

Abstract
We develop an electrostatic map for the vibrational NH stretch (amide A) of the protein backbone with a focus on vibrational chiral sum frequency generation spectroscopy (chiral SFG). Chiral SFG has been used to characterize protein secondary structure at interfaces using the NH stretch and to investigate chiral water superstructures around proteins using the OH stretch. Interpretation of spectra has been complicated because the NH stretch and OH stretch overlap spectrally. Although an electrostatic map for water OH developed by Skinner and coworkers was used previously to calculate the chiral SFG response of water structures around proteins, a map for protein NH that is directly responsive to biological complexity has yet to be developed. Here, we develop such a map, linking the local electric field to vibrational frequencies and transition dipoles. We apply the map to two protein systems and achieve much better agreement with experiment than was possible in our previous studies. We show that couplings between NH and OH vibrations are crucial to the lineshape, which informs the interpretation of chiral SFG spectra, and that the chiral NH stretch response is sensitive to small differences in structure. This work increases the utility of the NH stretch in biomolecular spectroscopy.
Keywords: electrostatic map, NH stretch, chiral sum frequency generation spectroscopy, amide A
Graphical Abstract

Introduction
Of the four key vibrational normal modes of the protein amide group, the NH stretch mode (amide A) is perhaps the least studied. This is most likely because the NH group and the OH group of water vibrate in the same frequency range (3000–4000 cm−1), and in most vibrational spectra of biological systems, the water signal overwhelms the signal from the NH stretch. However, because the NH group participates in hydrogen bonding interactions with both other parts of the protein during the formation of secondary structures as well as the water around the protein, the NH stretch contains valuable information about the local environment. Unfortunately, this information is hidden in conventional vibrational spectroscopy, and it cannot be brought out with the use of D2O to shift the water response because the hydrogen atom of the NH group exchanges with water and shifts in the same way.
The NH stretch can be isolated in higher-order spectroscopies. Vibrational chiral-selective sum frequency generation spectroscopy (chiral SFG) with homodyne detection can detect the NH stretch and help characterize protein secondary structures.1, 2 Ultrafast methods such as two-dimensional infrared spectroscopy (2D-IR) can pick up the NH stretch through its interaction with other modes as a cross-peak.3, 4 Methods to model 2D-IR and chiral SFG of large systems are closely related,5, 6 so being able to model one method computationally corresponds to being able to model the other method as well. The key task is to find a way to relate local environments seen in molecular dynamics (MD) simulations to a fluctuating exciton Hamiltonian, which contains vibrational frequencies and couplings,5, 7–15 and thereby extract structural and dynamic information about molecular systems from spectra.
Simpson and coworkers have established symmetry-based chiral SFG theory to describe vibrational chiral SFG in the absence of electronic resonance as surface-selective in uniaxial (C∞) systems.16, 17 Over the past decade, chiral SFG has developed into a powerful technique for probing the secondary structure of chiral biomacromolecules at interfaces and the solvent around them.1, 18–25 There are aspects of chiral SFG that are not yet understood. For example, we do not have a straightforward mapping from chiral phase (signal sign) to dipole orientation, unlike in conventional (achiral) SFG. However, the chiral SFG response of the NH stretch can provide vibrational signatures for identifying the secondary structure of proteins,2 and the phase of the response correlates with the absolute chirality of protein.20 Hence, being able to simulate the chiral SFG response of the protein NH stretch can help interpret chiral SFG spectra of proteins and extract information about protein structure.
In addition, Petersen’s group used chiral SFG to demonstrate the existence of a chiral spine of hydration in the minor groove of DNA.26 Later, we showed that chiral SFG can also detect hydration structures around proteins.20–22 In particular, we demonstrated that this technique is specifically sensitive to the first hydration shell of water around a protein.25 However, the chiral OH signals produced by water overlap with the chiral NH signals produced by the protein.8, 27 Although signal at 3600–3800 cm−1 can be unmistakably assigned to the OH stretch, signal at lower frequencies may be due to the NH or OH stretch. Previously, we showed that isotope labeling with H218O allows identification of OH stretch peaks by red-shifting water peaks by roughly 12 cm−1.20, 22, 25 However, NH and OH interact with each other. Thus, the spectral perturbation induced by the 18O substitution may also be due to a delocalized mode involving OH and NH. Furthermore, the NH stretch peaks provide useful structural information as well as information about hydrogen-bonding interactions within the protein and between the protein and water. Hence, the ability to model the NH stretch spectra will reveal information about not only protein structures but also local interactions of water with the protein.
Vibrational spectra can be modeled computationally using MD simulations with an electrostatic mapping approach or with a direct dipole-polarizability time-correlation function approach.6, 8–12, 14, 15, 28–38 Recently we used the Skinner group’s OH stretch electrostatic map6, 8–10, 39–41 to predict chiral SFG signals produced by water around proteins. This map links the electric field along the OH bond at the OH hydrogen to the vibrational frequency, transition dipole, and transition polarizability. Although the map was developed for pure water,6, 9, 10, 42, 43 it can be extended to aqueous systems that include biomolecules and their atomic partial charges, as shown in our previous work.19, 20, 25 Of particular interest is LK7β, which forms a stable antiparallel β-sheet at the air-water interface. This system is an ideal model system for SFG, which is inherently surface selective.44–48 Although qualitative agreement was achieved between our computational and experimental spectra of water hydrating LK7β at interfaces,19, 20, 25 the lack of NH peaks in the computational spectra made comparison between experiment and theory challenging. Any insights hidden in the NH response remained elusive.
The Mukamel group has developed maps for the vibrational modes of the peptide bond group, including the NH stretch using N-methyl acetamide (NMA).49, 50 Although NMA contains all the relevant vibrational modes, it is very different from an actual protein environment. The frequencies extracted from this map range from ~3400 to 3500 cm−1. This range does not match the full range of frequencies exhibited by the NH stretch in biological systems.24, 27 An alternative approach is needed to model lower frequencies and understand complex biological environments.
Here, we report the development of an electrostatic map for the NH stretch of the protein backbone. Previous work has demonstrated that the NH signal produced by proteins is primarily due to the backbone.1, 24 Our map applies to all NH groups in the backbone except C-terminal amide (NH2) capping groups that most likely contribute little to the total NH response. This map, together with previously developed maps for the OH stretch, enables computational modeling of the full chiral SFG spectra, including both OH and NH modes and the coupling between them. These full spectra contain information about the local hydrogen-bonding environment and the protein-solvent interactions. In the future, our map may allow simulation of 2D-IR and related spectra that directly probe energy exchange between a protein and the surrounding solvent. Our map is validated using a combination of previously published and new experimental data. We focus on two closely related protein systems – LK7β (Ac-LKLKLKL-NH2) and LE7β (Ac-LELELEL-NH2). Each is a seven-residue peptide consisting of alternating nonpolar and polar residues. The nonpolar residue is leucine and the polar residue is positively charged lysine in LK7β or negatively charged glutamate in LE7β. These protein systems form antiparallel β-sheets at the air-water interface. We find that the map reproduces the experimental chiral SFG spectra of these systems quite well. Moreover, analysis of the spectra provides insights into subtle differences in hydrogen bonding between the two apparently similar protein systems. This analysis demonstrates that the NH group is a useful probe for the local environment in biological systems when heterodyne chiral SFG attenuates the overwhelming OH stretch background.
Computational methods
Designing the NH stretch electrostatic map.
Our NH stretch electrostatic map was based on Skinner’s method of mapping electric fields around water molecules to spectroscopic quantities (i.e., vibrational frequency, transition dipole, transition polarizability, vibrational couplings). In brief, Skinner’s method6 consists of running an MD simulation of bulk water using a water model such as TIP4P,51 extracting small water clusters from the trajectory, performing density functional theory (DFT) calculations to determine local vibrational frequencies and other quantities, and correlating these properties to the local electric field (as calculated from local point charges – this is distinct from the long range electric field that is typically calculated using the Ewald method). Importantly, rather than optimizing the geometry of the water cluster and using the Hessian to compute the harmonic frequencies, Skinner’s group stretched the O–H bond and solved the vibrational Schrödinger equation for the resulting one-dimensional potential energy curve.6 This procedure results in more accurate vibrational frequencies because it accounts for anharmonic effects and does not involve relaxing (i.e., changing) the configuration for which the electric field is measured.
The first step of our development of an NH stretch electrostatic map was to generate configurations of a relatively small protein that was amenable to DFT calculations but also captured the essence of protein secondary structure. As both α-helices and β-sheets, the two most common secondary structures in proteins, involve hydrogen bonds (H-bonds) between NH groups and carbonyl groups, we focused on reproducing this interaction. We selected a tetraalanine peptide that is able to fold on itself to produce an intramolecular NH/carbonyl H-bond between the first (C=O) and fourth (NH) alanine residue (Figure 1). Because the NH stretch is highly local,52 the “folded” structure could effectively simulate NH interactions that would occur in both α-helices and β-sheets. Capping groups (acetyl on the N-terminal, –NH2 on the C-terminal) were used to avoid charged terminus effects. To overcome the significant thermodynamic barriers to this kind of folding, we conducted a metadynamics enhanced sampling simulation with the distance between the NH nitrogen and carbonyl oxygen as the collective variable and collected configurations sampling the H-bonded state.53 The simulation was performed in explicit TIP4P-Ew water54 with ~1000 water molecules. See the SI for details about the metadynamics simulation.
Figure 1.
