

Abstract

The physical factors governing the catalysis in Lewis acid-promoted carbonyl-ene reactions have been explored in detail quantum chemically. It is found that the binding of a Lewis acid to the carbonyl group directly involved in the transformation greatly accelerates the reaction by decreasing the corresponding activation barrier up to 25 kcal/mol. The Lewis acid makes the process much more asynchronous and the corresponding transition state less in-plane aromatic. The remarkable acceleration induced by the catalyst is ascribed, by means of the activation strain model and the energy decomposition analysis methods, mainly to a significant reduction of the Pauli repulsion between the key occupied π-molecular orbitals of the reactants and not to the widely accepted stabilization of the LUMO of the enophile.
Introduction
The Alder-ene reaction, named after its discoverer Alder in 1943,1 constitutes one of the most fundamental reactions in organic chemistry. This transformation belongs to the family of group transfer pericyclic reactions and forms a C–C bond with concomitant 1,5-hydrogen shift by the reaction of an alkene bearing an allylic hydrogen atom (ene) with a multiple bond (enophile).2 Owing to its compatibility with a number of functional groups attached either to the ene or enophile moieties, this process has been widely applied to the synthesis of complex molecules including natural products.3 A particular type of Alder-ene reaction is that involving a carbonyl group as an enophile, known as the carbonyl-ene reaction.4 This process has been also widely applied to the formation of homoallylic alcohols and, similarly, has been used in the synthesis of complex molecules such as, for instance, (+)-steenkrotin A5 or (±)-andrastin C,6 among others.
Similar to the parent ene reaction involving alkenes as enophiles, the analogous carbonyl-ene reactions also exhibit relatively high barriers, which is translated into high reaction temperatures (in the range of 150–300 °C) limiting in many instances the scope of the transformation. Despite this, the carbonyl moiety can be activated by complexation with a Lewis acid leading to a significant acceleration of the transformation, which makes these reactions feasible even at room temperature or below.7 For instance, whereas the intramolecular carbonyl-ene reactions involving the unactivated unsaturated aldehydes 1 produce alcohols 2 upon heating at 150 °C, the analogous process under Lewis acid catalysis produces the same reaction product in good yields at −78 °C (Scheme 1a).8 Similarly, good to excellent reaction yields of unsaturated alcohols 4 are achieved in the room temperature Me2AlCl-catalyzed intermolecular reaction involving aldehydes 3 and methylenecyclohexane (Scheme 1b).9
Scheme 1. Representative Examples of Lewis Acid-Catalyzed Carbonyl-Ene Reactions.

Typically, this remarkable acceleration is rationalized in terms of the stabilization of the LUMO of the enophile upon binding to the Lewis acid, which leads to a more favorable HOMO(ene)–LUMO(enophile) interaction.10 However, we recently found that this so-called LUMO-lowering concept in catalysis is rather incomplete for related Lewis acid-catalyzed Diels–Alder cycloaddition reactions.11 Instead, a remarkable decrease in the Pauli repulsion between the key occupied π-molecular orbitals and not an enhancement of orbital interactions constitutes the actual physical mechanism behind the acceleration induced by Lewis acids. This so-called Pauli repulsion lowering concept(12) seems general as it also operates in related Diels–Alder cycloaddition reactions where the catalyst establishes noncovalent interactions (hydrogen,13 halogen,14 or chalcogen bonds15) with the dienophile and even in slightly related dihalogen-catalyzed Michael-addition reactions16 and iminium-ion-catalyzed cycloadditions.17 The reduction in Pauli repulsion also plays a significant role in controlling the reactivity of uncatalyzed azadiene inverse-electron-demand Diels–Alder cycloadditions18 and even transition-metal-catalyzed reactions.19 This mechanism is also behind the acceleration observed in Lewis acid-catalyzed ene reactions involving α,β-unsaturated carbonyl compounds as enophiles (i.e., a parent ene reaction where the enophile is an alkene attached to an electron-withdrawing carbonyl group) as recently reported by Hamlin and co-workers (Scheme 2a).20 Despite this, it is unclear whether this Pauli repulsion lowering mechanism is also behind the acceleration induced by Lewis acids in carbonyl-ene reactions, where the carbonyl group is directly involved in the transformation, and therefore, the influence of the Lewis acid should be much more pronounced. This prompted us to carry out a detailed computational exploration of the factors governing the catalysis in these fundamental reactions. To this end, the intermolecular reaction involving 1-butene and acetaldehyde promoted by a wide variety of Lewis acids has been selected (Scheme 2b) and analyzed by applying the combination of the activation strain model (ASM)21 of reactivity with the energy decomposition analysis (EDA)22 method. In addition, issues such as the impact of aromaticity of the corresponding transition states (TSs) and the synchronicity on this particular process shall be also explored in detail.
Scheme 2. (a) Previous Work on Lewis Acid-Catalyzed Ene Reactions and (b) Carbonyl-Ene Reactions Considered in This Work.
Results and Discussion
We first focused on the parent uncatalyzed reaction involving 1-butene and acetaldehyde and the corresponding Lewis acid-catalyzed processes. Our calculations (PCM(dichloromethane)-ωB97xD/def2-TZVP//PCM(dichloromethane)-ωB97xD/def2-SVP level) indicate that in all cases, the reaction proceeds concertedly via an asynchronous, six-membered TS, which leads to the exergonic formation of the respective (E)-hex-4-en-2-ol (see Figure 1). Despite this, note that under different conditions (for instance, 2 equiv of Lewis acid reagent or change in the Lewis acid), a stepwise mechanism may operate.7,8 As expected, the barrier height computed for the uncatalyzed reaction is relatively high (ΔG≠ = 44.1 kcal/mol), which agrees with previous calculations23,24 and the available experimental data (see above) for related ene reactions. At variance, the analogous LA-catalyzed reactions exhibit lower activation barriers that systematically decrease with the relative Lewis acidity of the catalyst. For instance, whereas weak Lewis acids, such as Cl2 or Br2, bonded to the carbonyl group through weak halogen bonds, lead only to a slight reduction in the barrier (ΔG≠ = 43.8 and 41.7 kcal/mol, respectively), strong LAs such as BF3 or AlCl3 lead to markedly lower barriers (ΔG≠ = 22.4 and 19.3 kcal/mol, respectively, see Table 1). Therefore, a remarkable reduction of the barrier of up to ca. 25 kcal/mol can be achieved in the LA-mediated carbonyl-ene reactions, which is fully consistent with the temperatures used experimentally (see above). Our calculations confirm that the effect of the Lewis acid is much more significant than when placed remotely (Scheme 2a), where a much lower reduction of the barrier (up to 12 kcal/mol for AlCl3) was found.20 Similar values and reactivity trends were found when considering the formation of the (Z)-hex-4-en-2-ol isomer (see Table S1 in the Supporting Information).
Figure 1.

Optimized TSs involved in the considered carbonyl-ene reactions. Bond distances are given in angstroms. All data have been computed at the PCM(dichloromethane)-ωB97xD/def2-SVP level.
