

Abstract
Approximately 20% of rheumatoid arthritis (RA) patients have RA-related interstitial lung disease (RA-ILD). Stattic, an STAT3 inhibitor, has been confirmed to be relevant to both RA and ILD. Therefore, this study explored the effect of Stattic on the progression of joint disease and pulmonary fibrosis in zymosan-treated female SKG mice, an established model for autoimmune arthritis. The experimental mice developed pulmonary interstitial pneumonia, which is similar to human cellular and fibrotic nonspecific interstitial pneumonia. Oral gavage of Stattic (60 mg/kg/d) was initiated 10 weeks after zymosan injection. Arthritis and lung fibrosis outcome scores decreased significantly following Stattic treatment. An obvious decrease in lung collagen levels, measured using hydroxyproline level determination and collagen staining, was detected after 6 weeks in Stattic-exposed mice with established disease. Stattic also dramatically restricted arthritis progression, based on joint evaluation. Transforming growth factor beta 1 (TGF-β1) is a pivotal fibrosis-causing cytokine, used here to treat myofibroblasts, thereby establishing a lung fibrosis cell model. Stattic treatment can mitigate the TGF-β1-triggered inflammatory response, myofibroblast activation, oxidative stress, and hyperproliferation by modulating the JAK1/STAT3 pathway. Our observations support a direct role of Stattic-inhibited STAT3 activation in lung fibrosis, which may be particularly relevant in the RA-ILD context.
Keywords: Interstitial lung disease, lung fibrosis, rheumatoid arthritis, Stattic, zymosan, hyperproliferation
Impact Statement
Our research aimed to explore the effect of Stattic on the progression of joint disease and pulmonary fibrosis in zymosan-treated female SKG mice, an established model for autoimmune arthritis. We found that Stattic exhibited beneficial effects in rheumatoid arthritis (RA)-related interstitial lung disease (RA-ILD) SKG mice. Treatment with this compound during disease development could mitigate joint swelling and reduce pulmonary fibrosis in human patients. These findings potentially provide a new treatment option for patients with RA-ILD.
Introduction
Rheumatoid arthritis (RA)-related interstitial lung disease (RA-ILD) occurs in systemic autoimmune disease. It is characterized by diffuse interstitial pulmonary alterations and joint lesions 1 and has an unfavorable prognosis. 2 The chance of developing this complication increases with time following a diagnosis of RA, and the median survival is approximately 2.6–3.5 years.3,4 Based on its histological characteristics, RA-ILD is classified into two main subtypes: nonspecific interstitial pneumonitis (NSIP) and usual interstitial pneumonitis (UIP);5,6 there are also relatively uncommon subtypes such as desquamative interstitial pneumonitis, organizing pneumonia, respiratory bronchiolitis-relevant ILD, pleuroparenchymal fibroelastosis, lymphocytic interstitial pneumonitis, and diffuse alveolar injury. 7 The UIP pattern, characterized by heterogeneity, is more likely to produce a fibrotic disease process accompanied by subpleural and paraseptal matrix precipitation, 8 whereas the NSIP pattern, characterized by homogeneity, is more likely to result in inflammatory infiltration accompanied by alveolar wall thickening. 9
In RA-ILD patients, RA generally develops prior to the occurrence of ILD. 6 However, ILD can also present before10,11 or concurrently with RA. 6 The pathogenesis of RA-ILD remains unclear and effective treatments are needed. Thus, it is critical to identify potential targets for RA-ILD.
To address the issue of therapeutic interventions in RA-ILD, we used a model of arthritis-prone female SKG mice established by previous studies. The model showed many RA-ILD manifestations, including lung and joint disease with 20% and 100% penetrance, respectively. In this high-authenticity model, we measured the effectiveness of Stattic, a STAT3 inhibitor, as a treatment for RA-ILD.