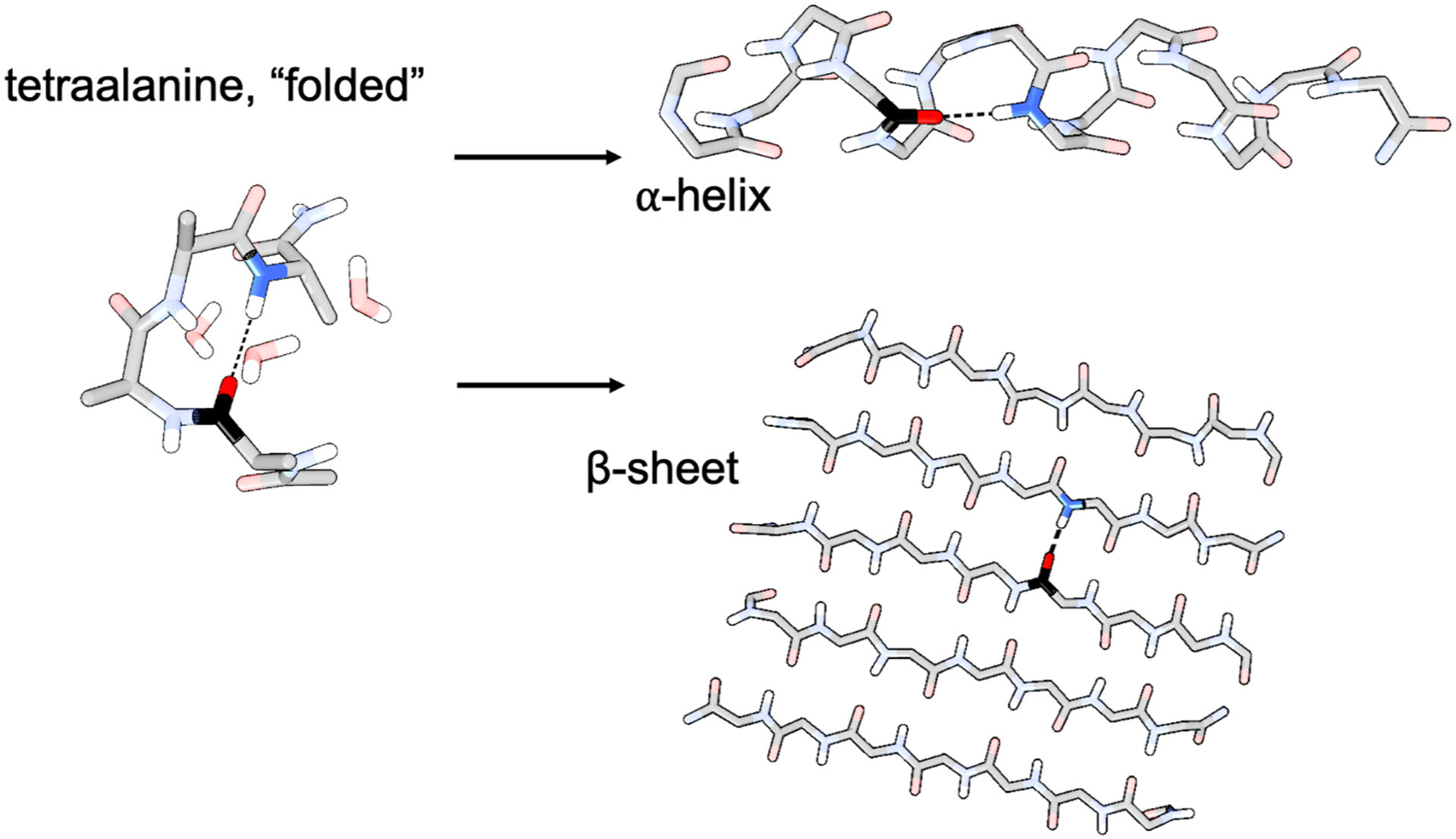
The mapping procedure relied on a small molecule (tetraalanine) mimic of the protein secondary structure environment, which is a H-bond between a backbone NH group and a backbone carbonyl group.
We also extracted configurations by sampling the non-H-bonded state and created a separate electrostatic map for that case. In total, we extracted 400 “H-bonded” and 400 “free” configurations. Note that in the free configurations, the NH group may still donate a H-bond to a water molecule. (Note that NH as an acceptor from water is vanishingly rare in this type of system.) We defined a H-bond using a heavy atom donor−acceptor distance cutoff of 3.2 Å and an angle greater than 135°. Because most free configurations of interest would involve NH groups on the outer edge of β-sheets or in unstructured loops, to ensure that the free configurations were not close to the H-bonded ones, we only considered free configurations where the donor−acceptor distance between the carbonyl oxygen and the amide nitrogen was greater than 4.5 Å, corresponding mostly to extended conformations of tetraalanine. Intermediate configurations (3.2–4.5 Å) were not considered for the maps.
After gathering these configurations, we extracted the entire peptide and the three closest water molecules to the fourth residue’s NH hydrogen. We then stretched the NH bond about the NH center of mass, keeping all other atoms frozen. The center of mass remained stationary during the stretch, so each atom moved in inverse proportion to its mass. The NH distance ranged from 0.58 Å to 1.70 Å over 15 evenly spaced configurations. We then performed single-point energy calculations on each configuration at the B3LYP/6–311G++(d,p)55–59 level of theory using Gaussian 1660 with default convergence criteria.6 We also included implicit water using the conductor-like polarizable continuum model (C-PCM) to account for polarization effects due to long range interactions with the bulk solvent.61–64 The inclusion of implicit water was necessary to bring the frequencies down to the experimental range (~3200–3500 cm−1). We then solved the one-dimensional Schrödinger equation for the resulting potential energy curve using the Fourier grid Hamiltonian method65 using 1,500 quadratically interpolated grid points for high accuracy. We used the reduced mass of NH as the effective mass because both atoms were moving about their center of mass. We used a scaling factor of 1.0006 for the frequency based on comparing an analogous calculation with NMA in vacuum to the experimental value cited by Mukamel and coworkers.49, 66 To estimate the NH dipole derivative, we also performed a constrained geometry optimization for each configuration, allowing only the hydrogen of the NH group to relax, followed by a dipole derivative calculation for the NH stretch mode.8
We then calculated the electric field at various points around the NH group. We found that computing the electric field at points slightly past the hydrogen atom along the NH bond led to better correlations. To maximize the accuracy of the map in complex environments, we sampled the electric field at 51 (3 × 17) points, with one group of 17 points centered at 1.0 Å past the N toward the H along the NH bond, a second group centered at 1.5 Å, and a third group centered at 2.0 Å (See Figure S1 for details). For each central point, the group of 17 points included 16 points located 0.1 and 0.2 Å in all four directions from the central point along y and z. Moreover, the electric field was sampled in three directions for a total of 153 electric field components. The directions were defined by the NH bond (unit vector x), the vector perpendicular to the NH bond vector and pointing toward the adjacent residue’s carbonyl carbon (unit vector y) and the cross product of these vectors, z = x × y. We used these directions to set up our 51 points at each center.
For both the design and application of this model, the electric fields were calculated from the solvent charges (i.e., water molecules and ions) and the charges of the bound carbonyl carbon and oxygen for the H-bonded configurations and from only the solvent charges for the free configurations. TIP4P-Ew54 charges were used for the water, standard parameters associated with the TIP4P-Ew model were used for the ions when present,67, 68 and the AMBER ff14SB force field69 was used for the carbonyl group charges. Solvent charges within 10 Å of the NH sampling points were included. The distance cutoff was applied to individual atoms. We found that including whole water molecules (if any atom was within 10 Å of the sampling point) produced essentially the same distribution of frequencies and transition dipoles as an individual atom cutoff (Figure S5).
During the model design stage, the calculated electric fields, as well as the frequencies and transition dipole derivatives, were used to train a random-forest regression model. The input is the 153 electric field components described above, and the output is the frequency and transition dipole derivative for the specified NH group. We chose this nonlinear model both because it offered better inference about the data than linear regression (measured by r2) and because it tends to give more reasonable predictions for rare cases where the input data falls outside the bounds of the training data. Such a model is robust to unexpected and complex environments. Random forest models tend to respond to an extreme input with an output close to the edge of that produced by the training data, which is a reasonable response for spectroscopy, where peaks do not generally shift past a certain point regardless of the extremes of the environment. In contrast, linear regression can produce wildly inaccurate predictions for even a small deviation from the training data bounds. However, the 153-point model was designed to capture as much local complexity as possible to avoid such edge cases. All r2 values from the model were above 0.90 (see the SI for details).
In addition, a linear regression model was designed to determine the transition, off-diagonal, position matrix element defined as
| (1) |
where ψ0 is the ground state vibrational wave function and ψ1 is the first excited vibrational state wave function. In the model, the input is the frequency of the specified NH mode, and the output is the transition position matrix element. The transition dipole could then be approximated to first order by multiplying the transition dipole derivative by the transition position matrix element. The combination of the random forest model and the linear regression model constitutes the NH mapping model developed herein. The SI provides more details about this model, including binaries that enable its direct use with Python.
To apply this model, an MD trajectory of the system is propagated using the same force fields as those used to train the model. For each configuration sampled, the electric fields induced by the solvent and ion charges, as well as the H-bonded carbonyl group charges for the H-bonded cases, are calculated for each NH group. The random forest regression map is used to determine the frequency and transition dipole derivatives from these electric fields, and the linear regression model is used to obtain the transition position matrix element from the frequency. These quantities can be used to compute IR, Raman, or SFG spectra. Note that the same map can also be used to compute higher-dimensional spectra from correlation functions, including 2D-IR. The development of the NH map is summarized in Figure 2.
Figure 2.
