

Abstract
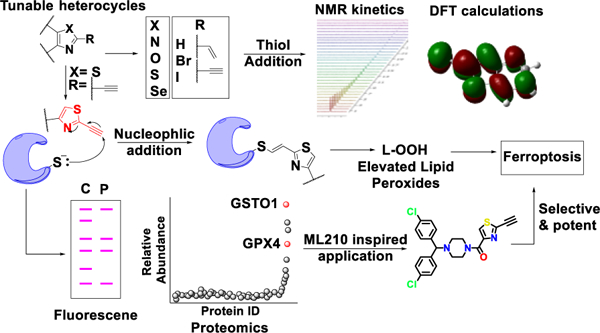
Once considered potential liabilities, the modern era witnesses a renaissance of interest in covalent inhibitors in drug discovery. The available toolbox of electrophilic warheads is limited with constraints on tuning reactivity and selectivity. Following our work on a class of ferroptotic agents termed CETZOLEs, we discovered new tunable heterocyclic electrophiles which are capable of inducing ferroptosis. Biological evaluation demonstrated that thiazoles with an alkyne electrophile at the 2-position selectively induce ferroptosis with high potency. Density functional theory calculations and NMR kinetic studies demonstrated the ability of our heterocycles to undergo thiol addition, an apparent prerequisite for cytotoxicity. Chemoproteomic analysis indicated several potential targets, the most prominent among them being GPX4 protein. These results were further validated by western blot analysis and Cellular Thermal Shift Assay. Incorporation of these heterocycles into appropriate pharmacophores generated highly cytotoxic agents such as the analog BCP-T.A, with low nM IC50 values in ferroptosis-sensitive cell lines.
Graphical Abstract
INTRODUCTION
Covalent inhibitors were considered outcasts by the pharmaceutical industry for a long time. This was due to an abiding fear stemming from studies performed in 20th century. Drugs like acetaminophen alarmed the scientific community due to possible induction of liver toxicity from the generation of metabolites capable of irreversible binding1. This and other studies2 led to the development of the reactive metabolite theory. According to this theory, several initially “inert” chemicals are converted in vivo to electrophilic metabolites, which can harm biological systems by reacting with several endogenous nucleophiles3, a process commonly referred to as “off-target effects”. This dogma has prevented the development of covalent inhibitors as drugs. However, several important exceptions show that covalent inhibition is not a barrier, but an opportunity to develop more selective and potent drugs. Aspirin, which acts as a covalent inhibitor of cyclooxygenase-1 (COX-1) enzyme is the most widely used medication in the world, with an estimated 80 billion tablets consumed annually in the United States alone4. Clopidogrel is a prodrug that is metabolized in vivo to its active form which then acts as a covalent inhibitor of P2Y12 purinergic receptor5 with application in antiplatelet therapy. Following the discovery of penicillin in 1928, β-lactam class of antibiotics revolutionized antibiotic treatment, signifying the origin of modern “antibiotic era”6. These compounds act by inhibiting bacterial DD-transpeptidase by covalent modification. Currently, about 30% of the marketed drugs are covalent inhibitors7 and recent years have seen a resurgence in the development of covalent inhibitors3. Covalent inhibition can provide highly potent drugs (low IC50 values) that produce prolonged therapeutic effects and can be administered at lower doses and less frequently. This leads to increased patient compliance and greater therapeutic outcomes. In addition, covalent inhibition has been associated with attenuated drug resistance8. A major requirement however, is achieving selectivity, which necessitates complete target identification and understanding of their mechanism of action.
There are two basic categories of covalent inhibitors. The first includes molecules which already have reactive electrophilic moieties, while the second category includes prodrugs requiring in vivo metabolic activation. The latter has limitations on the types of electrophilic moieties it can generate and is highly dependent on metabolic enzymes. On the other hand, the incorporation of tunable electrophilic warheads into a drug-like pharmacophore is more versatile and allows the tuning of reactivity of the electrophilic species, useful in targeted covalent inhibition (TCI). TCI is different from random reactivity of electrophiles with biological nucleophiles, since in TCI the drug with the reactive electrophile is first positioned at the appropriate site of the target protein by non-covalent interactions for subsequent nucleophilic addition. Thus, it is important that the electrophilic warheads should demonstrate optimum reaction kinetics; highly reactive electrophiles react in a nonspecific manner while weak electrophiles might not react with the desired target. In addition, the reaction must be both electronically and sterically favored. Modern drug discovery is limited by the availability of electrophilic warheads for incorporation into a drug-like pharmacophore. Despite many efforts to find optimized electrophiles, simple acrylamides are still the most frequent choice. They provide easy synthetic accessibility, selectivity for cysteine over other amino acids, and compatible ADMET (absorption, distribution, metabolism, excretion and toxicity) properties. A recent review by Gehringer et al. covers the available toolkit of electrophiles9. As with acrylamides, most of these electrophiles suffer from lack of available sites for tuning reactivity and selectivity. Analysis of 39 FDA approved covalent drugs by Singh et al.4 revealed that 33% of these drugs are anti-infectives (most notably the β-lactam class of antibiotics), 15% treat gastrointestinal disorders, and ~15% are used to treat central nervous system and cardiovascular indications. Cancer, which is still the second leading cause of death globally, accounted for 20%. They include aromatase10, thymidylate synthetase11, ribonucleotide reductase12 and kinase inhibitors (figure 1A) 13–15. There is the potential for irreversible binders to play even a greater role in cancer chemotherapy. However, these inhibitors must have a drug-like pharmacophore that directs the molecule to the active site. This is followed by a nucleophilic cysteine present in close proximity attacking the electrophilic warhead leading to covalent attachment of the drug to the target protein. In the case of kinases, this binding event can overcome competition by the high endogenous concentration of ATP, making irreversible kinase inhibitors an attractive approach. Recently, McAulay et al.16 reported alkynyl benzoxazines as novel irreversible kinase inhibitors13 (figure 1A). Currently, there is a heightened interest in the search for irreversible inhibitors capable of inducing ferroptosis17–21, an iron-dependent nonapoptotic cell death mechanism first reported in 2012 by Dixon et al.22. The hallmark of ferroptosis is accumulation of lipid peroxides, which leads to robust cell death due to cells failing to maintain a reductive environment. Two prototypical examples of processes that lead to ferroptotic cell death are inhibition of system-Xc− and inhibition of glutathione peroxidase-4 (GPX4). Erastin, the first ferroptotic inducer identified, is believed to inhibit the Xc− antiporter, but the exact mechanism remains unknown. Most of the electrophilic ferroptosis inducers such as RSL320,23, ML16224,25, ML21017, propiolamides21 and diacylfuroxans18 (figure 1B) are reported to inhibit GPX4 enzyme. GPX4 is responsible for conversion of lipid peroxides to alcohols, a process necessary for maintaining the reductive homeostasis of cells. Further, research confirmed that these electrophiles inhibit GPX4 by covalent interaction with a selenocysteine present in the enzyme active site24 (figure 1C). Although RSL3 and ML162 have a chloroacetmide (C.A) electrophilic warhead, ML210 and diacylfuroxans do not have any obvious electrophilic warhead capable of covalent inhibition. Excellent work by Schreiber and co-workers demonstrated that the latter two molecules act as masked nitrile oxides17,18 (figure 1B), which being highly electrophilic, covalently inhibit GPX4. Propiolamide (P.A) too can be included in the toolbox of electrophilic warheads to inhibit GPX4 and induce ferroptosis21. The active site of GPX4 is exposed (figure 1C) with a relatively flat surface in proximity to the catalytic selenocysteine residue. These characteristics provide broad substrate scope, but result in lack of a drug-like binding pocket and high reliance on the selenocysteine residue for its catalytic activity. Thus, covalent inhibition of GPX4 seems to be the only attractive choice for the design of inhibitors. Herein, we report novel hetero-aromatic electrophilic warheads capable of undergoing thiol addition, and inducing ferroptosis. Following up on our efforts to develop open chain epothilones (figure 1D)26, we discovered a new class of compounds capable of inducing ferroptosis, which we termed CETZOLEs27,28(figure 1D). Our subsequent studies showed that elimination of the cyclopentene ring did not have a significant effect on the cytotoxicity, identifying 2-alkynylthiazole moiety as the effective warhead. Although similar fragments have been reported to undergo thiol addition29–31, to our knowledge their ferroptosis-induction potential has not been reported. We used this moiety to rationally design a fragment library of heterocyclic electrophiles capable of inducing ferroptosis in a more selective manner. DFT analysis and NMR kinetic studies confirmed the electrophilicity of these warheads. Additionally, we designed and synthesized probes incorporating these fragments to perform cysteine labeling in cells. Proteomic analysis of pulldown experiments demonstrated enrichment of several proteins which are required for maintaining a reduced intracellular environment. Of these, GPX4 was notable, given its prominent role in detoxifying lipid peroxides. Our results expand the toolbox of electrophilic warheads available for covalent inhibition. These new warheads are capable of inducing ferroptosis and, when incorporated into appropriate pharmacophores, generate cytotoxic molecules with low nM potency. We expect these fragments to find a wider use in TCI or as part of hybrid molecule designs. In addition, they will be useful as tools in chemical biology applications.
Figure 1.
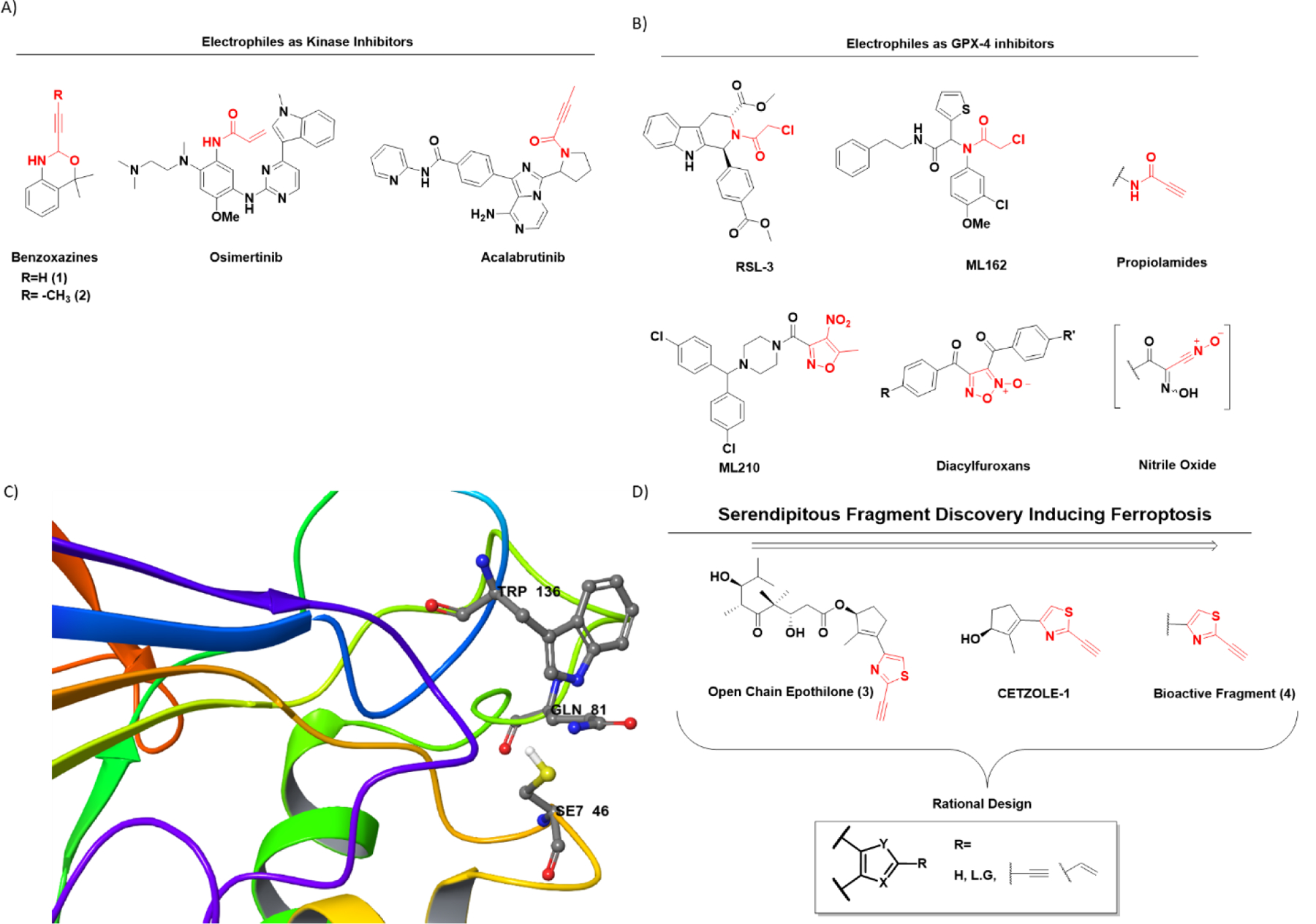
The heterocyclic warhead toolbox available for the design of covalent inhibitors. (A) Structures of some representative covalent kinase inhibitors. Typically, simple acrylamides have been used as electrophilic warheads of these molecules. Recently, McAulay et al.16 reported alkynyl benzoxazines (compounds 1 and 2) as potential kinase inhibitors. (B) Structures of several classical GPX4 inhibitors. Prototypical examples of covalent GPX4 inhibitors include RSL3 and ML162, both which have chloro-acetamide (C.A) as electrophilic warheads. New examples include ML210 and diacylfuroxans, which act as masked nitrile oxides (C) GPX4 active site representing the catalytic triad Se7 46, Gln81, and Trp136. The 3D model is generated using Maestro version 13.1 and protein data bank (PDB) entry with PDBid: 6ELW. The SeO2 residue in the original entry has been mutated in silico to SeH for more accurate representation. (D) Serendipitous discovery of thiazole-based electrophilic warheads following efforts to synthesize open-chain epothilones led to CETZOLE series of molecules. SAR studies reveal the thiazole fragment as the bioactive moiety.
RESULTS AND DISCUSSION
Design and Synthesis
We used compound (5) as the scaffold to design CETZOLE analogs in which the cyclopentene ring was replaced by an aromatic ring through simple peptide coupling as part of a general SAR investigation (scheme 1). The electronic properties were investigated by the positioning of EWG and EDG at para-position of the phenyl ring, as well as by extending conjugation between the thiazole ring and the phenyl ring. Sonogashira coupling of compound (5) with trimethylsilyl acetylene followed by hydrolysis provided compound (6) in moderate yields. Amide coupling under Steglich conditions provided analogs (8–10). NaBH4 reduction followed by Swern oxidation of (5) generated the aldehyde (11) in excellent yields. Wittig olefination of (11) with (12) provided the conjugated ester (13), which was subjected to hydrolysis and peptide coupling to provide analog (16).
Scheme 1.
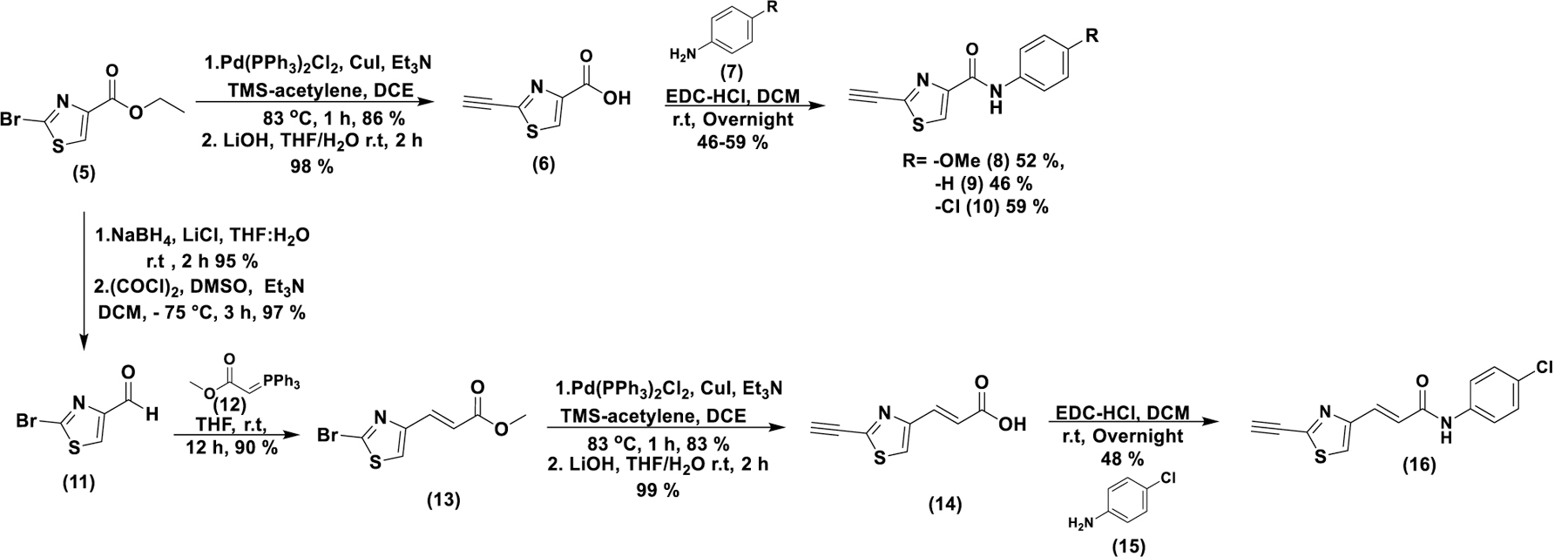
Synthesis of CETZOLE-1 analogs in which the cyclopentene ring has been replaced by para-substituted anilines.
These analogs lacking the cyclopentene ring not only retained activity, but, in some cases, showed an enhanced cytotoxic profile (table 3 and figure S9). We hypothesized that the 2-alkynylthiazole moiety constitutes the pharmacophore associated with ferroptosis activity. This was particularly captivating considering the small size of the pharmacophore. We hypothesized that the thiazole ring with the terminal alkyne behaves as a Michael acceptor and acts as a covalently interacting electrophilic warhead that induces ferroptosis. To test this hypothesis, we synthesized analogs of (5) without any additional group tethered at the ester functionality (scheme 2). We focused on steric and electronic modifications of the alkyne group. It was converted to the corresponding propyne (20) and (phenyl)acetylene analogs (24) and (22) via decarboxylative cross coupling and Sonogashira coupling, respectively. Conversion to the terminal alkene (18) was achieved through Suzuki cross coupling using vinylboronic acid pinacol ester (17).
Table 3.
IC50 ± SD (n = 3) values (μM) for CETZOLE-1, SAHA and the designed library of electrophilic heterocycles on HCT-116 and NCI-H522 cell lines.
| IC50 (μM) | Fold Change* | ||
|---|---|---|---|
| Compound | HCT-116 | NCI-H522 | |
| CETZOLE-1 | >40 | 3.99 ± 0.33 | > 10.02 |
| SAHA | 1.31 ± 0.08 | 1.72 ± 0.28 | 0.76 |
| 8 | 8.39 ± 0.53 | 0.67 ± 0.07 | 12.52 |
| 9 | 4.88 ± 1.28 | 0.66 ± 0.05 | 7.39 |
| 10 | 15.88 ± 1.02 | 0.54 ± 0.09 | 29.40 |
| 16 | 23.18 ± 1.96 | 0.37 ± 0.03 | 62.64 |
| 30 | >40 | >40 | N.A |
| 29 | >40 | >40 | N.A |
| 28 | >40 | >40 | N.A |
| 27 | >40 | >40 | N.A |
| 32 | >40 | >40 | N.A |
| 34 | >40 | >40 | N.A |
| 36 | >40 | >40 | N.A |
| 38 | >40 | >40 | N.A |
| 31 | 17.51 ± 1.59 | 1.02 ± 0.05 | 17.16 |
| 37 | >40 | >40 | N.A |
| 33 | >40 | 11.7 ± 2.08 | 3.41 |
| 35 | >40 | >40 | N.A |
| 39 | 9.5 ± 1.04 | 2.34 ± 0.27 | 4.05 |
| 40 | >40 | >40 | N.A |
| 41 | >40 | >40 | N.A |
| 42 | >40 | 12.2 ± 2.26 | > 3.27 |
| 43 | 19.36 ± 3.68 | 8.14 ± 1.10 | 2.37 |
| 44 | 11.61 ± 1.11 | 1.26 ± 0.08 | 9.12 |
| 45 | 19.61 ± 1.95 | 0.86 ± 0.06 | 22.80 |
| 47 | 24.96 ± 3.74 | 2.17 ± 0.13 | 11.50 |
| 46 | >40 | >40 | N.A |
| 48 | >40 | 5.65 ± 0.58 | > 7.07 |
| 24 | >40 | >40 | N.A |
| 22 | >40 | 2.67 ± 0.34 | > 14.98 |
| 18 | >40 | >40 | N.A |
| 20 | >40 | >40 | N.A |
| 51 | >40 | >40 | N.A |
| 49 | >40 | >40 | N.A |
| 57 | >40 | 3.53 ± 0.40 | 11.33 |
| 56 | >40 | >40 | N.A |
| 1 | >40 | 8.40 ± 1.81 | > 4.76 |
| 2 | 5.61 ± 0.52 | 0.89 ± 0.07 | 6.30 |
| 60 | >40 | >40 | N.A |
| 61 | >40 | >40 | N.A |
| 58 | >40 | >40 | N.A |
| 59 | >40 | >40 | N.A |
As fold change is the ratio of IC50 (HCT-116)/ IC50 (NCI-H522).
Scheme 2.
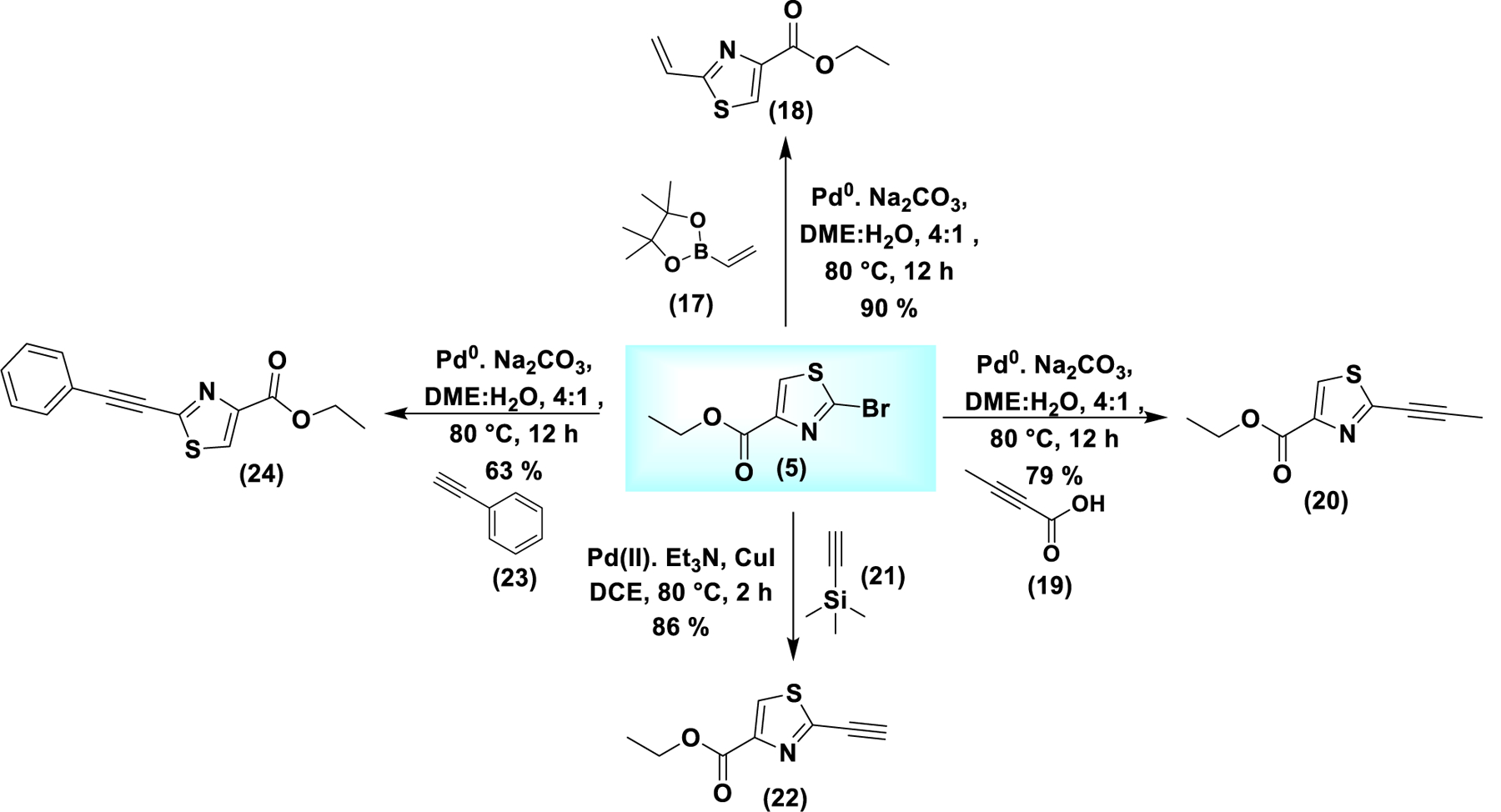
Synthesis of analogs of (5) with unaltered ester functionality. Modifications were in the steric and electronic properties of the alkyne moiety at the 2-position. Suzuki coupling generates the terminal alkene (18). Decarboxylative cross coupling provides the propyne analog (20). Sonogashira coupling provides the alkynes (22) and (24).
To investigate the effect of the nature of the heterocycle and the nature of the electrophile at 2-position, we synthesized a series of benzo[d]heterocylces with different electrophilic groups at the 2-position. Simple fused benzo[d]heterocycles were commercially available, and others were synthesized as shown in scheme 3. To synthesize Se-analog (27), 2-iodo aniline (25) was converted to the diselenide (26) by a two-step sequence32. Coupling of the diselenide with formic acid in the presence of PBu3, followed by intramolecular cyclization generated compound (27). Compounds 27–30 were subjected to halogenation at the 2-position using perfluorobutyl iodide or carbon tetrabromide to generate the corresponding iodo and bromo analogs (31–38)33. They were then subjected to Sonogashira, Suzuki and decarboxylative cross coupling to provide the corresponding terminal alkyne (43–45), terminal alkene (39–41) and propyne analogs (46), respectively. The corresponding alkene and alkyne selenium analogs ((42) and (47), respectively) were synthesized as shown in scheme 3 from intermediate (26) through peptide coupling with the corresponding carboxylate followed by deoxygenative cyclization with POCl334. To investigate the effect of a ring substituent at 5-position on the reactivity of the thiazole alkyne, the non-fused 5-phenyl thiazole analog (48) was synthesized by Sonogashira coupling of the corresponding halide.
Scheme 3.