The STAT family consists of seven members (STAT1, 2, 3, 4, 5a, 5b, and 6), which serve as transcriptional factors. 12 The Janus kinase (JAK)/STAT molecular pathway can be activated by a wide range of profibrotic/pro-inflammatory cytokines, which are elevated in diverse ILDs. 13 Specifically, JAK2/STAT3, which induces cell changes in ILDs, predominates among the different JAK/STAT isoforms. 13 In contrast, STAT-amplified interleukin (IL)-6 signaling and/or persistent infections are associated with the chronic inflammation observed in RA. 14 A previous study showed that STAT3 exerts its effects through a positive feedback loop that promotes the expression of inflammatory cytokines, resulting in osteoclastogenesis and concomitant inflammation, which are necessary precursors to joint destruction. 15 JAK/STAT pathway is an attractive target to be proven in future clinical trials of lung fibrotic disorders. 13 The JAK/STAT molecular pathway is activated under the interaction of a broad number of profibrotic/pro-inflammatory cytokines, such as IL-6, IL-11, and IL-13, among others, which are increased in different ILDs. 13 Wang et al. 16 reported that RNA sequencing of lung biopsies from patients with RA-ILD and idiopathic pulmonary fibrosis, and suggested STAT signaling pathway gene signature can distinguish RA-interstitial pneumonia from idiopathic pulmonary fibrosis. Ruan et al. 17 demonstrated that fedratinib inhibited inflammation and fibrosis processes during bleomycin-induced pulmonary fibrosis by inducing tumor growth factor (TGF)-β1 and IL-6 by targeting the JAK2 receptor. These reports have indicated that JAK/STAT pathway is essential for RA-ILD progression. However, further investigation is required to determine whether STAT3 is a potential target for RA-ILD therapy.
Homozygous SKG mice exhibit a spontaneous point mutation in the second SH2 domain of ZAP70, causing hypomorphic ZAP70 function in T-cells. 18 Decreased ZAP70 function results in damage to the negative and positive CD4+ T-cell selection process in the thymus, permitting the escape of self-reactive CD4+ T-cells to the periphery. 18 In mice residing in specific non-pathogenic conditions, intraperitoneal injection of zymosan, an innate β-glucan agonist, can activate this pathway, causing RA-ILD clinical phenotype development.19,20
Here, oral administration of Stattic was used to deactivate the STAT3 sensor in an in-vivo RA-ILD animal model and an in-vitro fibrosis cell model. Further study confirmed inhibitory effects on joint disease and pulmonary fibrosis progression in SKG mice with zymosan-induced arthritis, and on fibrosis activation in TGF-β1-induced mouse fibroblast NIH3T3 cells. Collectively, these results confirm that Stattic could act against fibrosis and may exhibit direct clinical effects.
Materials and methods
Arthritis and pulmonary disease induction
SKG mice (female, 8–10 weeks, n = 30) was purchased from Hunan Experimental Animal Center. Animals were randomly divided into three groups with 10 mice in each group. Except for negative control mice (n = 10) who received normal saline administration, SKG mice (n = 20) were intraperitoneally injected with zymosan (5 mg) to elicit ILD and arthritis, as described previously. 20 All experiments were approved by the Animal Care and Use Committee (The First Affiliated Hospital, Hengyang Medical School, University of South China). Fifty mice were injected per treatment condition to accommodate the 20% penetrance of ILD. The animal experiment was repeated for three times.
Stattic treatment
The ILD mice (n = 10) were subjected to daily gavage administration of Stattic (25 mg/kg in normal saline) for 6 weeks, starting 10 weeks after the zymosan injection. Control mice were administered saline by gavage.
Evaluation of lung pathologic change
A small-animal ventilator (SCIREQ, Montreal, QC, Canada) was used to evaluate lung mechanics and quasi-static compliance (Cst). 21
Changes in lung collagen were determined by measuring the hydroxyproline levels in the upper right lobe after phosphate-buffered saline (PBS) homogenization and hydrolysis in the same volume of HCl (12 N) at 120°C for 8 h. 22
A 10% formalin solution was used to fix each excised left lung, which was then embedded in paraffin and sectioned (5 μm). 22 Serial sections from three mice with a hydroxyproline score lower than 1 standard deviation (SD) below the average value in each group were subjected to hematoxylin and eosin (H&E) staining and observed using a microscope (BX51; Olympus, Tokyo, Japan).
Lung sections were immunostained for alpha smooth muscle actin (α-SMA) (1:1,000 dilution) and evaluated using a semi-quantitative stereology grid count, as described previously. 21 A blinded evaluation was conducted using 10 mice per group and 10 images per mouse.
Joint disease evaluation
The arthritis score was calculated weekly 18 based on visible joint swelling. The severity scores given were: 0, no joint swelling; 0.1, one-finger joint swelling; 0.5, mild wrist or ankle swelling; 0.75, moderate wrist or ankle swelling; 1.0, serious wrist or ankle swelling. The scores for the wrists, ankles, and fingers were totaled. Arthritis was detected and scored by two independent investigators who were blinded to the treatment groups.