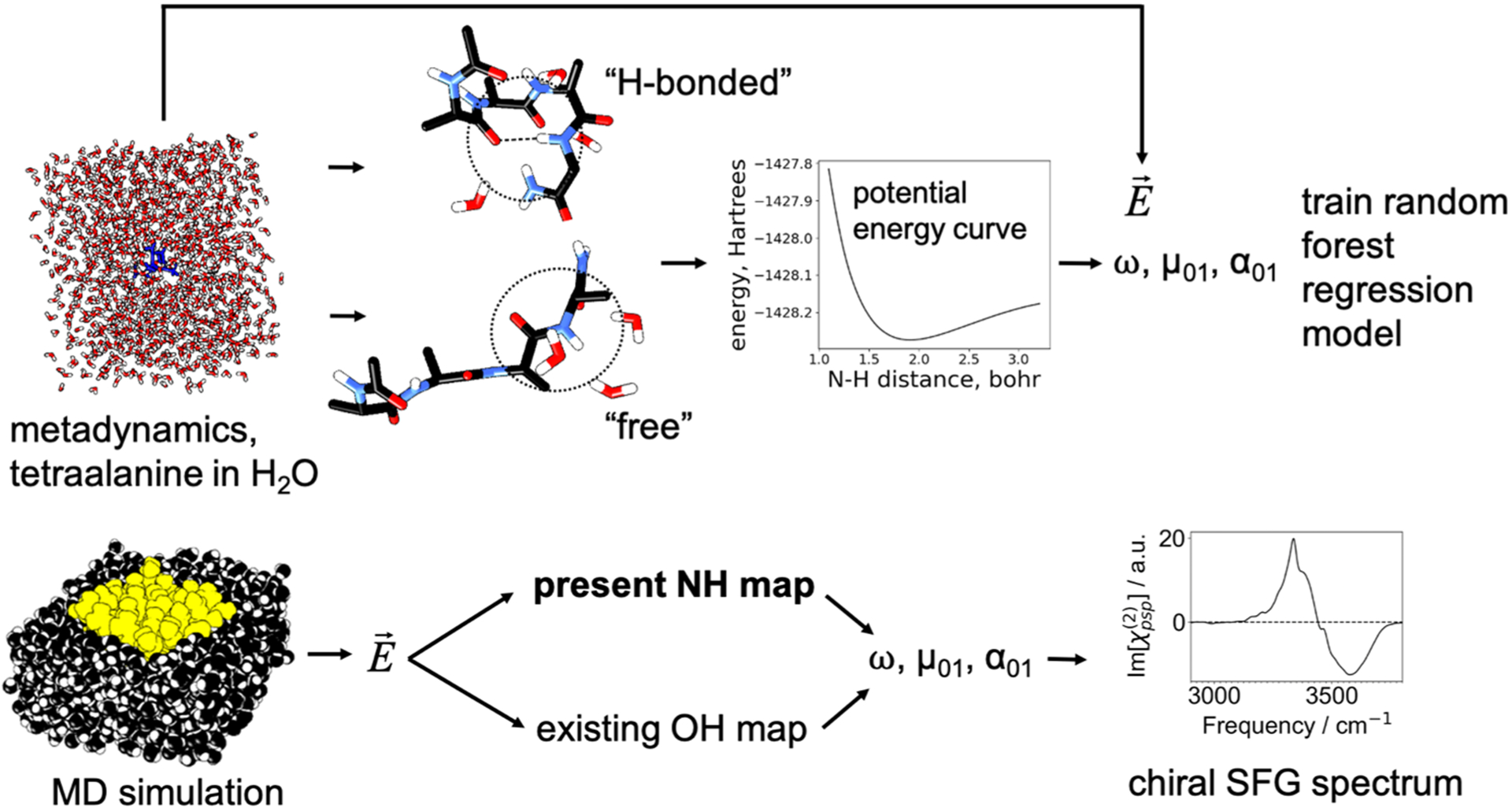
Top: H-bonded and free structures (400 each) were collected from a metadynamics simulation of tetraalanine in water, and each type of structure was used to produce a map for the corresponding type of environment. For a small cluster extracted from the system, the NH bond was stretched to map out a potential energy curve, and an anharmonic vibrational frequency was calculated. The transition dipole derivatives were estimated for a partially minimized structure of this cluster, and the transition dipole was approximated as the product of the dipole derivative and the transition position matrix element given in equation 1. The electric field was calculated near the NH group (Figure S1) from the MD simulation. The vibrational frequency and transition dipole were mapped to the electric field by a random forest regression model combined with a linear regression model. The transition polarizability was inferred partially from the transition position matrix element (equation 2). Bottom: The map was then used to estimate these quantities from an MD simulation of the protein (yellow) in aqueous solution (black), and a chiral SFG spectrum was generated.
Estimating the relative intensities of the NH and OH vibrational response.
When spectra due to both NH and OH responses are calculated, the relative magnitudes of quantities, such as the transition dipole and transition polarizability, are important. The polarizability was approximated as a bond polarizability, so that the transition polarizability is given by8, 70
| (2) |
where α01 is the transition polarizability tensor, x01 is given by equation 1, is the component of the bond polarizability parallel to the bond axis, is the component of the bond polarizability perpendicular to the bond axis, u is the bond unit vector, and i and j are indices or unit vectors representing the x, y, and z directions. The second term is zero for chiral SFG, where the relevant polarizability component is either zy or zx,12 so only the magnitude of the first term needs to be evaluated. For OH groups, Skinner and coworkers used an ratio of 5.6.8 The precise value of this ratio only controls the magnitude of the chiral spectrum and does not change the lineshape, so for NH we chose to use the same ratio. The ratio of the magnitudes of the polarizability derivative was estimated by calculating the transition polarizability derivative matrix associated with the OH stretch for water and the NH stretch for NMA in vacuum to obtain the isotropic transition polarizability derivative magnitude using43
| (3) |
where , , and are the diagonal components of the resulting tensor. This ratio (1.23/0.82) was then applied to the transition polarizability of NH as a prefactor in equation 2 to estimate the ratio of its magnitude to that of OH. Note that this ratio is quite close to unity, so the relative prominence of NH peaks in the combined NH and OH spectrum does not reflect a substantially larger transition dipole or polarizability, but rather the greater chiral ordering of the NH groups relative to the water and the contribution of only the first hydration shell to the water signal.25 If we had simply assumed the transition polarizability derivatives were the same, the predicted spectra would have been very similar.
Intermediate configurations.
The design of the map considered only groups that were very far from a recipient carbonyl group to be free NH. In real systems, however, occasionally there is a nearby carbonyl but the H-bond between it and an NH group does not quite conform to our definition of a H-bond (i.e., the heavy atom donor−acceptor distance is greater than 3.2 Å). In this case, we assigned the NH group in question to the H-bonded map as long as the heavy atom donor−acceptor distance was less than 3.4 Å.
Calculating chiral SFG spectra using MD trajectories with electrostatic maps.
Chiral SFG spectra were calculated from MD trajectories as previously reported by our group,19, 20, 25 using a method originally developed by Skinner and coworkers.41 In brief, the spectra were calculated using an inhomogeneous limit approximation, which neglects dynamical effects but is much more computationally efficient than the time-dependent approach.8, 35 In particular, the absorptive (imaginary) component of the second-order response tensor component is approximated as41
| (4) |
where ω is the response frequency, U is the eigenvector matrix of the exciton Hamiltonian, λ is the eigenvalue vector of the exciton Hamiltonian, α01 is the transition polarizability tensor, μ01 is the transition dipole vector, τ is the vibrational lifetime, and the indices a and b run over each OH and each NH group. The average is over all configurations sampled in the MD trajectory. The exciton Hamiltonian matrix in the local-mode basis contains vibrational frequencies on the diagonal and vibrational couplings on the off-diagonal. The technical details for the calculation of the couplings have been described in previous work.6, 8, 71 The transition dipole, transition polarizability, frequencies, and couplings are obtained from the electrostatic maps described above and in previous work.6, 8, 10, 19, 20, 25, 41, 43 To calculate chiral SFG spectra, the component must be obtained from a uniaxial system (i.e., a system with C∞ symmetry). To simulate the effect of a uniaxial system from an MD trajectory, the quantity was obtained as , as described in prior work.12
Experimental methods
The (L-) LK7β (Ac-Leu-Lys-Leu-Lys-Leu-Lys-Leu-NH2) (98.39% purity) and (L-) LE7β (Ac-Leu-Glu-Leu-Glu-Leu-Glu-Leu-NH2) (98.51% purity) peptides were obtained as lyophilized powders (GL Biochem Ltd., Shanghai). For heterodyne experiments on quartz substrate, stock solutions of (L-) LK7β at 1.0 mM and (L-) LE7β at 0.5 mM were made by dissolving the powders in H2O (Millipore Synergy UV-R system, 18.2 MΩ), or in H218O (97%, SIGMA-ALDRICH Co., St. Louis, MO). The quartz substrate used in these experiments was a right-handed z-cut α-quartz crystal (Conex Systems Technology inc., San Jose, CA), which was rinsed with copious H2O, dried with nitrogen, and plasma-cleaned (Harrick Plasma; PDC-32G) on “low” for 15 min. Samples were prepared by applying 5 – 10 μL of peptide solution directly onto the clean quartz substrate, which was subsequently dried in a sealed container, with desiccant. The quartz substrate was used as the phase reference to obtain internal heterodyne spectra using a broad-bandwidth SFG spectrometer72 following the procedure described previously.20, 25, 47 For the experiments at the air-water interface, 1 mM stock solutions of (L-) LK7β and (L-) LE7β were prepared. (L-) LE7β was dissolved in phosphate buffer (10 mM pH = 7.4) and (L-) LK7β was dissolved in H2O. Then, 100 μL of the stock solutions were added to 3.9 mL of 10 mM phosphate buffer (pH = 7.4) in a Teflon dish using a Hamilton syringe to give a final peptide concentration of 25 μM. The intensity of the main NH peak was monitored to determine when the system reached equilibrium, roughly two hours after addition of peptide. Homodyne spectra were then obtained (see the SI). All stock solutions of the peptides were prepared in batches and separated into aliquots, which were then flash frozen in liquid nitrogen and stored at –80 °C. Aliquots were thawed on the day of the experiment and discarded after use.
Results and discussion
Model protein systems.
After developing our NH map, we applied to it two related protein systems, LK7β and LE7β. LK7β is a seven-residue peptide consisting of alternating lysine residues (polar, hydrophilic) and leucine residues (nonpolar, hydrophobic).73 LK7β forms antiparallel β-sheets at the air-water interface, where the leucine residues point into the air and the lysine residues point into the water (Figure 3). LK7β is positively charged due to the lysines and has been used as a model system for SFG studies.44.74 LE7β is a closely related peptide that has negatively charged glutamate residues instead of lysines. LE7β is negatively charged but also forms β-sheets at the air-water interface. Folding at the interface makes LK7β and LE7β ideal model systems for chiral SFG, which is interface-selective. Computationally, we model LK7β and LE7β as pentamers consisting of antiparallel β-sheets surrounded by ~1350 water molecules at the vacuum-water interface. We also added 15 Cl− ions to the LK7β system and 15 Na+ ions to the LE7β system to neutralize the charged residues. We have previously published both experimental spectra and calculations of the OH stretch response for LK7β,20, 25 whereas LE7β is a new system. We chose LE7β as a second test system because although it is similar to LK7β, it has a different polar sidechain with an opposite charge. Our aim was to determine if we could detect any differences in the antiparallel β-sheet structure between the two systems experimentally and computationally. By so doing, we could generate experimental benchmarks to test our computational methods. We have achieved qualitative agreement with experiment for LK7β in the past.19, 20, 25 For example, the phase of our calculated spectra of water OH flips when the (D-) enantiomer is used instead of the native (L-) enantiomer, and the OH peaks appear approximately at the correct frequencies. However, the quantitative agreement was limited due to our inability to model the NH response of the protein backbone, which dominates the total chiral SFG response. Here, we test our new NH map with the LK7β and LE7β systems and compare to our experimental results.