Table 1. Computed Activation and Reaction Energies (in kcal/mol), Synchronicity (Sy), and NICS(3,+1) Values in the Corresponding TSs (in ppm) of the Considered Carbonyl-Ene Reactions.
| catalyst | ΔG≠a | ΔE≠a | ΔGRb | ΔERb | Sy | NICS(3,+1) |
|---|---|---|---|---|---|---|
| none | 44.1 | 31.3 | –2.6 | –14.7 | 0.87 | –19.9 |
| Cl2 | 43.8 | 28.7 | –1.9 | –16.1 | 0.72 | –16.9 |
| Br2 | 41.7 | 26.8 | –2.9 | –16.6 | 0.68 | –15.9 |
| BMe3 | 34.8 | 19.8 | –4.1 | –18.5 | 0.62 | –13.5 |
| BPh3 | 28.0 | 14.1 | –6.0 | –19.6 | 0.58 | –11.2 |
| SnCl4 | 27.4 | 13.8 | –3.4 | –17.8 | 0.61 | –9.9 |
| AlMe2Cl | 25.8 | 11.6 | –6.1 | –19.8 | 0.59 | –10.3 |
| FeF3 | 26.4 | 11.6 | –3.2 | –18.3 | 0.60 | –9.6 |
| FeCl3 | 24.0 | 9.5 | –3.8 | –18.4 | 0.58 | –8.2 |
| FeBr3 | 21.8 | 7.6 | –5.7 | –20.0 | 0.58 | –8.0 |
| InCl3 | 24.7 | 10.4 | –6.7 | –20.2 | 0.61 | –10.5 |
| TiCl4 | 24.0 | 10.2 | –3.8 | –17.9 | 0.55 | –6.5 |
| BF3 | 22.4 | 9.2 | –5.9 | –18.8 | 0.58 | –8.8 |
| AlCl3 | 19.3 | 6.1 | –5.7 | –19.0 | 0.56 | –7.8 |
Activation barriers computed as ΔE≠ = E(TS) – E(1-butene) – E(aldehyde).
Reaction energies computed as ΔER = E((E)-hex-4-en-2-ol) – E(1-butene) – E(aldehyde). All data have been computed at the PCM(dichloromethane)-ωB97xD/def2-TZVPP//PCM(dichloromethane)-ωB97xD/def2-SVP level.
Closer inspection of the optimized geometries of the TSs (Figure 1) indicates that although the formation of the new C–C and O–H bonds is accompanied by the C–H bond rupture, the LA-catalyzed reactions are much more asynchronous than the parent uncatalyzed reaction. This is confirmed by the calculation of the synchronicity (Sy) of the process (see Computational Details section), which shows that whereas the uncatalyzed reaction presents a high Sy value of 0.87 (close to the value for a synchronous process, Sy = 1), the LA-mediated reactions exhibit much lower values (Sy ranging from 0.72 to 0.55), which, for those cases involving strong Lewis acids (such as AlCl3), are actually in the limit for a concerted process. This asynchronicity is manifested in both the C···C bond-forming distance, which becomes shorter in the catalyzed reactions and particularly, in the O···H bond-forming distance, which becomes much longer. Interestingly, there appears to be a correlation between the computed barriers and these geometrical parameters in the sense that the transformation is kinetically easier (i.e., proceeds with a lower barrier) when the O···H bond-forming distance is longer. Indeed, when plotting both parameters, a very good linear relationship was found (correlation coefficient of 0.93, Figure 2), which suggests that these carbonyl-ene reactions follow the Hammond–Leffer postulate.25
Figure 2.
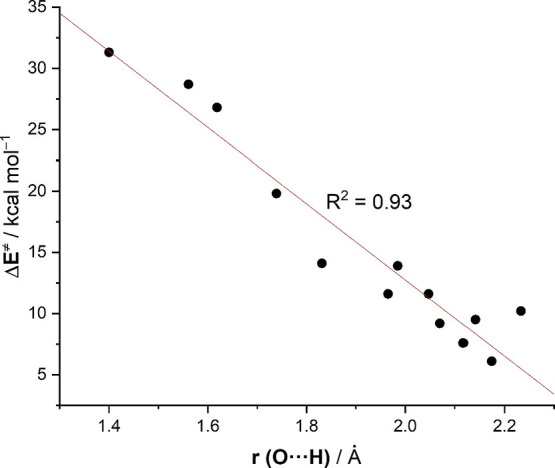
Correlation between the computed activation barriers (ΔE≠) and O···H bond-forming distances in the TSs associated with the considered carbonyl-ene reactions.
Similar to other pericyclic reactions,26 including the parent ene reaction involving an alkene as an enophile,24 the transition structure for the uncatalyzed carbonyl-ene reaction can be considered as aromatic according to the negative nuclear independent chemical shift (NICS)27 value computed at the (3,+1) ring critical point (RCP)28 of the six-membered cycle (NICS(3,+1) = −19.9 ppm, see Table 1). This highly negative NICS value results from the involvement of the six [σ2 + π2 + π2] electrons in the concerted process, which approximately lie in the molecular plane leading to a remarkable diamagnetic shielding at the RCP. Indeed, the delocalization of these six electrons becomes evident when applying the anisotropy of the induced current density (AICD)29 method, which clearly shows a diatropic (i.e., clockwise vectors) current within the six-membered ring (Figure 3, left). Moreover, the in-plane aromatic30 nature of this TS is further supported by the variation of the NICS values along the z-axis perpendicular to the molecular plane, which shows the usual bell-shaped plot having a maximum NICS value at z = 0 Å (i.e., at the RCP, see Figure S1 in the Supporting Information).
Figure 3.
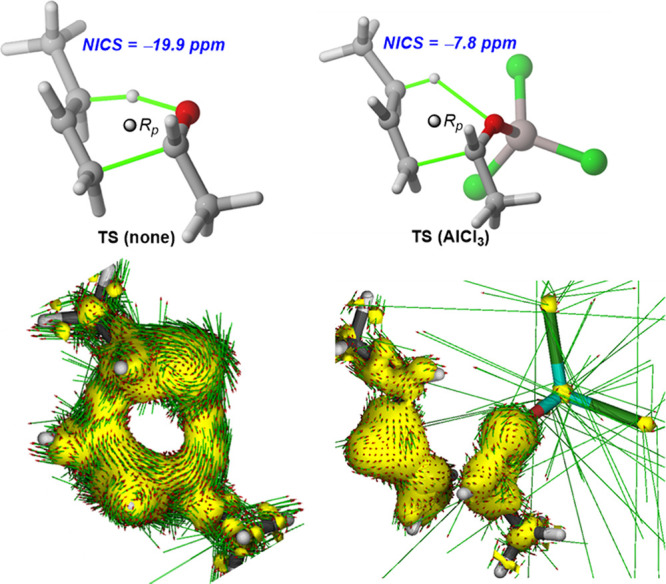
NICS values and AICD plots for the TSs associated with the uncatalyzed carbonyl-ene reaction (left) and the analogous reaction catalyzed by AlCl3 (right).
