

Abstract
Despite the extensive body of research, understanding the exact molecular mechanisms governing inflammatory bowel diseases (IBDs) still demands further investigation. Transforming growth factor–β1 (TGF-β1) signaling possesses a multifacial effect on a broad range of context-dependent cellular responses. However, long-term TGF-β1 activity may trigger epithelial–mesenchymal transition (EMT), followed by fibrosis. This study aimed to determine the role of epithelial TGF-β1 signaling in inflammatory bowel disease (IBD) pathogenesis. The expression of TGF-β1 signaling components and EMT-related and epithelial tight junction markers was examined in IBD patients (n = 60) as well as LPS-induced Caco-2/RAW264.7 co-culture model using quantitative real-time polymerase chain reaction (qRT-PCR), Western blotting, and immunofluorescence staining. Furthermore, the effect of A83-01, as a TGF-β receptor I (TβRI) inhibitor, on the inflamed epithelial cells was evaluated in vitro. To evaluate the cytotoxic effects of the TβRI inhibitor, a cell viability assay was performed by the MTS method. Considering the activation of canonical and non-canonical TGF-β1 signaling pathways in IBD patients, expression results indicated that administering A83-01 in inflamed Caco-2 cells substantially blocked the expression level of TGF-β1, SMAD4, and PI3K and the phosphorylation of p-SMAD2/3, p-AKT, and p-RPS6 as well as prevented downregulation of LncGAS5 and LncCDKN2B. Further analysis revealed that the inhibition of TGF-β1 signaling in inflamed epithelial cells by the small molecule could suppress the EMT-related markers as well as improve the expression of epithelial adherens and tight junctions. Collectively, these findings indicated that the inhibition of the TGF-β1 signaling could suppress the induction of EMT in inflamed epithelial cells as well as exert a protective effect on preserving tight junction integrity. There is a pressing need to determine the exact cellular mechanisms by which TGF-β1 exerts its effect on IBD pathogenesis.
Keywords: Inflammatory bowel disease, TGF-β signaling, Epithelial–mesenchymal transition, PI3K signaling, A83-01, Long noncoding RNAs
Impact Statement.
Considering previous efforts, the underlying mechanisms of inflammatory bowel disease (IBD) development are still not fully understood. According to the literature, the multifunctional transforming growth factor–β1 (TGF-β1) signaling is a complicated pathway associated with the development of inflammation diseases, including IBD, as well as virtually targeting all cell types residing in the gut, including leukocytes, stromal cells, and epithelial cells. Consequently, we assessed the TGF-β1 signaling activity in mucosal cells of active lesions in patients suffering from IBD, followed by the inhibition of TGF-β1 signaling in an in vitro co-culture model. We showed that the TGF-β receptor I (TβRI) inhibitor could suppress the canonical and PI3K/AKT (non-canonical) TGF-β1 pathways, leading to the prevention of the epithelial–mesenchymal transition (EMT) initiation as a hallmark of IBD. In addition, the TβRI inhibitor exerts a protective effect on the tight junction impairment. We suggested that TGF-β1 signaling suppression could be a novel therapeutic target to manage IBD.
Introduction
Inflammatory bowel disease (IBD), classified into two main entities, namely, ulcerative colitis (UC) and Crohn’s disease (CD), remains a lifelong disease predominantly observed in industrialized countries and is thought to be triggered by multiple environmental factors in genetically susceptible individuals. 1 An impaired mucosal barrier, along with disturbed commensal gut flora, results in the intestinal immune system dysregulation. Xenobiotic receptors are chemical-sensing transcription factors that play crucial roles in the transcriptional regulation of intestinal inflammation, and dysregulation of xenobiotic sensing is correlated to an increased risk of IBD. 2 Although notable progress has been made in understanding IBD development in recent decades, and a great number of investigations have been conducted to interfere with IBD, the exact molecular mechanism underlying IBD remains intractable. 3
Transforming growth factor–β (TGF-β), a multifunctional signaling protein whose receptors are widely expressed, has wide-ranging effects on a variety of cell types, especially, although not exclusively, immune cells and epithelial cells. 4 There are three TGF-β sub-family members, namely, TGF-β1, β2, and β3, among which TGF-β1 is the most abundant isoform in most tissues, particularly immune system. According to previous studies, TGF-β1 is generally increased in response to injury and stress and by inflammation induction, while TGF-β2 and TGF-β3 are mostly involved in the regulation of developmental processes. 5 Furthermore, TGF-β1 is strongly found in both developing embryos and adults, and involved in widespread multifocal inflammatory diseases, whereas both TGF-β2 null mice and TGF-β3 null mice could not survive after birth due to the severe developmental defects in the heart, lung, and ear.6 –8
There are contradictory studies about TGF-β1 function which is thought to depend on TGF-β1-secreting tissue.4,9 Accumulating evidence demonstrates that TGF exerts a dual effect on different types of cancers and inflammatory disorders. There are several controversial issues regarding the effect of TGF-β1 on IBD pathogenesis. Early work carried out by Babyatsky et al. 10 reported that gut mucosa from patients with UC and CD displays a high level of TGF-β1. Furthermore, TGF-β expression was shown to be higher in blood samples of active IBD patients. 11 Contrarily, Monteleone et al. 12 implicated that phospho-SMAD3 in mucosa lamina propria mononuclear cells are significantly downregulated than those found in normal controls. Indeed, TGF-β1 can activate both SMAD-dependent (canonical) and SMAD-independent (non-canonical) signaling pathways to exert its biological effects.13,14 More importantly, TGF-β1 is a crucial regulator to drive epithelial–mesenchymal transition (EMT) which could result in the induction of tight junction degradation in epithelial cells, activation of myofibroblasts, and excessive production of extracellular matrix (ECM). 15 EMT is defined by disintegration of cell–cell adhesion with the loss of epithelial markers and the rise of mesenchymal markers (also known as EMT markers). 16 TGF-induced EMT is mostly mediated by SMAD-level activities, leading to increased expression of E-cadherin transcriptional repressors such as SNAIL, ZEB, and TWIST that interact with other transcription regulators in the nucleus. Therefore, blockade of activated TGF pathways seems to be an appealing strategy to prevent the EMT onset in colitis, which may efficiently improve the damaged epithelial tight junction architecture in colitis.
