

Abstract
Several molecular mechanisms have been described in Alzheimer's disease (AD), including repressed gene transcription and mitochondrial and endoplasmic reticulum (ER) dysfunction. In this study, we evaluate the potential efficacy of transcriptional modifications exerted by inhibition or knockdown of class I histone deacetylases (HDACs) in ameliorating ER‐mitochondria cross‐talk in AD models. Data show increased HDAC3 protein levels and decreased acetyl‐H3 in AD human cortex, and increased HDAC2‐3 in MCI peripheral human cells, HT22 mouse hippocampal cells exposed to Aβ1–42 oligomers (AβO) and APP/PS1 mouse hippocampus. Tacedinaline (Tac, a selective class I HDAC inhibitor) counteracted the increase in ER‐Ca2+ retention and mitochondrial Ca2+ accumulation, mitochondrial depolarization and impaired ER‐mitochondria cross‐talk, as observed in 3xTg‐AD mouse hippocampal neurons and AβO‐exposed HT22 cells. We further demonstrated diminished mRNA levels of proteins involved in mitochondrial‐associated ER membranes (MAM) in cells exposed to AβO upon Tac treatment, along with reduction in ER‐mitochondria contacts (MERCS) length. HDAC2 silencing reduced ER‐mitochondria Ca2+ transfer and mitochondrial Ca2+ retention, while knockdown of HDAC3 decreased ER‐Ca2+ accumulation in AβO‐treated cells. APP/PS1 mice treated with Tac (30 mg/kg/day) also showed regulation of mRNA levels of MAM‐related proteins, and reduced Aβ levels. These data demonstrate that Tac normalizes Ca2+ signaling between mitochondria and ER, involving the tethering between the two organelles in AD hippocampal neural cells. Tac‐mediated AD amelioration occurs through the regulation of protein expression at MAM, as observed in AD cells and animal models. Data support transcriptional regulation of ER‐mitochondria communication as a promising target for innovative therapeutics in AD.
Keywords: amyloid beta peptide, calcium, histone deacetylases, mitochondria, mitochondrial‐associated ER membranes, tacedinaline
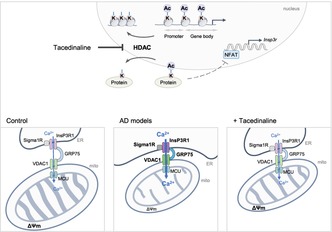
Inhibition or silencing of class I HDACs counteract ER‐mitochondria Ca2+ dyshomeostasis and mitochondrial function by regulating the expression of Ca2+‐handling proteins at MAM in AD cell and animal models. HDAC inhibition also decreases ER‐mitochondria contact length and NFAT transcriptional activity that regulates InsP3R expression, potentially controlling the cross‐talk between the two organelles in AD hippocampus.
Abbreviations
- acetyl‐H3
acetylated histone H3
- AD
Alzheimer's disease
- AβO
amyloid‐beta (Aβ1–42) oligomers
- Ca2+ i
intracellular free Ca2+
- CDR
clinical dementia rating
- ECF
enhanced chemifluorescence
- ER
endoplasmic reticulum
- FCCP
trifluoromethoxy carbonylcyanide phenylhydrazone
- GRP75
75 kDa glucose‐regulated protein
- HDACi
histone deacetylase (HDAC) inhibitor
- InsP3R
inositol 1,4,5‐trisphosphate receptor
- MAM
mitochondrial‐associated ER membranes
- MCI
mild cognitive impairment
- MCU
mitochondrial Ca2+ uniporter
- MERCS
mitochondria‐ER contact sites
- Mfn
mitofusin
- MICOS
mitochondrial contact site and cristae organizing system
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5diphenyltetrazolium bromide
- NFAT
nuclear factor of activated T cells
- NMDA
N‐methyl‐d‐aspartate
- NMDAR
NMDA receptor
- OMM
outer mitochondrial membrane
- PBMCs
peripheral blood mononuclear cells
- PDZD8
PDZ domain‐containing protein 8
- PI
propidium iodide
- PMSF
phenylmethylsulfonyl fluoride
- PEG
polyethylene glycol
- qRT‐PCR
quantitative real time (RT) PCR
- RMD3
regulator of microtubule dynamics protein 3
- SAHA
suberoylanilide hydroxamic acid
- SB
sodium butyrate
- SERCA
sarco/ER Ca2+‐ATPase
- Sigma1R
sigma‐1 receptor
- Tac
tacedinaline
- TBS‐T
tris‐buffered saline (TBS) plus Tween‐20
- TEM
transmission electron microscopy
- TMRM+
tetramethylrhodamine methyl ester
- VAPB
vesicle‐associated membrane protein‐associated protein B
- VDAC1
voltage dependent anion channel 1
- ΔΨm
mitochondrial transmembrane potential
1. INTRODUCTION
Alzheimer's disease (AD) is an age‐related neurodegenerative disease and the most common form of dementia, characterized by the deposition of insoluble aggregates of amyloid‐beta peptide (Aβ) and hyperphosphorylated tau, forming senile plaques and neurofibrillary tangles, respectively, particularly in the hippocampus (Knopman et al., 2021). Early diagnosis of AD ranging from mild cognitive impairment (MCI), in which patients have pure memory impairment, to dementia stages progressing to global cognitive decline and loss of autonomy has been defined in recent years (Jack et al., 2018).
Aβ1–42 oligomers (AβO), described as the most neurotoxic amyloid form, induce disruption of intracellular Ca2+ homeostasis mediated by N‐methyl‐d‐aspartate receptors (NMDAR) containing the GluN2B subunit (Ferreira et al., 2012). AβO‐induced immediate activation of NMDAR enhances mitochondrial Ca2+ accumulation in a mechanism involving endoplasmic reticulum (ER) Ca2+ release through inositol 1,4,5‐trisphosphate receptor (InsP3R) and mitochondrial depolarization (Ferreira et al., 2015), revealing increased susceptibility of glutamatergic synapses and mitochondrial dysfunction in AD.
Mitochondria‐associated ER membranes (MAM) are highly regulated dynamic contacts that sustain crucial neuronal function, namely Ca2+ and protein homeostasis, mitochondrial dynamics and ATP production (Resende et al., 2022 for review). Indeed, disturbances in mitochondrial function have been largely associated with upregulated MAM function, augmented cross‐talk between these two organelles (Area‐Gomez et al., 2012; Hedskog et al., 2013) and increased expression of Ca2+ channels (InsP3R and voltage dependent anion channel 1 [VDAC1]), which could be explained by abnormal Aβ interaction with mitochondria (Area‐Gomez et al., 2012; Chu et al., 2021; Hedskog et al., 2013). Furthermore, AβO downregulates the expression of the ER Ca2+ sensor STIM2 in a compensatory manner, impairing the neuronal store‐operated Ca2+ entry pathway, which further contributes to abnormal intracellular Ca2+ signaling observed in AD models (Zhang et al., 2015). Moreover, MAM proteins related with ER‐associated protein degradation, oxidative stress response, mitochondrial protein transport and ATP production were shown to be deregulated in the cortex of APP/PS1 mice prior to memory deficits (Völgyi et al., 2018).
Transcriptional deregulation has been described in AD pathogenesis, related to repressed expression of genes regulating mitochondrial biogenesis (Sheng et al., 2012), vesicle trafficking, cell adhesion, actin cytoskeleton dynamics (Sebollela et al., 2012) and antioxidant defense (Caldeira et al., 2013 for review). These observations may be associated with overexpression of histone deacetylases (HDACs), particularly nuclear HDAC2 and HDAC3 (class I) and cytosolic HDAC6 (class II) (Ding et al., 2008; Gräff et al., 2012; Zhu et al., 2017), although decreased HDAC levels were also reported in AD brains (Pascoal et al., 2022). Moreover, nuclear epigenome was shown to communicate with mitochondria in a bidirectional way, in which mitochondria mediates epigenetic processes and conversely, changes in epigenome regulate mitochondrial function (Matilainen et al., 2017).