Data in Table 1 also indicate that the analogous Lewis acid-catalyzed reactions also feature in-plane aromatic TSs. However, the computed NICS(3,+1) values are systematically lower (NICS ranging from −16.9 to −6.5 ppm) than that computed for the parent uncatalyzed reaction. Once again an evident relationship between the relative Lewis acidity of the catalyst and the computed aromaticity was found. This is not surprising because, as commented above, Lewis acids lead to more asynchronous TSs, which in turn results in a less efficient delocalization of the six electrons involved in the transformation and consequently, in a lower diamagnetic shielding at the corresponding RCP. This can be also confirmed by the visualization of the corresponding ring current by the AICD method, which clearly shows, for the reaction mediated by the strong Lewis acid AlCl3, an interrupted ring current that severely hampers the electronic delocalization in the transformation (Figure 3, right), which is reflected in the computed rather low NICS value.
Despite the clear impact of the Lewis acid on the (in-plane) aromaticity of the TSs, there is no straightforward, causal physical relationship between the above computed magnetic indicators and the energetics of the transformation. Indeed, one should expect that an increase in the aromaticity should result in a gain in stability and, therefore, in a lower barrier, which is not the case. A similar finding was observed in double-group transfer reactions, related pericyclic processes, which typically involve the concerted migration of two atoms or groups from one compound to another.30d For this reason, we then applied the activation strain model (ASM)21 of reactivity to understand, in a quantitative manner, the factors leading to the acceleration induced by the Lewis acids in these carbonyl-ene reactions. To this end, we focused on the parent uncatalyzed reaction and the analogous processes mediated by the strong Lewis acid AlCl3 and a weaker acid, SnCl4 (ΔE≠ decreases in the order: 31.3 kcal/mol, none >13.8 kcal/mol, SnCl4 >6.1 kcal/mol, AlCl3). The corresponding activation strain diagrams (ASDs) for these reactions, computed from the initial stages of the transformation up to the respective TSs and projected onto the C···C bond-forming distances,31 are shown in Figure 4. Data in Figure 4 indicate that the catalyzed reactions benefit from a less destabilizing strain energy (measured by the ΔEstrain term), in particular, at the TS region. The trend in ΔEstrain (none > SnCl4 > AlCl3) can be directly ascribed to the extent of the asynchronicity (Sy decreases in the order: 0.87, none >0.61, SnCl4 >0.56, AlCl3), since a higher asynchronicity implies that the energy penalty required by the reactants (mainly the alkene in this particular transformation) to reach the TS geometry is lower. Despite this, the main factor behind the remarkable reduction in the barrier height is a significant enhancement in the interaction energy between the increasingly deformed reactants. As depicted in Figure 4, the catalyzed reactions exhibit a much stronger ΔEint along the entire reaction coordinate, and once again, the enhancement in ΔEint is directly related to the relative Lewis acidity of the catalyst. Therefore, our ASM analysis indicates that the catalysis in these carbonyl-ene reactions originates mainly from an enhancement of the interaction between the deformed reactants and, also, from a less destabilizing geometrical distortion as a consequence of a greater asynchronicity (albeit to a lesser extent).
Figure 4.
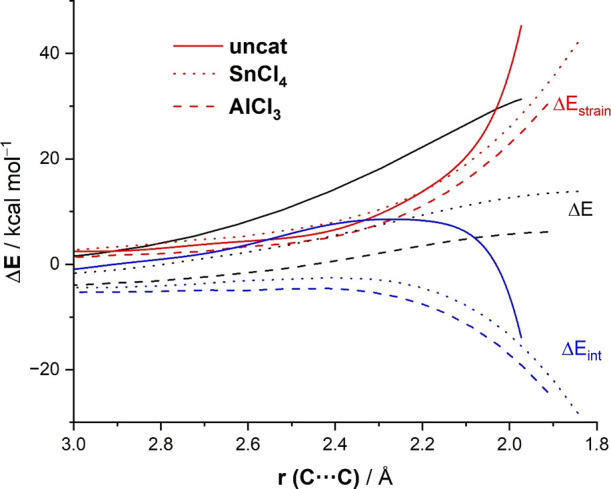
ASDs of the carbonyl-ene reactions between 1-butene and acetaldehyde (uncatalyzed, solid lines) and analogous catalyzed reactions by SnCl4 (dotted lines) and AlCl3 (dashed lines) projected onto the C···C bond-forming distance. All data have been computed at the PCM(dichloromethane)-ωB97xD/def2-TZVPP//PCM(dichloromethane)-ωB97xD/def2-SVP level.
The factors behind the stronger interaction between the deformed reactants computed for the Lewis acid-mediated reactions discussed above can be analyzed using the energy decomposition analysis (EDA).22 The change in the EDA terms along the reaction coordinate for the extreme situations represented by the uncatalyzed and AlCl3-catalyzed carbonyl-ene reactions is graphically shown in Figure 5. At the TS region, the attractive electrostatic, ΔVelstat, and orbital, ΔEorb, interactions are similar or even slightly more stabilizing for the uncatalyzed reaction, which suggests that the stronger interaction computed for the AlCl3-catalyzed reaction does not derive from either the electrostatic attractions or the orbital interactions. At variance, data in Figure 5 indicate that the catalyzed reaction clearly exhibits a much less destabilizing Pauli repulsion practically along the entire transformation. For instance, the difference in this crucial term is ΔΔEPauli = 38.0 kcal/mol (favoring the catalyzed reaction), when computed at the same consistent C···C bond-forming distance of 2.0 Å.32 This suggests that this significant reduction in the Pauli repulsion compensates the main attractive terms (ΔVelstat and ΔEorb) and becomes the exclusive factor behind the enhanced interaction computed for the catalyzed reaction. Therefore, our calculations confirm that similar to other catalyzed pericyclic reactions,11−15 the origin of the acceleration induced by Lewis acids in these carbonyl-ene reactions, where the carbonyl group is directly involved in the transformation, can be also rationalized in terms of the Pauli repulsion lowering concept.12
Figure 5.
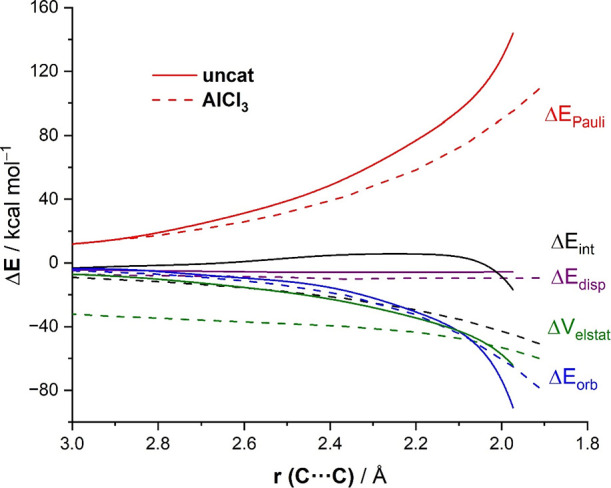
Energy decomposition analyses of the carbonyl-ene reactions between 1-butene and acetaldehyde (uncatalyzed, solid lines) and the analogous catalyzed reaction by AlCl3 (dotted lines) projected onto the C···C bond-forming distance. All data have been computed at the ZORA-ωB97xD/TZ2P//PCM(dichloromethane)-ωB97xD/def2-SVP level.