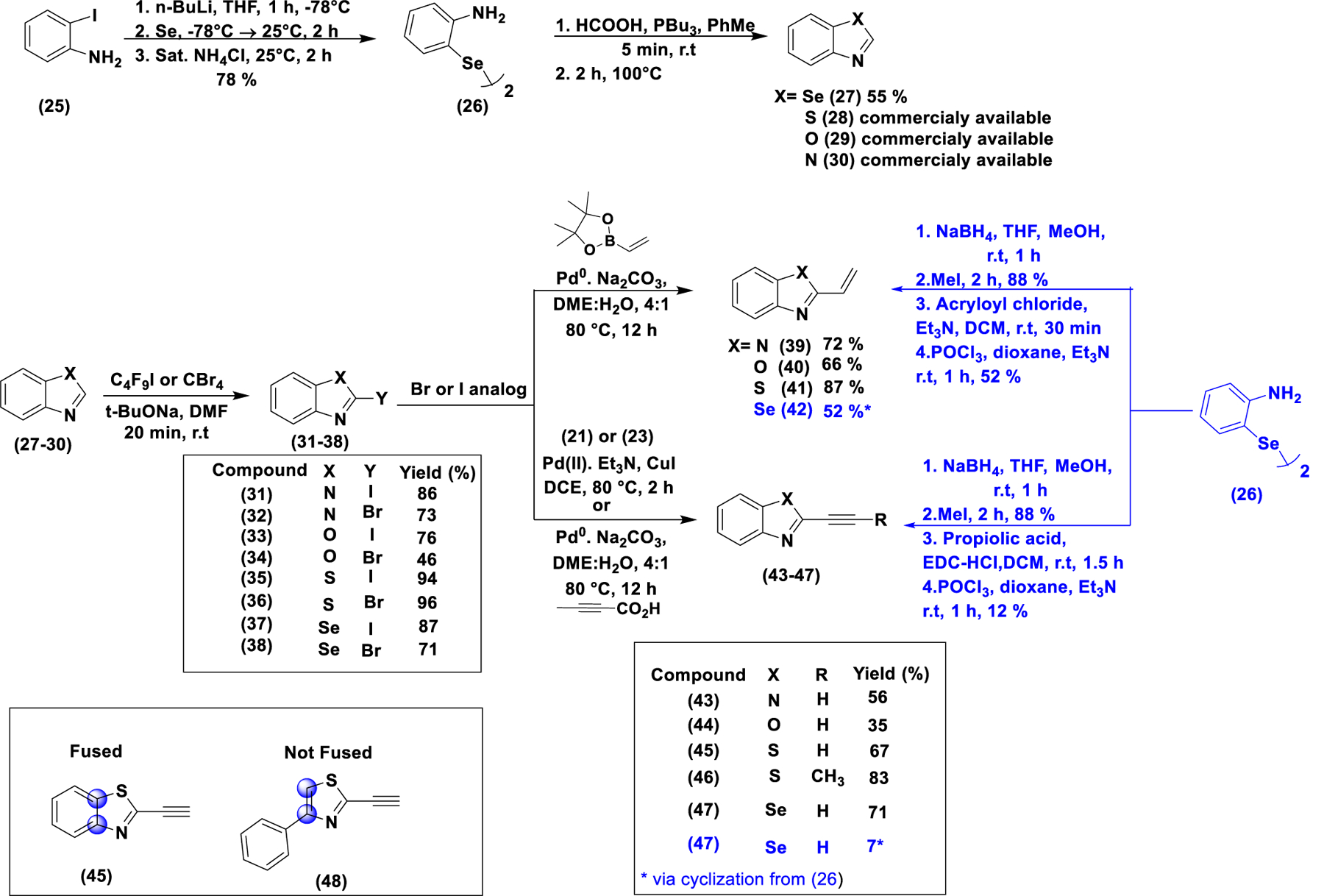
Synthesis of Benzo[d]heterocylces differently substituted at the 2-position with H, Br, I, alkyne, and alkene. For the unsubstituted precursors (27–30), only the benzoselnazole (27) was synthesized while the rest (28–30) were commercially available. They were then converted to the corresponding Br or I analogs (31–38), which were then converted to the alkyne or alkene analogs (39–47) by appropriate cross-coupling reaction. The corresponding alkene and alkyne selenium analogs (42) and (47), respectively) were synthesized as shown in scheme 3 from intermediate (26) through peptide coupling with the corresponding carboxylate, followed by deoxygenative cyclization with POCl3. In addition, a benzothiazole analog with an OTf group at the 2 position was synthesized and tested (figure S7A and scheme S7).
All analogs described so far have the electrophilic moiety at the 2-position of the heterocycle. In order to investigate the effect of substitution at other positions on reactivity and bioactivity, the 4-alkynyl and 5-alkynyl analogs (51) and (57) were synthesized. For the synthesis of the 4-alkynyl analog (51), the aldehyde (11) (scheme 4) was directly converted to the terminal alkyne through Seyferth-Gilbert homologation utilizing Ohira-Bestmann reagent (50) (synthesized as shown in scheme S1)35. In addition, the corresponding terminal alkene (49) too was synthesized via Wittig reaction (scheme 4). The bromine at the 2-position was retained to increase the electron deficiency of the thiazole ring, making the alkyne more electrophilic. The synthesis of the 5-alkynyl analog (57) was more challenging due to lack of appropriately substituted building blocks and the higher reactivity of 2-position compared to 5-position. Incorporation of a para-fluoro-phenyl ring served to block this position and also to reduce the electron density of the thiazole ring due to its strong electron withdrawing nature and thereby to increase the electrophilicity of the alkyne moiety. For this synthesis, the aminothiazole (52) was converted to the corresponding iodo derivative (53) through a Sandmeyer type iodination involving diazonium salt formation under strong acid conditions. Selective Suzuki coupling with para-fluoro-phenyl boronic acid under standard conditions gave (55). It was converted to 5-alkynyl (57) and 5-vinyl (56) analogs by Sonogashira and Suzuki coupling, respectively. Efforts to replace the ester with another aromatic ring generated highly hydrophobic compounds which showed no activity due to poor solubility (scheme S8 and figure S8).
Scheme 4.
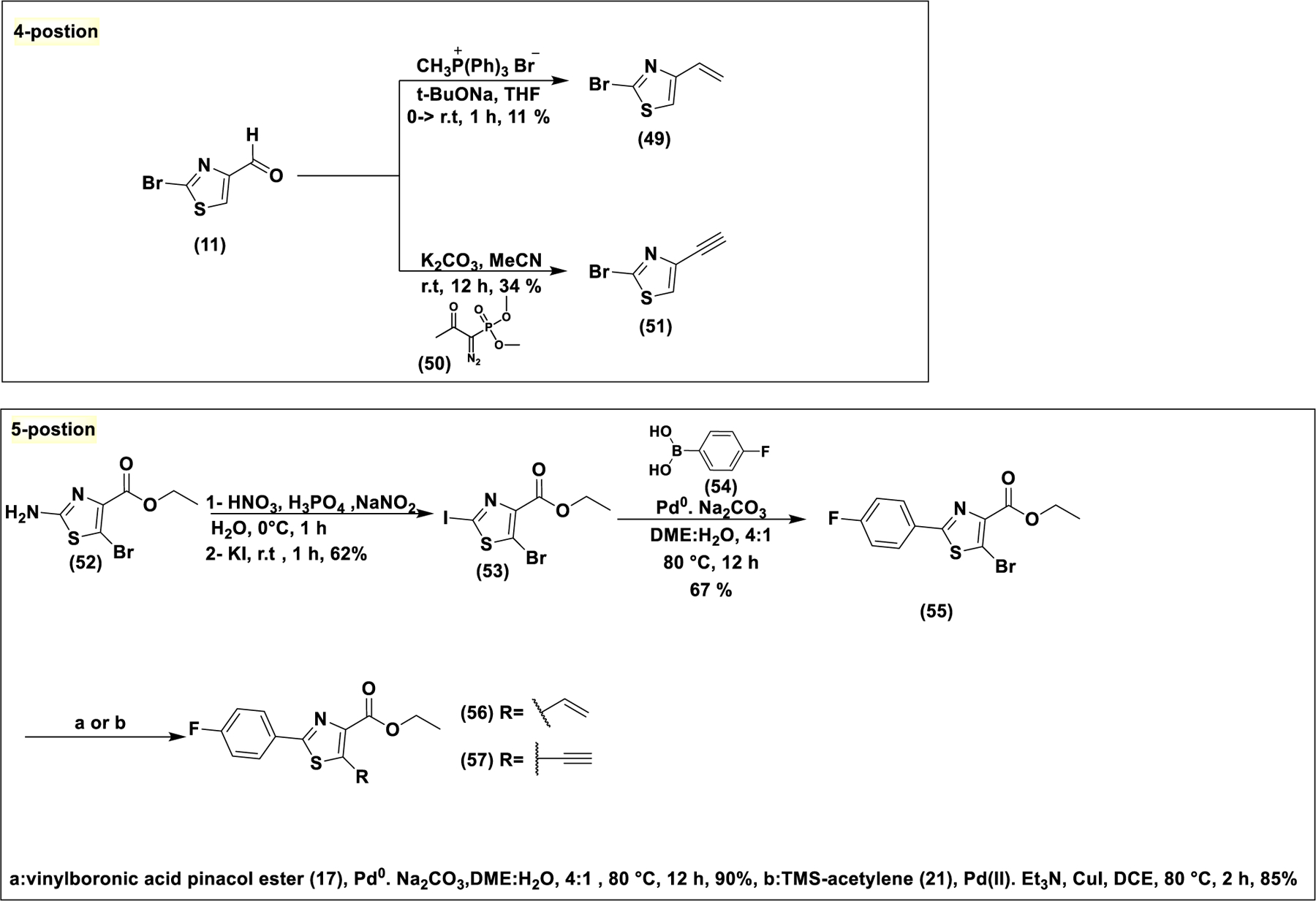
Synthesis of 4-and 5-substituted alkynyl analogs. For the 4-position, aldehyde (11) was converted to alkene (49) or alkyne (51) through Wittig reaction or through Seyferth-Gilbert homologation respectively. For the 5-substituted analogs, lack of available building blocks with appropriate functionality at the 5 position makes the synthesis a more challenging task. Blocking of the 2-position through a Suzuki reaction proved to be the optimum choice.
Analogs (58) and (59) were used to study the effect of alkyne vs isosteric nitrile at the thiazole 2-position. Analog (58) was synthesized via Sonogashira coupling of the corresponding bromide. The corresponding nitrile (59) is commercially available. To study the role of five-membered vs six-membered heterocycles, compounds (1) and (2) were synthesized. They were recently reported as potential kinase inhibitors bearing a new electrophilic warhead16. Although the electrophile is not a part of an aromatic system, these compounds have significant similarities with the fragments studied in this work and therefore, it was thought prudent to evaluate their ability to induce ferroptosis. They were synthesized as reported previously (Scheme S2)16. The commercially available pyridine (60) and pyrimidine (61) compounds were evaluated as additional representatives of 6-memberted ring electrophiles. All heterocycles reported and evaluated in this work are listed in figure 2.
Figure 2.
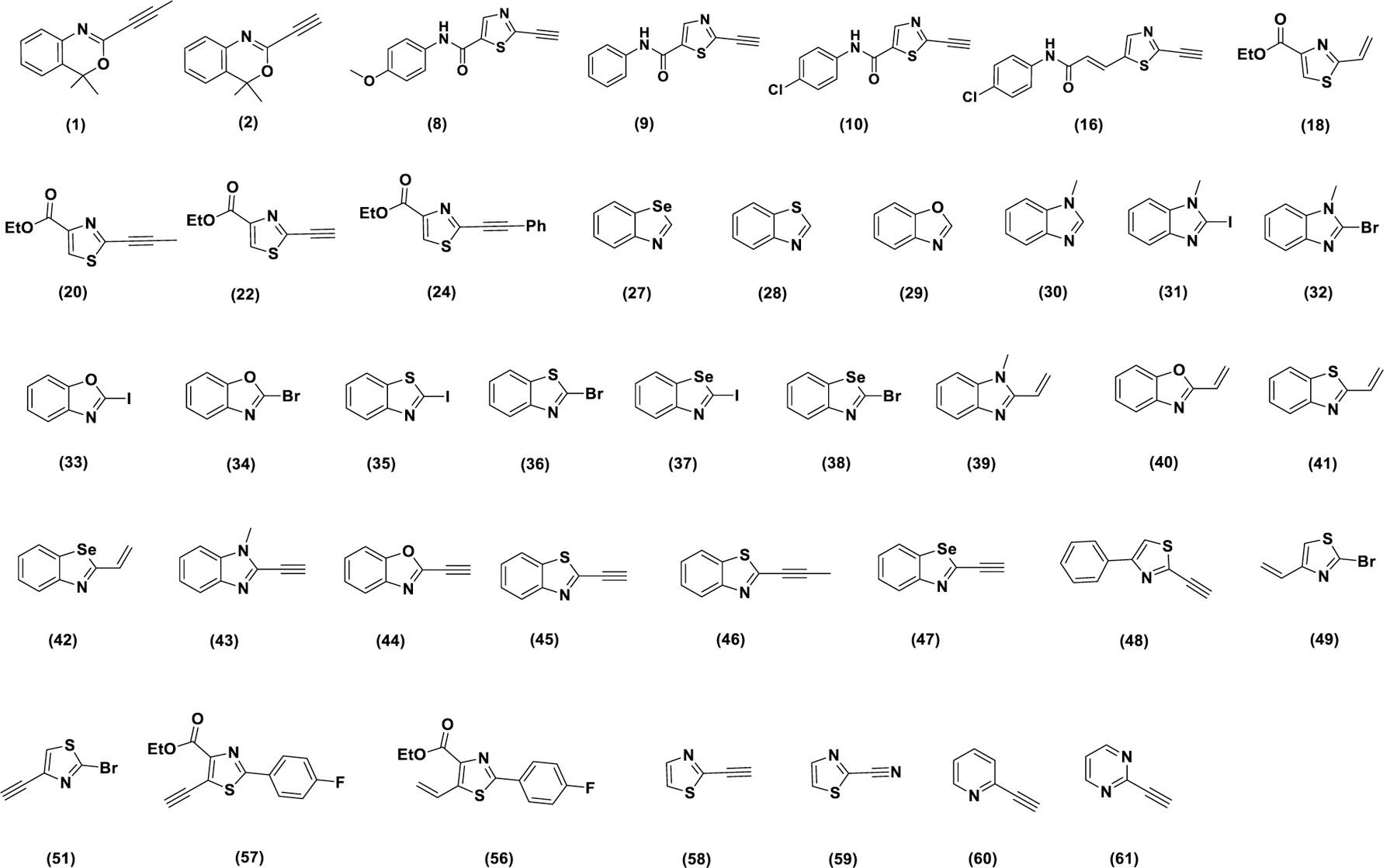
Library of heterocyclic electrophiles studied in this work. All Compounds were synthesized as shown in schemes 1–4 and scheme S2 with the exception of compounds (30), (29), (28), (59), (60) and (61), which were purchased from commercial sources.
DFT & Kinetic Studies
Prior to biological evaluation, we performed density functional theory (DFT) calculations on several representative analogs. Computational analysis was performed to test the tunability of the electrophilicity of the heterocycles. First, we analyzed the electrophilic sites for several representative analogs by calculating their Fukui functions36, confirming high electrophilicity for the 2-position on the heterocycle as well as the corresponding terminal alkyne/alkene positions (figure S3). Then, we estimated the adduct formation and transition state energies for the reaction of the analogs with methyl thiol. The results (table 1) show that the alkene analogs are less reactive than their alkyne counterparts (compare (56) vs (57), (51) vs (49), (18) vs (22) and (41) vs (45)). To our surprise, the bromo and iodo-analogs (tables 1 and 2) turned out to be highly reactive. This can be a major disadvantage in a biological system where it can lead to nonspecific reactions. For the benzo[d]heterocycles, we hypothesized that in addition to the nature of the electrophile at the 2-position, we can tune the reactivity by changing the nature of the heteroatom in the heterocycle itself. Heterocycles with strong lone pair-donating hetero atoms like in imidazole will have reduced reactivity, while those with hetero atoms which are weak lone pair donors such as Se will be more reactive electrophiles. Indeed, DFT calculations confirmed that the reactivity of the alkyne series inversely correlates with lone pair nucleophilicity. Thus, the imidazole analog (43) has the highest adduct formation energy (0.22 kcal/mol), while the selenazole analog (47) has the lowest (−10.43 kcal/mol). This hypothesis was further confirmed by calculations of the LUMO energy (figure 3 and table S2). Electron donation by neighboring heteroatoms increases the LUMO energy of the corresponding electrophile, making nucleophilic attack energetically more demanding (figure 3). As shown in table 1 and figure 3, substitution on the alkyne moiety (analog (46)) makes it a weaker electrophile by increasing both LUMO energy and presumably, steric interactions during thiol addition (high transition state energy of 6.06 kcal/mol).
Table 1.
Results from the DFT calculations to estimate adduct formation energies and transition state barriers at the M06-2X/6-31(p,d) level (with SCRF=(IEFPCM,solvent=water)). S-cis conformation energies were reported except in the case of (49).
| compound | Adduct formation energy (kcal/mol) |
Transition state energy kcal/mol |
|---|---|---|
| 56 | −7.39 | 1.10 |
| 57 | −12.04 | 3.21 |
| 49* | 18.47 | - |
| 51 | 6.51 | 12.00 |
| 18 | 0.84 | 3.31 |
| 22 | −4.46 | 2.29 |
| 28 | 9.30 | 9.96 |
| 36 | −43.59 | 5.32 |
| 41 | −2.40 | 2.14 |
| 45 | −7.68 | 3.54 |
| 46 | −2.42 | 6.06 |
| 44 | −7.71 | 3.23 |
| 43 | 0.22 | - |
| 47 | −10.43 | 2.12 |
Table 2.
Results from the DFT calculations of (36) and (35) to estimate adduct formation energies and transition state barriers at the M06-2X/ def2-TZVP level (with SCRF = (IEFPCM, solvent=water)).
| compound | Adduct formation energy (kcal/mol) | Transition state energy kcal/mol |
|---|---|---|
| 36 | −49.93 | 7.34 |
| 35 | −53.52 | 7.63 |
Figure 3.
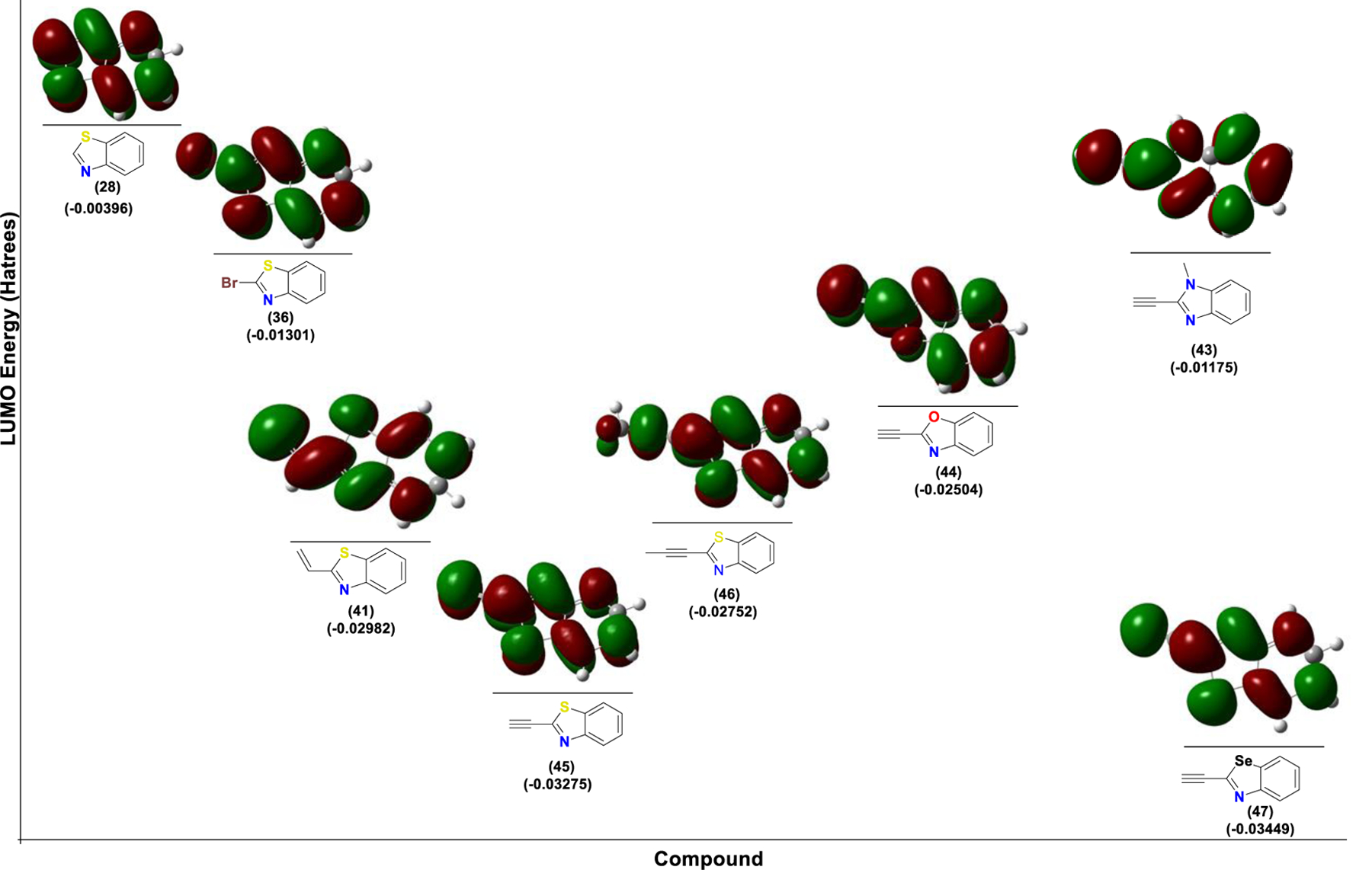
LUMO energies for different benzothiazole electrophiles. The LUMO energies depend on the nature of the electrophile as well as the nucleophilicity of lone pair of the heterocycle.
To compare the relative electrophilicity of the analogs described in figure 2, we performed an NMR-based kinetic study utilizing the methyl ester of N-Boc cysteine as a thiol-based nucleophile in the presence of DABCO as the base (figure 4). DMF was used as an internal standard. The reactions were performed in DMSO-d6 and NMR spectra were recorded at regular time intervals. The series of spectra was processed utilizing MestReNova software and the half-lives (t1/2) were calculated (figure 4B and table S1). For the terminal alkyne and alkene analogs, disappearance of the triple bond (as shown in representative example in figure 4B) and the double bond was monitored. For other types of electrophiles, shifting of appropriate aromatic proton signals was observed. Not surprisingly, for the alkyne series, as shown in figure 4A (and scheme S9), formation of only the double bond in E-configuration (based on J coupling) was observed. As shown in figure 4C, the heterocycles can be classified into four different groups based on their t1/2 values. Those with t1/2 < 5 min, those with 5 min < t1/2 < 1 hr, analogs with 1 < t1/2 < 6 hr and analogs with t1/2 > 6 hr. The alkyne analogs are more reactive than the corresponding alkenes, while alkyl-substituted alkynes had reduced reactivity. However, for bromo and iodo-analogs, despite being extremely reactive in the computational analysis for thiol additions, their reactivity in the NMR-based kinetic study was extremely slow (> 6 hr). Comparison of electrophilicity as determined by t1/2 for thiol addition in NMR studies with cytotoxicity as determined by IC50 values for growth inhibition of NCI H522 cells (represented by different color codes in figure 4C) reveals a direct correlation between the two; the most reactive electrophiles being typically the most biologically active as well.
Figure 4.
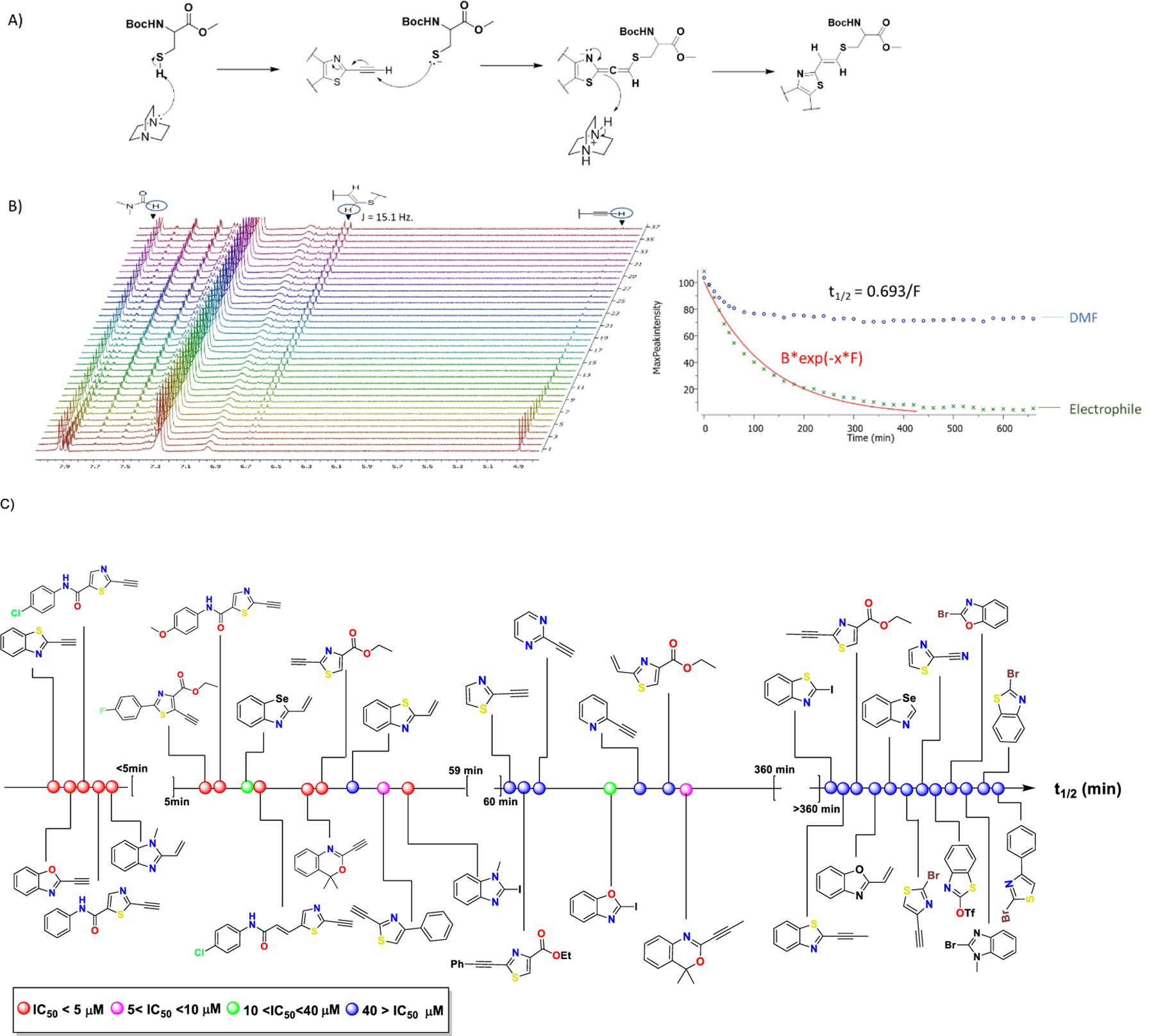
NMR kinetic analysis of thiol addition to our designed electrophiles. Methyl ester of N-Boc-cysteine was used as the thiol source. Reaction was facilitated by the addition of DABCO as a base. Reactions were performed in DMSO-d6. (A) Mechanism for addition of N-Boc-cysteine resulting in the trans-alkene. (B) Half-lives (t1/2) were calculated using the NMR processing software MestReNova. A representative NMR spectrum for alkyne analog (58) is shown. Disappearance of the terminal alkyne proton was monitored and used for the calculation of t1/2 . (C) t1/2 for the tested heterocycles are placed on the same axis, which increases from left to right. Ranges of IC50 values are represented by different color codes and increases (therefore, biological activity decreases) in the order red < pink < green < blue. There is a correlation of t1/2 with bioactivity where typically the faster reacting molecules are more potent as well. Consistent with previous reports, there is no observable correlation of LUMO energies and t1/216.
These molecules are highly cytotoxic at low μM range. If they exhibit similar reaction kinetics in biological systems, then they should react almost immediately with glutathione (GSH) and get deactivated (thiol adducts are inactive, figure S7B). However, GSH deprivation cannot be a sufficient explanation for the observed phenotypes as intracellular GSH levels can reach as high as 10 mM37 and cannot be significantly reduced by molecules with concentrations < 1 μM or in some cases low nM. Thus, it can be hypothesized that in a biological system these molecules are not as reactive towards thiol addition as they appear to be in the NMR study. This difference can be attributed to pH. The NMR kinetic study was performed in a basic environment generated by the addition DABCO. Indeed, if the same study is conducted without the addition of a base, the reaction half-lives increase significantly. For example, the half-lives of (45) (t1/2 < 5 min) and (22) (t1/2 18.9 min) in the presence of DABCO, increase to > 6 h and 4.43 h, respectively in the absence of DABCO. This observation is consistent with previous studies which suggest that the reaction of electrophiles with cysteines is highly dependent on the propensity for the thiol to be deprotonated38,39. We further validated the reactivity of our heterocycles by determining incubation time50 (IT50). IT50 is the time a drug treatment takes to cause irreversible damage leading to 50% cell death. We compared analog (45) (vide infra for biological studies) with the well-known suicide inhibitor of GPX4, RSL3. NCI-H522 cells were treated with the respective inhibitor and washed at different time points (figure 5A). Results (figure 5B and C) suggest that cells incubated with (45) and RSL-3 for 1–2 h cannot be rescued by washing off of the drug.