Hind joints displaying a mean arthritis score the same as the reported group average score (n = 10 mice/group; early treatment group [5–11 weeks after zymosan injection]) were scanned using micro computed tomography (μ-CT), generating cross-sectional images with an isotropic voxel size of 9 μm. The acquisition parameters for the μ-CT were as follows: 50 kV X-ray tube voltage, 500 μA current, 0.900 s exposure time, 0.5 mm aluminum filter, and 0.4° rotation step. The bone in the images was identified by setting an image threshold via a consistent value (pixel intensity of 75). The calcaneus and talus bones were manually separated from the threshold regions. 23 Bone volumes were obtained by multiplying the voxel number in each segmented area by the voxel volume.
Western blotting (WB)
Whole-cell lysates and tissue homogenates were obtained using RIPA buffer (1% Tween 20, 0.1% SDS, 150 mmol/L NaCl, 10 mmol/L Tris-HCl (pH 7.4), 0.25 mmol/L phenylmethylsulfonylfluoride, 1 mmol/L Na3VO4, 5 mmol/L NaF, 10 μg/mL aprotinin, and 10 μg/mL leupeptin). Equal quantities of homogenate were processed using SDS-PAGE and PVDF membrane transfer to isolate component proteins. These were identified using relevant primary and secondary antibodies. AlphaView SA software was used to scan the blots.
ELISA analysis
Lung tissue homogenates were centrifuged for 20 min at 1000g prior to ELISA analysis. The concentrations of inflammatory factors were measured using ELISA kits, as per relevant guidance.
qPCR
Total RNA was separated from NIH3T3 cells and lung tissue homogenates using TRIzol reagent and reverse transcribed to obtain cDNA. The qPCR analysis was conducted in accordance with the manufacturer’s instructions. Relative gene expression was determined using the comparative CT method with GAPDH as the reference.
Cell cultivation
NIH3T3 cells were bought from ATCC and cultivated in Dulbecco’s modified Eagle’s medium plus 10% fetal bovine serum. The cells were maintained at 37°C in a humidified atmosphere with 5% CO2. For fibrosis activation, NIH3T3 cells were cultured using 5 ng/mL TGF-β1 for 0.5 days. For Stattic treatment, NIH3T3 cells were cultivated using 2 μM Stattic and 5 ng/mL TGF-β1 for 0.5 days.
Reactive oxygen species determination
Reactive oxygen species (ROS) production was determined using a DCFH-DA fluorescence probe. Cells were cultivated using Propionibacterium acnes for the indicated times and subsequently further incubated with 10 μM DCFH-DA at 37°C for 0.5 h in the dark. Fluorescence intensity (Ex 488/Em 525 nm) was determined using a fluorescence microscope (Olympus BX51, Tokyo, Japan).
Cell survival
The MTT assay was used to measure cell viability. After inoculation onto 96-well plates, NIH3T3 cells were treated with Stattic (2 µM) for 1 day. MTT (10 µL, 5 mg/mL) was then added, and 4 h later, DMSO (120 µL) was added. Absorbance was measured at 570 nm.
Colony formation
After cultivation in 6-well plates for 7 days, the cells were fixed in 4% formaldehyde for 20 min and subjected to 1.0% crystal violet staining.
Flow cytometry
Apoptosis was determined through flow cytometry (FCM). Cells that had been cultivated for 2 h following treatment were harvested and washed three times with PBS. FCM was performed according to the relevant instructions to identify the proportions of cells in each cell cycle phase and of apoptotic cells. The proportion of apoptotic cells was approximated using double staining with annexin V-FITC/PI. The final proportion was determined using the FACSDiva software to analyze the flow cytometry results.
Data analysis
Quantitative results from at least three separate experiments were analyzed using GraphPad Prism 7 and are displayed as means ± SD. One-way analysis of variance (ANOVA) followed by the Tukey–Kramer test was used to identify significant differences between groups. Statistical significance was set at p < 0.05.
Results
Influence of Stattic on established joint swelling
In our animal model, following intraperitoneal injections of zymosan, joint swelling occurred after 2–3 weeks in 85% of mice, and interstitial pneumonia developed in 20% of mice, by 10 weeks. All mice per treatment group were evaluated to accommodate the low penetrance of lung disease.
To confirm the influence of Stattic on pulmonary and joint disease, mice were orally administered saline or Stattic starting 10 weeks after zymosan injection (Figure 1(A)). Stattic administration restored the individual weight and group survival percentage of mice compared with those in saline-treated mice (Figure 1(B) and (C)).
Figure 1.

Stattic treatment slowed the development of arthritis in SKG mice: (A) Daily oral administration of saline or Stattic began 10 weeks after zymosan injection. (B, C) Changes in weight and survival in mice. (D) The visual arthritis score decreased dramatically after 1 week of Stattic treatment relative to that in control (saline-exposed) animals.