We can further investigate the reasons leading to the reduction in the crucial ΔEPauli term for the Lewis acid-catalyzed Alder-ene reactions by performing a Kohn–Sham molecular orbital analysis. It is found that the four-electron interaction between the doubly occupied HOMO of the alkene (mainly centered in the reactive C=C and C–H bonds) and the filled π–(C=O) molecular orbital of the enophile (Figure 6) constitutes the main contributor to the Pauli repulsion. At a C···C bond-forming distance of 2.0 Å,32 the corresponding HOMO(alkene)-π(C=O) orbital overlap (S) is much larger, and therefore, much more destabilizing for the uncatalyzed reaction (S = 0.043 vs S = 0.026). This reduction in the overlap for the catalyzed reaction results from the polarization, induced by the Lewis acid, of the occupied π–(C=O) molecular orbital of the enophile. This translates into a noticeable depopulation in the reactive carbon atom of the carbonyl in the acetaldehyde-AlCl3 complex as compared to the parent CH3CHO as viewed from the increase of the positive charge at this carbon atom (computed NBO-charge of +0.46 in CH3CHO vs +0.59 in CH3CHO–AlCl3). Not surprisingly, the extent of this polarization of the key π(C=O) orbital directly depends on the relative Lewis acidity of the catalyst. For instance, the corresponding charge at the reactive carbon atom in the weak Lewis acid complex CH3CHO–Cl2 (+0.48) resembles that of the parent acetaldehyde, whereas the stronger CH3CHO–SnCl4 complex exhibits a more positive value (+0.54), which of course is lower than that computed for their AlCl3-counterpart. Although the decrease in the charges is small compared to the significant changes in the Pauli repulsion, these small changes in charges translated into a dramatic lowering of Pauli repulsion. Therefore, it can be concluded that the polarization of the key π(C=O) orbital of the carbonyl group induced by the Lewis acid, which is reflected in a clear reduction of ΔEPauli, is the ultimate factor controlling the catalysis in these carbonyl-ene reactions.
Figure 6.
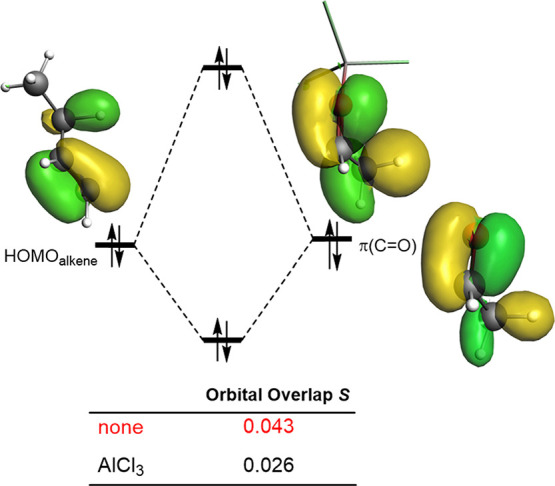
Schematic orbital interaction diagram of the main occupied orbital overlap involved in the carbonyl-ene reactions. All data have been computed at the ZORA-ωB97xD/TZ2P//PCM(dichloromethane)-ωB97xD/def2-SVP level.
Finally, we were curious to understand why the catalyzed reactions exhibit weaker orbital interactions although the corresponding enophile features a more stabilized π*(C=O) molecular orbital (for instance, 0.05 eV vs −0.73 eV, for CH3CHO and CH3CHO–AlCl3, respectively). According to the frontier molecular orbital (FMO) theory, this would result in a more favorable HOMO(alkene)−π*–(C=O) interaction and therefore, in stronger orbital interactions. However, our calculations indicate that the trend in the ΔEorb term is the opposite, i.e., slightly stronger for the uncatalyzed reaction (see above). One plausible explanation would be once again related to the marked asynchronicity of the catalyzed reactions in the sense that the longer H···O bond-forming distance should result in a less favorable HOMO−π* overlap. Indeed, our calculations confirm this hypothesis as viewed from the computed smaller overlap for the AlCl3-mediated reaction (S = 0.21) as compared to the analogous uncatalyzed process (S = 0.24). Moreover, we also applied the natural orbital for chemical valance (NOCV)33 extension of the EDA method, which allows not only the visualization but also the quantification of the main orbital interactions involved in the total ΔEorb term. As expected, the HOMO(alkene) → π*–(C=O) interaction (denoted as ρ1) method constitutes the main orbital interaction between the reactants, contributing ca. 90% to the total ΔEorb term (see Figure 7). Interestingly, and although the π*(C=O) molecular orbital becomes stabilized in the catalyzed system, the strength of this interaction is clearly higher (i.e., stronger) in the uncatalyzed reaction than in the AlCl3-catalyzed reaction (see stabilizing energies at a C···C bond-forming distance of 2.0 Å in Figure 7), which, in addition to the computed larger overlap, derives in the stronger orbital interactions ΔEorb computed for the uncatalyzed reaction. This result confirms that one should be cautious when using arguments solely derived from the energy of the FMO of the reactants, particularly in catalyzed pericyclic reactions.
Figure 7.
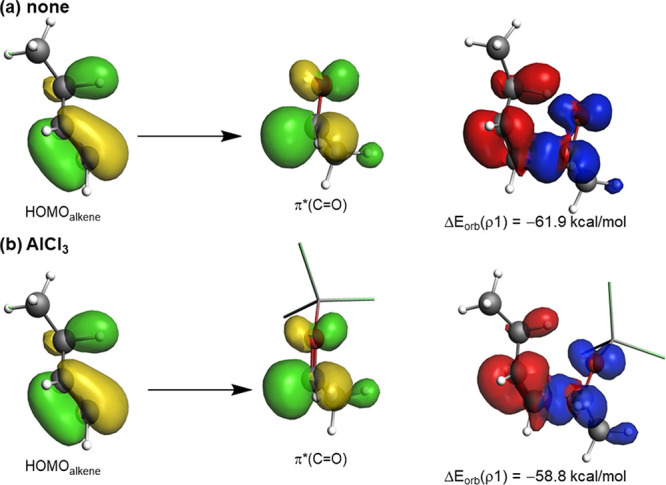
Plot of the deformation densities Δρ and associated molecular orbitals together with the corresponding stabilization energies ΔE(ρ) involved in the parent uncatalyzed carbonyl-ene reaction (a) and its AlCl3-catalyzed counterpart (b). Charge flows in the red → blue direction. All data have been computed at the ZORA-ωB97xD/TZ2P//PCM(dichloromethane)-ωB97xD/def2-SVP level.
