

Abstract
P2Y14 receptor (P2Y14R) is activated by extracellular UDP-glucose, a damage-associated molecular pattern that promotes inflammation in the kidney, lung, fat tissue, and elsewhere. Thus, selective P2Y14R antagonists are potentially useful for inflammatory and metabolic diseases. The piperidine ring size of potent, competitive P2Y14R antagonist (4-phenyl-2-naphthoic acid derivative) PPTN 1 was varied from 4- to 8-membered rings, with bridging/functional substitution. Conformationally and sterically modified isosteres included N-containing spirocyclic (6–9), fused (11–13), and bridged (14, 15) or large (16–20) ring systems, either saturated or containing alkene or hydroxy/methoxy groups. The alicyclic amines displayed structural preference. An α-hydroxyl group increased the affinity of 4-(4-((1R,5S,6r)-6-hydroxy-3-azabicyclo[3.1.1]heptan-6-yl)phenyl)-7-(4-(trifluoromethyl)-phenyl)-2-naphthoic acid 15 (MRS4833) compared to 14 by 89-fold. 15 but not its double prodrug 50 reduced airway eosinophilia in a protease-mediated asthma model, and orally administered 15 and prodrugs reversed chronic neuropathic pain (mouse CCI model). Thus, we identified novel drug leads having in vivo efficacy.
Graphical Abstract
INTRODUCTION
Activation of the Gi protein-coupled P2Y14 receptor (P2Y14R, previously designated GPR105) is proinflammatory in various cell types, and its principal native full agonist UDP-glucose (UDPG) is considered a damage-associated molecular pattern (DAMP).1–5 In classically activated M1 macrophages, the endogenous P2Y14R agonist UDPG results in transcription factor STAT1 activation, which induces the expression of proinflammatory mediators.1 In mouse epithelial cells, the receptor activates the production and secretion of interleukin-8 (IL-8) murine homologues (i.e., CXCL1 and CXCL2) following renal ischemia-reperfusion injury, which was attenuated with a P2Y14R antagonist. A marked elevation of IL-1β was almost completely abolished by the P2Y14R antagonist.2–4 The renal P2Y14R in collecting duct intercalated cells mediates chemokine expression, followed by neutrophil and monocyte infiltration. These neutrophils and monocytes “attack” proximal tubule cells that were weakened by ischemia and induce proximal tubule damage.4 In a protease-induced asthma model, P2Y14R activation on eosinophils increases their chemokinesis, resulting in the passage of eosinophils from the blood to the lungs and increased airway inflammation, which is reduced by antagonists.6
Selective P2Y14R antagonists are desired for their anti-inflammatory effect, given the proinflammatory activity of UDPG. Other UDP-sugars, i.e., UDP-galactose, UDP-glucuronic acid, UDP-N-acetylgalactosamine, and UDP-N-acetylglucosamine activated P2Y14R with lower potency,7 and UDP has been characterized as a potent partial agonist.8 Thus, P2Y14R antagonists could potentially be useful for the treatment of inflammation, kidney disease, asthma, diabetes, obesity, chronic pain, gout, and neurofibromatosis.6,9–13 On the other hand, P2Y14R activation in autologous cardiac progenitor cell therapy might benefit heart failure.14 An important pharmacological tool molecule for this receptor is the selective antagonist 4-phenyl-2-naphthoic acid derivative 1 (PPTN, Chart 1), originally identified in a patent by Belley et al.15 Compound 1 was shown by Harden and colleagues to bind selectively to the P2Y14R as a high-affinity antagonist with a Ki value of 0.43 nM in reversing agonist-induced inhibition of cAMP production.16 We later introduced a fluorescent binding assay that is suitable for routine drug screening at the P2Y14R, and 1 displayed IC50 values of 6–8 nM in a whole-cell fluorimetric assay.5,17
Chart 1.
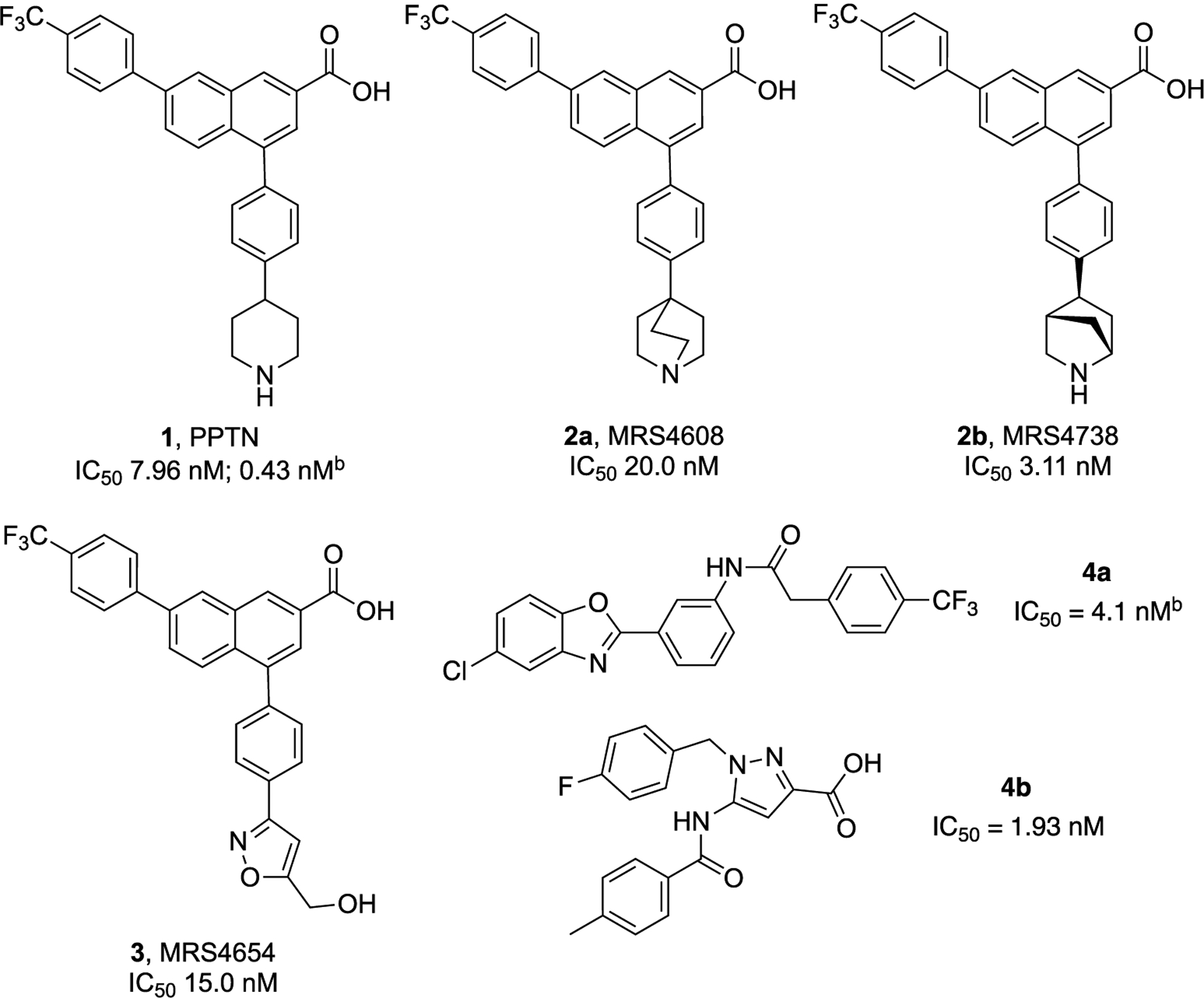
Stuctures and Reported Potencya of Representative P2Y14R Antagonists, Derived from Lead Piperidine 1 (Including C-Bridged 2 and Uncharged 3 Bioisosteres) and Recently Reported 2-Phenyl-benzoxazole Acetamide 4a and 5-Amido-1H-pyrazole-3-carboxylic Acid 4b Derivatives 20,21
bInhibitory activity in a cAMP assay.16,20aIC50 in a fluorescent binding assay, unless noted.
We are exploring the structure–activity relationship (SAR) of 4-phenyl-2-naphthoic acid derivatives related to 1 as P2Y14Rantagonists, both as pharmacological tool molecules and as potential translational compounds. Previous publications have focused on all regions of the zwitterionic 1 scaffold and have identified both charged and uncharged bioisosteres.5,17 Human (h) P2Y14R structural modeling based on the X-ray crystallo-graphic structure of the closely homologous ADP-responsive P2Y12R has aided in the selection of target molecules to synthesize based on their predicted binding to the P2Y14R orthosteric site.5,17 However, 1 is amphiphilic and has low oral bioavailability (5% F, in mouse).18 To address this short-coming, orally active prodrugs of 1 and its recently introduced analogues have been successfully identified using an N,N-dimethylamidomethyl esterification of the important naphthalene-carboxylic acid moiety.5,18,19
Recent SAR studies have focused on the modification or replacement of the piperidine moiety of 1, which is predicted to point outward from the P2Y14R orthosteric binding site.5,10,17 We have utilized that insight to make chain extensions from the piperidine N that still retain receptor affinity and in vivo efficacy in pain and asthma models. Charged amino derivatives, e.g., 2a and 2b and an uncharged isoxazole 3, were found to contain suitable isosteric replacements for the piperidine ring. Among the many charged derivatives we reported, quinclidine 2a and (1S,4S,5S)-5-phenyl-2-azabicyclo[2.2.1]heptane (a 2-azanorbornane) 2b derivatives displayed notably high affinity.5,17 Thus, constraining the piperidine ring of 1 in a receptor-preferred conformation was a productive means of increasing three-dimensionality and at the same time preserving or enhancing binding affinity. Other chemotypes have been reported to be P2Y14R antagonists, including the 2-phenyl-benzoxazole acetamide derivative 4a and 5-amide-1H-pyrazole-3-carboxyl derivative 4b.20,21
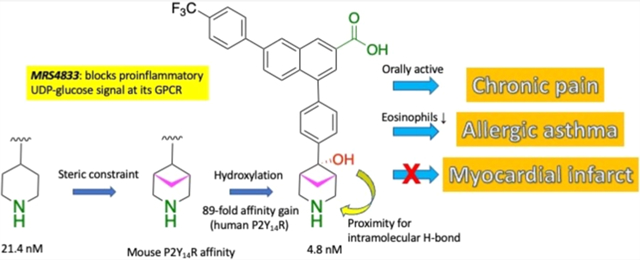
Here, we have varied the ring size of the alicyclic amine of 1 from four to eight atoms, with bridging and functional group substitution, including spiro and fused cyclo(aza)aliphatic rings. The occurrence, conformation, and utility of various cycloalkyl rings in experimental drugs have been reviewed.22–24 These new derivatives complement our recent study of analogues in which the piperidine moiety has been sterically constrained with one- and two-carbon bridges.5 Surprisingly, not only were prodrug derivatives protective in a chronic pain model (either mono-ester or mixed ester/carbamate double prodrugs), but a potent, sterically constrained zwitterionic parent drug 15 (with mP2Y14R affinity 4.5-fold higher affinity than reference antagonist 1) was highly efficacious upon oral administration.
RESULTS AND DISCUSSION
Selection of Compounds Prepared.
In an effort to explore other scaffolds reported to be P2Y14R antagonists as potential lead compounds, in addition to the piperidine series of 1, we considered recently reported antagonists 4a (Zhou et al.) and 4b (Wang et al.).20,21 We resynthesized the uncharged, small molecular P2Y14R antagonist 4a in the 2-phenyl-benzoxazole acetamide series (Scheme S1, Supporting information, compound 46 in Zhou et al.20). We evaluated the compound in a whole cell (hP2Y14R-expressing CHO cells) binding assay using 52 (Supporting Information), a fluorescent analogue of 1 used in our previous studies.5 However, no binding inhibition by 4a was observed up to 100 μM, indicating a lack of binding to the receptor orthosteric site (Table S1, Figure S1, Supporting Information). Therefore, it was not a suitable lead for further derivatization as a competitive antagonist. We did not perform a cAMP assay of 4a or test the possibility of allosteric receptor modulation.
We then returned our attention to the 4-phenyl-2-naphthoic acid series related to 1 and continued the SAR exploration of this high-affinity series.5,17 There is considerable steric and conformational freedom to vary the piperidine moiety, as noted earlier. By varying the ring size and introducing bridging and fused rings as well as functional groups (Table 1), we aimed to identify antagonists with nM affinity at the P2Y14R. Although we recently reported many bicyclic variations of piperidine within this chemical series and identified (S,S,S)-2-azanorbornane derivative 2b as a particularly potent P2Y14R antagonist, there were additional potential bioisosteres of piperidine that could be compared.
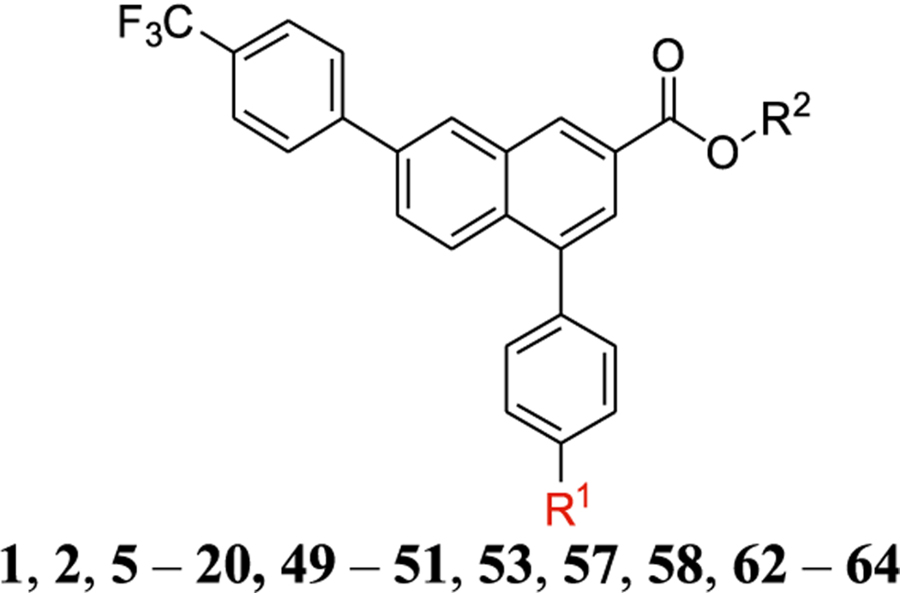
Table 1.
Structure and Affinity at hP2Y 14 R of Derivatives of Potent Antagonist 1 i
| ||||||
|---|---|---|---|---|---|---|
| No. | Structure, R1 = | IC50 (nM) | Ki (nM)d | Log D7.4e | cLogDf | log([brain]: [blood]f |
| 4-membered rings | ||||||
| 5 |
|
65.9±19.3 | 38.3 | 1.4±0.0 | 2.7 | −0.869 |
| 6 |
|
46.6±7.1 | 27.1 | 1.9±0.0 | 3.1 | −0.782 |
| 7 |
|
9.69±3.56 | 5.63 | 1.9±0.0 | 2.9 | −0.717 |
| 8 |

|
20.8±5.6 | 12.1 | 0.81±0.0 | 2.2 | −0.954 |
| 9 b |
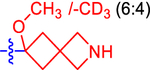
|
46.5±10.0 | 27.0 | 1.8±0.0 | 2.7 | −0.835 |
| 5-membered rings | ||||||
| 10 a |
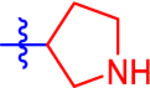
|
31.5±4.2 | 18.3 | 1.6±0.0 | 2.8 | −0.869 |
| 11 a |

|
9.48±1.66 | 5.51 | 1.1±0.0 | 2.4 | −1.08 |
| 12 a |
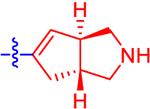
|
16.6±3.2 | 9.65 | 2.2±0.0 | 3.1 | −0.643 |
| 13 a |
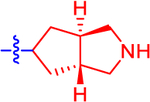
|
32.0±11.2 | 18.6 | 2.4±0.0 | 3.1 | −0.877 |
| 6-membered rings | ||||||
| 1 c |

|
7.96±0.35 | 4.63 | 1.7±0.0 | 2.9 | −0.867 |
| 2a c |
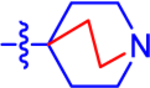
|
20.0±4.4 | 11.6 | 4.2±0.95 | 3.5 | −0.577 |
| 2b c |
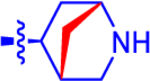
|
3.11±0.62 | 1.81 | 1.9±0.15 | 2.9 | −0.874 |
| 14 |
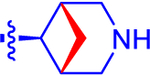
|
524±104 | 305 | 2.8±0.0 | 3.1 | −0.878 |
| 15 |
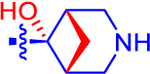
|
5.92±0.55 | 3.44 | 1.2±0.0 | 2.4 | −1.08 |
| 7-membered rings | ||||||
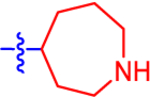
| 16 a |
|
9.58±5.14 | 5.57 | 2.1±0.0 | 3.0 | −0.861 |
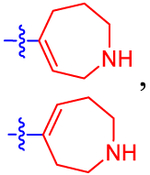
| 17 b |
|
11.9±3.6 | 6.92 | 2.2±0.0 | 2.9 | −0.612 |
| 8-membered rings | ||||||
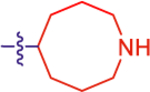
| 18 |
|
18.5±4.8 | 10.8 | 2.5±0.0 | 3.1 | −0.854 |
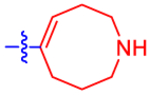
| 19 |
|
5 8.4± 16.4 | 34.0 | 2.9±0.0 | 3.0 | −0.616 |
| 20 |
|
1780±440 | 1030 | ND | 2.2 | −1.06 |
| prodrug derivatives of 1 and IS | ||||||
| 49 |
|
4810±1160 | 2800 | ND | 3.3 | 0.291 |
| 50 |
|
ND | ND | ND | 5.4 | −0.953 |
| 51 |
|
81.6±4.9 | 47.4 | ND | 3.3 | −1.24 |
| 62 |
|
ND | ND | ND | 6.4 | −0.595 |
| 63 c |
|
ND | ND | ND | 4.0 | 0.502 |
| 64 c |

|
ND | ND | ND | 6.5 | −0.664 |
| fluorescent derivative 53 and precursors | ||||||
| 53 | See Figure 1 | 25,900h | 9240h | ND | 3.6 | −1.46 |
| 57 | See Scheme S2 | 424±82 | 247 | ND | 4.3 | −1.06 |
| 58 | See Scheme S2 | 116±43g | 67.4 | ND | 3.7 | −1.08 |
Racemate.