Long noncoding RNAs (LncRNAs) are defined as RNA transcripts longer than 200 nucleotides and considered as novel regulators of gene expression. 17 Although there are few data on the mechanism of action of many LncRNAs, recent findings have indicated that they are involved in a variety of biological processes, including chromatin remodeling, regulation of gene expression, and protein activity modulation. 18 It has recently been proposed that LncRNAs may contribute to IBD pathophysiology. Dysregulation of 47 LncRNAs has been reported in IBD, highlighting their potential role in the control of inflammatory pathways. 19 Nowadays, a wide range of studies investigated the regulatory role of SMAD-dependent TGF-β signaling LncRNAs in fibrosis and inflammation. 20 Meanwhile, despite numerous studies on the role of LncRNAs in diseases, their contribution to TGF signaling activity in IBD remains largely unknown.
Therefore, the aim of this study is to determine the activity status of the TGF-β1 signaling in the inflamed tissues of IBD patients and examine whether the inhibition of TGF-β1 signaling through small molecules improves impaired epithelial adherens and junction in IBD with a focus on the control of EMT pathways. To this end, the expression of canonical and non-canonical TGF-β1 signaling genes as well as some TGF signaling-related LncRNAs was first evaluated in mucosal biopsies of IBD patients. Thereafter, the cytotoxicity assay was performed to achieve the optimum concentration of A83-01. Then, the effect of TGF-β receptor I inhibitor (TβRI inhibitor) on the expression of EMT-related, epithelial adherens and tight junction markers was assessed in the LPS-induced Caco-2/RAW264.7 co-culture model.
Materials and methods
Patients
A total of 60 cases and controls, including 16 CD patients, 20 UC patients, and 24 healthy controls, were enrolled in this study. Colonic biopsies were collected from patients with moderate/severe active disease based on the pathologic and endoscopic reports in 2019–2021 at the gastrointestinal and liver diseases clinic of Taleghani hospital associated with Shahid Beheshti University of Medical Sciences (SBMU). Healthy controls showed endoscopy with normal findings and no gastrointestinal disease. The study excluded patients suffering from other autoimmune diseases (including psoriasis, primary sclerosing cholangitis [PSC], autoimmune hepatitis [AIH], primary biliary cholangitis [PBC], and cirrhosis) or having a history of using antibiotics during the last three months. Written informed consent was obtained from all subjects. The ethics committee of SBMU approved the protocols used in this study. Samples were stored at ‒80°C. Clinical information and patient characteristics were retrieved from participants.
Cell line culture and reagents
Caco-2 and RAW264.7 cell lines were purchased from the Iranian Biological Resource Center (IBRC). They were cultured at 37°C and 5% CO2 in DMEM medium (Gibco) that was supplemented with 10% fetal bovine serum (FBS) (Gibco), 2 mM L-glutamine (Gibco), 10 mM HEPES, and 1% nonessential amino acids (Gibco).
The co-culture model of Caco-2/RAW264.7 cells
The co-culture model was established according to the previous study. 21 Cells were harvested to confirm the establishment of an in vitro IBD model. To evaluate the role of the TGF-β1 signaling pathway in IBD, 2 µM of A83-01, as a TGF-β signaling inhibitor, was used for 24 h on the apical side of the Transwell insert, while inflammation was induced in macrophage RAW264.7 cells. Wells treated with 0.1% dimethyl sulfoxide (DMSO) were used as controls.
Cell viability assay
MTS assay was used to assess the effect of TGF-β1 receptor I inhibitor, A83-01 on the viability of Caco2 cell viability, and determine the optimized dose, according to the manufacturer’s protocol (Promega). In this regard, 104 Caco-2 cells were plated into a 96-well plate and treated overnight with the A-83-01 small molecule at concentrations of 0, 0.5, 1, 1.5, 2, 3, and 4 μM. DMSO reagent was used in the control group. The absorbance at 490 nm was determined using a multi-well plate reader (ELx800; BioTek).
Real-time quantitative polymerase chain reaction
Total RNA from biopsy specimens and cultured cells was extracted using TRIzol (Invitrogen) according to the manufacturer’s instructions. For this purpose, genomic DNA was first decontaminated using RNase-free DNase (Qiagen), and RNA was eluted with 15–25 µL RNase-free water and was reverse-transcribed to cDNA. The expression levels of candidate genes were determined by real-time quantitative polymerase chain reaction (RT-qPCR) in a Rotor Gene Q System (Qiagen) as described before. 22 The relative expression of candidate genes was achieved using the comparative Ct (ΔΔCt) method. The expression of individual values was normalized to that of the GAPDH as a housekeeping gene. Supplementary Table 1 indicates the primer sequences used in this study.
Immunofluorescence analysis
After the fixation of Caco-2 cells in 4% paraformaldehyde solution for 20 min at room temperature (RT), the cells were blocked and incubated with primary antibodies, VIM diluted 1:100 and E-cadherin diluted 1:100, overnight, followed by treatment with fluorescence-conjugated secondary antibodies for 30 min. The nucleus of the cells was counterstained by 40,6-diamidino-2-phenylindole (DAPI) (D9542; Sigma-Aldrich). Fluorescent cells were visualized and digital images were captured using a microscopy camera (Olympus BioScapes). Supplementary Table 2 indicates the list of antibodies used in this study.
Immunoblotting assay
Protein extraction was performed using radioimmunoprecipitation assay (RIPA) buffer (BIO Basic, USA). Protein concentration was then measured by BCA assay kit (Thermo Scientific). Proteins (30 μg) were separated on 12% SDS-polyacrylamide gel and transferred to polyvinylidene difluoride (PVDF) membrane. They were then blocked for 2 h at RT and incubated overnight with primary antibodies. After washing, horseradish peroxidase (HRP)-conjugated secondary antibodies were used for 1 h. Chemiluminescent peroxidase substrate was used to visualize protein-specific bands by an imaging system (VILBER). Relative densities were determined as the ratio of sample signal intensity to α-actin intensity. Antibodies are listed in Supplementary Table 2.
Statistical analysis
Comparisons among multiple groups were performed using one-way analysis of variance (ANOVA) and Tukey’s post hoc tests. The Student’s t-test was used for two group comparisons. Statistical analyses were performed using GraphPad 6 Prism (GraphPad, La Jolla, CA, USA). The association between any two genes was obtained by calculating Pearson correlation coefficients (PCCs) using cor function in R, and cor.mtest from the corrplot package was utilized to estimate the P values of the expression levels of gene pairs in each condition. The correlation plots were also generated by R package corrplot based on the PCCs of gene pairs with a P value ⩽ 0.05. The results are presented as mean ± SEM (n = 3). P values ⩽ 0.05 were considered statistically significant.