Figure 5:
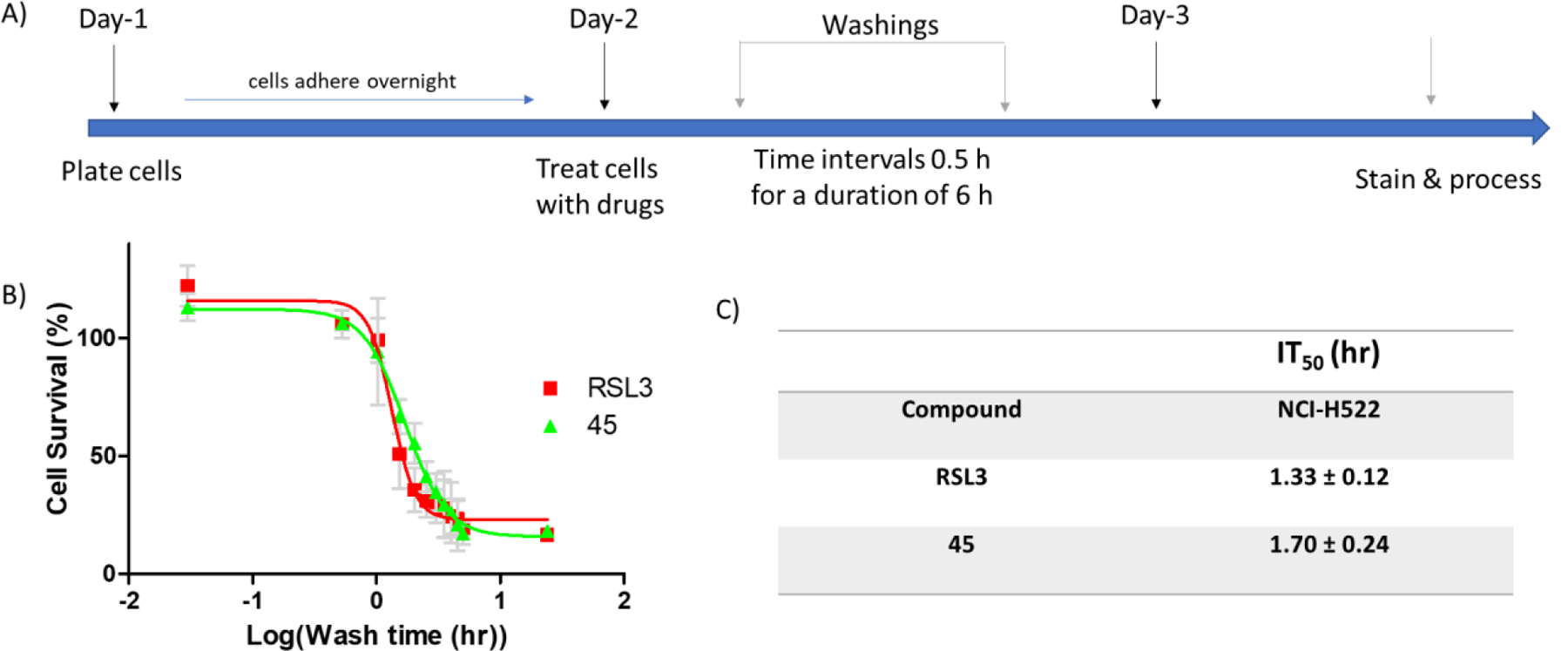
(A) Time line of the experiment to determine IT50. (B) wash time vs cell survival (%) of NCI-H522 cells treated with RSL3 (0.5 μM) and 45 (2 μM) (approximately 2–3 times the corresponding IC50s). (C) Calculated IT50s. To mitigate the effects of the process on cell proliferation, each data point is represented as % of the corresponding data point of DMSO treated cells. Data are mean ± SD (n=3).
Biological Evaluation
With the library of electrophilic fragments in our hands, we proceeded to evaluate their cytotoxicity on two cancer cell lines, the ferroptosis-sensitive mesenchymal cell line NCI-H522 and the ferroptosis-insensitive epithelial cell line HCT-116 (table 3 and figures S9 and S10). Previous studies in our laboratory have confirmed that HCT-116 cells are sensitive to erastin, but not CETZOLEs, sulfasalazine, or simple cystine starvation40. This may be due to erastin having several other targets including mitochondrial voltage dependent anion channels (VDAC2/3)41. Thus, this system of two cell lines is ideal for initial screening of the compounds for ferroptotic death. The thiazole anilides (8–10 and (16)) demonstrated superior cytotoxicity with IC50 values ranging from 0.37 ± 0.03 μM to 0.67 ± 0.07 μM (table 3 and figure S9A). As expected, (8) with a para-methoxy group was the least active analog of this series with IC50 0.67 ± 0.07 μM, whereas positioning an electron withdrawing group (EWG) such as chlorine at this position as in (10) enhanced cytotoxicity (IC50 0.54 ± 0.09 μM). Extension of conjugation in combination with the electron withdrawing effect further enhanced the sensitivity of NCI-H522 cells, with analog (16) demonstrating an IC50 value of 0.37 ± 0.03 μM. The attenuated effects of these compounds on the HCT-116 cells was demonstrated by approximately 8–62-fold increase in the IC50 values (table 3 and figure S9A). For the benzo-heterocyclic series, positioning of an electrophilic group at the 2-position proved to be crucial for activity since compounds (27–30) showed no effect (table 3 and figure S9B). The analogs such as (32), (34), (36) and (38) with a bromo substituent at the 2-position had no significant effect either (table 3 and figure S9C). Replacing bromine with the better leaving group iodine did not prove to be useful in the benzothiazole and benzoselenazole series, but the corresponding benzimidazole (31) and benzoxazole (33) analogs proved to be potent cytotoxic agents (table 3 and figure S9D). Incorporation of a double bond did not provide highly potent molecules. The benzoxazole and benzothiazole did not benefit from incorporation of a double bond as a softer electrophile, but the benzimidazole and benzoselenazole series provided the cytotoxic analogs (39) and (42), respectively (table 3 and figure S9E). Placing a terminal alkyne at the 2-position generated highly active analogs for all tested series (table 3 and figure S9F). The benziimidazole analog (43) showed the highest IC50 value. However, although the benzoselenazole alkyne (47) was expected to be the most active analog of this series, the best cytotoxic profile was demonstrated by the corresponding benzothiazole alkyne (45) with IC50 values of 0.86 ± 0.06 and 19.61 ± 1.95 μM for NCI-H522 and HCT-116, respectively. We attribute this to the potential instability of the selenium-based analog. The steric and electronic demand for a terminal alkyne is shown by the total loss of activity of the corresponding propyne analog (46). Ring fusion favors biological activity as shown by the comparison of activities of (45) and (48) (table 3 and figure S10D). We then investigated the effect of substitution of the alkyne in the thiazole ester series in (table 3 and figure S10A). Consistent with previous observations, substitution on alkyne (20 and 24) or replacement with terminal alkene (18) resulted in diminished activity. Only the corresponding terminal alkyne (22) showed significant cytotoxic effects with IC50 2.67 ± 0.34 μM on the H522 cell line with no observable effects on the HCT-116 cells even at 40 μM (IC50 > 40 μM), pointing to more selective ferroptosis induction. Positioning of the substituent at the 4-position of the thiazole ring generated inactive analogs whereas substitution at the 5-position produced the cytotoxic alkyne analog (57) (table 3 and figure S10B). Consistent with our observations, in the highly cytotoxic benzoxazine analogs reported by McAulay et al.16, the terminal alkyne (2) was more active than the propyne analog (1) (table 3 and figure S10C). The unsubstituted thiazole-alkyne (58) did not show any significant cytotoxicity, a phenomenon yet to be fully understood. Similarly, the corresponding 2-nitrile (59) was inactive. The pyridine (60) and pyrimidine (61) derivatives showed no significant activity. Table 3 summarizes the IC50 values for all of the tested compounds. In order to investigate biological effects which, stem from reaction only at the 2-position of heterocycles, analogs (22) and (45) were preferred in further biological assays.
Liproxstatin-1 rescue and C11-BODIPY lipid peroxide measurement
As HCT-116 cells are more “resistant” to ferroptosis than NCI-H522 cells, compounds that selectively induce ferroptosis would show significantly higher IC50 values in this cell line when compared to the NCI-H522 cell line. For example, the original compound CETZOLE-1, was not able to kill the HCT-116 cells at 40 μM, the highest concentration we had used, indicating that IC50 value on this cell line should be much higher than 10-fold when compared to the NCI-H522 (table 3). In contrast, the pan HDAC inhibitor SAHA, which induces cell death by mitotic arrest followed by apoptosis showed comparable IC50 values on both cell lines. Although this system of two different cell lines can provide some information on selective ferroptosis induction, it is not sufficient to prove that these differences are indeed due to ferroptosis. To associate the activity of the fragments with ferroptosis induction, we performed rescue experiments using the selective ferroptosis inhibitor Liproxstatin-1, which acts as a radical trap selectively on lipid bilayers42. Thus, we treated NCI-H522 cells with 2–3 times the IC50 of the cytotoxic analogs, in the presence (0.25 μM) and absence of Liproxstatin-1. All analogs of the amide series have significant cytotoxicity on the NCI-H522 cells, which was completely rescued in the presence of Liproxstatin-1 (figure 6). For the benzo[d]heterocycles, ferroptosis inhibition rescued cells only with the alkynes. The corresponding alkene (39) and iodo-derivative (31) kill NCI-H522 cells at the same rates both in the absence and presence of Liproxstatin-1, indicating that these molecules have the ability to induce other cell death mechanisms. Liproxstatin-1 treatment provided significant mitigation of cytotoxicity induced by the alkyne benzo[d]heterocycles (compounds 43–45 and (47)). Consistently, the promising cytotoxic profile of analog (22) is associated with selective ferroptosis induction, with Liproxstatin-1 treatment rescuing 100% of the cells. The investigation of positional effects demonstrated similar results with the benzo[d]heterocycles. For the alkynyl benzoxazine analogs, only (2) showed significant rescue of cells by Liproxstatin-1.
Figure 6.
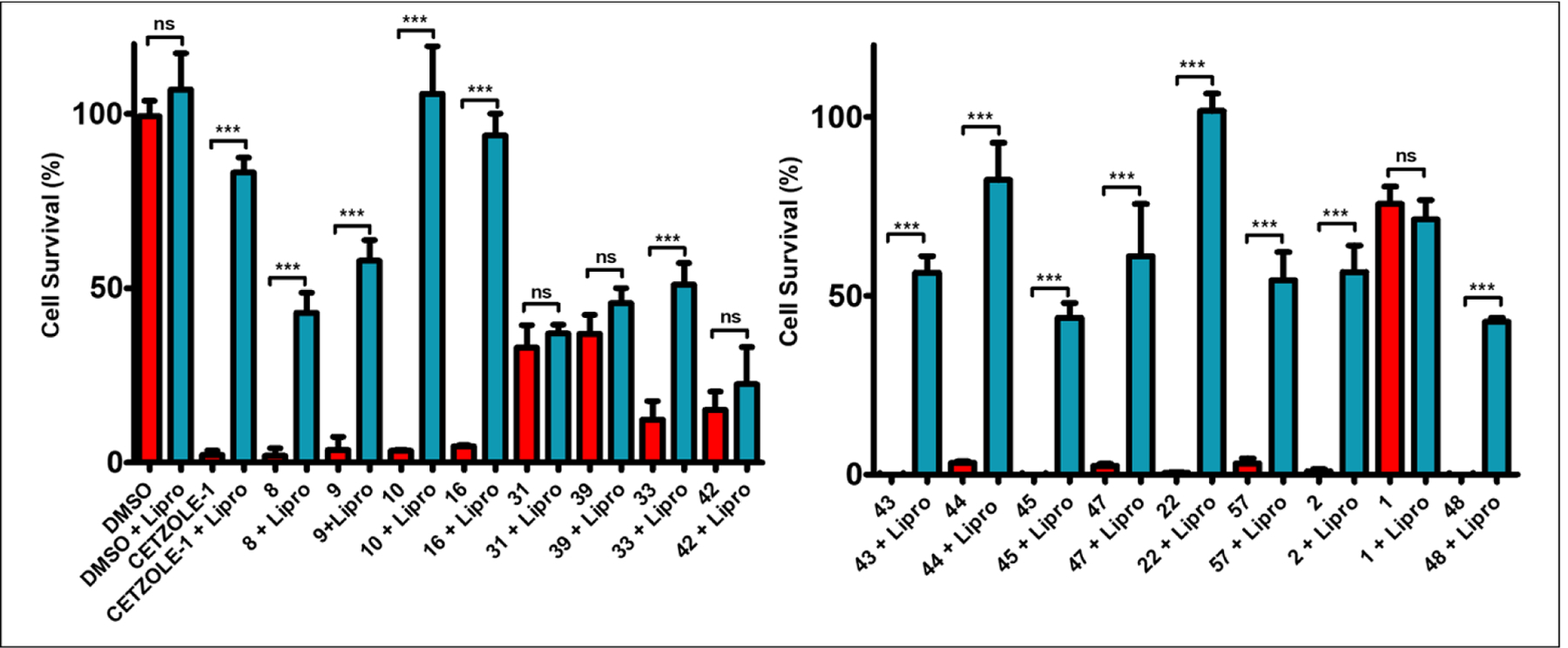
Liproxstatin-1 (0.25 μM) rescue experiments with the cytotoxic fragments on the NCI-H522 cell line; CETZOLE-1 (10 μM), (8) (2.5 μM), (9) (1.5 μM), (10) (2.5 μM), (16) (2.5 μM), (31) (2 μM), (39) (4.5 μM), (33) (30 μM), (42) (20 μM), (43) (20 μM), (44) (2 μM), (45) (2.5 μM), (47) (5 μM), (22) (10 μM), (57) (10 μM), (2) (2 μM), (1) (20 μM), and (48) (15 μM). Data are mean ± SD. (n = 3) and expressed as percentage decrease relative to the value for the control group (DMSO). Statistical analysis utilizing one-way ANOVA test *P < 0.05, **P < 0.01, ***P < 0.001.
Induction of ferroptosis is associated with accumulation of lipid peroxides. Therefore, we used C11-Bodipy as a surrogate marker for lipid peroxides (figure 7). C11-Bodipy inserts into cellular membranes and exhibits a shift in emission spectrum upon oxidation, widely considered to indicate the presence of reactive oxygen species in the vicinity of biological membranes43,44. After treatment with the imidazole iodine analog (31) C11-Bodipy oxidation was indistinguishable from the corresponding DMSO treatment (figure 7A). Thus, considering the data from the rescue experiments, analog (31) does not associate with ferroptosis induction. The same can be applied for the alkene analogs (42) and (39) (figures 7B and C), which marginally increase C11-Bodipy oxidation, but, failure to rescue NCI-H522 cells from Liproxstatin-1 co-treated with these compounds indicates cytotoxicity is not limited to the ferroptotic cell death mechanism. Interestingly, all alkyne analogs resulted in significant increase in C11-Bodipy oxidation (figures 7D–L and figure S4). As an example, compound (45) increased the C11-Bodipy oxidation more than 5-fold within 6 h of treatment. It should be noted that for compounds in figure 7F and J–L, the incubation time had to be reduced to 3 h due to accelerated cell death. In fact, for some of the tested compounds, cell death was so fast that most of the cells were dead by 3 h as shown in figure S4.
Figure 7.
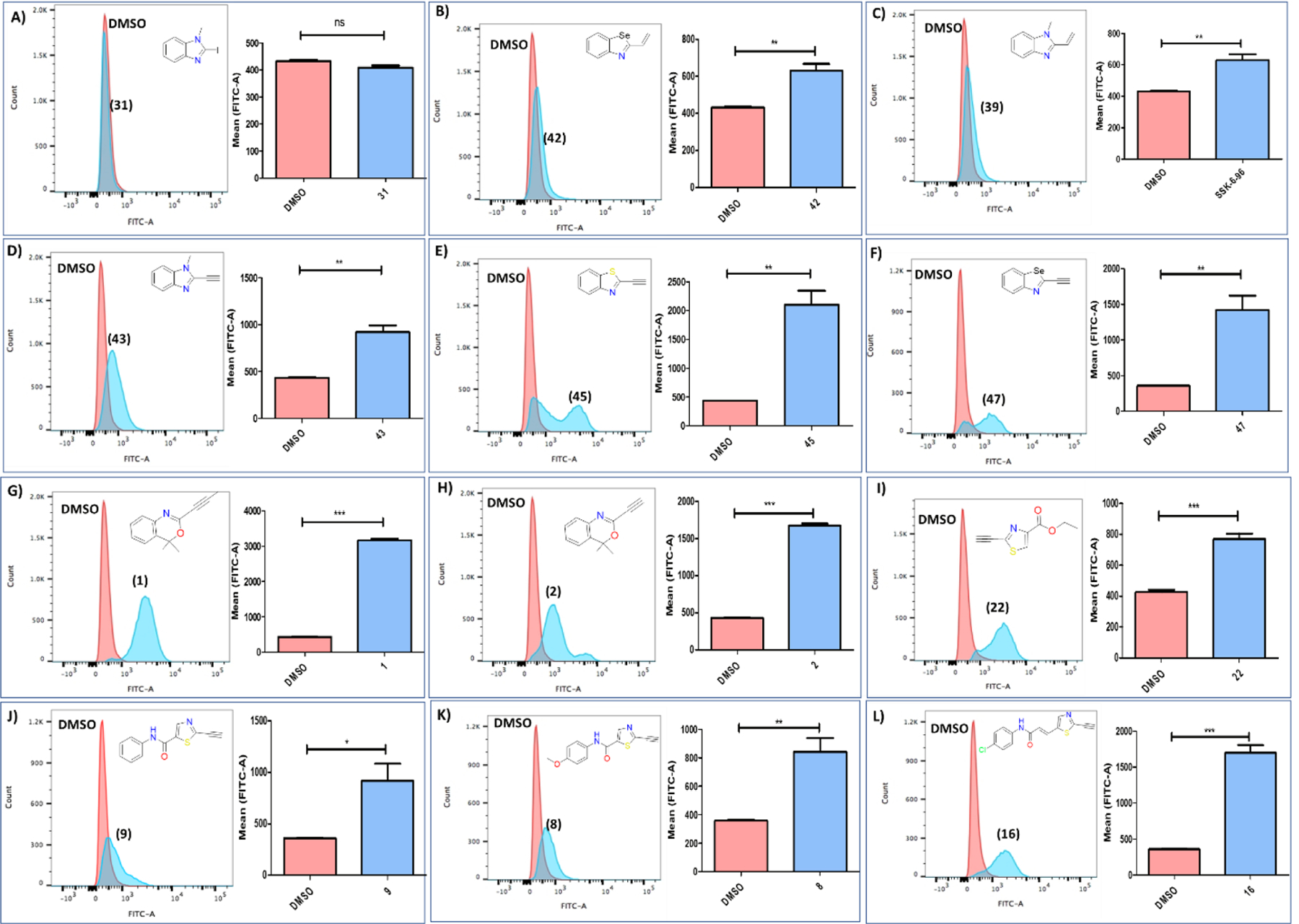
Lipid peroxide levels measured by flow cytometry using C11-BODIPY dye. (A) The benzimidazole iodo analog (31) does not increase levels of lipid peroxides. (B,C) The alkene analogs (39) and (42) 6 h after treatment marginally increase lipid peroxide levels. (D-L) The tested alkyne analogs led to significant increase of the lipid peroxides levels when compared to the corresponding DMSO treatment. The concentrations of the compounds used are: (31) (5 μM), (42) (60 μM), (39) (10 μM), (43) (40 μM), (45) (5 μM), (47) (10 μM), (1) (40 μM), (2) (5 μM), (22) (20 μM), (9) (5 μM), (8) (5 μM), (16) (5 μM). The treatment time for figures 9A–E and 9G-I is 6 h, while for figures 9F and 9J–L is 3h. Data are mean ± SD. (n = 3). Statistical analysis using t-test, *P < 0.05, **P < 0.01, ***P < 0.001.
Chemoproteomic Evaluation
Covalent inhibitors provide a powerful alternative for the development of potent therapeutic agents due to permanent inactivation of proteins. However, lack of selectivity is usually a major concern in such approaches. Thus, target elucidation is a crucial step in the development of covalent therapeutic agents. We have previously reported that induction of ferroptosis by CETZOLEs is consistent with inhibition of system Xc−. For example, glutamate secretion as well as uptake of cystine-FITC are reduced in treated cells28. However, these effects may be either direct or indirect and do not definitively identify this antiporter as a direct target. To investigate the potential biological targets of our fragments, we performed appropriate chemoproteomic experiments by activity-based protein profiling (ABPP) introduced by Cravatt and co-workers (figure 8A)45. We designed and synthesized (Schemes S3 and S4) appropriate probes containing a terminal alkyne as a bioorthogonal handle for incorporation of affinity or reporter tags (figure 8C)46. We used TAMRA-PEG3-N3 synthesized in our laboratory from TAMRA-acid (synthesized as previously reported47) and NH2-PEG3-N3 (synthesized as previously reported48) (Scheme S5 and figure S1) as the reporter tag, while commercially available Biotin-TAMRA-PEG3-N3 was used as the affinity tag (figure 8B). This allowed the monitoring of the proteins pulled down by in-gel fluorescence analysis (figure 10) and their identification by mass spectroscopy. Efforts to synthesize probes where the tags will be directly attached to the probe were not successful as shown in Scheme S6 and figure S2.
Figure 8.
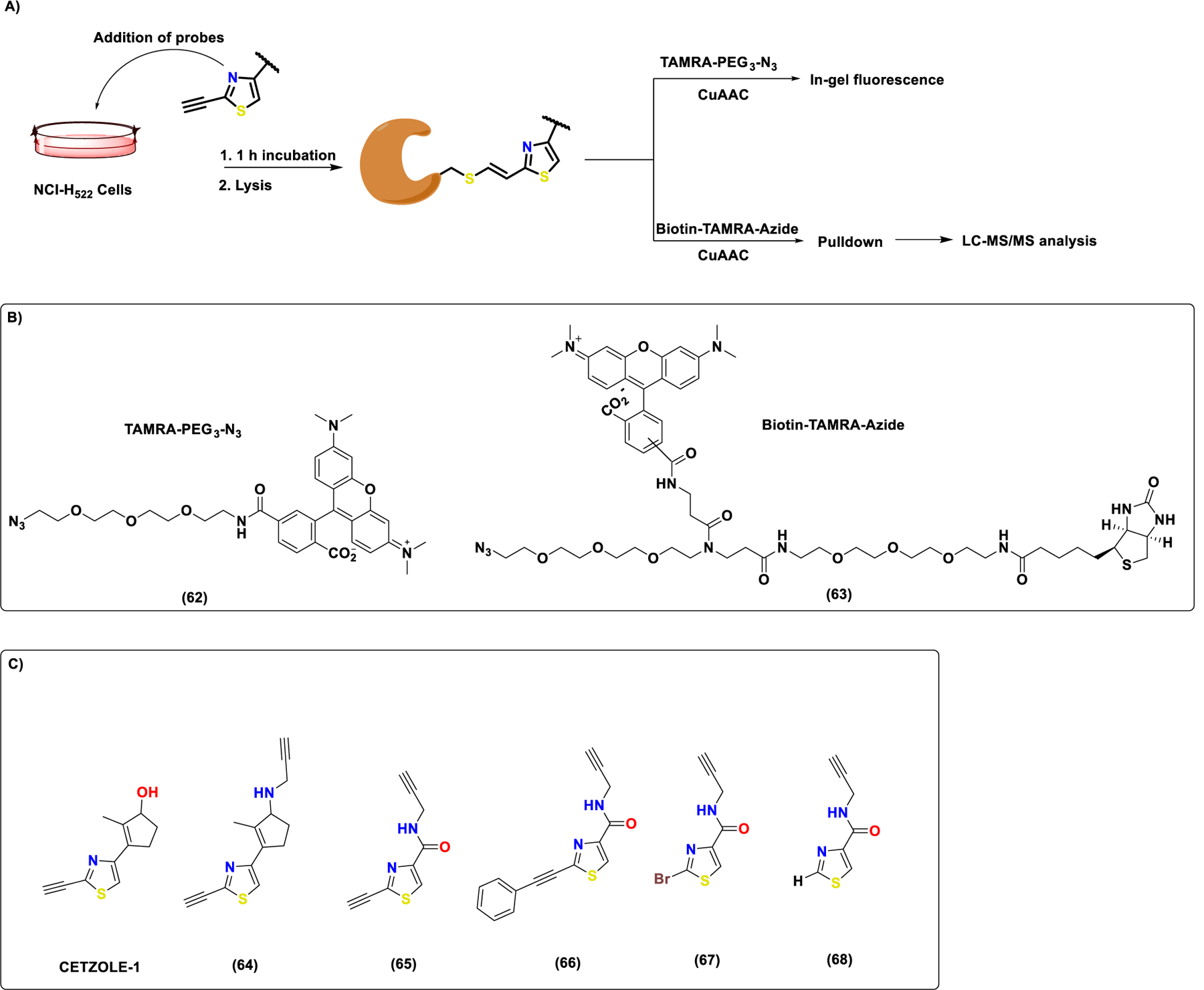
Protein pulldown protocol and chemical structures of compounds used for the chemoproteomic analysis following ABPP approach. (A) Simplified cartoon representation of the ABPP approach followed for chemoproteomic analysis of the heterocyclic warheads. Briefly, NCI-H522 cells were treated with appropriate probes for 1 h. Cells were collected and lysed. Then lysates were subjected to CuAAC with either TAMRA-PEG3-N3 (62) or Biotin-TAMRA-PEG3-N3 (63) under standard click conditions. (B) Chemical structures of the affinity and reporter tags used. (C) Library of the probes designed for the ABPP approach. Analog (64) serves as a probe that is structurally related to the original CETZOLE series. Probe (65) serves as the ferroptosis inducing heterocyclic fragment probe, while probes 66–68 serve as inactive controls.
Figure 10.

In-gel fluorescece labeling using the library of probes and TAMRA-PEG3-N3 as a reporter tag. (A) The CETZOLE-1 probe (64) (20 μM) and the warhead probe (65) (10 μM) show almost identical labeling profiles, indicating that the replacement of the cyclopentenyl ring does not alter the protein labeling profile. (B) The intensity of the bands observed with probe (65) by in-gel fluorescence increases in a dose dependent manner. (C) The labeling of almost all identified bands diminishes by preincubation with iodoacetamide (I.A) (0.2 and 2 mM). (D) RSL3 (10 μM), ML162 (10 μM) show overlapping small molecule interactome. As expected no competition was observed with erastin (20 μM). Left: optimum exposure of the competition gel, no band is saturated. Right: long exposure ( ~2 min) of the same gel.
The small heterocyclic probes were obtained by peptide coupling of the corresponding carboxylic acid with propargylamine (scheme S4). Compound (65) serves as the appropriate probe, because it is a derivative of (22), which induces ferroptosis with high selectivity when compared to the majority of the tested heterocycles. In addition, we tested three negative controls, the phenylacetylene analog (66), the bromine analog (67) and the unsubstituted analog (68). Among these, (66) could be considered the most appropriate control, because the corresponding heterocyclic ester (24), although undergoes thiol addition (vide supra for NMR kinetic studies), does not induce ferroptosis, a characteristic that will assist identification of proteins associated with the observed phenotype in subsequent fluorescent labeling and proteomic experiments. In addition, we designed compound (64) as a probe of the parent CETZOLE-1 molecule by reductive amination of the corresponding ketone (scheme S3) with propargylamine. This was to ensure that the labeling profile does not alter due to structural simplification of CETZOLE-1 to the thiazole heterocycles. We initially performed extensive biological investigations of the library of probes to confirm whether they retained the ability to induce the same phenotype (figure 9). Consistent with the cytotoxicity profile of the heterocycles, both probes (64) and (65) were more toxic to the ferroptosis sensitive NCI-H522 cells than the ferroptosis “resistant” HCT-116 cells. Although probe (65) showed some toxicity on the HCT-116 cells, the required concentration is relatively high to rule out its application in chemoproteomic experiments. Fortunately, the designed controls (66–68) did not have any effect on both cell lines (IC50 > 40 μM). Liproxstatin-1 rescue experiments and lipid peroxidation measurements confirmed the ability of the probes to induce ferroptosis in NCI-H522 cells.
Figure 9.