Mixture.
Calculated from IC50 values by dividing by 1.72 (see Figure S1, Supporting Information).
Distribution coefficient Log D7.4 calculated from high-performance liquid chromatography (HPLC) retention factors k of ligands using the calibration curve of Log D7.4 vs k generated from five standards with reported log D7.4. (HPLC column: Agilent Eclipse XDB-C18 (4.6 mm × 250 mm, 5 μm); Mobile phase: A: acetonitrile, B: 10 mM TEAA, 10%−100% A in 20 min; Flow rate: 1 mL/min).
cLog D and blood–brain barrier (BBB) ratio calculated using the StarDrop program (v. 7.3.2, https://www.optibrium.com/stardrop-installers).25
Fluorescent binding IC50 value for long-chain amino derivative 58 was reported previously as 133 ± 13 nM.26
n = 1.
The affinity was determined using a whole-cell fluorescent binding assay (fluorescent tracer 52).5,17 R2 = H, and n = 3–4, unless noted. The piperidine moiety of 1 is shown in blue, while the various bridging groups, added functional groups, and other ali(hetero)cyclic rings are shown in red. ND, not determined.
Chemical Synthesis.
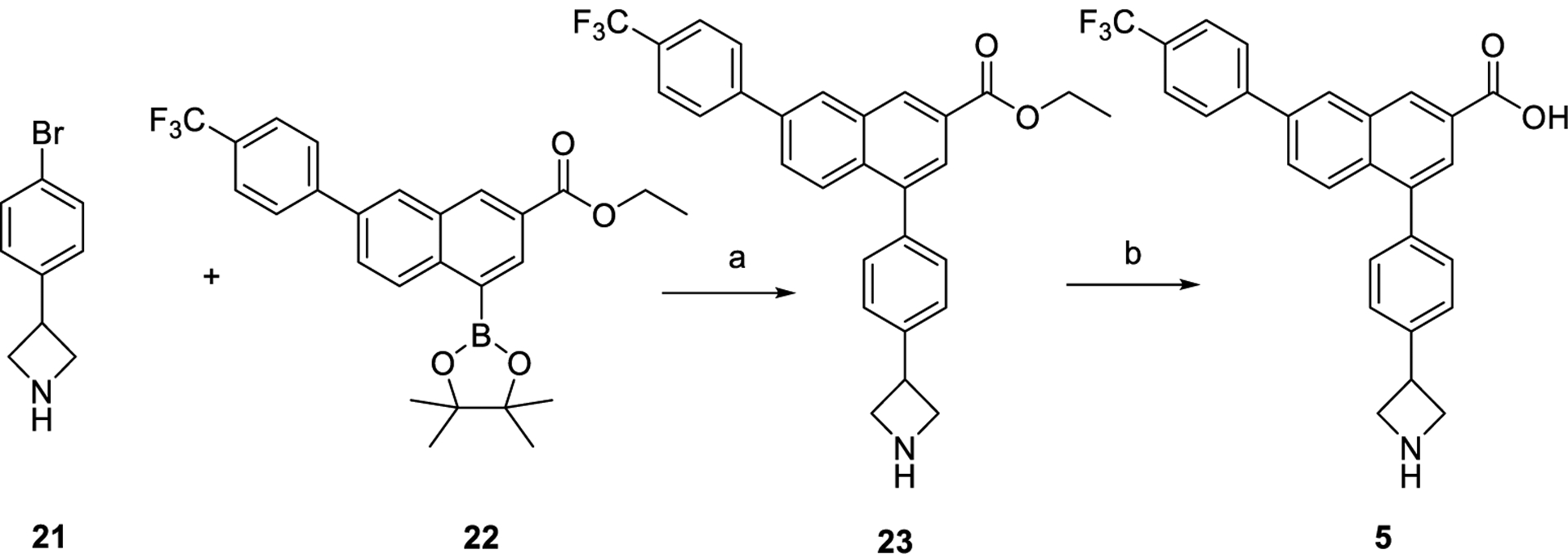
The preferred synthesis of P2Y14R antagonists based on 1 as a lead includes, sequentially, a Grignard reaction of a ketone-containing starting material for the generation of a bromophenyl intermediate, a Suzuki coupling of the bromophenyl intermediate, such as 21 (Scheme 1), with boronic acid pinacol ester 22 to obtain the 4-phenyl-2-naphthoic acid core structure, ester hydrolysis to a free carboxylic acid, Boc/benzyl deprotection to a free amine, and conversion of some of the analogues obtained to their related ligands. Thus, the smallest ring analogue, i.e., azetidine analogue 5, was prepared via Suzuki coupling between commercially available 3-(4-bromophenyl)azetidine 21, without N-protection, and a naphthalenyl boronic acid pinacol ester 2241 followed by ester hydrolysis. A small-scale hydrolysis reaction (6 mg of 23) was performed followed by HPLC purification, which gave a relatively low yield of 47%.
Scheme 1.
Synthesis of an Analogue of 1 Containing Azetidinea
aReagents and conditions: (a) Na2CO3, Pd(PPh3)4, 1,2-dimethoxyethane (DME)-H2O (4:1), 80 °C, overnight, 57%; (b) LiOH, tetrahydrofuran (THF)-MeOH-H2O (3:1:1), rt, 3 h, 47%.
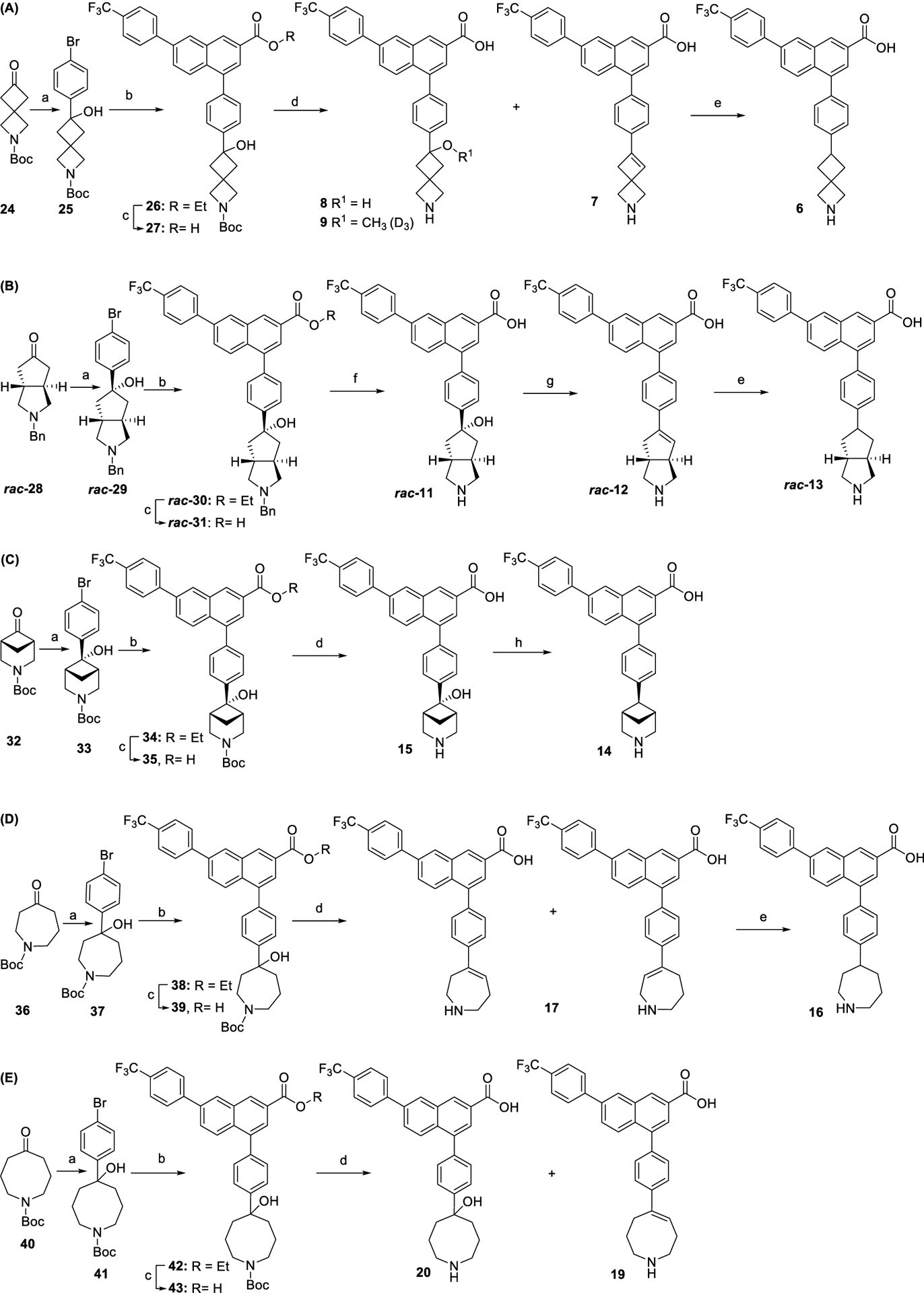
The preparation of analogues of 1 with piperidine substitutions consisting of 2-azaspiro[3.3]heptane (Scheme 2A), octahydrocyclopenta[c]pyrrole (B), 3-azabicyclo[3.1.1]-heptane (C), 1-azacycloheptane (D), and azocane (E) are summarized in Scheme 2. In a one-pot reaction, treatment of ketones 24, 28, 32, 36, and 40 with in situ generated Grignard reagent 4-bromophenylmagnesium bromide provided bromophenyl intermediates 25, 29, 33, 37, and 41, respectively. Derived from a racemic starting material 28, intermediate 29 is a racemic mixture of left-handed and right-handed helical enantiomers. A stereoselective Grignard reaction of 32 yielded enantiomer 33 with a hydroxyl group and one-carbon bridge identified as trans to each other.
Scheme 2.
Preparation of Analogues of 1 with 2-Azaspiro[3.3]heptane (A), Octahydrocyclopenta[c]pyrrole (B), 3-Azabicyclo[3.1.1]heptane (C), 1-Azacycloheptane (D), and Azocane (E) Moieties a
aReagents and conditions: (a) 4-bromophenylmagnesium bromide (in situ generated: dibromobenzene, magnesium, THF, sonication, rt, 2 h), THF, 0 °C, 4 h, 39–81%; (b) boronic acid pinacol ester 22, Na2CO3, Pd(PPh3)4, DME-H2O (4:1), 85 °C, overnight, 57–93%; (c) LiOH, THF MeOH-H2O (3:1:1), rt, 3 h, quantitative; (d) trifluoroacetic acid (TFA), dichloromethane (DCM), rt, 1 h, 18–52%; (e) H2, Pd/C, N,N-dimethylformamide (DMF), 3 h, 22–51%; (f) H2, Pd/C, DMF, 5 h, 35% overall yield from 30; (g) TFA, 90 °C, 2 h, 33% overall yield from 31; (h) NaBH4, THF, H2SO4, rt, 30 min, 11%.
The Suzuki coupling of naphthalenyl boronic acid pinacol ester 22 with the 4-(4-bromo-phenyl) intermediates 25, 29, 33, 37, and 41 gave their corresponding esters of analogues 26, 30, 34, 38, and 42, respectively. The ester hydrolysis of 26, 30, 34, 38, and 42 provided the derivatives 27, 31, 35, 39, and 43, respectively. Boc deprotection of 27 with TFA in DCM offered product 8 in addition to dehydrated product 7. Compound 8 was unstable in the reaction residue in the presence of methanol and partially reacted with methanol and methanol-d4 to give O-methylated ligand 9, and its isotopic ratio was estimated by NMR. Thus, methanol should be avoided if a methylated by-product is not needed. Similarly, Boc deprotection of 43 gave product 20 and dehydrated compound 19. Nevertheless, Boc deprotection of 39 meanwhile dehydrated the hydroxyl group to yield alkene mixture 17. Boc deprotection of compound 35 simply gave α-hydroxyl-3-azabicyclo[3.1.1]heptan-6-yl derivative 15. Pd-catalyzed de benzylation of 31 with H2 afforded ligand 11, which was further dehydrated by refluxing in TFA to give 12. Reduction of 7, 12, and 17 by Pd-catalyzed hydrogenation offered analogues of 1, i.e., 6 with 2-azaspiro[3.3]heptane (Scheme 2A), 13 with octahydrocyclopenta[c]pyrrole (B), and 16 with 1-azacycloheptane (D), respectively. Stereoselective deoxyge-nation5 of 15 with sodium borohydride in sulfuric acid afforded 14. Thus, hygroscopic sulfuric acid was employed to readily generate a tertiary benzylic carbocation, which was further reduced by sodium borohydride to yield deoxygenated product.
Compound 18 would be obtained from 19 via hydrogenation using hydrogen gas (condition e, Scheme 2). Nevertheless, the preparation of 19 from dehydration of 43 in TFA/DCM (Scheme 2E) suffered from a low yield. Thus, an alternative synthetic strategy was developed as shown in Scheme 3. Dehydration27 of 5-hydroxylazocane 41 using triphosgene and 4-dimethylaminopyridine (DMAP) under mild conditions gave alkene 44 in a quantitative yield. The Suzuki coupling of 22 with 44 gave ester 45. Reduction catalyzed by Pd/C28 of 45 was carried out using triethylsilane as the hydrogen source, in which hydrogen was generated and used for reduction in situ. Boc deprotection of 46 followed by ester hydrolysis provided the azocane analogue 18 with an overall yield of 54% from 45 after three reaction steps (steps c, d, and e), during which crude residues from steps c and d were used directly for the next steps without column purification.
Scheme 3.

Preparation of Azocane Analogue 18 Using Alternative Synthetic Strategiesa
aReagents and conditions: (a) triphosgene, DMAP, DCM, rt, 2 h, quantitative; (b) boronic acid pinacol ester 22, Na2CO3, Pd(PPh3)4, DME-H2O (4:1), 85 °C, overnight, quantitative; (c) Et3SiH, 10% Pd/C, MeOH/THF = 1:5, rt, 30 min, quantitative; (d) TFA, THF, rt, 20 min, quantitative; (e) LiOH, THF-MeOH-H2O (3:1:1), rt, 2 h, overall yield 54% from 45.
Our previous study5 demonstrated unanticipated potent in vivo activity in a mouse asthma model of single (ester or carbamate29) and double prodrugs of 2b. Also, a N,N-dimethylamidomethyl ester prodrug of 1 was previously shown to be orally bioavailable.18 Using an analogous strategy to prepare prodrugs of potent antagonist 15, a N,N-dimethylamidomethyl ester prodrug 49 was prepared in two steps from the amino-protected parent carboxylic acid 35 via esterification using 2-chloro-N,N-dimethylacetamide in the presence of base Cs2CO3 to give 48 (Scheme 4). The Boc deprotection of 48 gave ester prodrug 49. The reaction of 49 with ethyl chloroformate provided the double prodrug 50 bearing both ester and carbamate moieties. The carbamate prodrug 51 was synthesized from the treatment of the parent drug 15 with ethyl chloroformate. Table 1 also lists, for comparison, the corresponding double prodrug (ester/carbamate) of 1, i.e., compound 62, and two previously reported prodrugs of 2b, i.e., 63 and 64.
Scheme 4.

Preparation of Double (A) and Mono (A and B) Prodrugs of 15 (i.e., 49–51) as well as Double Prodrug 62 of 1 (C)a
aReagents and conditions: (a) 2-chloro-N,N-dimethylacetamide, Cs2CO3, DMF, rt, overnight, 56%-quantitative; (b) TFA, DCM, rt, 1 h, 59%; (c) ethyl chloroformate, N,N-diisopropylethylamine, DMF, rt, overnight, 17–62%.
Absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties were calculated using the StarDrop software (Tables 1 and S6, Supporting Information).25 Most compounds were predicted to be excluded from crossing the BBB, but active drugs 2a, 7, 12, 17, and 19, and prodrug 49 had profiles consistent with some bioavailability in the brain (≥19% of blood level). All of these active drugs, except 19, were of relatively high affinity (IC50 ≤ 20 nM) at the hP2Y14R. Compounds 5–8 and 10–20 each had only 5 rotatable bonds with MW ~500. Key antagonist 15 had Log D of 2.37, TPSA of 70 Å2, 3 H-bond donors, 4 H-bond acceptors, and ligand efficiency (LE) of 0.311, which is within a drug-like range.49
Pharmacological Evaluation.
The synthesized analogues were compared in hP2Y14R affinity in a fluorescent whole-cell competitive binding assay, by measuring displacement by flow cytometry using the antagonist fluorescent tracer 52 (20 nM), which contains an AlexaFluor488 fluorophore (Figure 1).30 Although a sub-nanomolar hP2Y14R affinity of 52 was previously determined in a cAMP assay,30 we reexamined its affinity in a fluorescent binding saturation study in whole CHO cells expressing the hP2Y14R. A KD value of 27.8 ± 9.3 nM (n = 5) was determined as shown in Figure 1. This KD value allowed using the Cheng-Prusoff equation31 to convert the IC50 values to Ki values, which we report here for both the newly prepared compounds and those from our previous SAR studies5 of this chemical series (Table 1). Except for monoester 49 and monocarbamate 51, most of the mono and double prodrugs were not measured in the binding assays, because previous experience indicates very low P2Y14R affinity for similar derivatives lacking a free carboxylate.30
Figure 1.