Results
SMAD-dependent TGF-β signaling is significantly activated in the inflamed colonic mucosa of IBD patients
A total of 60 patients were classified into three clinical phenotypes, where groups included 20 UC and 16 CD patients as well as 24 healthy controls. Supplementary Table 3 shows relevant characteristics of phenotypes and comparative outcome groups. There was no remarkable difference in age and gender across the three groups. Interestingly, no significant differences were observed in disease activity index (clinical or endoscopic scores) between CD and UC groups, representing similar levels of inflammation.
To further clarify the role of TGF-β1 signaling in the IBD disease, we first assessed the expression of TGF-β1 signaling–related genes in inflamed colon tissues of IBD patients using qRT-PCR. Results showed that the expression of TGF-β1 and SMAD4 transcripts was upregulated in the inflamed UC and CD tissues compared with healthy controls (Figure 1(A)).
Figure 1.

SMAD-dependent TGF-β1 signaling in IBD patients and Caco-2/RAW264.7 co-culture model. (A) Expression levels of TGF-β1, SMAD4, and LncGAS5 transcripts in CD and UC patients and healthy controls (B) and in the Caco-2/RAW264.7 co-culture model after treatment with 2 µM of A83-01. LPS-treated and LPS-untreated cells in the co-cultured system were considered as the control (Ctrl) and non-inflamed groups. (C) Protein levels for SMAD-dependent TGF-β1 signaling molecules, p-SMAD2 and p-SMAD3 in LPS-induced Caco-2/RAW264.7 co-culture model treated with A83-01. β-actin was used as a loading control. (D) The histogram represented the intensity of proteins for each group which was normalized to the β-actin band. Results are presented as the mean ± SEM. In vitro analyses were performed in three biological replicates. *, **, and *** refer to the adjusted P value < 0.05, <0.01, and <0.001, respectively.
According to the RNA immunoprecipitation (RIP) assays for SMAD protein–associated LncRNAs screening, LncGAS5 (GAS5) showed a high affinity in binding to SMAD3 and SMAD4 and physically associated with the endogenous SMAD3 in 10T1/2 cells. 23 Hence, GAS5 expression was also evaluated by qRT-PCR and showed a substantial decrease in active lesions of IBD patients compared with healthy controls (Figure 1(A)).
Considering the overexpression of canonical TGF-β1 signaling components in the active lesions derived from patients with UC and CD, this study examined the effect of the TβRI inhibitor, A83-01, on the inflamed Caco-2 cells. Cells treated with DMSO were used as a control group. We performed MTS assay to evaluate the viability of Caco-2 cells in the presence of various concentrations (0–4 µM) of A83-01 for 24 h. As the concentration of A83-01 increased, cell viability decreased in a dose-dependent pattern. A83-01 at 2 μM concentration had no significant cytotoxic effect on cells (Supplementary Figure 1); Therefore, 2 μM was selected as the optimum concentration for further analysis. According to previous reports, the effect of A83-01 on cell viability is dependent on the cell type and pathophysiological condition and different doses have been used in studies, such as 25 μM for A549 cells and MCF-7 cells 24 and 1 μM for human endometrial mesenchymal stem cells 25 and postnatal Nkx2.5 cardiomyoblast. 26 Thus, we applied the MTS test as a time- and cost-effective assay to select the optimum dose.
To decipher the role of TGF-β1 signaling in IBD pathogenesis, we used a previously established co-culture system including intestinal epithelial Caco-2 cells and RAW264.7 macrophage cells in a Transwell® plate. 21 Briefly, the intestinal physiological environment was simulated through differentiation of Caco-2 cells for 21 days into a monolayer of enterocyte-like cells, at which point RAW264.7 macrophages were pre-seeded on the basolateral side in Transwell plates, allowing interaction between the two cell monolayers. Subsequently, LPS (at a concentration of 1.2 μg/mL) was added to stimulate RAW264.7 macrophages on the basolateral side for 24 h, resulting in the overexpression of tumor necrosis factor–α (TNF-α) and interleukin-6 (IL-6) within 24 h (data not shown). LPS-treated and LPS-untreated RAW264.7 cells in the co-cultured system were considered as the control and non-inflamed groups, respectively. Then, qRT-PCR was performed to validate the increased expression of TGF-β1, SMAD4, and GAS5 in inflamed enterocyte-like cells (Figure 1(B)) . Over 24 h, A83-01 (at a 2 µM concentration) was added to the apical side of the LPS-induced Caco-2/RAW264.7 co-culture model. Figure 1(B) demonstrates that the small molecule A83-01 could prevent the overexpression of TGF-β1 in the inflamed epithelial cells in vitro. Furthermore, A83-01 significantly decreased SMAD4 expression induced in the inflamed epithelial cells at the transcript level (Figure 1(B)). Subsequently, the suppression effect of A83-01 was evaluated on the phosphorylation of SMAD2/3 family proteins, as downstream markers of the TGF-β1 pathway. Increased p-SMAD2/3 in the inflamed epithelial cells was completely blocked by A83-01, as illustrated in Figure 1(C) and (D). Because GAS5 can bind to SMAD3 and dephosphorylate, we sought to determine whether the suppression of the TGF-β1 pathway affects the expression level of GAS5 in the inflamed enterocyte-like cells. Interestingly, our results (Figure 1(B)) showed that suppression of TGF-β1 pathway could prominently lead to increased transcriptional levels of GAS5. Our findings demonstrated that TβRI inhibitors as a therapeutic option could decrease SMAD-dependent TGF-β1 signaling activated in IBD.
