

Abstract
Colorectal cancer is the third leading cause of cancer-related death and the third most common cause of cancer. As the five-year survival with advanced metastatic colorectal cancer (mCRC) is 14%, new treatment strategies are needed. Immune checkpoint blockade, which takes advantage of an individual’s immune system to fight cancer, has an impact in the clinic; however, for CRC, it is only effective and approved for treating mismatch repair (MMR)-deficient cancer. Moreover, long-term outcomes in MMR-deficient mCRC suggest that most patients are not cured and eventually develop therapy resistance. We hypothesized that targeting TGF-β signaling may enhance immune-mediated T-cell killing by MMR-deficient CRC cells. Using GLPG-0187, an inhibitor of multiple integrin receptors and TGF-β, we demonstrate minimal cytotoxicity against MMR-deficient HCT116 or p53null HCT116 human CRC cells. GLPG-0187 promoted significant immune cell killing of the CRC cells by TALL-104 T lymphoblast cells and reduced phosphoSMAD2 in HCT116 p53-null cells either in the absence or presence of exogenous TGF-β. We observed a reduction in CCL20, CXCL5, prolactin, and TRAIL-R3, while GDF-15 was increased in TALL-104 cells treated with a T-cell activating dose of GLPG-0187 (4 µM). Our results suggest that TGF-β signaling inhibition by a general integrin receptor inhibitor may boost T-cell killing of MMR-deficient colorectal cancer cells and suggest that a combination of anti-GDF-15 in combination with TGF-β blockade be further investigated in the treatment of MMR-deficient mCRC. Our results support the development of a novel immune-based therapeutic strategy to treat colorectal cancer by targeting the TGF-β signaling pathway through integrin receptor blockade.
Keywords: TGF-β, GLPG-0187, SMAD2, T-cell killing, cancer, immunotherapy, mismatch repair-deficiency
Introduction
Chemotherapy is an established modality for treating colorectal cancer (CRC) [1]. Median overall survival for metastatic colorectal cancer nearly tripled over three decades to approximately 24 months, as documented in phase 3 clinical trials [2]. Advances in treatment over the last 20 years include the addition of oxaliplatin and irinotecan to 5-Fluorouracil, which dates back over half a century, as well as more recent targeted agents, including anti-VEGF, anti-EGFR therapies, anti-BRAF, or anti-KRAS therapies [1]. One of the significant breakthroughs in recent years has been the discovery of responsiveness of mismatch repair (MMR)-deficient CRC to immune checkpoint blockade (ICB) therapy, with subsequent FDA approval in microsatellite-high MMR-deficient colorectal cancer cells, as well as MMR-deficient tumors from other tissue origins [3-6].
While overall mortality from CRC has been declining, the side effects of chemotherapy are significant, and survival rates for more advanced stages remain below 15%. There is a necessity for newer, more effective treatment options [7-11]. Immunotherapy was initially approved for the treatment of melanoma and since then has become the standard for many other types of cancer, including lung and bladder cancers with high tumor mutation burden [12,13]. As stated above, ICB therapy has also become the standard of care for various types of cancer, including gastrointestinal-related cancer with MMR deficiency, and ICB therapies have been well tolerated.
A significant challenge with ICB therapy is emergent therapy resistance or failure to respond to treatment [14,15]. There are several contributing factors, including the inability to recruit immune cells to so-called “cold” tumors or other immune suppressive signals or cell types within the tumor microenvironment (TME). Immune-suppressive cell types include T-regulatory cells, myeloid-derived suppressor cells (MDSCs), or “exhausted” T-cells [16]. There are also immune suppressive cytokines, such as TGF-β, which contribute to the evasion of immune surveillance or tumor cell killing by immune cells [17,18].
TGF-β binds to a serine/threonine kinase receptor, followed by receptor-mediated phosphorylation of SMAD2 and SMAD3 [19,20]. Activated SMAD2 and SMAD3 then bind to SMAD4 and enter the nucleus, where the protein complex binds to DNA and regulates the transcription of effector genes [21].
GLPG-0187 is an integrin inhibitor that prevents TGF-β’s transformation from its latent to active form [22,23]. Integrin receptors are transmembrane glycoproteins that are crucial in endothelial cell adhesion and migration. GLPG-0187 functions by binding to and blocking the activity of 5 RGD-integrin receptor subtypes which include αvβ1, αvβ3, αvβ5, αvβ6, and α5β1 [24]. These effects result in the inhibition of endothelial cell-cell interactions, cell-matrix interactions, prevention of angiogenesis, and metastasis of tumor cells that express such receptors [22].
In the present studies, we tested the hypothesis that targeting TGF-β signaling may enhance immune-mediated T-cell killing by MMR-deficient CRC cells. Our results suggest that GLPG-0187 inhibits TGF-β signaling and promotes significant immune cell killing of the CRC cells by T-cells.
Materials and methods
Cell culture
HCT116 p53-/-, wild type, and TALL-104 cells were maintained in a CO2 incubator at 37 degrees Celsius. HCT116 cells were kept in McCoy’s 5A medium with 1% penicillin/streptomycin and 10% FBS. TALL-104 cells were kept in ATCC RPMI media with 20% non-heat activated FBS and 1% penicillin/streptomycin. All cell lines were authenticated and tested to ensure they were free of mycoplasma infection.
Western blots
In a 6-well plate, 500,000 HCT116 p53-/- cells per well were plated in 2 mL of media. Cells were allowed to adhere overnight, and drug doses were added. 250,000 TALL-104 cells were plated per well in a 12-well plated and allowed to adhere overnight. They were then treated with TGF-β or GLPG-0187. After a 24-hour incubation, cell pellets were collected, and lysates were further analyzed by immunoblotting. Antibodies used were phospho-SMAD2 (Cell Signaling Technology, cat # 3102) and RAN (BD Biosciences, cat # 610341).
Tumor cell-immune cell co-culture
360,000 cancer and immune cells were separately suspended in 250 µL of media. 0.5 µL of blue CMAC dye was added to the cancer cells, and 0.25 µL of Green CMFDA was added to the immune cells. Both vials of cells were incubated for one hour, then spun down and washed three times before plating. The cancer cells were plated at a confluency of 10,000 cells per well and allowed to adhere overnight. The TALL-104 cells were then added to a third of the cancer cells at a 1:1 ratio. The appropriate wells were then treated at 0.5 µM GLPG-0187, 1 µM GLPG-0187, or 2 µM GLPG-0187, and the plate was then incubated. The first row of the 48-well-plate consisted of only cancer cells at the various doses of GLPG-0187, its second row was a 1:1 ratio of T-cells and cancer cells at the different doses of GLPG-0187, and the third row consisted solely of TALL-104 T-cells at the various drug doses (Figure 1A). The wells were then imaged at 10× magnification, with two fields of view per well, at 2 hours and 24 hours post immune cell addition (Figure 1B).
Figure 1.