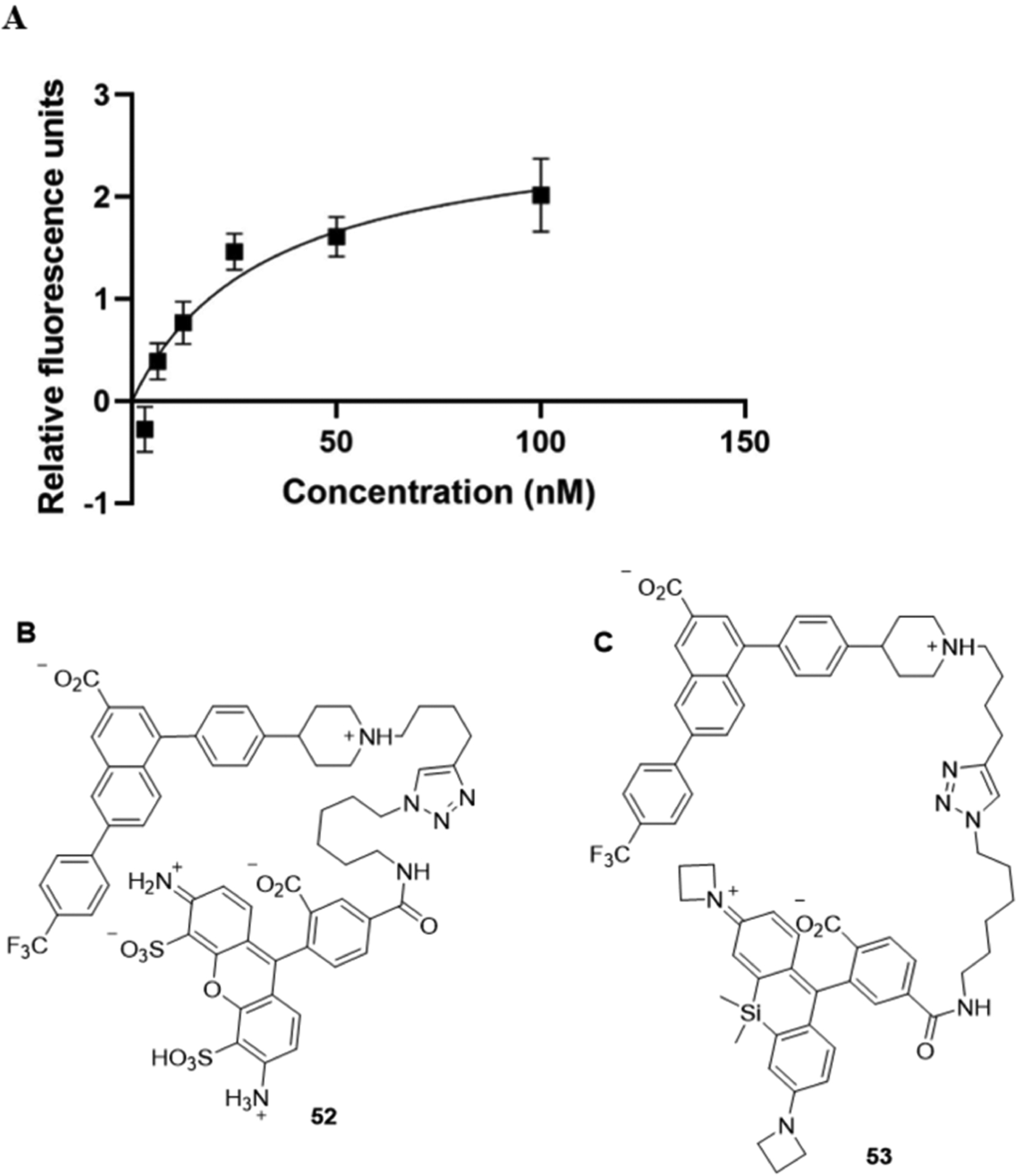
Representative saturation curve (A) for specific binding of 52 (B) to the hP2Y14R in hP2Y14R-expressing CHO cells, as determined using flow cytometry. (C) Structure of the newly prepared fluorescent conjugate 53 of JaneliaFluor 64632 with the same pharmacophore as 52.
We have used a bitopic fluorescent probe compound 52 as the basis of our receptor binding assay. In an effort to improve our in vitro assay, we attached a novel tricyclic fluorophore (JaneliaFluor 646) that has advantages for protein labeling32 to an amino-functionalized intermediate, i.e., 58, used previously to prepare 52 (Scheme S2, Supporting Information). However, this new fluorescent conjugate 53 did not demonstrate potent specific binding in whole CHO cells expressing the hP2Y14R under the similar conditions used for 52 within a range of final concentrations of 53 of 2, 100, 500, and 20,000 nM (Supporting Information) compared to a concentration of 52 of 20 nM. This surprising result emphasized that distal functional groups on the fluorophore coupled to the antagonist pharmacophore can have a major effect on the receptor interactions.
A piperidine moiety is commonly found in a wide range of pharmaceuticals and natural products.33 Systematic approaches to identify piperidine bioisosteres and bridged piperidines as phenyl bioisosteres have been reported for diverse targets34,35 and for P2Y R antagonists.5 We have substituted the piperidine moiety of 1 with nitrogen-containing spirocyclic (6–9), fused (11–13), and bridged (14, 15) or large (16–20) ring systems, either fully saturated or containing one alkene or hydroxy/methoxy group. The P2Y14R affinity varied considerably, from IC50 5.9 nM (15) to >500 nM, indicating that although this moiety binds in a relatively flexible receptor region as predicted in earlier modeling,5 there is still a steric and conformational preference for this moiety.
Four-membered and spiro ring substitution has been applied widely in medicinal chemistry.36 In the current series such substitution in 5–9 led to intermediate P2Y14R affinity, except for unsaturated analogue 7, which displayed an IC50 of 9.69 nM. The highest affinity among five-membered ring analogues was with racemic fused 5-hydroxyoctahydrocyclopenta[c]-pyrrol-5-yl derivative 11, with an IC50 of 9.48 nM. An 2-azaspiro[3.3]heptane analogue was only moderately potent in P2Y14R binding, but its affinity was enhanced 4.8-fold upon introduction of unsaturation in 7. 2-Azaspiro[3.3]heptane was previously identified as a piperidine bioisostere.37 However, it is to be noted that the success of a given bioisostere is target-dependent due to the constraints of each binding environment.
Given the high affinity of piperidine derivative 1 and its bicyclic analogues 2a and 2b,5,17 we introduced a fused four-membered ring on the piperidine ring of 1 in 14 and 15. This variation on a six-membered ring substituent displayed high affinity, only in the case of (1R,5S,6r)-6-aryl-3-azabicyclo-[3.1.1]heptan-6-ol derivative 15 (IC50 5.92 nM), containing a α-hydroxyl group trans to the carbon bridge. The seven-membered ring analogues 16 and 17 also diplayed relatively high affinity with IC50 values of ~10 nM at the P2Y14R. The benefits and conformational properties of seven-membered rings in medicinal chemistry have recently been reviewed.38 However, the eight-membered ring analogues 18–20 were found to have the lowest affinity among these derivatives (Table 1).
Selected compounds were additionally evaluated in binding to the mouse (m) P2Y14R expressed in HEK-293 cells (Table 2) using a whole-cell fluorescent binding assay with fluorescent tracer 52. Five out of the nine analogues compared at the two species had IC50 values within a factor of 2. Potent reference antagonist 2b was 11-fold weaker at the mP2Y14R, while racemic (3aR,6aR)-1,2,3,3a,4,6a-hexahydrocyclopenta[c]-pyrrol-5-yl derivative 12 was 3-fold more potent at the mP2Y14R.
Table 2.
Affinity of Selected Antagonists at the mP2Y14R, Including Published Data on 1 and 25,17
| compound | IC50 (nM) | IC50 ratio (mouse/human) |
|---|---|---|
| 1 | 21.6 ± 7.0 | 2.71 |
| 2a | 21.4 ± 7.9 | 1.07 |
| 2b | 33.9 ± 4.6 | 10.9 |
| 7 | 15.9 ± 6.0 | 1.64 |
| 11 | 6.78 ± 1.58 | 0.715 |
| 12 | 5.57 ± 1.51 | 0.336 |
| 15 | 4.80 ± 0.70 | 0.811 |
| 16 | 25.7 ± 4.6 | 2.68 |
| 18 | 11.6 ± 0.9 | 0.627 |
The starkest contrast of two closely related alicyclic amines substituted for the piperidine moiety of 1, among both α-hydroxy and non-α-hydroxy analogues, was seen with the 3-azabicyclo[3.1.1]heptan-6-yl system in 14 and 15. The presence of an α-hydroxyl group increased the affinity by roughly 100-fold. However, there was only a small effect of a hydroxyl group in B compared to piperidine analogue A (compound 1, Table 1). In our previous study (Table 3),5 a hydroxyl group present on the piperidine ring α-carbon fused to the phenyl ring had variable effects. In the case of nortropane derivative C (IC50 38.3 ± 2.0 nM), a hydroxyl group in D, cis-oriented with respect to the ethylene bridge, reduced affinity (IC50 117 ± 38 nM). However, a hydroxyl group (reversed stereochemistry) greatly increased the P2Y14R affinity of F (IC50 21.3 ± 1.1 nM) compared to E (IC50 696 ± 100 nM).
Table 3.
Previously Published Structures with Piperidine Ring Variation Showing the Effect on P2Y14R Binding Affinity Associated with an α-Hydroxyl Substituent
| R = | ICso (P2Y14R), nMa | Reference |
|---|---|---|
|
|
2.2b | Belley et al.15 |
|
|
6.3 b | Belley et al.15 |
|
|
38.3±2.0 | Wen et al.5 |
|
|
117±38 | Wen et al.5 |
|
|
696±100 | Wen et al.5 |
|
|
21.3±1.2 | Wen et al.5 |
Human P2Y14R, unless noted.
Nonhuman primate (chimpanzee) P2Y14R.
Off-target radioligand binding activity of five analogues (7, 11, and 15–17) at diverse receptors was examined by the NIMH Psychoactive Drug Screening Program (PDSP). Weak μM interactions were found, reflecting the relative selectivity of these antagonists for their target receptor (Supporting Information).39 Binding (Ki, μM) to the σ1 receptor (16, 0.89; 17, 0.84) and σ2 receptor (7, 2.2; 16, 2.8; 17, 3.9) was observed for several analogues. Additional interactions were observed at the serotonin 5-HT1B (7, 6.7; 11, 5.4; 15, 6.0; 16, 4.5; 17, 4.1), 5-HT5A (11, 2.4; 16, 4.8; 17, 4.0), muscarinic M5 (7, 5.0; 15, 6.0), histamine H1 (7, 5.6; 16, 2.8), adrenergic β3 (7, 6.8; 16, 6.6; 17, 6.3), α1B (7, 7.3; 11, 6.4; 17, 7.4), α2A (11,5.7) and κ-opioid (17, 0.46) receptors.
The ligands’ lipophilicities were determined based on their HPLC retention time in comparison to known compounds.5,40 Using the five standard compounds with reported distribution coefficient Log D7.4 (Table S2, Supporting Information), a calibration line of Log D7.4 vs retention factor k (Figure S3) was plotted for calculating HPLC-based Log D7.4 of the analogues. The Log D7.4 values are summarized in Figure S4 and Table 1. The lipophilicities of all of the ligands were consistent with their structures. The lipophilicities of ligands increased as the heterocyclic ring size increased from 4- to 8-membered (from compounds 5, 10, 1, and 16 to 18). Most of other compounds had higher lipophilicities compared to lead compound 1, except 8, 11, 15, and 20 that all had a hydroxyl group at the benzyl position of the alicyclic amino substituent. All of these less lipophilic active drugs, except 20, bound with relatively high hP2Y14R affinity (IC50 ≤ 20 nM). However, none of these more polar derivatives were predicted to cross the blood–brain barrier (BBB).
Experimental studies of the absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of compound 15 were carried out. Both in vitro and in vivo results indicated that the compound was suitable for further testing in animal models of disease (Table S3, Supporting Information). Four doses of 15 (1, 3, and 10 mg/kg, i.p. and 0.5 mg/kg, i.v.) were administered in male Wistar rats and compared to our previous data determined using the same methods for 2b.5 No mortality or morbidity was observed in any of the groups, and all animals were normal throughout the experimental period. Although oral bioavailability was not examined, the drug has a substantial half-life in plasma following i.p. administration (Figure 2). The t1/2 values of 2b and 15 (3 mg/kg) were 8.6 and 4.3 h, respectively. Strikingly, the bioavailability of 2b and 15 diverged with 43 and 114%F (3 mg/kg, i.p.), respectively. The drug exposure (AUC0-last) was proportional to the dose for 15 but not for 2b, and the clearance (Cl) was nearly identical across doses for 15 but not for 2b. The volume of distribution (Vd) was considerably lower for 15 compared to 2b, indicating a tendency to remain in the plasma. This parameter was also consistent with its lower lipophilicity (Log D7.4 of 1.2) compared to many of the other analogues, including 2b (Log D7.4 of 1.9).
Figure 2.

Plasma levels of compound 15 following administration to male Wistar rats. Pharmacokinetic (PK) parameters and conditions are shown in Table S3A,C,D (Supporting Information). Half-life values (i.p. dose, h) were: 1.0 mg/kg, 3.01; 3.0 mg/kg, 4.32; 10.0 mg/ kg, 4.15. Half-life at 0.5 mg/kg, i.v. was 2.36 h. Maximal plasma concentration (mg/kg i.p., μM): were: 1.0, 0.50; 3.0, 1.3; 10.0, 4.0.
Compound 2b was more stable than 15 in simulated gastric fluid, both were stable in simulated intestinal fluid, and neither compound displayed liver cell toxicity (Table 4). Compounds 2b and 15 showed IC50 values >30 μM for all 5 CYPs tested.The hERG activity was determined separately in a fluorescent assay (method in the Supporting Information), and both compounds showed weak hERG inhibition with IC50 > 30 μM.The plasma protein binding of the two antagonists was high in three species.
Table 4.
In Vitro ADMET Values for P2Y 14 R Antagonist 15 in This Study, Compared to Reference Antagonist 2b a
| test | 2bb | 15 |
|---|---|---|
| simulated intestinal fluid (% remaining at 120 min) | 99.1 | 93.3 |
| simulated gastric fluid (% remaining at 120 min) | 100a | 69.8 |
| CYP1A2 (IC50, μM) | >30 | >30 |
| CYP2C9 (IC50, μM) | >30 | >30 |
| CYP2C19 (IC50, μM) | >30 | >30 |
| CYP2D6 (IC50, μM) | >30 | >30 |
| CYP3A4 (IC50, μM) | >30 | >30 |
| plasma stability (3 species)c(% remaining at 120 min) | 82.8 (r); 100 (m) | 81.3 (r); 100 (m); 91.1 (h) |
| microsomal stability (t1/2, min) | 243 (m), 91.0 (r), 280 (h) | 355 (m), 342 (r), >400 (h) |
| hERG, IC50 (μM) | >30 | >30 |
| hepG2 (human hepatoma) cell toxicity, IC50 (μM)a | 11.0 | 32.6 ± 5.0 |
| plasma protein binding (3 species, % bound) | 99.76 (m), 99.79 (r), 87.98 (h) | 99.86 (m), 99.48 (r), 99.30 (h) |
| solubility, (μg/mL) | 1.90 (pH 7.4)d | 7.1 ± 0.4 (pH 4.0), 1.4 ± 0.1 (pH = 7.4)e |
Procedures are described in Jung et al.17 hERG inhibition was measured using a fluorescent dye binding method (Supporting Information).
Data determined in Wen et al.5
Species tested for plasma stability were human, rat, and mouse; species as indicated for microsomal stability.
Mean ± standard deviation (SD), pION method, determined by Jai Research Foundation (JRF) India of JRF Global (Gujarat, India).
Mean ± SD, pION method, determined at NIH.
The pharmacokinetic behavior of reference prodrugs 62 and 63 was characterized in mouse indicating modest oral bioavailability of 62 (Table S5, Supporting Information) and rapid cleavage of the ester group of 63. The in vivo pharmacokinetics of mono-ester prodrug 63, administered subcutaneously (10 mg/kg, s.c.) in female CD1 mice was studied (Table S4, Supporting Information). The product (active drug) was the 2-azanorbornyl derivative 2b, and the conversion in vivo to 2b was so rapid that the rate of loss of 63 could not be followed. 2b reached a peak concentration (~2 μg/mL) between 2 and 4 h, with a t1/2 of 2.63 h. The AUCinf value was 15,300 ng/mL*h. The total amount of 2b excreted in the urine was 1550 ng. For comparison with 63, the corresponding mono-ester of active drug 1 was studied for plasma stability in vitro. Both 63 and the mono-ester of 1 were rapidly hydrolyzed in mouse plasma (t1/2 ~1.5 min), but not human plasma (t1/2 >720 min). In a separate study, the in vivo pharmacokinetics of double prodrug 62 (containing both ester and carbamate prodrug moieties) was determined following administration by oral gavage (p.o.) in female CD1 mice (Table S5, Supporting Information). The resulting active drug was compound 1, and in this case both the prodrug and active drug were detectable, and an in vivo time course was established for the conversion. 62 reached a peak concentration (~2 μg/mL) at 1 h, with a t1/2 of 1.45 h (6.9% F), and 1 reached a peak concentration at 7 h, with a Cmax of 33 ng/mL. The AUCinf values were 1780 ng/mL*h for 62 and 515 ng/mL*h for 1. The species dependence of t1/2 values and the complexity of the 2-step unmasking of 62 suggest that additional pharmacokinetic studies of the various prodrugs will be needed.