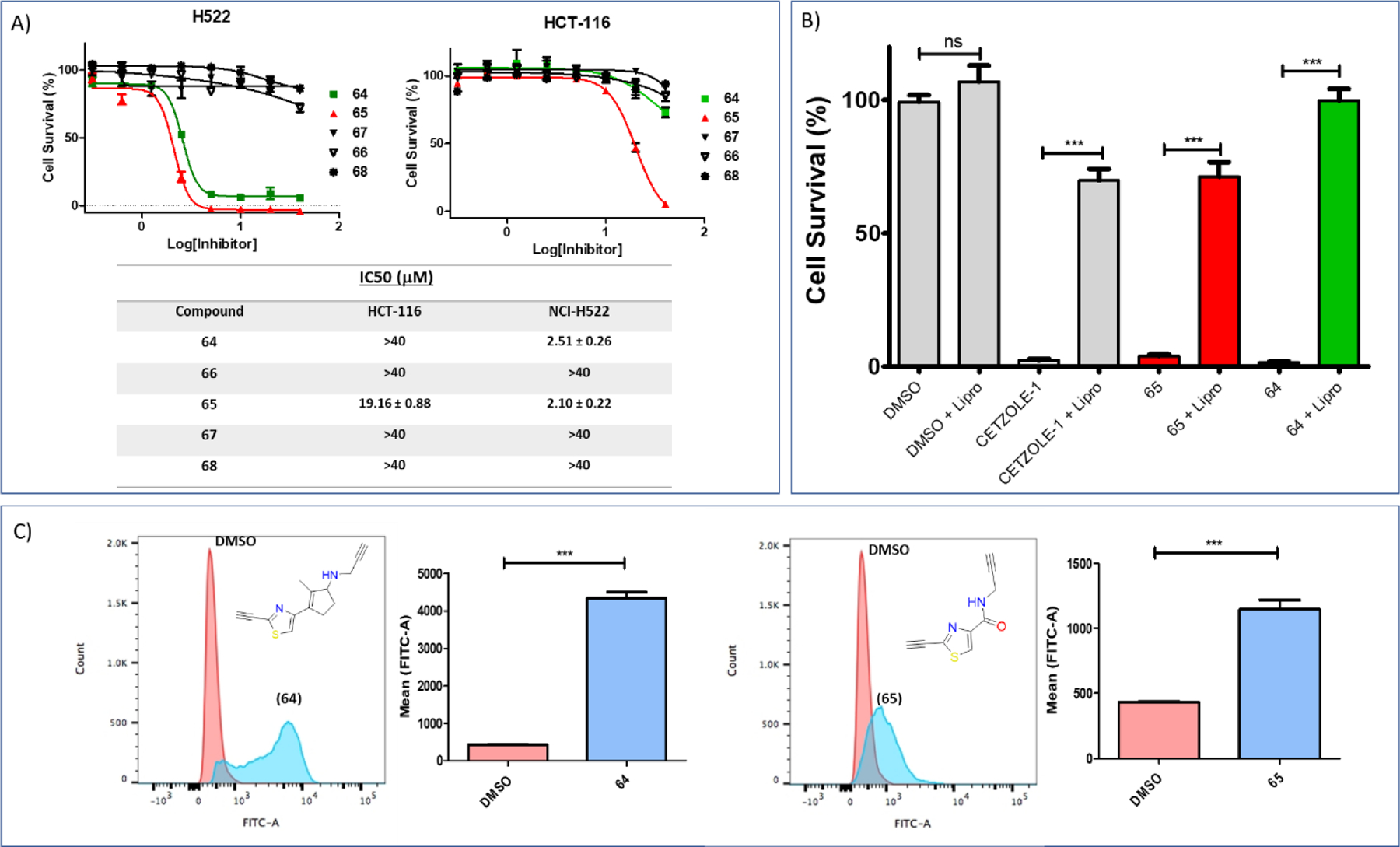
Biological evaluation of the library of designed probes. (A) Dose response graphs and IC50 values for several probes on the NCI-H522 and HCT-116 cell lines. (B) Liproxstatin-1 rescue experiments for the active probes (64) (20 μM) and (65) (10 μM). For both probes, their cytotoxicity can be associated with ferroptosis induction due to significant rescue by treatment with Liproxstatin-1 (0.25 μM). Data are mean ± SD. (n = 3) and are expressed as percentage decrease relative to the value for the control group (DMSO). (C) Ferroptosis was further confirmed by the increased levels of lipid peroxides induced by both probes (64) (20 μM) and (65) (10 μM). Data are mean ± SD. (n = 3) Statistical analysis utilizing one-way ANOVA test for figure 9B and t-test for figure 9C *P < 0.05, **P < 0.01, ***P < 0.001.
To confirm that the labeling profile of our small heterocyclic warheads is similar to that of the parent molecule CETZOLE-1, we performed fluorescent labeling experiments. NCI-H522 cells were treated with appropriate probes for covalent binding with their target proteins. Cells were lysed and copper-catalyzed azide-alkyne cycloaddition (CuAAC) was performed under standard conditions to append the fluorescent reporter TAMRA-PEG3-N3. The labeled proteins were visualized by in-gel fluorescence analysis. The results show that the probe, based on the parent molecule (64) and the probe of the heterocyclic warhead (65), produce almost identical labeling profiles (figure 10A). This confirmed the conclusion from our SAR studies that the cyclopentene ring of CETZOLE-1 can be replaced with other groups. In addition, the fluorescent intensity of all labeled proteins followed a dose-dependent pattern (figure 10B). The labeling of several prominent proteins (e.g., at ∼50 kDa, ∼30 kDa and ∼32 kDa) could be blocked by the pretreatment of cells with CETZOLE-1, suggesting specific binding interactions (figure S5A). The covalent nature of the labeling was further validated by competition with iodoacetamide (IA), which is a known nondiscriminatory electrophile that covalently attaches to nucleophilic residues of proteins such as cysteine (figure 10C). Pretreatment of NCI-H522 cells with IA diminished the labeling of most of the targeted proteins, suggesting that indeed the proteins of interest have nucleophilic residues that covalently bind to our probes. Competition experiments with a series of ferroptosis inducers (figure 10D) such as RSL3 (10 μM), ML162 (10 μM) show overlapping small molecule interactome (compare bands ∼32 kDa and ∼19 kDa in figure 10D). As expected, no competition was observed with erastin (20 μM) (figure 10D). Among the tested heterocycles, the terminal alkynes (45) and (2) (20 μM) demonstrated high competition, in contrast to the iodo (31) or terminal alkene (39) benzimidazole analogs (20 μM) (figure S5B).
We next performed pulldown assays to identify the labeled proteins by mass spectrometry. Bioorthogonal ligation with the TAMRA-Biotin-Azide tag allows for enrichment of the desired proteins utilizing streptavidin magnetic beads. Part of the enriched samples (30%) was analyzed for fluorescent labeling to confirm that the pulldown succeeded in enriching the same proteins that were visualized in the fluorescent labeling experiments (figure 11). The rest of the samples were analyzed by LC-MS/MS (by MS Bioworks). As shown in figure 11, enriched samples were separated on short SDS gel (2cm), which was then cut in to 12 equally sized bands, of which the last three bands were combined to give a total of 10 bands. Each band was processed by in-gel digestion providing peptide fragments, which were identified by mass spectroscopy using Mascot (Matrix Science) and further validated with Scaffold (Proteome Software). Following this process more than 2000 proteins were identified, but among them only 227 met the enrichment criteria (figure 11 and S.I proteomic excel file). Because large proteins usually tend to contribute more to the spectral count than small ones, the relative values of protein abundance were estimated by Normalized Spectral Abundance Factor (NSAF, = (SpC/MW)/Σ(SpC/MW)N) method (figure 11 and S.I proteomic data file). The NSAF method is useful to minimize the variations originated from protein length49,50.The majority of the proteins identified are involved in intracellular redox pathways. As shown in figure 12A subjecting the enriched proteins to protein analysis through evolutionary relationships (PANTHER)51 classification revealed that most of the enriched proteins are involved in catalytic processes. In addition, BLAST analysis (in the S.I) showed that all enriched proteins contain multiple cysteine residues, while three of them contained selenocysteine, both capable of covalently attaching to our heterocyclic fragments. Among, the proteins identified, GPX4 is the most notable contender whose relationship with induction of ferroptosis is undisputable17–21,52. However, the possible contribution of other proteins in generating the phenotype observed with our electrophiles cannot be overlooked. For example, glutathione S-transferase omega-1 (GSTO1), which has the highest relative abundance, is an atypical GST isoform whose functions are not fully understood. Although, as shown by Ramkumar et al.53, covalent inhibition (or knock-down) of GSTO1 by chloroacetamide based electrophiles proved to be lethal in cancer cells, its correlation with ferroptosis is not clear despite its role in cellular stress responses is now emerging54–56. To further validate the mass spectrometry results, we performed pulldown experiments and analyzed the enriched proteins by western blot. However, performing such analysis for 227 proteins will be extremely tedious. Thus, we compared the data for the enriched proteins using probe (65) with previous reports17–20 for other electrophilic probes ML162-yne, RSL3-yne and diacylfuroxan-di-yne (figure 12B). We considered enriched proteins which were confirmed either by mass spectroscopy data or western blot analysis. We have observed enrichment of GPX4, GTSO1 and TRXD1 proteins with all these probes, which is represented in the Venn diagram (figure 12C). Thus, we aimed to further validate the finding by western blot analysis. As shown in figure 12D, western blot results are consistent with mass spec results for GPX4 and GSTO1. Although TXRD1 was marginally enriched in the proteomics data, western blot analysis reveals that even inactive/less active compound (66) has the ability to label this protein. We further validated the ability of our compounds to bind to GPX4 by Cellular Thermal Shift Assay (CETSA)57. We used analog BCP-T.A (vide infra for “not all warheads are equal”), a bulkier molecule onto which we have incorporated our electrophilic fragment. Interaction of BCP-T.A with GPX-4 is indicated by the stabilizing effect our molecule has on GPX4 by shifting its aggregation temperature (Tagg) by ∼7 °C (figure 12E–G). This stabilizing effect on GPX4 is in line with previous reports on stabilization of GPX4 by ML21017, which has the same pharmacophore as BCP-T.A, but different electrophilic warhead. Thus, we have proven the ability of our electrophilic warheads to bind to GPX4 by mass spectrometry, western blot analysis and CETSA. Although, GPX4 inevitably becomes the focus of our work, inhibition of other proteins should not be overlooked when incorporating these fragments into potential drug candidates. In addition, the possibility of an altered small molecule interactome can emerge, warranting additional chemoproteomic analysis for potential drug candidates.
Figure 11.
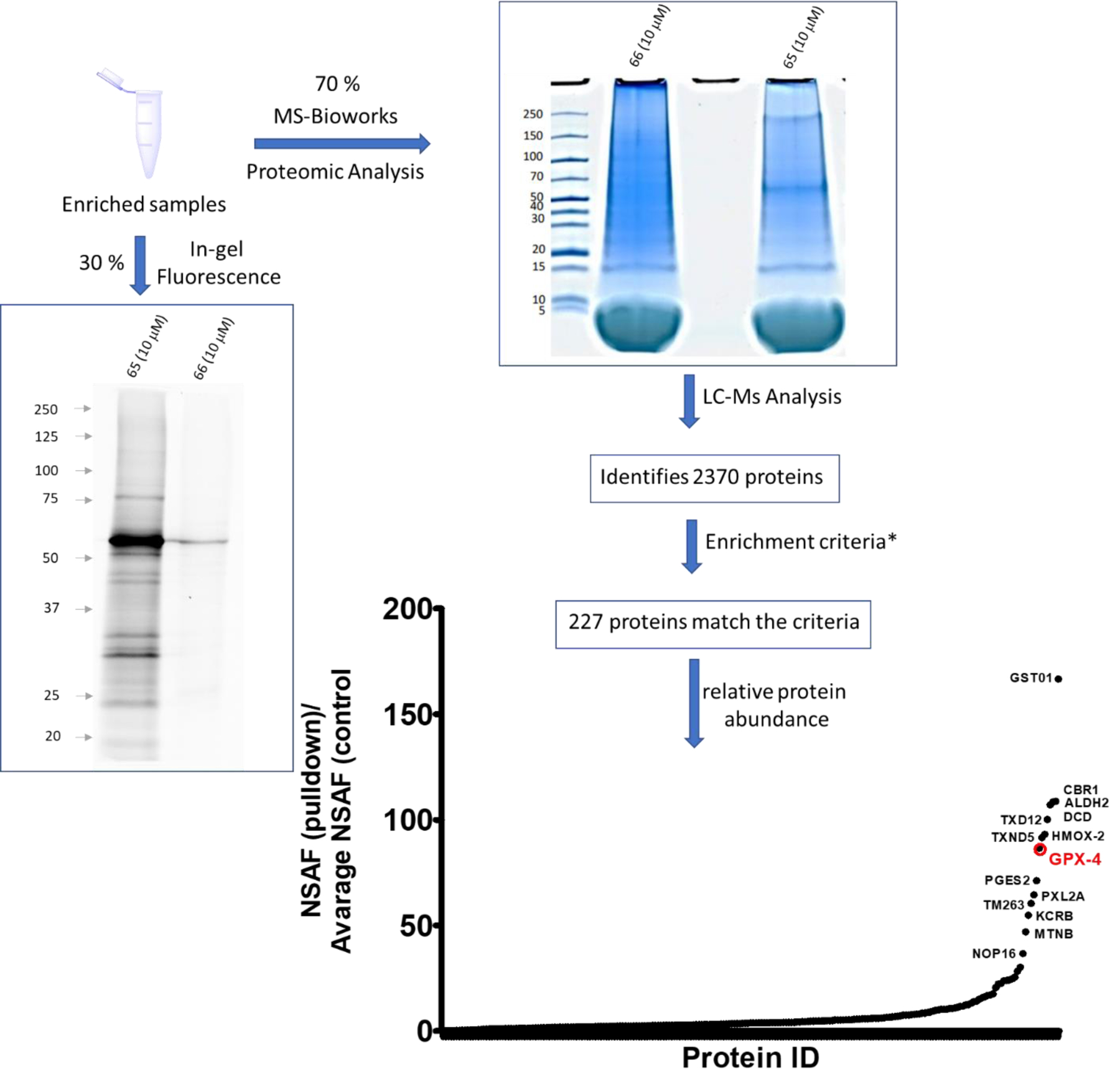
Pulldown experiments using the probe (65) (10 μM) and TAMRA-Biotin-Azide as an affinity and reporter tag. Prior to Mass Spectrometry analysis, enrichment was confirmed by analyzing 30% of the sample by in-gel fluorescence. The remaining 70% was analyzed by LC-MS/MS at MS Bioworks. A total of 2370 proteins were identified, but only 227 met the enrichment criteria (S.I proteomic excel file). Less than 20 stand out as the most enriched proteins. Most of them are associated with oxidative homeostasis such as GPX4, a key regulator in ferroptosis inhibition. Note that this data is qualitative and not quantitative, calculated using: spectral count (SpC), molecular weight (MW) and number of pulled down protein (N). *Enrichment criteria are: 1. Protein had at least 5 SpC in sample (65) 2. Protein was not detected in the (66) or 3. Protein was detected with a 4-fold or more increase based on dividing the NSAF values.
Figure 12.
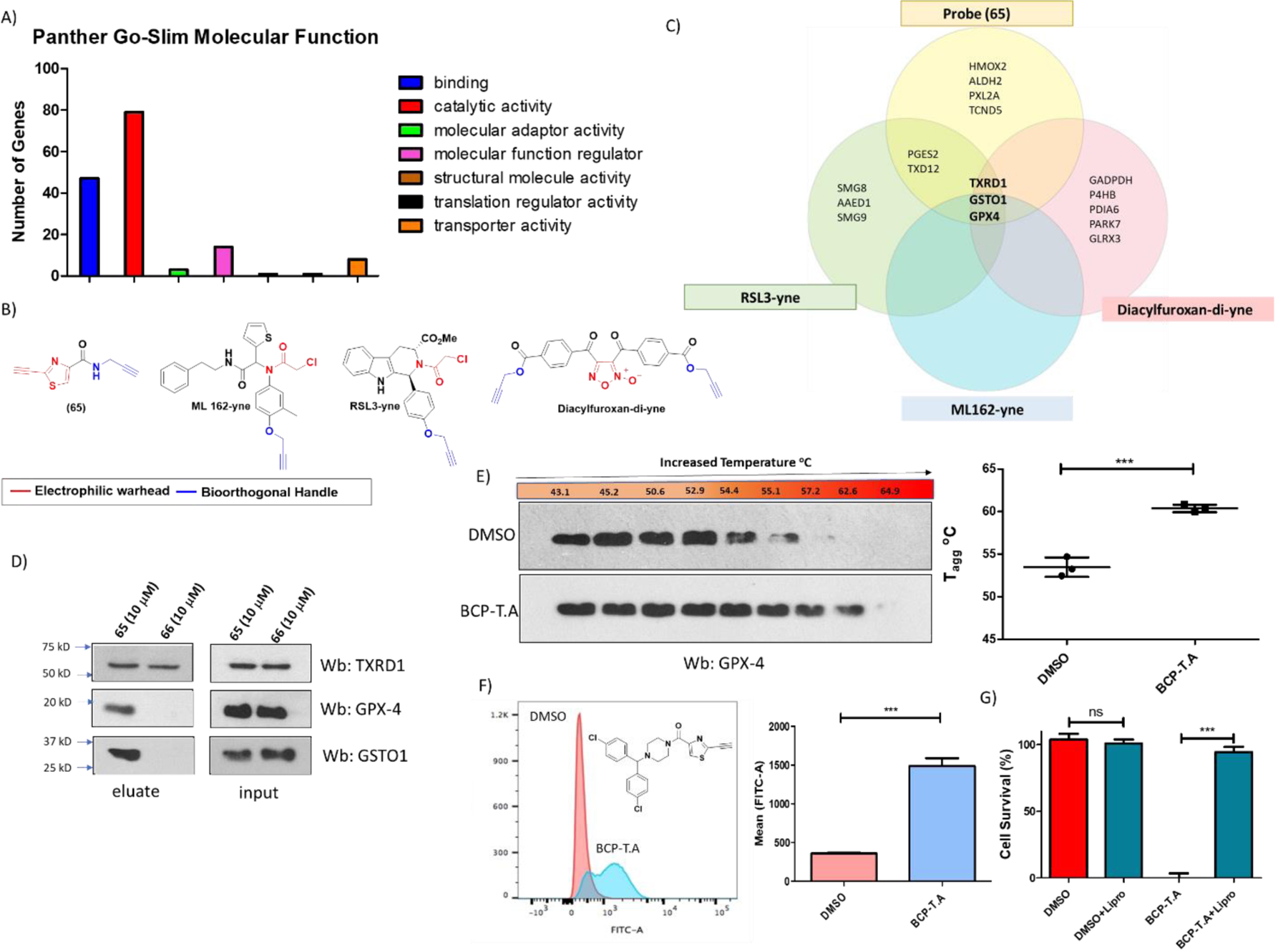
Analysis and evaluation of identified proteins from pulldown experiments. (A) Molecular function analysis utilizing PANTHER (Protein Analysis Through Evolutionary Relationships) software51. Most of the enriched proteins have enzymatic functions, which was expected as these proteins were obtained through ABPP chemoproteomic analysis. (B) Chemical structures of ABPP probes for ML-162, RSL-3 and diacylfuroxans for which proteomic data has been previously reported17–20 and confirmed either by mass spectroscopy or western blot analysis. The probe (65) consists of the electrophilic warhead (red) and a terminal alkyne as a bioorthogonal handle (blue)46, following a similar approach as with ML-162, RSL-3 and diacylfuroxans. (C) Venn diagram for some of the proteins enriched by probes in figure 12B shows that these molecules have in common three targets, GPX4, GTSO1 and TXRD1. (D) Evaluation of pulldown experiments by western blot analysis. Probe (65) (10 μM), consistent with proteomics data, selectively enriched GPX4 and GSTO1. Western blot analysis shows that even inactive probe (66) has the ability to interact with TXRD1, which was only marginally enriched in the proteomic data of (65). “Long” exposures (> 20 min) for the enriched GPX4 and GTSO1 proteins can be found on figure S6. (E) CETSA for compound BCP-T.A. Analog BCP-T.A (0.5 μM) has the ability to stabilize GPX4 protein. As a result, the aggregation temperature (Tagg) for GPX4 increases by ∼7 °C when compared to DMSO treatment. (data are mean ± SD. (n = 3)). Statistical analysis t-test, *P < 0.05, **P < 0.01, ***P < 0.001). (F) At the same concentration BCP-T.A increases lipid peroxides (0.5 μM, 3 h treatment), (G) Liproxstatin-1 (0.25 μM) rescues cells from cytotoxicity induced by BCP-T.A (0.5 μM), indicating selective ferroptosis induction and lack of toxicity originating from off-target effects.
Not all electrophilic warheads are equal
In order to demonstrate that our novel heterocyclic warheads are capable of generating highly cytotoxic molecules, we synthesized a series of analogs based on the pharmacophore of 1-(bis(4-chlorophenyl)methyl)piperazine (BCP) of ML210, by altering the electrophilic warhead (scheme 5). In addition to the masked nitrile oxide ML210 (commercially available), C.A and P.A warheads were incorporated into BCP generating compounds BCP-C.A and BCP-P.A (same molecules have been previously reported19). Following the same approach, our novel electrophilic warhead thiazole alkyne was incorporated to form BCP-T.A. We investigated the selective ferroptosis induction ability of the BCP -series by calculating their IC50 values in the presence and absence of Liproxtastin-1 on the NCI-H522 cell line (table 4 and figure S11). ML210 showed a mean IC50 value of 44 nM, which in the presence of Liproxstatin-1 (0.25 μM) was about 800-fold higher. Consistent with previous reports, C.A and P.A warheads proved to have poor potency and selectivity. To our amazement, BCP-T.A which incorporated the novel thiazole alkyne electrophile demonstrated an astonishing mean IC50 value of 17 nM, which was increased approximately 500-fold by Liproxstatin-1 (0.25 μM). The same cytotoxicity profile was demonstrated on multiple cancer cell lines (table 4 and figure S11). The enhancement of cytotoxicity by the BCP pharmacophore intrigued us to further investigate how this pharmacophore synergizes with our fragments and lowers their IC50 from low μM to low nM. Our NMR kinetic studies suggested that the presence of a base is crucial for thiol addition. Thus, we hypothesized that the basic nitrogen of the piperazine may be crucial for cytotoxicity enhancement. Indeed, incorporation of morpholine or piperidine pharmacophores increased the IC50 back to low μM (scheme 5B and Scheme S10). In addition, replacement of the BCP moiety with the less basic phenyl-piperazines (scheme 5C), resulted in analogs with attenuated IC50 values (2–3 fold), but still in the low nM range. Introduction of aliphatic substitution on the piperazine ring such as in the case of the ethyl-group, resulted in the unstable analog (75) which showed no sign of cytotoxicity (IC50 > 1 μM). These data suggest a potential role for the presence of aromatic groups on generating low nM agents, possibly by participating in secondary interactions with target proteins and/or by stabilizing the corresponding analogs. Previous SAR studies in our laboratory, as well as comparison of the activity of amide series analogs 8-10 and 16 suggest that extended conjugation of the thiazole alkyne moiety results in analogs with enhanced cytotoxicity. Accordingly, combination of the piperazine pharmacophore BCP with π-extended thiazole alkyne fragment results in a highly potent analog (78) with single digit IC50 value (6 nM) (scheme 5D and Scheme S10). This observation is crucial for targeted covalent inhibition, because in some cases, thiol addition can be affected by the presence of a base in proximity to the site of action, since cysteines surrounded by basic residues have lower pKa values than the ones surrounded by acidic residues38. Although, this can result in introduction of some degree of selectivity, it limits the cysteines that can be targeted. Our preliminary observations suggest the possibility of incorporating the “right” base into a pharmacophore as a means of enhancing thiol addition and selectivity. Similar observations have been previously suggested for acrylamide warheads bearing a dimethylamino group at the β position58,59. In the case of the GPX4, the catalytic cycle of selenocysteine oxidation in glutathione peroxidase catalysis was recently studied using a quantum chemistry approach60,61. According to these studies selenol deprotonation occurs via long-range proton shuttling to the imino-nitrogen of Trp136 (figure 1C). Thus, the mechanism of GPX4 catalysis generates selenolate anion which is capable of covalent attachment, suggesting some degree of selectivity for covalent inhibition. These are preliminary observations that need further investigation and confirmation. As shown in table 4, electrophilic warheads that have the ability to induce ferroptosis can generate highly cytotoxic molecules with low nM IC50 values. However, one drawback of ferroptosis inducers that is typically overlooked is their poor selectivity for cancer cells over normal cells, especially mesenchymal normal cells like mouse embryonic fibroblasts (MEFS) and human lung fibroblasts (WI-38) (table 4 and figure S10). This may be one of the reasons for the lack of any ferroptosis inducers in clinical use, leading to a need for novel approaches such as hybrid molecules and combinatorial treatments to exploit their remarkable anticancer effects.
Scheme 5.
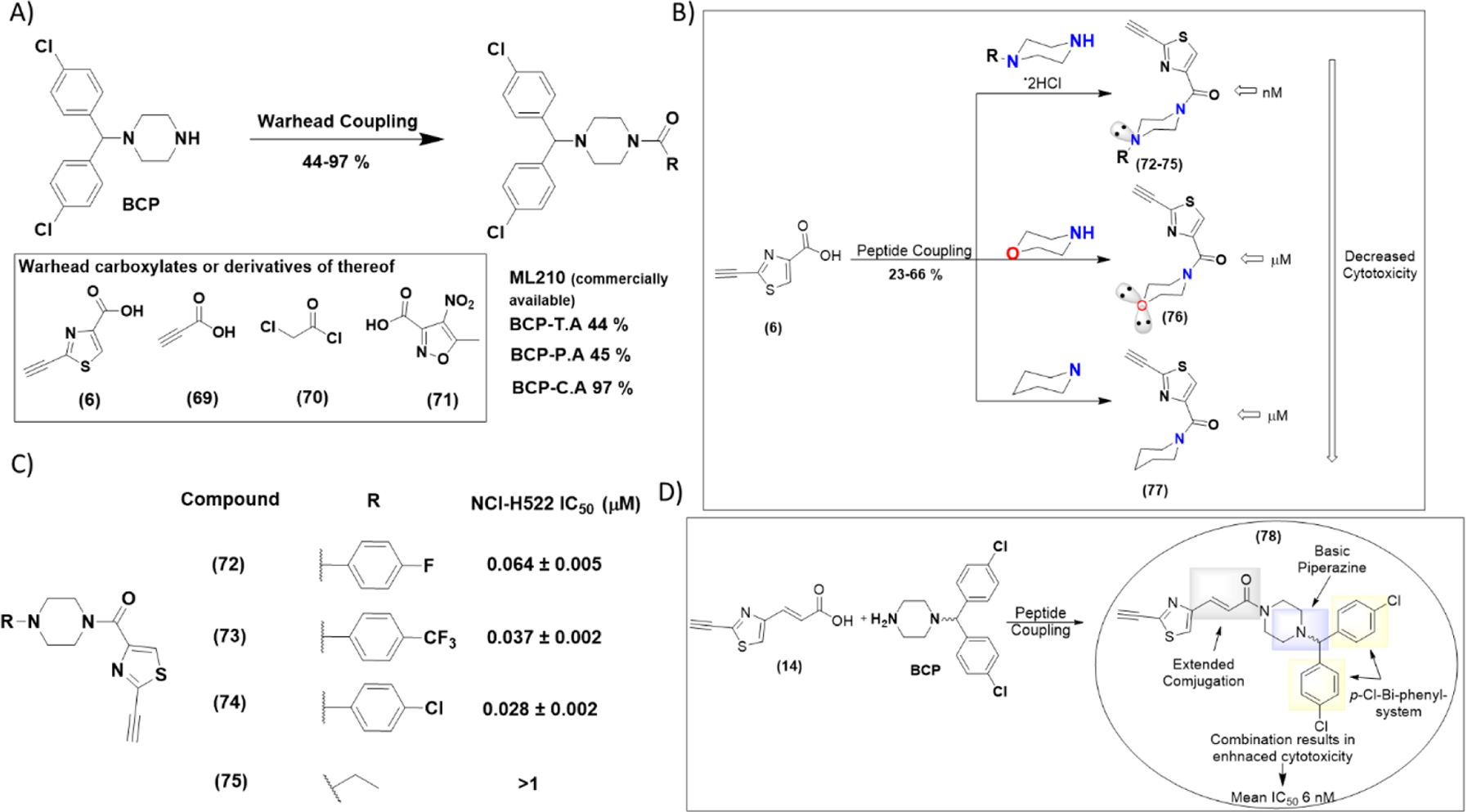
Thiazole alkyne fragments can generate highly cytotoxic ferroptosis inducers with low nM IC50 values when incorporated onto appropriate pharmacophores. (A) Synthesis of analogs with different electrophilic warheads incorporated into 1-(bis(4-chlorophenyl)methyl)piperazine (BCP) pharmacophore. Conditions: a = (6) or (69), EDC-HCl, Et3N, DCM, r.t, 12 h, 44 % for BCP-T.A, 45 % for BCP.P.A. b= (70), Et3N, DCM, r.t, 12 h, 97 % for BCP-C.A. c=analog ML210 with fragment (71) was purchased. (B) Investigation of the other cyclohexyl-based pharmacophores suggests that BCP pharmacophore is crucial for enhanced cytotoxicity. Replacement with morpholine or piperidine moieties results in analogs with attenuated cytotoxicity. (C) Replacement of the BCP moiety with the less basic phenyl-piperazines resulted in analogs (72–75) with attenuated IC50 values (2–3 fold), but still in the low nM range. (D). Combination of extended conjugation with a piperazine pharmacophore enhanced cytotoxicity even more. More information on figures 5B and 5C can be found in scheme S10.
Table 4.