HDACs inhibitors (HDACis), namely sodium butyrate (SB) and suberoylanilide hydroxamic acid (SAHA), which inhibit both class I and II HDACs, were shown to be effective in reverting Aβ pathology and the underlying cognitive decline (Fernando et al., 2020; Kilgore et al., 2010). Nevertheless, the influence of transcriptional modulation on mitochondrial abnormalities still remains to be elucidated in AD, as most of the studies have been focused on neuroprotection against oxidative stress (Choi et al., 2015; Gaisina et al., 2018; Rivieccio et al., 2009). Moreover, we have previously observed that SB rescued mitochondrial bioenergetic deficits (Naia et al., 2017) and improved mitochondrial‐dependent Ca2+ handling (Oliveira et al., 2006) in cell and mouse models of Huntington's disease. Tacedinaline (Tac; CI‐994), a synthetic benzamide‐based HDACi selective to class I HDACs (Beckers et al., 2007), was shown to improve cognitive and memory dysfunction in an intellectual disability mouse model (Cooper et al., 2020) and in C57BL/6 aged mice (McClarty et al., 2021) and exerted neuroprotective effects in mice spinal cord and brain injury (Sada et al., 2020; Zhang et al., 2018). Furthermore, a crucial physiologic function of class I HDACs in modulating mitochondrial homeostasis was described in Caenorhabditis elegans (Shao et al., 2020).
Therefore, we hypothesized that reduction of class I HDACs activity plays a protective role in AD pathogenesis by ameliorating transcriptional activity related with mitochondrial function and inter‐organelle communication. Here we report, for the first time, that Tac or class I HDACs knockdown in AD hippocampal neural cell models counteract ER‐mitochondria Ca2+ dyshomeostasis by regulating the expression of Ca2+‐handling proteins that control the cross‐talk between the two organelles. Tac‐induced regulation of ER/MAM proteins is further observed in the hippocampus of AD animals. These findings support ER‐mitochondrial‐related transcriptional regulation as a promising target for innovative therapeutics in AD.
2. RESULTS
2.1. Tac and SB, but not SAHA, protect against AβO‐induced cytotoxicity
Class I HDACs (Figure 1a), particularly HDAC2 and HDAC3, regulate synaptic plasticity and memory formation and are abundantly expressed in AD human and mouse brains (e.g., Gräff et al., 2012; Zhu et al., 2017). Accordingly, we found increased HDAC3 levels in the cortex of AD patients at Braak stages III–IV, as compared to control individuals (Figure 1b), along with a significant reduction in acetylated histone H3 (acetyl‐H3) levels (Figure 1c). Because HDACs are also expressed in PBMCs (Zhang et al., 2011) and these peripheral cells were previously shown by us to be affected in early AD human and mouse stages, correlating with changes in cerebral cortex (Mota et al., 2015), we determined the levels of HDAC2 and HDAC3 in PBMCs derived from MCI subjects (Clinical Dementia Rating (CDR) of 0.5, as described in Mota et al., 2015) or AD patients with increasing degrees of cognitive impairment. Both HDAC2 and HDAC3 protein levels were significantly increased in MCI, as compared with age‐matched control individuals; however, no differences were observed in PBMCs from mild, moderate, or severe AD stages (Figure 1d). Notably, exposure of HT22 cells to AβO for 23 h caused a significant increase in both HDAC2 and HDAC3 proteins levels (Figure 1e), suggesting a potential role of these deacetylases as transcriptional regulators due to their nuclear localization. These results support the hypothesis that class I HDACs (particularly HDAC2 and HDAC3) are possible therapeutic targets in AD, which can be mimicked by AβO exposure.
FIGURE 1.
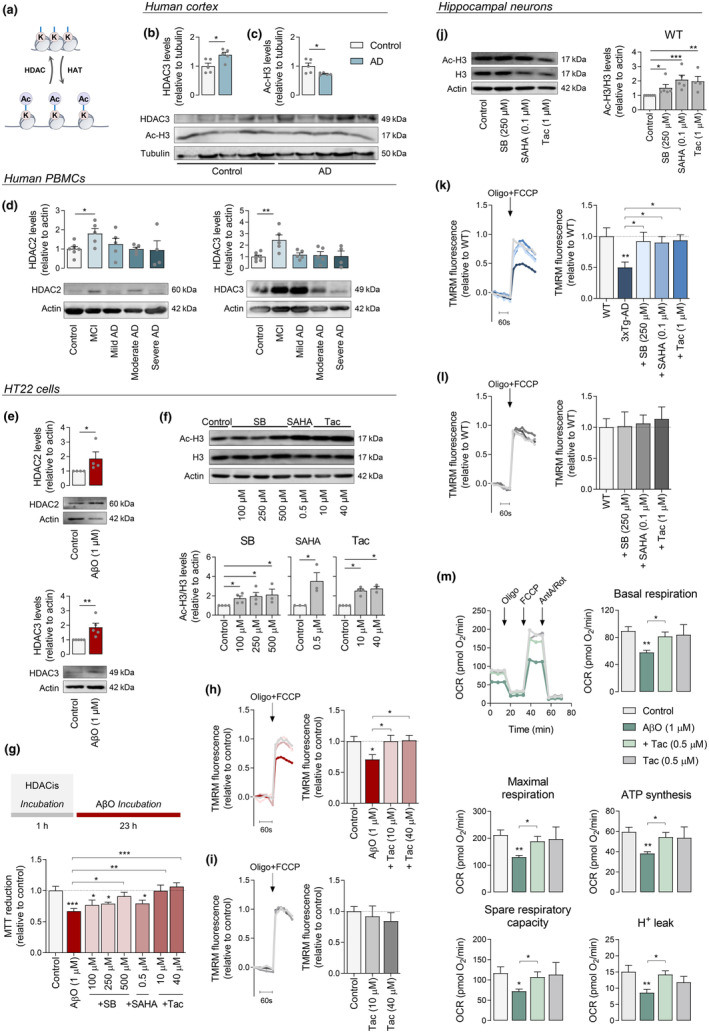
Levels of HDAC2 and HDAC3 and effect of HDACis on acetyl‐H3 levels, AβO‐induced toxicity, and mitochondrial function. Scheme of histone acetylation/deacetylation (a) Western blotting analysis of HDAC3 (b) and acetyl‐H3 (c) protein levels in postmortem brain cortex derived from control individuals or AD patients. Data are the mean ± SEM of 5 individuals per group. Western blotting analysis of HDAC2 and HDAC3 protein levels in PBMCS derived from control individuals, MCI or AD patients. Data are the mean ± SEM of 4–10 individuals per group (d). HT22 cells were treated for 23 h with 1 μM AβO. Western blotting analysis of HDAC2 and HDAC3 protein levels (e). Cells were treated for 24 h with HDACis (SB, SAHA and Tac at the indicated concentrations). Western blotting analysis of acetyl‐H3 protein levels (f). Cells were pre‐exposed for 1 h with HDACis and then co‐treated with 1 μM AβO for the remaining 23 h of incubation. Cell metabolic activity was measured using the MTT assay (g). Representative TMRM+ fluorescence trace and peak amplitude after oligo plus FCCP stimulation in the presence (h) or absence (i) of AβO. Hippocampal neurons were treated for 24 h with HDACis (250 μM SB; 0.1 μM SAHA or 1 μM Tac). Western blotting analysis of acetyl‐H3 protein levels (j). Representative TMRM+ fluorescence trace and peak amplitude in response to maximal mitochondrial depolarization induced by oligo plus FCCP (2 μg/mL; 2 μM) in 3xTg‐AD (k) and WT (l) neurons. WT hippocampal neurons were pre‐treated for 1 h with Tac (0.5 μM) and then co‐incubated with 1 μM AβO for the remaining 23 h. Basal respiration, maximal respiration, ATP production (oligomycin‐sensitive respiration), spare respiratory capacity, and H+ leak were quantified using a Seahorse analyzer (m). HT22 cells (f–i) were pre‐treated for 1 h with Tac (10 or 40 μM) and then co‐incubated with 1 μM AβO for the remaining 23 h. Representative F340/380 fluorescence trace and peak amplitude after oligo plus FCCP stimulation in the presence (h) or absence (i) of AβO. Data are the mean ± SEM of 3–7 independent experiments, run in triplicates to quadruplicates. Statistical analysis: Kruskal–Wallis followed by uncorrected Dunn's multiple comparison test, one‐way ANOVA followed by uncorrected Fisher's LSD multiple comparison test, Mann–Whitney test and unpaired Student's t‐test; *p < 0.05; **p < 0.01; ***p < 0.001 when compared to the control.
