

Abstract
Multiple acyl-CoA dehydrogenase deficiency (MADD) is a metabolic disorder due to dysfunction of electron transfer flavoprotein (ETF) or ETF-ubiquinone oxidoreductase (ETF-QO). Mutations in ETFDH, encoding ETF-QO have been associated with both riboflavin-responsive and non-responsive MADD as well as a myopathic form of CoQ10 deficiency, although pathomechanisms responsible for these different phenotypes are not well-defined. We performed mutation analysis in four Taiwanese MADD patients. Three novel ETFDH mutations were identified in four patients and all harbored the p.A84T mutation. Muscle CoQ10 levels and respiratory chain activities measured in two patients were normal. Three patients improved on riboflavin together with carnitine. Our results show that not all MADD patients have CoQ10 deficiency. Based upon our data, riboflavin and carnitine may be the first-line treatment for MADD.
Keywords: Multiple acyl-CoA dehydrogenase deficiency, CoQ10 deficiency, Riboflavin, Electron transfer flavoprotein ubiquinone oxidoreductase
1. Introduction
Multiple acyl-CoA dehydrogenase deficiency (MADD), also known as glutaric aciduria type II, is an autosomal-recessive inherited, organic acid disorder caused by defects in electron transfer flavoprotein (ETF), ETF-ubiquinone oxidoreductase (ETF-QO), or an, as yet, unidentified abnormality in flavin metabolism or transport [1]. In mitochondria, ETF, which is located in the matrix, receives electron from several dehydrogenases involved in fatty acid oxidation. ETF transfers electrons to ETF-QO, located in the inner mitochondrial membrane, and subsequently, electrons are passed to ubiquinone in the respiratory chain (RC). ETF, ETF-QO, and most mitochondrial enzymes involved in electron transfer system are flavoproteins, which contain flavin adenine dinucleotide prosthetic groups.
The clinical phenotypes of MADD has been classified as neonatal onset forms with (type I) or without (type II) congenital anomalies, and mild and/or later onset form (type III). Neonatal onset forms usually present with hypotonia, hepatomegaly, hyperammonemia, non-ketotic hypoglycemia, metabolic acidosis and poor prognosis. Milder type III patients manifest proximal myopathy often with hepatomegaly, encephalopathy, and episodic lethargy, vomiting and hypoglycemia [1]; these episodes can be lethal [2,3]. Importantly, riboflavin treatment has been shown to strikingly ameliorate the symptoms and metabolic profiles in many MADD patients, particularly type III patients [4,5].
To date, mutations in ETFA, ETFB and ETFDH, which encode α- and β-subunits of ETF and ETF-QO, respectively [1], have been identified in patients with MADD. Recently, ETFDH mutations were reported to be major causes of riboflavin-responsive MADD (RR-MADD) [6]. About the same time, ETFDH mutations were also reported to cause the myopathic form of coenzyme Q10 (CoQ10) deficiency [7]. However, the relationships between riboflavin responsiveness, CoQ10 levels and ETFDH gene mutations are not well-defined. In RR-MADD patients, reduced biochemical activities of other mitochondrial enzymes including flavin-dependent and RC enzymes, and decreased flavin concentration in mitochondria have been reported [5–9]. However, it is still unknown if this mitochondrial dysfunction is a general finding in RR-MADD and if this mitochondrial dysfunction is secondary to ETFDH mutations or caused by additional abnormalities of mitochondrial flavin metabolism or transport.
Herein, we report the results of mutation analysis and variable clinical manifestations in four Taiwanese MADD patients from three unrelated families, as well as CoQ10 levels and RC activities in two. Among these four patients, three improved with riboflavin treatment.
2. Patients (Table 1)
Table 1.