IC50 ± SD (n = 3) values (μM) for ML210, BCP-T.A, BCP-C.A and BCP-P.A on NCI-H522 (in the presence or absence of Liproxstatin-1 (0.25 μM)), HT-1080, MDA-MB-468, MDA-MB-231, HeLa, HCT-116, U2OS, WI-38 and MEFS cell lines Data are mean ± SD (n=3).
| Cell Line╲Compound | ML-210 | BCP-T.A | BCP-C.A | BCP-P.A |
|---|---|---|---|---|
|
|
|
|
|
| NCI-H522 | 0.044 ± 0.005 | 0.017 ± 0.002 | 0.752 ± 0.083 | 1.129 ± 0.180 |
| NCI-H522 + Lirpoxstatin-1 | 34.515 ± 6.182 | 7.894 ± 0.811 | 3.098 ± 0.243 | 2.879 ± 0.251 |
| Fold Change* | 784.431 | 464.353 | 4.120 | 2.550 |
| HT-1080 | 0.022 ± 0.003 | 0.019 ± 0.003 | 0.302 ± 0.017 | 0.376 ± 0.008 |
| MDA-MB-468 | 0.066 ± 0.006 | 0.084 ± 0.005 | 1.746 ± 0.153 | 0.065 ± 0.076 |
| MDA-MB-231 | 0.020 ± 0.004 | 0.021 ± 0.003 | 0.494 ± 0.054 | 0.038 ± 0.004 |
| HeLa | 0.253 ± 0.041 | 0.242 ± 0.019 | 2.111 ± 0.110 | 0.303 ± 0.020 |
| HCT-116 | > 1.000 | > 1.000 | 1.641 ± 0.528 | 3.801 ± 0.447 |
| U2OS | 0.822 ± 0.249 | 0.367 ± 0.055 | 1.708 ± 0.278 | 0.638 ± 0.016 |
| WI-38 | 0.060 ± 0.004 | 0.022 ± 0.004 | 1.742 ± 0.222 | 0.500 ± 0.036 |
| MEFS | 0.016 ± 0.003 | 0.010 ± 0.001 | 0.352 ± 0.032 | 0.082 ± 0.008 |
Fold change is the ratio of IC50 (NCI-H522 + Lirpoxstatin-1)/ IC50 (NCI-H522).
CONCLUSIONS
In this work we report the rational design of heterocyclic electrophiles which can act as warheads capable of inducing ferroptosis. We evaluated the cytotoxicity of our electrophiles in two cell lines, the ferroptosis-sensitive NCI-H522 cell line and the ferroptosis “resistant” HCT-116 cell line to delineate ferroptosis induction from other cell death mechanisms. We further confirmed the ability of our molecules to induce ferroptosis by rescue experiments utilizing the selective ferroptosis inhibitor Liproxstatin-1 and by measuring the levels of lipid peroxides. Biological evaluation demonstrated that thiazoles with an alkyne group at the 2-position are the most potent and selective ferroptosis inducers. One characteristic that makes our warheads unique when compared to existing alternatives is the ability to tune their reactivity in multiple ways. NMR kinetic studies showed that our heterocycles have the ability to undergo thiol addition at different rates, depending on steric and electronic factors as well as other environmental factors such as pH. DFT calculations confirmed the ability to tune reactivity by changing the nature of the electrophile, the nature of the heterocycle and by ring fusion. Interestingly, there is direct correlation of the rate of thiol addition with bioactivity, the fastest reacting electrophiles being typically the most cytotoxic as well. In order to have a detailed understanding of the proteins targeted by our electrophiles, we designed appropriate ABPP probes for chemoproteomic analysis. Initially, we confirmed by in-gel fluorescent analysis that our heterocyclic fragments have almost identical interacting proteins with the parent CETZOLE molecules. In addition, we showed that fluorescent labeling of target proteins is covalent in nature and intersects with other covalent ferroptosis inducers such RSL3, ML162. Detailed fluorescence analysis enabled us to perform pulldown experiments. Enriched samples were analyzed by LC-MS/MS leading to the identification of more than 2000 proteins, of which only 227 were enriched by our ferroptosis inducing probe. GPX4 emerges as the most prominent target due to its high correlation with ferroptosis induction. Considering data from previous proteomic studies with similar warheads, GSTO1 can be considered another important target, although its relation to ferroptosis remains unclear. Proteomic data were further validated by western blot analysis of pulldown samples. Due to the importance of targeting GPX4, we additionally evaluated interaction of our fragments with GPX4 by CETSA. Incorporation of heterocyclic fragments into ferroptosis-related pharmacophores such as the one from ML210 demonstrated the ability of our fragments to generate highly potent molecules. Analog BCP-T.A, demonstrated low nM IC50 on a series of ferroptosis sensitive cell lines, while on the NCI-H522 cell line, co-treatment with Liproxtatin-1 increased the IC50 value by approximately 500-fold, indicating high selectivity. In conclusion, the discovery of CETZOLE molecules as ferroptotic agents culminated in the identification of tunable heterocyclic electrophiles that induce ferroptosis. Their reactivity can be tuned in a myriad of ways, with only the basic ones being studied in this work. Our goal was to demonstrate the ability of our fragments to undergo thiol addition and induce ferroptosis in a selective manner. Our previous work uncovered reduced activity of system Xc- in CETZOLE-treated cells; however direct binding was not determined. Our current work illustrates the importance of a chemoproteomics approach to identify direct targets. In fact, the current work indicates that GPX4 is a direct CETZOLE target consistent with ferroptosis induction. Effects on system Xc- function may be indirect; however direct binding to this transporter cannot be ruled out. Ferroptosis is a recently discovered cell death mechanism, of which the vast majority of interacting pathways remains unknown. As to how the inactivation of system Xc− and covalent inhibition of proteins such as GPX4 and GSTO1 intersect is unclear at this stage and will be the subject of our future studies. However, the tunable electrophilic fragments reported in this work induce ferroptosis in a selective manner and, when incorporated into appropriate pharmacophores, can generate highly cytotoxic molecules. They expand the available toolbox of ferroptosis-inducing electrophiles that can find applications in TCI, synthesis of hybrid molecules and design of chemical biology probes.
Experimental
Chemistry
Material and Methods.
All chemicals and solvents were purchased from commercial sources and used without further purification, unless stated otherwise. Anhydrous tetrahydrofuran was freshly distilled from sodium and benzophenone before use. 1H and 13C NMR spectra were recorded on Brucker Avance 600MHz, INOVA 600 MHz and Varian VXRS 400 MHz NMR spectrometers in deuterated solvents using residual undeuterated solvents as internal standard. High-resolution mass spectra (HRMS) were recorded on a Waters Synapt high definition mass spectrometer (HDMS) equipped with nano-ESI source. Melting points were determined on a Fisher-Johns melting point apparatus. Purification of crude products was performed by either flash chromatography on silica gel (40–63 µ) from Sorbent Technologies or on a Teledyne ISCO CombiFlash Companion chromatography system on RediSep prepacked silica cartridges. Thin layer chromatography (TLC) plates (20 cm x 20 cm) were purchased from Sorbent Technologies (catalog #4115126) and were viewed under Model UVG-54 mineral light lamp UV-254 nm. A Shimadzu Prominence HPLC with an LCT20AT solvent delivery system coupled to a Shimadzu Prominence SPD 20AV Dual wavelength UV/Vis absorbance Detector, a Shimadzu C18 column (1.9 m, 2.1 mm x 50 m) and HPLC grade solvents (MeOH, H2O with 0.1% formic acid) were used to determine the purity of compounds by HPLC.
General procedure 1: Hydrolysis of esters
To a stirred solution of the carboxylate (1 equiv.) in THF: H2O (3:1,3 mL/mmol) was added 2 N NaOH. The resulting mixture was stirred for 3 h at room temperature, after which it was extracted with DCM. The aqueous layer was acidified to pH = 4 and extracted with EtOAc. The organic extract was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to obtain pure product.
General Procedure 2: amide coupling
To a stirred solution of the carboxylic acid (1 equiv.) in anhydrous DCM (5 mL), were added N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (1.5 equiv.), triethylamine (2 equiv.), and the amine (1.2 equiv.). The mixture was stirred overnight at room temperature, after which it was diluted with brine and extracted with DCM. The organic extract was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica in EtOAc/hexanes to yield the pure product.
General procedure 3: Iodination at 2-position of heteroarenes
To a stirred solution of the heteroarene (1 equiv.) in anhydrous DMF (1 mL) was added perfluorobutyl iodide (1.1 equiv.), followed by sodium t-butoxide (0.5 equiv.). The resulting mixture was stirred for 30 min at room temperature, after which it was diluted with brine and extracted with Et2O. The organic extract was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica in EtOAc/hexanes to yield the pure product.
General procedure 4: bromination at 2-position heteroarenes
To a stirred solution of a heteroarene (1 equiv.) in anhydrous DMF (1 mL) was added carbon tetrabromide (1.1 equiv.), followed by sodium t-butoxide (3.6 equiv.). The resulting mixture was stirred for 30 min at room temperature, after which it was diluted with brine and extracted with Et2O. The organic extract was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica in EtOAc/hexanes to yield the pure product.
General procedure 5: Sonogashira coupling
To a stirred solution of the bromo/iodo-heteroarene (1 equiv.) in anhydrous DCE (2 mL/mmol) were added bis(triphenylphosphine)palladium (II) dichloride (5 mol%) copper(I) iodide (6 mol%), alkyne (1.2 equiv.), and triethylamine (2 equiv.). The resulting mixture was stirred for 3 h at 83 °C, after which it was allowed to cool to room temperature, and concentrated under reduce pressure in the presence of silica gel. The product loaded on silica gel was purified by flash chromatography on silica in EtOAc/hexanes to yield the pure product.
General procedure 6: Suzuki cross-coupling
To a stirred solution of the bromo/iodo-heteroarene (1 equiv.) in anhydrous DME/H2O (4:1, 1 mL/mmol) were added tetrakis(triphenylphosphine)palladium (5 mol%), boronic acid (2 equiv.), and Na2CO3 (3 equiv.), respectively. The resulting mixture was stirred overnight at 83 °C, after which it was concentrated under reduced pressure to remove DME and extracted with DCM. The organic extract was concentrated under reduced pressure and the crude product was purified by flash chromatography on silica in EtOAc/hexanes to yield the pure product.
General procedure 7: Decarboxylative Cross-Coupling
To a stirred solution of the bromo/iodo-heteroarene (1 equiv.) in anhydrous DME/H2O (4:1, 1 mL/mmol) were added tetrakis(triphenylphosphine)palladium (5 mol%), 2-butynoic acid (1.1 equiv.) and Na2CO3 (3 equiv.). The resulting mixture was stirred for 4 h at 83 °C, after which it was concentrated under reduced pressure to remove DME, the reaction mixture extracted with DCM. The organic extract was concentrated under reduced pressure and the crude product was purified by flash chromatography on silica in EtOAc/hexanes to yield the pure product.
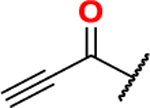
ethyl 2-ethynylthiazole-4-carboxylate (22):
The titled compound was synthesized according to the general procedure 5 to obtain (22) as a brown solid (160 mg, 86%). 1H NMR (600 MHz, CDCl3) δ 8.21 (s, 1H), 4.48 – 4.43 (m, 2H), 3.53 (s, 1H), 1.45 – 1.41 (m, 3H). 13C NMR (151 MHz, CDCl3) δ 160.73, 148.13, 147.65, 128.76, 83.38, 75.65, 61.81, 14.31. HRMS (ESI+) calcd for C8H8NO2S [M+H]+ 182.0275, found 182.0276.
2-ethynylthiazole-4-carboxylic acid (6):
The titled compound was synthesized according to the general procedure 1 to obtain (6) as a brown solid (1.5 g, 98%). Spectroscopic data was consistent with those reported previously.62
2-ethynyl-N-(4-methoxyphenyl)thiazole-5-carboxamide (8):
The titled compound was synthesized according to the general procedure 2 to obtain (8) as a yellow solid (78 mg, 52%). 1H NMR (600 MHz, CDCl3) δ 9.05 (s, 1H), 8.24 (s, 1H), 7.69 – 7.57 (m, 2H), 6.99 – 6.88 (m, 2H), 3.84 (s, 3H), 3.58 (s, 1H). 13C NMR (151 MHz, CDCl3) δ 157.8, 156.7, 150.7, 147.4, 130.6, 125.6, 121.5, 114.3, 83.4, 75.6, 55.5. HRMS (ESI+) calcd for C13H11N2O2S [M+H]+ 259.0541, found 259.0541. 93% HPLC purity.
2-ethynyl-N-phenylthiazole-5-carboxamide (9):
The titled compound was synthesized according to the general procedure 2 to obtain (9) as a yellow solid (70.3 mg, 46%). 1H NMR (600 MHz, CDCl3) δ 9.15 (s, 1H), 8.26 (s, 1H), 7.73 (dd, J = 8.5, 0.9 Hz, 2H), 7.42 – 7.38 (m, 2H), 7.20 – 7.17 (m, 1H), 3.61 (s, 1H). 13C NMR (151 MHz, CDCl3) δ 158.0, 150.7, 147.4, 137.5, 129.1, 125.8, 124.7, 119.9, 83.5, 75.6. HRMS (ESI+) calcd for C12H9N2OS [M+H]+ 229.0435, found 229.0430. 96% HPLC purity.
N-(4-chlorophenyl)-2-ethynylthiazole-5-carboxamide (10):
The titled compound was synthesized according to the general procedure 2 to obtain (10) as a yellow solid (88.5 mg, 59%). 1H NMR (600 MHz, CDCl3) δ 9.15 (s, 1H), 8.26 (s, 1H), 7.71 – 7.67 (m, 2H), 7.37 – 7.33 (m, 2H), 3.62 (s, 1H). 13C NMR (151 MHz, CDCl3) δ 157.9, 150.2, 147.4, 136.0, 129.6, 129.2, 126.0, 121.1, 83.6, 75.5. HRMS (ESI+) calcd for C12H8ClN2OS [M+H]+ 263.0045, found 262.9987. >99% HPLC purity.
2-bromothiazole-4-carbaldehyde (11):
To a stirred solution of oxalyl chloride (3.4 mL, 39 mmol, 1.1 equiv.) in anhydrous DCM (72 mL) under nitrogen atmosphere at −75 °C was added a solution of dimethyl sulfoxide (5.6 ml, 79.4 mmol, 2.2 equiv.) in anhydrous DCM (18 mL). The resulting mixture was stirred at −75 °C for 30 min, after which a solution of (2-bromothiazol-4-yl)methanol (S10) (7 g, 36 mmol, 1 equiv.) in anhydrous DCM (36 mL) was added, stirred at −75 °C for additional 2 h. Then triethylamine (25 mL, 180 mmol, 5 equiv.) was added to the reaction mixture. The resulting solution was stirred at room temperature for 1 h, after which it was diluted with sat. solution of NaHCO3. It was extracted with EtOAc and the organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica in EtOAc/hexanes to yield (11) as white solid (6.7 g, 97%). Spectroscopic data was consistent with those reported previously.63
methyl (E)-3-(2-bromothiazol-4-yl)acrylate (13):
To a stirred solution of triphenyl phosphine (2.3 g, 9 mmol, 1.05 equiv.) in EtOAc (14 mL) was slowly added a solution of bromomethyl acetate (1.3 g, 8.5 mmol, 1 equiv.) in EtOAc (2.5 mL). The resulting mixture was stirred at room temperature for 12 h. The precipitated white solid was collected by filtration, washed with Et2O and air-dried overnight. It was resuspended in 1 N NaOH (16 mL) and stirred for 20 min, at which point DCM was added. The organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to provide the corresponding ylide, which was used in the next step without further purification. The synthesized ylide (12) (9 mmol) was added to a stirring solution of 2-bromothiazole-4-carbaldehyde (11) (1.7 g, 9 mmol, 1 equiv.) in THF (52 mL) at room temperature and the mixture was stirred at this temperature for 12 h. It was treated with brine and extracted with DCM. The organic extract was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by silica gel chromatography in EtOAc/hexanes to yield (13) as a white solid (1.5 g, 90%). 1H NMR (600 MHz, CDCl3) δ 7.53 (d, J = 15.5 Hz, 1H), 7.37 (s, 1H), 6.77 (d, J = 15.5 Hz, 1H), 3.82 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 167.3, 152.0, 137.3, 135.0, 124.6, 121.4, 51.9.
Methyl (E)-3-(2-ethynylthiazol-5-yl)acrylate (S1):
The titled compound was synthesized according to the general procedure 5 to obtain (S1) as a brown solid (412 mg, 83%). 1H NMR (600 MHz, CDCl3) δ 7.60 (d, J = 15.6 Hz, 1H), 7.46 (s, 1H), 6.84 (d, J = 15.6 Hz, 1H), 3.82 (s, 3H), 3.54 (s, 1H). 13C NMR (151 MHz, CDCl3) δ 167.4, 152.1, 148.2, 135.4, 122.8, 121.6, 83.0, 76.0, 51.9.
(E)-3-(2-ethynylthiazol-5-yl)acrylic acid (14):
The titled compound was synthesized according to the general procedure 1 to obtain (14) as a brown solid (395.5 mg, 96%). 1H NMR (600 MHz, DMSO-d6) δ 12.56 (s, 1H), 8.21 (s, 1H), 7.57 (t, J = 14.8 Hz, 1H), 6.56 (d, J = 15.6 Hz, 1H), 5.05 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ 167.8, 151.7, 148.1, 136.0, 125.9, 122.1, 87.3, 76.7.
(E)-N-(4-chlorophenyl)-3-(2-ethynylthiazol-5-yl)acrylamide (16):
The titled compound was synthesized according to the general procedure 2 to obtain (16) as a yellow solid (71 mg, 48%). 1H NMR (600 MHz, Acetone-d6) δ 9.60 (s, 1H), 7.97 (s, 1H), 7.84 (dd, J = 9.3, 2.4 Hz, 2H), 7.65 (d, J = 15.1 Hz, 1H), 7.39 – 7.35 (m, 2H), 7.11 (d, J = 15.1 Hz, 1H), 4.41 (s, 1H). 13C NMR (151 MHz, Acetone-d6) δ 163.50, 152.9, 147.7, 138.2, 132.4, 128.5, 127.8, 125.0, 123.4, 120.6, 83.6, 75.9. HRMS (ESI+) calcd for C14H10ClN2OS [M+H]+ 289.0202, found 289.0222. 95% HPLC purity.
ethyl 2-vinylthiazole-4-carboxylate (18):
The titled compound was synthesized according to the general procedure 6 to obtain (18) as a brown oil (90 mg, 90%). The spectroscopic data was consistent with those reported previously.64
ethyl 2-(prop-1-yn-1-yl)thiazole-4-carboxylate (20):
The titled compound was synthesized according to the general procedure 7 to obtain (20) as a brown solid (79.2 mg, 79%). 1H NMR (600 MHz, CDCl3) δ 8.11 (s, 1H), 4.44 (q, J = 7.1 Hz, 2H), 2.13 (s, 3H), 1.42 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 161.0, 149.6, 147.1, 127.6, 93.3, 72.9, 61.5, 14.3, 4.6. HRMS (ESI+) calcd for C9H10NO2S [M+H]+ 196.0432, found 196.0449.
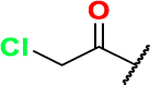
ethyl 2-(phenylethynyl)thiazole-4-carboxylate (24):
The titled compound was synthesized according to the general procedure 5 to obtain (24) as a yellow solid (630 mg, 63%). 1H NMR (600 MHz, CDCl3) δ 8.22 (s, 1H), 7.61 (dt, J = 8.4, 1.8 Hz, 2H), 7.46 – 7.39 (m, 3H), 4.48 (q, J = 7.1 Hz, 2H), 1.45 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 161.0, 149.4, 147.8, 132.1, 129.9, 128.6, 128.5, 121.0, 95.0, 81.7, 61.7, 14.3. HRMS (ESI+) calcd for C14H12NO2S [M+H]+ 258.0588, found 258.0576.
2,2’-diselanediyldianiline (26):
To a stirred solution of 2-iodoaniline (25) (4.4 g, 20 mmol, 1 equiv.) in anhydrous THF (80 mL) under nitrogen atmosphere at −75 °C was added n-butyllithium (24 mL of a 2.5 M solution in hexanes, 60 mmol, 3 equiv.). The resulting mixture was stirred at −75 °C for 1 h. The cooling bath was removed and selenium powder (1.58 g, 20 mmol, 1 equiv.) was added to the reaction mixture while a stream of nitrogen gas was passed through it. The reaction mixture was stirred at room temperature for 1 h, after which it was diluted with sat. solution of NH4Cl and stirred for 2 h. It was extracted with EtOAc and the organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica in EtOAc/hexanes to yield (26) as an orange oil (5.1, 74%). Spectroscopic data was consistent with those reported previously.32
benzo[d][1,3]selenazole (27):
To sealed microwave vial containing a stirred solution of 2,2’-diselanediyldianiline (26) (135 mg, 0.4 mmol, 1 equiv.) in anhydrous toluene (3 mL) under nitrogen atmosphere, at room temperature was added n-tributylphosphine (0.23 mL, 0.9 mmol, 3 equiv.) and stirred for 5 min. Formic acid (0.03, ml 0.6 mmol, 1.5 equiv.) was added and the resulting mixture was heated in the microwave synthesizer for 2 h at 100 °C. It was diluted with sat. NaHCO3 solution and extracted with DCM. The organic extract was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica in EtOAc/hexanes to yield (27) as a transparent oil (41.2 mg, 54%). Spectroscopic data was consistent with those reported previously.32
benzo[d]thiazole (28):
The titled compound was purchased from Ambeed (CAS number: 95–16-9) and used without further purification.
benzo[d]oxazole (29):
The titled compound was purchased from Ambeed (CAS number 273–53-0) and used without further purification.
1-methyl-1H-benzo[d]imidazole (30):
The titled compound was purchased from Ambeed (CAS number 1632–83-3) and used without further purification.
2-iodo-1-methyl-1H-benzo[d]imidazole (31):
The titled compound was synthesized according to the general procedure 3 to obtain (31) as a yellow solid (487.2 mg, 86%). Spectroscopic data was consistent with those reported previously.33
2-bromo-1-methyl-1H-benzo[d]imidazole (32):
The titled compound was synthesized according to the general procedure 4 to obtain (32) as a brown solid (87.6mg, 73%). Spectroscopic data was consistent with those reported previously.33
2-iodobenzo[d]oxazole (33):
The titled compound was synthesized according to the general procedure 3 to obtain (33) as a yellow solid (608 mg, 76%). Spectroscopic data was consistent with those reported previously.33
2-bromobenzo[d]oxazole (34):
The titled compound was synthesized according to the general procedure 4 to obtain (34) as a brown solid (300 mg, 46%) Spectroscopic data was consistent with those reported previously.33
2-iodobenzo[d]thiazole (35):
The titled compound was synthesized according to the general procedure 3 to obtain (35) as a yellow solid (189 mg, 94%). Spectroscopic data was consistent with those reported previously.33 98% HPLC purity.
2-bromobenzo[d]thiazole (36):
The titled compound was synthesized according to the general procedure 4 to obtain (36) as a yellow solid (145 mg, 96%) Spectroscopic data was consistent with those reported previously.33
2-iodobenzo[d][1,3]selenazole (37):
The titled compound was synthesized according to the general procedure 3 to obtain (37) as a yellow oil (17.4 mg, 87%). 1H NMR (600 MHz, CDCl3) δ 8.11 (dd, J = 8.1, 0.8 Hz, 1H), 7.92 (dd, J = 8.0, 0.8 Hz, 1H), 7.46 – 7.42 (m, 1H), 7.39 – 7.35 (m, 1H). 13C NMR (600 MHz, CDCl3) δ 155.6, 142.8, 126.5, 125.9, 124.2, 124.0, 103.2. HRMS (ESI+) calcd for C7H5INSe [M+H]+ 309.8631, found 309.8669.
2-bromobenzo[d][1,3]selenazole (38):
The titled compound has been synthesized according to the general procedure 4 which yields (38) as a transparent oil (15 mg, 71%). 1H NMR (600 MHz, CDCl3) δ 8.07 – 8.04 (m, 1H), 7.87 – 7.85 (m, 1H), 7.50 – 7.45 (m, 1H), 7.40 – 7.36 (m, 1H). 13C NMR (151 MHz, CDCl3) δ 153.5, 141.0, 137.9, 126.7, 126.0, 124.5, 124.3. HRMS (ESI+) calcd for C7H5BrNSe [M+H]+ 261.8770, found 261.8773.
1-methyl-2-vinyl-1H-benzo[d]imidazole (39):
The titled compound was synthesized according to the general procedure 6 to obtain (39) as a yellow oil (351 mg, 72%). Spectroscopic data was consistent with those reported previously.65 95% HPLC purity.
2-vinylbenzo[d]oxazole (40):
The titled compound was synthesized according to the general procedure 6 to obtain (40) as a brown oil (231 mg, 66%). Spectroscopic data was consistent with those reported previously.66
2-vinylbenzo[d]thiazole (41):
The titled compound was synthesized according to the general procedure 6 to obtain (41) as a brown oil (87 mg, 87%). Spectroscopic data was consistent with those reported previously.67
2-(methylselanyl)aniline (S2):
To a degassed stirred solution of 2,2’-diselanediyldianiline (26) (2 g, 6 mmol, 1 equiv.) in anhydrous THF (140 mL) under nitrogen atmosphere were added NaBH4 (0.68 g, 18 mmol, 3 equiv.) and MeOH (1.2 mL, 30 mmol, 5 equiv.). The resulting mixture was stirred at room temperature for 1 h. Iodomethane (0.8 mL, 12 mmol, 2 equiv.) was then added. The reaction mixture was stirred for an additional 1.5 h, after which it was diluted with brine and extracted with EtOAc. The organic extract was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica in DCM/hexanes to yield (S2) as a brown oil (1.8 g, 88%). The spectroscopic data was consistent with those reported previously.34
2-vinylbenzo[d][1,3]selenazole (42):
To a stirred solution of 2-(methylselanyl) aniline (S2) (500 mg, 2.7 mmol, 1 equiv.) in anhydrous DCM (7 mL) were added acryloyl chloride (0.23 mL, 3.24 mmol, 1.2 equiv.) and triethylamine (0.75 mL, 5.4 mmol, 2 equiv.). The resulting mixture was stirred at room temperature for 30 min, after which it was diluted with brine and extracted with DCM. The organic extract was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting product N-(2-(methylselanyl)phenyl)acrylamide was used in the next step without further purification. To a stirred solution of N-(2-(methylselanyl)phenyl)acrylamide in anhydrous 1,4-dioxane (40 mL) under nitrogen atmosphere were added a solution of POCl3 (1.1 mL, 10.8 mmol, 4 equiv.) and triethylamine (4.5 mL, 32.4 mmol, 12 equiv.) in 1,4-dioxane (40 mL). The resulting mixture was stirred at 105 °C for 1 h, after which it was diluted with aqueous sat. NaHCO3 and EtOAc. The two layers were separated, and the organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica in EtOAc/hexanes to yield (42) as a yellow oil (83 mg, 52% over two steps).34 1H NMR (600 MHz, CDCl3) δ 8.04 (dd, J = 8.2, 0.4 Hz, 1H), 7.92 – 7.90 (m, 1H), 7.50 – 7.46 (m, 1H), 7.36 – 7.30 (m, 1H), 7.02 (dd, J = 17.3, 10.6 Hz, 1H), 6.10 (d, J = 17.3 Hz, 1H), 5.84 (d, J = 10.6 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 171.9, 155.2, 137.1, 134.3, 126.4, 125.8, 124.8, 124.5. HRMS (ESI+) calcd for C9H8NSe [M+H]+ 209.9821, found 209.9845.
2-ethynyl-1-methyl-1H-benzo[d]imidazole (43):
The titled compound was synthesized according to the general procedure 5 to obtain (43) as a brown solid (273 mg, 56%). Spectroscopic data was consistent with those reported previously.68 98% HPLC purity.
2-ethynylbenzo[d]oxazole (44):
The titled compound was synthesized according to the general procedure 5 to obtain (44) as a brown solid (40.3 mg, 35%). Spectroscopic data was consistent with those reported previously.69 >99% HPLC purity.
2-ethynylbenzo[d]thiazole (45):
The titled compound was synthesized according to the general procedure 5 to obtain (45) as a brown solid (80.4 mg, 67%). Spectroscopic data was consistent with those reported previously.69 90% HPLC purity.