Therefore, we evaluated the neuroprotective efficacy of class I HDACis in AD hippocampal neural cells. Hippocampal neurons obtained from WT or 3xTg‐AD mice or mouse hippocampal‐derived HT22 cells were exposed to HDACis, SB, SAHA, and Tac for 24 h, and screened for cell metabolic activity, cell proliferation, and apoptotic/necrotic cell death (Figure S1B,E–G). Exposure to HDACis caused a decrease in MTT reduction in a concentration‐dependent manner in WT hippocampal neurons (Figure S1B) and HT22 cells (Figure S1E), while non‐toxic concentrations tested in WT neurons caused no major effects in 3xTg‐AD hippocampal neurons either (Figure S1B). Because HDACis can arrest cell growth (e.g., Marks et al., 2000), we analyzed whether 24 h treatment with SB, SAHA or Tac affects cell proliferation; however, no differences were observed between control and HDACis‐treated cells (Figure S1F). Moreover, treatment with HDACis increased the levels of acetyl‐H3 in both hippocampal neurons (Figure 1j, Figure S1C) and HT22 cells (Figure 1f).
A decrease in MTT reduction was observed in HT22 cells subjected to 1 μM AβO (Figure 1g), indicating a compromised metabolic activity, which was not accompanied by changes in cell death, as evaluated by double Hoechst/PI staining (Figure S1G). AβO‐induced metabolic compromise in HT22 cells was completely abolished in the presence of Tac (at 10 and 40 μM) (Figure 1g). In addition, cell viability remained unchanged when HT22 cells were incubated with Tac alone (Figure S1G). SB at 500 μM was able to revert cytotoxic effects exerted by AβO (Figure 1g), a concentration shown to cause a significant decrease in cell metabolic activity (about 17%; Figure S1E). In contrast, SAHA (0.5 μM) did not protect HT22 cells against AβO‐induced cell dysfunction (Figure 1g).
2.2. HDACis decrease mitochondrial Ca2+ retention in 3xTg‐AD hippocampal neurons subjected to NMDAR activation and AβO‐treated HT22 cells
Mitochondrial transmembrane potential (ΔΨm), a key indicator of mitochondrial activity required for ATP production, is reported to be reduced in several AD cell models (e.g., Jadiya et al., 2019). Our results evidence decreased ΔΨm in 3xTg‐AD hippocampal neurons (Figure 1k) and in HT22 cells following AβO treatment (Figure 1h), as revealed by the decrease in TMRM+ fluorescence following complete mitochondrial depolarization, when compared with WT neurons. HDACis, namely SB (250 μM), SAHA (0.1 μM), and Tac (1 μM), completely reverted ΔΨm dissipation in 3xTg‐AD neurons (Figure 1k). Moreover, Tac (10, 40 μM) prevented mitochondrial depolarization induced by exposure to AβO in HT22 cells (Figure 1h). Evaluation of O2 consumption rate (OCR), another readout of mitochondrial function, revealed that the basal and maximal respiration, ATP synthesis, spare respiratory capacity, and H+ leak were decreased in hippocampal neurons exposed to AβO and that Tac prevented these effects (Figure 1m). However, no changes in coupling efficiency or non‐mitochondrial respiration were observed in AβO‐treated neurons in the absence or in the presence of Tac, as compared to control (Figure S1D). Importantly, ΔΨm, mitochondrial respiration and ATP synthesis remained unchanged in WT neurons and control cells after inhibition of class I HDAC's activity (Figure 1i,l,m).
Another feature of early neuronal dysfunction in AD is Aβ‐mediated impairment of glutamatergic neurotransmission and subsequent mitochondrial Ca2+ dyshomeostasis (Ferreira et al., 2015). Considering this, hippocampal neurons derived from 3xTg‐AD (which produce and secrete Aβ [Vale et al., 2010]) and WT mice were directly subjected to selective activation of NMDAR by stimulation with 100 μM NMDA plus glycine in Mg2+‐free pyruvate‐based medium and mitochondrial Ca2+ levels measured directly or indirectly using the mitochondrial‐Ca2+ sensitive probes Rhod2‐AM (Figure S2A) or Fura2‐AM (Figure 2a), respectively. Our results show increased mitochondrial Ca2+ retention in 3xTg‐AD compared with WT neurons (Figure 2a, Figure S2A) in response to Ca2+ entry involving GluN2B‐containing NMDAR (Figure S2B). Interestingly, we observed no significant differences in Ca2+ i levels after NMDA stimulation in 3xTg‐AD compared to WT neurons (Figure 2a), suggesting that increased mitochondrial Ca2+ retention occurs independently of changes in Ca2+ influx through the NMDAR. We further ascertained if mitochondrial Ca2+ dyshomeostasis could occur independently of NMDAR activation. We found that under basal conditions (in the absence of NMDAR activation), mitochondrial‐Ca2+ levels in 3xTg‐AD neurons were similar to WT neurons (Figure S2C). These observations point to a mechanism whereby NMDAR activation leads to increased Ca2+ retention in mitochondria in 3xTg‐AD hippocampal neurons.
FIGURE 2.
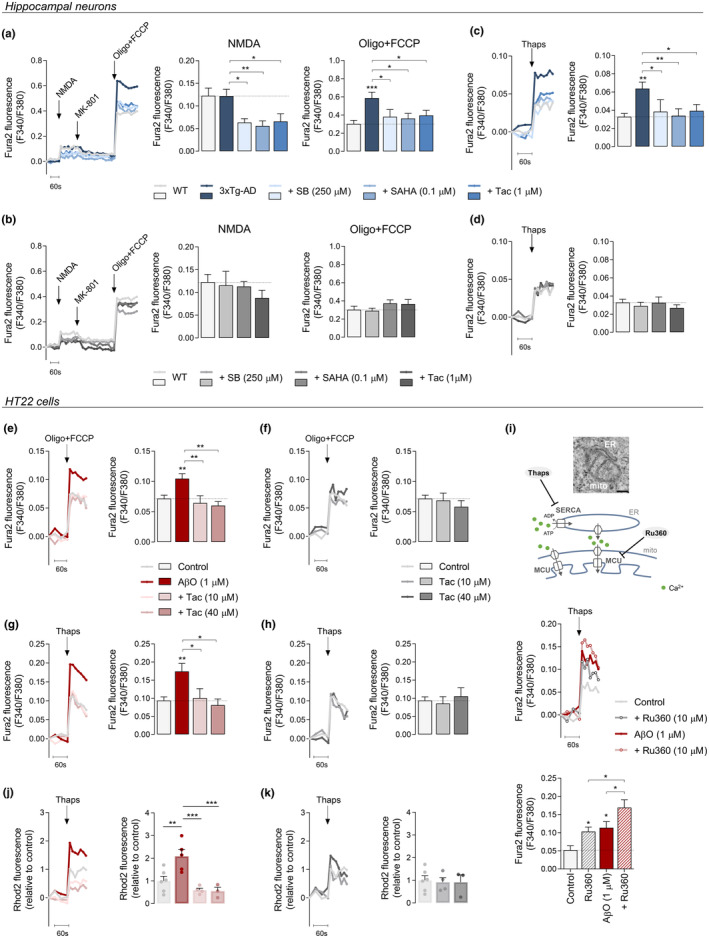
Effect of HDACis on ER‐mitochondria Ca2+ transfer in hippocampal neurons and AβO‐treated cells. Hippocampal neurons (a–d) were treated for 24 h with HDACis (250 μM SB; 0.1 μM SAHA or 1 μM Tac). Representative F340/380 fluorescence trace and peak amplitude in response to 100 μM NMDA stimulation and to maximal mitochondrial depolarization induced by oligo plus FCCP (2 μg/mL; 2 μM) in 3xTg‐AD (a) and WT (b) neurons following HDACis pre‐treatment. Representative F340/380 fluorescence trace and peak amplitude in response to ER‐Ca2+ storage depletion induced by thapsigargin (1 μM) in 3xTg‐AD (c) and WT (d) neurons. HT22 cells (e–k) were pre‐treated for 1 h with Tac (10 or 40 μM) and then co‐incubated with 1 μM AβO for the remaining 23 h. Representative F340/380 fluorescence trace and peak amplitude after oligo plus FCCP stimulation in the presence (e) or absence (f) of AβO. Representative F340/380 fluorescence trace and peak amplitude following thapsigargin stimulus in the presence (g) or absence (h) of AβO. Representative F340/380 fluorescence trace and peak amplitude in response to thapsigargin stimulation following a pre‐incubation with Ru360 (MCU inhibitor; 10 μM) (i). Representative Rhod2 fluorescence trace and peak amplitude after thapsigargin stimulation in the presence (j) or absence (k) of AβO. Data are the mean ± SEM of 3–8 independent experiments, run in triplicates to quadruplicates. Statistical analysis: Kruskal–Wallis followed by uncorrected Dunn's multiple comparison test, one‐way ANOVA followed by uncorrected Fisher's LSD multiple comparison test; *p < 0.05; **p < 0.01; ***p < 0.001 when compared to control.