Figure 3.
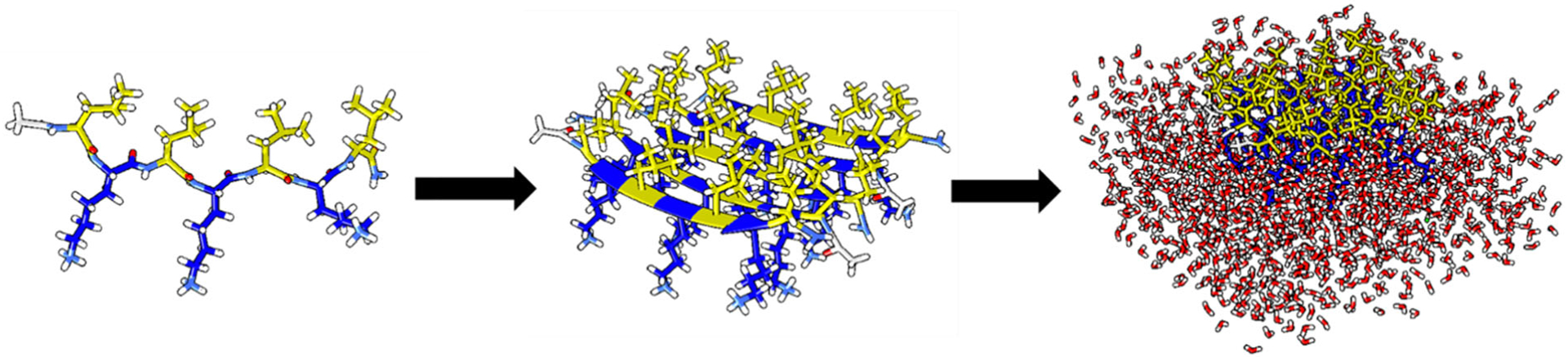
LK7β is a peptide (Ac-LKLKLKL-NH2) that assembles into antiparallel β-sheets at the air-water interface. Leucine residues are in yellow, lysine residues are in blue, and water molecules in the computational model are red. The polar lysine residues face into the aqueous phase, while the hydrophobic leucine residues face into the vacuum. LK7β is modeled as a five-strand β-sheet with ~1350 water molecules around it. LE7β forms the same structure (Figure S7), but with negatively charged glutamate residues facing the solvent instead of positively charged lysines.
Infrared (linear) response.
Although the NH response dominates chiral SFG, it is but a small component of the total vibrational response of these systems. Figure 4 shows the calculated IR spectrum of the LK7β system, for water alone (black) and for the whole system (NH and OH), including vibrational couplings between and among NH and OH groups (red). The two spectra are almost identical. Therefore, the NH stretch contributes very little to the IR signal. This is the central problem with using the NH group as a probe of local hydrogen bonding structure – it is obscured by the water response. Chiral SFG solves this problem because the water signal is heavily suppressed due to the small number of water molecules in the first hydration shell. In addition, water molecules are not intrinsically chiral, and their chirality is a higher-order and therefore more subtle effect.25 In contrast, the NH groups on the protein backbone are directly bonded to a chiral center, the α-carbon. Heterodyne detection further resolves NH and OH peaks from each other.
Figure 4.
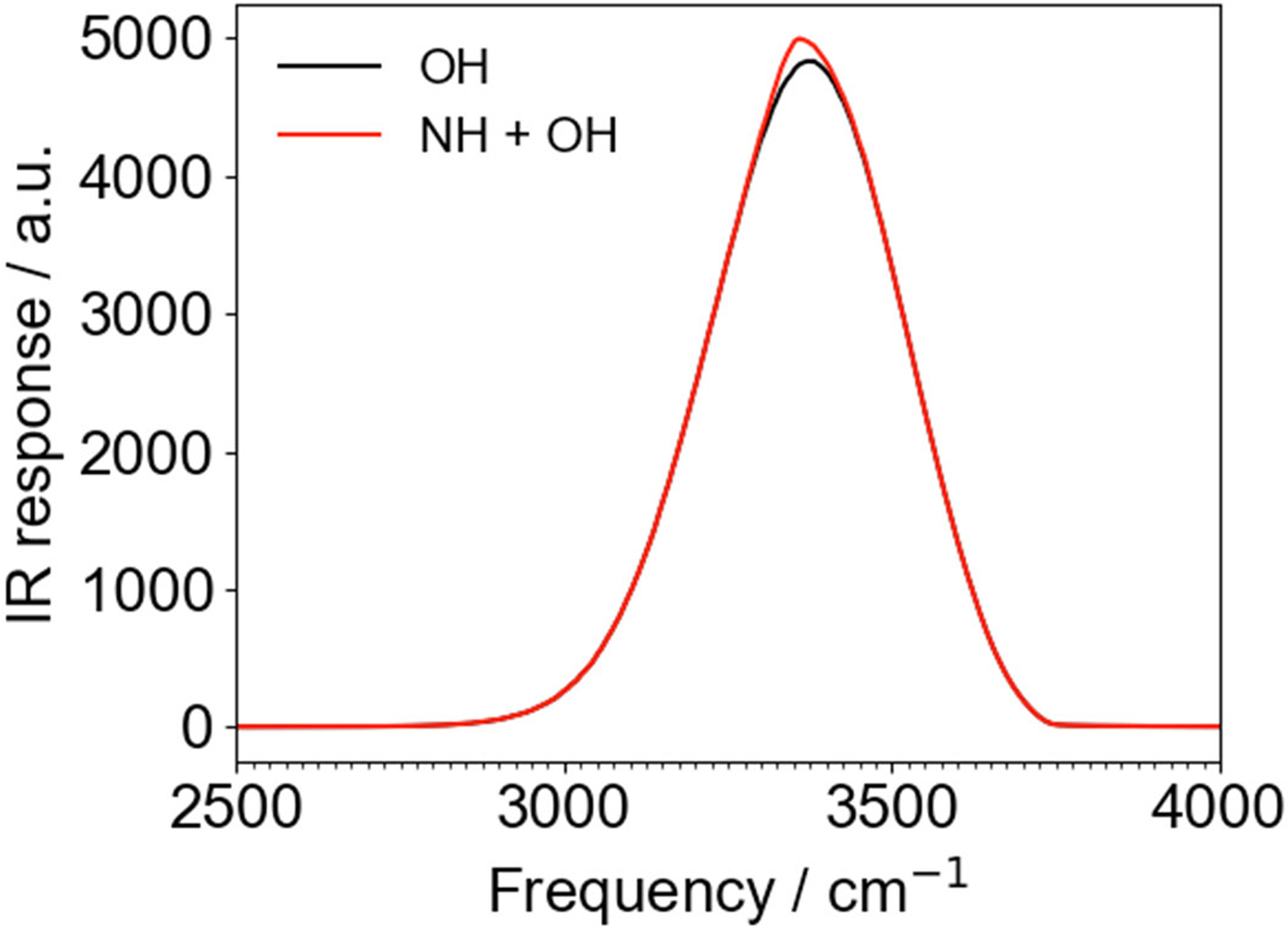
Computational IR spectra of the LK7β system for water alone (OH, black) and for NH + OH (red), including all vibrational couplings between and within each type of stretch. The NH stretch contributes very little to the total IR spectrum. Spectra were obtained from 1 ns of sampling (100,000 configurations) using Skinner’s time averaging approximation71 to achieve a realistic peak width. All vibrational couplings were included. a.u. = arbitrary units.
Comparing computational and experimental results.
After calculating the IR response with a combination of Skinner’s OH map and our new NH map, we moved on to chiral SFG. Figure 5 shows the results of our calculations for the heterodyne response of LK7β and LE7β. The calculated spectrum for LK7β (Figure 5a, i) consists of a positive, main peak at ~3336 cm−1, a negative peak at ~3575 cm−1, a small but distinct negative feature at ~3457 cm−1, and a positive shoulder at ~3387 cm−1. The calculated LE7β spectrum (Figure 5a, ii) consists of a small low-frequency negative peak at ~3243 cm−1, a shoulder at ~3303 cm−1, a main peak at ~3346 cm−1, a distinct but small negative feature at ~3467 cm−1, and a high-frequency negative feature at ~3610 cm−1. Comparison to heterodyne SFG experiments (Figure 5a, iii-iv) illustrates that we have achieved quite good agreement between experiment and theory for both systems, with both the main peaks and the subtler features. Almost every peak in the calculated spectra has a corresponding peak of the same phase and description in the experimental spectra. For example, the “main” positive peak in the LK7β spectra at 3336 cm−1 (Figure 5a, i) corresponds to a similar peak in the experiment at 3277 cm−1 (Figure 5a, iii). Similarly, the calculated spectra capture the subtler features, for example the small negative feature at 3457 cm−1 in the LK7β calculated spectra (Figure 5a, i) corresponds to a very similar feature at 3463 cm−1 in the experiment (Figure 5a, iii). The peak at ~3336 cm−1 in the LK7β spectrum is assigned to NH because it does not shift in the presence of H218O.25 The shoulder to the blue of the main peak in the LK7β spectrum and the small negative, kink-like, feature in both the LK7β and LE7β spectra are due to complex interactions between a broad OH peak and the main NH peak, as revealed in Figure 5b, i-iv. The high-frequency negative peak is due mostly to the OH stretch in both systems. The shoulder and the smaller, kink-like feature reveal how crucial it is to calculate the NH response as well as the OH response even if one is not interested in the protein response – some OH-related peaks appear only due to an NH/OH interaction. It is important to note that while the sign of a chiral SFG peak may depend in some way on orientation, there is currently no clear way to infer orientation from the sign of a peak, as is possible with some forms of conventional (achiral) SFG (for example, the ssp polarization setting).
Figure 5.

