

Abstract
目的
比较左肺切除、颈静脉注射脱氢野百合碱、左肺切除联合颈静脉注射脱氢野百合碱三种方法构建肺动脉高压(PAH)小鼠模型的差异和肺血管重构的特点,寻找与临床发病机制接近的PAH动物模型,为深入研究重度PAH的发病机制提供良好的模型基础。
方法
取59只C57BL/6雄性小鼠(10~12周,24~30 g),随机分为对照组(n=9)、左肺切除组(PE组,n=15)、颈静脉注射脱氢野百合碱组(MCTP组,n=15)和左肺切除联合颈静脉注射脱氢野百合碱组(P+M组,n=20),通过检测右心室收缩压(RVSP)、右心室肥厚指数(RV/LV+S)、肺动脉中膜厚度百分比(WT%)、非肌性肺动脉肌化程度、新生内膜形成情况和血管阻塞评分(VOS)评价造模效果及肺血管重构的特点。
结果
①与对照组比较,P+M组的RVSP、RV/LV+S、WT%、小肺动脉肌化程度均增加(P<0.01);MCTP组表现为轻度的RVSP升高、右心室肥厚、中膜增厚和小肺动脉肌化程度增加(P<0.05);PE组上述指标并无明显改变(P>0.05)。②P+M组腺泡内肺动脉可观察到新生内膜形成导致管腔明显狭窄,VOS为(1.25±0.80)分(P<0.001),MCTP组和PE组均未观察到新生内膜形成,VOS为0分(P>0.05)。
结论
左肺切除联合颈静脉注射脱氢野百合碱可诱导小鼠形成重度PAH,该模型能较好模拟临床重度PAH的特征性病理改变,即新生内膜形成。
Keywords: 肺动脉高压, 肺血管重构, 动物模型, 小鼠
Abstract
Objective
To compare and investigate the differences and characteristics of pulmonary vascular remodeling in three mouse models of pulmonary arterial hypertension (PAH) constructed by left pneumonectomy, jugular vein injection of monocrotaline pyrrole, and left pneumonectomy combined with jugular vein injection of monocrotaline pyrrole, to explore for a PAH animal model that approximates the clinical pathogenesis of PAH, and to create a model that will provide sound basis for thorough investigation into the pathogenesis of severe PAH.
Methods
59 male C57/BL mice (10-12 weeks, 24-30 g) were randomized into four groups, a control group (n=9), a group that had left pneumonectomy (PE, n=15), a group that had jugular vein injection of monocrotaline pyrrole (MCTP, n=15), and the last group that had left pneumonectomy combined with jugular injection of monocrotaline pyrrole (P+M, n=20). To evaluate the effect of modeling and the characteristics of pulmonary vascular remodeling, hemodynamic and morphological parameters, including right ventricular systolic pressure (RVSP), right ventricle/(left ventricle plus septum) (RV/LV+S), percent of wall thickness in the pulmonary artery (WT%), muscularization of non-muscular arteries, neointima formation, and vascular obstruction score (VOS), were measured in each group.
Results
1) Compared with those of the control group, the RVSP, RV/LV+S, WT%, and the degree of small pulmonary arteries muscularization in the P+M group were significantly increased (P<0.01). The MCTP group had just slightly higher findings for these indicators (P<0.05), while no significant change in these indicators was observed in the PE group (P>0.05). 2) Neointima formation in the acinus pulmonary arteries, which caused obvious stenosis of the lumen, was observed in the P+M group, the VOS being 1.25±0.80 points (P<0.001). In contrast, neointima formation was not observed in the MCTP group or the PE groups, the VOS being 0 point (P>0.05).
Conclusion
Left pneumonectomy combined with jugular intravenous injection of MCTP could induce severe PAH formation in mouse. The model provides a good simulation of neointima formation, the characteristic pathological change of clinical severe PAH.