Thus, we examined the effect of HDACis on intracellular Ca2+ homeostasis in hippocampal neurons subjected to NMDAR activation. Ca2+ i levels evoked by NMDA plus glycine were shown to be decreased following SB, SAHA, and Tac incubation in 3xTg‐AD neurons (Figure 2a); in contrast, no differences in NMDAR‐evoked Ca2+ entry were found in HDACis‐treated WT neurons (Figure 2b). These data suggest an inhibitory effect of HDACis at NMDAR level in 3xTg‐AD neurons. Importantly, the increase in mitochondrial Ca2+ levels upon NMDA stimulation observed in 3xTg‐AD neurons was significantly counteracted by HDACis pre‐exposure (Figure 2a), whereas no effect was observed in WT neurons (Figure 2b). Taken together, these results suggest that HDACis regulate NMDA‐induced Ca2+ i levels as a compensatory mechanism to prevent mitochondrial Ca2+ overload promoted by NMDAR activation in 3xTg‐AD hippocampal neurons. In contrast, basal mitochondrial Ca2+ levels remained unaltered following incubation with SB, SAHA, or Tac in both WT and 3xTg‐AD neurons (Figure S2C,D).
To better understand the effect of class I HDACs inhibition on Aβ‐mediated mitochondrial Ca2+ dyshomeostasis, mitochondrial Ca2+ measurements were also performed in HT22 cells exposed to AβO. Data show that AβO significantly enhanced mitochondrial Ca2+ retention, when compared to control cells, which was completely reverted upon treatment with Tac (Figure 2e). Conversely, SB or SAHA had no effect on mitochondrial Ca2+ levels in AβO‐treated HT22 cells (Figure S2E). In addition, we found no differences in control cells after Tac (Figure 2f), SB or SAHA treatment (Figure S2F).
2.3. Tac prevents enhanced mitochondrial Ca2+ accumulation from ER in AβO‐treated HT22 cells
Because ER is one of the largest Ca2+ stores in cells, ER‐Ca2+ levels were evaluated by Fura2 fluorescence in hippocampal neurons and HT22 cells subjected to thapsigargin, a selective non‐competitive inhibitor of sarco/ER Ca2+‐ATPase (SERCA) that disrupts Ca2+ homeostasis in this cellular compartment. ER‐Ca2+ levels were significantly increased in 3xTg‐AD hippocampal neurons (Figure 2c), as compared to WT neurons, as well as in HT22 cells upon AβO treatment (Figure 2g). Although Tac and the pan‐HDACis (SB and SAHA) treatment counteracted the enhanced ER‐Ca2+ accumulation in 3xTg‐AD hippocampal neurons (Figure 2c), no differences were found on HT22 ER‐Ca2+ levels in the presence of AβO plus pan‐HDACis (Figure S2G). In contrast, Tac, which effect is described to be selective to class I HDACs, decreased Ca2+ retention into ER in AβO‐treated cells (Figure 2g), again suggesting a specific regulation of AβO‐induced abnormalities in intracellular Ca2+ buffering. Moreover, no effect on ER‐Ca2+ levels was observed in WT neurons (Figure 2d) nor in HT22 control cells (Figure 2h, Figure S2H) following HDACis treatment.
Taking into account that ER interacts with mitochondria at MAM to control intracellular Ca2+ signaling, we assessed Ca2+ communication between the two organelles with Ru360 (10 μM), a selective inhibitor of mitochondrial Ca2+ uniporter (MCU) and thus of mitochondrial Ca2+ uptake, in cells subjected to ER‐Ca2+ release, in the presence of thapsigargin. Remarkably, Ru360 increased cytosolic Ca2+ levels after ER‐Ca2+ release as compared with control cells (Figure 2i), indicating Ca2+ transfer into mitochondria. Furthermore, Ca2+ i rise was significantly higher in AβO‐treated cells (Figure 2i), indicating enhanced mitochondrial Ca2+ uptake from ER in cells subjected to AβO exposure. These findings suggest that Aβ promotes mitochondrial Ca2+ overload associated with mitochondrial dysfunction via enhanced ER‐Ca2+ release, potentially triggered by excessive Ca2+ stored in the ER.
To determine the influence of HDACis in precluding intracellular Ca2+ dyshomeostasis, we measured mitochondrial Ca2+ levels with Rhod2‐AM in response to thapsigargin stimulation in HT22 cells. Our results show that AβO exposure elicited an increase in Ca2+ retained in mitochondria following ER‐Ca2+ release, which was significantly prevented in HT22 cells treated with Tac (Figure 2j). No alterations in mitochondrial Ca2+ were observed in control cells subjected to Tac (Figure 2k), indicating that HDACis (specifically Tac) control ER‐mitochondria cross‐talk and function.
2.4. Tac ameliorates the expression of MAM‐related proteins and MAM morphology in AβO‐treated HT22 cells
In order to dissect transcriptional modifications exerted by Tac on MAM‐related proteins in AβO‐treated HT22 cells, mRNA and related protein levels were evaluated by qRT‐PCR and Western blotting analysis, respectively. InsP3R, a major ER Ca2+‐release channel, which is stabilized by sigma‐1 receptor (Sigma1R), enables Ca2+ transfer into mitochondria through a complex with 75 kDa glucose‐regulated protein (GRP75) and mitochondrial VDAC1 (Figure 3a). Considering that disturbances in mitochondrial function have been mainly associated with upregulated MAM function and augmented cross‐talk between ER and mitochondria (Area‐Gomez et al., 2012; Hedskog et al., 2013), we evaluated whether the alterations in Ca2+ signaling observed in AβO‐treated cells correlated with changes in the expression of Ca2+‐handling proteins present at MAM, including InsP3R1, Sigma1R, GRP75, VDAC1, PDZ domain‐containing protein 8 (PDZD8), and MCU. Our results evidence that exposure to AβO led to increased mRNA levels of Insp3r (Figure 3b), Sigmar1 (Figure 3c), Grp75 (Figure 3d), and Vdac1 (Figure 3e). However, we did not observe changes in mRNA levels of mitochondrial Ca2+ uniporter (Mcu) (Figure 3f), known to be largely responsible for mitochondrial Ca2+‐uptake in AD neurons (e.g., Ferreira et al., 2015), nor in the ER‐mitochondria tethering protein Pdzd8 (Figure 3g), which regulates cytoplasmic Ca2+ dynamics and consequently Ca2+ uptake by mitochondria (Hirabayashi et al., 2017), in response to AβO treatment. Interestingly, we found upregulated expression of other MERCS components with a physiological structural function, like vesicle‐associated membrane protein‐associated protein B (Vapb) (Figure 3h) and mitofusin 2 (Mfn2) (Figure 3j), in AβO‐treated cells, while regulator of microtubule dynamics protein 3 (Rmdn3, also known as protein tyrosine phosphatase interacting protein 51, PTPIP51) (Figure 3i) and Mfn1 (Figure 3k), that interact with VAPB and Mfn2, respectively, to form MAM‐tethering complexes, showed similar mRNA levels to those found in control cells. Concordantly with mRNA levels, InsP3R1, GRP75, and VDAC1 protein levels were found to be increased in HT22 cells treated with AβO (Figure 3l–n). Upregulated expression of ER‐mitochondria Ca2+‐transfer proteins was completely prevented in AβO‐treated cells following Tac treatment (Figure 3b–n), but was not affected in control cells (Figure 3b–n).
FIGURE 3.