*p < 0.05.
Joint swelling, evaluated by means of a visual arthritis score, was first observed 2–4 weeks after zymosan injection and worsened progressively up to a severity peak after 6–12 weeks. Thereafter, the joint swelling severity decreased but was still evident after 16 weeks. Stattic administration led to less joint swelling than was seen in saline-treated mice (Figure 1(D)).
Influence of Stattic on established interstitial pneumonia in SKG mice
Cst was modestly, but significantly, lower in zymosan-exposed mice relative to that in control mice, but despite this, Stattic administration resulted in significant improvement in Cst in treated, but not control, mice (Figure 2(A)). The collagen level analysis in lungs, obtained using hydroxyproline determination, showed that daily zymosan treatment for 6 weeks did produce an obvious increase in lung fibrosis. The hydroxyproline levels in the lungs of Stattic-exposed mice were lower than those in saline-treated mice (Figure 2(B)).
Figure 2.

Effect of Stattic treatment on lung compliance, collagen levels, pathological lung fibrosis, and inflammatory cytokine levels. (A) Quasi-static compliance in negative control mice, and in mice injected with zymosan then treated with either Stattic or saline (positive controls). (B) Hydroxyproline levels in negative control mice, and in mice injected with zymosan then treated with either Stattic or saline (positive control). * (C) hematoxylin- and eosin-stained lung sections showed patchy regions of disease and inflammation, Scale bar, 200 μm. (D, E) Immunohistochemical staining showing α-SMA-positive fibroblasts (final magnification, ×400) and semi-quantitative analysis of α-SMA-positive cells, scale bar, 200 μm. (F) Protein levels of α-SMA, Fn, Col1, STAT3, and phosphorylated STAT3 in lung tissue. (G to J) BAL fluid levels of TNF-α, IFN-γ, IL-1β, and IL-6 in mice, as assessed by ELISA.
*p < 0.05.
**p < 0.01.
Effect of Stattic on lung fibrosis in SKG mice
The lung histology of animals treated with zymosan displayed a typical pattern of patchy subpleural and peribronchovascular inflammatory cell infiltration, as assessed by H&E staining. In Stattic-treated mice, the pathogenic changes of established lung fibrosis were ameliorated compared with those in the saline-treated group (Figure 2(C)).
Lung tissue staining and semi-quantitation of the pan-fibroblast marker α-SMA showed an obvious decrease in fibroblasts following Stattic administration, relative to that seen under saline treatment (Figure 2(D)). The proportion of α-SMA-positive cells in mice treated with Stattic was lower than that in control mice (Figure 2(E)). WB assays also confirmed that zymosan treatment increased α-SMA, Fn, and Col1 expression levels in lung tissue, while Stattic administration led to the downregulation of these three fibrotic markers (Figure 2(F)).
Influence of Stattic on established pulmonary inflammation
The relevance of Stattic to lung inflammation has been confirmed in the pulmonary fibrosis bleomycin model. 24 We hypothesized that Stattic would have a similar effect in our SKG model. IL-1β, IL-6, tumor necrosis factor (TNF)-α, and interferon (IFN)-γ levels of the four inflammatory molecules were dramatically elevated in the zymosan-only group, based on analysis of the pro-inflammatory cytokine signature in bronchoalveolar lavage (BAL) fluid, whereas the Stattic-treated group displayed alleviation of cytokine overproduction (Figure 2(G) to (J)).
Influence of Stattic on joint disease development
To further evaluate the effect of Stattic on joint disease development, the hind calcaneus and talus bones were examined using μ-CT and bone volume and remodeling were investigated. The bone volumes in Stattic-exposed mice were dramatically greater than those in control mice (Figure 3(A) and (B)).
Figure 3.
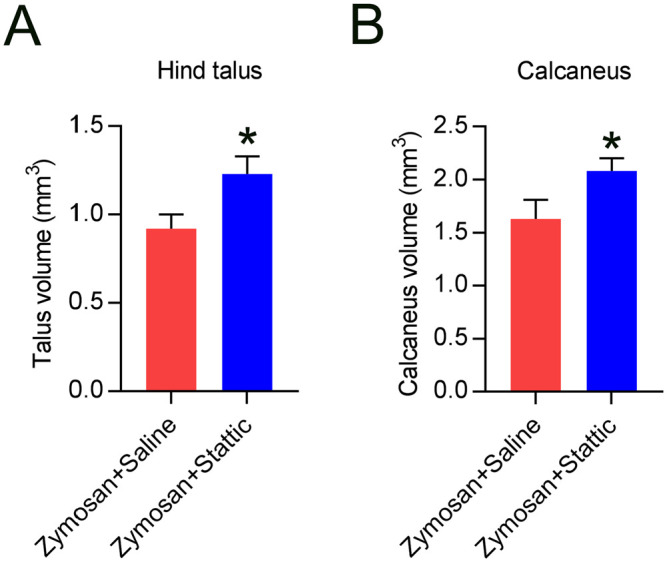
Stattic treatment can alleviate bone loss and remodeling in SKG mice. (A, B) The volumes of hind calcaneus and talus bones were notably greater following Stattic treatment. The results were obtained at week 10.