Plate setup for co-cultures and experimental timeline. A. Image represents the plating for the co-culture experiments. The blue represents the plating of tumor cells, while the green represents immune cells. There is a 1:1 ratio of tumor and immune cells in row B. Cells were plated in duplicates, and various doses of GLPG-0187 are listed. B. Timeline used for all the co-culture experiments.
Cytokine profiling
Using a Luminex 200 Instrument (Luminex Corporation, Austin, TX), a custom R&D systems Human Premixed Multi-Analyte Kit (R&D Systems, Inc., Minneapolis, MN; LXSAHM-60) was run according to the manufacturer’s instructions. Cell culture supernatant levels were measured, and analyte values were reported in picograms per milliliter (ρg/mL).
Results
GLPG-0187 mediated immune-killing effects of TALL-104 T-cells towards CRC cells
Co-cultures of tumor and immune cells were assessed by viability assays and fluorescence microscopy to examine the effects of GLP-0187 on each cell line and, more importantly, test whether there is a significant difference in the effect when immune cells and cancer cells were cultured together. At the different doses of the integrin inhibitor, the effect on tumor cells alone and immune cells alone was observed, and then in combination. The cells in each well were counted to determine whether there was a significant decrease in tumor cell count at the higher doses of GLPG (Figure 2A). Additionally, a one-way ANOVA was performed to compare the test wells with the control wells (Figure 2B). Representative images were taken of each well of interest to display what was taking place - where blue represents cancer cells, green immune cells, and red dead cells (Figures 3, 4). We observed a decrease in the tumor cell count and a significant increase in percent cell death, suggesting that integrin inhibitor GLPG-0187 has a potent effect on increasing immune T-cell killing of CRC cells. Similar results were observed in HCT116 p53-/- cells (Figures 5, 6), indicating that the effects of GLPG-0187 to increase T-cell killing of tumor cells was p53-independent.
Figure 2.