(a) Comparison of calculated chiral SFG spectra with heterodyne experimental results for LK7β and LE7β. Note the blue-shift of the main peak in LE7β vs. LK7β in both the experiment and the theory. All possible vibrational couplings are included. (b) NH-only and OH-only spectra, including only vibrational couplings within these groups. All peaks are labeled. Calculated spectra are averaged over 10,000,000 configurations spanning 100 ns. Data for LK7β in panel 5a, iii was obtained from Konstantinovsky, Perets et al.25 a.u. = arbitrary units.
The only major features missing in the calculated spectra are the low-frequency experimental signals at around 3125 cm−1 in the LK7β spectrum (Figure 5a, iii) and 3100 and 3175 cm−1 in the LE7β spectrum (Figure 5a, iv). This overall feature is common to both systems in the experiment and is perturbed when H218O is used as solvent, indicating a contribution from water.25 This feature may be due to a Fermi resonance or other interaction not considered in the computational model. However, it is unlikely to originate from the Fermi resonance between the OH stretch and the water bend, because the resonance usually leads to features with higher frequency (around 3200 cm−1).75 Indeed, we tried including this Fermi resonance phenomenologically as a vibrational coupling with the OH stretch following Skinner’s recent work.75 However, we could not computationally reproduce these low-frequency peaks (Figure S6). Our group continues to investigate the complex origins of these features.
The main peak in the theoretical and experimental heterodyne spectra (Figure 5), which is mostly due to the NH stretch, as revealed by comparison to the NH-only and OH-only peaks in Figure 5b, shifts by 10−15 cm−1 between LK7β and LE7β in both the experimental and calculated spectra. In the experiment, it shifts from 3277 cm−1 for LK7β to 3290 cm−1 for LE7β. In the computed spectra, the shift is from 3336 cm−1 to 3346 cm−1. This suggests that at least some NH groups in LE7β may experience weaker or fewer hydrogen bonds than the NH groups of LK7β. This is somewhat unexpected because both systems form stable antiparallel β-sheet structures as revealed by Fourier-transform infrared (FTIR) characterization (Figure S7) and MD simulation. However, we analyzed the average number of backbone H-bonds in the two simulated systems, and found that an LK7β pentamer typically contains ~20 “strong” H-bonds (r < 3.4 Å, angle > 135°) out of a possible ~24, whereas an LE7β pentamer contains only ~17 on average. There are roughly 24 possible H-bonds between the strands because there are roughly six possible H-bonds between each pair of strands and there are four inter-strand spaces in a pentamer. The average “strong” H-bond length in LE7β is also slightly longer (only by 0.005 Å, although this is statistically significant, p < 0.001 from a two-sided t-test). It seems that the lysine residues are a greater force for stabilizing the antiparallel β-sheet structure than are glutamate residues. Alternatively, the shift may be due to a more complicated interaction between the organizing forces of the nonpolar and polar residues. In any case, this finding illustrates how the NH stretch can be used as a sensitive probe of hydrogen bonding environments in proteins when its response is unmasked from the OH water stretches, even when the proteins are very closely related. These results demonstrate that it is possible to extract local structural information from the chiral SFG response of the protein NH stretch.
The chiral SFG spectra suggest that LE7β has a weaker antiparallel β-sheet structure than LK7β, but the differences do not stop there. Figure 5b shows that in the calculated OH-only spectra, the peaks due to the OH stretch are significantly red-shifted in the LE7β spectrum (Figure 5b, iv) compared to the LK7β spectrum (Figure 5b, iii) (from 3328 cm−1 to 3290 cm−1 for the positive peak, and from 3590 cm−1 to 3560 cm−1 for the negative peak). This suggests, perhaps paradoxically, that although LE7β has a weaker β-sheet structure, its interactions with and chirality transfer to water may be somewhat stronger than the analogous LK7β interactions. LE7β possibly gives up some of its inter-strand interactions in favor of interactions with water molecules. In any case, it is clear how, together, the OH and NH stretch responses reveal a detailed picture of the structural similarities and differences of the two related biological systems.
Theory matches experiments at the air-water interface.
The heterodyne experiments were done with a quartz surface as a phase reference. This is somewhat of a different environment compared to the computational model, where the antiparallel β-sheet pentamers of LK7β and LE7β are at the vacuum-water interface. We wanted to see if a true air-water interface experiment would agree with our theoretical predictions as to the difference between LK7β and LE7β. We obtained the homodyne chiral SFG spectra at the air-water interface with both LK7β and LE7β in the spectral region of 3000–3800 cm−1 and compared to the homodyne chiral SFG spectra predicted by the OH and NH maps (Figure 6). We find that the computational results match the experimental blue shift of the main peak for LE7β relative to LK7β (from 3340 to 3350 in the calculated spectra and from 3280 to 3305 in the experimental spectra) and correctly predict the lower-frequency peak to be by far the dominant feature. In the experiment the high-frequency response is very small, especially for LK7β, although it is not zero (see inset, Figure 6). In the calculated spectrum, the high-frequency features are larger relative to the main peak, although they are still much smaller than the main peak. Overall, the calculated and experimental results exhibit good qualitative agreement.
Figure 6.
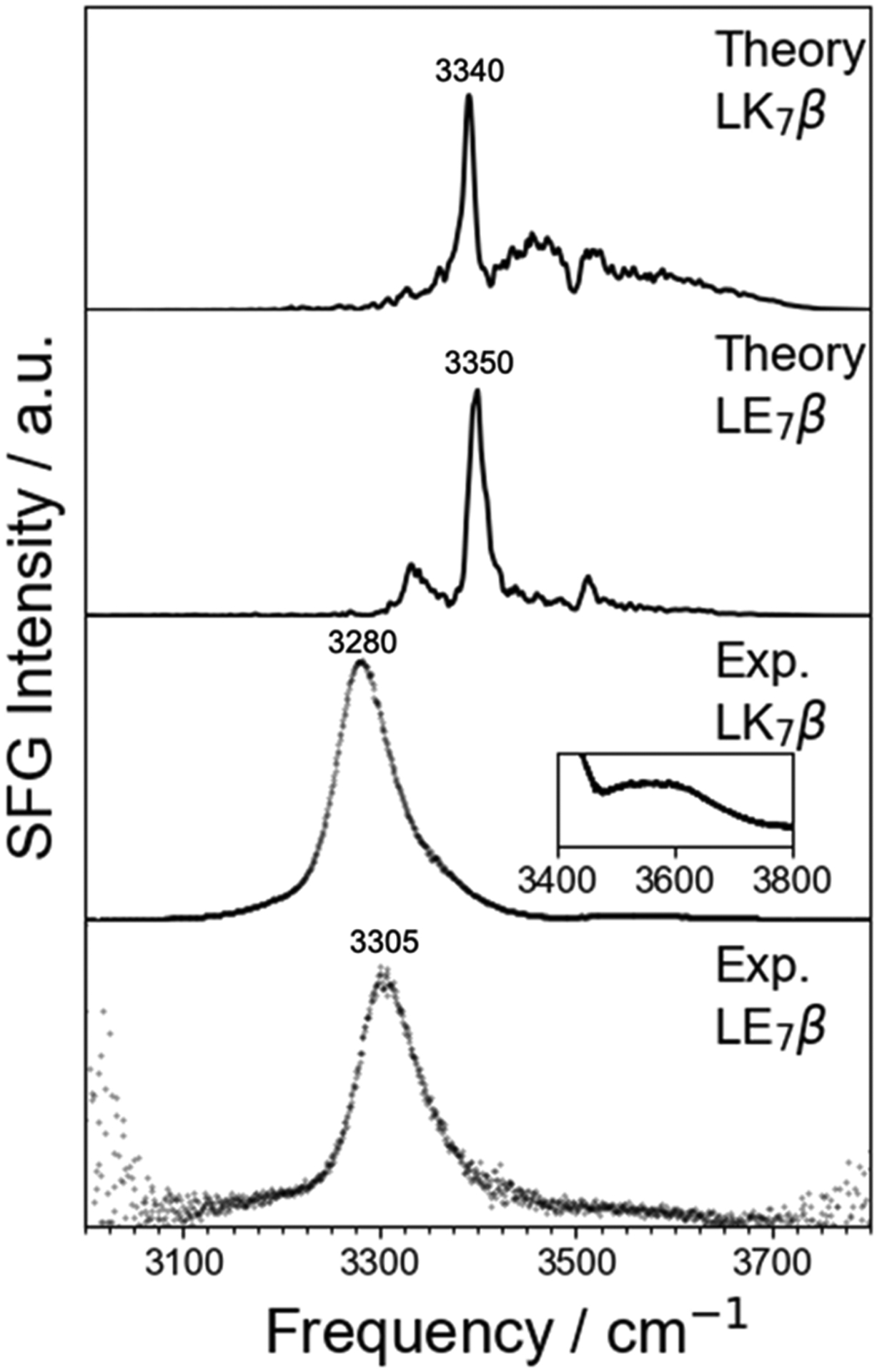
Comparison of calculated homodyne spectra and experimental homodyne results for LK7β and LE7β. Note the blue shift in both theory and experiment between LK7β and LE7β. The inset in the second plot from the bottom shows a detailed view of the high-frequency peak in the spectrum of LK7β that is in relatively low in intensity but undergoes some fluctuation (see Figure S12). Key peaks are labeled. The computational spectra are not smoothed because the key peaks are very sharp. Calculated spectra are averaged over 10,000,000 configurations spanning 100 ns. a.u. = arbitrary units.
Although our map was designed to be robust and sensitive to complex biological environments, there are features that may affect its accuracy. The map was designed to work primarily with solvent-adjacent secondary structures. As a result, deeply buried structures, for which water is far away, may not perform as well as structures closer to the solvent. On the other hand, the electric field in the H-bonded configurations was dominated by the H-bonded carbonyl group, so buried NH groups interacting with carbonyl bonds could be expected to perform well using this map. Our observation of a disagreement between experiment and theory for the lower-frequency peaks in the homodyne spectra (Figure 6) as well as in the heterodyne spectra (Figure 5) suggests that the differences are due to a deficiency of the map rather than a difference in experimental and computational setups. The map was derived using a small, computationally tractable peptide surrounded by water molecules and implicit water solvent. It is possible that this environment was too aqueous and did not faithfully reproduce the environment experienced by secondary structures with more hydrophobic surroundings. Future applications of the NH map to larger proteins with true hydrophobic cores will test this assertion further. It may be necessary to develop an extended NH map using less polar implicit solvents to mimic hydrophobic cores. Another possibility is that the tetraalanine peptide used to develop the map does not adequately sample strongly hydrogen-bonded configurations as much as an actual protein backbone samples such configurations. Future refinements of the map may weight strongly bound configurations more in the training of the model to capture low frequencies more faithfully. In any case, the agreement with experiment found in the present work represents a significant step forward in the modeling of chiral SFG. This map may also be extendable to model other kinds of vibrational spectroscopy, including higher-order techniques such as 2D-IR.