Several antagonists, potent active drugs 11, 15, and 16, mono-ester prodrug 63 and double prodrugs 50, 62, and 64, were subjected to an in vivo mouse model of chronic neuropathic pain (chronic constriction injury, CCI),41 as applied previously to various P2Y14R antagonists in the same chemical series.10 The reversal of established chronic neuropathic pain by all analogues is shown in Figure 3. Both administration by oral gavage (Figure 3A,B) and by i.p. injection (for prodrugs 62 and 64 (Figure 3C,D); for active drugs 11, 15, and 16, Figure S8, Supporting Information) provided benefit. Compound 15 achieved full reversal of mechano-allodynia within 0.5 h post-injection (i.p.), as well as nearly complete protection at 1 h post oral gavage. A monoester prodrug 63 of previously reported constrained antagonist 2b was fully efficacious within 1 h post oral gavage. The protection by 11 (i.p.) was maintained over 3 h, although the maximal efficacy was not as complete as for 15. We then compared 1 and 15 administered by oral gavage in the same CCI experiment (Figure 3E). Both antagonists reached full reversal of mechano-allodynia, but the duration of the protection by 15 was longer than protection by 1. Therefore, we have shown a more favorable in vivo profile for 15 compared to the original lead compound 1. Even with a low oral bioavailability of 1 in the mouse, a sufficient concentration of 1 was reached to demonstrate efficacy in the CCI model.
Figure 3.
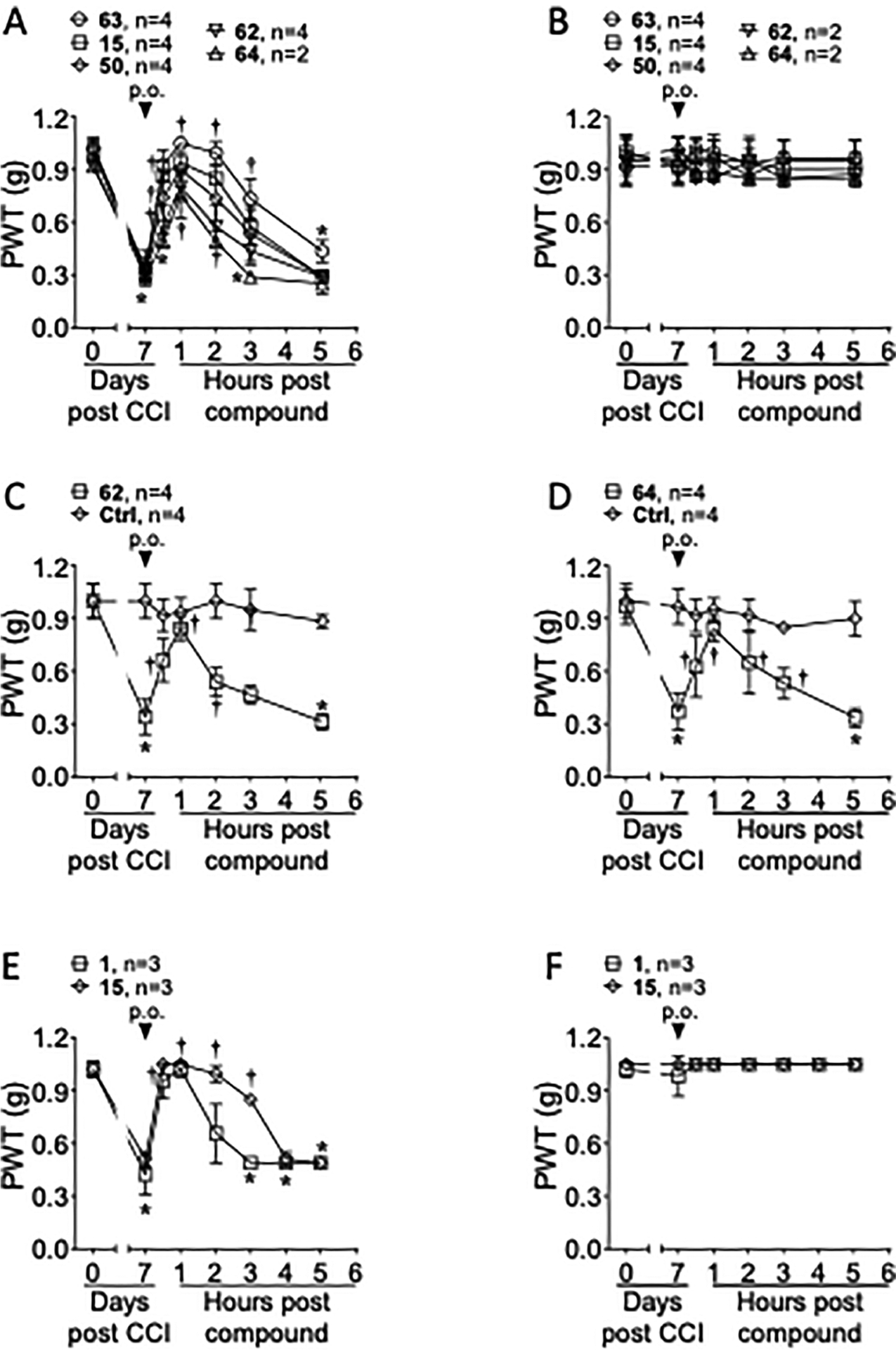
Representative time and dose dependence for reversal of established neuropathic pain in adult male mice by five newly synthesized P2Y14R antagonists and reference compound 1. The drugs were injected 7 days post-sciatic nerve constriction. The paw withdrawal threshold (PWT) was determined using von Frey filaments applied to the postsurgical hindpaw.41 (A–D) Single injection (10 μmol/kg, p.o.) of a P2Y14 R antagonist (active drug 15 or prodrugs 50 and 62–64) reversed the mechano-allodynia on the ipsilateral, nerve-injured hindpaw. (E, F) Reference antagonist 1 was compared to the potent antagonist 15, with both administered by oral gavage. A single p.o. administration (10 μmol/kg) of 1 or 15 reversed the mechano-allodynia. For all experiments, the drug injection had no effect on the contralateral hindpaw (B, F). Data were analyzed by twoway analysis of variance (ANOVA) with Dunnett’s comparisons, *P <0.05 versus day 0 and †P < 0.05 versus day 7. Data represent the mean ± SD. The vehicle used for the oral dosing consisted of 5% dimethyl sulfoxide (DMSO) + 10% (5% Kolliphor HS 15:DMSO, 5:95 by volume) in 0.5% methyl cellulose (0.2 mL dose). The vehicle used for the i.p. injection: 10% (5% Kolliphor HS 15:DMSO, 5:95 by volume) in saline (0.2 mL dose). Results with other antagonists (11, 15, and 16; i.p.) in the mouse CCI model are shown in Figure S8 (Supporting Information).
The unexpected oral activity of 15 in the mouse CCI model might be a function of the spatial proximity of its hydroxyl group to the secondary amine, which would effectively reduce the zwitterionic character (Figure S9, Supporting Information). Intramolecular H-bonds have been reported to increase the permeability, including oral bioavailability, of polar compounds.42,43 The hydroxyl group H is 2.4–4.1 Å away from the amine N, while H-bond distances are typically 2.7–3.3 Å, suggesting a possible H-bond between the OH and N at the bridged piperidine.
Two compounds, 3-azabicyclo[3.1.1]heptan-6-yl active drug 15 and its double prodrug 50 (Figure 4) were examined in an in vivo mouse model of allergic asthma, which was used previously to demonstrate the efficacy of compounds 1, 2b and others to reduce eosinophilic lung inflammation.5,6 Compound 15, but not the prodrug 50, demonstrated significant protection in this asthma model, with fewer eosinophils in the bronchoalveolar lavage (BAL) fluid. There were no statistically significant reductions of lymphocytes, macrophages, or neutrophils in the BAL fluid for any of the antagonists (Figure S7, Supporting Information), although there was a trend toward lower lymphocytes with 1 and 15. The reason for the lack of activity of the double prodrug is not evident, given that the analogous double (ester and carbamate) prodrug of antagonist 2b was highly efficacious in the same protease model of asthma.5
Figure 4.

Ability of compound 15, but not its double (ester and carbamate) prodrug 50, and the reference antagonist 1 to reduce airway eosinophilia in a protease-mediated mouse model of allergic asthma. Results with other BAL fluid cells are shown in Figure S7 (Supporting Information).
Myocardial infarction (MI) denotes the death of cardiac myocytes due to extended ischemia, leading to the invasion of inflammatory neutrophils and macrophages,44 both of which highly express the P2Y14 R.1 Myocardial reperfusion is the restoration of coronary blood flow after a period of coronary occlusion. Reperfusion has the potential to salvage ischemic myocardium but paradoxically can cause injury. Myocardial ischemia and reperfusion lead to an inflammatory response that causes further damage to viable tissue around the infarct. Acute and chronic immune responses elicited by myocardial ischemia have an important role in the functional deterioration of the heart.
Compound 15 was tested in a model of protection from cardiac ischemia-reperfusion (I/R) injury in the mouse (Figure S10, Supporting Information). 15 was administered at 2 mg/kg/day in a minipump (1 mL/h) prior to 30 min of myocardial ischemia and 48 h of reperfusion in an established mouse model.45,46 The results showed similar AAR/TTA (area at risk/total area of left ventricle) of 33.7 and 32.6% in 15- and vehicle-treated animals, respectively. 15 did not change the infarct size with similar IF/AAR at 31.8% in the drug-treated group (n = 12) vs 26.4% in the control group (n-12, P =0.124). Although this antagonist appears to have good potency and plasma concentration in rodents, 15 provided no protection against heart I/R injury in the mouse, raising the possibility of no role for P2Y14R antagonists in this disease model. However, it is possible that different dosing would demonstrate efficacy.
The potential use of P2Y14R antagonists in disease treatment has motivated us to continue the SAR probing of high-affinity naphthalene-based antagonists. There are envisioned applications of such antagonists at peripheral sites, such as the kidney, but their use in the CNS has been underexplored. Although several studies have indicated pain as a viable target to explore, there is a lack of BBB-penetrant antagonists, which will be addressed in future studies. Recently, a putative UDPG vesicular transporter, SLC35D3, has been identified in the brain,47 thus reinforcing the need for a broader selection of antagonists.
In conclusion, as an extension of our previous SAR studies, we have synthesized P2Y14R antagonists that are modified with alternative alicyclic and heteroalicyclic substituents in place of the piperidine moiety of the lead antagonist 1. The rank order of affinity at hP2Y14R (IC50 of 3–19 nM) was: 15 > 11, 16, 7 > 17 > 12 > 18. At mP2Y14R, IC50 values of <12 nM were observed for 11, 12, 15, and 18. We have seen that in various cases, achieving the highest affinity in a fluorescent whole-cell binding assay requires the presence of an α-hydroxyl group on the alicyclic ring attached to a 1,4-disubstituted phenyl moiety. The structural basis for the preference of certain constrained alicyclic structures can later be explored using computational modeling. Preliminary ADMET studies indicated that 6-hydroxy-3-azabicyclo[3.1.1]heptan-6-yl derivative 15 was well tolerated in vivo in rat, and the exposure of the drug was considerable, which is predictive of potential target engagement with the receptor. Among the compounds with the highest affinity, protective efficacy was observed in mouse models of chronic disease conditions. We demonstrate favorable in vivo activity of compound 15 and other P2Y14R antagonists in chronic pain and asthma models. Surprisingly, active zwitterionic 15 and 1 were both efficacious in reducing chronic pain when administered orally in the mouse. However, there was no protection by 15 infused post-MI in the mouse on the size of damaged area or cardiac function compared to control. These compounds can be further tested in vivo, such as in kidney inflammation models,48 to determine their suitability as candidates for disease treatment.
EXPERIMENTAL SECTION
Chemical Synthesis.
General Information. Materials and Methods.
All chemicals and anhydrous solvents were obtained directly from commercial sources. Bromo (21) and ketone (24, 28, 32, 36, and 40) starting materials were purchased from Enamine (Kyiv, Ukraine). All reactions were carried out under an argon atmosphere using anhydrous solvents unless specified otherwise. Room temperature (rt) refers to 25 ± 5 °C. Silica gel precoated with F254 on aluminum plates was used for thin-layer chromatography (TLC). The spots were examined under ultraviolet light at 254 nm and further visualized, where needed, by anisaldehyde or cerium ammonium molybdate stain solution. Column chromatography was performed on silica gel (40–63 μm, 60 Å). 1H NMR spectra were recorded on a Bruker 400 MHz spectrometer, and 13C NMR spectra were recorded on a Brucker 500 MHz spectrometer. NIM spectra for selected compounds are shown in the Supporting Information. Chemical shifts are given in ppm (δ), calibrated to the residual solvent signal peaks of CDCl3 (7.26 ppm), CD3OD (3.31 ppm), or DMSO-d6 (2.50 ppm) for 1H NMR, and CDCl3 (77.16 ppm) or CD3OD (49.00 ppm) for 13C NMR, with coupling constant (J) values reported in Hz. High-resolution mass (HRMS) measurements were performed on a proteomics optimized Q-TOF-2 (Micromass-Waters). The reversed-phase HPLC (RP-HPLC) was performed using Phenomenex Luna 5 μm C18(2)100A, AXIA, 21.2 mm × 250 mm column. Antagonist purity was determined as ≥95% using Agilent ZORBAX SB-Aq, 5 μm,4.6 mm × 150 mm column attached to Agilent 1100 HPLC system. The HPLC traces for compounds tested in vivo (11, 15, 16, 50, and 62) are included in the Supporting Information. The peak at around 3.5 min was the solvent peak, while the peak at around 29 min was eluent perturbation signal during the washing stage. 11, 15, and 16 had 100% purity. Compounds 50 and 62 had purities of 95 and 99%, respectively. The HPLC traces for compounds 63 and 64 were reported in Wen et al., 20225 while the same batches of samples were used for the in vivo tests in this work.
4-(4-(Piperidin-4-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid (1).
Prepared as reported.26 1H NMR (500 MHz, MeOD) δ 8.78−8.74 (m, 1H), 8.43 (d, J = 1.9 Hz, 1H), 8.05−7.97 (m, 4H), 7.93 (dd, J = 8.9, 2.0 Hz, 1H), 7.82 (d, J = 8.1 Hz, 2H), 7.54 (d, J = 8.3 Hz, 2H), 7.50 (d, J = 8.2 Hz, 2H), 3.62−3.55 (m, 2H), 3.29−3.19 (m, 2H), 3.13−3.03 (m, 1H), 2.26−2.19 (m, 2H), 2.11−1.99 (m, 2H). 13C NMR (126 MHz, MeOD) δ 169.65, 145.25, 145.11, 141.55, 139.84, 138.89, 134.93, 134.46, 132.20, 131.39, 129.50, 129.17, 128.91, 128.62, 127.99, 127.79, 127.69, 126.91, 45.70,40.88, 31.20.
4-(4-((1S,4S,5S)-2-Azabicyclo[2.2.1]heptan-5-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid (2b).
Prepared as reported.5 1H NMR (500 MHz, MeOD) δ 8.76 (t, J = 1.1 Hz, 1H), 8.44 (d, J = 1.9 Hz, 1H), 8.06−7.97 (m, 4H), 7.94 (dd, J = 8.9, 2.0 Hz, 1H), 7.82 (d, J = 8.2 Hz, 2H), 7.57−7.49 (m, 4H), 4.30−4.25 (m, 1H), 3.32−3.28 (m, 4H), 3.00−2.96 (m, 1H), 2.47−2.38 (m, 1H), 2.25−2.16 (m, 1H), 2.13 (d, J = 11.6 Hz, 1H), 1.87−1.80 (m, 1H); 13C NMR (126 MHz, MeOD) δ 169.72, 145.26, 144.53, 141.46, 139.53, 138.90, 134.94, 134.43, 132.20, 131.36, 129.62, 129.19, 128.91, 128.61, 128.21, 127.77, 127.72, 126.96, 126.92, 59.39, 51.97,45.51, 43.27, 35.51, 35.27.
4-(4-(Azetidin-3-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid (5).
Procedure A.
LiOH (7.4 mg, 0.3 mmol) was added to a solution of 23 (6.25 mg, 0.013 mmol) in THF-H2O MeOH (3:1:1, 1 mL). The resulting mixture was sonicated for 30 s and stirred at rt for 3 h. The reaction was quenched and neutralized by 1 M HCl. Volatiles were evaporated, and the residue was purified by RP-HPLC (C18, A: ACN, B: 10 mM TEAA, 45% → 60% A in 40 min, flow rate = 5 mL/min, tR = 17.2 min) to give 5 as a white powder (2.73 mg, 47%): 1H NMR (400 MHz, CD3OD) δ 8.76 (s, 1H), 8.44 (d, J = 2.0 Hz, 1H), 8.04−7.96 (m, 4H), 7.93 (dd, J = 8.8, 1.9 Hz, 1H), 7.81 (d, J = 8.1 Hz, 2H), 7.60 (s, 4H), 4.53−4.31 (m, 5H). HRMS m/z [M + H]+ for C27H21NO2F3 calculated 448.1524, found 448.1520.
4-(4-(2-Azaspiro[3.3]heptan-6-yl)phenyl)-7-(4-(trifluoromethyl)-phenyl)-2-naphthoic Acid (6).
Procedure B.