Keywords: Pulmonary arterial hypertension, Pulmonary vascular remodeling, Animal model, Mouse
肺动脉高压(pulmonary arterial hypertension, PAH)是一种多因素引起的肺血管疾病,尽管发病率不高,但因其病因复杂、自然病程短、预后差、发病机制不清楚等特点而受到广泛关注。一直以来,对于PAH发病机制和治疗药物药理机制的研究是该领域的热点及重点。目前,常用的PAH动物模型建模方法有慢性缺氧[1]、药物注射如野百合碱(monocrotaline, MCT)[2]和血管内皮生长因子受体拮抗剂Sugen[2]、单纯左肺切除(pneumonectomy, PE)[3]以及上述方法联合造模等;其中,左肺切除联合MCT注射诱导SD大鼠PAH模型[4]是目前最常用的动物模型之一,该模型可造成重度PAH,能观察到肺血管丛状损害和新生内膜形成[5],可较好地模拟人类重度PAH的病理生理改变。然而,随着疾病研究逐渐进入分子水平,以及基因编辑技术的快速发展,相对于目前已在人类疾病研究中广泛应用的转基因小鼠,大鼠中基因编辑技术发展落后。当前构建小鼠肺高压模型最常用的方法是慢性缺氧[6],但该法通常仅造成轻度肺高压,缺乏新生内膜形成和丛状损害,且从临床分类来讲,低氧引起的肺高压属于第三大类,不属于PAH,因此不能满足重度PAH的研究需求。鉴于左肺切除联合MCT注射可成功诱导SD大鼠重度PAH模型的优点,以及小鼠肝脏中缺乏将MCT转变为其活性形式脱氢野百合碱(monocrotaline pyrrole, MCTP)的肝酶[7]这一特点,本研究将药物注射方法改良为直接经颈静脉注射MCT的活性形式MCTP,拟在相同实验条件下比较左肺切除联合MCTP注射(left pneumonectomy combined with monocrotaline pyrrole injection, P+M)、MCTP注射和左肺切除3种方法构建小鼠PAH模型的差异,为PAH在基因层面的深入研究提供一种稳定可靠且可重复的小鼠模型构建方法。
1. 材料与方法
1.1. 动物分组及处理
采用59只SPF级C57BL/6雄性小鼠,10~12周龄,体质量24~30 g,由北京市维通利华实验动物技术有限公司提供。将59只小鼠随机分为4组,正常对照组(n=9):不做任何处理;左肺切除组(PE组,n=15):以2%异氟烷做空气诱导和维持麻醉,然后将小鼠仰卧位固定,左侧胸壁备皮消毒,止血钳沿左侧第5肋间隙顿性分离皮肤和肌肉,进入胸腔,轻轻夹住、牵拉并完全暴露左肺,结扎肺门处的肺动脉、肺静脉和支气管后切除左肺,将残端送回胸腔,缝合胸壁,碘伏消毒切口,术后常规饲养直至处死;脱氢野百合碱注射组(MCTP组,n=15):参照文献[8]在使用前将适量MCTP溶解于DMF中,配成质量浓度20 μg/μL工作液,用1%戊巴比妥钠腹腔麻醉小鼠后,在显微镜引导下用Hamilton微量注射器经右侧颈静脉缓慢注射MCTP 20 μg/g;左肺切除+脱氢野百合碱注射组(P+M组,n=20):左肺切除术后7 d经右侧颈静脉注射MCTP 20 μg/g。本动物实验获得四川大学华西第二医院实验动物伦理委员会的批准,所有操作均遵循《北京实验动物管理条理(1996)》和美国国立卫生院(National Institutes of Health, NIH)《实验动物饲养管理和使用指南》(Guide for the Care and Use of Laboratory Animals)的相关规定。所有实验动物术前术后均在四川大学华西第二医院SPF级实验动物中心饲养。
1.2. 检测指标
1.2.1. 右心室收缩压(right ventricular systolic pressure, RVSP)测定
所有动物均在肺切除术后第21天、35天和49天进行测压。用1%戊巴比妥钠(0.01 mL/g)腹腔麻醉小鼠后仰卧位固定,剪开颈部皮肤,充分暴露气管,做T形切口,气管插管后连接呼吸机维持通气。开胸暴露心脏,测压穿刺针经心尖缓慢穿刺进入右心室,采用BL-420N生物信号采集与分析系统记录波形及数据,待压力波形稳定至少10 s后拔出穿刺针,记录压力数据和波形图。
