming proteins in immune defense. Annu Rev Immunol. 2020;38:455–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ivanov AI, Rana N, Privitera G, Pizarro TT. The enigmatic roles of epithelial gasdermin B: Recent discoveries and controversies. Trends Cell Biol. 2023;33(1):48–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rana N, Privitera G, Kondolf HC, et al. GSDMB is increased in IBD and regulates epithelial restitution/repair independent of pyroptosis. Cell. 2022;185(2):283–298.e217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kambara H, Liu F, Zhang X, et al. Gasdermin D exerts anti-inflammatory effects by promoting neutrophil death. Cell Rep. 2018;22(11):2924–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Burgener SS, Leborgne NGF, Snipas SJ, Salvesen GS, Bird PI, Benarafa C. Cathepsin G inhibition by Serpinb1 and Serpinb6 prevents programmed necrosis in neutrophils and monocytes and reduces GSDMD-driven inflammation. Cell Rep. 2019;27(12):3646–3656.e3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen KW, Demarco B, Broz P. Beyond inflammasomes: emerging function of gasdermins during apoptosis and NETosis. EMBO J. 2020;39(2):e103397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen KW, Demarco B, Ramos S, et al. RIPK1 activates distinct gasdermins in macrophages and neutrophils upon pathogen blockade of innate immune signaling. Proc Natl Acad Sci USA. 2021;118(28):e2101189118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Oh C, Verma A, Hafeez M, Hogland B, Aachoui Y. Shigella OspC3 suppresses murine cytosolic LPS sensing. iScience. 2021;24(8):102910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ratitong B, Marshall M, Pearlman E. β-Glucan-stimulated neutro-phil secretion of IL-1α is independent of GSDMD and mediated through extracellular vesicles. Cell Rep. 2021;35(7):109139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Silva CMS, Wanderley CWS, Veras FP, et al. Gasdermin D inhibition prevents multiple organ dysfunction during sepsis by blocking NET formation. Blood. 2021;138(25):2702–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Karmakar M, Minns M, Greenberg EN, et al. N-GSDMD trafficking to neutrophil organelles facilitates IL-1beta release independently of plasma membrane pores and pyroptosis. Nat Commun. 2020;11(1):2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xie WJ, Xia S, Warshel A, Wu H. Electrostatic influence on IL-1 transport through the GSDMD pore. Proc Natl Acad Sci USA. 2022;119(6):e2120287119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Abu Khweek A, Amer AO. Pyroptotic and non-pyroptotic effector functions of caspase-11. Immunol Rev. 2020;297(1):39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Downs KP, Nguyen H, Dorfleutner A, Stehlik C. An overview of the non-canonical inflammasome. Mol Asp Med. 2020;76:100924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sarhan J, Liu BC, Muendlein HI, et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci USA. 2018;115(46):e10888–e10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Demarco B, Grayczyk JP, Bjanes E, et al. Caspase-8-dependent gasdermin D cleavage promotes antimicrobial defense but confers susceptibility to TNF-induced lethality. Sci Adv. 2020;6(47):eabc3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gram AM, Booty LM, Bryant CE. Chopping GSDMD: caspase-8 has joined the team of pyroptosis-mediating caspases. EMBO J. 2019;38(10):e102065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Orning P, Weng D, Starheim K, et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science. 2018;362(6418):1064–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Taabazuing CY, Okondo MC, Bachovchin DA. Pyroptosis and apoptosis pathways engage in bidirectional crosstalk in monocytes and macrophages. Cell Chem Biol. 2017;24(4):507–514.e504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhang Z, Zhang Y, Xia S, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature. 2020;579(7799):415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang C, Yang T, Xiao J, et al. NLRP3 inflammasome activation triggers gasdermin D-independent inflammation. Sci Immunol. 2021;6(64):eabj3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhou B, Abbott DW. Gasdermin E permits interleukin-1 beta release in distinct sublytic and pyroptotic phases. Cell Rep. 2021;35(2):108998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Qu Y, Franchi L, Nunez G, Dubyak GR. Nonclassical IL-1 beta secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J Immunol. 2007;179(3):1913–1925. [DOI] [PubMed] [Google Scholar]