Conclusions
From the above computational results, it is found that the binding of a Lewis acid to the carbonyl group directly involved in the considered carbonyl-ene reactions results in a remarkable reduction of the activation barrier (up to 25 kcal/mol) in comparison to the parent uncatalyzed reaction. Although the process occurs concertedly, via a six-membered, in-plane aromatic TS, the catalyzed reactions are much more asynchronous, being the extent of the asynchronicity directly related to the relative Lewis acidity of the catalyst. Interestingly, there exists a clear correlation between the barrier heights and the synchronicity in the sense that more asynchronous processes (i.e., mediated by stronger Lewis acids) are associated with lower barriers. In addition, the TSs for the catalyzed carbonyl-ene reactions are less aromatic than their analogous uncatalyzed saddle points. Moreover, their relative aromaticity strength also correlates with the asynchronicity of the process. Although the key π*(C=O) molecular orbital of the enophile becomes stabilized in the enophile-LA complexes, the observed acceleration in the catalyzed processes does not result from a more favorable HOMO(alkene)−π*(C=O) interaction, as traditionally viewed. Instead, our ASM-EDA calculations indicate that the catalysis finds its origin in a lower strain energy (as a consequence of the induced asynchronicity) and, more importantly, in a stronger interaction between the deformed reactants, which mainly results from a remarkable decrease of the destabilizing Pauli repulsion between the key π-molecular orbitals of the reagents. This confirms the general applicability of the Pauli repulsion lowering concept in catalysis.
Experimental Section
Computational Details
Geometry optimizations were performed without symmetry constraints using the Gaussian-16 (RevB.01)34 suite of programs at the dispersion-corrected ωB97xD35/def2-SVP36 level. Solvent effects (solvent = dichloromethane) were considered during the geometry optimizations by means of the polarization continuum model (PCM) method.37 This level is denoted as the PCM(dichloromethane)-ωB97xD/def2-SVP level. Reactants and adducts were characterized by frequency calculations and have positive definite Hessian matrices. TSs show only one negative eigenvalue in their diagonalized force constant matrices, and their associated eigenvectors were confirmed to correspond to the motion along the reaction coordinate under consideration using the intrinsic reaction coordinate (IRC) method.38 Natural bond order (NBO) calculations were performed with the NBO6.0 program39 at the PCM(dichloromethane)-ωB97xD/def2-SVP level. Additional single-point energy refinements were carried out at same DFT level using the much larger triple-ζ def2-TZVPP basis set.36 This level is denoted as the PCM(dichloromethane)-ωB97xD/def2-TZVPP//PCM(dichloromethane)-ωB97xD/def2-SVP level.
The aromaticity of the transition structures was assessed by the computation of the NICS27 values using the gauge invariant atomic orbital (GIAO) method40 at the B3LYP41/def2-SVP//PCM(dichloromethane)-ωB97xD/def2-SVP level. Ring currents were computed using the AICD method.29
The synchronicity42,43 of the reactions was quantified following a previously described approach.44,45 For a concerted reaction, synchronicity is defined as46
where n is the number of bonds involved in the reaction (in this case, n = 6) and δBi measures the relative variation of a given bond index Bi at the TS, according to the following formula:
where the superscripts R and P refer to the reactants and the product, respectively. The average value of δBi, denoted as δBAV is therefore
The Wiberg bond indices Bi were computed using the NBO method.39
ASM of Reactivity and EDA
Within the ASM method,21 the potential energy surface ΔE(ζ) is decomposed along the reaction coordinate, ζ, into two contributions, the strain ΔEstrain(ζ) associated with the deformation (or distortion) required by the individual reactants during the process and the interaction ΔEint(ζ) between these increasingly deformed reactants:
The EDA method22 can be used to further decompose the interaction energy into the following chemically meaningful terms:
The term ΔVelstat corresponds to the classical electrostatic interaction between the unperturbed charge distributions of the deformed reactants and is usually attractive. The Pauli repulsion ΔEPauli comprises the destabilizing interactions between occupied orbitals and is responsible for any steric repulsion. The orbital interaction ΔEorb accounts for bond pair formation, charge transfer (interactions between occupied orbitals on one moiety with unoccupied orbitals on the other, including HOMO–LUMO interactions), and polarization (empty-occupied orbital mixing on one fragment due to the presence of another fragment). Finally, the ΔEdisp takes into account those interactions coming from dispersion forces. Moreover, the NOCV (natural orbital for chemical valence)33 extension of the EDA method has been also used to further partition the ΔEorb term. The EDA-NOCV approach provides pairwise energy contributions for each pair of interacting orbitals to the total bond energy. The ASM-EDA(NOCV) approach has proven to provide detailed quantitative insight into the ultimate factors controlling fundamental processes in organic, main group, and organometallic chemistry.47
The program package ADF48 was used for EDA calculations using the optimized PCM(dichloromethane)-ωB97xD/def2-SVP geometries at the same DFT level in conjunction with a triple-ζ-quality basis set using uncontracted Slater-type orbitals (STOs) augmented by two sets of polarization functions with a frozen-core approximation for the core electrons.49 Auxiliary sets of s, p, d, f, and g STOs were used to fit the molecular densities and to represent the Coulomb and exchange potentials accurately in each SCF cycle.50 Scalar relativistic effects were incorporated by applying the zeroth-order regular approximation (ZORA).51 This level of theory is denoted as ZORA-ωB97xD/TZ2P//PCM(dichloromethane)-ωB97xD/def2-SVP.
Acknowledgments
This work was supported by the Spanish MCIN/AEI/10.13039/501100011033 (Grants PID2019-106184GB-I00 and RED2018-102387-T to I.F. and Grant PID2021-126747NB-I00 to J.I.P.), Fondo Europeo de Desarrollo Regional (FEDER), and “ERDF A way of making Europe”.
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.3c01059.