2-(prop-1-yn-1-yl)benzo[d]thiazole (46):
The titled compound was synthesized according to the general procedure 7 to obtain (46) as a brown solid (58.1 mg, 83%). 1H NMR (600 MHz, CDCl3) δ 8.06 – 8.03 (m, 1H), 7.86 – 7.84 (m, 1H), 7.52 (ddd, J = 7.2, 5.3, 1.2 Hz, 1H), 7.46 – 7.43 (m, 1H), 2.20 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 152.7, 149.2, 135.0, 126.5, 126.0, 123.5, 121.3, 94.5, 73.8, 4.7. HRMS (ESI+) calcd for C10H7NS [M+H]+ 174.0377, found 174.0377.
2-ethynylbenzo[d][1,3]selenazole (47):
To a stirred solution of 2-(methylselanyl) aniline (S2) (320 mg, 1.7 mmol, 1 equiv.) in anhydrous DCM (30 mL) were added propiolic acid (0.12 mL, 1.9 mmol, 1.1 equiv.), and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (520 mg, 2.7 mmol, 1.5 equiv.), respectively. The resulting mixture was stirred at room temperature for 1.5 h, after which it was diluted with brine and extracted with DCM. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The product N-(2-(methylselanyl)phenyl)propiolamide was used in the next step without further purification. To a stirred solution of N-(2-(methylselanyl)phenyl)propiolamide in anhydrous 1,4-dioxane (100 mL), under nitrogen atmosphere a solution of POCl3 (0.67 mL, 7.2 mmol, 4 equiv.), and triethylamine (3.2 mL, 21.6 mmol, 12 equiv.) in 1,4-dioxane (100 mL) were added. The resulting mixture was stirred at 105 °C for 1 h, after which it was diluted with aqueous sat. NaHCO3 solution and extracted with EtOAc. The organic extract was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica in EtOAc/hexanes to yield (47) as a brown oil (22 mg, 7%) over two steps.34 1H NMR (600 MHz, CDCl3) δ 8.16 (d, J = 8.2 Hz, 1H), 7.93 (d, J = 8.0 Hz, 1H), 7.55 – 7.52 (m, 1H), 7.42 (ddd, J = 7.2, 2.0, 1.0 Hz, 1H), 3.81 (s, 1H). 13C NMR (151 MHz, CDCl3) δ 154.2, 150.4, 139.3, 126.9, 126.6, 125.6, 124.7, 85.6, 79.3. HRMS (ESI+) calcd for C9H6NSe [M+H]+ 207.9665, found 207.9734.
2-ethynylbenzo[d][1,3]selenazole (47-a):
The titled compound was synthesized according to the general procedure 5 to obtain (47-A) as a brown solid (75 mg, 71 %). Spectroscopic data was consistent with previous (vide supra) method (47).
2-ethynyl-4-phenylthiazole (48):
The titled compound was synthesized according to the general procedure 5 to obtain (48) as a yellow solid (152.5 mg, 65%). 1H NMR (600 MHz, CDCl3) δ 7.95 – 7.93 (m, 2H), 7.51 (s, 1H), 7.47 – 7.44 (m, 2H), 7.40 – 7.37 (m, 1H), 3.54 (s, 1H). 13C NMR (151 MHz, CDCl3) δ 156.3, 147.3, 133.6, 128.8, 128.6, 126.6, 114.8, 82.3, 76.6. HRMS (ESI+) calcd for C11H8NS [M+H]+ 186.0377, found 186.0403.
2-bromo-4-vinylthiazole (49):
To a stirred solution of 2-bromothiazole-4-carbaldehyde (11) (192.03 mg, 1 mmol, 1 equiv.) and methyltriphenylphosphonium bromide (535.5 g, 1.5 mmol, 1.5 equiv.) in DCM (5 mL) was added dropwise a solution of NaOH (2 mL of 50% solution in H2O). The resulting biphasic mixture was stirred at room temperature for 2 h and partitioned between DCM and brine. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product mixture was separated on silica in EtOAc/hexanes, to yield (49) as a yellow oil (15 mg, 11%). 1H NMR (600 MHz, CDCl3) δ 7.05 (s, 1H), 6.64 (dd, J = 17.2, 10.8 Hz, 1H), 6.11 – 6.07 (m, 1H), 5.42 (dd, J = 10.8, 1.3 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 154.6, 136.1, 128.6, 118.5, 117.8. HRMS (ESI+) calcd for C5H5BrNS [M+H]+ 189.9326, found 189.0838.
4-methylbenzenesulfonyl azide (S4):
To a stirred solution of tosyl chloride (S3) (8.0 g, 42.0 mmol, 1.0 equiv.) in acetone/ H2O (1:1, 240 mL), was added NaN3 (3 g, 46 mmol, 1.2 equiv.). The resulting mixture was stirred at 0 °C for 2 h, after which it was concentrated under reduced pressure. The reaction mixture was partitioned between Et2O and brine. The two layers were separated, and the organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to yield (S4) as a colorless oil (8 g, 97%). It was handled with special care as it is potentially explosive material. Spectroscopic data was consistent with those reported previously.35
dimethyl (1-diazo-2-oxopropyl)phosphonate (50):
To a stirred solution of dimethyl (2-oxopropyl)phosphonate (S5) (1.66 g, 10 mmol, 1 equiv.) in anhydrous MeCN (20 mL), were added tosyl azide (2.4 g, 12 mmol, 1.2 equiv.) and K2CO3 (2.1 g, 15 mmol, 1.5 equiv.). The resulting mixture was stirred overnight at room temperature, after which it was filtered on celite and washed with EtOAc. The organic layer was concentrated under reduced pressure. The crude product mixture was separated on silica in EtOAc/hexanes to yield the pure product (50) (1.6 g, 83%). Spectroscopic data was consistent with those reported previously.35
2-bromo-4-ethynylthiazole (51):
To a stirred solution of 2-bromothiazole-4-carbaldehyde (11) (8 g, 4.3 mmol, 1 equiv.) in anhydrous MeOH (20 mL) were added dimethyl (1-diazo-2-oxopropyl) phosphonate (50) (1, g, 5.2 mmol, 1.2 equiv.), and K2CO3 (1.6 g, 5.2 mmol, 1.2 equiv.). The resulting mixture was stirred at room temperature overnight after which it was concentrated under reduced pressure. The reaction mixture was then diluted with brine and extracted with DCM. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica in EtOAc/hexanes to yield (51) as brown solid (281 mg, 34%). 1H NMR (600 MHz, CDCl3) δ 7.47 (s, 1H), 3.16 (s, 1H). 13C NMR (151 MHz, CDCl3) δ 136.9, 135.7, 127.4, 78.5, 76.3. HRMS (ESI+) calcd for C5H3BrNS [M+H]+ 187.9169, found 187.9198. >99% HPLC purity.
ethyl 5-bromo-2-iodothiazole-4-carboxylate (53):
To a stirred solution of ethyl 2-amino-5-bromothiazole-4-carboxylate (52) (12 g, 48 mmol, 1 equiv.) in phosphoric acid (100 mL) and nitric acid (100 mL) was added dropwise at 0 °C, an aqueous solution of NaNO2 (80 mL of a 1.38 M solution in H2O, 58 mmol, 1.2 equiv.). The resulting mixture was stirred at 0 °C for 1 h. KI (32 g, 192 mmol, 4 equiv.) was added and the reaction mixture was allowed to warm to room temperature gradually (1–2 h), after which it was diluted with aqueous sat. Na2S2O3, aqueous sat. NaHCO3, brine and extracted with EtOAc each time respectively. The combined organic extract was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica in DCM/hexanes to yield (53) as a yellow solid (7.4 g, 61%). Spectroscopic data was consistent with those reported previously.70
ethyl 5-bromo-2-(4-fluorophenyl)thiazole-4-carboxylate (55):
The titled compound was synthesized according to the general procedure 6 to obtain (55) as a pure product (243.3 mg, 67%). 1H NMR (600 MHz, CDCl3) δ 7.94 – 7.91 (m, 2H), 7.19 – 7.15 (m, 2H), 4.49 (q, J = 7.1 Hz, 2H), 1.47 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 166.6, 164.4 (d, J = 252.4 Hz), 161.1, 144.4, 128.7(d, J = 8.7 Hz), 128.6 (d, J = 3.3 Hz), 116.3 (d, J = 22.2 Hz), 116.3, 61.9, 14.3.
ethyl 2-(4-fluorophenyl)-5-vinylthiazole-4-carboxylate (56):
The titled compound was synthesized according to the general procedure 6 to obtain (56) as a yellow solid (144 mg, 89%). 1H NMR (600 MHz, CDCl3) δ 7.99 – 7.97 (m, 2H), 7.70 (dd, J = 17.4, 10.9 Hz, 1H), 7.18 – 7.14 (m, 2H), 5.77 (d, J = 17.4 Hz, 1H), 5.53 (d, J = 11.0 Hz, 1H), 4.48 (q, J = 7.1 Hz, 2H), 1.47 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 164.3, (d, J = 251.7 Hz), 163.3, 162.3, 145.5, 141.7, 129.1 (d, J = 3.3 Hz), 128.9 (d, J = 8.6 Hz), 127.5, 121.0, 116.1 (d, J = 22.1 Hz), 61.6, 14.3. HRMS (ESI+) calcd for C14H13FNO2S [M+H]+ 278.0651, found 278.0663.
ethyl 5-ethynyl-2-(4-fluorophenyl)thiazole-4-carboxylate (57):
The titled compound was synthesized according to the general procedure 5 to obtain (57) as a brown solid (67.1 mg, 85%). 1H NMR (600 MHz, CDCl3) δ 8.00 – 7.97 (m, 2H), 7.20 – 7.15 (m, 2H), 4.50 (q, J = 7.2 Hz, 2H), 3.89 (s, 1H), 1.47 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 165.8, 164.6 (d, J = 252.6 Hz), 160.9, 149.1, 129.1 (d, J = 8.8 Hz), 128.6 (d, J = 3.4 Hz), 124.1, 116.3 (d, J = 22.2 Hz), 90.5, 73.0, 61.9, 14.3. HRMS (ESI+) calcd for C14H11FNO2S [M+H]+ 276.0494, found 276.0506. 95% HPLC purity.
2-ethynylthiazole (58):
The titled compound was synthesized according to the general procedure 5 to obtain (58) as a brown oil (270 mg, 27%). Spectroscopic data was consistent with those reported previously.71
thiazole-2-carbonitrile (59):
The titled compound was purchased from Ambeed (CAS number 1452–16-0) and used without further purification.
2-(2-aminophenyl)propan-2-ol (S7):
To a stirred solution of 1-(2-aminophenyl)ethan-1-one (S6) (2.7 g, 20 mmol, 1 equiv.) in anhydrous THF (150 mL) under nitrogen atmosphere at 0 °C, was added a solution of methylmagnesium chloride ( 20 mL of 3 M solution in THF, 60 mmol, 3 equiv.). The resulting mixture was stirred overnight at room temperature, after which it was diluted with brine and extracted with Et2OAc. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica in EtOAc/DCM to yield the (S7) (2.3 g, 77%). Spectroscopic data was consistent with those reported previously.16
4,4-dimethyl-2-(prop-1-yn-1-yl)-4H-benzo[d][1,3]oxazine (1):
To a stirred solution of 2-butynoic acid (168 mg, 2 mmol, 1 equiv.) in anhydrous DCM (5 mL) were added N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (575 mg, 3 mmol, 1.5 equiv.), and 2-(2-aminophenyl) propan-2-ol (S7) (302 mg, 2mmol, 1 equiv.). The resulting mixture was stirred at room temperature for 2 h, after which it was diluted with brine and extracted with EtOAc. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The product N-(2-(2-hydroxypropan-2-yl)phenyl)but-2-ynamide (S9) was used in the next step without further purification. To a stirred solution of N-(2-(2-hydroxypropan-2-yl)phenyl)but-2-ynamide in anhydrous DCM (5 mL) was added methanesulfonic acid (0.65 mL, 10 mmol, 5 equiv.) under nitrogen atmosphere. The resulting mixture was stirred at 45 °C for 1 h, after which it was diluted with aqueous sat. NaHCO3, brine and extracted with EtOAc. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica in EtOAc/hexanes to yield (1) as a yellow solid (110 mg, 55% over two steps). Spectroscopic data was consistent with those reported previously.16
2-ethynyl-4,4-dimethyl-4H-benzo[d][1,3]oxazine (2):
To a stirred solution of 2-propynoic acid (140 mg, 2 mmol, 1 equiv.) in anhydrous DCM (5 mL) were added N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (575 mg, 3 mmol, 1.5 equiv.), and 2-(2-aminophenyl) propan-2-ol (S7) (302 mg, 2 mmol, 1 equiv.). The resulting mixture was stirred at room temperature for 2 h, after which it was diluted with brine and extracted with EtOAc. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The product N-(2-(2-hydroxypropan-2-yl)phenyl)propiolamide (S8) was used in the next step without further purification. To a stirred solution of N-(2-(2-hydroxypropan-2-yl)phenyl)propiolamide in anhydrous DCM (5 mL) was added methanesulfonic acid (0.65 mL, 10 mmol, 5 equiv.) under nitrogen atmosphere. The resulting mixture was stirred at 45 °C for 1 h, after which it was diluted with aqueous sat. NaHCO3 solution, brine and extracted with EtOAc. The organic extract was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica in EtOAc/hexanes to yield (2) as a brown solid (167 mg, 45%. over two steps). Spectroscopic data was consistent with those reported previously.16
2-ethynylpyridine (60):
The titled compound was purchased from Ambeed (CAS number 1945–84-2) and used without further purification.
2-ethynylpyrimidine (61):
The titled compound was purchased from Ambeed (CAS number 37972–24-0) and used without further purification.
(2-bromothiazol-4-yl)methanol (S10):
To stirred solution of ethyl 2-bromothiazole-4-carboxylate (5) (3 g, 12.7 mml, 1 equiv.) in THF (60 mL) were added sodium borohydride, (1.4 g, 38 mmol, 3 equiv.), lithium chloride (1.6 g, 38 mmol, 3 equiv.) and H2O (15 mL). The resulting mixture was stirred at room temperature for 2 h, after which was diluted with 1 M HCl and extracted with EtOAc. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica in EtOAc/hexanes to yield (S10) as a brown oil (2.8 mg, 95%). Spectroscopic data was consistent with those reported previously.72
1-(2-bromothiazol-4-yl)hexane-1,4-dione (S13):
To a stirred solution of 2-bromothiazole-4-carbaldehyde (11) (192 mg, 1 mmol, 1 equiv.) in anhydrous THF (5 mL) were added thiazolium, 3-ethyl-5-(hydroxymethyl)-4-methyl-, bromide (S12) (20 mg, 0.08 mmol, 8 mol%), triethylamine (0.044 mL, 0.32 mmol, 8 mol%), and ethyl vinyl ketone (S11) (0.1 mL, 1 mmol, 1 equiv.). The resulting mixture was stirred at 70 °C for 2 h, after which it was concentrated under reduced pressure. The reaction mixture was then diluted with brine and extracted with DCM. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by column chromatography on neutralized silica in DCM/hexanes to yield (S13) as white solid (151 mg, 80%). 1H NMR (600 MHz, CDCl3) δ 8.09 (s, 1H), 3.38 – 3.34 (m, 2H), 2.88 – 2.84 (m, 2H), 2.55 (q, J = 7.3 Hz, 2H), 1.10 (t, J = 7.3 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 209.7, 192.6, 154.1, 136.1, 128.6, 35.9, 35.6, 34.0, 7.8.
3-(2-bromothiazol-4-yl)-2-methylcyclopent-2-en-1-one (S14):
To a stirred solution of 1-(2-bromothiazol-4-yl)hexane-1,4-dione (S13) (3.8 g, 13.8 mmol, 1 equiv.) in ethanol (200 mL) was added sodium hydroxide (1.5 g, 41. mmol, 3 equiv.). The resulting mixture was stirred at room temperature for 3 h, after which it was concentrated under reduced pressure. The reaction mixture was then extracted with DCM. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to yield (S14) as pure yellow solid. 1H NMR (600 MHz, CDCl3) δ 7.58 (s, 1H), 2.97 (m, 2H), 2.58 – 2.55 (m, 2H), 2.14 (t, J = 2.1 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 209.7, 156.7, 152.7, 137.6, 136.2, 123.4, 33.7, 27.8, 10.1.
3-(2-ethynylthiazol-4-yl)-2-methylcyclopent-2-en-1-one (S15):
The titled compound was synthesized according to the general procedure 5 to obtain (S15) as a brown solid (3 g, 85%). 1H NMR (600 MHz, CDCl3) δ 7.67 (s, 1H), 3.57 (s, 1H), 3.05 – 3.01 (m, 2H), 2.59 – 2.56 (m, 2H), 2.16 (t, J = 2.0 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 209.9, 157.4, 152.9, 147.3, 137.7, 121.7, 83.1, 76.1, 33.7, 28.1, 10.2.
3-(2-ethynylthiazol-4-yl)-2-methyl-N-(prop-2-yn-1-yl)cyclopent-2-en-1-amine (64):
To a stirred solution of 3-(2-ethynylthiazol-4-yl)-2-methylcyclopent-2-en-1-one (S15) (150 mg, 0.73 mmol, 1 equiv.) in anhydrous THF (10mL) under nitrogen atmosphere was added titanium (IV) isopropoxide (0.22 mL, 0.73 mmol, 1 equiv.) and the mixture was stirred for 15 min. Propargyl amine (0.1 mL, 1.5 mmol, 2 equiv.) was added and the reaction mixture was stirred at room temperature for additional 2 h, after which NaBH4 (30 mg, 0.8 mmol, 1.1 equiv.) and MeOH (0.3 mL, 10 mmol, 10 equiv.) were added at 0 °C. The resulting mixture was stirred at room temperature for 1 h. It was then diluted with aqueous sat. NaHCO3 and extracted with EtOAc. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica in EtOAc/DCM to yield (64) as a brown oil (90.3 mg, 60% brsm). 1H NMR (600 MHz, MeOD-d4) δ 7.44 (s, 1H), 4.01 (t, J = 6.3 Hz, 1H), 3.50 – 3.39 (m, 2H), 3.33 (s, 1H), 2.84 (dddd, J = 15.6, 8.8, 4.3, 2.0 Hz, 1H), 2.75 – 2.67 (m, 1H), 2.64 (t, J = 2.5 Hz, 1H), 2.28 – 2.21 (m, 1H), 2.11 (d, J = 1.0 Hz, 3H), 1.78 (ddt, J = 13.2, 9.0, 5.7 Hz, 1H). 13C NMR (151 MHz, MeOD-d4) δ 153.6, 146.7, 138.5, 130.6, 117.6, 82.9, 80.9, 71.7, 67.3, 34.4, 32.9, 27.8, 12.5. HRMS (ESI+) calcd for C14H15N2S [M+H]+ 243.0955, found 243.1093.
2-ethynyl-N-(prop-2-yn-1-yl)thiazole-4-carboxamide (65):
The titled compound was synthesized according to the general procedure 2 to obtain (65) as a white solid (140.5 mg, 46%). 1H NMR (600 MHz, CDCl3) δ 8.18 (s, 1H), 7.49 (s, 1H), 4.26 (dd, J = 5.5, 2.6 Hz, 2H), 3.56 (s, 1H), 2.29 (t, J = 2.6 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 159.8, 149.9, 147.5, 125.6, 83.2, 79.0, 75.6, 71.9, 29.1. HRMS (ESI+) calcd for C9H7N2OS [M+H]+ 191.0279, found 191.0275. 98% HPLC purity.
2-bromothiazole-4-carboxylic acid (S17):
The titled compound was synthesized according to the general procedure 1 to obtain (S17) as a white solid (2 g, 99%). 1H NMR (600 MHz, DMSO-d6) δ 13.33 (s, 1H), 8.47 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ 161.4, 147.6, 137.1, 133.6.
2-(phenylethynyl)thiazole-4-carboxylic acid (S18):
The titled compound was synthesized according to the general procedure 1 to obtain (S18) as a yellow solid (633.41 mg, 97%). 1H NMR (600 MHz, DMSO-d6) δ 8.58 (s, 1H), 7.70 – 7.67 (m, 2H), 7.55 – 7.49 (m, 3H). 13C NMR (151 MHz, DMSO-d6) δ 162.0, 148.4, 148.1, 132.3, 131.3, 130.9, 129.5, 120.5, 94.4, 82.2.
2-(phenylethynyl)-N-(prop-2-yn-1-yl)thiazole-4-carboxamide (66):
The titled compound was synthesized according to the general procedure 2 to obtain (66) as a yellow solid (355.3 mg, 62%). 1H NMR (600 MHz, CDCl3) δ 8.19 (s, 1H), 7.65 – 7.62 (m, 2H), 7.55 (s, 1H), 7.48 – 7.41 (m, 3H), 4.28 (dd, J = 5.5, 2.6 Hz, 2H), 2.29 (t, J = 2.5 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 160.0, 149.9, 148.8, 132.1, 130.0, 128.6, 125.3, 120.8, 95.0, 81.3, 79.0, 71.8, 29.1. HRMS (ESI+) calcd for C15H11N2OS [M+H]+ 267.0592, found 267.0596. >99% HPLC purity.
2-bromo-N-(prop-2-yn-1-yl)thiazole-4-carboxamide (67):
The titled compound was synthesized according to the general procedure 2 to obtain (67) as a white solid (96.0 mg, 64%). 1H NMR (600 MHz, CDCl3) δ 8.10 (s, 1H), 7.39 (s, 1H), 4.26 (dd, J = 5.6, 2.6 Hz, 2H), 2.30 (t, J = 2.5 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 159.2, 149.3, 136.1, 127.4, 78.9, 72.0, 29.1. HRMS (ESI+) calcd for C7H6BrN2OS [M+H]+ 244.9384, found 244.9380. 92% HPLC purity.
N-(prop-2-yn-1-yl)thiazole-4-carboxamide (68):
The titled compound was synthesized according to the general procedure 2 to obtain (68) as a brown solid (265 mg, 53%). 1H NMR (600 MHz, CDCl3) δ 8.77 (d, J = 2.1 Hz, 1H), 8.20 (d, J = 2.1 Hz, 1H), 7.62 (s, 1H), 4.26 (dd, J = 5.6, 2.6 Hz, 2H), 2.28 (t, J = 2.6 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 160.5, 152.9, 150.5, 123.7, 79.3, 71.7, 29.0. HRMS (ESI+) calcd for C7H7N2OS [M+H]+ 167.0279, found 167.0426.
(4-(bis(4-chlorophenyl)methyl)piperazin-1-yl)(5-methyl-4-nitroisoxazol-3-yl)methanone(ML-210):
The titled compound was purchased from Cayman Chemical Company (CAS number 1360705–96-9) and used without further purification.
(4-(bis(4-chlorophenyl)methyl)piperazin-1-yl)(2-ethynylthiazol-4-yl)methanone (BCP-T. A):
The titled compound was synthesized according to the general procedure 2 to obtain (BCP-T. A) as a white solid (62 mg, 44%). 1H NMR (600 MHz, CDCl3) δ 7.97 (s, 1H), 7.34 (d, J = 8.5 Hz, 4H), 7.28 (d, J = 8.5 Hz, 4H), 4.25 (s, 1H), 3.94 (s, 2H), 3.80 (s, 2H), 3.50 (s, 1H), 2.45 (d, J = 28.5 Hz, 4H). 13C NMR (151 MHz, CDCl3) δ 161.5, 151.2, 146.4, 140.2, 133.1, 129.1, 129.0, 126.8, 82.8, 76.0, 74.5, 52.2, 51.5, 47.2, 42.9. HRMS (ESI+) calcd for C23H20Cl2N3OS [M+H]+ 456.0704, found 456.0691. 95% HPLC purity.
1-(4-(bis(4-chlorophenyl)methyl)piperazin-1-yl)-2-chloroethan-1-one (BCP-C. A):
The titled compound was synthesized according to the general procedure 2 to obtain (BCP-C. A) as a transparent waxy solid (120 mg, 97%). 1H NMR (600 MHz, CDCl3) δ 7.34 (d, J = 8.5 Hz, 4H), 7.29 (d, J = 8.2 Hz, 4H), 4.24 (s, 1H), 4.05 (s, 2H), 3.67 – 3.62 (m, 2H), 3.57 – 3.50 (m, 2H), 2.47 – 2.35 (m, 4H). 13C NMR (151 MHz, CDCl3) δ 165.0, 140.0, 133.2, 129.0, 74.4, 51.7, 51.2, 46.4, 42.2, 40.8.
1-(4-(bis(4-chlorophenyl)methyl)piperazin-1-yl)prop-2-yn-1-one (BCP-P. A):
The titled compound was synthesized according to the general procedure 2 to obtain (BCP-P. A) as a white solid (52 mg, 45%). 1H NMR (600 MHz, CDCl3) δ 7.34 – 7.32 (m, 4H), 7.30 – 7.28 (m, 4H), 4.25 (s, 1H), 3.81 – 3.76 (m, 2H), 3.68 – 3.63 (m, 2H), 3.11 (s, 1H), 2.45 – 2.36 (m, 4H). 13C NMR (151 MHz, CDCl3) δ 151.7, 140.0, 133.2, 129.0, 79.3, 75.3, 74.4, 51.8, 51.1, 47.0, 41.5.
2-((trimethylsilyl)ethynyl)thiazole-4-carbaldehyde (S19):
The titled compound was synthesized according to the general procedure 5 to obtain (S19) as a pure product (485mg, 70%). 1H NMR (600 MHz, CDCl3) δ 10.04 (s, 1H), 8.16 (s, 1H), 0.30 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 184.5, 154.7, 149.6, 127,3, 103.0, 95.3, −0.6.
N-((2-ethynylthiazol-4-yl)methyl)prop-2-yn-1-amine (S20):
To a stirred solution of 2-((trimethylsilyl)ethynyl)thiazole-4-carbaldehyde (S19) (137 mg, 1 mmol, 1 equiv.) in anhydrous THF (10 mL) under nitrogen atmosphere was added titanium (IV) isopropoxide (0.3 mL, 1 mmol, 1 equiv.) and the mixture stirred for 15 min. Propargyl amine (0.13 mL, 2 mmol, 2 equiv.) was added and the mixture stirred at room temperature for additional 2 h, after which NaBH4 (41 mg, 1.1 mmol, 1.1 equiv.) and MeOH (0.4 mL, 10 mmol, 10 equiv.) were added at 0 °C. The resulting mixture was stirred at room temperature for 1 h. It was diluted with aqueous sat. NaHCO3 and extracted with EtOAc. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica in EtOAc/DCM to yield (S20) as a brown oil (24.66 mg, 18%). 1H NMR (600 MHz, CDCl3) δ 7.21 (s, 1H), 4.03 (d, J = 0.7 Hz, 2H), 3.49 – 3.47 (m, 3H), 2.26 (t, J = 2.4 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 155.9, 147.6, 117.3, 82.1, 81.5, 76.4, 71.9, 47.9, 37.6.
TAMRA-PEG3-N3:
To a stirred solution of TAMRA-acid (0.86 gr, 2 mmol) in anhydrous DMF (6 mL) were added HATU (0.76 gr, 2 mmol, 1 equiv.), DIPEA (1.74 mL, 10 mmol, 5 equiv.) and NH2-PEG3-N3 (2.20 gr, 10 mmol, 5 equiv.). The resulting mixture was stirred at r.t for 2 h, at which point most of the DMF was removed under reduced pressure. The resulting crude mixture was diluted with brine and DCM. The organic layer was collected and washed with aqueous solutions in the following order 1 M HCl followed by sat. NaHCO3 and brine. After the last wash the organic layer was collected dried over Na2SO4, filtered and concentrated under reduced pressure. The crude mixture was purified on silica gel chromatography in H2O/MeCN (H2O 0->15%). The fractions which contained the desired product where collected and condensed under reduced pressure. Then they were dissolved in MeOH/DCM (10% MeOH) and filtered to remove any collected silica. The filtrate was collected and condensed under reduced pressure to yield TAMRA-PEG3-N3 as dark purple solid (0.82 g, 65% yield). 1H NMR (600 MHz, MeOD-d4) δ 8.17 (d, J = 8.2 Hz, 1H), 8.12 (dd, J = 8.2, 1.7 Hz, 1H), 7.77 (d, J = 1.5 Hz, 1H), 7.25 (d, J = 9.5 Hz, 2H), 6.99 (dd, J = 9.5, 2.4 Hz, 2H), 6.87 (d, J = 2.4 Hz, 2H), 3.68 – 3.56 (m, 14H), 3.35 – 3.32 (m, 2H), 3.25 (s, 12H). 13C NMR (151 MHz, MeOD-d4) δ 170.7, 167.2, 160.2, 157.5, 157.1, 142.7, 135.0, 132.8, 131.3, 129.7, 128.3, 128.1, 113.6, 113.5, 96.0, 70.2, 70.2, 70.1, 69.9, 69.7, 69.1, 50.3, 39.8, 39.5.