Summary of clinical, pathological, biochemical and molecular analyses
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | |
|---|---|---|---|---|
|
| ||||
| Age of onset (years) | 11 | 12 | 19 | 10 |
| Sex | Female | Female | Male | Female |
| Episodes of metabolic crisis | None | None | None | One |
| Episodes of severe weakness | Two, together with body weight loss | Two, together with body weight loss and frequent diarrhea | Three, together with body weight loss and frequent abdominal pain and diarrhea | Two |
| Episodes of other symptoms | Pancreatitis, six times | None | None | None |
| Symptoms between episodes | Exercise intolerance Minimal weakness |
Exercise intolerance | Mild weakness | Mild weakness |
| Muscle histology | Lipid accumulation | ND | Lipid accumulation | Lipid accumulation |
| Serum acylcarnitine (*high) | C6*1.5, C8*1.2, C10:1*3.4, C10*10.5, C14:2*2.7 | C8*4.2, C10*2.4, C14:2*1.3 | C6*1.1, C8*8, C10:1*1.5, C10*5, C14:2*5.2 | C8*2, C10*2.8, C14: 1*3.6, C16:1*3 |
| Urine organic acid profile | Elevation of glutaric, ethylmaloric, suberic, 3-methyladipic, 2- hydroxylglutaric acids | Elevation of glutaric, ethylmaloric, suberic, 2-hydroxylglutaric, 3-methylglutaconic, lactic acids | Elevation of glutaric, methylmalonic, ethylmalonic, 3-methylglutaconic, pyruvic acids | Elevation of glutaric, ethylmalonic, suberic, 2-hydroxyglutaric acids |
| CoQ10 level (normal 32.1 ± 6.8 μg/g-tissue) | ND | ND | Normal | Normal |
| Respiratory chain activity: | ND | ND | ||
| Complex I + III (normal 1.02 ± 0.38) | 0.71 | 0.71 | ||
| Complex IV (normal 2.8 ± 0.52) | 2.35 | 2.38 | ||
| Citrate synthase (normal 9.88 ± 2.55) | 9.85 | 9.91 | ||
| Riboflavin responsiveness | Yes | Yes | Yes | Uncertain |
| Mutation | p.A84T/p.R175L | p.A84T/p.R175L | p.A84T/p.A84T | p.A84T/ p.L127H |
The enzymatic activities of respiratory chain are normalized to citrate synthase; 1 unit means 1 μmol/min.
The number of controls for acylcarnitine and organic acid analyses is 100. ND: not done.
Patient 1 has been reported [10]. This 27-year-old woman has experienced exercise intolerance since early childhood. At age 11, she has suffered the first of six bouts of pancreatitis; after each episode, she recovered completely. At age 19, she lost weight and developed dysphagia with progressive weakness of neck and proximal limb muscles (manual muscle testing (MMT) score 2 out of 5). Hepatomegaly and elevated serum transaminases levels prompted a liver biopsy, which showed foamy hepatocytes suggesting lipid storage disease. Her symptoms were partially relieved by conservative medical treatment. A few months later, she suffered a more severe episode of muscle weakness with acute respiratory failure, but no metabolic acidosis and hypoketotic hypoglycemia. Serum creatine kinase (CK) level was elevated to 1509 IU/l (normal < 269). Muscle biopsy showed increased lipid droplets predominantly in type 1 fibers (Fig. 1A). Urine organic acid analysis and low serum free carnitine level were consistent with the diagnosis of MADD. With riboflavin (120 mg/day) and carnitine (1 g/day) treatment, she recovered completely, regained body weight, and has not suffered recurrence of pancreatitis or generalized muscle weakness for 8 years even though the metabolic profiles were not fully corrected. Serum CK level has also remained normal.
Fig. 1.