Evolution of the NICS values (Figure S1), activation and reaction energies, synchronicity, and NICS(3,+1) values (Table S1), and Cartesian coordinates (in Å) and total energies of all the stationary points discussed in the text (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Alder K.; Pascher F.; Schmitz A. Über die Anlagerung von Maleinsäure-anhydrid und Azodicarbonsäure-ester an einfach ungesättigte Koh an einfach ungesättigte Kohlenwasserstoffe. Zur Kenntnis von Substitutionsvorgängen in der Allyl-Stellung. Chem. Ber. 1943, 76B, 27–53. 10.1002/cber.19430760105. [DOI] [Google Scholar]; b Alder K.; Noble T. Über die Anlagerung von Azodicarbonsäure-ester an Aldehyde. Chem. Ber. 1943, 76B, 54–57. 10.1002/cber.19430760106. [DOI] [Google Scholar]
- Sankararaman S. in Pericyclic Reactions-A Textbook: Reactions, Applications and Theory; Wiley: Weinheim, 2005. [Google Scholar]
- For selected reviews on the application of Alder-ene reactions in synthesis, see:; a Hoffmann H. M. R. The Ene reaction. Angew. Chem., Int. Ed. 1969, 8, 556–577. 10.1002/anie.196905561. [DOI] [Google Scholar]; b Mikami K.; Shimizu M. Asymmetric ene reactions in organic synthesis. Chem. Rev. 1992, 92, 1021–1050. 10.1021/cr00013a014. [DOI] [Google Scholar]
- Clarke M. L.; France M. B. The carbonyl ene reaction. Tetrahedron 2008, 64, 9003–9031. 10.1016/j.tet.2008.06.075. [DOI] [Google Scholar]
- Pan S.; Gao B.; Hu J.; Xuan J.; Xie H.; Ding H. Enantioselective total synthesis of (+)-Steenkrotin A and determination of its absolute configuration. Chem. – Eur. J. 2016, 22, 959–970. 10.1002/chem.201503831. [DOI] [PubMed] [Google Scholar]
- Okamoto R.; Takeda K.; Tokuyama H.; Ihara M.; Toyota M. Toward the total synthesis of (±)-Andrastin C. J. Org. Chem. 2013, 78, 93–103. 10.1021/jo301948h. [DOI] [PubMed] [Google Scholar]
- Snider B. B. Lewis-acid catalyzed ene reactions. Acc. Chem. Res. 1980, 13, 426–432. 10.1021/ar50155a007. [DOI] [Google Scholar]
- Snider B. B.; Karras M.; Price R. T.; Rodini D. J. Alkylaluminum halide induced cyclization of unsaturated carbonyl compounds. J. Org. Chem. 1982, 47, 4538–4545. 10.1021/jo00144a027. [DOI] [Google Scholar]
- Snider B. B.; Rodini D. J. Dialkylaluminium chloride catalyzed ene reactions of aldehydes. Synthesis of ipsenol. Tetrahedron Lett. 1980, 21, 1815–1818. 10.1016/S0040-4039(00)92787-4. [DOI] [Google Scholar]
- a Fleming I. in Molecular Orbitals and Organic Chemical Reactions; Wiley: Hoboken, 2009. [Google Scholar]; b Clayden J.; Greeves N.; Warren S.. Organic Chemistry, 2nd ed.; Oxford University Press: Oxford, 2013. [Google Scholar]
- a Vermeeren P.; Hamlin T. A.; Fernández I.; Bickelhaupt F. M. How Lewis acids catalyze Diels-Alder reactions. Angew. Chem., Int. Ed. 2020, 59, 6201–6206. 10.1002/anie.201914582. [DOI] [PMC free article] [PubMed] [Google Scholar]; See also,; b Vermeeren P.; Tiezza M. D.; van Dongen M.; Fernández I.; Bickelhaupt F. M.; Hamlin T. A. Lewis acid-catalyzed Diels-Alder reactions: reactivity trends across the periodic Table. Chem. – Eur. J. 2021, 27, 10610–10620. 10.1002/chem.202100522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamlin T. A.; Bickelhaupt F. M.; Fernández I. The Pauli repulsion-lowering concept in catalysis. Acc. Chem. Res. 2021, 54, 1972–1981. 10.1021/acs.accounts.1c00016. [DOI] [PubMed] [Google Scholar]
- Vermeeren P.; Hamlin T. A.; Bickelhaupt F. M.; Fernández I. Bifunctional hydrogen bond donor-catalyzed Diels-Alder reactions: origin of stereoselectivity and rate enhancement. Chem. – Eur. J. 2021, 27, 5180–5190. 10.1002/chem.202004496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Portela S.; Cabrera-Trujillo J. J.; Fernández I. Catalysis by Bidentate iodine(iii)-based halogen donors: surpassing the activity of strong Lewis acids. J. Org. Chem. 2021, 86, 5317–5326. 10.1021/acs.joc.1c00534. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Portela S.; Fernández I. Nature of C-I···π halogen bonding and its role in organocatalysis. Eur. J. Org. Chem. 2021, 6102–6110. 10.1002/ejoc.202101244. [DOI] [Google Scholar]
- Portela S.; Fernández I. Understanding the catalysis by bis-selenonium cations as bidentate chalcogen bond donors. Tetrahedron Chem. 2022, 1, 100008 10.1016/j.tchem.2022.100008. [DOI] [Google Scholar]
- Hamlin T. A.; Fernández I.; Bickelhaupt F. M. How dihalogens catalyze Michael addition reactions. Angew. Chem., Int. Ed. 2019, 58, 8922–8926. 10.1002/anie.201903196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeeren P.; Hamlin T. A.; Fernández I.; Bickelhaupt F. M. Origin of rate enhancement and asynchronicity in iminium catalyzed Diels-Alder reactions. Chem. Sci. 2020, 11, 8105–8112. 10.1039/D0SC02901G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot A.; Devarajan D.; Gustafson S. J.; Fernández I.; Bickelhaupt F. M.; Ess D. H. Activation-Strain Analysis reveals unexpected origin of fast reactivity in heteroaromatic azadiene inverse-electron-demand Diels-Alder cycloadditions. J. Org. Chem. 2015, 80, 548–558. 10.1021/jo5025514. [DOI] [PubMed] [Google Scholar]