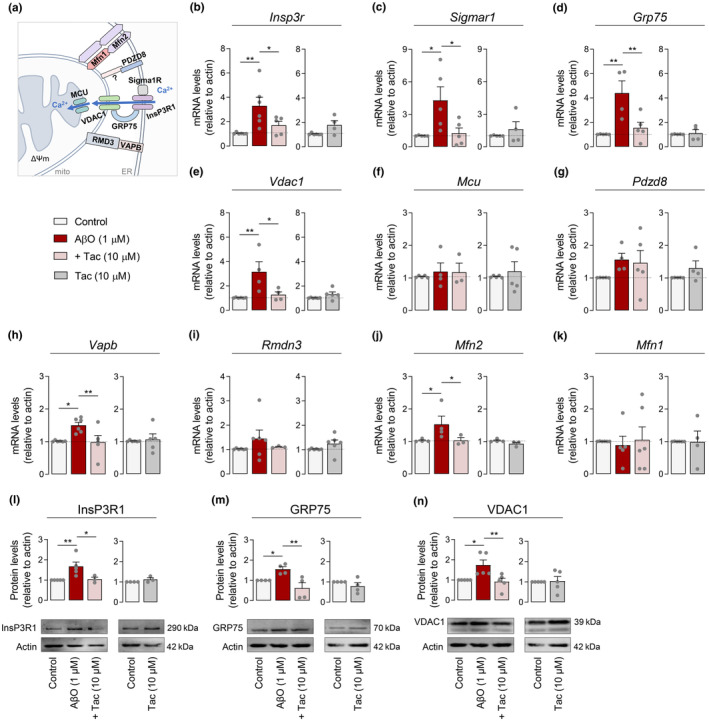
Tac effect on MAM‐related proteins in AβO‐treated cells. Schematic representation of proteins at MAM (a). HT22 cells were pre‐treated for 1 h with Tac (10 μM) and then co‐incubated with 1 μM AβO for the remaining 23 h or treated solely with Tac for 24 h. Relative expression of Insp3r (b), Sigmar1 (c), Grp75 (d), Vdac1 (e), Mcu (f), Pdzd8 (g), Vapb (h), Rmdn3 (i), Mfn2 (j), and Mfn1 (k) mRNA. Western blotting analysis of InsP3R1 (l), GRP75 (m), and VDAC1 (n) protein levels. Actin was used as the housekeeping messenger and the loading control. Data are the mean ± SEM of 3–6 independent experiments, run in triplicates to quadruplicates. Statistical analysis: Kruskal–Wallis test followed by uncorrected Dunn's multiple comparisons test, one‐way ANOVA followed by uncorrected Fisher's LSD multiple comparison test, Mann–Whitney test and unpaired Student's t‐test; *p < 0.05; **p < 0.01 when compared to control.
To test the hypothesis that close contacts between InsP3R1 and VDAC1 underlie augmented ER‐mitochondria Ca2+ transfer elicited by AβO, we examined the InsP3R1‐VDAC1 interaction by PLA. Our results showed that AβO triggered InsP3R1‐VDAC1 direct interactions, which were prevented by Tac (Figure 4a). However, Tac did not affect the formation of InsP3R1‐VDAC1 complexes in control cells (Figure 4a). These data provide the first evidence for a role of selective class I HDACis (namely Tac) in interfering with the transcription of genes relevant for MAM connectivity and concomitant ER‐mitochondria Ca2+ signaling in AD.
FIGURE 4.
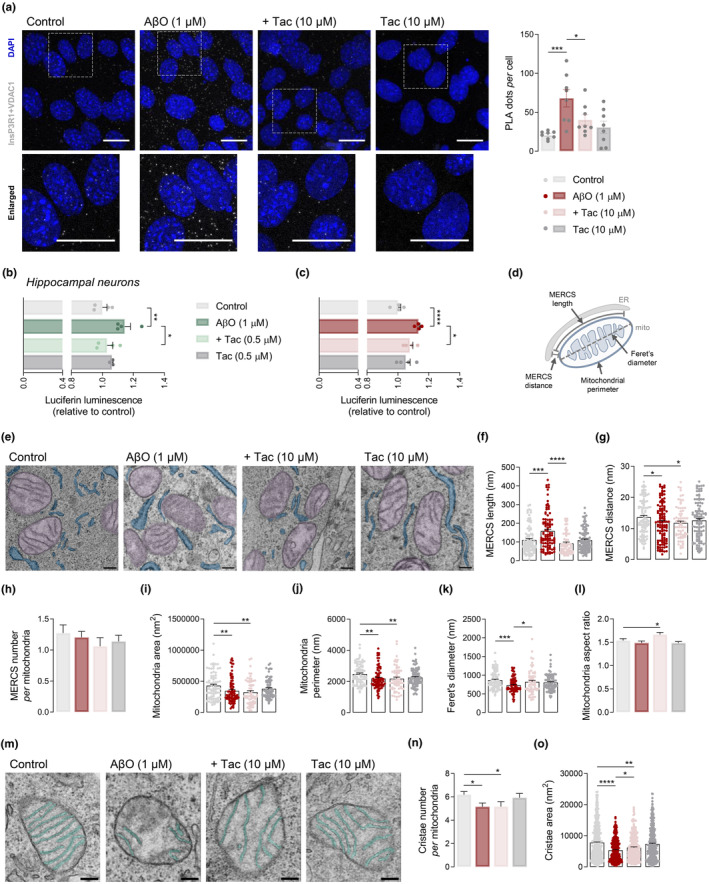
Tac effect on InsP3R1‐VDAC1 interaction and MERCS morphology in AβO‐treated cells. HT22 cells were pre‐treated for 1 h with Tac (10 μM) and then co‐incubated with 1 μM AβO for the remaining 23 h or treated solely with Tac for 24 h. Representative confocal images of in situ PLA signal (gray) indicating a physical interaction between InsP3R1 and VDAC1 (scale bar = 20 μm). The lower panels represent enlargements of the boxed areas in the upper panels. Quantification of number of PLA puncta in about 200–240 cells from 8 image stacks per condition (a). WT hippocampal neurons were pre‐treated for 1 h with Tac (0.5 μM) and then co‐incubated with 1 μM AβO for the remaining 23 h or treated solely with Tac for 24 h. NFAT transcriptional activity evaluated by luciferase reporter assay in hippocampal neurons (b) and HT22 cells (c). Data are the mean ± SEM of 4 independent experiments, run in triplicates to quadruplicates. Schematic representation of MERCS structural parameters: MERCS length and distance, mitochondrial perimeter and Feret's diameter (d). HT22 cells were pre‐treated for 1 h with Tac (10 μM) and then co‐incubated with 1 μM AβO for the remaining 23 h. Representative TEM images of mitochondria (in purple) in close contact with ER (in blue) (scale bar = 200 nm) in which distances ≤25 nm were considered as contacts (e). MERCS length (f) and distance (g), number of MERCS per mitochondria (h), mitochondrial area (i) and perimeter (j), Feret's diameter (k) and mitochondrial aspect ratio (l). Representative TEM images of mitochondrial cristae (in green) (scale bar = 200 nm) (m). Number of cristae per mitochondria (n), and cristae area (o). About 70–90 individual mitochondria from 10 randomly selected cells obtained from approximately 30 images were assessed in 26–38 independent TEM images. Statistical analysis: Kruskal–Wallis test followed by uncorrected Dunn's multiple comparisons test, one‐way ANOVA followed by uncorrected Fisher's LSD multiple comparison test; *p < 0.05; **p < 0.01; ***p < 0.0001; ****p < 0.0001 when compared to control.
Thus, the question arises as to how the ER‐mitochondria Ca2+‐transfer complexes are mechanistically regulated by Tac‐induced acetylation of histone (as shown in Figure 1f,j) and of non‐histone proteins (data not shown). Recent reports have shown that deacetylation‐activated Vav1 stimulates nuclear factor of activated T cells (NFAT) pathway (Rodríguez‐Fdez et al., 2020), which mediates InsP3R upregulation in an AD animal model (Shao et al., 2022). Therefore, to explore whether Tac modulates InsP3R1 expression by regulating NFAT, we measured NFAT transcriptional activity using a luciferase reporter plasmid in our AD cell models. We found a slight but significant increase in NFAT transcriptional activity in AβO‐treated hippocampal neurons (Figure 4b) and HT22 cells (Figure 4c), which was abolished by Tac (Figure 4b,c). No changes in NFAT activity were observed in Tac‐treated neurons (Figure 4b) or HT22 cells (Figure 4c).