TβRI inhibitor prevents the activation of SMAD-independent TGF-β signaling in inflamed intestinal epithelial cells
To investigate the role of the SMAD-independent TGF-β signaling in the development of colitis, the activity of phosphatidylinositol-3-kinase (PI3K)/AKT pathway as one of the SMAD-independent pathways was assessed. PI3K/AKT signaling is involved in the regulation of cell growth, cell proliferation, and inflammation through downstream signal transduction molecules such as p-70S6K and p-RPS6. 27 PI3K mRNA was found to be highly expressed in the inflamed epithelial tissue of UC and CD patients compared with healthy controls (Figure 2(A)), while LncCDKN2B (CDKN2B) as a key regulatory LncRNA of the PI3K/AKT pathway was decreased (Figure 2(A)). Likewise, there was a similar trend toward increased PI3K and decreased CDKN2B transcripts in the LPS-induced Caco-2/RAW264.7 co-culture model. Treatment of inflamed epithelial cells by A83-01 led to a decrease in PI3K and an increase in CDKN2B, showing to be statistically significant (Figure 2(B)). However, no changes in AKT were found in both patients and in vitro IBD model. Therefore, the phosphorylation level of AKT and its downstream target, p-RPS6, was analyzed by Western blotting in the in vitro co-culture model. As shown in Figure 2(C) and (D)), treatment with A83-01 suppressed the phosphorylation of AKT and subsequently RPS6 in the inflamed group compared with the control.
Figure 2.

PI3K/AKT signaling in IBD patients and Caco-2/RAW264.7 co-culture model. (A) Expression levels of PI3K, AKT, and LncCDKN2B transcripts in CD and UC patients and healthy controls (B) and in the Caco-2/RAW264.7 co-culture model after treatment with 2 µM of A83-01. LPS-treated and LPS-untreated cells in the co-cultured system were considered as the control (Ctrl) and non-inflamed groups. (C) Protein changes of p-AKT and p-RPS6 after treatment with A83-01. β-actin was used as a loading control. (D) The histogram represented the intensity of proteins for each group which was normalized to the β-actin band. Results are presented as the mean ± SEM. In vitro analyses were performed in three biological replicates. *, **, and *** refer to the adjusted P value < 0.05, <0.01, and <0.001, respectively.
TβRI inhibitor suppresses EMT induced in inflamed intestinal epithelial cells
To understand the contribution of TGF-β1 signaling to promote EMT in the inflamed mucosal biopsies of IBD patients, the expression of EMT markers was investigated. As illustrated in Figure 3(A), a broad panel of mesenchymal markers, including VIM, SNAI1, SNAI2, CTNNB1, ACTA2, and CDH2, were upregulated in active lesions of UC and CD patients compared with the healthy control, while the expression of CDH1 was downregulated. These data suggested that increased activity of TGF-β1 signaling in inflamed colonic tissues may trigger the expression of mesenchymal marker inducers, thereby causing reduction of epithelial markers. The R package corrplot was used to visualize correlations across genes. We intended to remove the nonsignificant correlations according to the confidence interval of 0.95. None of the correlations were removed due to the significant P value of less than 0.05. Figure 3(B) and (C)) demonstrates the correlation of any gene pairs in UC and CD conditions. In these plots, color and ellipticity represent the correlations. The correlation pattern between genes was similar due to the interconnected nature of these conditions. The strongest negative correlations were between ZO-1-SNAI1 and OCCLUDIN-LncH19 in UC and CD, respectively. The most positively correlated genes in UC were PI3K and CTNNB1, while they were CDH2 and SNAI1 in CD.
Figure 3.

TGF-β1 signaling correlates with the EMT pathway in IBD. (A) Changes in the levels of VIM, SNAI1, SNAI2, CTNNB1, α-SMA, CDH2, CDH1, and LncH19 mRNA in CD and UC patients were detected by qRT-PCR. Correlation analysis of gene pairs of (B) UC and (C) CD conditions. Correlations are indicated by the ellipticity and color of the ellipses as well as the numbers. The bar on the right represents the correlation values of PCCs. All the correlations have a significant P value of less than 0.05. Results are presented as the mean ± SEM. In vitro analyses were performed in three biological replicates. *, **, and *** refer to the adjusted P value < 0.05, <0.01, and <0.001, respectively.
Afterward, we sought whether the TβRI inhibitor affects the induction of EMT pathway in the LPS-induced Caco-2/RAW264.7 co-culture model. Our results revealed that A83-01 treatment could markedly decrease mRNA expression levels of VIM, SNAI1, SNAI2, CTNNB1, ACTA2, and CDH2 (Figure 4(A)). Similarly, decrease in the level of Vim, β-CATENIN, and α-SMA proteins was detected in the inflamed epithelial cells in response to the A83-01 treatment (Figure 4(B) and (C))). The immunostaining analysis also confirmed lower expression of VIM (green), a main marker of EMT, in the A83-01-treated cells (Figure 5(A)). Contrary to mesenchymal markers, the expression level of CDH1, a well-established marker of cell adhesion, remarkably increased at both transcript and protein levels in the A83-01-treated epithelial cells compared with the control group (Figure 4(A) and (B)), consistent with results obtained from the E-CADHERIN immunostaining (red) (Figure 5(B)). Based on increasing levels of LncRNAH19 (H19) in many inflammatory diseases and organ fibrosis, as well as its novel regulatory role in the EMT progression,28,29 we first examined the mRNA expression of H19 in patient groups. The expression level of H19 exhibited the greatest increment in active lesions of patients with UC and CD compared with the healthy control (Figure 3(A)). Furthermore, the expression level of H19 was assessed in the LPS-induced Caco-2/RAW264.7 co-culture model upon treatment with A83-01. Intriguingly, H19 was downregulated following A83-01 treatment (Figure 4(A)), indicating its involvement in the TGF-β1-induced EMT in the inflamed epithelial cells. Overall, these results suggested that the TβRI inhibitor maintains the epithelial characteristics of intestinal cells in inflammatory conditions by limiting the EMT induction.
Figure 4.

TGF-β1 signaling affected the EMT pathway in the Caco-2/RAW264.7 co-culture model. (A) Expression changes of EMT-related markers at the transcript level in the Caco-2/RAW264.7 co-culture model after treatment with 2 µM of A83-01. LPS-treated and LPS-untreated cells in the co-cultured system were considered as the control (Ctrl) and non-inflamed groups. (B) Protein expression levels of VIM, β-CATENIN, α-SMA, and E-CADHERIN in the Caco-2/RAW264.7 co-culture model. β-actin was used as a loading control. (C) The histogram represented the intensity of proteins for each group which was normalized to the β-actin band. Results are presented as the mean ± SEM. In vitro analyses were performed in three biological replicates. *, **, and *** refer to the adjusted P value < 0.05, <0.01, and <0.001, respectively.
Figure 5.