1.2.2. 右心室肥厚指数(right ventricle hypertrophy index, RHI)测定
每次测压完成后取出心脏(每个组每个时间点至少保证处死3只小鼠),分离出右心室(right ventricle, RV)、左心室和室间隔(left ventricle and ventricular septum, LV+S),洗净血液,用滤纸吸净表面水分后分别称重,根据公式RHI=RV/LV+S计算得到RHI值以反映右心室肥厚程度。
1.2.3. 肺组织的准备
每次取出心脏的同时取出右肺,用2 mL生理盐水通过肺动脉冲洗剩余的血液,然后用体积分数为4%多聚甲醛固定液在10 cm H2O(1 cm H2O=0.098 kPa)压力下经气管缓慢注入肺内,使肺逐渐充盈膨胀,约2 min后切除右肺,置于20 mL体积分数为4%多聚甲醛中在4 ℃条件下固定24~48 h;常规石蜡包埋、切片,进行HE、Elastin Van Gieson(EVG)和α-SMA IHC染色。
1.2.4. 肺小动脉中膜厚度百分比
光镜(×400)下观察EVG染色切片,每只小鼠随机观察10~15个高倍视野并采集图片。在Image-Pro plus图像采集处理系统中,用光标勾划出血管内、外弹力板的轮廓,分别测量内、外弹力板的平均直径,计算肺小动脉的中膜厚度(内、外弹力板之间的厚度,wall thickness, WT)和血管外径(外弹力板的平均直径,diameter of elastic plate, ED),然后根据公式计算中膜厚度百分比:WT%=(2×WT/ED)×100%[9]。每只小鼠连续测量15~30根直径30~200 μm肺动脉,计算WT%平均值。
1.2.5. 非肌性肺小动脉的肌化程度
光镜(×400)下观察α-SMA IHC染色切片,每只小鼠随机观察10~15个高倍视野并采集图片,统计15~30根直径15~50 μm的腺泡内小动脉。根据α-SMA染色程度,统计完全肌化(中膜有完整的肌层)、部分肌化(中膜肌层不完整)和无肌化的小动脉(没有明显的肌层)数量[10],根据公式计算非肌性肺小动脉的肌化程度:非肌性肺小动脉的肌化程度=(完全肌化+部分肌化的血管数)/观察血管总数×100%。
1.2.6. 肺小动脉新生内膜形成情况及管腔阻塞程度评分(vascular obstruction score, VOS)
光镜(×400)下观察EVG染色切片,每只小鼠随机观察10~15个高倍视野并采集图片,统计15~30根直径15~50 μm的腺泡内小动脉,观察新生内膜形成情况;并进行血管腔阻塞程度评分:无新生内膜形成为0分,管腔阻塞<50%为1分,管腔阻塞>50%为2分[11]。
1.3. 统计学方法
呈正态分布的计量资料采用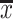 ±s表示,多组间比较采用单因素方差分析,组间两两比较采用LSD法。不服从正态分布的计量资料采用M(P25~P75)表示,多组间比较采用 Kruskal-Wallis H秩和检验, 组间两两比较采用bonfferoni法。计数资料用率表示,组间比较采用χ2检验。P<0.05为差异有统计学意义。
±s表示,多组间比较采用单因素方差分析,组间两两比较采用LSD法。不服从正态分布的计量资料采用M(P25~P75)表示,多组间比较采用 Kruskal-Wallis H秩和检验, 组间两两比较采用bonfferoni法。计数资料用率表示,组间比较采用χ2检验。P<0.05为差异有统计学意义。
2. 结果
2.1. 大体表现
4组小鼠在整个实验过程中均未出现明显呼吸急促、活动减少、毛发耸立和脱落等表现。在切肺或者给药后1周内,3个模型组小鼠体质量与实验前自身体质量比较均有下降,随着时间推移,体质量逐渐增加。到实验结束(第35天)时,与实验前自身体质量比较,PE组小鼠体质量增长良好,而MCTP组和P+M组小鼠体质量增加不明显甚至有轻微降低,低于同期对照组小鼠体质量(P<0.001,图1)。
图 1.