Pd/C (10%, 3 mg) was added to a solution of 7 (0.0016 mmol) in DMF (0.3 mL). The resulting mixture was bubbled with H2 at rt for 5 h. The Pd/C was filtered. Volatiles were evaporated, and the residue was purified by RP-HPLC (C18, A: ACN, B: 10 mM TEAA, 45% → 60% A in 40 min, flow rate = 5 mL/min, tR = 20 min) to give 6 as a white powder (0.4 mg, 51%): 1H NMR (400 MHz, CD3OD) δ 8.74 (s, 1H), 8.43 (d, J = 2.0 Hz, 1H), 8.01 (dd, J = 8.7, 3.6 Hz, 3H), 7.96−7.89 (m, 2H), 7.81 (d, J = 8.1 Hz, 2H), 7.48 (d, J = 8.0 Hz, 2H), 7.42 (d, J =8.0 Hz, 2H), 4.30 (s, 2H), 4.07 (s, 2H), 3.62−3.60 (m, 1H), 2.83−2.75 (m, 2H), 2.51 (td, J = 9.7, 2.9 Hz, 2H). HRMS m/z [M + H]+ for C30H25NO2F3 calculated 488.1837, found 488.1833.
4-(4-(2-Azaspiro[3.3]hept-5-en-6-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid (7), 4-(4-(6-Hydroxy-2-azaspiro[3.3]heptan-6-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid (8), and 4-(4-(6-Methoxy-2-azaspiro[3.3]heptan-6-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid, and 4-(4-(6-(Methoxy-d3)-2-azaspiro[3.3]heptan-6-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid (6:4, 9).
A TFA solution in THF (67%, 4.5 mL) was added to 27 (entire residue from hydrolysis of 26, 0.057 mmol). The resulting mixture was stirred at rt for 1 h. The stir bar was washed with MeOH. Volatiles were evaporated, and the residue was dissolved in CD3OD for crude NMR.CD3OD was removed and the residue was purified by RP-HPLC (C18, A: ACN, B: 10 mM TEAA, 45% → 60% A in 40 min, flow rate = 5 mL/min) to give 7 (1.8 mg, 7%, tR = 42.4 min), 8 (2.54 mg, 9%, tR = 33.0 min), and 9 (10.5 mg, 36%, tR = 40.6 min). 7 has: 1H NMR (400 MHz, CD3OD) δ 8.76 (s, 1H), 8.43 (d, J = 1.9 Hz, 1H), 8.04−7.97 (m, 4H), 7.97−7.91 (m, 1H), 7.81 (d, J = 8.1 Hz, 2H), 7.62 (d, J = 7.9 Hz, 2H), 7.56 (d, J = 8.0 Hz, 2H), 6.55 (s, 1H), 4.42−4.31 (m, 4H), 3.20 (s, 2H). HRMS m/z [M + H]+ for C30H23NO2F3 calculated 486.1681, found 486.1682. 8 has: 1H NMR (400 MHz, CD3OD) δ 8.72 (s, 1H), 8.41 (d, J = 1.9 Hz, 1H), 7.99 (d, J = 8.7 Hz, 4H), 7.90 (dd, J = 8.9, 1.9 Hz, 1H), 7.80 (d, J = 8.1 Hz, 2H), 7.62 (d, J = 8.0 Hz, 2H), 7.54 (d, J = 7.9 Hz, 2H), 4.30 (s, 2H), 4.09 (s, 2H), 2.94 (d, J =13.0 Hz, 2H), 2.66 (d, J = 12.5 Hz, 2H). HRMS m/z [M + H]+ for C30H25NO3F3 calculated 504.1787, found 504.1796. 9 has: 1H NMR (600 MHz, CD3OD) δ 8.77−8.74 (m, 1H), 8.44 (d, J = 1.9 Hz, 1H),8.05−8.00 (m, 4H), 7.94 (dd, J = 8.8, 2.0 Hz, 1H), 7.83 (d, J = 8.2 Hz, 2H), 7.63−7.55 (m, 4H), 4.27 (s, 2H), 4.08 (s, 2H), 3.07 (s, 2H), 2.91−2.84 (m, 2H), 2.80−2.74 (m, 2H). HRMS m/z [M + H]+ for C31H27NO3F3 calculated 518.1943, found 518.1939; for C31H24D3NO3F3 calculated 521.2117, found 521.2126.
rac-4-(4-((3aR,6aR)-5-Hydroxyoctahydrocyclopenta[c]pyrrol-5-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid (11).
Pd/C (10%, 53 mg) was added to a solution of 31 (10.7 mg, 0.0177 mmol) in DMF (5 mL). The resulting mixture was bubbled with H2 at rt for 5 h. The Pd/C was filtered. The filtrate was purified by RP-HPLC (C18, A: ACN, B: 10 mM TEAA, 30% → 50% A in 40 min, flow rate = 5 mL/min, tR = 35 min) to give 11 as a white powder (3.18 mg, 35% overall yield from 30): 1H NMR (400 MHz, CD3OD) δ 8.75 (s, 1H), 8.43 (d, J = 2.0 Hz, 1H), 8.05−7.96 (m, 4H), 7.93 (dd, J = 9.0, 2.0 Hz, 1H), 7.81 (d, J = 8.1 Hz, 2H), 7.69 (d, J = 8.2 Hz, 2H), 7.54 (d, J = 8.0 Hz, 2H), 3.56−3.46 (m, 2H), 2.98 (t, J = 11.2 Hz, 2H), 2.76−2.65 (m, 1H), 2.60−2.45 (m, 2H), 2.22 (dd, J = 12.6, 5.4 Hz, 1H), 2.06–1.95 (m, 2H). 13C NMR (126 MHz, MeOD) δ 149.68, 145.31, 141.54, 139.73, 138.88, 134.98, 134.44, 132.11, 130.90, 129.18, 128.92, 128.54, 127.80, 126.93, 125.82, 116.40, 90.29, 52.72, 50.48, 47.94, 47.45, 45.07, 44.61, 40.43. HRMS m/z [M + H]+ for C31H27NO3F3 calculated 518.1943, found 518.1944.
rac-4-(4-((3aR,6aR)-1,2,3,3a,4,6a-Hexahydrocyclopenta[c]-pyrrol-5-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid (12).
A solution of 11 (0.012 mmol, crude reaction residue without HPLC purification) in TFA (2 mL) was refluxed in a 90 °C oil bath for 2 h. Volatiles were evaporated, and the residue was purified by RPHPLC (C18, A: ACN, B: 10 mM TEAA, 45% → 60% A in 40 min, flow rate = 5 mL/min, tR = 21 min) to give 12 as a white powder (1.9 mg, 33% overall yield from 31): 1H NMR (400 MHz, CD3OD) δ 8.75 (s, 1H), 8.43 (d, J = 2.0 Hz, 1H), 8.07−7.96 (m, 4H), 7.94 (dd, J =8.9, 2.0 Hz, 1H), 7.81 (d, J = 8.1 Hz, 2H), 7.68 (d, J = 8.1 Hz, 2H), 7.53 (d, J = 8.0 Hz, 2H), 6.61 (s, 1H), 3.62 (d, J = 3.3 Hz, 1H), 3.53 (dd, J = 10.2, 6.5 Hz, 1H), 3.26−3.15 (m, 1H), 3.04 (d, J = 7.0 Hz, 2H), 2.99 (dd, J = 14.4, 6.0 Hz, 1H), 2.68−2.55 (m, 1H), 2.40 (dq, J = 11.9, 5.9 Hz, 1H). HRMS m/z [M + H]+ for C31H25NO2F3 calculated 500.1837, found 500.1834.
rac-4-(4-((3aR,6aR)-Octahydrocyclopenta[c]pyrrol-5-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid (13).
Treatment of 12 (0.021 mmol, crude reaction residue without HPLC purification) by using Procedure B gave 13 (0.57 mg, 5% overall yield from 31) as a white powder: 1H NMR (400 MHz, CD3OD) δ 8.74 (s, 1H), 8.42 (d, J = 1.9 Hz, 1H), 8.06−7.95 (m, 4H), 7.93 (d, J = 8.4 Hz, 1H),7.81 (d, J = 8.0 Hz, 2H), 7.49 (s, 4H), 4.01 (d, J = 9.2 Hz, 1H), 3.48 (p, J = 6.4 Hz, 2H), 3.03−2.86 (m, 2H), 2.59 (dd, J = 12.8, 6.4 Hz, 1H), 2.47−2.29 (m, 2H), 2.17−1.98 (m, 2H), 1.64 (q, J = 11.0 Hz, 1H). HRMS m/z [M + H]+ for C31H27NO2F3 calculated 502.1994, found 502.1986.
4-(4-((1R,5S,6r)-3-Azabicyclo[3.1.1]heptan-6-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid (14).
Concentrated sulfuric acid (95−98%, 0.9 mL) was added to a mixture of 15 (9 mg, 0.015 mmol) and NaBH4 (5.7 mg, 0.15 mmol) in THF (0.1 mL) under argon (caution, running the reaction under argon is required to avoid fire/explosion). The resulting solution was stirred at rt for 30 min. The mixture was basified with cold 6 N NaOH aqueous solution and extracted with ethyl acetate three times. The organic layer was dried over anhydrous Na2SO4. Volatiles were evaporated, and the residue was purified by RP-HPLC (C18, A: ACN, B: 10 mM TEAA, 35% → 55% A in 40 min, flow rate = 5 mL/min, tR = 38.8 min) to give 14 as a white powder (0.8 mg, 11%): 1H NMR (400 MHz, CD3OD) δ 8.76 (s, 1H), 8.44 (d, J = 2.0 Hz, 1H), 8.07−7.96 (m, 4H), 7.96−7.90 (m, 1H), 7.81 (d, J = 8.1 Hz, 2H), 7.56 (d, J = 7.8 Hz, 2H), 7.38 (d, J = 7.7 Hz, 2H), 4.09 (q, J = 9.6 Hz, 1H), 3.62 (t, J = 7.2 Hz, 1H), 3.42−3.33 (m, 3H), 3.23 (dd, J = 16.7, 7.1 Hz, 2H), 2.74 (q, J = 9.1 Hz, 1H), 2.33 (q, J = 10.5 Hz, 1H). HRMS m/z [M +H]+ for C30H24F3NO2 calculated 488.1837, found 488.1829.
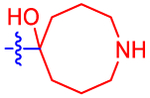
4-(4-((1R,5S,6r)-6-Hydroxy-3-azabicyclo[3.1.1]heptan-6-yl)-phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid (15).
Procedure C.
A TFA solution in THF (67%, 2 mL) was added to 35(0.022 mmol, reaction residue). The resulting mixture was stirred at rt for 1 h. Volatiles were evaporated, and the residue was purified by RPHPLC (C18, A: ACN, B: 10 mM TEAA, 35% → 55% A in 40 min, flow rate = 5 mL/min, tR = 27.7 min) to give 15 (4.6 mg, 42%) as a white solid: 1H NMR (400 MHz, DMSO) δ 8.76 (s, 1H), 8.65 (d, J =2.0 Hz, 1H), 8.06 (t, J = 8.2 Hz, 3H), 7.98 (d, J = 8.9 Hz, 1H), 7.92−7.85 (m, 3H), 7.73 (d, J = 7.9 Hz, 2H), 7.59 (d, J = 7.8 Hz, 2H), 3.69 (d, J = 11.7 Hz, 2H), 3.47 (d, J = 11.9 Hz, 2H), 2.89 (d, J = 4.5 Hz, 2H), 1.71 (s, 2H). 13C NMR (126 MHz, MeOD) δ 168.27, 143.84, 142.24, 139.99, 139.55, 137.54, 133.56, 132.98, 130.93, 129.88, 128.21, 127.82, 127.52, 127.27, 126.39, 126.32, 125.56, 125.53, 73.18, 44.35, 39.91, 20.75. HRMS m/z [M + H]+ for C30H25NO3F3 calculated 504.1787, found 504.1793.
4-(4-(Azepan-4-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid (16).
Treatment of 17 (0.038 mmol, crude reaction residue without HPLC purification) by using Procedure B gave 16 (4 mg, 22% overall yield from 38) as a white powder: 1H NMR (400 MHz, CD3OD) δ 8.77−8.72 (m, 1H), 8.42 (d, J = 1.9 Hz, 1H), 8.05−7.95 (m, 4H), 7.92 (dd, J = 8.9, 1.9 Hz, 1H), 7.81 (d, J = 8.1 Hz, 2H), 7.48 (q, J = 8.1 Hz, 4H), 3.54−3.35 (m, 4H), 3.02 (t, J = 10.9 Hz, 1H),2.22 (d, J = 15.9 Hz, 4H), 2.09−1.86 (m, 2H). HRMS m/z [M + H]+ for C30H27NO2F3 calculated 490.1994, found 490.1992.
Mixture of 4-(4-(2,3,6,7-Tetrahydro-1H-azepin-4-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid and 4-(4-(2,5,6,7-Tetra-hydro-1H-azepin-4-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid (17).
Treatment of 39 (all residue from hydrolysis of 38, 0.057 mmol) by using Procedure C gave 17 (5.08 mg, 18%) as a mixture of two constitutional isomers (1:1): 1H NMR (400 MHz, CD3OD) δ 8.76 (d, J = 2.8 Hz, 1H), 8.43 (d, J = 2.4 Hz, 1H), 8.05−7.97 (m, 4H), 7.972−7.90 (m, 1H), 7.81 (d, J = 8.1 Hz, 2H), 7.66−7.49 (m, 4H), 6.35 (t, J = 6.2 Hz, 0.5H), 6.21 (t, J = 6.5 Hz, 0.5H),3.99 (d, J = 6.5 Hz, 1H), 3.59−3.49 (m, 1H), 3.48−3.41 (m, 1H),3.39−3.32 (m, 1H), 3.14−3.05 (m, 1H), 3.06−2.99 (m, 1H), 2.74 (d, J = 5.5 Hz, 1H), 2.14 (d, J = 5.7 Hz, 1H). HRMS m/z [M + H] + for C30H25NO2F3 calculated 488.1837, found 488.1835.
4-(4-(Azocan-5-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid (18).
Treatment of 47 (6 mg, 0.0112 mmol) by using Procedure A gave 18 (3 mg, overall yield 54% from 45): 1H NMR (400 MHz, MeOD) δ 8.73 (s, 1H), 8.41 (d, J = 1.9 Hz, 1H), 8.06−7.95 (m, 4H), 7.92 (dd, J = 8.9, 2.0 Hz, 1H), 7.80 (d, J = 8.2 Hz, 2H), 7.47 (d, J = 8.2 Hz, 2H), 7.43 (d, J = 8.2 Hz, 2H), 3.46−3.37 (m, 2H), 3.36−3.32 (m, 2H), 3.06−2.95 (m, 1H), 2.27−2.09 (m, 4H), 2.07−1.96 (m, 4H). 13C NMR (126 MHz, MeOD) δ 168.58, 148.12, 143.92, 140.30, 137.68, 137.43, 133.56, 133.08, 130.61, 129.85, 127.74, 127.51, 127.10, 126.63, 126.46, 126.35, 125.54, 114.99, 46.54, 45.17, 32.49, 23.09. HRMS m/z [M + H]+ for C31H29NO2F3 calculated 504.2150, found 504.2141.
4-(4-(1,2,3,4,7,8-Hexahydroazocin-5-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid (19) and 4-(4-(5-Hydroxyazocan-5-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naph thoic Acid (20).
Treatment of 43 (all residue from hydrolysis of 42,0.049 mmol) by using Procedure C gave 19 (4.52 mg, 18%) in addition to 20 (4.67 mg, 18%). 19 has: 1H NMR (400 MHz, CD3OD) δ 8.72 (s, 1H), 8.40 (d, J = 2.0 Hz, 1H), 8.03–7.95 (m, 5H), 7.90 (dd, J = 9.0, 2.0 Hz, 1H), 7.78 (d, J = 8.1 Hz, 3H), 7.66 (d, J = 8.0 Hz, 3H), 7.51 (d, J = 8.0 Hz, 2H), 6.28 (t, J = 8.3 Hz, 1H),3.35−3.28 (m, 2H), 3.25−3.21 (m, 2H), 2.90 (t, J = 6.3 Hz, 2H),2.72 (dd, J = 12.0, 7.5 Hz, 2H), 2.08 (s, 2H). HRMS m/z [M + H]+ for C31H26NO2F3 calculated 502.1994, found 502.2001. 20 has: 1HNMR (400 MHz, CD3OD) δ 8.75 (s, 1H), 8.42 (d, J = 1.9 Hz, 1H),8.00−7.94 (m, 4H), 7.91 (dd, J = 8.9, 1.9 Hz, 1H), 7.78 (d, J = 8.1 Hz, 2H), 7.70 (d, J = 8.4 Hz, 2H), 7.65 (d, J = 8.5 Hz, 2H), 3.85 (dt, J = 13.3, 7.0 Hz, 2H), 3.41 (dt, J = 12.5, 6.7 Hz, 2H), 2.70 (dt, J = 13.5,6.8 Hz, 2H), 2.47 (dt, J = 13.5, 6.7 Hz, 2H), 2.35 (dp, J = 13.7, 6.8 Hz, 2H), 2.24 (dq, J = 13.5, 6.8 Hz, 2H). HRMS m/z [M–OH−]+ for C31H27NO2F3 calculated 502.1994, found 502.1993.
Ethyl 4-(4-(Azetidin-3-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoate (23).
Procedure D.