To further characterize the effect caused by class I HDACs inhibition on MAM morphology, we performed ultrastructural analysis of HT22 cells using TEM. MERCS were defined by the ER‐mitochondria proximity within 25 nm and the MERCS length was quantified as the length of ER segments within 25 nm distance from the outer mitochondrial membrane (OMM) (Figure 4d). Electron micrographs analysis in AβO‐treated cells showed increased length of the ER‐mitochondria interface (Figure 4e,f) and a narrower gap between the OMM and ER membrane (Figure 4e,g), when compared to control cells. In contrast, the number of contacts between the two organelles, in relation to the number of mitochondria, remained unaltered (Figure 4e,h), indicating that the close apposition between ER and mitochondria relies on expression of intracellular Ca2+ signaling‐regulating proteins. Importantly, treatment with Tac in cells exposed to AβO induced a redistribution of ER in close contact with mitochondria by decreasing the MERCS length (Figure 4e,f), without affecting the distance between the organelles (Figure 4e,g) or the MERCS number per mitochondria (Figure 4e,h). ER‐mitochondria interactions in Tac‐treated cells, not exposed to AβO, were similar to control cells (Figure 4e–h). These results suggest that class I HDACis decrease the frequency of tighter interactions between ER and mitochondria, while ameliorates Ca2+ transfer into the mitochondrial matrix in AβO‐treated cells.
Additionally, we assessed if Tac treatment could also modulate mitochondrial morphology, specifically mitochondrial size (perimeter and surface area), shape (aspect ratio, defined as the ratio between major and minor mitochondrial axis), fragmentation (Feret's diameter, defined as longest distance between any two points in the mitochondria; Gillmore et al., 2022; Figure 4d), and cristae organization (number and surface area). Mitochondria from AβO‐treated cells exhibited decreased mitochondrial area (Figure 4e,i) and perimeter (Figure 4e,j), as well as reduced Feret's diameter (Figure 4e,k), indicating mitochondrial fragmentation. In contrast with the morphologic evidence of enhanced fission, no significant changes were observed in mitochondrial aspect ratio (Figure 4e,l). Additionally, mitochondria were shown to have a reduced number of cristae (Figure 4m,n) with reduced surface area (Figure 4m,o) in the presence of AβO. Interestingly, we found that while mitochondria were smaller as evaluated by area and perimeter values following Tac treatment in the presence of AβO (Figure 4e,i,j), when compared to control cells, Tac was able to prevent mitochondrial fragmentation (Figure 4e,k), which was accompanied by a significant increase in aspect ratio (Figure 4e,l). Furthermore, Tac treatment caused mitochondrial cristae remodeling by increasing the surface area (Figure 4m,o) without affecting the number of cristae per mitochondria (Figure 4m,n). None of the mitochondrial morphological features were altered following treatment with Tac alone (Figure 4e,i–o).
2.5. HDAC2‐siRNA prevents enhanced ER‐mitochondria Ca2+ transfer while HDAC3‐siRNA decreases ER‐Ca2 + retention in AβO‐treated HT22 cells
Neuroprotective effects observed following Tac treatment may arise from the combined inhibition of HDAC1, 2, 3, and 8 (class I HDACs). As both nuclear HDAC2 and HDAC3 are upregulated in the hippocampus of AD mouse models and human patients (Gräff et al., 2012; Zhu et al., 2017), we further investigated the impact of these HDACs on mitochondrial function and ER‐mitochondria cross‐talk. Thus, HDAC2 or HDAC3 expression were downregulated by using specific HDAC‐siRNAs in HT22 cells subjected to AβO (Figure 5a). Nontarget siRNA was used as control. In these conditions, quantitative analysis showed a 75% selective reduction in HDAC2 levels and a 67% decrease in HDAC3 levels in cells transfected with HDAC2‐ or HDAC3‐siRNA, respectively (Figure 5b).
FIGURE 5.
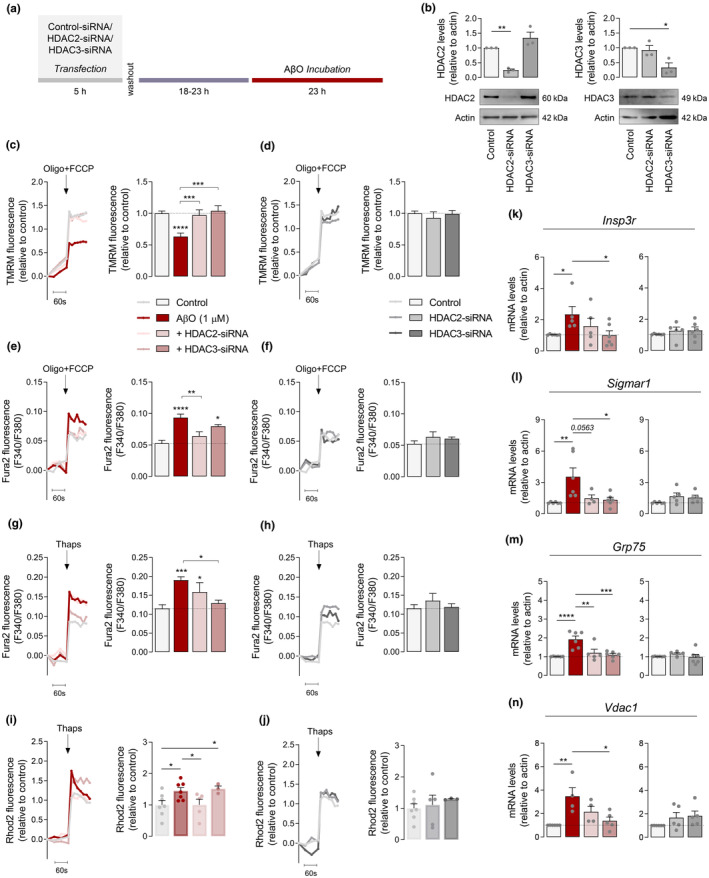
HDAC2/3 knockdown effect on mitochondrial membrane potential, Ca2+ i levels and MAM proteins in AβO‐treated cells. HT22 cells were transfected with either HDAC2‐ or HDAC3‐siRNA. Scrambled siRNA was used as a control. After transfection, cells were treated with 1 μM AβO and incubated for additional 24 h (a). Western blotting analysis of HDAC2 and HDAC3 proteins levels 48 h after transfection with HDAC2‐ or HDAC3‐siRNA (b). Representative trace of TMRM+ fluorescence and peak amplitude relative to control in response to maximal mitochondrial depolarization induced by oligo (2 μg/mL) plus FCCP (2 μM) in the presence (c) or absence (d) of AβO. Representative F340/380 fluorescence trace and peak amplitude after oligo plus FCCP stimulus in the presence (e) or absence (f) of AβO. Representative F340/380 fluorescence trace and peak amplitude in response to ER‐Ca2+ storage depletion induced by thapsigargin in the presence (g), or absence (h) of AβO. Representative Rhod2 fluorescence trace and peak amplitude after thapsigargin stimulation in the presence (i) or absence (j) of AβO. Relative expression of Insp3r (k), Sigmar1 (l), Grp75 (m), and Vdac1 (n) mRNA. Actin was used as the housekeeping messenger and the loading control. Data are the mean ± SEM of 3–7 independent experiments, run in triplicates to quadruplicates. Statistical analysis: Kruskal–Wallis test followed by uncorrected Dunn's multiple comparisons test and one‐way ANOVA followed by uncorrected Fisher's LSD multiple comparison test; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 when compared to control.