Changes in body mass in each group before and after the experiment initiated on day 35 (n=9)
各组动物实验前及实验第35天时体质量(n=9)
*** P<0.001, vs. control group before experiment; ☆ P<0.001, vs. control group after experiment.
2.2. 血流动力学指标
在实验期间,PE组RVSP均值波动在20.5~25 mmHg(1 mmHg=0.133 kPa),较对照组均值(16.17~17.67 mmHg)略有升高,但差异无统计学意义(P>0.05);MCTP组RVSP均值波动在25.5~29.5 mmHg,较对照组升高(P<0.05);P+M组RVSP均值在29~41 mmHg之间,较对照组和PE组均升高(P<0.05),其中以肺切除术后第35天RVSP升高最为显著(P<0.05,图2A)。
图 2.

RVSP and RHI values in each group (n=3)
各组小鼠RVSP和RHI值(n=3)
* P<0.05, ** P<0.01, vs. control group; △ P<0.01 , vs. PE group; ★ P<0.05 , vs. MCTP group.
2.3. 右心室肥厚程度
从肺切除术后第21天开始,3个模型组的RHI均有不同程度升高,PE组RHI均值范围为0.28~0.29,与对照组(0.26~0.27)比较,差异无统计学意义(P>0.05)。MCTP组RHI均值波动在0.30~0.35,但仅第35天(0.35±0.04)较对照组(0.26±0.02)升高有统计学意义(P<0.01)。P+M组RHI均值在0.35~0.42之间,3个时间点均较对照组和PE组升高(P<0.01)。P+M组RHI随着时间推移有进行性升高趋势,但进一步分析发现差异并无统计学意义(P>0.05,图2B)。
2.4. 肺小动脉中膜增厚程度
实验中发现肺切除术后第35天各组小鼠RVSP最高,且MCTP组和P+M组显示有不同程度右心室肥厚,故选择该时间点观察肺动脉中膜厚度。通过镜下观察(图3)到对照组肺动脉管壁薄,血管周围无炎症细胞。PE组肺动脉形态结构和管壁厚度与对照组无直观视觉上差异,血管周围未见炎性细胞。MCTP组肺动脉轻微增厚,血管周围可见少许炎性细胞。P+M组肺动脉管壁呈明显偏心性或同心圆性增厚,血管中膜平滑肌层增生,管腔变窄,血管周围可见较多炎性细胞浸润。
图 3.

HE and α-SMA IHC staining images
HE和α-SMA IHC染色图片
A-D: HE staining, ×400; E-H: α-SMA IHC staining, ×400.
PE组WT%中位数为23.79%,较对照组(23.42%)无增厚(P>0.05)。MCTP组WT%中位数为33.56%,较对照组和PE组均升高 (P<0.001);P+M组肺动脉增厚最为明显,WT%中位数高达50.18%,高于对照组、PE组和MCTP组(P<0.001,表1)。
表 1. Comparison of pulmonary artery media thickness percentage, muscularization degree and VOS.
肺动脉中膜厚度百分比、肌化程度和VOS比较
| Group | n | WT% (median [P25-P75]) | Muscularization degree/%, 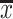 ±s ±s
|
VOS (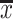 ±s) ±s) |
| *** P<0.001, vs. control group; # P<0.001, vs. PE group; & P<0.001, vs. MCTP group. | ||||
| Control | 3 | 23.42 (18.44-29.74) | 34.09±2.27 | 0 |
| PE | 4 | 23.79 (19.65-29.64) | 38.64±1.53 | 0 |
| MCTP | 3 | 33.56 (26.70-38.40)***, # | 63.73±1.03***, # | 0 |
| P+M | 3 | 50.18 (39.38-66.85)***, #, & | 94.50±6.19***, #, & | 1.25±0.80***, #, & |
2.5. 非肌性肺动脉肌化程度比较
对照组肺动脉肌化程度为(34.09±2.27)%,PE组为(38.64±1.53)%,两组比较无明显差异(P>0.05);MCTP组为(63.73±1.03)%,高于对照组和PE组(P<0.001);P+M组肺动脉肌化程度高达(94.50±6.19)%,高于对照组、PE组和MCTP组(P<0.001,表1)。与对照组和PE组比较,MCTP组和P+M组中完全肌化肺动脉所占比例显著增加(图4)。
图 4.