2-(6-(dimethylamino)-3-(dimethyliminio)-3H-xanthen-9-yl)-4-(((2-ethynylthiazol-4-yl)methyl)(prop-2-yn-1-yl)carbamoyl)benzoate (S21):
The titled compound was synthesized according to the general procedure 2 to obtain (S21) as a purple fluorescent solid, which was identified by HRMS (ESI+) calcd for C34H29N4O4S [M+H]+ 589.1909, found 589.1909.
methyl 5-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanoate (S22):
To a stirred solution of 5-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanoic acid (biotin) (1 g, 4.1 mmol, 1 equiv.) in methanol (20 mL) was added thionyl chloride (2 mL, 20 mmol, 5 equiv.) at 0 °C. The resulting mixture was allowed to warm up to room temperature and stirred overnight. It was concentrated under reduced pressure, diluted with aqueous sat. NaHCO3 and extracted with DCM. The organic extract was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to yield (S22) as a white solid (970 mg, 94%). 1H NMR (600 MHz, DMSO-d6) δ 6.45 (s, 1H), 6.38 (s, 1H), 4.34 – 4.28 (m, 1H), 4.16 – 4.10 (m, 1H), 3.59 (s, 3H), 3.10 (dt, J = 8.6, 6.1 Hz, 1H), 2.83 (dd, J = 12.4, 5.1 Hz, 1H), 2.58 (d, J = 12.4 Hz, 1H), 2.31 (dd, J = 15.5, 8.1 Hz, 2H), 1.65 – 1.27 (m, 6H). 13C NMR (151 MHz, DMSO-d6) δ 173.8, 163.2, 61.5, 59.6, 55.8, 51.7, 33.6, 28.5, 28.4, 24.9.
benzo[d]thiazol-2-yl trifluoromethanesulfonate (S24):
To a stirred solution of benzothiazolone (S23) (152 mg, 1 mmol, 1 equiv.) in anhydrous DCM (5 mL) under nitrogen atmosphere at −75 °C were added triethylamine (0.3 mL, 2 mmol, 2 equiv.) and trifluoromethanesulfonic anhydride (0.25 mL, 1.5 mmol, 1.5 equiv.). The resulting mixture was stirred at −75 °C for 10 min, and an additional 40 min at room temperature, after which it was diluted with brine and extracted with DCM. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica in EtOAc/hexanes to yield (S24) as a gray oil (203 mg, 72%). Spectroscopic data was consistent with those reported previously.73
2,4-bis(4-fluorophenyl)thiazole (S26):
The titled compound was synthesized according to the general procedure 6 to obtain (S26) as a white solid (1.1 g, 67%). 1H NMR (600 MHz, CDCl3) δ 8.06 – 8.03 (m, 2H), 8.00 – 7.97 (m, 2H), 7.42 (s, 1H), 7.20 – 7.13 (m, 4H). 13C NMR (151 MHz, CDCl3) δ 166.8, 163.9 (d, J = 250.5 Hz), 162.8 (d, J = 247.6 Hz), 155.3, 130.7 (d, J = 3.2 Hz), 130.0 (d, J = 3.3 Hz), 128.5 (d, J = 8.4 Hz), 128.2 (d, J = 8.2 Hz), 116.0 (d, J = 22.1 Hz), 115.7 (d, J = 21.6 Hz) 112.1.
5-bromo-2,4-bis(4-fluorophenyl)thiazole (S27):
To a stirred solution of 2,4-bis(4-fluorophenyl)thiazole (S26) (273 mg, 1 mmol, 1.0 equiv.) in THF (5 mL), was added N-bromosuccinimide (195.7mg, 1.1 mmol, 1.1 equiv.). The resulting mixture was stirred overnight at room temperature, after which it was concentrated under reduced pressure. It was diluted with brine and extracted with DCM. The organic extract was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica in EtOAc/hexanes to yield (S27) as yellow solid (260.2 mg, 95%). 1H NMR (600 MHz, CDCl3) δ 8.04 – 8.01 (m, 2H), 7.96 – 7.90 (m, 2H), 7.22 – 7.14 (m, 4H). 13C NMR (151 MHz, CDCl3) δ 166.1, 164.1 (d, J = 251.4 Hz), 162.8 (d, J = 248.8 Hz), 152.3, 130.5 (d, J = 8.3 Hz), 129.5 (d, J = 3.2 Hz), 129.3 (d, J = 3.3 Hz), 128.3 (d, J = 8.6 Hz), 116.2 (d, J = 22.2 Hz), 115.3 (d, J = 21.6 Hz), 103.0.
2,4-bis(4-fluorophenyl)-5-vinylthiazole (S28):
The titled compound was synthesized according to the general procedure 6 to obtain (S28) as a yellow solid (844 mg, 84%). 1H NMR (600 MHz, CDCl3) δ 8.02 – 7.97 (m, 2H), 7.71 – 7.66 (m, 2H), 7.20 – 7.13 (m, 4H), 6.90 (dd, J = 17.2, 10.9 Hz, 1H), 5.64 (d, J = 17.2 Hz, 1H), 5.35 (d, J = 10.9 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ 164.17 (d, J = 204.5 Hz), 163.2, 162.76 (d, J = 248.1 Hz), 152.1, 132.4, 131.00 (d, J = 8.2 Hz), 130.82 (d, J = 3.2 Hz), 129.80 (d, J = 3.2 Hz), 128.51 (d, J = 8.5 Hz), 127.4, 117.5, 116.06 (d, J = 22.1 Hz), 115.45 (d, J = 6.5 Hz). HRMS (ESI+) calcd for C17H12F2NS [M+H]+ 300.0658, found 300.0655.
5-ethynyl-2,4-bis(4-fluorophenyl)thiazole (S29):
The titled compound was synthesized according to the general procedure 5 to obtain (S29) as a brown solid (1.3 g, 63%). 1H NMR (600 MHz, CDCl3) δ 8.33 – 8.29 (m, 2H), 8.02 – 7.97 (m, 2H), 7.20 – 7.15 (m, 4H), 3.72 (s, 1H). 13C NMR (151 MHz, CDCl3) δ 165.3, 164.3 (d, J = 251.7 Hz), 163.0 (d, J = 249.2 Hz), 157.1, 130.1 (d, J = 3.2 Hz), 129.9 (d, J = 8.3 Hz), 129.3 (d, J = 3.3 Hz), 128.7 (d, J = 8.6 Hz), 116.2 (d, J = 22.2 Hz), 115.4 (d, J = 21.6 Hz), 110.9, 87.23, 75.24. HRMS (ESI+) calcd for C17H10F2NS [M+H]+ 298.0502, found 298.0518.
Ethyl(R,E)-2-(2-((2-((tert-butoxycarbonyl)amino)-3-methoxy-3-oxopropyl)thio)vinyl)thiazole-4-carboxylate (S30):
To a stirred solution of ethyl 2-ethynylthiazole-4-carboxylate (22) (100 mg, 0.55 mmol, 1.0 equiv.) in MeCN (5 mL) were added N-Boc-L-cysteine methyl ester (1.3 g, 5.5 mmol, 10 equiv.), and 1,4-diazabicyclo[2.2.2]octane (61.6 mg, 0.55 mmol, 1 equiv.). The resulting mixture was stirred at room temperature overnight, after which it was concentrated under reduced pressure. The residue was partitioned between brine and DCM. The two layers were separated, and the organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The precipitated solid product was collected by filtration, filtered, washed with hexanes to yield (S30) (207 mg, 91%). 1H NMR (600 MHz, CDCl3) δ 8.01 – 8.01 (m, 1H), 7.26 (d, J = 15.5 Hz, 1H), 6.75 (d, J = 15.5 Hz, 1H), 4.69 (d, J = 6.5 Hz, 1H), 4.43 (q, J = 7.1 Hz, 2H), 3.80 (s, 3H), 3.34 (m, 2H), 1.45 (s, 9H), 1.42 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 170.6, 166.0, 161.4, 155.0, 147.5, 134.9, 125.7, 119.3, 80.5, 61.5, 53.3, 52.9, 35.6, 28.3, 14.4. HRMS (ESI+) calcd. for C17H25N2O6S2 [M+H]+ 417.1154, found 417.1156.
(2-Ethynylthiazol-5-yl)(morpholino)methanone (76):
The titled compound was synthesized according to the general procedure 2 to obtain (76) as a yellow solid (56 mg, 56%). 1H NMR (600 MHz, DMSO-d6) δ 8.26 (s, 1H), 5.04 (s, 1H), 3.64 (d, J = 8.0 Hz, 6H), 3.35 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 161.8, 150.5, 146.7, 127.4, 87.2, 76.4, 66.8, 66.5, 47.7, 42.9.
(4-ethylpiperazin-1-yl)(2-ethynylthiazol-5-yl)methanone (S75):
The titled compound was synthesized according to the general procedure 2 to obtain (75) as a yellow oil (40 mg, 42%). 1H NMR (600 MHz, Acetone-d6) δ 8.12 (s, 1H), 4.40 (s, 1H), 3.75 (d, J = 46.9 Hz, 4H), 2.51 – 2.39 (m, 6H), 1.07 (t, J = 7.2 Hz, 3H). 13C NMR (151 MHz, Acetone-d6) δ 161.2, 151.6, 146.3, 126.0, 83.9, 75.9, 53.1, 52.4, 51.8, 46.8, 42.3, 11.5.
(2-ethynylthiazol-5-yl)(4-(4-fluorophenyl)piperazin-1-yl)methanone(72):
The titled compound was synthesized according to the general procedure 2 to obtain (72) as a yellow solid (45 mg, 43%). 1H NMR (600 MHz, CDCl3) δ 8.06 (s, 1H), 7.05 – 7.01 (m, J = 4.8 Hz, 4H), 4.11 (dd, J = 55.1, 48.0 Hz,4H), 3.54 (s, 1H), 3.19 (dd, J = 21.8, 16.6 Hz, 4H). 13C NMR (151 MHz, CDCl3) δ 161.5, 151.0, 146.6, 129.2, 118.4, 115.9, 82.9, 76.0, 50.6, 49.4, 45.4, 39.8.
(2-Ethynylthiazol-5-yl)(4-(4-(trifluoromethyl)phenyl)piperazin-1-yl)methanone (73):
The titled compound was synthesized according to the general procedure 2 to obtain (S34) as a yellow solid (57 mg, 57%). 1H NMR (600 MHz, CDCl3) δ 8.08 (s, 1H), 7.53 (d, J = 8.9 Hz, 2H), 6.98 (d, J = 8.7 Hz, 2H), 4.08 (m, 4H), 3.55 (s, 1H), 3.39 (m, 4H). 13C NMR (151 MHz, CDCl3) δ 161.5, 152.8, 150.9, 146.6, 127.6, 126.6 (q, J = 3.6 Hz), 124.6 (d, J = 270.8 Hz), 121.5 (q, J = 32.6 Hz), 115.1, 83.0, 76.0, 48.8, 48.2, 46.6, 42.6.
(4-(4-chlorophenyl)piperazin-1-yl)(2-ethynylthiazol-5-yl)methanone (74):
The titled compound was synthesized according to the general procedure 2 to obtain (S35) as a yellow solid (65 mg, 66%). 1H NMR (600 MHz, CDCl3) δ 8.06 (s, 1H), 7.26 (d, J = 8.9 Hz, 2H), 6.92 (s, 2H), 4.07 (m, 4H), 3.54 (s, 1H), 3.26 (d, J = 20.7 Hz, 4H). 13C NMR (151 MHz, CDCl3) δ 161.5, 151.0 146.6, 129.2, 127.4, 118.0 82.9, 76.0, 50.2, 49.6, 46.8, 42.6.
(E)-1-(4-(4-chlorophenyl)piperazin-1-yl)-3-(2-ethynylthiazol-5-yl)prop-2-en-1-one (S31):
The titled compound was synthesized according to the general procedure 2 to obtain (S31) as a yellow solid (93 mg, 62%). 1H NMR (600 MHz, Acetone-d6) δ 7.95 (s, 1H), 7.59 (d, J = 14.9 Hz, 1H), 7.42 (d, J = 15.0 Hz, 1H), 7.28 – 7.25 (m, 2H), 7.06 – 7.02 (m, 2H), 4.41 (s, 1H), 3.87 (m, 4H), 3.27 (m, 4H). 13C NMR (151 MHz, Acetone-d6) δ 164.1, 153.1, 150.2, 147.7, 133.4, 128.8, 123.9, 122.7, 120.9, 117.6, 84.0, 76.1, 49.5, 48.7, 45.3, 41.6.
(E)-1-(4-(bis(4-chlorophenyl)methyl)piperazin-1-yl)-3-(2-ethynylthiazol-5-yl)prop-2-en-1-one (78):
The titled compound was synthesized according to the general procedure 2 to obtain (78) as a yellow solid (93 mg, 62%).1H NMR (600 MHz, Acetone-d6) δ 7.90 (s, 1H), 7.56 – 7.52 (m, 5H), 7.37 (dd, J = 8.8, 2.2 Hz, 4H), 7.33 (d, J = 14.9 Hz, 1H), 4.46 (s, 1H), 4.39 (s, 1H), 3.74 (m, 4H), 2.45 (m, 4H). 13C NMR (151 MHz, Acetone-d6) δ 164.0, 153.0, 147.6, 141.3, 133.2, 132.4, 129.5, 128.6, 122.6, 120.9, 83.9, 76.1, 73.8, 52.2, 51.4, 45.5, 41.8.
(2-ethynylthiazol-5-yl)(piperidin-1-yl)methanone (77):
The titled compound was synthesized according to the general procedure 2 to obtain (S38) as a yellow oil (100 mg, 59%). 1H NMR (600 MHz, CDCl3) δ 7.84 (s, 1H), 3.70 – 3.65 (m, 5H), 3.52 (s, 1H), 1.68 – 1.62 (m, 5H). 13C NMR (151 MHz, CDCl3) δ 161.9, 151.6, 146.3, 125.3, 82.6, 76.1, 48.3, 43.8, 26.6, 25.6.
GSH addition kinetic experiment by NMR spectroscopy:
Two stock solutions (stock A and stock B) in DMSO-d6 were prepared as follows: Stock A contained a heterocyclic compound with final concentration of 6 mM. Stock B contained, DABCO (14 mg) as base and DMF (33 μL) as internal standard with final concentration of 124.8 mM, and 426.2 mM respectively, in 1 mL of DMSO-d6. NMR experiments were carried out by vortexing 690 μL of a heterocycle compound stock A with 10 μL of stock B in an NMR tube, affording a final heterocycle compound concentration of 6 mM in a total volume of ~700 μL. N-Boc-L-cysteine methyl ester (9.88 mg, 6.9 μL, 42 mmol, 10 equiv.) was added and the mixture was vortexed to ensure the homogeneity of the NMR solution. Acquisition of 1H NMR data was started immediately after the addition of the N-Boc-L-cysteine methyl ester. The time for the completion of the first 1H spectrum was recorded and included in the final half-life calculations. The rest of the spectra were recorded at standard time intervals.16
D-Biotin-Kinetic experiment based on NMR:
Two stock solutions (stock A and stock B) in DMSO-d6 were prepared as follows: stock A contained ethyl 2-ethynylthiazole-4-carboxylate (22) with final concentration of 6 mM, and DABCO (4.7 mg, 42 mmol, 10 equiv.). Stock B contained DMF (33 μL), as internal standard with final concentration of 426.2 mM in 1 mL of DMSO-d6. NMR experiments were carried out by vortexing 690 μL of ethyl 2-ethynylthiazole-4-carboxylate and DABCO solution with 10 μL of DMF stock solution, affording ethyl 2-ethynylthiazole-4-carboxylate with final concentration of 6 mM in a total volume of 700 μL. After that methyl 5-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanoate (S22) (1.1 mg, 4.2 mmol, 1 equiv.) was added, vortexed to ensure the homogeneity of the NMR solution. Acquisition of 1H NMR data was started immediately. The time for the completion of the first 1H spectrum was recorded and included in the final half-life calculations. The rest of the spectra were recorded at standard time intervals.
Biology
Cell Culture and Cell Viability Assays.
All tested cell lines were maintained in Dulbecco’s Modified Eagle’s medium (Mediatech, Inc.) supplemented with 10% fetal bovine serum (Gemini Bio-Products # 100–106) and 1000 U/ml of both Penicillin and Streptomycin (Mediatech, Inc.) at 37 °C with 5% CO2. Cell viability was assessed using methylene blue staining: cells were plated at 5000/well in 96 well plates (n=3) and treated the next day. Three days after treatment, cells were fixed/stained in methylene blue saturated in 50% ethanol for 30 min at RT. Plates were washed with excess water to wash off extra dye. Retained dye was dissolved in 0.1N HCl and absorbance was measured at 668 nm.
Incubation time50 (IT50) assay.
NCI-H522 cells (5*103 cells/well) were plated on 96 well plates (n=3) and let overnight to adhere. The next day, cells were treated with the corresponding compounds or DMSO. Then the first set of wells were washed immediately after treatment, while the rest were washed after 30 min time points, for a total of 6 h. The last wells were not washed. Cells were fixed/stained in methylene blue saturated in 50% ethanol for 30 min at RT. Plates were washed with excess water to wash off extra dye. Retained dye was dissolved in 0.1N HCl and absorbance was measured at 668 nm. Note, this process can affect cells, thus it is important to convert each data point as a % survival of the corresponding DMSO treatment.
Liproxstatin-1 rescue experiments.
NCI-H522 cells (25*103 cells/well) were plated on 24 well plates (n=3) and let overnight to adhere. The next day cells were treated with the corresponding inhibitors in the presence (0.25 μM) or absence of Liproxstatin-1. Three days after treatment, cells were fixed/stained in methylene blue saturated in 50% ethanol for 30 min at RT. Plates were washed with excess water to wash off extra dye. Retained dye was dissolved in 0.1N HCl and absorbance was measured at 668 nm.
C11-BODIPY lipid peroxidation assay.
NCI-H522 cells (~70% density) were plated on 7cm dishes and let overnight to adhere. The next day cells were treated with DMSO or Inhibitors (corresponding concentration). Bodipy 581/591 C11 (1 μM) (ThermoFischer) was added at the time of treatment. After the corresponding time of treatment (3 h or 6 h), cells were washed with 1xPBS and collected by trypsinization and centrifugation. The cells were washed once with 1xPBS and resuspended in PBS containing 2% FBS. Cells were analyzed using a BD LSR Fortessa FACScanner and FlowJo software. Preferably 20*103 cells were analyzed unless accelerated cell death occurred.
Western blotting.
NCI-H522 cell pellets obtained after the corresponding treatment conditions were lysed using lysis buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 0.5% NP-40, (supplemented with 1 µg/ml aprotinin, 2 µg/ml leupeptin, 1 µg/ml pepstatin A, 1 mM DTT, and 0.1 M PMSF) for 30 minutes on ice and centrifuged at 13*103 g for 25 minutes at 4 °C. The protein levels of the lysates obtained were normalized using BCA Protein Assay Kit (Pierce) and separated by SDS-polyacrylamide gel electrophoresis (12.5% acrylamide). Transfer to polyvinylidene difluoride membranes (Millipore) was followed by blocking of membranes with blocking buffer containing 5% (w/v) non-fat dry milk dissolved in PBST [1X PBS containing 0.05% (v/v) Tween 20] for 1 hour at room temperature. Membranes were then incubated with corresponding primary antibodies overnight at 4 °C. The membranes were then washed (3 X 15 min each) with PBST and incubated with secondary antibodies conjugated to horse-radish peroxidase, obtained from Biorad and used at a dilution of 1:10,000. Bound antibodies were detected using enhanced chemiluminescence (Biorad).
Fluorescent Labeling using TAMRA-PEG3-N3.
NCI-H522 cells (~70% density) were plated on 6 cm dishes and left overnight to adhere. The next day, the cells were treated with probe (65) (10 mM) or control (66) (10 mM) for 1 h at 37 °C. Then cells were washed with 1xPBS, collected and lysed using lysis buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 0.5% NP-40, (supplemented with 1 µg/ml aprotinin, 2 µg/ml leupeptin, 1 µg/ml pepstatin A, and 0.1 M PMSF) for 30 minutes on ice and centrifuged at 13*103 g for 25 minutes at 4 °C. The protein levels of the lysates obtained were normalized to 2 mg/mL using BCA Protein Assay Kit (Pierce). Then the normalized lysates were subjected to CuAAC as follows. To each 50 μL of lysate (2 mg/mL), the following reagents were added; 100 μL of 1xPBS, 2 μL of TAMRA-PEG3-N3 (stock: 1 mM in DMSO), 10 μL of THPTA (stock: 100 mM in H2O), 10 μL of CuSO4-5.H2O (stock: 20 mM in H2O) and last, to initiate the reaction, 10 μL of sodium ascorbate (stock: 300 mM in H2O). The reaction mixtures were vortexed briefly after the addition of each reagent, covered with aluminum foil and left react for 30 min at room temperature. After the completion of the reaction, 40 μL of 5X loading buffer was added to stop the reaction. The samples were boiled for 7 min and separated by SDS-polyacrylamide gel electrophoresis (12.5% acrylamide). After completion of gel electrophoresis, gels were stored into fixing solution (50% H2O, 40% MeOH and 10% AcOH), until visualization by Biorad imager ChemiDoc MP. During electrophoresis, the gel should be run long enough for unreacted TAMRA-PEG3-N3 and loading dye to be not visible on the gel. Or else, the gel area with TAMRA-PEG3-N3 and loading dye should either be removed, or the gel washed with fixing solution multiple times till clean image is obtained. After fluorescence visualization, each gel was stained with Coomassie staining to ensure equal protein loading.
Fluorescent Labeling using TAMRA-PEG3-N3 and competition.
For the competition experiments, similar protocol was followed with the modification that cells are first treated with the competitors (corresponding concentrations) and then with probe (65).
Pulldown experiments using Biotin-TAMRA for proteomic analysis.
NCI-H522 cells (~70% density) were plated on 10 cm dishes (5x per treatment) and let overnight to adhere. The next day, the cells were treated with probe (65) (10 mM) or control (66) (10 mM) for 1 h at 37 °C. Then cells were washed with 1xPBS, collected and lysed using lysis buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 0.5% NP-40, (supplemented with 1 µg/ml aprotinin, 2 µg/ml leupeptin, 1 µg/ml pepstatin A, and 0.1 M PMSF) for 30 minutes on ice and centrifuged at 13*103 g for 25 minutes at 4 °C. The protein levels of the lysates obtained were normalized using BCA Protein Assay Kit (Pierce). Then to each sample, prewashed streptavidin magnetic beads (20 μL) were added and the samples were rotated at 4 °C for 2 h. Then lysates were collected using magnetic separation rack and subjected to CuAAC as follows: For each 50 μL of lysate, the following reagents were added; 100 μL of 1xPBS, 2 μL of TAMRA-PEG3-N3 (stock: 1 mM in DMSO), 10 μL of THPTA (stock: 100 mM in H2O), 10 μL of CuSO4-5.H2O (stock: 20 mM in H2O) and finally, to initiate the reaction, 10 μL of sodium ascorbate (stock: 300 mM in H2O). The reaction mixtures were vortexed briefly after the addition of each reagent, covered with aluminum foil and left react for 30 min at room temperature. After the completion of the reaction, proteins were precipitated using MeOH: CHCl3, the resulting protein pellets were centrifuged, washed with MeOH (3x) and left to air-dry for 20 min. Each of the samples was resuspended in 1 mL RIPA buffer and sonicated for 30 sec. Then, prewashed streptavidin magnetic beads (50 μL) were added and rotated at 4 °C for overnight. At that point, beads were collected using magnetic rack and washed (3x 1 mL) with RIPA buffer. Beads were resuspended in 40 μL RIPA and 10 μL loading buffer and boiled for 10 min. Then beads were centrifuged, 15 μL of the supernatant was analyzed by in-gel fluorescence as described above. The rest (35 μL) was sent for LC-MS/MS analysis.
LC-MS/MS analysis.
Proteomic analysis was perfomed by MS-Bioworks as follows: Half of each sample was processed by SDS-PAGE using 10% Bis-Tris NuPage Mini-gel (Invitrogen) with the MES buffer system. The gel was run 1cm and the mobility region excised into 10 equally sized bands. Each band was processed by in-gel digestion with trypsin using a robot (ProGest, DigiLab) with the following protocol: Washed with 25mM ammonium bicarbonate followed by acetonitrile. Reduced with 10mM dithiothreitol at 60°C followed by alkylation with 50 mM iodoacetamide at RT. Digested with sequencing grade trypsin (Promega) at 37°C for 4h. Quenched with formic acid and the supernatant was analyzed directly without further processing. Half of the digested sample was analyzed by nano LC-MS/MS with a Waters M-Class HPLC system interfaced to a ThermoFisher Fusion Lumos mass spectrometer. Peptides were loaded on a trapping column and eluted over a 75µm analytical column at 350 nL/min; both columns were packed with Luna C18 resin (Phenomenex). The mass spectrometer was operated in data-dependent mode, with the Orbitrap operating at 60,000 FWHM and 15,000 FWHM for MS and MS/MS respectively. APD was enabled and the instrument was run with a 3s cycle for MS and MS/MS. 5hrs of instrument time was used for the analysis of each sample. Data were searched using a local copy of Mascot (Matrix Science) with the following parameters: Enzyme: Trypsin/P Databases: SwissProt Human (concatenated forward and reverse plus common contaminants) Fixed modifications: Carbamidomethyl (C) Variable modifications: Acetyl (N-term), Deamidation (N,Q), Oxidation (M), Pyro-Glu (N-term Q) Mass values: Monoisotopic Peptide Mass Tolerance: 10 ppm Fragment Mass Tolerance: 0.02 Da Max Missed Cleavages: 2 Mascot DAT files were parsed into Scaffold (Proteome Software) for validation, filtering and to create a non-redundant list per sample. Data were filtered at 1% protein and peptide FDR and requiring at least two unique peptides per protein.
Pulldown experiments using Biotin-TAMRA for western blot analysis.
NCI-H522 cells (~70% density) were plated on 10 cm dishes (2x per treatment) and left overnight to adhere. The next day, cells were treated with probe (65) (10 mM) or control (66) (10 mM) for 1 h at 37 °C. Then, cells were washed with 1xPBS, collected and lysed using lysis buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 0.5% NP-40, (supplemented with 1 µg/ml aprotinin, 2 µg/ml leupeptin, 1 µg/ml pepstatin A, and 0.1 M PMSF) for 30 minutes on ice and centrifuged at 13*103 g for 25 minutes at 4 °C. The protein levels of the obtained lysates were normalized using BCA Protein Assay Kit (Pierce). Then to each sample, prewashed streptavidin magnetic beads (20 μL) were added and the samples were rotated at 4 °C for 2 h. Then lysates were collected using magnetic separation rack and subjected to CuAAC as follows: For each 50 μL of lysate, the following reagents were added; 100 μL of 1xPBS, 4 μL of TAMRA-PEG3-N3 (stock: 1 mM in DMSO), 10 μL of THPTA (stock: 100 mM in H2O), 10 μL of CuSO4-5.H2O (stock: 20 mM in H2O), and last to initiate the reaction, 10 μL of sodium ascorbate (stock: 300 mM in H2O). The reaction mixtures were vortexed briefly after the addition of each reagent, covered with aluminum foil and left to react for 20 min at room temperature and for 10 min at 37 °C. After the completion of the reaction, proteins were precipitated using MeOH: CHCl3, the resulting protein pellets were centrifuged, washed with MeOH and left to air-dry for 20 min. Each of the samples was resuspended in 1 mL RIPA buffer and sonicated for 30 sec. Then prewashed streptavidin magnetic beads (50 μL) and 0.5 mL of PBS were added and rotated at 4 °C for overnight. At that point, beads were collected using magnetic rack and washed (3x 1 mL) with RIPA buffer and (1x 1mL) PBS buffer. Beads were resuspended in 30 μL RIPA and 7.5 μL of (5x) loading buffer and boiled for 10 min. The beads were centrifuged and proteins were separated by SDS-polyacrylamide gel electrophoresis (12.5% acrylamide). Transfer to polyvinylidene difluoride membranes (Millipore) was followed by blocking of membranes with blocking buffer containing 5% (w/v) non-fat dry milk dissolved in PBST [1X PBS containing 0.05% (v/v) Tween 20] for 1 hour at room temperature. Membranes were then incubated with corresponding primary antibodies overnight at 4 °C. The membranes were then washed (3 X 15 min each) with PBST and incubated with secondary antibodies conjugated to horse-radish peroxidase, obtained from Biorad and used at a dilution of 1:10,000. Bound antibodies were detected using enhanced chemiluminescence (Biorad).
Cellular thermal shift assay.