- 120.Qu Y, Ramachandra L, Mohr S, et al. P2X7 receptor-stimulated secretion of MHC class II-containing exosomes requires the ASC/NLRP3 inflammasome but is independent of caspase-1. J Immunol. 2009;182(8):5052–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dubyak GR. P2X7 receptor regulation of non-classical secretion from immune effector cells. Cell Microbiol. 2012;14(11):1697–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sitia R, Rubartelli A. The unconventional secretion of IL-1beta: Handling a dangerous weapon to optimize inflammatory responses. Semin Cell Dev Biol. 2018;83:12–21. [DOI] [PubMed] [Google Scholar]
- 123.Nauseef WM. Human neutrophils ≠ murine neutrophils: does it matter? Immunol Rev. 2022. doi: 10.1111/imr.13154. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cui J, Oehrl S, Ahmad F, et al. Detection of in vivo inflammasome activation for predicting sepsis mortality. Front Immunol. 2020;11:613745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Aymonnier K, Ng J, Fredenburgh LE, et al. Inflammasome activation in neutrophils of patients with severe COVID-19. Blood Adv. 2022;6(7):2001–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Coudereau R, Gossez M, Py BF, et al. Monitoring NLRP3 inflammasome activation and exhaustion in clinical samples: a refined flow cytometry protocol for ASC speck formation measurement directly in whole blood after ex vivo stimulation. Cell. 2022;11(20):3306. doi: 10.3390/cells11203306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Münzer P, Negro R, Fukui S, et al. NLRP3 inflammasome assembly in neutrophils is supported by PAD4 and promotes NETosis under sterile conditions. Front Immunol. 2021;12:683803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Holzinger D, Gieldon L, Mysore V, et al. Staphylococcus aureus Panton-Valentine leukocidin induces an inflammatory response in human phagocytes via the NLRP3 inflammasome. J Leukoc Biol. 2012;92(5):1069–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Migita K, Izumi Y, Jiuchi Y, et al. Serum amyloid A induces NLRP-3-mediated IL-1β secretion in neutrophils. PLoS One. 2014;9(5):e96703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pérez-Figueroa E, Torres J, Sánchez-Zauco N, Contreras-Ramos A, Alvarez-Arellano L, Maldonado-Bernal C. Activation of NLRP3 inflammasome in human neutrophils by Helicobacter pylori infection. Innate Immun. 2016;22(2):103–112. [DOI] [PubMed] [Google Scholar]
- 131.Yokose K, Sato S, Asano T, et al. TNF-α potentiates uric acid-induced interleukin-1β (IL-1β) secretion in human neutrophils. Mod Rheumatol. 2018;28(3):513–517. [DOI] [PubMed] [Google Scholar]
- 132.Demirel I, Persson A, Brauner A, Särndahl E, Kruse R, Persson K. Activation of NLRP3 by uropathogenic Escherichia coli is associated with IL-1β release and regulation of antimicrobial properties in human neutrophils. Sci Rep. 2020;10(1):21837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rigby KM, DeLeo FR. Neutrophils in innate host defense against Staphylococcus aureus infections. Semin Immunopathol. 2012;34(2):237–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Li M, Diep BA, Villaruz AE, et al. Evolution of virulence in epidemic community-associated methicillin-resistant Staphylococcus aureus. Proc Natl Acad Sci USA. 2009;106(14):5883–5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cho JS, Guo Y, Ramos RI, et al. Neutrophil-derived IL-1beta is sufficient for abscess formation in immunity against Staphylococcus aureus in mice. PLoS Pathog. 2012;8(11):e1003047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kremserova S, Nauseef WM. Frontline Science: Staphylococcus aureus promotes receptor-interacting protein kinase 3-and protease-dependent production of IL-1β in human neutrophils. J Leukoc Biol. 2019;105(3):437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Greenlee-Wacker MC, Kremserová S, Nauseef WM. Lysis of human neutrophils by community-associated methicillin-resistant Staphylococcus aureus. Blood. 2017;129(24):3237–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.DeLeo FR, Otto M, Kreiswirth BN, Chambers HF. Community-associated meticillin-resistant Staphylococcus aureus. Lancet. 2010;375(9725):1557–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Coll RC, Schroder K, Pelegrin P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol Sci. 2022;43(8):653–668. [DOI] [PubMed] [Google Scholar]