Of relevance, AβO‐mediated mitochondrial depolarization was completely precluded after HDAC2 and HDAC3 silencing (Figure 5c), but not in control cells (Figure 5d). Further mitochondrial functional assays showed that HDAC2‐siRNA substantially attenuated the increased Ca2+ levels accumulated in mitochondria in AβO‐treated cells (Figure 5e). However, HDAC3 knockdown did not significantly alter mitochondria Ca2+ retention induced by AβO (Figure 5e). These observations suggest that in the presence of AβO, HDAC3‐siRNA is able to prevent mitochondrial dysfunction independently of mitochondrial Ca2+ overload. Next, we determined whether selective silencing of nuclear HDACs altered ER‐Ca2+ accumulation. HDAC3‐siRNA abolished the augmented ER‐Ca2+ retention induced by AβO, whereas HDAC2‐siRNA did not cause alterations in the ER‐Ca2+ levels in AβO‐treated cells (Figure 5g). In agreement with reduced mitochondrial Ca2+ accumulation, Ca2+ transfer from ER to mitochondria was markedly decreased in HDAC2 knockdown cells treated with AβO, but not in cells exposed to HDAC3‐siRNA (Figure 5i). Together, these results suggest a differential role of nuclear HDACs in regulating ER and mitochondria Ca2+ signaling under AβO exposure. No significant differences were found in ER or mitochondrial Ca2+ levels following selective HDAC knockdown in cells non‐exposed to AβO (Figure 5f,h,j).
In addition, we assessed the influence of HDAC2 and HDAC3 silencing on mRNA levels of Insp3r, Sigmar1, Grp75, and Vdac1. HDAC3‐siRNA led to reduced expression of Insp3r (Figure 5k), Sigmar1 (Figure 5l), Grp75 (Figure 5m), and Vdac1 (Figure 5n) in AβO‐treated cells. Selective HDAC2 knockdown in the presence of AβO significantly decreased mRNA levels of Grp75 (Figure 5m) and showed a tendency to reduce Sigmar1 mRNA levels (Figure 5l), but did not induce significant changes in the expression of Insp3r (Figure 5k) or Vdac1 (Figure 5n). These data suggest that HDAC2 and HDAC3 act in a cooperative manner in regulating mitochondrial function and ER‐mitochondria cross‐talk.
2.6. Tac decreases Aβ levels and alters the expression of MAM‐related proteins in the hippocampus of APP/PS1 mice
Our in vitro results indicate that both pharmacologically inhibiting class I HDACs with Tac and silencing both HDAC2 and HDAC3 constitute relevant neuroprotective strategies in AD hippocampal neural cells. Hence, we sought to determine if the alterations in ER‐mitochondrial cross‐talk induced by Tac could be recapitulated in vivo by using the APP/PS1 mouse model. We initially observed an increase in the levels of nuclear HDAC2 and HDAC3 in the hippocampus of 10 month old APP/PS1 mice, compared to WT mice (Figure 6a), similarly as observed in human AD cortex and MCI PBMCs and HT22 cells exposed to AβO (Figure 1). To examine the neuroprotective effect of class I HDACis in vivo, APP/PS1 mice versus WT aged 9 month‐old were treated with 30 mg/kg/day Tac by intraperitoneal injection for 30 days (until 10 months of age) (Figure 6b). Mice body weight was maintained throughout the experiment (data not shown). Both genotypes exhibited significantly increased acetyl‐H3 levels in the hippocampus upon Tac treatment in comparison with mice treated with vehicle (Figure 6c).
FIGURE 6.
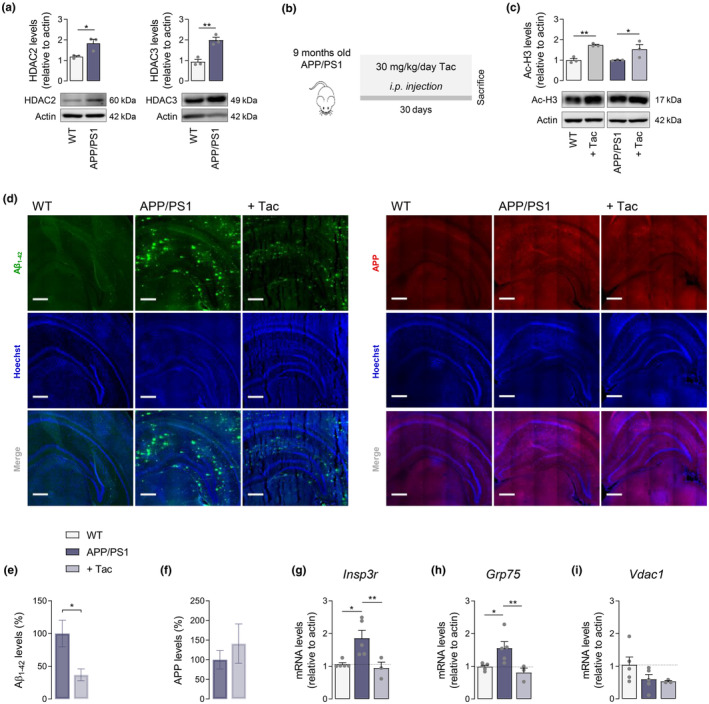
Effect of Tac on Aβ levels and expression of genes involved in ER‐mitochondria Ca2+ communication in the hippocampus of APP/PS1 mice. Western blotting analysis of HDAC2 and HDAC3 protein levels in 10 months old APP/PS1 and WT mice (a). APP/PS1 versus WT mice were treated with Tac (30 mg/kg/day) for 30 days (b). Western blotting analysis of acetyl‐H3 protein levels (c). Representative immunofluorescent images (d; scale bar = 100 μm; 25–30 coronal sections) and quantification of Aβ (e) and APP (f) levels. Relative expression of Insp3r (g), Grp75 (h), and Vdac1 (i) mRNA. Actin was used as the housekeeping messenger and the loading control. Data are the mean ± SEM of 3–5 mice per group. Statistical analysis: Kruskal–Wallis test followed by uncorrected Dunn's multiple comparisons test, one‐way ANOVA followed by uncorrected Fisher's LSD multiple comparison test and unpaired Student's t‐test; *p < 0.05; **p < 0.01 when compared to control.
An ELISA‐based assay indicated increased levels (~17‐fold) of soluble Aβ1–42 in APP/PS1 mice aged 6–13 month‐old as compared to WT (data not shown). We further assessed the effect of Tac treatment on Aβ levels in APP/PS1 mouse hippocampus by immunofluorescence. A significant reduction in Aβ was observed following Tac treatment, when compared with vehicle‐treated APP/PS1 (Figure 6d,e), suggesting alterations in either Aβ formation or clearance. No Aβ accumulation was found in WT mice (Figure 6d,e). Furthermore, no significant differences in APP levels were observed in the hippocampus of Tac‐treated APP/PS1, compared to APP/PS1 mice, while the presence of APP was scarce in WT mice (Figure 6d,f). Tac‐mediated unaltered APP levels preclude the hypothesis of potential changes in APP expression.
Additionally, we analyzed the expression of genes involved in Ca2+ signaling at MAM. Increased mRNA levels of Insp3r (Figure 6g) and Grp75 (Figure 6h) were detected in the hippocampus of APP/PS1 as compared to WT mice, which was completely reverted by Tac (Figure 6g,h). In addition, no significant differences in the expression of Vdac1 were observed (Figure 6i). These results suggest that inhibition of class I HDACs abolishes early transcriptional dysregulation of key protein players involved in ER‐mitochondria Ca2+ cross‐talk, concomitantly with reducing Aβ pathology, in APP/PS1 transgenic mice.