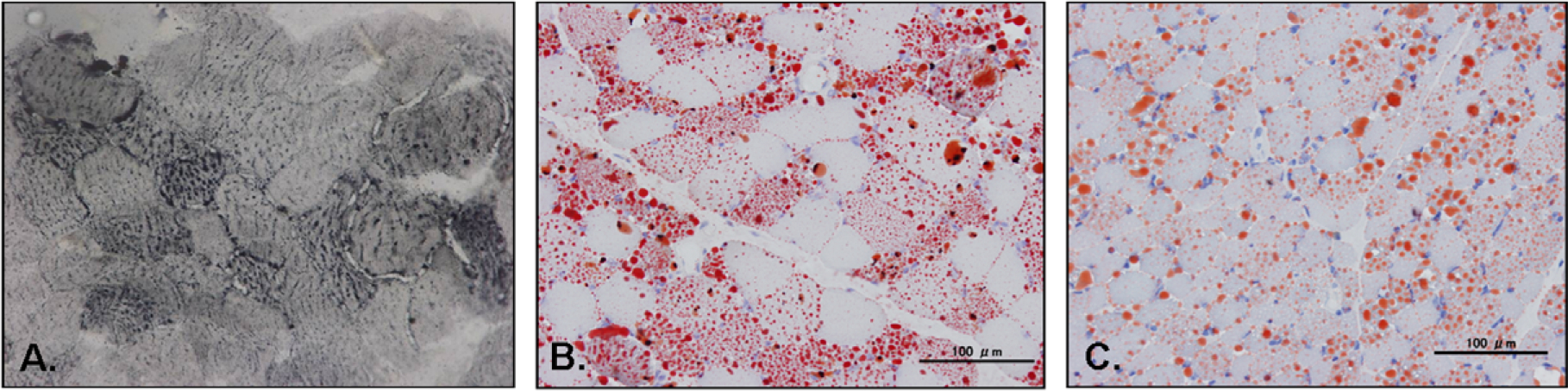
Histological findings of muscle in patient 1 (A. Sudan black stain 400×), patient 3 (B. Oil Red-O stain 200×) and patient 4 (C. Oil Red-O stain 200×): lipid storage in vacuolar muscle fibers, predominantly in type 1 fibers.
Patient 2, a 42 year-old woman, is the elder sister of patient 1. Since age 12, she has had two bouts of muscle weakness without specific precipitants and manifesting as with difficulty in climbing upstairs and combing her hair. She has never had metabolic crises, hypoketotic hypoglycemia or respiratory failure. Between the episodes, she had normal muscle strength but mild exercise intolerance. Due to her affected sister, at age 35, she underwent laboratory tests, which showed low serum carnitine, increased serum acylcarnitine levels and elevated glutaric, ethylmalonic, 2-hydroxylglutaric, 3-methylglutaconic and lactic acids in urine. She was given the diagnosis of MADD and treated with riboflavin (120 mg/day) and carnitine (1 g/day). On treatment, her exercise intolerance improved and she has had no recurrence of muscle weakness over the last 7 years.
Patient 3, a 22 year-old man with non-consanguineous parents, was healthy until age 14 when he lost weight (7 kg/6 months) and developed exercise intolerance, dysphagia, poor head control, and limb weakness. At age 16, he was virtually wheelchair-bound as he could walk only 10 m with assistance. Physical and neurological examinations showed neck and proximal muscle weakness (MMT score 2 in neck and thigh, 3 in upper arm) with wasting, lordosis, winged scapula and absent tendon reflexes. Serum CK level was mildly elevated and venous lactate level was normal. No metabolic acidosis and hypoketotic hypoglycemia was found. Nerve conduction studies (NCS) were normal and electromyography (EMG) showed myopathic changes. Pulmonary function tests demonstrated severe restrictive ventilatory defect. Muscle biopsy revealed markedly increased lipid droplets, predominantly in type 1 fibers (Fig. 1B), but urine organic acid analysis was normal. Mitochondrial disease was suspected and subsequently, 30 mg/day of CoQ10 was prescribed. After starting coenzyme Q 30 mg/day, he became able to walk more than 100 m. Nevertheless, on CoQ10 treatment, his muscle strength and body weight did not return to normal although he showed some improvement, and during infectious episodes, the muscle weakness worsened and he developed diarrhea and lost weight. At age 20, an abnormal urine organic acid profile and increased serum acylcarnitine indicated MADD. Riboflavin (120 mg/day) and carnitine (1 g/day) supplementation was started. Since then, he has gradually gained weight (from 35 kg to 50 kg) and shown marked improvement in muscle strength (MMT score 3+ in neck and 4+ in proximal muscles), although the metabolic abnormalities have not been corrected completely.