TGF-β1 signaling affected the EMT pathway, epithelial adherens, and tight junctions in IBD. (A, B) Immunofluorescence analyses of EMT-related proteins, VIM (green) and E-CADHERIN (red) in the Caco-2/RAW264.7 co-culture model after treatment with 2 µM of A83-01. (Scale bar: 50 µm). (C) The OCCLUDIN, CLAUDIN7, and ZO-1 transcript levels in CD and UC patients and healthy controls (D) and in the Caco-2/RAW264.7 co-culture model after treatment with 2 µM of A83-01. LPS-treated and LPS-untreated cells in the co-cultured system were considered as the control (Ctrl) and non-inflamed groups.
The protective effect of TβRI inhibitor on the impaired epithelial barrier
We assessed the expression of epithelial-related markers, including CLAUDIN7, ZO-1, and OCCLUDIN, in the inflamed colon tissues. In agreement with previous studies, 30 we indicated that expression levels of CLAUDIN7, ZO-1, and OCCLUDIN decreased in active tissue lesions from patients with UC and CD compared with the healthy control (Figure 5(C)). We, therefore, aimed to assess the possible association of epithelial adherens and tight junction markers with the suppression of TGF-β1 signaling in the co-culture model. Intriguingly, mRNA levels of OCCLUDIN, CLAUDIN7 and ZO-1 in the LPS-induced Caco-2/RAW264.7 co-culture model upon treatment with A83-01 sharply enhanced compared with those in the control (Figure 5(D)). These findings revealed that the TGF-β1 signaling suppression may play an effective role in preventing the disruption of tight junctions and maintaining the epithelial barrier in inflammatory conditions.
Discussion
The results obtained from this study offers an insight into the role of SMAD-dependent and SMAD-independent TGF-β1 pathways in intestinal epithelial cells undergoing inflammation conditions which may reconcile some of the apparently contradicting results in patients suffering from IBD. We revealed how TGF-β1 signaling suppression leads to targeting the EMT process as well as preventing the impairment of epithelial tight junctions in the in vitro IBD model (Figure 6).
Figure 6.
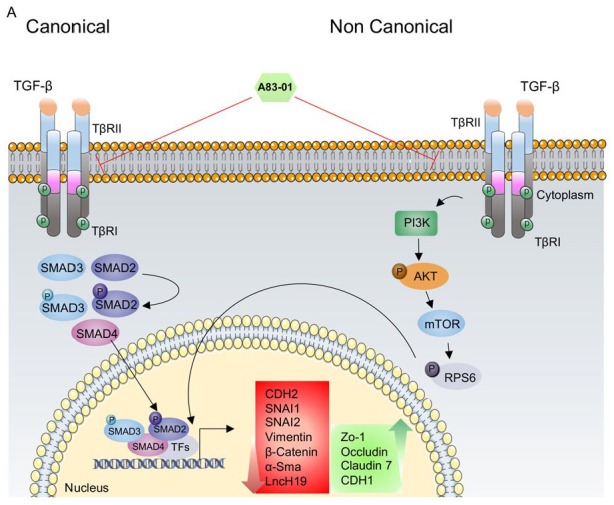
Schematic of SMAD-independent and SMAD-independent TGF-β1 signaling pathways in IBD. (A) In the canonical pathway, the TGF-β1 ligand binds to the TβRII receptor. Activated TβRI can phosphorylate a group of transcription factors, including SMAD2 and SMAD3 on C-terminal serine. Consequently, a trimeric complex containing SMAD2, SMAD3, and SMAD4 was formed. This complex translocates into the nucleus, where it induces or inhibits target gene expression. In addition to the canonical TGF-β1 signaling pathway, TGF-β type I and II receptors can also activate downstream non-canonical pathways, including PI3K/AKT signaling pathway, in an SMAD-independent manner.
According to previous studies, the TGF-β1 pathway plays a vital role in the pathogenesis of inflammatory disorders. 4 The upregulation of TGF-β1, SMAD4, and PI3K transcripts in inflamed biopsies of IBD patients confirmed previously published results,31,32 while the expression level of AKT transcript did not alter because AKT serves as a serine/threonine kinase, which is induced by the phosphorylation. In addition, expression trends of TGF-β1, SMAD4, AKT, and PI3K in our LPS-induced Caco-2/RAW264.7 co-culture model were similar to those observed in patients. In agreement with our study, a study conducted by Huang et al. 32 reported that the development of UC is closely related to the activation of the PI3K/AKT signal transduction pathway, an SMAD-independent TGF-β1 signaling pathway, in the colon mucosa of UC patients and colitis mice. Therefore, overexpression of these transcripts as well as p-SMAD2, p-SMAD3, p-AKT, and p-RPS6 at the protein level highlighted the activation of both SMAD-dependent and SMAD-independent TGF-β1 pathways in response to the immune activation. Furthermore, our results demonstrated a noticeable decrease in GAS5 and CDKN2B expression, known as regulators of TGF-β1 pathway important mediators, in active lesions from patients with CD and UC. In this line, findings conducted by Lucafò et al. 33 were also based on the dramatically lower expression of GAS5 in inflamed CD and UC tissues compared with the adjacent normal part. Interestingly, as reported in the literature, GAS5 is involved in the regulation of TGF/SMAD signaling pathway. It has been shown that GAS5 suppresses TGF/SMAD3 signaling and inhibits TGF-β-induced smooth muscle cell (SMC) differentiation. Indeed, Smad3 phosphorylation/dephosphorylation turnover is affected through directly binding GAS5 into the Smad3 promoter in 3T3 cells. 23
Our results revealed that upregulation of the TGF-β1 pathway in IBD patients was negatively correlated with the GAS5 expression. In addition, according to the study performed by Mirza et al., 34 CDKN2B expression was reduced in inflamed CD and UC patients compared with the healthy control, which was also in agreement with our findings. A substantial decrease in CDKN2B expression has been also found when Caco-2 and HT29 colonic epithelial cells were stimulated with TNF-α or IL-1β.35,36 Furthermore, Nanda et al. 37 illustrated that CDKN2B deficiency is accompanied by an increase in the TGF-β1 production in the endothelial and smooth muscle cells. enzyme-linked immunosorbent assay (ELISA)-based assays showed that knockdown of CDKN2B in hypoxic cells resulted in an increase in the TGF-β1 expression compared with the control group. Furthermore, the enhancement of SMAD3 as a downstream factor in the TGF-β1 pathway has been shown in CDKN2B-deficient cells. Presumably, TGF-β1 pathway activation may act as a molecular brake for the GAS5 and CDKN2B in the inflamed epithelial tissue as well as the LPS-induced Caco-2/RAW264.7 co-culture model.