- 140.Lahoz-Beneytez J, Elemans M, Zhang Y, et al. Human neutrophil kinetics: modeling of stable isotope labeling data supports short blood neutrophil half-lives. Blood. 2016;127(26):3431–3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Luo HR, Loison F. Constitutive neutrophil apoptosis: mechanisms and regulation. Am J Hematol. 2008;83(4):288–295. [DOI] [PubMed] [Google Scholar]
- 142.Lawrence SM, Corriden R, Nizet V. How neutrophils meet their end. Trends Immunol. 2020;41(6):531–544. [DOI] [PubMed] [Google Scholar]
- 143.Geering B, Simon HU. Peculiarities of cell death mechanisms in neutrophils. Cell Death Differ. 2011;18(9):1457–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yamashita K, Takahashi A, Kobayashi S, et al. Caspases mediate tumor necrosis factor-alpha-induced neutrophil apoptosis and downregulation of reactive oxygen production. Blood. 1999;93(2):674–685. [PubMed] [Google Scholar]
- 145.Murphy BM, O’Neill AJ, Adrain C, Watson RW, Martin SJ. The apoptosome pathway to caspase activation in primary human neutrophils exhibits dramatically reduced requirements for cyto-chrome C. J Exp Med. 2003;197(5):625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Maianski NA, Geissler J, Srinivasula SM, Alnemri ES, Roos D, Kuijpers TW. Functional characterization of mitochondria in neutrophils: a role restricted to apoptosis. Cell Death Differ. 2004;11(2):143–153. [DOI] [PubMed] [Google Scholar]
- 147.Maianski NA, Roos D, Kuijpers TW. Bid truncation, bid/bax targeting to the mitochondria, and caspase activation associated with neutrophil apoptosis are inhibited by granulocyte colony-stimulating factor. J Immunol. 2004;172(11):7024–7030. [DOI] [PubMed] [Google Scholar]
- 148.Akgul C, Moulding DA, Edwards SW. Molecular control of neutro-phil apoptosis. FEBS Lett. 2001;487(3):318–322. [DOI] [PubMed] [Google Scholar]
- 149.Akgul C, Edwards SW. Regulation of neutrophil apoptosis via death receptors. Cell Mol Life Sci. 2003;60(11):2402–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Liles WC, Kiener PA, Ledbetter JA, Aruffo A, Klebanoff SJ. Differential expression of Fas (CD95) and Fas ligand on normal human phagocytes: implications for the regulation of apoptosis in neutrophils. J Exp Med. 1996;184(2):429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Geering B, Gurzeler U, Federzoni E, Kaufmann T, Simon HU. A novel TNFR1-triggered apoptosis pathway mediated by class IA PI3Ks in neutrophils. Blood. 2011;117(22):5953–5962. [DOI] [PubMed] [Google Scholar]
- 152.Moulding DA, Akgul C, Derouet M, White MR, Edwards SW. BCL-2 family expression in human neutrophils during delayed and accelerated apoptosis. J Leukoc Biol. 2001;70(5):783–792. [PubMed] [Google Scholar]
- 153.Moulding DA, Quayle JA, Hart CA, Edwards SW. Mcl-1 expression in human neutrophils: regulation by cytokines and correlation with cell survival. Blood. 1998;92(7):2495–2502. [PubMed] [Google Scholar]
- 154.Dzhagalov I, St John A, He YW. The antiapoptotic protein Mcl-1 is essential for the survival of neutrophils but not macrophages. Blood. 2007;109(4):1620–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Fecho K, Bentley SA, Cohen PL. Mice deficient in fas ligand (gld) or fas (lpr) show few alterations in granulopoiesis. Cell Immunol. 1998;188(1):19–32. [DOI] [PubMed] [Google Scholar]
- 156.Fecho K, Cohen PL. Fas ligand (gld)-and Fas (lpr)-deficient mice do not show alterations in the extravasation or apoptosis of inflammatory neutrophils. J Leukoc Biol. 1998;64(3):373–383. [DOI] [PubMed] [Google Scholar]
- 157.Adrover JM, Del Fresno C, Crainiciuc G, et al. A neutrophil timer coordinates immune defense and vascular protection. Immunity. 2019;51(5):966–967. [DOI] [PubMed] [Google Scholar]
- 158.Lum JJ, Bren G, McClure R, Badley AD. Elimination of senescent neutrophils by TNF-related apoptosis-inducing [corrected] ligand. J Immunol. 2005;175(2):1232–1238. [DOI] [PubMed] [Google Scholar]