The degree of pulmonary artery muscularization in each group
各组实验动物肺动脉肌化程度
2.6. 小肺动脉新生内膜形成情况及VOS比较
对照组(图5A)、PE组和MCTP组均未观察到新生内膜形成,VOS均为0分。P+M组中可见小肺动脉的内弹力板内侧与血管内皮之间有少量新生内膜形成,造成管腔明显狭窄(图5B),VOS为1.25±0.80,与对照组比较差异有统计学意义(P<0.001,表1)。进一步做α-SMA IHC染色,结果示新生内膜染色为阳性(图5C)。
图 5.

Neointima formation in small pulmonary arteries
小肺动脉新生内膜形成
A: Control gourp; B: The red arrow indicated that the neointima formation in the small pulmonary artery in the P+M group, which subsequently caused obvious stenosis of the lumen; C: The red arrow indicated the positive α-SMA IHC staining in the neointima of the small pulmonary artery in the P+M group. A and B: EVG staining, ×400; C: α-SMA IHC staining, ×400.
3. 讨论
PAH是一种多因素以及可在多种临床状态下发生的肺血管疾病,肺血管重构是其核心病理改变,随着疾病进展肺循环阻力进行性增加,最终引起右心衰竭而出现不良结局[12]。在PAH的早期,其组织学改变并无特异性,以肺动脉中膜平滑肌细胞的肥大增厚,外膜增厚为主要表现,同时可见非肌性肺动脉的部分肌化或完全肌化。随着疾病进展到更严重的阶段,则出现重度PAH的特征性病理改变:丛状损害和新生内膜,导致中小肺动脉严重管腔狭窄甚至阻塞,引起流经肺动脉的血流严重受限[13-14]。丛状病灶通常发生于直径小于300 μm的小动脉分支的远端,是由小而扁平的内皮细胞和肌成纤维细胞构成的裂隙样通道状管腔丛[15-16]。尽管丛状损害是重度PAH的特征性改变,但也有研究发现在PAH的晚期阶段,丛状损害并不总是存在[17]。新生内膜是严重PAH的肺血管内皮与内弹力板之间形成的一层新内膜,是进行性的、不可逆的重度PAH区别于可逆的轻中度PAH的特征性病理学改变。从病理学角度上讲,相对于丛状病变,新生内膜对于明确重度PAH的特异性更高,指向性更强,因此我们认为新生内膜形成是重度PAH的标志,也是判断PAH动物模型是否达到重度的标准。目前国内鲜有报道重度PAH小鼠模型的构建方法,基于以小鼠为实验动物的优势众多,本研究在同等条件下采用左肺切除、MCTP颈静脉注射和左肺切除联合MCTP注射三种方法构建小鼠PAH模型,并观察肺血管重构等组织病理学特点,结果发现:单纯左肺切除并不能诱导小鼠PAH形成,经颈静脉注射MCTP可引起以肺动脉中膜增厚为特点的轻度PAH,而将两者结合则可诱导以新生内膜形成为特征的重度PAH。
左肺切除法诱导构建PAH的理论基础是:单纯一侧肺切除后可导致剩余肺的血流量增加。在本实验中观察到的结果是切除左肺后仅仅会引起RVSP的轻微升高,并无右心室肥厚和中膜增厚、非肌性肺动脉肌化程度增加等组织病理学改变,说明单纯切除左肺的方法不能诱导PAH小鼠模型的建立。该模型中的高肺血流与人类左向右分流型先天性心脏病的血流动力学变化一致,而本实验得到的结果也与临床观察到的实际情况较为吻合:来自荷兰的一项研究报道,6000名先天性心脏病患者中,PAH的患病率仅4.2%;以房间隔缺损和室间隔缺损为代表的间隔缺损性先天性心脏病,因长期左向右分流导致右肺血流量增加,仅6.1%会发生PAH,而绝大多数肺动脉压力均正常或仅表现为暂时性轻微升高,经手术关闭缺损后,肺动脉压力逐渐恢复正常[18]。上述动物实验结果和临床数据均表明,单纯肺血流量增多引起的血流动力学改变可引起肺动脉压力的轻微升高,但引起以肺血管重构为病理特征的PAH的几率较小。