Patient 4, a 10-year-old girl was a slow runner since early childhood. One month prior to hospitalization, she had an upper respiratory tract infection followed by progressive proximal muscle weakness. On admission, strength by MMT was 2 in neck and thigh, and 3 in upper arm. Serum CK and venous lactate levels were mildly elevated. NCS was normal and EMG revealed myopathic changes. Due to hepatomegaly and elevation of serum transaminases levels, a liver biopsy was performed and showed marked fat accumulation, suggesting a lipid storage disease. Muscle biopsy revealed vacuoles with positive oil red-O staining, predominantly in type 1 fibers (Fig. 1C). After supportive care, all symptoms were improved. A few days after discharge from the hospital, she complained poor appetite and general malaise without clear precipitating factors. Her condition rapidly deteriorated; she became comatose and developed cardiopulmonary failure. On evaluation, marked metabolic acidosis, hyperammonemia and hypoglycemia were noticed. The diagnosis of MADD was made based on elevated C5–10 dicarboxylic aciduria shown in urine organic acid analysis. Despite immediate riboflavin and carnitine prescription, she expired due to multiple organ failure.
3. Methods
3.1. Mutation analysis
With informed consent, genomic DNA was extracted from peripheral blood lymphocytes using standard methods. The primer sequences for ETFDH gene are available on request. We directly sequenced all exons and their flanking intronic regions of ETFDH using an automated 3100 DNA sequencer (Applied Biosystems, Foster, CA) with the BigDye Terminator cycle sequencing system following the manufacturer’s protocol. DNA sequences were analyzed with the SeqScape program (Applied Biosystems). We performed direct sequencing or restriction enzyme digestion analyses on 200 chromosomes from 100 healthy Taiwanese controls to determine the prevalence of identified mutations in normal population.
3.2. Haplotype analysis
We used three microsatellite markers (D4S1785i, D4S0024i, D4S1014i) and nine single nucleotide polymorphisms (rs11559290, rs1140065, rs17843967, rs3210749, rs12644851, rs12640862, rs7679753, rs11737481, rs4690909) for haplotype analysis. PCR products were analyzed by Gene mapper and/or direct sequencing (All primer pair sequences are available upon request).
3.3. RC activities and CoQ10 measurement
Frozen muscle specimens were used for biochemical analyses. RC and citrate synthase activities were determined spectro-photometrically using muscle homogenate and CoQ10 level was measured by high performance liquid chromatography as previously described [7]. RC enzyme activities were normalized to citrate synthase. We studied 135 normal skeletal muscle samples to establish the normal range of RC activities and CoQ10 level. Sample size allowed these analyses in patients 3 and 4 only.
4. Results
4.1. Mutation analysis
Sequence analysis of ETFDH revealed compound heterozygous missense mutations, c.250G > A (p.A84T) and c.524G > T (p.R175L), in patient 1 and 2; a homozygous missense mutation, c.250G > A (p.A84T), in patient 3; and compound heterozygous missense mutations, c.250G > A (p.A84T) and c.380T > A (p.L127H), in patient 4. All mutations have been tested in parental DNA in order to confirm the autosomal-recessive inheritance. These three novel mutations affect highly conserved amino acid residues in FAD-binding domain of ETF-QO. One of 200 control chromosomes had c.250G > A, which was shared by all patients, while none carried c.380T > A and c.524G > T substitutions. Sequence analyses of ETFA and ETFB showed no mutation in these four patients.
4.2. Haplotype analysis
No specific haplotype was associated with c.250G > A mutation.
4.3. RC activity
Activities of respiratory chain complexes I + III (NADH – cytochrome c reductase), IV (cytochrome c oxidase) and citrate synthase were measured in only patient 3 and 4 due to limited amounts of muscle specimens. All the activities of enzymes tested are within normal range (Table 1).
4.4. CoQ10 measurement
CoQ10 levels measured in frozen muscle samples were normal, 32.5 μg/g-tissue in patient 3 and 36.0 in patient 4 (control: 32.1 ± 6.8) (Table 1.).