- a Hu L.; Gao H.; Hu Y.; Lv X.; Wu Y.-B.; Lu G. Computational study of silver-catalyzed stereoselective hydroalkylation of alkynes: Pauli repulsion controlled Z/E selectivity. Chem. Commun. 2021, 57, 6412–6415. 10.1039/D1CC01917A. [DOI] [PubMed] [Google Scholar]; b Gao H.; Hu L.; Hu Y.; Lv X.; Wu Y.-B.; Lu G. Origins of Lewis acid acceleration in nickel-catalysed C–H, C–C and C–O bond cleavage. Catal. Sci. Technol. 2021, 11, 4417–4428. 10.1039/D1CY00660F. [DOI] [Google Scholar]; c Gao H.; Hu L.; Hu Y.; Lv X.; Wu Y.-B.; Lu G. Origins of regioselectivity in Ni-catalyzed hydrofunctionalization of alkenes via ligand-to-ligand hydrogen transfer mechanism. Chem. Commun. 2022, 58, 8650–8653. 10.1039/D2CC02691K. [DOI] [PubMed] [Google Scholar]; d Hu L.; Gao H.; Hu Y.; Wu Y.-B.; Lv X.; Lu G. Origins of regioselectivity in CuH-catalyzed hydrofunctionalization of alkenes. J. Org. Chem. 2023, 88, 2750–2757. 10.1021/acs.joc.2c02296. [DOI] [PubMed] [Google Scholar]
- Tiekink E. H.; Vermeeren P.; Bickelhaupt F. M.; Hamlin T. A. How Lewis acids catalyze ene reactions. Eur. J. Org. Chem. 2021, 2021, 5275–5283. 10.1002/ejoc.202101107. [DOI] [Google Scholar]
- a Fernández I.; Bickelhaupt F. M. The activation strain model and molecular orbital theory: understanding and designing chemical reactions. Chem. Soc. Rev. 2014, 43, 4953–4967. 10.1039/C4CS00055B. [DOI] [PubMed] [Google Scholar]; b Bickelhaupt F. M.; Houk K. N. Analyzing reaction rates with the distortion/interaction-activation strain model. Angew. Chem., Int. Ed. 2017, 56, 10070–10086. 10.1002/anie.201701486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bickelhaupt F. M.; Baerends E. J. in Reviews in Computational Chemistry; Lipkowitz K. B., Boyd D. B., Eds.; Wiley-VCH: Weinheim, 2000; 15, 1–86. [Google Scholar]; and references therein; b Zhao L.; von Hopffgarten M.; Andrada D. M.; Frenking G. Energy decomposition analysis. WIREs Comput. Mol. Sci. 2018, 8, e1345 10.1002/wcms.1345. [DOI] [Google Scholar]
- a Loncharich R. J.; Houk K. N. Transition structures of ene reactions of ethylene and formaldehyde with propene. J. Am. Chem. Soc. 1987, 109, 6947–6952. 10.1021/ja00257a008. [DOI] [Google Scholar]; b Thomas B. E. IV; Loncharich R. J.; Houk K. N. Force field modeling of transition structures of intramolecular ene reactions and ab initio transition structures for an activated enophile. J. Org. Chem. 1992, 57, 1354–1362. 10.1021/jo00031a011. [DOI] [Google Scholar]; c Houk K. N.; Beno B. R.; Nendel M.; Black K.; Yoo H. Y.; Wilsey S.; Lee J. K. Exploration of pericyclic reaction transition structures by quantum mechanical methods: competing concerted and stepwise mechanisms. J. Mol. Struct.: THEOCHEM 1997, 398-399, 169–179. 10.1016/S0166-1280(96)04970-6. [DOI] [Google Scholar]
- Fernández I.; Bickelhaupt F. M. Alder-Ene reaction: aromaticity and activation-strain analysis. J. Comput. Chem. 2012, 33, 509–516. 10.1002/jcc.22877. [DOI] [PubMed] [Google Scholar]
- a Leffler J. E. Parameters for the description of transition states. Science 1953, 117, 340–341. 10.1126/science.117.3039.340. [DOI] [PubMed] [Google Scholar]; b Hammond G. S. A. A Correlation of reaction rates. J. Am. Chem. Soc. 1955, 77, 334–338. 10.1021/ja01607a027. [DOI] [Google Scholar]
- Schleyer P. v. R.; Wu J. I.; Cossío F. P.; Fernández I. Aromaticity in transition structures. Chem. Soc. Rev. 2014, 43, 4909–4921. 10.1039/C4CS00012A. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Wannere C. S.; Corminboeuf C.; Puchta R.; Schleyer P. v. R. Nucleus-Independent Chemical Shifts (NICS) as an aromaticity criterion. Chem. Rev. 2005, 105, 3842–3888. 10.1021/cr030088+. [DOI] [PubMed] [Google Scholar]
- This point was recommended due to its high sensitivity to diamagnetic effects and its unambiguous character, which is crucial for the considered asynchronous transition structures. See references 26 and 30.
- a Herges R.; Geuenich D. Delocalization of electrons in molecules. J. Phys. Chem. A 2001, 105, 3214–3220. 10.1021/jp0034426. [DOI] [Google Scholar]; b Geuenich D.; Hess K.; Köhler F.; Herges R. Anisotropy of the Induced Current Density (ACID), a general method to quantify and visualize electronic delocalization. Chem. Rev. 2005, 105, 3758–3772. 10.1021/cr0300901. [DOI] [PubMed] [Google Scholar]
- a Cossío F. P.; Morao I.; Jiao H.; Schleyer P. v. R. In-Plane Aromaticity in 1,3-Dipolar Cycloadditions. Solvent Effects, Selectivity, and Nucleus-Independent Chemical Shifts. J. Am. Chem. Soc. 1999, 121, 6737–6746. 10.1021/ja9831397. [DOI] [Google Scholar]; b Morao I.; Cossío F. P. A Simple Ring Current Model for Describing In-Plane Aromaticity in Pericyclic Reactions. J. Org. Chem. 1999, 64, 1868–1874. 10.1021/jo981862+. [DOI] [PubMed] [Google Scholar]; c Fernández I.; Sierra M. A.; Cossío F. P. In-Plane aromaticity in double group transfer reactions. J. Org. Chem. 2007, 72, 1488–1491. 10.1021/jo062310r. [DOI] [PubMed] [Google Scholar]; d Fernández I.; Bickelhaupt F. M.; Cossío F. P. Double group transfer reactions: role of activation strain and aromaticity in reaction barriers. Chem. – Eur. J. 2009, 15, 13022–13032. 10.1002/chem.200902024. [DOI] [PubMed] [Google Scholar]
- This critical reaction coordinate undergoes a well-defined change throughout the reaction and has successfully been used in the past for the analysis of other pericyclic reactions. See, for instance, references 11–17.
- Performing this analysis at a consistent point along the reaction coordinate (near all transition structures), rather than the TS alone, ensures that the results are not skewed by the position of the TS.