MCT是一种吡咯啶型生物碱,引起PAH的机制尚不十分清楚。目前普遍认同的观点是:MCT在肝脏中经P450单氧化酶转化为其活性形式——MCTP,MCTP随血液循环到达肺,沉积于肺血管,直接引起血管内皮损伤,损伤的内皮细胞释放多种细胞因子和炎性介质,诱导肺动脉平滑肌细胞的增殖和血管周围炎症,促进PAH的形成[19-20]。由于小鼠肝脏相对缺乏将MCT转化为MCTP的关键酶P450,因此小鼠对MCT并不敏感,故本实验直接使用MCT的活性形式MCTP造模。结果发现经右侧颈静脉注射MCTP后,小鼠的体质量增长不如正常小鼠良好,RVSP亦有轻微升高,右心室略为肥厚,组织学上有肺动脉中膜轻微增厚和非肌性小肺动脉肌化程度增加等轻度PAH改变,但无新生内膜形成,说明经右侧颈静脉注射MCTP可诱导形成轻度PAH,与直接用MCT诱导大鼠PAH模型所观察到的结果一致[3]。该结果说明药物造成的肺血管内皮损伤可以引起PAH的重要病理改变之一:中膜增厚,该病变主要是由肺动脉平滑肌细胞肥厚、增殖和血管外基质沉积引起。
左肺切除联合MCTP注射组动物的血流动力学改变为RVSP显著升高伴明显右心室肥厚,组织学上出现肺动脉中膜显著增厚、小肺动脉肌化程度增加、血管周围炎症、新生内膜形成导致管腔明显狭窄等一系列典型病理改变,尤其是新生内膜形成足以说明该方法诱导的PAH小鼠模型成功模拟了重度PAH的病理学特征,该模型可满足不同层次的实验需求。进一步对新生内膜做α-SMA IHC染色,结果显示染色阳性,提示新生内膜细胞具有平滑肌细胞的一些特性。但关于新生内膜细胞的来源问题,至今尚有争议。既往认为新生内膜细胞来自肺动脉平滑肌细胞,是平滑肌细胞向内皮下迁移和增殖的结果[21]。但最近研究发现在重度PAH小鼠模型上,内皮遗传谱系细胞在新生内膜中占主导,这些细胞同时表达平滑肌细胞的α-SMA和平滑肌肌球蛋白重链,由此认为PAH的新生内膜细胞主要来源于内皮基因谱系,但同时激活部分平滑肌基因的表达,具体机制尚不十分清楚,推测可能与肺血管内皮损伤后启动平滑肌相关基因的表达程序有关[8]。
本研究结果提示仅有肺血流动力学改变或血管内皮损伤均不能诱导新生内膜形成,无法构建重度PAH小鼠模型。有文献报道,MCT处理后的大鼠,无新生内膜形成,经锁骨下动脉和肺动脉吻合术造成肺动脉血流量增多后,则出现了新生内膜[22]。结合本实验研究结果,提示只有将两种因素联合使用才能诱导出以新生内膜形成为特征的重度PAH。这一结果支持OKADA等[3]在1997年提出的PAH中肺血管重构模式的形成理论:正常肺血管在发生内皮损伤后可促进平滑肌细胞肥厚增生引起中膜增厚,在此基础上若发生肺血管血流动力学改变,才会进一步引起新生内膜形成。因此,重度PAH中新生内膜形成的必要条件是:肺血管剪切应力增加和血管内皮损伤,两者缺一不可。肺血管剪切应力增加可激活细胞内多种信号调节因子的释放和表达,如血小板源性生长因子[23-24]、丝裂原激活蛋白激酶、基质金属蛋白酶[25]等。而血管内皮损伤后,屏障功能受损,释放5-羟色胺、内皮素-1、血管紧张素Ⅱ等多种细胞因子和炎性介质[26-27]。两种因素联合,在细胞因子和炎症介质的相互作用以及内皮细胞和平滑肌细胞、成纤维细胞的相互影响下,逐渐发生血管重构,促进重度PAH形成。
总之,本研究发现了一种可靠的构建重度PAH小鼠模型的方法:左肺切除联合颈静脉注射MCTP,该模型能较好模拟人重度PAH的特征性病理改变:新生内膜形成。结合用小鼠做疾病模型的优势在于可做转基因小鼠,进行基因过表达和基因敲除,因此该模型为更深入地探讨严重PAH的分子发病机制,为进一步寻找新的治疗靶点提供更为合适且优越的动物模型,值得推广。
* * *
利益冲突 所有作者均声明不存在利益冲突
Funding Statement
国家自然科学基金面上项目(No. 82170067)和四川省科技计划项目(No. 2019YJ0024)资助
Contributor Information
丽 古 (Li GU), Email: 459208715@qq.com.