We further identified that targeting TβRI by A83-01 not only inhibited LPS-induced overexpression of TGF-β1 signaling components and the phosphorylation of SMAD2, SMAD3, AKT, and RPS6 proteins but also enhanced the expression of GAS5 and CDKN2B in inflamed intestinal epithelial cells. The inhibition of the PI3K/AKT pathway was found to be an ideal therapeutic option in a variety of inflammatory disorders. 38 Anti-colitis effect of Arctigenin, a pharmaceutical compound, has been reported through the phosphorylation blockade of p-RPS6 and p-70S6K in dextran sulfate sodium (DSS)-induced mice 39 which is in agreement with our findings.
Considering the downregulation of CDKN2B and GAS5 in the inflamed epithelial cells and the recovery effect of A83-01 on their expression, it seems preserving their expression by small molecules, oligonucleotide, or drug candidates could be a proper therapeutic strategy,40,41 although more systematic studies are needed. In this line, Shi et al. 42 showed lower expression of GAS5 in patients with type 2 diabetes mellitus (T2DM) and introduced a peptidomimetic ligand, as a binding probe to GAS5, which highlighted the importance of the proper GAS5 level in glucose uptake and metabolism.
Due to increased TGF-β1 signaling in both IBD patients and the in vitro model, we decided to assess the EMT process and markers responsible for preserving epithelial characteristics of epithelial-like cells undergoing inflammation conditions. EMT-related markers were upregulated in active CD and UC patients as well as the LPS-induced Caco-2/RAW264.7 co-culture model and correlated with the activated TGF-β signaling. In a seminal study aiming to elucidate the molecular process underlying UC development, Zhao et al. 43 found an increase in the EMT-related markers in active lesions of UC patients. Our findings support the hypothesis that TGF-β1 activation may contribute to the induction of EMT in inflamed intestinal tissues. Interestingly, we revealed that treatment with A83-01 could suppress the EMT pathway as well as improve the epithelial tight junction markers. In this context, a decrease in the TGF-β1 signaling pathway members has been shown to prevent EMT induction and decrease fibrosis in the kidney nephropathies. 44 Furthermore, the higher expression of H19 in inflamed epithelial cells was completely suppressed after the A83-01 treatment. In a study, Chen et al. 28 also demonstrated that inflamed UC patients exhibit overexpression of H19 and decreased expression of tight junction markers, including ZO-1 and Occludin. On the contrary, we demonstrated that upregulation of H19 could be associated with the induced-EMT pathway in inflamed epithelial cells, which is consistent with the previous observations that the higher expression of H19 is connected to the EMT process in carcinogenesis and embryogenesis. 45 Yang and colleagues illustrated that TGF-β1 enhances H19 expression through PI3K/AKT pathway. They established an stable mammary alveolar cell-T (MAC-T) cell clones to assess the relationship between H19- and TGF-induced EMT in MAC-T cells and found that overexpression of H19 sharply promotes TGF-β1-induced EMT in MAC-T cells. 29
TGF-β, activin, nodal, and bone morphogenetic proteins (BMPs) are members of the TGF-β superfamily and as multifunctional cytokines play important regulatory roles in a wide range of cellular processes. These ligands bind to type II serine/threonine kinase receptors at the cell surface, leading to the phosphorylation of type I receptors (also, termed activin receptor-like kinases (ALKs)). 46 A83-01 small molecule serves directly as a potent selective inhibitor of TβRI (ALK5), activin/nodal type I receptor (ALK4), and nodal type I receptor (ALK7).47,48 In this study, we focused on the inhibitory effect of A83-01 on the TGF-β1 pathway, although it can influence signaling pathways induced by other ligands that share receptors TβRI and Alk4 and Alk7, which needs further investigation.
These in vitro results strongly highlighted the function of A83-01 in preventing the EMT initiation in inflamed epithelial cells by repression of the SMAD-dependent and SMAD-independent TGF-β1 signaling pathways, as well as noted the contribution of three LncRNAs, namely, LncGAS5, CDKN2B and H19, in IBD pathogenesis.
Conclusions
The function of TGF-β1 signaling is a complicated phenomenon depending on the cell type. However, insights into its pathological effect during IBD are currently unclear. In this study, we indicated that both canonical and non-canonical TGF-β1 signaling pathways are activated in inflamed intestinal epithelial cells resulting in EMT activation, along with the epithelial tight junction disruption, as a hallmark of IBD. Based on the results, TGF-β1 signaling inhibition could attenuate the induction of EMT in inflamed epithelial cells and exert a protective effect on the tight junction integrity. In this scenario, attempts to suppress epithelial TGF-β1 signaling function for remodeling intestinal epithelial cells would be an ideal candidate to control this disease. Furthermore, we highlighted the possible important role of three EMT-related LncRNAs, namely, H19, GAS5, and CDKN2B, in the IBD pathogenesis, which may have implications for better IBD management, although further in vitro and in vivo mechanistic studies are necessary.
Supplemental Material
Supplemental material, sj-pdf-1-ebm-10.1177_15353702231151959 for TGF-β receptor I inhibitor may restrict the induction of EMT in inflamed intestinal epithelial cells by Mahsa Ghorbaninejad, Meghdad Abdollahpour-Alitappeh, Shabnam Shahrokh, Sara Fayazzadeh, Hamid Asadzadeh-Aghdaei and Anna Meyfour in Experimental Biology and Medicine
Footnotes
Authors’ Contributions: MG contributed to investigation, methodology and writing—original draft; MA contributed to review & editing; SS contributed to resources and consultation; SF contributed to review and bioinformatics analysis; HA contributed to resources and consultation; and AM contributed to conceptualization, investigation, supervision, and writing—review & editing. All authors reviewed and approved the manuscript.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval: Tissue biopsies were obtained with patients’ consent and the study was confirmed by the Research Ethics Committee of the Research Institute for Gastroenterology and Liver Diseases, SBMU.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was granted by the Research Institute for Gastroenterology and Liver Diseases, SBMU.
ORCID iD: Anna Meyfour  https://orcid.org/0000-0003-3694-8912
https://orcid.org/0000-0003-3694-8912
Supplemental Material: Supplemental material for this article is available online.