- Mitoraj M. P.; Michalak A.; Ziegler T. A. A combined charge and energy decomposition scheme for bond analysis. J. Chem. Theory Comput. 2009, 5, 962–975. 10.1021/ct800503d. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford CT, 2016. [Google Scholar]
- Chai J.-D.; Head-Gordon M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. 10.1039/b810189b. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- a Miertuš S.; Scrocco E.; Tomasi J. Electrostatic interaction of a solute with a continuum. A direct utilization of ab-initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. 10.1016/0301-0104(81)85090-2. [DOI] [Google Scholar]; b Pascual-Ahuir J. L.; Silla E.; Tuñón I. GEPOL: An improved description of molecular surfaces. III. A new algorithm for the computation of a solvent-excluding surface. J. Comput. Chem. 1994, 15, 1127–1138. 10.1002/jcc.540151009. [DOI] [Google Scholar]; c Barone V.; Cossi M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. 10.1021/jp9716997. [DOI] [Google Scholar]
- a Gonzalez C.; Schlegel H. B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. 10.1021/j100377a021. [DOI] [Google Scholar]; b Ishida K.; Morokuma K.; Komornicki A. The intrinsic reaction coordinate. An ab initio calculation for HNC→HCN and H– + CH4→ CH4 + H–. J. Chem. Phys. 1977, 66, 2153–2156. 10.1063/1.434152. [DOI] [Google Scholar]
- Glendening E. D.; Landis C. R.; Weinhold F. NBO 6.0: Natural bond orbital analysis program. J. Comput. Chem. 2013, 34, 1429–1437. 10.1002/jcc.23266. [DOI] [PubMed] [Google Scholar]
- Wolinski K.; Hilton J. F.; Pulay P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. 10.1021/ja00179a005. [DOI] [Google Scholar]
- a Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem.Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]; b Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]; c Vosko S. H.; Wilk L.; Nusair M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can. J. Phys. 1980, 58, 1200–1211. 10.1139/p80-159. [DOI] [Google Scholar]
- a Dewar M. J. S. Multibond reactions cannot normally be synchronous. J. Am. Chem. Soc. 1984, 106, 209–219. 10.1021/ja00313a042. [DOI] [Google Scholar]; b Borden W. T.; Loncharich R. J.; Houk K. N. Synchronicity in multibond reactions. Annu. Rev. Phys. Chem. 1988, 39, 213–236. 10.1146/annurev.pc.39.100188.001241. [DOI] [Google Scholar]
- Leroy has proposed the term “asynchronism” in similar contexts. See:; Leroy G.; Sana M. Etude theorique de la cycloaddition dipolaire 1–3. Tetrahedron 1975, 31, 2091–2097. 10.1016/0040-4020(75)80200-6. [DOI] [Google Scholar]
- Moyano A.; Pericás M. A.; Valentí E. A theoretical study on the mechanism of the thermal and the acid-catalyzed decarboxylation of 2-oxetanones (.beta.-lactones). J. Org. Chem. 1989, 54, 573–582. 10.1021/jo00264a014. [DOI] [Google Scholar]
- For an alternative method to compute synchronicity based on the Electron Localization Function (ELF) method, see:; Merino P.; Chiacchio M. A.; Legnani L.; Delso I.; Tejero T. Introducing topology to assess the synchronicity of organic reactions. Dual reactivity of oximes with alkenes as a case study. Org. Chem. Front. 2017, 4, 1541–1554. 10.1039/C7QO00233E. [DOI] [Google Scholar]
- a Lecea B.; Arrieta A.; Lopez X.; Ugalde J. M.; Cossío F. P. On the stereochemical outcome of the catalyzed and uncatalyzed cycloaddition reaction between activated ketenes and aldehydes to form cis- and trans-2-oxetanones. an ab initio study. J. Am. Chem. Soc. 1995, 117, 12314–12321. 10.1021/ja00154a034. [DOI] [Google Scholar]; b Fernández I.; Sierra M. A.; Cossío F. P. DFT study on the diels-alder cycloaddition between alkenyl-M(0) (M = Cr, W) carbene complexes and neutral 1,3-dienes. J. Org. Chem. 2008, 73, 2083–2089. 10.1021/jo702257s. [DOI] [PubMed] [Google Scholar]
- For recent reviews showing different applications of the ASM-EDA approach see:; a Fernández I. Understanding the Reactivity of Polycyclic Aromatic Hydrocarbons and Related Compounds. Chem. Sci. 2020, 11, 3769–3779. 10.1039/D0SC00222D. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Vermeeren P.; Hamlin T. A.; Bickelhaupt F. M. Chemical reactivity from an activation strain perspective. Chem. Commun. 2021, 57, 5880–5896. 10.1039/D1CC02042K. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Fernández I. Understanding the reactivity of frustrated Lewis pairs with the help of the activation strain model–energy decomposition analysis method. Chem. Commun. 2022, 58, 4931–4940. 10.1039/D2CC00233G. [DOI] [PubMed] [Google Scholar]; d Sengupta A.; Li B.; Svatunek D.; Liu F.; Houk K. N. Cycloaddition reactivities analyzed by Energy Decomposition Analyses and the Frontier Molecular Orbital model. Acc. Chem. Res. 2022, 55, 2467–2479. 10.1021/acs.accounts.2c00343. [DOI] [PubMed] [Google Scholar]
- a te Velde G.; Bickelhaupt F. M.; Baerends E. J.; Fonseca Guerra C.; van Gisbergen S. J. A.; Snijders J. G.; Ziegler T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. 10.1002/jcc.1056. [DOI] [Google Scholar]; b Baerends E. J.; Ziegler T.; Atkins A. J.; Autschbach J.; Baseggio O.; Bashford D.; Bérces A.; Bickelhaupt F. M.; Bo C.; Boerrigter P. M.; Cappelli C.; Cavallo L.; Daul C.; Chong D. P.; Chulhai D. V.; Deng L.; Dickson R. M.; Dieterich J. M.; Egidi F.; Ellis D. E.; van Faassen M; Fan L.; Fischer T. H.; Förster A.; Fonseca Guerra C.; Franchini M.; Ghysels A.; Giammona A.; van Gisbergen S. J. A.; Goez A.; Götz A. W.; Groeneveld J. A.; Gritsenko O. V.; Grüning M; Gusarov S.; Harris F. E.; van den Hoek P.; Hu Z.; Jacob C. R.; Jacobsen H.; Jensen L.; Joubert L.; Kaminski J. W.; van Kessel G.; König C.; Kootstra F.; Kovalenko A.; Krykunov M. V.; Lafiosca P.; van Lenthe E.; McCormack D. A.; Medves M.; Michalak A.; Mitoraj M.; Morton S. M.; Neugebauer J.; Nicu V. P.; Noodleman L.; Osinga V. P.; Patchkovskii S.; Pavanello M.; Peeples C. A.; Philipsen P. H. T.; Post D.; Pye C. C.; Ramanantoanina H.; Ramos P.; Ravenek W.; Reimann M.; Rodríguez J. I.; Ros P.; Rüger R.; Schipper P. R. T.; Schlüns D.; van Schoot H.; Schreckenbach G.; Seldenthuis J. S.; Seth M.; Snijders J. G.; Solà M.; Stener M.; Swart M.; Swerhone D.; Tognetti V.; te Velde G.; Vernooijs P.; Versluis L.; Visscher L.; Visser O.; Wang F.; Wesolowski T. A.; van Wezenbeek E. M.; Wiesenekker G.; Wolff S.K.; Woo T. K.; Yakovlev A. L.. ADF 2020, SCM, Theoretical Chemistry; Vrije Universiteit: Amsterdam, The Netherlands. [Google Scholar]
- Snijders J. G.; Vernooijs P.; Baerends E. J.. Roothaan-Hartree-Fock-Slater atomic wave functions. single-zeta, double-zeta, and extended Slater-type basis sets for 87Fr-103Lr. Atomic Data and Nuclear Data Tables, 1981, 483–509.
- Krijn J.; Baerends E. J.. Fit Functions in the HFS-method: internal report; Vrije Univ: Amsterdam, Netherlands: 1984. [Google Scholar]
- a van Lenthe E.; Baerends E. J.; Snijders J. G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. 10.1063/1.466059. [DOI] [Google Scholar]; b van Lenthe E.; Baerends E. J.; Snijders J. G. Relativistic total energy using regular approximations. J. Chem. Phys. 1994, 101, 9783–9792. 10.1063/1.467943. [DOI] [Google Scholar]; c van Lenthe E.; Ehlers A.; Baerends E. J. Geometry optimizations in the zero-order regular approximation for relativistic effects. J. Chem. Phys. 1999, 110, 8943–8953. 10.1063/1.478813. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.