莉 余 (Li YU), Email: yulischuaxi@scu.edu.cn.
瀚旻 刘 (Han-min LIU), Email: liuhm@scu.edu.cn.
References
- 1.JIANG X, LI T, SUN J, et al SETD3 negatively regulates VEGF expression during hypoxic pulmonary hypertension in rats. Hypertens Res. 2018;41(9):691–698. doi: 10.1038/s41440-018-0068-7. [DOI] [PubMed] [Google Scholar]
- 2.BOURGEOIS A, LAMBERT C, HABBOUT K, et al FOXM1 promotes pulmonary artery smooth muscle cell expansion in pulmonary arterial hypertension. J Mol Med (Berl) 2018;96(2):223–235. doi: 10.1007/s00109-017-1619-0. [DOI] [PubMed] [Google Scholar]
- 3.OKADA K, TANAKA Y, BERNSTEIN M, et al Pulmonary hemodynamics modify the rat pulmonary artery response to injury. A neointimal model of pulmonary hypertension. Am J Pathol. 1997;151(4):1019–1025. [PMC free article] [PubMed] [Google Scholar]
- 4.NISHIMURA T, FAUL J L, BERRY G J, et al 40-O-(2-hydroxyethyl)-rapamycin attenuates pulmonary arterial hypertension and neointimal formation in rats. Am J Respir Crit Care Med. 2001;163(2):498–502. doi: 10.1164/ajrccm.163.2.2006093. [DOI] [PubMed] [Google Scholar]
- 5.FAUL J L, NISHIMURA T, BERRY G J, et al Triptolide attenuates pulmonary arterial hypertension and neointimal formation in rats. Am J Respir Crit Care Med. 2000;162(6):2252–2258. doi: 10.1164/ajrccm.162.6.2002018. [DOI] [PubMed] [Google Scholar]
- 6.CHEN T, ZHOU G, ZHOU Q, et al Loss of microRNA-17 approximately 92 in smooth muscle cells attenuates experimental pulmonary hypertension via induction of PDZ and LIM domain 5. Am J Respir Crit Care Med. 2015;191(6):678–692. doi: 10.1164/rccm.201405-0941OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.GOTO J, ISHIKAWA K, KAWAMURA K, et al Heme oxygenase-1 reduces murine monocrotaline-induced pulmonary inflammatory responses and resultant right ventricular overload. Antioxid Redox Signal. 2002;4(4):563–568. doi: 10.1089/15230860260220058. [DOI] [PubMed] [Google Scholar]
- 8.QIAO L, NISHIMURA T, SHI L, et al Endothelial fate mapping in mice with pulmonary hypertension. Circulation. 2014;129(6):692–703. doi: 10.1161/CIRCULATIONAHA.113.003734. [DOI] [PubMed] [Google Scholar]
- 9.HOSHIKAWA Y, ONO S, SUZUKI S, et al Generation of oxidative stress contributes to the development of pulmonary hypertension induced by hypoxia. J Appl Physiol (1985) 2001;90(4):1299–1306. doi: 10.1152/jappl.2001.90.4.1299. [DOI] [PubMed] [Google Scholar]
- 10.SCHERMULY R T, KREISSELMEIER K P, GHOFRANI H A, et al Chronic sildenafil treatment inhibits monocrotaline-induced pulmonary hypertension in rats. Am J Respir Crit Care Med. 2004;169(1):39–45. doi: 10.1164/rccm.200302-282OC. [DOI] [PubMed] [Google Scholar]
- 11.NISHIMURA T, VASZAR L T, FAUL J L, et al Simvastatin rescues rats from fatal pulmonary hypertension by inducing apoptosis of neointimal smooth muscle cells. Circulation. 2003;108(13):1640–1645. doi: 10.1161/01.CIR.0000087592.47401.37. [DOI] [PubMed] [Google Scholar]
- 12.MONTANI D, JAÏS X, SITBON O, et al Pulmonary arterial hypertension. Rev Mal Respir. 2005;22(4):651–666. doi: 10.1016/S0761-8425(05)85616-5. [DOI] [PubMed] [Google Scholar]
- 13.MEYRICK B The pathology of pulmonary artery hypertension. Clin Chest Med. 2001;22(3):393–404. doi: 10.1016/S0272-5231(05)70279-3. [DOI] [PubMed] [Google Scholar]