References
- 1. Kaplan GG, Ng SC. Understanding and preventing the global increase of inflammatory bowel disease. Gastroenterology 2017;152:313–21 [DOI] [PubMed] [Google Scholar]
- 2. Hudson GM, Flannigan KL, Erickson SL, Vicentini FA, Zamponi A, Hirota CL, Alston L, Altier C, Ghosh S, Rioux KP, Mani S, Chang TK, Hirota SA. Constitutive androstane receptor regulates the intestinal mucosal response to injury. Br J Pharmacol 2017;174:1857–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, Panaccione R, Ghosh S, Wu JC, Chan FK. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet 2017;390:2769–78 [DOI] [PubMed] [Google Scholar]
- 4. Ihara S, Hirata Y, Koike K. TGF-β in inflammatory bowel disease: a key regulator of immune cells, epithelium, and the intestinal microbiota. J Gastroenterol 2017;52:777–87 [DOI] [PubMed] [Google Scholar]
- 5. Roberts AB, Sporn MB. Differential expression of the TGF-β isoforms in embryogenesis suggests specific roles in developing and adult tissues. Mol Reprod Dev 1992;32:91–8 [DOI] [PubMed] [Google Scholar]
- 6. Han G, Li F, Singh TP, Wolf P, Wang XJ. The pro-inflammatory role of TGFβ1: a paradox. Int J Biol Sci 2012;8:228–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Salgado RM, Cruz-Castañeda O, Elizondo-Vázquez F, Pat L, De la Garza A, Cano-Colín S, Baena-Ocampo L, Krötzsch E. Maltodextrin/ascorbic acid stimulates wound closure by increasing collagen turnover and TGF-β1 expression in vitro and changing the stage of inflammation from chronic to acute in vivo. J Tissue Viability 2017;26:131–7 [DOI] [PubMed] [Google Scholar]
- 8. Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature 1992;359:693–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nat Rev Mol Cell Biol 2014;15:178–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Babyatsky MW, Rossiter G, Podolsky DK. Expression of transforming growth factors alpha and beta in colonic mucosa in inflammatory bowel disease. Gastroenterology 1996;110:975–84 [DOI] [PubMed] [Google Scholar]
- 11. Sambuelli A, Diez R, Sugai E, Boerr L, Negreira S, Gil A, Camartino G, Huernos S, Doldán I, Felstiner D. Serum transforming growth factor-β1 levels increase in response to successful anti-inflammatory therapy in ulcerative colitis. Aliment Pharmacol Ther 2000;14:1443–9 [DOI] [PubMed] [Google Scholar]
- 12. Monteleone G, Kumberova A, Croft NM, McKenzie C, Steer HW, MacDonald TT. Blocking Smad7 restores TGF-β1 signaling in chronic inflammatory bowel disease. J Clin Invest 2001;108:601–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Macias MJ, Martin-Malpartida P, Massagué J. Structural determinants of Smad function in TGF-β signaling. Trends Biochem Sci 2015;40:296–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang YE. Non-Smad pathways in TGF-β signaling. Cell Res 2009;19: 128–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xu J, Lamouille S, Derynck R. TGF-β-induced epithelial to mesenchymal transition. Cell Res 2009;19:156–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Djediai S, Gonzalez Suarez N, Cheikh-Hussein E, Rodriguez Torres S, Gresseau L, Dhayne S, Joly-Lopez Z, Annabi B. MT1-MMP cooperates with TGF-β receptor-mediated signaling to trigger SNAIL and induce epithelial-to-mesenchymal-like transition in U87 glioblastoma cells. Int J Mol Sci 2021;22:13006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell 2009;136:629–41 [DOI] [PubMed] [Google Scholar]
- 18. Zhao D, Wang C, Yan S, Chen R. Advances in the identification of long non-coding RNA binding proteins. Anal Biochem 2021:114520. [DOI] [PubMed] [Google Scholar]
- 19. Yarani R, Mirza AH, Kaur S, Pociot F. The emerging role of lncRNAs in inflammatory bowel disease. Exp Mol Med 2018;50:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gu YY, Dou JY, Huang XR, Liu XS, Lan HY. Transforming growth factor-β and long non-coding RNA in renal inflammation and fibrosis. Front Physiol 2021;12:684236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ghorbaninejad M, Meyfour A, Maleknia S, Shahrokh S, Abdollahpour-Alitappeh M, Asadzadeh-Aghdaei H. Inhibition of epithelial SHH signaling exerts a dual protective effect against inflammation and epithelial–mesenchymal transition in inflammatory bowel disease. Toxicol in Vitro 2022;82:105382. [DOI] [PubMed] [Google Scholar]
- 22. Meyfour A, Ansari H, Pahlavan S, Mirshahvaladi S, Rezaei-Tavirani M, Gourabi H, Baharvand H, Salekdeh GH. Y chromosome missing protein, TBL1Y, may play an important role in cardiac differentiation. J Proteome Res 2017;16:4391–402 [DOI] [PubMed] [Google Scholar]
- 23. Tang R, Zhang G, Wang Y-C, Mei X, Chen S-Y. The long non-coding RNA GAS5 regulates transforming growth factor β (TGF-β)–induced smooth muscle cell differentiation via RNA Smad–binding elements. J Biol Chem 2017;292:14270–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bydoun M, Sterea A, Weaver IC, Bharadwaj AG, Waisman DM. A novel mechanism of plasminogen activation in epithelial and mesenchymal cells. Sci Rep 2018;8:1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gurung S, Deane JA, Darzi S, Werkmeister JA, Gargett CE. In vivo survival of human endometrial mesenchymal stem cells transplanted under the kidney capsule of immunocompromised mice. Stem Cells Dev 2018;27:35–43 [DOI] [PubMed] [Google Scholar]