To a vacuum-dried mixture of 21 (5.8 mg, 0.023 mmol), 2241 (11 mg, 0.023 mmol), Pd(PPh3)4 (2.7 mg, 0.0023 mmol), and Na2CO3 (6 mg, 0.057 mmol) was added sonicated 1,2-dimethoxyethane-water (4:1, 1 mL). The reaction mixture was degassed with argon for 10 min and then stirred at 80 °C overnight. Volatiles were evaporated, and the residue was column-chromatographed (CH2Cl2/MeOH/TEA, 100:0:1 → 90:10:1) to give 23 (6.25 mg, 57%): 1H NMR (400 MHz, CDCl3) δ 8.68 (s, 1H), 8.22 (d, J = 1.9 Hz, 1H), 8.01 (d, J = 1.7 Hz, 1H), 7.97 (d, J = 8.8 Hz, 1H),7.86−7.77 (m, 3H), 7.75 (d, J = 8.3 Hz, 2H), 7.61 (d, J = 7.9 Hz, 2H), 7.55 (d, J = 8.0 Hz, 2H), 4.57−4.41 (m, 5H), 3.15 (q, J = 7.3 Hz, 2H), 1.44 (td, J = 7.2, 4.1 Hz, 3H). HRMS m/z [M + H]+ for C32H29NO2F3 calculated 516.2150, found 516.2145. HRMS m/z [M + H]+ for C29H25NO2F3 calculated 476.1837, found 476.1837.
tert-Butyl 6-(4-Bromophenyl)-6-hydroxy-2-azaspiro[3.3]-heptane-2-carboxylate (25).
Procedure E.
A mixture of 1,4-dibromobenzene (2.9 g, 12.5 mmol) and magnesium turnings (60.8 mg, 2.5 mmol) in THF (5 mL) was sonicated for 2 h. The solution was cooled to 0 °C. Into the Grignard reagent was added the solution of 24 (105.6 mg, 0.5 mmol) in THF (10 mL). The resulting mixture was stirred at 0 °C to rt overnight. The residue was partitioned between ethyl acetate and aqueous NaHCO3. The organic layer was dried over anhydrous Na2SO4. Volatiles were evaporated, and the residue was column-chromatographed (hexane/ethyl acetate = 80:20 → 40:60) to give 25 (72.3 mg; 39%): 1H NMR (400 MHz, CDCl3) δ 7.50−7.44 (m, 2H), 7.29−7.24 (m, 2H), 4.05 (s, 2H), 3.78 (s, 2H),2.72−2.66 (m, 2H), 2.52 (d, J = 12.7 Hz, 2H), 1.41 (s, 9H). HRMS m/z [M - t-butyl + 2H]+ for C13H15N79BrO3 calculated 312.0235, found 312.0234.
tert-Butyl 6-(4-(3-(Ethoxycarbonyl)-6-(4-(trifluoromethyl)-phenyl)naphthalen-1-yl)phenyl)-6-hydroxy-2-azaspiro[3.3]-heptane-2-carboxylate (26).
Treatment of 25 (25.6 mg, 0.07 mmol) by using Procedure D gave 26 (36.12 mg, 82%): 1H NMR (400 MHz, CDCl3) δ 8.68 (s, 1H), 8.22 (d, J = 1.8 Hz, 1H), 8.06−7.96 (m, 2H),7.81 (d, J = 8.1 Hz, 2H), 7.76 (td, J = 6.8, 3.3 Hz, 3H), 7.57 (d, J =8.0 Hz, 2H), 7.52 (d, J = 8.1 Hz, 2H), 4.46 (q, J = 7.1 Hz, 2H), 4.13 (s, 2H), 3.90 (s, 2H), 2.87 (d, J = 12.8 Hz, 2H), 2.64 (d, J = 12.6 Hz, 2H), 1.44 (d, J = 9.7 Hz, 12H). HRMS m/z [M + Na]+ for C37H36NO5F3Na calculated 654.2443, found 654.2446.
4-(4-(2-(tert-Butoxycarbonyl)-6-hydroxy-2-azaspiro[3.3]heptan-6-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid (27)
Treatment of 26 (36 mg, 0.057 mmol) by using Procedure A (without HPLC purification) gave 27 of sufficient purity to be used in next step: 1H NMR (400 MHz, CD3OD) δ 8.50 (s, 1H), 8.21 (s, 1H),7.91–7.80 (m, 4H), 7.67 (dd, J = 14.7, 9.8 Hz, 3H), 7.52−7.45 (m, 2H), 7.37 (t, J = 5.3 Hz, 2H), 3.98 (s, 2H), 3.76 (s, 2H), 2.73 (d, J =12.1 Hz, 2H), 2.52−2.45 (m, 2H), 1.32 (d, J = 3.8 Hz, 9H). HRMS m/z [M + Na]+ for C35H32NO5F3Na calculated 626.2130, found 626.2136.
rac-(3aR, 6aR)-2-Benzyl-5-(4- bromophenyl)-octahydrocyclopenta[c]pyrrol-5-ol (29).
Treatment of 28 (125.9 mg,0.5 mmol) by using Procedure E (column chromatography; DCM/ MeOH, 100:0 → 95:5) gave 29 (101.18 mg, 54%): 1H NMR (400MHz, CDCl3) δ 7.48−7.40 (m, 2H), 7.40−7.20 (m, 7H), 3.94−3.78 (m, 2H), 2.88 (dq, J = 9.6, 4.5 Hz, 2H), 2.80 (s, 1H), 2.50 (tdt, J =16.7, 11.3, 8.9 Hz, 3H), 2.29 (dd, J = 12.7, 7.1 Hz, 1H), 2.18 (dtd, J =16.1, 11.4, 5.6 Hz, 1H), 1.97 (dd, J = 12.6, 4.7 Hz, 1H), 1.76−1.61 (m, 2H). HRMS m/z [M + H]+ for C20H23NO79Br calculated 372.0963, found 372.0966.
rac-Ethyl4-(4-((3aR,6aR)-2-benzyl-5-hydroxyoctahydrocyclopenta[c]pyrrol-5-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoate (30).
Treatment of 29 (26.1 mg, 0.07 mmol) by using Procedure D gave 30 (33.7 mg, 76%): 1HNMR (400 MHz, CDCl3) δ 8.65 (s, 1H), 8.20 (d, J = 1.9 Hz, 1H),8.04−7.95 (m, 2H), 7.79 (d, J = 8.2 Hz, 2H), 7.76−7.68 (m, 3H),7.60 (d, J = 8.1 Hz, 2H), 7.54 (d, J = 7.4 Hz, 2H), 7.48 (d, J = 8.0 Hz, 2H), 7.36 (dt, J = 13.7, 7.2 Hz, 3H), 4.44 (q, J = 7.1 Hz, 2H), 4.14 (s, 2H), 3.32−3.17 (m, 2H), 2.99−2.76 (m, 3H), 2.57−2.42 (m, 2H),2.20 (dd, J = 12.6, 5.2 Hz, 1H), 1.94 (dt, J = 17.9, 12.1 Hz, 2H), 1.44 (t, J = 7.1 Hz, 3H). HRMS m/z [M + H] + for C40H37NO3F3 calculated 636.2726, found 636.2724.
rac-4-(4-((3aR,6aR)-2-Benzyl-5-hydroxyoctahydrocyclopenta[c]-pyrrol-5-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid (31).
Treatment of 30 (33.7 mg, 0.053 mmol) by using Procedure A (without HPLC purification) gave 31 of sufficient purity to be used in next step: 1H NMR (400 MHz, CD3OD) δ 8.59 (s, 1H), 8.30 (s, 1H),8.00 (s, 1H), 7.92 (dd, J = 8.5, 5.3 Hz, 3H), 7.74 (d, J = 7.5 Hz, 3H),7.65 (dd, J = 15.8, 6.5 Hz, 4H), 7.51−7.39 (m, 5H), 4.62−4.50 (m, 2H), 3.54 (d, J = 12.9 Hz, 2H), 3.22 (q, J = 13.9 Hz, 2H), 2.94−2.66 (m, 2H), 2.47 (dd, J = 13.2, 7.1 Hz, 1H), 2.18 (dd, J = 12.8, 5.0 Hz, 1H), 1.97 (q, J = 11.3 Hz, 2H). HRMS m/z [M + H]+ for C38H33NO3F3 calculated 608.2413, found 608.2408.
tert-Butyl (1R,5S,6r)-6-(4-Bromophenyl)-6-hydroxy-3-azabicyclo-[3.1.1]heptane-3-carboxylate (33).
Treatment of 32 (105.65 mg, 0.5 mmol) by using Procedure E gave 33 (99.36 mg, 54%): 1H NMR (400 MHz, CDCl3) δ 7.50 (dd, J = 8.7, 2.3 Hz, 2H), 7.39 (d, J = 8.5 Hz, 2H), 3.82−3.66 (m, 4H), 2.82−2.76 (m, 1H), 2.71−2.63 (m, 1H), 1.55 (dt, J = 11.4, 6.2 Hz, 1H), 1.45 (d, J = 2.5 Hz, 9H), 1.33 (d, J = 10.1 Hz, 1H). HRMS m/z [M - t-butyl + 2H]+ for C13H15N79BrO3 calculated 312.0235, found 312.0234.
tert-Butyl (1R,5S,6r)-6-(4-(3-(Ethoxycarbonyl)-6-(4-(trifluoromethyl)phenyl)naphthalen-1-yl)phenyl)-6-hydroxy-3-azabicyclo[3.1.1]heptane-3-carboxylate (34).
Treatment of 33 (25.8 mg, 0.07 mmol) by using Procedure D gave 34 (41 mg, 93%): 1H NMR (400 MHz, CDCl3) δ 8.69 (d, J = 1.6 Hz, 1H), 8.23 (d, J = 1.9 Hz, 1H), 8.07−8.01 (m, 2H), 7.81 (d, J = 8.2 Hz, 2H),7.79−7.73 (m, 3H), 7.71 (d, J = 8.0 Hz, 2H), 7.56 (d, J = 8.0 Hz, 2H), 4.45 (q, J = 7.1 Hz, 2H), 3.94−3.77 (m, 4H), 3.02−2.94 (m, 1H), 2.88 (d, J = 6.7 Hz, 1H), 1.74 (dt, J = 11.4, 6.1 Hz, 1H), 1.52 (s, 9H), 1.48−1.40 (m, 4H). HRMS m/z [M–Boc–O]+ for C32H27NO2F3 calculated 514.1994, found 514.2001.
4-(4-((1R,5S,6r)-3-(tert-Butoxycarbonyl)-6-hydroxy-3-azabicyclo-[3.1.1]heptan-6-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid (35).
Treatment of 34 (41 mg, 0.065 mmol) by using Procedure A (without HPLC purification) gave 35 of sufficient purity to be used in next step: 1H NMR (400 MHz, CD3OD) δ 8.58 (s, 1H),8.31 (d, J = 1.9 Hz, 1H), 8.04 (d, J = 1.6 Hz, 1H), 7.97 (dd, J = 8.6,3.2 Hz, 3H), 7.81−7.74 (m, 3H), 7.72 (d, J = 7.9 Hz, 2H), 7.53 (t, J =6.6 Hz, 2H), 3.84−3.69 (m, 4H), 2.96−2.82 (m, 2H), 1.69 (dt, J =11.1, 6.0 Hz, 1H), 1.36 (d, J = 9.7 Hz, 1H). MS m/z [M–Boc–O]+ for C30H23NO2F3 calculated 486.2, found 486.2.
tert-Butyl 4-(4-Bromophenyl)-4-hydroxyazepane-1-carboxylate (37).
Treatment of 36 (106.65 mg, 0.5 mmol) by using Procedure E gave 37 (135 mg, 73%): 1H NMR (400 MHz, CDCl3) δ 7.45 (d, J = 8.0 Hz, 2H), 7.32 (d, J = 8.3 Hz, 2H), 3.92–3.47 (m, 2H), 3.32 (d, J = 11.3 Hz, 2H), 2.27−1.67 (m, 7H), 1.47 (d, J = 4.1 Hz, 9H).
tert-Butyl 4-(4-(3-(Ethoxycarbonyl)-6-(4-(trifluoromethyl)-phenyl)naphthalen-1-yl)phenyl)-4-hydroxyazepane-1-carboxylate (38).
Treatment of 37 (38.9 mg, 0.105 mmol) by using Procedure D gave 38 (36.4 mg, 82%): 1H NMR (400 MHz, CDCl3) δ 8.67 (s, 1H), 8.22 (s, 1H), 8.03 (d, J = 9.4 Hz, 2H), 7.81 (d, J = 8.1 Hz, 2H),7.74 (d, J = 8.0 Hz, 3H), 7.62 (d, J = 8.1 Hz, 2H), 7.55−7.45 (m, 2H), 4.46 (q, J = 7.1 Hz, 2H), 3.93−3.53 (m, 2H), 3.45−3.34 (m, 2H), 2.35−2.05 (m, 3H), 1.99−1.68 (m, 4H), 1.51 (s, 9H), 1.45 (t, J=7.2 Hz, 3H). HRMS m/z [M + Na]+ for C37H38NO5F3Na calculated 656.2600, found 656.2589.
4-(4-(1-(tert-Butoxycarbonyl)-4-hydroxyazepan-4-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid (39).
Treatment of 38(36.4 mg, 0.057 mmol) by using Procedure A (without HPLC purification) gave 39 of sufficient purity to be used in next step: 1HNMR (400 MHz, CD3OD) δ 8.34 (s, 1H), 8.08 (s, 1H), 7.80 (s, 1H),7.74 (d, J = 8.1 Hz, 3H), 7.55 (d, J = 8.0 Hz, 3H), 7.39 (d, J = 8.1 Hz, 2H), 7.25 (d, J = 7.2 Hz, 2H), 3.46 (s, 2H), 3.23−3.16 (m, 2H),2.16−1.51 (m, 6H), 1.27 (s, 9H). MS m/z [M–Boc–O]+ for C30H25NO2F3 calculated 488.1837, found 488.1840.
tert-Butyl 5-(4-Bromophenyl)-5-hydroxyazocane-1-carboxylate (41).
Treatment of 40 (113.65 mg, 0.5 mmol) by using Procedure E gave 41 (155 mg, 81%): 1H NMR (400 MHz, CDCl3) δ 7.44−7.39 (m, 2H), 7.37−7.32 (m, 2H), 3.46−3.24 (m, 4H), 2.06−1.77 (m, 6H), 1.58−1.45 (m, 2H), 1.44 (s, 9H). HRMS m/z [M–t-butyl+H– OH−]+ for C14H17N79BrO2 calculated 310.0443, found 310.0437.
tert-Butyl 5-(4-(3-(Ethoxycarbonyl)-6-(4-(trifluoromethyl)-phenyl)naphthalen-1-yl)phenyl)-5-hydroxyazocane-1-carboxylate (42).
Treatment of 41 (26.9 mg, 0.07 mmol) by using Procedure D gave 42 (35.5 mg, 78%): 1H NMR (400 MHz, CDCl3) δ 8.68 (s, 1H), 8.22 (d, J = 1.9 Hz, 1H), 8.072−8.00 (m, 2H), 7.81 (d, J = 8.1 Hz, 2H), 7.78−7.71 (m, 3H), 7.66 (d, J = 8.1 Hz, 2H), 7.54−7.47 (m, 2H), 4.46 (q, J = 7.1 Hz, 2H), 3.56−3.37 (m, 5H), 2.27−2.06 (m, 2H), 2.00 (d, J = 14.6 Hz, 4H), 1.71 (s, 2H), 1.50 (s, 9H), 1.45 (t, J =7.1 Hz, 3H). MS m/z [M–Boc–O]+ for C33H31NO2F3 calculated 530.2, found 530.2.
4-(4-(1-(tert-Butoxycarbonyl)-5-hydroxyazocan-5-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid (43).
Treatment of 42 (32 mg, 0.049 mmol) by using Procedure A (without HPLC purification) gave 43 of sufficient purity to be used in next step: 1HNMR (400 MHz, CD3OD) δ 8.57 (s, 1H), 8.32−8.28 (m, 1H),8.05−8.01 (m, 1H), 7.96 (d, J = 8.3 Hz, 3H), 7.82−7.73 (m, 3H),7.67 (d, J = 7.9 Hz, 2H), 7.46 (d, J = 7.9 Hz, 2H), 3.56 (q, J = 11.3 Hz, 2H), 3.45–3.28 (m, 2H), 2.27−1.87 (m, 6H), 1.67−1.53 (m, 2H), 1.49 (s, 9H). MS m/z [M–Boc–O]+ for C31H27NO2F3 calculated 502.1994, found 502.1990.
tert-Butyl 5-(4-Bromophenyl)-3,4,7,8-tetrahydroazocine-1(2H)-carboxylate (44).
Triphosgene (34.7 mg, 0.117 mmol) was added into a solution of 41 (30 mg, 0.078 mmol) and DMAP (28.6 mg,0.234 mmol) in DMF (2 mL). The resulting solution was stirred at rt for 2 h. Volatiles were evaporated, and the residue was column-chromatographed (hexane/ethyl acetate = 70:30 → 60:40) to give 44 (28 mg, quantitative): 1H NMR (400 MHz, CDCl3) δ 7.41 (d, J = 8.1 Hz, 2H), 7.29−7.22 (m, 2H), 6.13−6.02 (m, 1H), 3.50−3.35 (m, 2H), 3.21 (dt, J = 12.2, 6.0 Hz, 2H), 2.54 (hept, J = 6.1 Hz, 2H), 2.35 (dq, J = 13.6, 6.4 Hz, 2H), 1.99−1.72 (m, 2H), 1.44 (s, 9H). HRMS m/z [M–Boc+2H]+ for C13H17N79Br calculated 266.0544, found 266.0543.
tert-Butyl 5-(4-(3-(Ethoxycarbonyl)-6-(4-(trifluoromethyl)-phenyl)naphthalen-1-yl)phenyl)-3,4,7,8-tetrahydroazocine-1(2H)-carboxylate (45).