Co-culture of HCT116 WT cells. A. Graph of the cell count for the various treatment groups. B. One-way ANOVA graph of the percent cell death for each treatment group. The x-axis displays whether there are immune and tumor cells in the well, and if treated with GLPG-0187, the dose is indicated.
Figure 3.
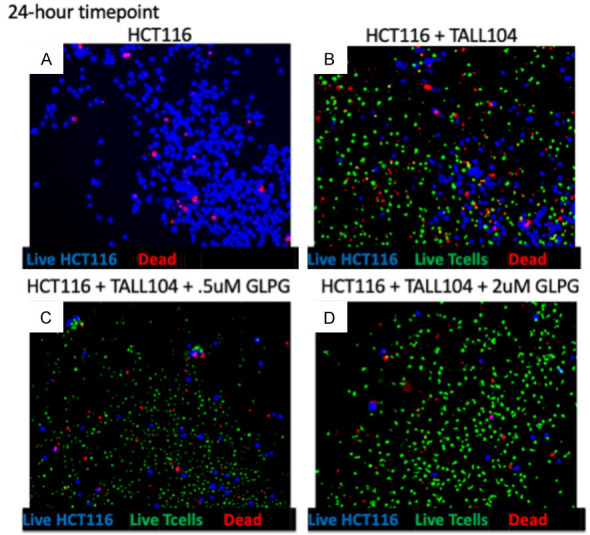
Co-culture of HCT116 WT cells: images. Representative images of treatment groups post-24-hour incubation of HCT116 WT cancer cells with GLPG-0187 and TALL-104 immune cells. A. Image of tumor cells alone. B. Image of control of tumor cells and immune cells. C. Image of low-dose GLPG-0187 treatment. D. Image of high dose GLPG-0187 treatment. Blue corresponds to live tumor cells, red signifies dead cells, and green corresponds to immune cells.
Figure 4.

Co-culture of HCT116 WT cells. This is a replicate to confirm the results of Figure 3.
Figure 5.

Co-culture of HCT116 p53-/- cells: images. A. Graph of the cell count for the various treatment groups. B. One-way ANOVA graph of the percent cell death for each treatment group. The x-axis displays whether there are immune and tumor cells in the well, and if treated with GLPG-0187, the dose is indicated.
Figure 6.

Co-culture of HCT116 p53-/- cells. Representative images of treatment groups post-24-hour incubation of HCT116 p53-/- cancer cells with GLPG-0187 and TALL-104 immune cells. A. Image of tumor cells alone. B. Image of control of tumor cells and immune cells. C. Image of low-dose GLPG-0187 treatment. D. Image of high dose GLPG-0187 treatment. Blue corresponds to live tumor cells, red signifies dead cells, and green corresponds to immune cells.
Suppression of TGF-β signaling by GLPG-0187 in CRC and TALL-104 cells
Western blots were then performed on colorectal cancer cells and immune cells to evaluate the effects of GLPG-0187 on TGF-β and p-SMAD signaling. Cell lysates, following treatment with different doses of GLPG-0187, were analyzed for alterations in pSMAD2 levels. RAN was used as a protein loading control in both cell lines’ immune blots. We evaluated a range of doses, including 1 µM, 2 µM, 4 µM, 6 µM, and 8 µM GLPG-0187 on p-SMAD2 levels. We observed that the higher the dose of GLPG-0187, the lower the intensity of the band of p-SMAD2 (Figure 7A). In the TALL-104 immune cells, lysates consisted of control and treatment groups of 100 ρM of TGF-β and 2 µM, 4 µM, and 8 µM of GLPG-0187. In the lysates treated with TGF-β, there was a significant increase in the band intensity at the molecular weight of p-SMAD2, and at higher doses of GLPG-0187, the p-SMAD2 bands were not visualized (Figure 7B).
Figure 7.

The effect of varying doses of GLPG-0187 on TGF-β pathway in HCT116 p53-/- Cells or TALL-104 Immune Cells post-24-hour Incubation. These immunoblots include cancer cells (A) or immune cells (B) at various doses of GLPG-0187. This experiment probes for pSMAD2 and uses RAN as a protein loading control.
Modulation of cytokine profiling in GLPG-0187 treated tumor and immune cells
Cytokine profiling was performed on control and 4 µM GLPG-0187 lysates to determine whether cytokine production was altered at higher doses of GLPG-0187. Heat maps of cytokine levels and the full change of 4 µM GLPG-0187 versus the control (Figure 8). Cytokines CCL4, Angiopoietin 1, and IL6 showed the most significant differences in the CRC cells (Figure 9A), and in the TALL-104 cells, cytokines CCL20, GDF-15, CXCL5, TRAILR3, prolactin, and IL2 showed the greatest alterations (Figure 9B).
Figure 8.