*p < 0.05.
Stattic repressed TGF-β1-elicited myofibroblast activation, inflammation, proliferation, and apoptosis resistance
To confirm whether Stattic affects the proliferation of fibroblasts induced by TGF-β1, NIH3T3 cells cultured with and without 5 ng/mL TGF-β1 and Stattic for 12 h. We observed an inhibitory effect of Stattic on TGF-β1-elicited myofibroblast hyperactivation. These findings indicate that Stattic could ameliorate the TGF-β1-induced increases in the mRNA and protein levels of the main profibrotic factors. This suggests the possibility of decreased extracellular matrix (ECM) precipitation (Figures 4(A) and 6(B)). Meanwhile, the administration of Stattic contributed to the deactivation of STAT3 (Figure 4(B)).
Figure 4.

Stattic incubation inhibited TGF-β1-elicited myofibroblast activation in NIH3T3 cells. (A to C) Levels of α-SMA, Fn, and Col1 mRNA in cells, from the qPCR analysis. (D) Levels of α-SMA, Fn, Col1, and STAT3 (with and without phosphorylation) in cells, from western blotting analysis.
*p < 0.05.
**p < 0.01.
Figure 6.

Stattic treatment suppressed TGF-β1-elicited hyperproliferation and apoptosis resistance in NIH3T3 cells. (A) The cell survival rate as determined by MTT assay. (B) The cell growth rate was assessed using a colony formation assay. (C) Flow cytometry with annexin V and propidium iodide stain showing the apoptotic cell proportion in populations of NIH3T3 cells. (D) Levels of cleaved caspase-3, Bcl2, and Bax proteins under each treatment.
*p < 0.05.
Inflammatory responses accelerate the development of fibrosis. Thus, the influence of Stattic on TGF-β1-elicited inflammation and oxidative stress was determined. ELISA data showed that Stattic administration inhibited the release of TNF-α, IL-1β, and IL-6, which were induced by TGF-β1 (Figure 5(A) to (C)). Meanwhile, we found that TGF-β1 treatment elicited oxidative stress in cells, as evidenced by upregulated ROS levels, whereas ROS levels in the Stattic treatment group were significantly lower (Figure 5(D)). Nrf2/HO-1 pathway is a well-documented anti-oxidative axis in many diseases;25 –29 therefore, we chose them as representative of antioxidant genes. WB analysis revealed that levels of both proteins were lower in cells exposed to TGF-β1, while Stattic treatment increased the expression levels of Nrf2 and HO-1 (Figure 5(E)).
Figure 5.

Stattic treatment counteracted TGF-β1-elicited inflammation and oxidative stress in NIH3T3 cells. (A to C) ELISA results showing the quantities of inflammatory cytokines released in cells. (D) DC-FDA staining showing cellular reactive oxygen species levels in NIH3T3 cells under each treatment. (E) Levels of Nrf2 and HO-1 proteins as assessed by western blotting.
*p < 0.05.
**p < 0.01.
We then examined whether Stattic administration plays a role in TGF-β1-stimulated hyperproliferation and apoptosis repression. The MTT assay showed a lower cell survival rate in the Stattic group than in TGF-β1-stimulated cells (Figure 6(A)). Colony-generation assay data showed that TGF-β1 induced a high myofibroblast growth rate, which was abolished by Stattic treatment (Figure 6(B)). The effect of Stattic on apoptosis was also investigated. TGF-β1 stimulation decreased the proportion of apoptotic cells, whereas Stattic administration dramatically increased the proportion of apoptotic cells (Figure 6(C)). TGF-β1-exposed NIH3T3 cells exhibited an anti-apoptotic phenotype, indicated by an elevated Bcl-2 level and lower cleaved caspase-3 and Bax levels; this trend was counteracted by co-administration of Stattic (Figure 6(D)). These findings indicate that Stattic is capable of repressing TGF-β1-elicited myofibroblast activation, inflammation, hyperproliferation, and apoptosis resistance by suppressing STAT3 signaling.