It should be noted that although the lower frequencies are less accurate than higher frequencies using the current NH map, the map correctly predicts the qualitative effect of a strong H-bonding environment. Figure 7 shows the average NH stretch frequency predicted by the current map for each NH group in LK7β. NH groups directly facing the solvent are shown as blue bars, and those facing inward are shown as yellow bars. It is clear, with a few exceptions, that the NH groups facing inward have a lower frequency than the NH groups facing the solvent. This is a result of a more stable H-bonding environment “inside” the structure compared to the exterior surface of the protein. The lowest average frequencies are found in the very center of the structure, as expected. Note that there are actually two different maps being used here (see Computational Methods), one for NH groups experiencing H-bonds with the protein and one for those not experiencing such H-bonds. Future studies using this map might reproduce Figure 7 for globular proteins and other structures with highly buried NH groups.
Figure 7.
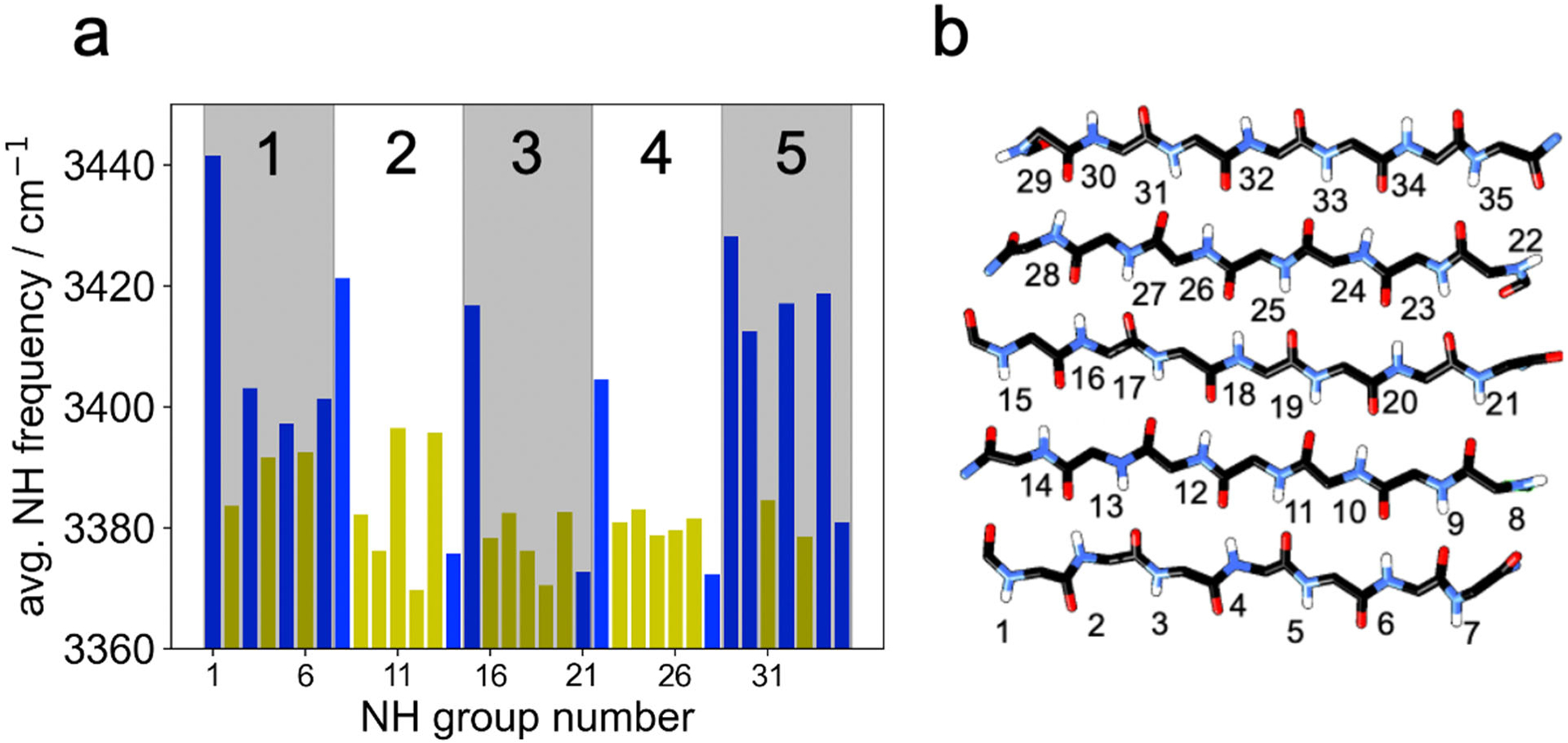
(a) Average NH frequencies in LK7β by NH group number computed using our map. The shading highlights the different β-strands, which are numbered at the top. The blue bars correspond to NH groups pointing into the solvent, and the yellow bars correspond to NH groups on the inside of the β-sheet. The average frequencies were based on analysis of 100,000 configurations spanning 100 ns. For the most part, the NH groups pointing toward the core of the LK7β pentamer (yellow) experience stronger H-bonding interactions than those facing the solvent (blue), corresponding to lower vibrational frequencies. (b) An illustration of the numbering scheme.
The role of vibrational couplings.
One of the central advantages of being able to model the NH stretch response is the newfound ability to model interactions between the NH and OH stretches, here modeled as transition dipole vibrational couplings.42 The data in Figure 8 shows the unexpected importance of these interactions for both systems we studied. The NH-only spectra for both systems look very little like the NH-derived components of the full spectra and exhibit poor agreement with experimental spectra. In particular, the NH-only spectra have a large negative peak at ~3352 cm−1 (Figure 5b,i-ii and Figure 8) that is not present in the full spectra. Similarly, the OH-only spectra have a large negative peak at ~3590 cm−1 (LK7β) and ~3560 cm−1 (LE7β) (Figure 5b,iii-iv and Figure 8) that is severely attenuated and reshaped in the full spectra, even though there is no corresponding positive NH peak that could attenuate these peaks by simple addition. In fact, adding the NH-only and OH-only responses produces “total” spectra that are much farther from the experimental spectra than the modeled spectra that include vibrational couplings (Figure 8, uncoupled spectra). This suggests that vibrational couplings between NH and OH groups are a key component of the chiral SFG response, which is perhaps not surprising given that vibrational couplings play a formative role in the chiral SFG response of water by itself for fundamental, symmetry-related, reasons.19 Although these findings do not negate the assignment of chiral SFG peaks as “NH” or “OH” (the couplings are much smaller in magnitude than the vibrational frequencies), they do suggest that an OH peak may contain the influence of an NH oscillation, and vice versa. This may not be surprising, as we previously found that the majority of the chiral SFG signals of water arises from the first hydration shell of the protein.25 In general, the true picture of these condensed-phase systems is one of highly delocalized vibrational excitons including many different molecular species. These findings show that vibrational chiral SFG is more than a way to enumerate molecular environments experienced by OH and NH groups. It is a way to study and document the complex interactions between these species. Thus, interactions that would produce a slight frequency shift (if that) in linear (e.g., IR) spectroscopy radically reshape chiral SFG spectra and are more visible as a result.
Figure 8.

The effect of vibrational coupling between NH and OH groups on the calculated chiral SFG spectra and comparison with experimental spectra. (a) Spectra for LK7β and (b) spectra for LE7β. Experimental data for LK7β in (a) was obtained from Konstantinovsky, Perets et al.25 a.u. = arbitrary units.
Treatment of hydrogen bonds of intermediate length.
When we gathered tetraalanine configurations for the H-bonded and free NH maps, we used a H-bond cutoff of 3.2 Å between the heavy atom donor and acceptor. This cutoff was chosen to ensure that the H-bonded configurations accurately represented the typical situation of an NH group inside a β-sheet or α-helix. Free configurations were gathered exclusively from unfolded tetraalanine molecules for the same reason. In real systems, however, H-bonds within a secondary structure are occasionally slightly longer but in all other ways resemble the H-bonded tetraalanine configurations more than the free configurations. These situations were treated using the H-bonded map rather than the free map, and the H-bond distance cutoff used for the calculation of the SFG spectra was 3.4 Å to capture these edge cases. We found that assigning the edge cases to the H-bonded versus free map did not significantly change the chiral SFG spectrum, except by making the OH-related shoulder at 3380 cm−1 more pronounced in the 3.4 Å case (Figure S2). This similarity also shows that the H-bonded and free maps generally agree quite well on the edge cases, where an inter-residue H-bond is nearly broken. This analysis suggests that the two maps cover the entire range of likely environments around an NH group with some degree of overlap.
The NH group as a useful vibrational probe.
Although a great deal of effort has been directed toward developing electrostatic maps for water and the amide modes,6, 8, 11, 12, 14, 15, 71 few groups have pursued the NH stretch.49 The main reason for the smaller number of models developed for the NH stretch is that this mode is difficult to observe experimentally due to the overwhelming OH stretch of water. This problem is largely solved by chiral SFG, where the NH signal is prominent due to the chiral nature of proteins and the enhanced peak resolution afforded by heterodyne detection. In contrast, the water signal is relatively small because only the water molecules in the first hydration shell contribute significantly to the chiral SFG signal from OH groups.25 Thus, chiral SFG is an ideal technique to study the environments experienced by NH groups in complex biological systems with the caveat that “NH” peaks may contain solvent contributions as well if they are not buried in a hydrophobic pocket. The presence of NH-like groups on amino acid sidechains is unlikely to cause problems for chiral SFG. The fact that we were able to reproduce almost all spectral features computationally for LK7β while completely ignoring the lysine NH3+ groups suggests that the high degree of chiral ordering of the backbone eclipses any signal from the flexible sidechains. We have also shown previously that the water molecules near the NH3+ groups produce minimal chiral SFG signal, possibly due to the ability of positive charge to disrupt water structure.25
Conclusion
The NH stretch map developed in this paper enables the simulation of full chiral SFG spectra of aqueous proteins in the 3000–4000 cm−1 range. The NH maps developed herein have distinct advantages over previously developed maps for modeling vibrational responses. First, they were designed to model the protein environment around the NH group using a tetraalanine peptide. Second, the design of these maps included the effect of solvent both explicitly (through the inclusion of nearby water molecules in the tetraalanine configurations) and implicitly (through C-PCM implicit solvation in the DFT calculations of the NH frequencies), thereby driving the NH frequency down to the experimental range. Third, the map is based on a complete survey of the local environment through the electric field at 51 points around the NH, including points beyond the NH bond. Finally, the dependence of the transition dipole moment on the local environment is included by mapping the transition dipole to the local electric field. However, one limitation of the current map is that it was created with the AMBER ff14SB force field69 and TIP4P-Ew54 water and therefore must be used with these force fields to ensure accuracy.