Cytokine levels of TALL-104 immune cells and HCT116 p53-/- cells post 24-hour incubation with GLPG-0187. A. Heat map of the various cytokines. B. Full change from the control to the high dose of GLPG treatment.
Figure 9.

Full-change of cytokines in cancer cells and immune cells treated with GLPG-0187. A. Analyte concentrations of CCL4, Angiopoietin 1, and IL6 in control versus high treatment dose of GLPG-0187 in HCTp53-/- colorectal cancer cells. B. Analyte concentrations of CCL20, GDF-15, CXCL5, TRAIL R3, Prolactin, and IL2 in control versus high treatment dose of GLPG-0187 in immune cells.
Discussion
The results all point to the conclusion that at higher doses of an integrin inhibitor in CRC cells and immune cells, there is suppression of phospho-SMAD signaling. Western blots confirmed that TGF-β signaling inhibition by a general integrin receptor inhibitor boosts T-cell killing of MMR-deficient colorectal cancer cells. Furthermore, in immune cells, by using a controlled dose and a lysate treated with TGF-β, the suppression of the phospho-SMAD signaling was observed. In the cytokine profiles, the higher dose of GLPG-0187 significantly reduced the levels of cytokines such as CCL4, Angiopoietin 1, and IL6 - which at high levels all typically correlate to a poor prognosis in CRC. The results showed that the strategy holds promise even in p53-deficient CRC cells.
Limitations of our study include the lack of in vivo experiments, the number of cell lines, and the types of ways in which TGF-β was inhibited. Given this, the future direction of this study would include experiments with CRC cell lines beyond HCT116, such as MMR-proficient cells.
Our results suggest that inhibition of integrins and TGF-β may have beneficial therapeutic effects within the immune tumor microenvironment. Our results support the development of a novel immune-based therapeutic strategy to treat colorectal cancer by targeting the TGF-β signaling through integrin receptor blockade.
Disclosure of conflict of interest
None.
References
- 1.Biller LH, Schrag D. Diagnosis and treatment of metastatic colorectal cancer: a review. JAMA. 2021;325:669–685. doi: 10.1001/jama.2021.0106. [DOI] [PubMed] [Google Scholar]
- 2.Shen C, Tannenbaum D, Horn R, Rogers J, Eng C, Zhou S, Johnson B, Kopetz S, Morris V, Overman M, Parseghian C, Chang GJ, Lopez-Olivo MA, Kanwal R, Ellis LM, Dasari A. Overall survival in phase 3 clinical trials and the surveillance, epidemiology, and end results database in patients with metastatic colorectal cancer, 1986-2016: a systematic review. JAMA Netw Open. 2022;5:e2213588. doi: 10.1001/jamanetworkopen.2022.13588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Duffy SM, Goldberg RM, de la Chapelle A, Koshiji M, Bhaijee F, Huebner T, Hruban RH, Wood LD, Cuka N, Pardoll DM, Papadopoulos N, Kinzler KW, Zhou S, Cornish TC, Taube JM, Anders RA, Eshleman JR, Vogelstein B, Diaz LA Jr. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cercek A, Lumish M, Sinopoli J, Weiss J, Shia J, Lamendola-Essel M, El Dika IH, Segal N, Shcherba M, Sugarman R, Stadler Z, Yaeger R, Smith JJ, Rousseau B, Argiles G, Patel M, Desai A, Saltz LB, Widmar M, Iyer K, Zhang J, Gianino N, Crane C, Romesser PB, Pappou EP, Paty P, Garcia-Aguilar J, Gonen M, Gollub M, Weiser MR, Schalper KA, Diaz LA Jr. PD1 blockade in mismatch repair-deficient, locally advanced rectal cancer. N Engl J Med. 2022;386:2363–2376. doi: 10.1056/NEJMoa2201445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.André T, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, Smith D, Garcia-Carbonero R, Benavides M, Gibbs P, de la Fouchardiere C, Rivera F, Elez E, Bendell J, Le DT, Yoshino T, Van Cutsem E, Yang P, Farooqui MZH, Marinello P, Diaz LA Jr KEYNOTE-177 Investigators. Pembrolizumab in microsatellite-instability-high advanced colorectal cancer. N Engl J Med. 2020;383:2207–2218. doi: 10.1056/NEJMoa2017699. [DOI] [PubMed] [Google Scholar]