NCI-H522 cells in 6 well plates (~80% confluent) were treated with BCP-T.A (0.5 µM) or DMSO (0.1% v/v) for 1 h. Cells were washed with 1xPBS (pH.7.4) and detached from the flask with trypsin-EDTA (0.2 mL), collected with 1 mL of FBS media and pelleted by centrifugation (2000g, 5 min). The medium was removed, and the cell pellet was washed with 1xPBS (pH 7.4). Cells were resuspended in PBS and aliquoted into PCR tubes. Each tube was heated to corresponding temperature between 43–67 °C for 3 min. After cooling to ambient temperature, cells were pelleted by centrifugation (2000g, 5 min) and lysed using lysis buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 0.5% NP-40, (supplemented with 1 µg/ml aprotinin, 2 µg/ml leupeptin, 1 µg/ml pepstatin A, and 0.1 M PMSF) for 30 minutes on ice and centrifuged at 13*103 g for 25 minutes at 4 °C. The soluble fraction was diluted with 5x SDS loading buffer, heated to 95 °C for 10 min, and subjected to SDS-PAGE and western blotting analysis.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by NIH grant R15CA213185 to L.M.V.T and grant R15GM120712 to W.R.T. and grant Z01 ES043010 of the intramural research program of the National Institute of Environmental Health Sciences to L.P. The authors would like to thank the NMR director at The University of Toledo Dr. Yong-Wah Kim for his direction and help with the NMR kinetic studies.
ABBREVIATIONS
- ABPP
Activity Based Protein Profiling
- ADMET
absorption, distribution, metabolism, excretion and toxicity
- ANOVA
Analysis of Variance
- BCP
1-(bis(4-chlorophenyl)methyl)piperazine
- BLAST
Basic Local Alignment Search Tool
- C.A
Chloroacetamide
- CETSA
Cellular Thermal Shift Assay
- COX-1
cyclooxygenase-1
- CuAAC
Copper(I)-Catalyzed Alkyne-Azide Cycloaddition
- d
doublet (spectral)
- DABCO
1,4-diazabicyclo[2.2.2]octane
- DCM
dichloromethane
- DFT
Density functional theory
- DMF
Dimthylformamide
- DMSO
Dimethyl sulfoxide
- EDG
electron donating group
- ESI
Electrospray Ionization
- et al.
and others
- EWG
electron withdrawing group
- FDA
Food and Drug Administration
- GPX4
Glutathione Peroxidase 4
- GST
Glutathione S-Transferase
- GSTO1
Glutathione S-Transferase Omega-1
- HPLC
High Performance Liquid Chromatography
- HRMS
High-Resolution Mass Spectra
- I.A
Iodoacetamide
- IC50
half maximal inhibitory concentration
- IT50
half maximal Incubation Time
- LUMO
Lowest Unoccupied Molecular Orbital
- MEFS
mouse embryonic fibroblasts
- MW
Molecular Weight
- NCI
National Cancer Insitute
- NMR
Nuclear Magnetic Resonance
- NSAF
Normalized Spectral Abundance Factor
- P.A
Propiolamide
- PANTHER
Protein Analysis Through Evolutionary Relationships
- PDB
Protein Data Bank
- PEG
Polyethylene glycol
- q
quartet (spectral)
- s
singlet (spectral)
- S.I
Supporting Information
- SAR
Structure Activity Relationship
- SD
Standard Deviation
- SpC
Spectral Count
- t
triplet (spectral)
- T.A
Thiazole alkyne
- t1/2
Half Live
- Tagg
Aggregation Temperature
- TAMRA
Carboxytetramethylrhodamine
- TCI
targeted covalent inhibition
- TLC
Thin Layer Chromatography
- TMS
Trimethylsilyl
- TNBC
Triple-Negative Breast Cancer
- TXNRD1
Thioredoxin reductase 1
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at Additional synthetic schemes; 1H and 13C NMR spectra for all compounds, HPLC traces; results from additional biology experiments, and BLAST analysis (supporting information file)
Proteomics data (excel file)
Molecular formula strings for compounds (CSV) PDB file for GPX4 crystal structure with PDBid: 6ELW
The authors declare no competing financial address
REFERENCES
- (1).Jollow DJ; Mitchell JR; Potter WZ; Davis DC; Gillette JR; Brodie BB Acetaminophen Induced Hepatic Necrosis. II. Role of Covalent Binding in Vivo. J. Pharmacol. Exp. Ther 1973, 187 (1), 195–202. [PubMed] [Google Scholar]
- (2).Koen YM; Hanzlik RP Identification of Seven Proteins in the Endoplasmic Reticulum as Targets for Reactive Metabolites of Bromobenzene. Chem. Res. Toxicol 2002, 15 (5), 699–706. 10.1021/tx0101898. [DOI] [PubMed] [Google Scholar]
- (3).Bauer RA Covalent Inhibitors in Drug Discovery: From Accidental Discoveries to Avoided Liabilities and Designed Therapies. Drug Discov. Today 2015, 20 (9), 1061–1073. 10.1016/j.drudis.2015.05.005. [DOI] [PubMed] [Google Scholar]
- (4).Singh J; Petter RC; Baillie TA; Whitty A The Resurgence of Covalent Drugs. Nat. Rev. Drug Discov 2011, 10 (4), 307–317. 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- (5).Taubert D; Kastrati A; Harlfinger S; Gorchakova O; Lazar A; von Beckerath N; Schömig A; Schömig E Pharmacokinetics of Clopidogrel after Administration of a High Loading Dose. Thromb. Haemost 2004, 92 (2), 311–316. 10.1160/th04-02-0105. [DOI] [PubMed] [Google Scholar]
- (6).Aminov RI A Brief History of the Antibiotic Era: Lessons Learned and Challenges for the Future. Front. Microbiol 2010, 1 (DEC). 10.3389/fmicb.2010.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Sutanto F; Konstantinidou M; Dömling A Covalent Inhibitors: A Rational Approach to Drug Discovery. RSC Med. Chem 2020, 11 (8), 876–884. 10.1039/d0md00154f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Walter AO; Sjin RTT; Haringsma HJ; Sun J; Ohashi K; Lee K; Dubrovskiy A; Labenski M; Wang Z; Zhu Z; Sheets M; Martin TS; Karp R; van Kalken D; Chaturvedi P; Niu D; Nacht M; Petter RC; Lin K; Westlin W; Jaw-Tsai S; Raponi M; Van Dyke T; Etter J; Pao W; Weaver Z; Singh J; Simmons AD; Harding TC; Allen A Discovery of a Mutant-Selective Covalent Inhibitor of EGFR That Overcomes T790M Mediated Resistance in NSCLC. Cancer Discov 2013, 3 (12), 1404–1415. 10.1158/2159-8290.CD-13-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Gehringer M; Laufer SA Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem 2019, 62 (12), 5673–5724. 10.1021/acs.jmedchem.8b01153. [DOI] [PubMed] [Google Scholar]
- (10).Dixon JM Exemestane: A Potent Irreversible Aromatase Inactivator and a Promising Advance in Breast Cancer Treatment. Expert Rev. Anticancer Ther 2002, 2 (3), 267–275. 10.1586/14737140.2.3.267. [DOI] [PubMed] [Google Scholar]
- (11).Thomas DM; Zalcberg JR 5-Fluorouracil: A Pharmacological Paradigm in the Use of Cytotoxics. Clin. Exp. Pharmacol. Physiol 1998, 25 (11), 887–895. 10.1111/j.1440-1681.1998.tb02339.x. [DOI] [PubMed] [Google Scholar]
- (12).Xu H; Faber C; Uchiki T; Racca J; Dealwis C Structures of Eukaryotic Ribonucleotide Reductase I Define Gemcitabine Diphosphate Binding and Subunit Assembly. Proc. Natl. Acad. Sci. U. S. A 2006, 103 (11), 4028–4033. 10.1073/pnas.0600440103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zhao Z; Bourne PE Progress with Covalent Small-Molecule Kinase Inhibitors. Drug Discov. Today 2018, 23 (3), 727–735. 10.1016/j.drudis.2018.01.035. [DOI] [PubMed] [Google Scholar]
- (14).Butterworth S; Cross DAE; Finlay MRV; Ward RA; Waring MJ The Structure-Guided Discovery of Osimertinib: The First U.S. FDA Approved Mutant Selective Inhibitor of EGFR T790M. Medchemcomm 2017, 8 (5), 820–822. 10.1039/c7md90012k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Barf T; Covey T; Izumi R; Van De Kar B; Gulrajani M; Van Lith B; Van Hoek M; De Zwart E; Mittag D; Demont D; Verkaik S; Krantz F; Pearson PG; Ulrich R; Kaptein A Acalabrutinib (ACP-196): A Covalent Bruton Tyrosine Kinase Inhibitor with a Differentiated Selectivity and in Vivo Potency Profile. J. Pharmacol. Exp. Ther 2017, 363 (2), 240–252. 10.1124/jpet.117.242909. [DOI] [PubMed] [Google Scholar]
- (16).McAulay K; Hoyt EA; Thomas M; Schimpl M; Bodnarchuk MS; Lewis HJ; Barratt D; Bhavsar D; Robinson DM; Deery MJ; Ogg DJ; Bernardes GJL; Bernardes GJL; Ward RA; Waring MJ; Kettle JG Alkynyl Benzoxazines and Dihydroquinazolines as Cysteine Targeting Covalent Warheads and Their Application in Identification of Selective Irreversible Kinase Inhibitors. J. Am. Chem. Soc 2020, 142 (23), 10358–10372. 10.1021/jacs.9b13391. [DOI] [PubMed] [Google Scholar]
- (17).Eaton JK; Furst L; Ruberto RA; Moosmayer D; Hilpmann A; Ryan MJ; Zimmermann K; Cai LL; Niehues M; Badock V; Kramm A; Chen S; Hillig RC; Clemons PA; Gradl S; Montagnon C; Lazarski KE; Christian S; Bajrami B; Neuhaus R; Eheim AL; Viswanathan VS; Schreiber SL Selective Covalent Targeting of GPX4 Using Masked Nitrile-Oxide Electrophiles. Nat. Chem. Biol 2020, 16 (5), 497–506. 10.1038/s41589-020-0501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Eaton JK; Ruberto RA; Kramm A; Viswanathan VS; Schreiber SL Diacylfuroxans Are Masked Nitrile Oxides That Inhibit GPX4 Covalently. J. Am. Chem. Soc 2019, 141 (51), 20407– 20415. 10.1021/jacs.9b10769. [DOI] [PubMed] [Google Scholar]
- (19).Eaton JK; Furst L; Cai LL; Viswanathan VS; Schreiber SL Structure–Activity Relationships of GPX4 Inhibitor Warheads. Bioorganic Med. Chem. Lett 2020, 30 (23), 127538. 10.1016/j.bmcl.2020.127538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Yang WS; SriRamaratnam R; Welsch ME; Shimada K; Skouta R; Viswanathan VS; Cheah JH; Clemons PA; Shamji AF; Clish CB; Brown LM; Girotti AW; Cornish VW; Schreiber SL; Stockwell BR Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156 (1–2), 317–331. 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Cordon MB; Jacobsen KM; Nielsen CS; Hjerrild P; Poulsen TB Forward Chemical Genetic Screen for Oxygen-Dependent Cytotoxins Uncovers New Covalent Fragments That Target GPX4. ChemBioChem 2021, cbic.202100253. 10.1002/cbic.202100253. [DOI] [PubMed]
- (22).Dixon SJ; Lemberg KM; Lamprecht MR; Skouta R; Zaitsev EM; Gleason CE; Patel DN; Bauer AJ; Cantley AM; Yang WS; Morrison B; Stockwell BR Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149 (5), 1060–1072. 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Yang WS; Stockwell BR Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol 2008, 15 (3), 234–245. 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Moosmayer D; Hilpmann A; Hoffmann J; Schnirch L; Zimmermann K; Badock V; Furst L; Eaton JK; Viswanathan VS; Schreiber SL; Gradl S; Hillig RC Crystal Structures of the Selenoprotein Glutathione Peroxidase 4 in Its Apo Form and in Complex with the Covalently Bound Inhibitor ML162. Acta Crystallogr. Sect. D Struct. Biol 2021, 77 (2), 237–248. 10.1107/S2059798320016125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Weïwer M; Bittker JA; Lewis TA; Shimada K; Yang WS; MacPherson L; Dandapani S; Palmer M; Stockwell BR; Schreiber SL; Munoz B Development of Small-Molecule Probes That Selectively Kill Cells Induced to Express Mutant RAS. Bioorganic Med. Chem. Lett 2012, 22 (4), 1822–1826. 10.1016/j.bmcl.2011.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Alhamadsheh MM; Hudson RA; Tillekeratne LMV Design, Total Synthesis, and Evaluation of Novel Open-Chain Epothilone Analogues. Org. Lett 2006, 8 (4), 685–688. 10.1021/ol0528787. [DOI] [PubMed] [Google Scholar]
- (27).Fedorka SR; So K; Al-Hamashi AA; Gad I; Shah R; Kholodovych V; Alqahtani HD; Taylor WR; Tillekeratne LMV Small-Molecule Anticancer Agents Kill Cancer Cells by Harnessing Reactive Oxygen Species in an Iron-Dependent Manner. Org. Biomol. Chem 2018, 16 (9), 1465–1479. 10.1039/c7ob03086j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Taylor WR; Fedorka SR; Gad I; Shah R; Alqahtani HD; Koranne R; Kuganesan N; Dlamini S; Rogers T; Al-Hamashi A; Kholodovych V; Barudi Y; Junk D; Rashid MS; Jackson MW; Tillekeratne LMV Small-Molecule Ferroptotic Agents with Potential to Selectively Target Cancer Stem Cells. Sci. Rep 2019, 9 (1), 5926. 10.1038/s41598-019-42251-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Oballa RM; Truchon JF; Bayly CI; Chauret N; Day S; Crane S; Berthelette C A Generally Applicable Method for Assessing the Electrophilicity and Reactivity of Diverse Nitrile-Containing Compounds. Bioorganic Med. Chem. Lett 2007, 17 (4), 998–1002. 10.1016/j.bmcl.2006.11.044. [DOI] [PubMed] [Google Scholar]
- (30).Keeley A; Ábrányi-Balogh P; Keseru GM Design and Characterization of a Heterocyclic Electrophilic Fragment Library for the Discovery of Cysteine-Targeted Covalent Inhibitors. Medchemcomm 2019, 10 (2), 263–267. 10.1039/c8md00327k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Keeley A; Ábrányi-Balogh P; Hrast M; Imre T; Ilaš J; Gobec S; Keserű GM Heterocyclic Electrophiles as New MurA Inhibitors. Arch. Pharm. (Weinheim) 2018, 351 (12), 1800184. 10.1002/ardp.201800184. [DOI] [PubMed] [Google Scholar]
- (32).Schwartz Radatz C; Rampon DS; Balaguez RA; Alves D; Schneider PH Synthesis of 2-Substituted 1,3-Benzoselenazoles from Carboxylic Acids Promoted by Tributylphosphine. European J. Org. Chem 2014, 2014 (31), 6945–6952. 10.1002/ejoc.201402808. [DOI] [Google Scholar]
- (33).Liu X; Zhao X; Liang F; Ren BT -BuONa-Mediated Direct C-H Halogenation of Electron-Deficient (Hetero)Arenes. Org. Biomol. Chem 2018, 16 (6), 886–890. 10.1039/c7ob03081a. [DOI] [PubMed] [Google Scholar]
- (34).Kremer A; Aurisicchio C; Deleo F; Ventura B; Wouters J; Armaroli N; Barbieri A; Bonifazi D Walking Down the Chalcogenic Group of the Periodic Table: From Singlet to Triplet Organic Emitters. Chem. - A Eur. J 2015, 21 (43), 15377–15387. 10.1002/chem.201501260. [DOI] [PubMed] [Google Scholar]
- (35).Bowen JI; Wang L; Crump MP; Willis CL Ambruticins: Tetrahydropyran Ring Formation and Total Synthesis. Org. Biomol. Chem 2021, 19 (28), 6210–6215. 10.1039/d1ob00883h. [DOI] [PubMed] [Google Scholar]
- (36).Parr RG; Yang W Density Functional Approach to the Frontier-Electron Theory of Chemical Reactivity. J. Am. Chem. Soc 1984, 106 (14), 4049–4050. 10.1021/ja00326a036. [DOI] [Google Scholar]
- (37).Forman HJ; Zhang H; Rinna A Glutathione: Overview of Its Protective Roles, Measurement, and Biosynthesis. Mol. Aspects Med 2009, 30 (1–2), 1–12. 10.1016/j.mam.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Awoonor-Williams E; Rowley CN How Reactive Are Druggable Cysteines in Protein Kinases? J. Chem. Inf. Model 2018, 58 (9), 1935–1946. 10.1021/acs.jcim.8b00454. [DOI] [PubMed] [Google Scholar]
- (39).Ferrer-Sueta G; Manta B; Botti H; Radi R; Trujillo M; Denicola A Factors Affecting Protein Thiol Reactivity and Specificity in Peroxide Reduction. Chem. Res. Toxicol 2011, 24 (4), 434–450. 10.1021/tx100413v. [DOI] [PubMed] [Google Scholar]
- (40).Kuganesan N; Dlamini S; McDaniel J; Tillekeratne VLM; Taylor WR Identification and Initial Characterization of a Potent Inhibitor of Ferroptosis. J. Cell. Biochem 2021, 122 (3–4), 413– 424. 10.1002/jcb.29870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Yagoda N; von Rechenberg M; Zaganjor E; Bauer AJ; Yang WS; Fridman DJ; Wolpaw AJ; Smukste I; Peltier JM; Boniface JJ; Smith R; Lessnick SL; Sahasrabudhe S; Stockwell BR RAS–RAF–MEK-Dependent Oxidative Cell Death Involving Voltage-Dependent Anion Channels. Nature 2007, 447 (7146), 865–869. 10.1038/nature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Zilka O; Shah R; Li B; Friedmann Angeli JP; Griesser M; Conrad M; Pratt DA On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci 2017, 3 (3), 232–243. 10.1021/acscentsci.7b00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Naguib YMA A Fluorometric Method for Measurement of Peroxyl Radical Scavenging Activities of Lipophilic Antioxidants. Anal. Biochem 1998, 265 (2), 290–298. 10.1006/abio.1998.2931. [DOI] [PubMed] [Google Scholar]
- (44).Pap EHW; Drummen GPC; Winter VJ; Kooij TWA; Rijken P; Wirtz KWA; Op den Kamp JAF; Hage WJ; Post JA Ratio-Fluorescence Microscopy of Lipid Oxidation in Living Cells Using C11-BODIPY 581/591. FEBS Lett 1999, 453 (3), 278–282. 10.1016/S0014-5793(99)00696-1. [DOI] [PubMed] [Google Scholar]
- (45).Cravatt BF; Wright AT; Kozarich JW Activity-Based Protein Profiling: From Enzyme Chemistry to Proteomic Chemistry. Annu. Rev. Biochem 2008, 77 (1), 383–414. 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- (46).Karaj E; Sindi SH; Viranga Tillekeratne LM Photoaffinity Labeling and Bioorthogonal Ligation: Two Critical Tools for Designing “Fish Hooks” to Scout for Target Proteins. Bioorg. Med. Chem 2022, 116721. 10.1016/j.bmc.2022.116721. [DOI] [PubMed]
- (47).Mudd G; Pi IP; Fethers N; Dodd PG; Barbeau OR; Auer M A General Synthetic Route to Isomerically Pure Functionalized Rhodamine Dyes. Methods Appl. Fluoresc 2015, 3 (4), 045002. 10.1088/2050-6120/3/4/045002. [DOI] [PubMed] [Google Scholar]
- (48).Goswami LN; Houston ZH; Sarma SJ; Jalisatgi SS; Hawthorne MF Efficient Synthesis of Diverse Heterobifunctionalized Clickable Oligo(Ethylene Glycol) Linkers: Potential Applications in Bioconjugation and Targeted Drug Delivery. Org. Biomol. Chem 2013, 11 (7), 1116–1126. 10.1039/c2ob26968f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Park JH; Jin JH; Lim MS; An HJ; Kim JW; Lee GM Proteomic Analysis of Host Cell Protein Dynamics in the Culture Supernatants of Antibody-Producing CHO Cells. Sci. Rep 2017, 7 (1), 44246. 10.1038/srep44246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).McIlwain S; Mathews M; Bereman MS; Rubel EW; MacCoss MJ; Noble WS Estimating Relative Abundances of Proteins from Shotgun Proteomics Data. BMC Bioinformatics 2012, 13 (1), 308. 10.1186/1471-2105-13-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Mi H; Ebert D; Muruganujan A; Mills C; Albou LP; Mushayamaha T; Thomas PD PANTHER Version 16: A Revised Family Classification, Tree-Based Classification Tool, Enhancer Regions and Extensive API. Nucleic Acids Res 2021, 49 (D1), D394–D403. 10.1093/nar/gkaa1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Dixon SJ; Stockwell BR The Hallmarks of Ferroptosis. Annu. Rev. Cancer Biol 2019, 3 (1), 35– 54. 10.1146/annurev-cancerbio-030518-055844. [DOI] [Google Scholar]
- (53).Ramkumar K; Samanta S; Kyani A; Yang S; Tamura S; Ziemke E; Stuckey JA; Li S; Chinnaswamy K; Otake H; Debnath B; Yarovenko V; Sebolt-Leopold JS; Ljungman M; Neamati N Mechanistic Evaluation and Transcriptional Signature of a Glutathione S-Transferase Omega 1 Inhibitor. Nat. Commun 2016, 7 (1), 13084. 10.1038/ncomms13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Burmeister C; LÜersen K; Heinick A; Hussein A; Domagalski M; Walter RD; Liebau E Oxidative Stress in Caenorhabditis Elegans : Protective Effects of the Omega Class Glutathione Transferase ( GSTO‐1 ) . FASEB J 2008, 22 (2), 343–354. 10.1096/fj.06-7426com. [DOI] [PubMed] [Google Scholar]
- (55).Kim K; Kim SH; Kim J; Kim H; Yim J Glutathione S-Transferase Omega 1 Activity Is Sufficient to Suppress Neurodegeneration in a Drosophila Model of Parkinson Disease. J. Biol. Chem 2012, 287 (9), 6628–6641. 10.1074/jbc.M111.291179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Kodym R; Calkins P; Story M The Cloning and Characterization of a New Stress Response Protein: A Mammalian Member of a Family of θ Class Glutathione s-Transferase-like Proteins. J. Biol. Chem 1999, 274 (8), 5131–5137. 10.1074/jbc.274.8.5131. [DOI] [PubMed] [Google Scholar]
- (57).Jafari R; Almqvist H; Axelsson H; Ignatushchenko M; Lundbäck T; Nordlund P; Molina DM The Cellular Thermal Shift Assay for Evaluating Drug Target Interactions in Cells. Nat. Protoc 2014, 9 (9), 2100–2122. 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]
- (58).Wissner A; Overbeek E; Reich MF; Floyd MB; Johnson BD; Mamuya N; Rosfjord EC; Discafani C; Davis R; Shi X; Rabindran SK; Gruber BC; Ye F; Hallett WA; Nilakantan R; Shen R; Wang Y-F; Greenberger LM; Tsou H-R Synthesis and Structure−Activity Relationships of 6,7-Disubstituted 4-Anilinoquinoline-3-Carbonitriles. The Design of an Orally Active, Irreversible Inhibitor of the Tyrosine Kinase Activity of the Epidermal Growth Factor Receptor (EGFR) and the Human Epi. J. Med. Chem 2003, 46 (1), 49–63. 10.1021/jm020241c. [DOI] [PubMed] [Google Scholar]
- (59).Jackson PA; Widen JC; Harki DA; Brummond KM Covalent Modifiers: A Chemical Perspective on the Reactivity of α,β-Unsaturated Carbonyls with Thiols via Hetero-Michael Addition Reactions. J. Med. Chem 2017, 60 (3), 839–885. 10.1021/acs.jmedchem.6b00788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Orian L; Mauri P; Roveri A; Toppo S; Benazzi L; Bosello-Travain V; De Palma A; Maiorino M; Miotto G; Zaccarin M; Polimeno A; Flohé L; Ursini F Selenocysteine Oxidation in Glutathione Peroxidase Catalysis: An MS-Supported Quantum Mechanics Study. Free Radic. Biol. Med 2015, 87, 1–14. 10.1016/j.freeradbiomed.2015.06.011. [DOI] [PubMed] [Google Scholar]
- (61).Maiorino M; Bosello-Travain V; Cozza G; Miotto G; Orian L; Roveri A; Toppo S; Zaccarin M; Ursini F Glutathione Peroxidase 4. In Selenium; Springer International Publishing: Cham, 2016; pp 223–234. 10.1007/978-3-319-41283-2_18. [DOI] [Google Scholar]
- (62).Nicolaou KC; Buchman M; Bellavance G; Krieger J; Subramanian P; Pulukuri KK Syntheses of Cyclopropyl Analogues of Disorazoles A1 and B1 and Their Thiazole Counterparts. J. Org. Chem 2018, 83 (20), 12374–12389. 10.1021/acs.joc.8b02137. [DOI] [PubMed] [Google Scholar]
- (63).Sinenko VO; Slivchuk SR; Mityukhin OP; Brovarets VS Synthesis of New 1,3-Thiazolecarbaldehydes. Russ. J. Gen. Chem 2017, 87 (12), 2766–2775. 10.1134/S1070363217120039. [DOI] [Google Scholar]
- (64).Shibue T; Hirai T; Okamoto I; Morita N; Masu H; Azumaya I; Tamura O Total Syntheses of Tubulysins. Chem. - A Eur. J 2010, 16 (38), 11678–11688. 10.1002/chem.201000963. [DOI] [PubMed] [Google Scholar]
- (65).Soares MIL; Pinho e Melo TMVD Synthesis and Thermal Reactivity of Thiazolo[3,4-a]Benzimidazole-2,2-Dioxides: Approach to 1H-Benzo[d]Imidazoles via Novel Benzo-2,5-Diazafulvenium Methides. Tetrahedron 2015, 71 (24), 4227–4235. 10.1016/j.tet.2015.04.079. [DOI] [Google Scholar]
- (66).Minami T; Isonaka T; Okada Y; Ichikawa J Copper (I) Salt-Mediated Arylation of Phosphinyl-Stabilized Carbanions and Synthetic Application to Heterocyclic Compounds. J. Org. Chem 1993, 58 (25), 7009–7015. 10.1021/jo00077a018. [DOI] [Google Scholar]
- (67).Xiao J; Huang Y; Song Z; Feng W Facile Catalyst-Free Synthesis of 2-Vinylquinolines via a Direct Deamination Reaction Occurring during Mannich Synthesis. RSC Adv 2015, 5 (120), 99095–99098. 10.1039/c5ra21304e. [DOI] [Google Scholar]
- (68).Zornik D; Meudtner RM; Ela Malah T; Thiele CM; Hecht S Designing Structural Motifs for Clickamers: Exploiting the 1,2,3-Triazole Moiety to Generate Conformationally Restricted Molecular Architectures. Chem. - A Eur. J 2011, 17 (5), 1473–1484. 10.1002/chem.201002491. [DOI] [PubMed] [Google Scholar]
- (69).Dong X-Y; Zhang Y-F; Ma C-L; Gu Q-S; Wang F-L; Li Z-L; Jiang S-P; Liu X-Y A General Asymmetric Copper-Catalysed Sonogashira C(Sp3)–C(Sp) Coupling. Nat. Chem 2019, 11 (12), 1158–1166. 10.1038/s41557-019-0346-2. [DOI] [PubMed] [Google Scholar]
- (70).Teshima Y; Saito M; Mikie T; Komeyama K; Yoshida H; Osaka I Dithiazolylthienothiophene Bisimide-Based π-Conjugated Polymers: Improved Synthesis and Application to Organic Photovoltaics as P-Type Semiconductor. Bull. Chem. Soc. Jpn 2020, 93 (4), 561–567. 10.1246/BCSJ.20200016. [DOI] [Google Scholar]
- (71).Neenan TX; Whitesides GM Synthesis of High Carbon Materials from Acetylenic Precursors. Preparation of Aromatic Monomers Bearing Multiple Ethynyl Groups. J. Org. Chem 1988, 53 (11), 2489–2496. 10.1021/jo00246a018. [DOI] [Google Scholar]
- (72).Colombo R; Wang Z; Han J; Balachandran R; Daghestani HN; Camarco DP; Vogt A; Day BW; Mendel D; Wipf P Total Synthesis and Biological Evaluation of Tubulysin Analogues. J. Org. Chem 2016, 81 (21), 10302–10320. 10.1021/acs.joc.6b01314. [DOI] [PubMed] [Google Scholar]
- (73).Langille NF; Dakin LA; Panek JS Sonogashira Coupling of Functionalized Trifloyl Oxazoles and Thiazoles with Terminal Alkynes: Synthesis of Disubstituted Heterocycles. Org. Lett 2002, 4 (15), 2485–2488. 10.1021/ol026099r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.