Discussion
This study aimed to conduct a preclinical assessment of Stattic, an inhibitor of STAT3, in a well-documented murine model of RA-ILD and a fibrosis cell model. We observed that daily oral Stattic administration in RA-ILD mice ameliorated joint swelling, joint disease, pulmonary fibrosis, and inflammation in zymosan-exposed SKG mice by deactivating STAT3 phosphorylation (Figure 7). In-vitro experiments showed that Stattic represses TGF-β1-triggered myofibroblast activation, inflammation, hyperproliferation, and apoptosis resistance via STAT3 modulation.
Figure 7.

Schematic diagram. Daily Stattic administration in RA-ILD mice ameliorated joint swelling, joint disease, pulmonary fibrosis, and inflammation in zymosan-exposed SKG mice by deactivating STAT3 phosphorylation.
Mice suffering from lung disease were exposed daily to Stattic for 6 weeks; by the end of this treatment, fibrosis and inflammation were dramatically decreased. Lung compliance, as represented by Cst, also improved after Stattic treatment. A previous study has shown that the use of nintedanib can affect the proportions of certain cell subpopulations. The numbers of lymphocytes, BAL neutrophils, and inflammatory macrophages in lung tissues were elevated in nintedanib-exposed mice, showing decreased fibrosis. 30 Reduced fibrosis was relevant to a decrease in an M2/alternative macrophage phenotype.22,31 Our results are limited, because the proportions of cell types, such as CD4+ and CD8+ T-cells, and macrophages were not recorded. However, it was clear that administration of Stattic alleviated bleomycin-induced fibrosis, causing a decrease in α-SMA positive (myo) fibroblasts. Similar results were obtained in the WB analysis. In the BAL fluid of mice, administration of Stattic was related to an obvious decrease in the production of inflammatory cytokines.
In addition to its influence on lung function, we also evaluated the influence of Stattic on arthritis development and joint swelling in zymosan-exposed SKG mice. Joint swelling in mice with established disease exposed to Stattic for 6 weeks showed obvious improvement. Evaluation of joints using μ-CT revealed clearly that reduction in bone volume was alleviated in the hind calcaneus and talus bones by Stattic treatment. These results indicate that Stattic treatment could prevent deterioration and alleviate disease in patients with RA. Clinical trials involving newly diagnosed RA patients with or without high-resolution CT are warranted to confirm whether Stattic could prevent joint and lung disease development.
JAK2/STAT3 signaling is generally thought to be essential for aberrant, TGF-β1-elicited fibroblast-myofibroblast transition and epithelial cell wound healing. 24 Here, our in-vitro cell-based experiment showed that Stattic inhibited abnormal TGF-β1-elicited myofibroblast activation, inflammation, and apoptosis resistance. TGF-β1 activation has been reported to play a major part in modulating tissue fibrosis. TGF-β1-elicited myofibroblast activation and anti-apoptotic phenotypes are involved in the generation of fibrotic foci and in ECM precipitation. 32 In our study, Stattic incubation repressed TGF-β1-elicited myofibroblast activation, as evidenced by downregulated α-SMA, Fn, and Col1 expression at both the mRNA and protein levels. ELISA and DH-FDA staining showed that TGF-β1 induced inflammation and oxidative stress in NIH3T3 cells, while Stattic abolished this effect. MTT and colony formation assays showed that Stattic treatment reduced the TGF-β1-elicited abnormal proliferation of cells. This phenomenon could be attributed to Stattic-associated apoptosis repression, as evaluated by FCM and the expression of Bcl-2, Bax, and cleaved caspase-3. These data confirm the role of Stattic in treating fibrotic diseases at the cellular level.
In summary, Stattic exhibited beneficial effects in RA-ILD SKG mice. Treatment with this compound during disease development could mitigate joint swelling and reduce pulmonary fibrosis in human patients. Stattic and its target STAT3, are therefore promising targets for preventing lung function decline and alleviating joint swelling, potentially providing a new treatment option for patients with RA-ILD.
Footnotes
Authors’ Contributions: LHX and XPL designed experiments. YYL and WTT carried out experiments. QXZ and CL analyzed experimental results. LHX wrote the manuscript. XPL revised the manuscript. All authors approved the final manuscript.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by The Instructional Planning Project of Hengyang City (grant nos. 202121034626 and 202121034627); The Construction of an Innovative Province with a Special Emergency Response Topic (grant no. 2020SK3010) in 2020; Fund Project of University of South China for Prevention and Control of COVID-19 (grant no. 2020-27) and Scientific Research Project of Hunan Provincial Health Commission (grant no. 20200509).