- 14.PIETRA G G, CAPRON F, STEWART S, et al Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):25S–32S. doi: 10.1016/j.jacc.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 15.SMITH P, HEATH D Electron microscopy of the plexiform lesion. Thorax. 1979;34(2):177–186. doi: 10.1136/thx.34.2.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.SMITH P, HEATH D, YACOUB M, et al The ultrastructure of plexogenic pulmonary arteriopathy. J Pathol. 1990;160(2):111–121. doi: 10.1002/path.1711600204. [DOI] [PubMed] [Google Scholar]
- 17.PIETRA G G, EDWARDS W D, KAY J M, et al Histopathology of primary pulmonary hypertension. A qualitative and quantitative study of pulmonary blood vessels from 58 patients in the National Heart, Lung, and Blood Institute, Primary Pulmonary Hypertension Registry. Circulation. 1989;80(5):1198–1206. doi: 10.1161/01.CIR.80.5.1198. [DOI] [PubMed] [Google Scholar]
- 18.DUFFELS M G, ENGELFRIET P M, BERGER R M, et al Pulmonary arterial hypertension in congenital heart disease: An epidemiologic perspective from a Dutch registry. Int J Cardiol. 2007;120(2):198–204. doi: 10.1016/j.ijcard.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 19.GOMEZ-ARROYO J G, FARKAS L, ALHUSSAINI A A, et al The monocrotaline model of pulmonary hypertension in perspective. Am J Physiol Lung Cell Mol Physiol. 2012;302(4):L363–369. doi: 10.1152/ajplung.00212.2011. [DOI] [PubMed] [Google Scholar]
- 20.KUANG T, WANG J, PANG B, et al Combination of sildenafil and simvastatin ameliorates monocrotaline-induced pulmonary hypertension in rats. Pulm Pharmacol Ther. 2010;23(5):456–464. doi: 10.1016/j.pupt.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.NISHIMURA T, FAUL J L, BERRY G J, et al Simvastatin attenuates smooth muscle neointimal proliferation and pulmonary hypertension in rats. Am J Respir Crit Care Med. 2002;166(10):1403–1408. doi: 10.1164/rccm.200203-268OC. [DOI] [PubMed] [Google Scholar]
- 22.TANAKA Y, SCHUSTER D P, DAVIS E C, et al The role of vascular injury and hemodynamics in rat pulmonary artery remodeling. J Clin Invest. 1996;98(2):434–442. doi: 10.1172/JCI118809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MITSUMATA M, FISHEL R S, NEREM R M, et al Fluid shear stress stimulates platelet-derived growth factor expression in endothelial cells. Am J Physiol. 1993;265(1 Pt 2):H3–H8. doi: 10.1152/ajpheart.1993.265.1.H3. [DOI] [PubMed] [Google Scholar]
- 24.HSIEH H J, LI N Q, FRANGOS J A Shear-induced platelet-derived growth factor gene expression in human endothelial cells is mediated by protein kinase C. J Cell Physiol. 1992;150(3):552–558. doi: 10.1002/jcp.1041500316. [DOI] [PubMed] [Google Scholar]
- 25.SUN H W, LI C J, CHEN H Q, et al Involvement of integrins, MAPK, and NF-kappaB in regulation of the shear stress-induced MMP-9 expression in endothelial cells. Biochem Biophys Res Commun. 2007;353(1):152–158. doi: 10.1016/j.bbrc.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 26.BUDHIRAJA R, TUDER R M, HASSOUN P M Endothelial dysfunction in pulmonary hypertension. Circulation. 2004;109(2):159–165. doi: 10.1161/01.CIR.0000102381.57477.50. [DOI] [PubMed] [Google Scholar]
- 27.FARBER H W, LOSCALZO J Pulmonary arterial hypertension. N Engl J Med. 2004;351(16):1655–1665. doi: 10.1056/NEJMra035488. [DOI] [PubMed] [Google Scholar]