- 6.Boyiadzis MM, Kirkwood JM, Marshall JL, Pritchard CC, Azad NS, Gulley JL. Significance and implications of FDA approval of pembrolizumab for biomarker-defined disease. J Immunother Cancer. 2018;6:35. doi: 10.1186/s40425-018-0342-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xie YH, Chen YX, Fang JY. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct Target Ther. 2020;5:22. doi: 10.1038/s41392-020-0116-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gustavsson B, Carlsson G, Machover D, Petrelli N, Roth A, Schmoll HJ, Tveit KM, Gibson F. A review of the evolution of systemic chemotherapy in the management of colorectal cancer. Clin Colorectal Cancer. 2015;14:1–10. doi: 10.1016/j.clcc.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 9.Zhang B, Fang C, Deng D, Xia L. Research progress on common adverse events caused by targeted therapy for colorectal cancer (review) Oncol Lett. 2018;16:27–33. doi: 10.3892/ol.2018.8651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Braun MS, Seymour MT. Balancing the efficacy and toxicity of chemotherapy in colorectal cancer. Ther Adv Med Oncol. 2011;3:43–52. doi: 10.1177/1758834010388342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72:7–33. doi: 10.3322/caac.21708. [DOI] [PubMed] [Google Scholar]
- 12.Esfahani K, Roudaia L, Buhlaiga N, Del Rincon SV, Papneja N, Miller WH Jr. A review of cancer immunotherapy: from the past, to the present, to the future. Curr Oncol. 2020;27(Suppl 2):S87–S97. doi: 10.3747/co.27.5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20:651–668. doi: 10.1038/s41577-020-0306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bai R, Chen N, Li L, Du N, Bai L, Lv Z, Tian H, Cui J. Mechanisms of cancer resistance to immunotherapy. Front Oncol. 2020;10:1290. doi: 10.3389/fonc.2020.01290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang S, Xie K, Liu T. Cancer immunotherapies: from efficacy to resistance mechanisms - not only checkpoint matters. Front Immunol. 2021;12:690112. doi: 10.3389/fimmu.2021.690112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chow A, Perica K, Klebanoff CA, Wolchok JD. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat Rev Clin Oncol. 2022;19:775–790. doi: 10.1038/s41571-022-00689-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nixon BG, Gao S, Wang X, Li MO. TGFβ control of immune responses in cancer: a holistic immuno-oncology perspective. Nat Rev Immunol. 2022;23:346–362. doi: 10.1038/s41577-022-00796-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Batlle E, Massagué J. Transforming growth factor-β signaling in immunity and cancer. Immunity. 2019;50:924–940. doi: 10.1016/j.immuni.2019.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakao A, Imamura T, Souchelnytskyi S, Kawabata M, Ishisaki A, Oeda E, Tamaki K, Hanai JI, Heldin CH, Miyazono K, ten Dijke P. TGF-β receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997;16:5353–5362. doi: 10.1093/emboj/16.17.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Massagué J. TGFbeta in cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Massagué J, Wotton D. Transcriptional control by the TGF-β/Smad signaling system. EMBO J. 2000;19:1745–1754. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Slack RJ, Macdonald SJF, Roper JA, Jenkins RG, Hatley RJD. Emerging therapeutic opportunities for integrin inhibitors. Nat Rev Drug Discov. 2022;21:60–78. doi: 10.1038/s41573-021-00284-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huntington KE, Carlsen L, So EY, Piesche M, Liang O, El-Deiry WS. Integrin/TGF-β1 inhibitor GLPG-0187 blocks SARS-CoV-2 delta and omicron pseudovirus infection of airway epithelial cells in vitro, which could attenuate disease severity. Pharmaceuticals (Basel) 2022;15:618. doi: 10.3390/ph15050618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cirkel GA, Kerklaan BM, Vanhoutte F, Van der Aa A, Lorenzon G, Namour F, Pujuguet P, Darquenne S, de Vos FY, Snijders TJ, Voest EE, Schellens JH, Lolkema MP. A dose escalating phase I study of GLPG0187, a broad spectrum integrin receptor antagonist, in adult patients with progressive high-grade glioma and other advanced solid malignancies. Invest New Drugs. 2016;34:184–192. doi: 10.1007/s10637-015-0320-9. [DOI] [PMC free article] [PubMed] [Google Scholar]