Treatment of 44 (8.9 mg, 0.024 mmol) by using Procedure D gave 45 (15 mg, quantitative): 1H NMR (400 MHz, CDCl3) δ 8.70−8.66 (m, 1H), 8.23 (d, J = 1.9 Hz, 1H), 8.06 (dd, J =5.1, 3.2 Hz, 2H), 7.83 (d, J = 8.2 Hz, 2H), 7.80−7.73 (m, 3H), 7.58 (dd, J = 8.3, 2.2 Hz, 2H), 7.48 (d, J = 7.9 Hz, 2H), 6.25 (q, J = 8.2 Hz, 1H), 4.47 (q, J = 7.1 Hz, 2H), 3.56−3.42 (m, 2H), 3.30 (d, J = 13.7Hz, 2H), 2.67 (dt, J = 9.7, 6.1 Hz, 2H), 2.50–2.37 (m, 2H), 2.07−1.89 (m, 2H), 1.49–1.42 (m, 12H). HRMS m/z [M–Boc+2H] + for C33H31NO2F3 calculated 530.2307, found 530.2303.
tert-Butyl 5-(4-(3-(Ethoxycarbonyl)-6-(4-(trifluoromethyl)-phenyl)naphthalen-1-yl)phenyl)azocane-1-carboxylate (46).
To a mixture of 45 (7 mg, 0.0112 mmol) and 10% Pd-C (21 mg) in MeOH/THF (2.1 mL, 1:2) was added triethylsilane (21 μL, 15.3 mg,0.13 mmol). The resulting mixture was stirred at rt for 5 min. Another portion of triethylsilane (28 μL, 20.4 mg, 0.175 mmol) diluted in 1 mL of THF was added dropwise within 5 min. The mixture was stirred for 5 min. Pd-C was removed by filtration and volatiles were evaporated, and the residue was dried under high vacuum to give 46 (7 mg, quantitative), which was used for the next step without further purification. 46 has: 1H NMR (400 MHz, CDCl3) δ 8.67 (d, J = 1.6 Hz, 1H), 8.23 (d, J = 1.9 Hz, 1H), 8.10−8.02 (m, 2H), 7.83 (d, J =8.1 Hz, 2H), 7.80−7.72 (m, 3H), 7.44 (d, J = 8.0 Hz, 2H), 7.30 (d, J = 7.9 Hz, 2H), 4.46 (q, J = 7.1 Hz, 2H), 3.74 (q, J = 7.6 Hz, 2H), 3.62 (d, J = 14.4 Hz, 1H), 3.30−3.17 (m, 2H), 2.95−2.79 (m, 1H), 2.02−1.80 (m, 6H), 1.53 (s, 9H), 1.45 (t, J = 7.1 Hz, 3H), 1.33−1.26 (m,2H). HRMS m/z [M + H] + for C38H40NO4F3Na calculated 654.2807, found 654.2811.
Ethyl 4-(4-(Azocan-5-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoate (47).
Treatment of 46 (all residue from reduction of 45, 0.0112 mmol) by using Procedure C gave 47 (volatiles were removed and the residue was used for next step without purification; 6 mg, quantitative): 1H NMR (400 MHz, CDCl3) δ 8.67 (s, 1H), 8.22 (d, J = 2.0 Hz, 1H), 8.07−8.01 (m, 2H), 7.82 (d, J = 8.2 Hz, 2H), 7.80−7.73 (m, 3H), 7.47 (d, J = 7.3 Hz, 2H), 7.38 (d, J = 7.5 Hz, 2H), 4.46 (q, J = 7.1 Hz, 2H), 3.56−3.21 (m, 4H), 3.08−2.97 (m, 1H), 2.28−2.00 (m, 8H), 1.47−1.43 (m, 3H). MS m/z [M + H]+ for C33H33NO2F3 calculated 532.2, found 532.2.
tert-Butyl (1R,5S,6r)-6-(4-(3-((2-(Dimethylamino)-2-oxoethoxy)-carbonyl)-6-(4-(trifluoromethyl)phenyl)naphthalen-1-yl)phenyl)-6-hydroxy-3-azabicyclo[3.1.1]heptane-3-carboxylate (48).
To a solution of 35 (62 mg, 0.103 mmol) and Cs2CO3 (66.93 mg, 0.205 mmol) in DMF (5 mL) under an argon atmosphere was added 2-chloro-N,N-dimethylacetamide (19 μL, 22.5 mg, 0.186 mmol). The reaction mixture was stirred at rt overnight. The reaction mixture was partitioned between ethyl acetate (5 mL) and water (5 mL), and the aqueous phase was extracted with ethyl acetate (2 × 5 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated under pressure. The residue was purified by silica gel chromatography (hexane/ethyl acetate = 1:1 → 0:10) to afford compound 48 (40 mg, 56%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 8.79 (s, 1H), 8.23 (s, 1H), 8.11 (s, 1H), 8.04 (d, J = 8.8 Hz, 1H), 7.85−7.73 (m, 5H), 7.70 (d, J = 7.8 Hz, 2H), 7.56 (d, J = 7.8 Hz, 2H), 5.05 (s, 2H), 3.93−3.70 (m, 4H), 3.08 (s, 3H), 3.05−2.98 (m, 4H), 2.90−2.85 (m, 1H), 1.79−1.70 (m, 1H), 1.45−1.41 (m, 1H). HRMS m/z [M–Boc+H–OH−]+ for C34H30N2O3F3 calculated 571.2209, found 571.2205.
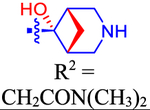
2-(Dimethylamino)-2-oxoethyl 4-(4-((1R,5S,6r)-6-hydroxy-3-azabicyclo[3.1.1]heptan-6-yl)phenyl)-7-(4-(trifluoromethyl)-phenyl)-2-naphthoate (49).
A TFA solution in THF (67%, 4.5 mL) was added to 48 (40 mg, 0.058 mmol). The resulting mixture was stirred at rt for 1 h. Volatiles were evaporated, and the residue was purified by RP-HPLC (C18, A: ACN, B: 0.2% TFA, 50% → 100% A in 40 min, flow rate = 5 mL/min, tR = 21.6 min) to give 49 as a white powder (20 mg, 59%): 1H NMR (400 MHz, DMSO) δ 8.83 (s, 1H),8.72 (d, J = 1.9 Hz, 1H), 8.30 (s, 1H), 8.12−8.05 (m, 3H), 7.98 (d, J = 8.9 Hz, 1H), 7.94−7.86 (m, 3H), 7.74 (d, J = 7.9 Hz, 2H), 7.61 (d, J = 7.9 Hz, 2H), 6.15 (s, 1H), 5.11 (s, 2H), 3.70 (d, J = 11.7 Hz, 2H),3.49 (d, J = 11.9 Hz, 2H), 3.00 (s, 3H), 2.90 (d, J = 6.0 Hz, 2H), 2.85 (s, 3H), 1.77−1.66 (m, 1H), 1.62 (d, J = 11.1 Hz, 1H). 13C NMR(126 MHz, MeOD) δ 169.06, 167.46, 145.15, 143.71, 141.52, 140.79, 139.09, 134.88, 134.56, 132.63, 131.28, 129.30, 128.93, 128.47, 127.80, 127.59, 126.98, 74.57, 63.09, 45.75, 41.31, 36.25, 35.88, 22.12. HRMS m/z [M + H]+ for C34H32N2O4F3 calculated 589.2314, found 589.2321.
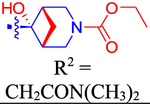
Ethyl (1R,5S,6r)-6-(4-(3-((2-(Dimethylamino)-2-oxoethoxy)-carbonyl)-6-(4-(trifluoromethyl)phenyl)naphthalen-1-yl)phenyl)-6-hydroxy-3-azabicyclo[3.1.1]heptane-3-carboxylate (50).
To a solution of 49 (34 mg, 0.058 mmol) and N,N-diisopropylethylamine (40 μL, 30 mg, 0.232 mmol) in DMF (5 mL) was added ethyl chloroformate (9 μL, 10.4 mg, 0.1 mmol). The resulting mixture was stirred at rt overnight. The volatiles were removed, and the residue was column-chromatographed (hexane/EtOAc, 60:40 → 0:100) to give 50 (23.8 mg, 62%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 8.80 (s, 1H), 8.24 (s, 1H), 8.12 (s, 1H), 8.04 (d, J = 8.8 Hz, 1H), 7.85−7.74 (m, 5H), 7.71 (d, J = 7.8 Hz, 2H), 7.58 (d, J = 7.8 Hz, 2H), 5.05 (s, 2H), 4.22 (q, J = 7.2 Hz, 2H), 3.97−3.83 (m, 4H),3.08 (s, 3H), 3.05−2.99 (m, 4H), 2.92 (s, 1H), 1.76 (t, J = 5.2 Hz, 1H), 1.46 (d, J = 10.5 Hz, 1H), 1.32 (t, J = 7.1 Hz, 3H). 13C NMR(126 MHz, MeOD) δ 167.64, 166.06, 157.23, 143.90, 143.73, 140.28, 138.79, 137.55, 133.43, 133.19, 131.11, 129.66, 127.82, 127.51, 127.01, 126.45, 126.39, 126.16, 125.55, 125.52, 73.37, 61.68, 61.06,46.23, 45.89, 40.33, 40.21, 34.86, 34.48, 22.57, 13.74. HRMS m/z [M + Na]+ for C37H35F3N2O6Na calculated 683.2345, found 683.2344.
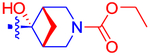
4-(4-((1R,5S,6r)-3-(Ethoxycarbonyl)-6-hydroxy-3-azabicyclo-[3.1.1]heptan-6-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid (51).
To a solution of 15 (11 mg, 0.02 mmol) and TEA(8.4 μL, 6.1 mg, 0.06 mmol) in DMF (0.5 mL) was added ethyl chloroformate (2.1 μL, 2.39 mg, 0.022 mmol). The resulting mixture was stirred at rt overnight. Volatiles were evaporated, and the residue was purified by RP-HPLC (C18, A: ACN, B: 10 mM TEAA, 50% →100% A in 40 min, flow rate = 5 mL/min, tR = 18.8 min) to give 51 as a white powder (2.00 mg, 17%): 1H NMR (400 MHz, MeOD) δ 8.60 (s, 1H), 8.37 (d, J = 2.0 Hz, 1H), 8.07−7.96 (m, 4H), 7.84 (dd, J =8.8, 1.9 Hz, 1H), 7.82−7.73 (m, 4H), 7.59 (d, J = 7.8 Hz, 2H), 4.18 (q, J = 7.1 Hz, 2H), 3.94−3.75 (m, 4H), 3.00−2.99 (m, 1H), 2.95−2.91 (m, 1H), 1.82−1.72 (m, 1H), 1.43 (d, J = 10.2 Hz, 1H), 1.32 (t, J = 7.1 Hz, 3H). HRMS m/z [M + H]+ for C33H29F3NO5 calculated 576.1998, found 576.2004.
4-(4-(1-(Ethoxycarbonyl)piperidin-4-yl)phenyl)-7-(4-(trifluoromethyl)phenyl)-2-naphthoic Acid (61).
To a solution of 1 (570.6 mg, 1.2 mmol), TEA (501 μL, 363.6 mg, 3.6 mmol), and LiOH (28.8 mg, 1.2 mmol) in DMF (30 mL) was added ethyl chloroformate (114.7 μL, 130.2 mg, 1.2 mmol). The resulting mixture was stirred at rt overnight. Volatiles were evaporated, and the residue was purified by RP-HPLC (C18, A: ACN, B: 10 mM TEAA, 50% →70% A in 40 min, flow rate = 5 mL/min, tR = 36 min) to give 61 as a white powder (414 mg, 63%): 1H NMR (400 MHz, MeOD) δ 8.57 (s, 1H), 8.30 (d, J = 2.0 Hz, 1H), 7.98−7.89 (m, 4H), 7.80−7.70 (m, 3H), 7.40 (d, J = 7.9 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 4.26 (d, J =13.1 Hz, 2H), 4.12 (q, J = 7.1 Hz, 2H), 2.99−2.84 (m, 2H), 2.84−2.73 (m, 1H), 1.91−1.84 (m, 2H), 1.73−1.58 (m, 2H), 1.28−1.26 (m, 3H). HRMS m/z [M + H]+ for C32H29F3NO4 calculated 548.2049, found 548.2042.
Ethyl 4-(4-(3-((2-(Dimethylamino)-2-oxoethoxy)carbonyl)-6-(4-(trifluoromethyl)phenyl)naphthalen-1-yl)phenyl)piperidine-1-carboxylate (62).
To a solution of 61 (667 mg, 1.22 mmol) and Cs2CO3 (795 mg, 2.44 mmol) in DMF (20 mL) under argon atmosphere was added 2-chloro-N,N-dimethylacetamide (151 μL, 178 mg, 1.464 mmol). The reaction mixture was stirred at rt overnight. The reaction mixture was partitioned between ethyl acetate (20 mL) and water (20 mL), and the aqueous phase was extracted with ethyl acetate (2 × 20 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated under pressure. The residue was purified by silica gel chromatography (hexane/ethyl acetate = 70:30 → 35:65) to afford compound 62 (480 mg, 62%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 8.74 (s, 1H), 8.19 (d, J = 1.9 Hz, 1H), 8.07 (d, J =1.7 Hz, 1H), 8.02 (d, J = 8.8 Hz, 1H), 7.82−7.69 (m, 5H), 7.44 (d, J = 7.8 Hz, 2H), 7.32 (d, J = 7.8 Hz, 2H), 5.01 (s, 2H), 4.40−4.26 (m, 2H), 4.15 (q, J = 7.1 Hz, 2H), 3.04 (s, 3H), 2.99 (s, 3H), 2.96−2.83 (m, 2H), 2.80−2.70 (m, 1H), 1.96−1.88 (m, 2H), 1.77−1.64 (m, 2H), 1.31−1.24 (m, 3H). 13C NMR (126 MHz, CDCl3) δ 166.41, 166.35, 155.75, 145.32, 144.00, 140.60, 137.86, 137.79, 133.62, 133.40, 131.42, 130.28, 128.19, 127.78, 127.20, 126.99, 126.04, 62.11,61.45, 44.61, 42.47, 36.07, 35.76, 33.31, 14.89. HRMS m/z [M + H]+ for C36H36F3N2O5 calculated 633.2576, found 633.2574.
Characterization of Lipophilicity.
pION Solubility Tests: pION Buffer and Blank Buffer Preparation.
pION solubility tests were performed with pION system solution (pION, Inc., Billerica, MA, P/ N 110151). pH 4.0 and 7.4 pION buffers were prepared by adding 2.5 mL of pION system solution to 97.5 mL of Milli-Q water and adjusting the pH to 4.0 and 7.4 with 0.5 N NaOH, respectively. Blank buffers were prepared by mixing 15 mL of pION buffers (pH 4.0 and 7.4) with 14 mL of n-propanol.
Control and Sample UV Plate Preparation.
All ligands were stored as 5 mM stock solutions in DMSO. 3 μL of each ligand stock solution (including DMSO control) was added to 300 μL of pH 4.0 and 7.4 pION buffers, mixed, and incubated for 20 h. The undissolved compound particles were removed by centrifuge at 14,000 rpm for 20 min. 100 μL of supernatant was added to a 96-well plate containing 100 μL of n-propanol and mixed well with a pipette.
Reference UV Plate Preparation.
4 μL of each ligand stock solution was added to 76 μL of n-propanol. 10 μL of resulting solution was added to 290 μL of blank buffer, mixed, and incubated for 20 h. 200 μL of the reference solution with a final concentration of ligand as 8.33 μM was transferred to a 96-well plate.
UV Absorption Tests.
The Greiner UV-Star 96-well plates have low UV absorption background and excellent resistance to general organic solvents. The UV absorption of control (Acontrol), ligands (Aligand), and references (Areference) were collected in triplicate at the ligands’maximum absorption at 270 nm.
Solubility Calculation.
Solubility(ligand) = 2 × average (Asample –Acontrol)/average (Areference – Acontrol) × 8.33 μM = 2 × average (Asample– Acontrol)/average (Areference – Acontrol) × 8.33 × M × 10−3μg/mL.
Relative Lipophilicities of Analogues of 1 Compared to 1.
All ligands were stored as 250 μM stock solutions in DMSO. HPLC-based measurements of relative lipophilicities of analogues of 1 compared to 1 were carried out with an Agilent Eclipse XDB-C18(4.6 mm × 250 mm, 5 μm) and a 20 min gradient of 10 to 100% acetonitrile in 10 mM triethylammonium acetate with a flow rate as 1.0 mL/min.
The HPLC-based lipophilicity of each ligand was calculated from a calibration curve using five standards and plotting their respective measured retention factor (k) versus reported Log D7.4 from the literature (Table S2, Figure S3). The correlation of Log D7.4 to k has a correlation coefficient r2 of 0.986. The lipophilicities Log D7.4 of modified piperidine analogues of piperidine 1 are exhibited in Table 1 and Figure S4.
Mouse Models of Mechano-Allodynia and Protease-Induced Asthma.
Mouse mechano-allodynia in Hsd:ICR (CD-1) outbred adult males (20–30 g, Envigo, Indianapolis, IN) was determined as described.10 The drugs were administered i.p. on the day of peak pain (7 days, post-surgery). The methods conformed to the guidelines of the International Association for the Study of Pain and the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee (IACUC) of St. Louis University.
Compound 15 and one antagonist double prodrug 50 were tested in mice (C57BL/6J, Jackson Laboratory, Bar Harbor, ME) displaying protease-induced asthma.6 There were two sensitizations, one week apart, consisting of oropharyngeal instillation of 50 μg of LPS-free ovalbumin (OVA, Worthington Biomedical, CA) and 20 μg of Aspergillus oryzae (Sigma-Aldrich, St. Louis, MO) in 50 μL of phosphate-buffered saline (PBS). The antagonist injection was performed on the day of ovalbumin challenge (day 14 post-initial sensitization using ovalbumin/Aspergillus), intraperitoneally at a dose of 10 mg/kg with 10 mL/kg vehicle (10% DMSO, 30% PEG-400, 60% water). Following 30 min, the mice inhaled 1% OVA aerosol in PBS for 1 h. 48 h later, BALF was collected, and the inflammatory cells were counted. The animal protocol was approved by the NIEHS IACUC (Research Triangle Park, NC).