ORCID iD: Xiaoping Long  https://orcid.org/0000-0001-6159-5544
https://orcid.org/0000-0001-6159-5544
References
- 1. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet 2016; 388:2023–38 [DOI] [PubMed] [Google Scholar]
- 2. Solomon JJ, Chung JH, Cosgrove GP, Demoruelle MK, Fernandez-Perez ER, Fischer A, Frankel SK, Hobbs SB, Huie TJ, Ketzer J, Mannina A, Olson AL, Russell G, Tsuchiya Y, Yunt ZX, Zelarney PT, Brown KK, Swigris JJ. Predictors of mortality in rheumatoid arthritis-associated interstitial lung disease. Eur Respir J 2016;47:588–96 [DOI] [PubMed] [Google Scholar]
- 3. Bongartz T, Nannini C, Medina-Velasquez YF, Achenbach SJ, Crowson CS, Ryu JH, Vassallo R, Gabriel SE, Matteson EL. Incidence and mortality of interstitial lung disease in rheumatoid arthritis: a population-based study. Arthritis Rheum 2010;62:1583–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hakala M. Poor prognosis in patients with rheumatoid arthritis hospitalized for interstitial lung fibrosis. Chest 1988;93:114–8 [DOI] [PubMed] [Google Scholar]
- 5. Oliveira DS, Araújo Filho JA, Paiva AFL, Ikari ES, Chate RC, Nomura CH. Idiopathic interstitial pneumonias: review of the latest American Thoracic Society/European Respiratory Society classification. Radiol Bras 2018;51:321–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee HK, Kim DS, Yoo B, Seo JB, Rho JY, Colby TV, Kitaichi M. Histopathologic pattern and clinical features of rheumatoid arthritis-associated interstitial lung disease. Chest 2005;127:2019–27 [DOI] [PubMed] [Google Scholar]
- 7. Tansey D, Wells AU, Colby TV, Ip S, Nikolakoupolou A, du Bois RM, Hansell DM, Nicholson AG. Variations in histological patterns of interstitial pneumonia between connective tissue disorders and their relationship to prognosis. Histopathology 2004;44:585–96 [DOI] [PubMed] [Google Scholar]
- 8. Veeraraghavan S, Nicholson AG, Wells AU. Lung fibrosis: new classifications and therapy. Curr Opin Rheumatol 2001;13:500–4 [DOI] [PubMed] [Google Scholar]
- 9. Katzenstein AL, Myers JL. Nonspecific interstitial pneumonia and the other idiopathic interstitial pneumonias: classification and diagnostic criteria. Am J Surg Pathol 2000;24:1–3 [DOI] [PubMed] [Google Scholar]
- 10. Kolfenbach JR, Deane KD, Derber LA, O’Donnell CI, Gilliland WR, Edison JD, Rosen A, Darrah E, Norris JM, Holers VM. Autoimmunity to peptidyl arginine deiminase type 4 precedes clinical onset of rheumatoid arthritis. Arthritis Rheum 2010;62:2633–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brusca SB, Abramson SB, Scher JU. Microbiome and mucosal inflammation as extra-articular triggers for rheumatoid arthritis and autoimmunity. Curr Opin Rheumatol 2014;26:101–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Owen KL, Brockwell NK, Parker BS. JAK-STAT signaling: a double-edged sword of immune regulation and cancer progression. Cancers 2019;11:2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Montero P, Milara J, Roger I, Cortijo J. Role of JAK/STAT in interstitial lung diseases; molecular and cellular mechanisms. Int J Mol Sci 2021;22:6211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Murakami M, Hirano T. A four-step model for the IL-6 amplifier, a regulator of chronic inflammations in tissue-specific MHC class II-associated autoimmune diseases. Front Immunol 2011;2:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mori T, Miyamoto T, Yoshida H, Asakawa M, Kawasumi M, Kobayashi T, Morioka H, Chiba K, Toyama Y, Yoshimura A. IL-1β and TNFα-initiated IL-6-STAT3 pathway is critical in mediating inflammatory cytokines and RANKL expression in inflammatory arthritis. Int Immunol 2011;23:701–12 [DOI] [PubMed] [Google Scholar]
- 16. Wang S, Liu M, Li X, Zhang J, Wang F, Zhang C, Roden A, Ryu JH, Warrington KJ, Sun J, Matteson EL, Tschumperlin DJ, Vassallo R. Canonical and noncanonical regulatory roles for JAK2 in the pathogenesis of rheumatoid arthritis-associated interstitial lung disease and idiopathic pulmonary fibrosis. FASEB J 2022;36:e22336 [DOI] [PubMed] [Google Scholar]