Most importantly, this work has revealed the NH stretch to be a potent reservoir of information about the local environment of the amide group, on par with the more-studied amide I mode. Chiral SFG is ideally suited to probing this mode, as the problems of OH interference almost disappear with this technique. One aspect of the OH stretch response that does not disappear, however, is the coupling of this response to the NH response. We show that coupling between the OH and NH stretch modes is vital to the creation of the chiral SFG lineshape. This finding has implications for the ability of researchers to label peaks as “OH” or “NH” via fitting or even with H218O labeling. This work paves the way for the NH stretch to become a more useful, informative probe in the biological spectroscopy community. Although it can be probed easily by chiral SFG, the NH stretch can also serve as a probe for other vibrational spectroscopy methods such as 2D-IR, which can be modeled with the same type of NH electrostatic maps. Thus, the NH maps developed in this work have implications for a wide range of vibrational spectroscopies applied to diverse chemical and biological systems.
Supplementary Material
Acknowledgements.
The authors thank Matthew Tremblay and Dr. Pablo E. Videla at Yale University and Prof. Thomas la Cour Jansen at the University of Groningen for helpful discussions. This work was supported by the National Institutes of Health Grant R35 GM139449 (S.H.-S.) and the NSF Grant CHE-2108690 (E.C.Y.Y.). D.K. was supported by these grants. T.S. was supported by CHE-2108690 (to E.C.Y.Y.). E.A.P. was supported by the NIH (5T32GM008283-31) and a John C. Tully Chemistry Research Fellowship from Yale University. K.O. was supported by a Donald Crothers Chemistry Research Fellowship from Yale University.
Footnotes
Supporting Information. Computational and experimental methods details, the effect of H-bond distance cutoff on the chiral SFG spectrum, information about the Fourier smoothing process, effect of transition dipole placement on chiral SFG, infrared amide I spectra of LK7β and LE7β, the effect of including Fermi resonance with the bending mode. Python scripts that show the correct ordering of the electric fields for the random forest regression models and binaries of the trained random forest regression models ready for use in Python can be found at https://github.com/dkonstan/NH_map.
References
- 1.Yan ECY; Fu L; Wang Z; Liu W Biological Macromolecules at Interfaces Probed by Chiral Vibrational Sum Frequency Generation Spectroscopy. Chem. Rev 2014, 114, 8471–8498. [DOI] [PubMed] [Google Scholar]
- 2.Fu L; Liu J; Yan ECY Chiral Sum Frequency Generation Spectroscopy for Characterizing Protein Secondary Structures at Interfaces. J. Am. Chem. Soc 2011, 133, 8094–8097. [DOI] [PubMed] [Google Scholar]
- 3.Rubtsov IV; Wang J; Hochstrasser RM Dual frequency 2D-IR of peptide amide-A and amide-I modes. J. Chem. Phys 2003, 118, 7733–7736. [Google Scholar]
- 4.Rubtsov IV; Wang J; Hochstrasser RM Vibrational Coupling between Amide-I and Amide-A Modes Revealed by Femtosecond Two Color Infrared Spectroscopy. J. Phys. Chem. A 2003, 107, 3384–3396. [Google Scholar]
- 5.Jansen T. l. C.; Knoester J Nonadiabatic Effects in the Two-Dimensional Infrared Spectra of Peptides: Application to Alanine Dipeptide. J. Phys. Chem. B 2006, 110, 22910–22916. [DOI] [PubMed] [Google Scholar]
- 6.Corcelli SA; Lawrence CP; Skinner JL Combined electronic structure/molecular dynamics approach for ultrafast infrared spectroscopy of dilute HOD in liquid H2O and D2O. J. Chem. Phys 2004, 120, 8107–8117. [DOI] [PubMed] [Google Scholar]
- 7.Schmidt JR; Corcelli SA; Skinner JL Ultrafast vibrational spectroscopy of water and aqueous N-methylacetamide: Comparison of different electronic structure/molecular dynamics approaches. J. Chem. Phys 2004, 121, 8887. [DOI] [PubMed] [Google Scholar]
- 8.Auer BM; Skinner JL IR and Raman spectra of liquid water: Theory and interpretation. J. Chem. Phys 2008, 128, 224511. [DOI] [PubMed] [Google Scholar]
- 9.Auer BM; Skinner JL Vibrational Sum-Frequency Spectroscopy of the Water Liquid/Vapor Interface. J. Phys. Chem. B 2009, 113, 4125–4130. [DOI] [PubMed] [Google Scholar]
- 10.Pieniazek PA; Tainter CJ; Skinner JL Interpretation of the water surface vibrational sum-frequency spectrum. J. Chem. Phys 2011, 135, 044701. [DOI] [PubMed] [Google Scholar]
- 11.Wang L; Middleton CT; Zanni MT; Skinner JL Development and Validation of Transferable Amide I Vibrational Frequency Maps for Peptides. J. Phys. Chem. B 2011, 115, 3713–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carr JK; Wang L; Roy S; Skinner JL Theoretical Sum Frequency Generation Spectroscopy of Peptides. J. Phys. Chem. B 2015, 119, 8969–8983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayashi T; la Cour Jansen T; Zhuang W; Mukamel S Collective Solvent Coordinates for the Infrared Spectrum of HOD in D2O Based on an ab Initio Electrostatic Map. J. Phys. Chem. A 2005, 109, 64–82. [DOI] [PubMed] [Google Scholar]
- 14.Jansen T. l. C.; Knoester J A transferable electrostatic map for solvation effects on amide I vibrations and its application to linear and two-dimensional spectroscopy. J. Chem. Phys 2006, 124, 044502. [DOI] [PubMed] [Google Scholar]
- 15.Jansen T. l. C.; Dijkstra AG; Watson TM; Hirst JD; Knoester J Modeling the amide I bands of small peptides. J. Chem. Phys 2006, 125, 044312. [DOI] [PubMed] [Google Scholar]
- 16.Moad AJ; Simpson GJ A Unified Treatment of Selection Rules and Symmetry Relations for Sum-Frequency and Second Harmonic Spectroscopies. J. Phys. Chem. B 2004, 108, 3548–3562. [Google Scholar]
- 17.Simpson GJ Molecular Origins of the Remarkable Chiral Sensitivity of Second-Order Nonlinear Optics. ChemPhysChem 2004, 5, 1301–1310. [DOI] [PubMed] [Google Scholar]
- 18.Perets EA; Olesen KB; Yan ECY Chiral Sum Frequency Generation Spectroscopy Detects Double-Helix DNA at Interfaces. Langmuir 2022, 38, 5765–5778. [DOI] [PubMed] [Google Scholar]
- 19.Konstantinovsky D; Perets EA; Yan ECY; Hammes-Schiffer S Simulation of the Chiral Sum Frequency Generation Response of Supramolecular Structures Requires Vibrational Couplings. J. Phys. Chem. B 2021, 125, 12072–12081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perets EA; Konstantinovsky D; Fu L; Chen J; Wang H-F; Hammes-Schiffer S; Yan ECY Mirror-image antiparallel β-sheets organize water molecules into superstructures of opposite chirality. Proc. Natl. Acad. Sci. USA 2020, 117, 32902–32909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perets EA; Videla PE; Yan ECY; Batista VS Chiral inversion of amino acids in antiparallel beta sheets at interfaces probed by vibrational sum frequency generation spectroscopy. J. Phys. Chem. B 2019, 123, 5769–5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perets EA; Yan ECY Chiral Water Superstructures around Antiparallel β-Sheets Observed by Chiral Vibrational Sum Frequency Generation Spectroscopy. J. Phys. Chem. Lett 2019, 10, 3395–3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan ECY; Wang Z; Fu L Proteins at Interfaces Probed by Chiral Vibrational Sum Frequency Generation Spectroscopy. J. Phys. Chem. B 2015, 119, 2769–2785. [DOI] [PubMed] [Google Scholar]
- 24.Fu L; Wang Z; Yan ECY N-H Stretching Modes Around 3300 Wavenumber From Peptide Backbones Observed by Chiral Sum Frequency Generation Vibrational Spectroscopy. Chirality 2014, 26, 521–524. [DOI] [PubMed] [Google Scholar]
- 25.Konstantinovsky D; Perets EA; Santiago T; Velarde L; Hammes-Schiffer S; Yan ECY Detecting the First Hydration Shell Structure around Biomolecules at Interfaces. ACS Cent. Sci 2022, 8, 1404–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McDermott ML; Vanselous H; Corcelli SA; Petersen PB DNA’s Chiral Spine of Hydration. ACS Cent. Sci 2017, 3, 708–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee S-H; Mirkin NG; Krimm S A quantitative anharmonic analysis of the amide A band in α-helical poly (L-alanine). Biopolymers 1999, 49, 195–207. [Google Scholar]
- 28.Nagata Y; Mukamel S Vibrational Sum-Frequency Generation Spectroscopy at the Water/Lipid Interface: Molecular Dynamics Simulation Study. J. Am. Chem. Soc 2010, 132, 6434–6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagata Y; Hsieh C-S; Hasegawa T; Voll J; Backus EHG; Bonn M Water Bending Mode at the Water–Vapor Interface Probed by Sum-Frequency Generation Spectroscopy: A Combined Molecular Dynamics Simulation and Experimental Study. J. Phys. Chem. Lett 2013, 4, 1872–1877. [DOI] [PubMed] [Google Scholar]