5. Discussion
MADD is mainly caused by the defects in ETF or ETF-QO and, in recent years, has been diagnosed by enzymatic assay and mutation analysis [11,12]. Here, we identified three novel missense ETFDH mutations in four Taiwanese MADD patients. All three mutated residues are located in FAD-binding domain and are highly conserved in Caenorhabditis elegans, Drosophila, Xenopus, Gallus, Swine, Rattus and humans; therefore, the mutations are likely to change the affinity of FAD to ETF:QO and affect the electron transfer in mitochondria [13]. A mutation at c.524G has been reported this year [14], however, we identified different nucleotide substitution that translated to different amino acid in our patients (c.524G > A, p.R175H V.S. c.524G > T, p.R175L). The previously reported patient and our patients with c.524 mutations all have late onset, but the former patient showed vomiting, hypotonia and liver dysfunction without reports of muscle weakness and riboflavin responsiveness, which differed from our patients. The different symptoms may be due to the differences in the amino acid substitutions and mutations in the second allele. However, more functional analyses are needed to discuss the pathogenic nature of the c.524G mutation. Interestingly, all four patients share one common mutation, c.250G > A, p.A84T in exon 3 which has never been described in other populations [14]. Except for patient 1 and 2, who are siblings, the patients are unrelated, and their families are from different regions of Taiwan. We did not detect the mutation in 100 control chromosomes from Japanese, Korean and Thai each, whereas this mutation was identified in one of 200 Taiwanese control chromosomes. This mutation might be due to a founder in Taiwan, but we could not link a specific haplotype to this mutation. Although we have not excluded the possibility that this mutation is a neutral polymorphism, p.A84 is well-conserved among different species (data not shown) and located in the important FAD-binding domain.
Due to its wide variety of clinical symptoms, MADD is difficult to diagnose. Recurrent pancreatitis was the initial symptom in patient 1, and subsequently, during an episode of severe, muscle weakness caused respiratory failure prompting the diagnostic muscle biopsy and metabolic screening. Patient 2 in the same family, harboring identical mutations, had much milder symptoms. Patient 3 showed severe progressive muscle weakness causing loss of ability to walk, without notable metabolic crises or respiratory distress. Patient 4 had a lethal metabolic crisis with respiratory failure and encephalopathy. The intra- and extra-familial phenotypic heterogeneity suggests that additional factors, such as diet, may modulate the disease severity [6,15]. Noteworthily, neck flexor and extensor weakness has been described as a characteristic clinical feature in MADD patients [6] and was also observed in all of our patients. Since MADD is often amenable to treatment, expanded use of mass spectrometry to detect the characteristic pattern of urine organic acids or blood acylcarnitine profiling combined with early mutation analysis are recommended to identify patients [6,16–18].
Last year, MADD and the myopathic form of CoQ10 deficiency were reported to be possibly allelic disease according to the same causative gene [7]. However, in our series, the CoQ10 levels in skeletal muscles of patient 3 and 4 were normal and patient 3 showed partial improvement with low-dose CoQ10 supplementation. In addition, normal CoQ10 level was also detected in another Japanese MADD patient with ETFDH mutation [19], indicating that CoQ10 is not deficient in all MADD patients with ETFDH mutations. Interestingly, in patient 3 and 4 with normal CoQ10 levels, activities of RC complexes I + III and IV were normal, unlike previous reports in RR-MADD patients [5,7–9]. This year, a report suggested that total muscle CoQ10 was the best predictor of an electron transport chain complex abnormality [20]; our results support their conclusion. However, previous studies showed that CoQ10 level varied widely from patient to patient with mtDNA mutations [21]. Thus, more patients should be studied to determine the causal relationship between CoQ10 and RC activities in MADD patients. Because decreased RC activities are also seen in other metabolic diseases, including mitochondrial enzyme defects, and not present in all RR-MADD patients, the phenomenon may be secondary [22].