- 17. Ruan H, Luan J, Gao S, Li S, Jiang Q, Liu R, Liang Q, Zhang R, Zhang F, Li X. Fedratinib attenuates bleomycin-induced pulmonary fibrosis via the JAK2/STAT3 and TGF-β1 signaling pathway. Molecules 2021; 26:4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sakaguchi N, Takahashi T, Hata H, Nomura T, Tagami T, Yamazaki S, Sakihama T, Matsutani T, Negishi I, Nakatsuru S, Sakaguchi S. Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature 2003;426:454–60 [DOI] [PubMed] [Google Scholar]
- 19. Keith RC, Powers JL, Redente EF, Sergew A, Martin RJ, Gizinski A, Holers VM, Sakaguchi S, Riches DW. A novel model of rheumatoid arthritis-associated interstitial lung disease in SKG mice. Exp Lung Res 2012;38:55–66 [DOI] [PubMed] [Google Scholar]
- 20. Yoshitomi H, Sakaguchi N, Kobayashi K, Brown GD, Tagami T, Sakihama T, Hirota K, Tanaka S, Nomura T, Miki I, Gordon S, Akira S, Nakamura T, Sakaguchi S. A role for fungal {beta}-glucans and their receptor Dectin-1 in the induction of autoimmune arthritis in genetically susceptible mice. J Exp Med 2005;201:949–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Redente EF, Jacobsen KM, Solomon JJ, Lara AR, Faubel S, Keith RC, Henson PM, Downey GP, Riches DW. Age and sex dimorphisms contribute to the severity of bleomycin-induced lung injury and fibrosis. Am J Physiol Lung Cell Mol Physiol 2011;301:L510–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Redente EF, Keith RC, Janssen W, Henson PM, Ortiz LA, Downey GP, Bratton DL, Riches DW. Tumor necrosis factor-α accelerates the resolution of established pulmonary fibrosis in mice by targeting profibrotic lung macrophages. Am J Respir Cell Mol Biol 2014;50:825–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yushkevich PA, Piven J, Hazlett HC, Smith RG, Ho S, Gee JC, Gerig G. User-guided 3D active contour segmentation of anatomical structures: significantly improved efficiency and reliability. Neuroimage 2006;31: 1116–28 [DOI] [PubMed] [Google Scholar]
- 24. Ruan H, Luan J, Gao S, Li S, Jiang Q, Liu R, Liang Q, Zhang R, Zhang F, Li X, Zhou H, Yang C. Fedratinib attenuates bleomycin-induced pulmonary fibrosis via the JAK2/STAT3 and TGF-β1 signaling pathway. Molecules 2021;26:4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Amata E, Pittalà V, Marrazzo A, Parenti C, Prezzavento O, Arena E, Nabavi SM, Salerno L. Role of the Nrf2/HO-1 axis in bronchopulmonary dysplasia and hyperoxic lung injuries. Clinical Science 2017;131:1701–12 [DOI] [PubMed] [Google Scholar]
- 26. Loboda A, Damulewicz M, Pyza E, Jozkowicz A, Dulak J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci 2016;73: 3221–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Minj E, Yadav RK, Mehan S. Targeting abnormal Nrf2/HO-1 signaling in amyotrophic lateral sclerosis: current insights on drug targets and influences on neurological disorders. Curr Mol Med 2021;21:630–44 [DOI] [PubMed] [Google Scholar]
- 28. Saha S, Buttari B, Panieri E, Profumo E, Saso L. An overview of Nrf2 signaling pathway and its role in inflammation. Molecules 2020;25:5474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang Y, Gao L, Chen J, Li Q, Huo L, Wang Y, Wang H, Du J. Pharmacological modulation of Nrf2/HO-1 signaling pathway as a therapeutic target of Parkinson’s disease. Front Pharmacol 2021;12:757161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Redente EF, Aguilar MA, Black BP, Edelman BL, Bahadur AN, Humphries SM, Lynch DA, Wollin L, Riches DWH. Nintedanib reduces pulmonary fibrosis in a model of rheumatoid arthritis-associated interstitial lung disease. Am J Physiol Lung Cell Mol Physiol 2018;314:L998–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 2016;44:450–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, Pradere JP, Schwabe RF. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun 2013;4:2823. [DOI] [PMC free article] [PubMed] [Google Scholar]