- 26. Chen WP, Wu SM. Small molecule regulators of postnatal Nkx 2.5 cardiomyoblast proliferation and differentiation. J Cell Mol Med 2012;16:961–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yi YW, You KS, Park J-S, Lee S-G, Seong Y-S. Ribosomal protein S6: a potential therapeutic target against cancer? Int J Mole Sci 2021;23:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen SW, Wang PY, Liu YC, Sun L, Zhu J, Zuo S, Ma J, Li TY, Zhang JL, Chen GW, Wang X, Zhu QR, Zheng YW, Chen ZY, Yao ZH, Pan YS. Effect of long noncoding RNA H19 overexpression on intestinal barrier function and its potential role in the pathogenesis of ulcerative colitis. Inflamm Bowel Dis 2016;22:2582–92 [DOI] [PubMed] [Google Scholar]
- 29. Yang W, Li X, Qi S, Li X, Zhou K, Qing S, Zhang Y, Gao MQ. lncRNA H19 is involved in TGF-β1-induced epithelial to mesenchymal transition in bovine epithelial cells through PI3K/AKT signaling pathway. PeerJ 2017;5:e3950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bürgel N, Bojarski C, Mankertz J, Zeitz M, Fromm M, Schulzke JD. Mechanisms of diarrhea in collagenous colitis. Gastroenterology 2002;123:433–43 [DOI] [PubMed] [Google Scholar]
- 31. Zhu L, Shen H, Gu PQ, Liu YJ, Zhang L, Cheng JF. Baicalin alleviates TNBS-induced colitis by inhibiting PI3K/AKT pathway activation. Exp Ther Med 2020;20:581–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang XL, Xu J, Zhang XH, Qiu BY, Peng L, Zhang M, Gan HT. PI3K/Akt signaling pathway is involved in the pathogenesis of ulcerative colitis. Inflamm Res 2011;60:727–34 [DOI] [PubMed] [Google Scholar]
- 33. Lucafò M, Pugnetti L, Bramuzzo M, Curci D, Di Silvestre A, Marcuzzi A, Bergamo A, Martelossi S, Villanacci V, Bozzola A. Long non-coding RNA GAS5 and intestinal MMP2 and MMP9 expression: a translational study in pediatric patients with IBD. Int J Mole Sci 2019;20:5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mirza AH, Berthelsen CH, Seemann SE, Pan X, Frederiksen KS, Vilien M, Gorodkin J, Pociot F. Transcriptomic landscape of lncRNAs in inflammatory bowel disease. Genome Med 2015;7:39–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Haberman Y, BenShoshan M, Di Segni A, Dexheimer PJ, Braun T, Weiss B, Walters TD, Baldassano RN, Noe JD, Markowitz J. Long ncRNA landscape in the ileum of treatment-naive early-onset Crohn disease. Inflamm Bowel Dis 2018;24:346–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wu F, Huang Y, Dong F, Kwon JH. Ulcerative colitis-associated long noncoding RNA, BC012900, regulates intestinal epithelial cell apoptosis. Inflamm Bowel Dis 2016;22:782–95 [DOI] [PubMed] [Google Scholar]
- 37. Nanda V, Downing KP, Ye J, Xiao S, Kojima Y, Spin JM, DiRenzo D, Nead KT, Connolly AJ, Dandona S. CDKN2B Regulates TGF β signaling and smooth muscle cell investment of hypoxic neovessels. Circ Res 2016;118:230–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Banham-Hall E, Clatworthy MR, Okkenhaug K. Suppl 2: the therapeutic potential for PI3K inhibitors in autoimmune rheumatic diseases. Open Rheumatol J 2012;6:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wu X, Dou Y, Yang Y, Bian D, Luo J, Tong B, Xia Y, Dai Y. Arctigenin exerts anti-colitis efficacy through inhibiting the differentiation of Th1 and Th17 cells via an mTORC1-dependent pathway. Biochemical Pharmacol 2015;96:323–36 [DOI] [PubMed] [Google Scholar]
- 40. Zeng B, Li Y, Jiang F, Wei C, Chen G, Zhang W, Zhao W, Yu D. LncRNA GAS5 suppresses proliferation, migration, invasion, and epithelial-mesenchymal transition in oral squamous cell carcinoma by regulating the miR-21/PTEN axis. Exp Cell Res 2019;374:365–73 [DOI] [PubMed] [Google Scholar]
- 41. Li F, Sun J, Huang S, Su G, Pi G. LncRNA GAS5 overexpression reverses LPS-induced inflammatory injury and apoptosis through up-regulating KLF2 expression in ATDC5 chondrocytes. Cell Physiol Biochem 2018;45:1241–51 [DOI] [PubMed] [Google Scholar]
- 42. Shi Y, Patel NA, Cai J. Discovery of a macrocyclic γ-AApeptide binding to lncRNA GAS5 and its therapeutic implication in type 2 diabetes. Future Med Chem 2019;11:2233–35 [DOI] [PubMed] [Google Scholar]
- 43. Zhao X, Fan J, Zhi F, Li A, Li C, Berger AE, Boorgula MP, Barkataki S, Courneya J-P, Chen Y. Mobilization of epithelial mesenchymal transition genes distinguishes active from inactive lesional tissue in patients with ulcerative colitis. Hum Mol Genet 2015;24:4615–24 [DOI] [PubMed] [Google Scholar]
- 44. Li J, Qu X, Yao J, Caruana G, Ricardo SD, Yamamoto Y, Yamamoto H, Bertram JF. Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes 2010;59:2612–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Oviedo-Boyso J, Valdez-Alarcón JJ, Valdez-Alarcón JJ, Ochoa-Zarzosa A, López-Meza JE, Bravo-Patiño A, Baizabal-Aguirre VM. Innate immune response of bovine mammary gland to pathogenic bacteria responsible for mastitis. J Infect 2007;54:399–409 [DOI] [PubMed] [Google Scholar]
- 46. Shi Y, Massagué J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003;113:685–700 [DOI] [PubMed] [Google Scholar]
- 47. Wu Y, Tran T, Dwabe S, Sarkissyan M, Kim J, Nava M, Clayton S, Pietras R, Farias-Eisner R, Vadgama JV. A83-01 inhibits TGF-β-induced upregulation of Wnt3 and epithelial to mesenchymal transition in HER2-overexpressing breast cancer cells. Breast Cancer Res Treat 2017;163:449–60 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48. Tojo M, Hamashima Y, Hanyu A, Kajimoto T, Saitoh M, Miyazono K, Node M, Imamura T. The ALK-5 inhibitor A-83-01 inhibits Smad signaling and epithelial-to-mesenchymal transition by transforming growth factor-β. Cancer Sci 2005;96:791–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-pdf-1-ebm-10.1177_15353702231151959 for TGF-β receptor I inhibitor may restrict the induction of EMT in inflamed intestinal epithelial cells by Mahsa Ghorbaninejad, Meghdad Abdollahpour-Alitappeh, Shabnam Shahrokh, Sara Fayazzadeh, Hamid Asadzadeh-Aghdaei and Anna Meyfour in Experimental Biology and Medicine