To date, there is no established therapeutic strategy for MADD patients. There are accumulating reports of MADD patients or cell lines demonstrating the efficacy of riboflavin [5,8,17,23] although the mechanism of riboflavin efficacy in MADD patients is not yet completely understood. The emerging consensus of these publications is that riboflavin prescription is the first-line treatment for MADD patients. Most reports have advocated the combination therapy of riboflavin and carnitine as more effective although a few reports noted ineffectiveness of carnitine [2,6,9,24]. Interestingly, the clinical manifestations of primary carnitine deficiency are sometimes similar to those in MADD, especially during metabolic crisis [25,26]. Accordingly, supplementation of riboflavin together with carnitine is reasonable for the patients who suspected of having MADD, but not yet diagnosed. A more recent study suggested that combination therapy of riboflavin and CoQ10 resulted in better long-term outcome rather than riboflavin plus carnitine in patients with the myopathic form of CoQ10 deficiency due to ETFDH mutations [7]. Our patient 3 showed some improvement after starting coenzyme Q supplementation, raising a possibility that CoQ10 supplementation may be effective even when CoQ10 levels are normal. However, his muscle strength and body weight showed only partial improvement and did not return to normal, and during infectious episodes, the muscle weakness still got worsened, questioning whether this ‘‘improvement” is truly due to this relatively low dose of CoQ10 or just reflecting fluctuation of clinical severity. Since not all MADD patients have CoQ10 deficiency and the response to CoQ10 treatment is still uncertain, further studies are necessary to confirm this notion. Other regimens, such as glycine, prednisolone and insulin, have also been studied, but their effectiveness remains uncertain [27,28]. Additional clinical and therapeutic data on MADD patients are needed for better clinical management of this complicated disorder.
Footnotes
6. Conflicts of interest
The authors report no conflicts of interest.
References
- [1].Frerman FE, Goodman SI. Defects of electron transfer flavoprotein and electron transfer flavoprotein–ubiquinone oxidoreductase: glutaric acidemia type II. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. p. 2357–65. [Google Scholar]
- [2].Di Donato S, Frerman FE, Rimoldi M, Rinaldo P, Taroni F, Wiesmann UN. Systemic carnitine deficiency due to lack of electron transfer flavoprotein:ubiquinone oxidoreductase. Neurology 1986;36:957–63. [DOI] [PubMed] [Google Scholar]
- [3].Angle B, Burton BK. Risk of sudden death and acute life-threatening events in patients with glutaric academia type II. Mol Genet Metab 2008;93:36–9. [DOI] [PubMed] [Google Scholar]
- [4].Gregersen N, Rhead W, Christensen E. Riboflavin responsive glutaric aciduria type II. Prog Clin Biol Res 1990;321:477–94. [PubMed] [Google Scholar]
- [5].Vergani L, Barile M, Angelini C, et al. Riboflavin therapy: biochemical heterogeneity in two adult lipid storage myopathies. Brain 1999;122:2401–11. [DOI] [PubMed] [Google Scholar]
- [6].Olsen RK, Olpin SE, Andresen BS, et al. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain 2007;130:2045–54. [DOI] [PubMed] [Google Scholar]
- [7].Gempel K, Topaloglu H, Talim B, et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 2007;130:2037–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gianazza E, Vergani L, Wait R, et al. Coordinated and reversible reduction of enzymes involved in terminal oxidative metabolism in skeletal muscle mitochondria from a riboflavin-responsive, multiple acyl-CoA dehydrogenase deficiency patient. Electrophoresis 2006;27:1182–98. [DOI] [PubMed] [Google Scholar]
- [9].Antozzi C, Garavaglia B, Mora M, et al. Late-onset riboflavin-responsive myopathy with combined multiple acyl-coenzyme A dehydrogenase and respiratory chain deficiency. Neurology 1994;4:2153–8. [DOI] [PubMed] [Google Scholar]