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the NIH, National Institute of Diabetes and Digestive and Kidney Diseases Intramural Research Program (ZIADK031116) and Kantum Pharma, Inc. (Concord, NH) for support through NIDDK CRADA 17-0824. They also thank Tocris (Bristol, U.K.) for the gift of reactive fluorophore intermediates (JaneliaFluor dyes), and Dr. Bryan L. Roth (Univ. North Carolina at Chapel Hill) and National Institute of Mental Health’s Psychoactive Drug Screening Program (Contract # HHSN-271-2008-00025-C) for screening data.
ABBREVIATIONS USED
- ADME
absorption, distribution, metabolism, and excretion
- AAR/TTA
area at risk/total area of left ventricle
- BAL
bronchoalveolar lavage
- CCI
chronic constriction injury
- CHO
Chinese hamster ovary
- CXCL
chemokine (C-X-C motif) ligand
- DCM
dichloromethane
- DMAP
4-dimethylaminopyridine
- DMF
N,N-dimethylformamide
- IF
infarct size
- IL
interleukin
- I/R
ischemia/reperfusion
- MI
myocardial infarction
- PDSP
NIMH Psychoactive Drug Screening Program
- PWT
paw withdrawal threshold
- SAR
structure–activity relationship
- SLC
solute carrier
- UDPG
uridine-5′-diphosphoglucose
- TEA
triethylamine
- TEAA
triethylammonium acetate
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c00664.
Schemes S1 and S2 and synthetic methods, additional binding data, determination of lipophilicity by HPLC, off-target activities, ADMET properties, results from asthma, chronic pain, cardiac ischemia models, NMR spectra and HPLC purity, and calculated ADMET properties (PDF)
Molecular strings P2Y14 (CSV)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.3c00664
The authors declare no competing financial interest.
Contributor Information
Zhiwei Wen, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, United States.
Asmita Pramanik, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, United States.
Sarah A. Lewicki, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, United States
Young-Hwan Jung, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, United States.
Zhan-Guo Gao, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, United States.
John C. R. Randle, Random Walk Ventures, LLC, Boston, Massachusetts 02111, United States
Chunxia Cronin, Pat and Jim Calhoun Cardiology Center, University of Connecticut Health Center, Farmington, Connecticut 06030, United States.
Zhoumou Chen, Department of Pharmacology and Physiology and the Henry and Amelia Nasrallah Center for Neuroscience, Saint Louis University School of Medicine, St. Louis, Missouri 63104, United States.
Luigino A. Giancotti, Department of Pharmacology and Physiology and the Henry and Amelia Nasrallah Center for Neuroscience, Saint Louis University School of Medicine, St. Louis, Missouri 63104, United States
Gregory S. Whitehead, Immunity, Inflammation and Disease Laboratory, National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, North Carolina 27709, United States
Bruce T. Liang, Pat and Jim Calhoun Cardiology Center, University of Connecticut Health Center, Farmington, Connecticut 06030, United States
Sylvie Breton, Centre de Recherche du CHU de Québec, Département d’Obstétrique, de Gynécologie et Reproduction, Faculté de Médecine, Université Laval, Laval, Québec G1V 4G2, Canada.
Daniela Salvemini, Department of Pharmacology and Physiology and the Henry and Amelia Nasrallah Center for Neuroscience, Saint Louis University School of Medicine, St. Louis, Missouri 63104, United States.
Donald N. Cook, Immunity, Inflammation and Disease Laboratory, National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, North Carolina 27709, United States
Kenneth A. Jacobson, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892, United States
REFERENCES
- (1).Klaver D; Thurnher M Control of macrophage inflammation by P2Y purinergic receptors. Cells 2021, 10, 1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Arase T; Uchida H; Kajitani T; Ono M; Tamaki K; Oda H; Nishikawa S; Kagami M; Nagashima T; Masuda H; Asada H; Yoshimura Y; Maruyama T The UDP-glucose receptor P2RY14 triggers innate mucosal immunity in the female reproductive tract by inducing IL-8. J. Immunol 2009, 182, 7074–7084. [DOI] [PubMed] [Google Scholar]
- (3).Müller T; Bayer H; Myrtek D; Ferrari D; Sorichter S; Ziegenhagen MW; Zissel G; Virchow JC Jr.; Luttmann W; Norgauer J; Di Virgilio F; Idzko M The P2Y14 receptor of airway epithelial cells: Coupling to intracellular Ca2+ and IL-8 secretion. Am. J. Respir. Cell Mol. Biol 2005, 33, 601–609. [DOI] [PubMed] [Google Scholar]
- (4).Battistone MA; Mendelsohn AC; Spallanzani RG; Allegretti AS; Liberman RN; Sesma J; Kalim S; Wall SM; Bonventre JV; Lazarowski ER; Brown D; Breton S Proinflammatory P2Y14 receptor inhibition protects against ischemic acute kidney injury in mice. J. Clin. Invest 2020, 130, 3734–3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Wen Z; Salmaso V; Jung Y-H; Phung NB; Gopinatth V; Shah Q; Patterson AT; Randle JCR; Chen Z; Salvemini D; Lieberman DI; Whitehead GS; Karcz TP; Cook DN; Jacobson KA Bridged piperidine analogues of a high affinity naphthalene-based P2Y14R antagonist. J. Med. Chem 2022, 65, 3434–3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Karcz TP; Whitehead GS; Nakano K; Nakano H; Grimm SA; Williams JG; Deterding LJ; Jacobson KA; Cook DN UDP-glucose and P2Y14 receptor amplify allergen-induced airway eosinophilia. J. Clin. Invest 2021, 131, No. e140709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ko H; Das A; Carter RL; Fricks IP; Zhou Y; Ivanov AA; Melman A; Joshi BV; Kovác P; Hajduch J; Kirk KL; Harden TK; Jacobson KA Molecular recognition in the P2Y14 receptor: Probing the structurally permissive terminal sugar moiety of uridine-5′-diphosphoglucose. Bioorg. Med. Chem 2009, 17, 5298–5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hamel M; Henault M; Hyjazie H; Morin N; Bayly C; Skorey K; Therien AG; Mancini J; Brideau C; Kargman S Discovery of novel P2Y14 agonist and antagonist using conventional and nonconventional methods. SLAS Discovery 2011, 16, 1098–1105. [DOI] [PubMed] [Google Scholar]
- (9).Jain S; Pydi SP; Jung YH; Scortichini M; Kesner EL; Karcz TP; Cook DN; Gavrilova O; Wess J; Jacobson KA Adipocyte P2Y14 receptors play a key role in regulating whole-body glucose and lipid homeostasis. JCI Insight 2021, 6, No. e146577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Mufti F; Jung YH; Giancotti LA; Yu J; Chen Z; Phung NB; Jacobson KA; Salvemini D P2Y14 receptor antagonists reverse chronic neuropathic pain in a mouse model. ACS Med. Chem. Lett 2020, 11, 1281–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Patritti Cram J; Wu J; Coover RA; Rizvi TA; Chaney KE; Ravindran R; Cancelas JA; Spinner RJ; Ratner N P2RY14 cAMP signaling regulates Schwann cell precursor self-renewal, proliferation, and nerve tumor initiation in a mouse model of neurofibromatosis. eLife 2022, 11, No. e73511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Liu C; Zhou M; Jiang W; Ye S; Tian S; Jiang C; Hao K; Li H; Hu Q GPR105-targeted therapy promotes gout resolution as a switch between NETosis and apoptosis of neutrophils. Front. Immunol 2022, 13, No. 870183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Xu J; Morinaga H; Oh D; Li P; Chen A; Talukdar S; Mamane Y; Mancini JA; Nawrocki AR; Lazarowski E; Olefsky JM; Kim JJ GPR105 ablation prevents inflammation and improves insulin sensitivity in mice with diet-induced obesity. J. Immunol 2012, 189, 1992–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Khalafalla FG; Kayani W; Kassab A; Ilves K; Monsanto MM; Alvarez R, Chavarria M Jr; Norman B; Dembitsky WP; Sussman MA Empowering human cardiac progenitor cells by P2Y14 nucleotide receptor overexpression. J. Physiol 2017, 595, 7135–7148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Belley M; Deschenes D; Fortin R; Fournier J-F; Gagne S; Gareau Y; Gauthier JY; Li L; Robichaud J; Therien M; Tranmer GK; Wang Z Substituted 2-naphthoic acids as antagonists of GPR105 activity. WO2009070873A1, June 11, 2009.
- (16).Barrett MO; Sesma JI; Ball CB; Jayasekara PS; Jacobson KA; Lazarowski ER; Harden TK A selective high affinity antagonist of the P2Y14 receptor inhibits UDP-glucose-stimulated chemotaxis of human neutrophils. Mol. Pharmacol 2013, 84, 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Jung YH; Yu J; Wen Z; Salmaso V; Phung NB; Karcz T; Chen Z; Duca S; Bennett JM; Dudas S; Cook DN; Salvemini D; Gao ZG; Jacobson KA Exploration of alternative scaffolds for P2Y14 receptor antagonists containing a biaryl core. J. Med. Chem 2020, 63, 9563–9589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Robichaud J; Fournier J-F; Gagné S; Gauthier JY; Hamel M; Han Y; Hénault M; Kargman S; Levesque JF; Mamane Y; Mancini J; Morin N; Mulrooney E; Wu J; Black WC Applying the pro-drug approach to afford highly bioavailable antagonists of P2Y14. Bioorg. Med. Chem. Lett 2011, 21, 4366–4368. [DOI] [PubMed] [Google Scholar]
- (19).Jung YH; Salmaso V; Wen Z; Bennett JM; Phung NB; Lieberman DI; Gopinatth V; Randle JCR; Chen Z; Salvemini D; Karcz TP; Cook DN; Jacobson KA Structure-activity relationship of heterocyclic P2Y14 receptor antagonists: Removal of the zwitterionic character with piperidine bioisosteres. J. Med. Chem 2021, 64, 5099–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zhou M; Wang W; Wang Z; Wang Y; Zhu Y; Lin Z; Tian S; Huang Y; Hu Q; Li H Discovery and computational studies of 2-phenyl-benzoxazole acetamide derivatives as promising P2Y14R antagonists with anti-gout potential. Eur. J. Med. Chem 2022, 227, No. 113933. [DOI] [PubMed] [Google Scholar]
- (21).Wang YH; Zhou MZ; Ye T; Wang PP; Lu R; Wang YL; Liu CX; Xiao W; Li JY; Meng ZB; Xu LL; Hu QH; Jiang C Discovery of a series of 5-amide-1H-pyrazole-3-carboxyl derivatives as potent P2Y14R antagonists with anti-inflammatory characters. J. Med. Chem 2022, 65, 15967–15990. [DOI] [PubMed] [Google Scholar]
- (22).Kalgutkar AS; Driscoll JP Is there enough evidence to classify cycloalkyl amine substituents as structural alerts? Biochem. Pharmacol 2020, 174, No. 113796. [DOI] [PubMed] [Google Scholar]
- (23).van der Kolk MR; Jansen MACH; Rutjes FPJT; Blanco-Ania D Cyclobutanes in small molecule drug candidates. ChemMedChem 2022, 17, No. e202200020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Shearer J; Castro JL; Lawson ADG; MacCoss M; Taylor RD Rings in clinical trials and drugs: Present and future. J. Med. Chem 2022, 65, 8699–8712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Segall MD Multi-parameter optimization: identifying high quality compounds with a balance of properties. Curr. Pharm. Des 2012, 18, 1292–1310. [DOI] [PubMed] [Google Scholar]
- (26).Yu J; Ciancetta A; Dudas S; Duca S; Lottermoser J; Jacobson KA Structure-guided modification of heterocyclic antagonists of the P2Y14 receptor. J. Med. Chem 2018, 61, 4860–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ganiu MO; Cleveland AH; Paul JL; Kartika R Triphosgene and DMAP as mild reagents for chemoselective dehydration of tertiary alcohols. Org. Lett 2019, 21, 5611–5615. [DOI] [PubMed] [Google Scholar]
- (28).Mandal PK; McMurray JS Pd–C-induced catalytic transfer hydrogenation with triethylsilane. J. Org. Chem 2007, 72, 6599–6601. [DOI] [PubMed] [Google Scholar]
- (29).Ghosh AK; Brindisi M Organic carbamates in drug design and medicinal chemistry. J. Med. Chem 2015, 58, 2895–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kiselev E; Barrett M; Katritch V; Paoletta S; Weitzer CD; Brown KA; Hammes E; Hammes E; Yin AL; Yin AL; Zhao Q; Zhao Q; Stevens RC; Stevens RC; Harden TK; Harden TK; Jacobson KA Exploring a 2-naphthoic acid template for the structure-based design of P2Y14 receptor antagonist molecular probes. ACS Chem. Biol 2014, 9, 2833–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Cheng Y; Prusoff WH Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- (32).Grimm JB; Muthusamy AK; Liang Y; Brown TA; Lemon WC; Patel R; Lu R; Macklin JJ; Keller PJ; Ji N; Lavis LD A general method to fine-tune fluorophores for live-cell and in vivo imaging. Nat. Methods 2017, 14, 987–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Abdelshaheed MM; Fawzy IM; El-Subbagh HI; Youssef KM Piperidine nucleus in the field of drug discovery. Future J. Pharm. Sci 2021, 7, No. 188. [Google Scholar]
- (34).Ratni H; Baumann K; Bellotti P; Cook XA; Green LG; Luebbers T; Reutlinger M; Stepan AF; Vifian W Phenyl bioisosteres in medicinal chemistry: discovery of novel γ-secretase modulators as a potential treatment for Alzheimer’s disease. RSC Med. Chem 2021, 12, 758–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Sheffler DJ; Nedelcovych MT; Williams R; Turner SC; Duerk BB; Robbins MR; Jadhav SB; Niswender CM; Jones CK; Conn PJ; Daniels RN; Lindsley CW Novel GlyT1 inhibitor chemotypes by scaffold hopping. Part 2: development of a [3.3.0]-based series and other piperidine bioisosteres. Bioorg. Med. Chem. Lett 2014, 24, 1062–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Carreira EM; Fessard TC Four-membered ring-containing spirocycles: Synthetic strategies and opportunities. Chem. Rev 2014, 114, 8257–8322. [DOI] [PubMed] [Google Scholar]
- (37).Burkhard JA; Wagner B; Fischer H; Schuler F; Müller K; Carreira EM Synthesis of azaspirocycles and their evaluation in drug discovery. Angew. Chem., Int. Ed 2010, 49, 3524–3527. [DOI] [PubMed] [Google Scholar]
- (38).Ouvry G Recent applications of seven-membered rings in drug design. Bioorg. Med. Chem 2022, 57, No. 116650. [DOI] [PubMed] [Google Scholar]
- (39).Besnard J; Ruda GF; Setola V; Abecassis K; Rodriguiz RM; Huang XP; Norval S; Sassano MF; Shin AI; Webster LA; Simeons FR; Stojanovski L; Prat A; Seidah NG; Constam DB; Bickerton GR; Read KD; Wetsel WC; Gilbert IH; Roth BL; Hopkins AL Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Valkó K Application of high-performance liquid chromatography based measurements of lipophilicity to model biological distribution. J. Chromatogr. A 2004, 1037, 299–310. [DOI] [PubMed] [Google Scholar]
- (41).Bennett GJ; Xie YK A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988, 33, 87–107. [DOI] [PubMed] [Google Scholar]
- (42).Kuhn B; Mohr P; Stahl M Intramolecular hydrogen bonding in medicinal chemistry. J. Med. Chem 2010, 53, 2601–2611. [DOI] [PubMed] [Google Scholar]
- (43).Ashwood VA; Field MJ; Horwell DC; Julien-Larose C; Lewthwaite RA; McCleary S; Pritchard MC; Raphy J; Singh L Utilization of an intramolecular hydrogen bond to increase the CNS penetration of an NK1 receptor antagonist. J. Med. Chem 2001, 44, 2276–2285. [DOI] [PubMed] [Google Scholar]
- (44).Duncan SE; Gao S; Sarhene M; Coffie JW; Linhua D; Bao X; Jing Z; Li S; Guo R; Su J; Fan G Macrophage activities in myocardial infarction and heart failure. Cardiol. Res. Pract 2020, 2020, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Bohl S; Medway DJ; Schulz-Menger J; Schneider JE; Neubauer S; Lygate CA Refined approach for quantification of in vivo ischemia and reperfusion injury in the mouse heart. Am. J. Physiol. Heart Circ. Physiol 2009, 297, H2054–H2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Liu W; Cronin C; Cao Z; Wang C; Ruan J; Pulikkot S; Hall A; Sun H; Groisman A; Chen Y; Vella AT; Hu L; Liang BT; Fan Z Nexinhib20 inhibits neutrophil adhesion and β2 integrin activation by antagonizing Rac-1-Guanosine 5′-Triphosphate Interaction. J. Immunol 2022, 209, 1574–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Qian C; Wu Z; Sun R; Yu H; Zeng J; Rao Y; Li Y Localization, proteomics, and metabolite profiling reveal a putative vesicular transporter for UDP-glucose. eLife 2021, 10, No. e65417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Breton S; Battistone MA Unexpected participation of intercalated cells in renal inflammation and acute kidney injury. Clinical Practice: Mini-Review. Nephron 2021, DOI: 10.1159/000519265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Leeson PD; Bento AP; Gaulton A; Hersey A; Manners EJ; Radoux CJ; Leach AR Target-based evaluation of “drug-like” properties and ligand efficiencies. J. Med. Chem 2021, 64, 7210–7230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.