- 159.Martin C, Burdon PC, Bridger G, Gutierrez-Ramos JC, Williams TJ, Rankin SM. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity. 2003;19(4):583–593. [DOI] [PubMed] [Google Scholar]
- 160.Uhl B, Vadlau Y, Zuchtriegel G, et al. Aged neutrophils contribute to the first line of defense in the acute inflammatory response. Blood. 2016;128(19):2327–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Cowland JB, Borregaard N. Granulopoiesis and granules of human neutrophils. Immunol Rev. 2016;273(1):11–28. [DOI] [PubMed] [Google Scholar]
- 162.Kettritz R Neutral serine proteases of neutrophils. Immunol Rev. 2016;273(1):232–248. [DOI] [PubMed] [Google Scholar]
- 163.Sanrattana W, Maas C, de Maat S. SERPINs-from trap to treatment. Front Med (Lausanne). 2019;6:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Benarafa C, Simon HU. Role of granule proteases in the life and death of neutrophils. Biochem Biophys Res Commun. 2017;482(3):473–481. [DOI] [PubMed] [Google Scholar]
- 165.Loison F, Xu Y, Luo HR. Proteinase 3 and Serpin B1: a novel pathway in the regulation of caspase-3 activation, neutrophil spontaneous apoptosis, and inflammation. Inflamm Cell Signal. 2014;1(6):e462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Loison F, Zhu H, Karatepe K, et al. Proteinase 3-dependent caspase-3 cleavage modulates neutrophil death and inflammation. J Clin Invest. 2014;124(10):4445–4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Baumann M, Pham CT, Benarafa C. SerpinB1 is critical for neutro-phil survival through cell-autonomous inhibition of cathepsin G. Blood. 2013;121(19):3900–3907. S3901-3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ruhl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science. 2018;362(6417):956–960. [DOI] [PubMed] [Google Scholar]
- 169.Antonopoulos C, Russo HM, El Sanadi C, et al. Caspase-8 as an effector and regulator of NLRP3 inflammasome signaling. J Biol Chem. 2015;290(33):20167–20184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun. 2019;10(1):1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.de Vasconcelos NM, Van Opdenbosch N, Van Gorp H, Parthoens E, Lamkanfi M. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ. 2019;26(1):146–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.DiPeso L, Ji DX, Vance RE, Price JV. Cell death and cell lysis are separable events during pyroptosis. Cell Death Dis. 2017;3:17070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zeng CY, Li CG, Shu JX, et al. ATP induces caspase-3/gasdermin E-mediated pyroptosis in NLRP3 pathway-blocked murine macrophages. Apoptosis. 2019;24(9–10):703–717. [DOI] [PubMed] [Google Scholar]
- 174.Evavold CL, Hafner-Bratkovič I, Devant P, et al. Control of gasdermin D oligomerization and pyroptosis by the Ragulator-Rag-mTORC1 pathway. Cell. 2021;184(17):4495–4511.e4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zanoni I, Tan Y, Di Gioia M, et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science. 2016;352(6290):1232–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zanoni I, Tan Y, Di Gioia M, Springstead JR, Kagan JC. By capturing inflammatory lipids released from dying cells, the receptor CD14 induces inflammasome-dependent phagocyte hyperactivation. Immunity. 2017;47(4):697–709.e693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zhivaki D, Borriello F, Chow OA, et al. Inflammasomes within hyperactive murine dendritic cells stimulate long-lived T cell-mediated anti-tumor immunity. Cell Rep. 2020;33(7):108381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zhivaki D, Kagan JC. Innate immune detection of lipid oxidation as a threat assessment strategy. Nat Rev Immunol. 2022;22(5):322–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Naqvi N, Ahuja K, Selvapandiyan A, Dey R, Nakhasi H, Puri N. Role of Mast Cells in clearance of Leishmania through extracellular trap formation. Sci Rep. 2017;7(1):13240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ueki S, Melo RC, Ghiran I, Spencer LA, Dvorak AM, Weller PF. Eosinophil extracellular DNA trap cell death mediates lytic release of free secretion-competent eosinophil granules in humans. Blood. 