- 30.Ohto T; Usui K; Hasegawa T; Bonn M; Nagata Y Toward ab initio molecular dynamics modeling for sum-frequency generation spectra; an efficient algorithm based on surface-specific velocity-velocity correlation function. J. Chem. Phys 2015, 143, 124702. [DOI] [PubMed] [Google Scholar]
- 31.Strunge K; Madzharova F; Jensen F; Weidner T; Nagata Y Theoretical Sum Frequency Generation Spectra of Protein Amide with Surface-Specific Velocity–Velocity Correlation Functions. J. Phys. Chem. B 2022, 126, 8571–8578. [DOI] [PubMed] [Google Scholar]
- 32.Khatib R; Backus EHG; Bonn M; Perez-Haro M-J; Gaigeot M-P; Sulpizi M Water orientation and hydrogen-bond structure at the fluorite/water interface. Sci. Reports 2016, 6, 24287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khatib R; Hasegawa T; Sulpizi M; Backus EHG; Bonn M; Nagata Y Molecular Dynamics Simulations of SFG Librational Modes Spectra of Water at the Water–Air Interface. J. Phys. Chem. C 2016, 120, 18665–18673. [Google Scholar]
- 34.Khatib R; Sulpizi M Sum Frequency Generation Spectra from Velocity–Velocity Correlation Functions. J. Phys. Chem. Lett 2017, 8, 1310–1314. [DOI] [PubMed] [Google Scholar]
- 35.Morita A; Hynes JT A Theoretical Analysis of the Sum Frequency Generation Spectrum of the Water Surface. II. Time-Dependent Approach. J. Phys. Chem. B 2002, 106, 673–685. [Google Scholar]
- 36.Nihonyanagi S; Ishiyama T; Lee T.-k.; Yamaguchi S; Bonn M; Morita A; Tahara T Unified Molecular View of the Air/Water Interface Based on Experimental and Theoretical χ(2) Spectra of an Isotopically Diluted Water Surface. J. Am. Chem. Soc 2011, 133, 16875–16880. [DOI] [PubMed] [Google Scholar]
- 37.Ishiyama T; Takahashi H; Morita A Molecular dynamics simulations of surface-specific bonding of the hydrogen network of water: A solution to the low sum-frequency spectra. Phys. Rev. B 2012, 86, 035408. [Google Scholar]
- 38.Ishiyama T; Morita A Nuclear Quantum Effect on the χ(2) Band Shape of Vibrational Sum Frequency Generation Spectra of Normal and Deuterated Water Surfaces. J. Phys. Chem. Lett 2019, 10, 5070–5075. [DOI] [PubMed] [Google Scholar]
- 39.Gruenbaum SM; Tainter CJ; Shi L; Ni Y; Skinner JL Robustness of Frequency, Transition Dipole, and Coupling Maps for Water Vibrational Spectroscopy. J. Chem. Theory Comput 2013, 9, 3109–3117. [DOI] [PubMed] [Google Scholar]
- 40.Pieniazek PA; Tainter CJ; Skinner JL Surface of Liquid Water: Three-Body Interactions and Vibrational Sum-Frequency Spectroscopy. J. Am. Chem. Soc 2011, 133, 10360–10363. [DOI] [PubMed] [Google Scholar]
- 41.Auer BM; Skinner JL Dynamical effects in line shapes for coupled chromophores: Time-averaging approximation. J. Chem. Phys 2007, 127, 104105. [DOI] [PubMed] [Google Scholar]
- 42.Auer BM; Skinner JL IR and Raman spectra of liquid water: Theory and interpretation. J. Chem. Phys 2008, 128, 224511. [DOI] [PubMed] [Google Scholar]
- 43.Corcelli SA; Skinner JL Infrared and Raman Line Shapes of Dilute HOD in Liquid H2O and D2O from 10 to 90 °C. J. Phys. Chem. A 2005, 109, 6154–6165. [DOI] [PubMed] [Google Scholar]
- 44.Phillips DC; York RL; Mermut O; McCrea KR; Ward RS; Somorjai GA Side Chain, Chain Length, and Sequence Effects on Amphiphilic Peptide Adsorption at Hydrophobic and Hydrophilic Surfaces Studied by Sum-Frequency Generation Vibrational Spectroscopy and Quartz Crystal Microbalance. J. Phys. Chem. C 2007, 111, 255–261. [Google Scholar]
- 45.Mermut O; Phillips DC; York RL; McCrea KR; Ward RS; Somorjai GA In Situ Adsorption Studies of a 14-Amino Acid Leucine-Lysine Peptide onto Hydrophobic Polystyrene and Hydrophilic Silica Surfaces Using Quartz Crystal Microbalance, Atomic Force Microscopy, and Sum Frequency Generation Vibrational Spectroscopy. J. Am. Chem. Soc 2006, 128, 3598–3607. [DOI] [PubMed] [Google Scholar]
- 46.Wang Z; Fu L; Yan ECY C–H Stretch for Probing Kinetics of Self-Assembly into Macromolecular Chiral Structures at Interfaces by Chiral Sum Frequency Generation Spectroscopy. Langmuir 2013, 29, 4077–4083. [DOI] [PubMed] [Google Scholar]
- 47.Fu L; Chen S-L; Wang H-F Validation of Spectra and Phase in Sub-1 cm–1 Resolution Sum-Frequency Generation Vibrational Spectroscopy through Internal Heterodyne Phase-Resolved Measurement. J. Phys. Chem. B 2016, 120, 1579–1589. [DOI] [PubMed] [Google Scholar]
- 48.York RL; Browne WK; Geissler PL; Somorjai GA Peptides Adsorbed on Hydrophobic Surfaces—A Sum Frequency Generation Vibrational Spectroscopy and Modeling Study. Isr. J. Chem 2007, 47, 51–58. [Google Scholar]
- 49.Hayashi T; Zhuang W; Mukamel S Electrostatic DFT Map for the Complete Vibrational Amide Band of NMA. J. Phys. Chem. A 2005, 109, 9747–9759. [DOI] [PubMed] [Google Scholar]
- 50.Hayashi T; Mukamel S Vibrational−Exciton Couplings for the Amide I, II, III, and A Modes of Peptides. J. Phys. Chem. B 2007, 111, 11032–11046. [DOI] [PubMed] [Google Scholar]
- 51.Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- 52.Wang J; Hochstrasser RM Anharmonicity of Amide Modes. J. Phys. Chem. B 2006, 110, 3798–3807. [DOI] [PubMed] [Google Scholar]
- 53.Barducci A; Bussi G; Parrinello M Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett 2008, 100, 020603. [DOI] [PubMed] [Google Scholar]
- 54.Horn HW; Swope WC; Pitera JW; Madura JD; Dick TJ; Hura GL; Head-Gordon T Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys 2004, 120, 9665–9678. [DOI] [PubMed] [Google Scholar]
- 55.Becke AD Density‐functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- 56.Stephens PJ; Devlin FJ; Chabalowski CF; Frisch MJ Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem 1994, 98, 11623–11627. [Google Scholar]
- 57.Lee C; Yang W; Parr RG Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- 58.McLean AD; Chandler GS Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys 1980, 72, 5639–5648. [Google Scholar]
- 59.Krishnan R; Binkley JS; Seeger R; Pople JA Self‐consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys 1980, 72, 650–654. [Google Scholar]
- 60.Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H, et al. Gaussian 16 Rev. C.01, Wallingford, CT, 2016. [Google Scholar]
- 61.Klamt A; Schüürmann G COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc, Perkin trans. 1993, 799–805. [Google Scholar]
- 62.Andzelm J; Kölmel C; Klamt A Incorporation of solvent effects into density functional calculations of molecular energies and geometries. J. Chem. Phys 1995, 103, 9312–9320. [Google Scholar]
- 63.Barone V; Cossi M Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar]
- 64.Cossi M; Rega N; Scalmani G; Barone V Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem 2003, 24, 669–681. [DOI] [PubMed] [Google Scholar]
- 65.Marston CC; Balint-Kurti GG The Fourier grid Hamiltonian method for bound state eigenvalues and eigenfunctions. J. Chem. Phys 1989, 91, 3571–3576. [Google Scholar]
- 66.Mayne LC; Hudson B Resonance Raman spectroscopy of N-methylacetamide: overtones and combinations of the carbon-nitrogen stretch (amide II’) and effect of solvation on the carbon-oxygen double-bond stretch (amide I) intensity. J. Phys. Chem 1991, 95, 2962–2967. [Google Scholar]
- 67.Joung IS; Cheatham TE III Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Joung IS; Cheatham TE III Molecular Dynamics Simulations of the Dynamic and Energetic Properties of Alkali and Halide Ions Using Water-Model-Specific Ion Parameters. J. Phys. Chem. B 2009, 113, 13279–13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Belch AC; Rice SA The OH stretching spectrum of liquid water: A random network model interpretation. J. Chem. Phys 1983, 78, 4817–4823. [Google Scholar]
- 71.Auer BM; Skinner JL Dynamical effects in line shapes for coupled chromophores: Time-averaging approximation. J. Chem. Phys 2007, 127, 104105. [DOI] [PubMed] [Google Scholar]
- 72.Ma G; Liu J; Fu L; Yan ECY Probing Water and Biomolecules at the Air—Water Interface with a Broad Bandwidth Vibrational Sum Frequency Generation Spectrometer from 3800 to 900 cm−1. Appl. Spectrosc 2009, 63, 528–537. [DOI] [PubMed] [Google Scholar]
- 73.DeGrado WF; Lear JD Induction of peptide conformation at apolar water interfaces. 1. A study with model peptides of defined hydrophobic periodicity. J. Am. Chem. Soc 1985, 107, 7684–7689. [Google Scholar]
- 74.Weidner T; Breen NF; Drobny GP; Castner DG Amide or Amine: Determining the Origin of the 3300 cm−1 NH Mode in Protein SFG Spectra Using 15N Isotope Labels. J. Phys. Chem. B 2009, 113, 15423–15426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kananenka AA; Skinner JL Fermi resonance in OH-stretch vibrational spectroscopy of liquid water and the water hexamer. J. Chem. Phys 2018, 148, 244107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.