- [10].Liang WC, Tsai KB, Lai CL, Chen LH, Jong YJ. Riboflavin-responsive glutaric aciduria type II with recurrent pancreatitis. Pediatr Neurol 2004;31:218–21. [DOI] [PubMed] [Google Scholar]
- [11].Goodman SI, Binard RJ, Woontner MR, Frerman FE. Glutaric acidemia type II: gene structure and mutations of the electron transfer flavoprotien:ubiquinone oxidoreductase (ETF:QO) gene. Mol Genet Metab 2002;77:86–90. [DOI] [PubMed] [Google Scholar]
- [12].Olsen RK, Andresen BS, Christensen E, Bross P, Skovby F, Gregersen N. Clear relationship between ETF/ETFDH genotype and phenotype in patients with multiple acyl-CoA dehydrogenation deficiency. Hum Mutat 2003;22: 12–23. [DOI] [PubMed] [Google Scholar]
- [13].Zhang J, Frerman FE, Kim JJ. Structure of electron transfer flavoprotein–ubiquinone oxidoreductase and electron transfer to the mitochondrial ubiquinone pool. Proc Natl Acad Sci USA 2006;103:16212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yotsumoto Y, Hasegawa Y, Fukuda S, et al. Clinical and molecular investigations of Japanese cases of glutaric acidemia type 2. Mol Genet Metab 2008;94:61–7. [DOI] [PubMed] [Google Scholar]
- [15].Moat SJ, Ashfield-Watt PA, Powers HJ, Newcombe RG, McDowell IF. Effect of riboflavin status on the homocysteine-lowering effect of folate in relation to the MTHFR (C677T) genotype. Clin Chem 2003;49:295–302. [DOI] [PubMed] [Google Scholar]
- [16].Köppel S, Gottschalk J, Hoffmann GF, Waterham HR, Blobel H, Kölker S. Late-onset multiple acyl-CoA dehydrogenase deficiency: a frequently missed diagnosis? Neurology 2006;67:1519. [DOI] [PubMed] [Google Scholar]
- [17].Beresford MW, Pourfarzam M, Davidson JE. So doctor, what exactly is wrong with my muscles? Glutaric aciduria type II presenting in a teenager. Neuromuscular Disord 2006;16:269–73. [DOI] [PubMed] [Google Scholar]
- [18].Curcoy A, Olsen RKJ, Ribes A, et al. Late-onset form of β-electron transfer flavoprotien deficiency. Mol Genet Metab 2003;78:247–9. [DOI] [PubMed] [Google Scholar]
- [19].Ohkuma A, Noguchi S, Sugie H et al. Clinical and genetic analysis of lipid storage myopathies. Muscle Nerve, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Miles HV, Miles L, Tang PH, et al. Systematic evaluation of muscle coenzyme Q10 content in children with mitochondrial respiratory chain enzyme deficiencies. Mitochondrion 2008;8:170–80. [DOI] [PubMed] [Google Scholar]
- [21].Matsuoka T, Maeda H, Goto Y, Nonaka I. Muscle coenzyme Q10 in mitochondrial encephalomyopathy. Neuromuscular Disord 1991;1:443–7. [DOI] [PubMed] [Google Scholar]
- [22].Schwab MA, Sauer SW, Okun JG, et al. Secondary mitochondrial dysfunction in propionic aciduria: a pathogenic role for endogeneous mitochondrial toxins. Biochem J 2006;398:107–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rhead W, Roettger V, Marshall T, Amendt B. Multiple acyl-coenzyme A dehydrogenation disorders responsive to riboflavin: substrate oxidation, flavin metabolism, and flavoenzyme activities in fibroblasts. Pediatr Res 1993;33:129–35. [DOI] [PubMed] [Google Scholar]
- [24].deVisser M, Scholte HR, Schutgens RB, et al. Riboflavin responsive lipid-storage myopathy and glutaric aciduria type II of early adult onset. Neurology 1986;36:267–72. [DOI] [PubMed] [Google Scholar]
- [25].Hou JW. Primary systemic carnitine deficiency presenting as recurrent Reye-like syndrome and dilated cardiomyopathy. Chang Gung Med J 2002;25:832–7. [PubMed] [Google Scholar]
- [26].Evangeliou A, Vlassopoulos D. Carnitine metabolism and deficit—when supplementation is necessary? Curr Pharm Biotechnol 2003;4:211–9. [DOI] [PubMed] [Google Scholar]
- [27].Turnbull DM, Bartlett K, Eyre JA, et al. Lipid storage myopathy due to glutaric aciduria type II: treatment of a potentially fatal myopathy. Dev Med Child Neurol 1988;30:667–72. [DOI] [PubMed] [Google Scholar]
- [28].Mooy PD, Przyrembel H, Giesberts MA, Scholte HR, Blom W, van Gelderen HH. Glutaric aciduria type II: treatment with riboflavin, carnitine and insulin. Eur J Pediatr 1984;143:92–5. [DOI] [PubMed] [Google Scholar]