2013;121(11):2074–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Miao EA, Leaf IA, Treuting PM, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11(12):1136–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Jorgensen I, Lopez JP, Laufer SA, Miao EA. IL-1β, IL-18, and eicosanoids promote neutrophil recruitment to pore-induced intracellular traps following pyroptosis. Eur J Immunol. 2016;46(12):2761–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Jorgensen I, Zhang Y, Krantz BA, Miao EA. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med. 2016;213(10):2113–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 Receptor in Infection and Inflammation. Immunity. 2017;47(1):15–31. [DOI] [PubMed] [Google Scholar]
- 185.McNeela EA, Burke A, Neill DR, et al. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog. 2010;6(11):e1001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Rabes A, Suttorp N, Opitz B. Inflammasomes in pneumococcal infection: innate immune sensing and bacterial evasion strategies. Curr Top Microbiol Immunol. 2016;397:215–227. [DOI] [PubMed] [Google Scholar]
- 187.Aachoui Y, Kajiwara Y, Leaf IA, et al. Canonical inflammasomes drive IFN-gamma to prime caspase-11 in defense against a cytosol-invasive bacterium. Cell Host Microbe. 2015;18(3):320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Aachoui Y, Leaf IA, Hagar JA, et al. Caspase-11 protects against bacteria that escape the vacuole. Science. 2013;339(6122):975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Minns MS, Lima TS, Liboro K, et al. Neutrophil IL-1β secretion induced by ExoS expressing Pseudomonas aeruginosa is dependent on NLRP3 and Gasdermin D. bioRxiv. 2022. doi: 10.1101/April7.2022.487503. [DOI] [Google Scholar]
- 190.Schubert KA, Xu Y, Shao F, Auerbuch V. The yersinia type III secretion system as a tool for studying cytosolic innate immune surveil-lance. Annu Rev Microbiol. 2020;74:221–245. [DOI] [PubMed] [Google Scholar]
- 191.Xu H, Yang J, Gao W, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature. 2014;513(7517):237–241. [DOI] [PubMed] [Google Scholar]
- 192.Shannon JG, Hasenkrug AM, Dorward DW, Nair V, Carmody AB, Hinnebusch BJ. Yersinia pestis subverts the dermal neutrophil response in a mouse model of bubonic plague. MBio. 2013;4(5):e00170–00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Spinner JL, Winfree S, Starr T, et al. Yersinia pestis survival and replication within human neutrophil phagosomes and up-take of infected neutrophils by macrophages. J Leukoc Biol. 2014;95(3):389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhang Z, Venditti R, Ran L, et al. Distinct changes in endosomal composition promote NLRP3 inflammasome activation. Nat Immunol. 2022;24:30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Seoane PI, Lee B, Hoyle C, et al. The NLRP3-inflammasome as a sensor of organelle dysfunction. J Cell Biol. 2020;219(12):e202006194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Takei H, Araki A, Watanabe H, Ichinose A, Sendo F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J Leukoc Biol. 1996;59(2):229–240. [DOI] [PubMed] [Google Scholar]
- 197.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–1535. [DOI] [PubMed] [Google Scholar]
- 198.Nauseef WM, Kubes P. Pondering neutrophil extracellular traps with healthy skepticism. Cell Microbiol. 2016;18(10):1349–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Hu JJ, Liu X, Xia S, et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol. 2020;21(7):736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Jiang K, Tu Z, Chen K, et al. Gasdermin D inhibition confers antineutrophil-mediated cardioprotection in acute myocardial infarction. J Clin Invest. 2022;132(1):e151268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Toldo S, Abbate A. The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol. 2018;15(4):203–214. [DOI] [PubMed] [Google Scholar]
- 202.Xie J, Zhu CL, Wan XJ, et al. GSDMD-mediated NETosis promotes the development of acute respiratory distress syndrome. Eur J Immunol. 2023;53(1):e2250011. [DOI] [PubMed] [Google Scholar]
- 203.Liu F, Ghimire L, Balasubramanian A, et al. Neutrophil-specific depletion of gasdermin D does not protect against sepsis. Blood. 2022. doi: 10.1182/blood.2022016931. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.