3. DISCUSSION
This work is the first to support evidence for epigenetic regulation exerted by selective nuclear class I HDACs, namely HDAC2 and HDAC3, as a relevant neuroprotective strategy for ER and mitochondrial dysfunction and deregulated Ca2+ handling in AD pathology. HDAC2 and HDAC3 have been mostly implicated in regulating neuroplasticity and cognitive function. As such, they were shown to be enhanced in AD mouse models, as well as in postmortem brain samples from sporadic AD patients (Gräff et al., 2012; Zhu et al., 2017), as shown in our study. Conversely, a significant reduction of class I HDACs (HDACs 1, 2, and 3) was recently reported in brains from MCI and AD patients, as evaluated by PET analysis using [11C]Martinostat tracer, as well as in transgenic rats with both amyloid and tau pathology, but not in aged transgenic rats (McGill‐R‐Thy1‐APP rats with the APP Swedish and Indiana mutations) expressing single amyloid pathology (Pascoal et al., 2022). In addition, HDAC1 activity was shown to be reduced in the hippocampus of aged 5XFAD transgenic mice (Pao et al., 2020), a mouse model characterized by the presence of thioflavin‐positive tau aggregates apart from amyloid plaque accumulation (Shin et al., 2021). These findings suggest HDACs reduction as a possible epigenetic AD signature downstream tau pathology, along disease progression. Of relevance, our results demonstrate that PBMCs from MCI individuals exhibited elevated HDAC2 and HDAC3 levels, but no alterations were observed in AD patients already in the stage of dementia, suggesting that peripheral epigenetic dysregulation associated with histone acetylation imbalance occurs in early pre‐dementia phase of AD (MCI) where patients develop a pure memory deficit while preserving full autonomy. Moreover, we found increased HDAC3 levels, concomitantly with diminished H3 acetylation, in postmortem cortex of AD patients at Braak stages III–IV, which was associated with CDR of 0.5 and very mild dementia (Therriault et al., 2022). Consistent with this study, Drosophila larvae, modeling early human AD neurodegeneration, exhibited enhanced HDAC2 in the brain before Aβ deposition (Beaver et al., 2022). Importantly, we also observed increased HDAC2 and HDAC3 protein levels following AβO treatment in HT22 cells and in APP/PS1 transgenic mice, supporting the importance of targeting class I HDACis in ameliorating cognitive function and preventing neuronal dysfunction. SB treatment, which acts through inhibition of both class I and II HDACs, was shown to improve memory in APP/PS1 mice (Govindarajan et al., 2013; Kilgore et al., 2010), to reduce amyloid burden in 5xFAD mouse model (Fernando et al., 2020), and to restore learning behavior and enhance neurogenesis in PS1/2 cDKO mice, an Aβ‐independent mouse model of AD (Cao et al., 2018). Tac, a novel benzamide‐based HDACi that completely inhibits class I HDACs and to a smaller extent HDAC6 (Beckers et al., 2007), was shown to improve cognition in an intellectual disability syndrome mouse model (Cooper et al., 2020), to increase neuronal plasticity after traumatic brain injury (Sada et al., 2020), to promote functional recovery following spinal cord injury (Zhang et al., 2018) and to reduce antipsychotic haloperidol‐induced motor side effects and memory impairment in aged mice (McClarty et al., 2021). However, the role of transcription modulation elicited by Tac has not been addressed in neurodegenerative disorders like AD yet.
Neuronal dysfunction in AD has been described to be mediated by AβO‐induced intracellular Ca2+ dyshomeostasis through mGluR5 overactivation, leading to elevated ER‐Ca2+ accumulation (Zhang et al., 2015), and through GluN2B‐composed NMDAR (Ferreira et al., 2012, 2015), which leads to mitochondrial Ca2+ overload as a consequence of ER‐Ca2+ release through InsP3R (Ferreira et al., 2015). Accordingly, our data evidence elevated ER‐Ca2+ levels and Ca2+ accumulation in mitochondria elicited by NMDAR activation in 3xTg‐AD hippocampal neurons, exhibiting increased levels of endogenous Aβ (Vale et al., 2010), as compared to WT neurons. However, unaltered NMDAR‐mediated Ca2+ entry and mitochondrial Ca2+ levels in non‐stimulated cells were observed, suggesting that increased Ca2+ retention into mitochondria, under NMDA stimulation, occurs after Ca2+ release from ER independently of Ca2+ influx through NMDAR. Interestingly, it was previously shown that Aβ production can occur at MAM, facilitating its regulatory role on ER‐mitochondria communication (Schreiner et al., 2015). We found that HDACis (SB, SAHA, and Tac) reverted excessive ER‐Ca2+ levels and subsequent mitochondrial Ca2+ retention following NMDAR activation in 3xTg‐AD neurons, which was preceded by a reduction in NMDA‐evoked Ca2+ entry, suggesting a compensatory mechanism to prevent intracellular Ca2+ dyshomeostasis.
Previously, it was reported that HT22 cells exposed to extracellular Aβ underwent a deleterious effect on mitochondrial morphology and function similar to that observed using mitochondria‐targeted Aβ (Cha et al., 2012). Here, we describe that AβO promotes excessive ER‐Ca2+ retention, favoring ER‐mitochondria Ca2+ transfer in HT22 cells, pointing out mitochondrial dysfunction as a consequence of the impaired ER function. However, this effect was counteracted by Tac, but not by SB and SAHA, suggesting that inhibition of selective class I HDACs, rather than that exerted by pan‐HDACis (as SB and SAHA), regulates Ca2+ signaling between ER and mitochondria promoted by AβO in the absence of an excitotoxic stimulus.
The impairment of ER‐mitochondria Ca2+ signaling observed in AβO‐treated cells might be explained by upregulation of mRNA levels of proteins involved in Ca2+ signaling at MAM, namely InsP3R, Sigma1R, GRP75, and VDAC1, InsP3R1‐VDAC1 direct interaction and associated augmented ER‐mitochondria coupling, in accordance with previous studies performed in AD cell and animal models before amyloid plaque formation and postmortem AD brains (Chu et al., 2021; Hedskog et al., 2013). Likewise, we found upregulated mRNA levels of MAM‐associated proteins in the hippocampus of presymptomatic APP/PS1 mice exhibiting increased Aβ levels. Interestingly, D'Eletto et al. (2018) demonstrated increased InsP3R‐GRP75 interaction associated with a reduction in MERCS number and ER‐mitochondria Ca2+ transfer and augmented distance between organelles, indicating that MAM structure and ER‐mitochondria cross‐talk have a complex non‐linear regulation. In fact, disturbances in ER–mitochondria cross‐talk in AD have been mainly associated with an increased interaction between both organelles (Area‐Gomez et al., 2012), for example, through binding of mutant PS2 with Mfn2, which promotes mitochondrial Ca2+ overload, ΔΨm collapse, and apoptosis (Filadi et al., 2016). In line with increased MERCS length upon AβO treatment observed in the present study, other findings showed that ER undergoes significant extension, promoting the formation of additional contacts with mitochondria and Ca2+ transfer between the two organelles under pathological conditions caused by DNA damage (Zheng et al., 2018). Interestingly, Bartok et al. (2019) evidenced that tight interactions between ER and mitochondria occur independently of Ca2+ flux through InsP3Rs. Notably, we observed that, through regulation of histone acetylation imbalance, Tac ameliorated the expression of MAM proteins involved in ER‐mitochondria Ca2+‐transfer, thereby reducing InsP3R1‐VDAC1 interaction, MAM connectivity, and excessive mitochondrial Ca2+ uptake in AβO‐treated cells. Recent data evidence that ablation of InsP3Rs in HEK293 cell line abrogated ER‐mitochondrial Ca2+ feedback and cell death (Booth et al., 2021). In addition, InsP3R1 silencing rescued exaggerated Ca2+ signaling in cortical and hippocampal neurons, and aberrant hippocampal long‐term potentiation and memory deficits in FAD mouse models (Shilling et al., 2014). Moreover, knockdown of GRP75 impaired ER–mitochondrial coupling, provided protection against mitochondrial dysfunction and cell death, and attenuated mitochondrial Ca2+ overload in HT22 cells submitted to glutamate‐induced oxidative stress (Honrath et al., 2017). Overall, these observations validate the neuroprotective role exerted by Tac on ER‐mitochondria cross‐talk through modulation of tethering and Ca2+ signaling between the two organelles. However, loss of ER‐mitochondria tethering has been described in temporal cortex from AD patients at the final stages of the disease (Lau et al., 2020), and the enhancement of ER‐mitochondria apposition as a neuroprotective mechanism in a Drosophila AD model (Garrido‐Maraver et al., 2020). Contrarily, we observed decreased expression of proteins involved in Ca2+ signaling at MAM in APP/PS1 hippocampus upon Tac treatment, concomitantly with decreased Aβ deposition.
Given the well‐established mechanism of HDACs inhibition in gene transcription, we surprisingly found that Tac negatively regulates the expression of MAM‐related proteins such as InsP3R1‐VDAC1 complexes in our AD cell and animal models. These changes might be due to Tac‐induced downregulation of NFAT transcriptional activity in AβO‐treated cells, as observed by us, as activated NFAT was shown to be required for InsP3R upregulation in a Drosophila AD model (Shao et al., 2022). This is in line with a study reporting that Ca2+‐dependent stimulation of NFAT is downmodulated by acetylated VAV1 and promoted by Lys deacetylation of VAV1 (Rodríguez‐Fdez et al., 2020), suggesting Lys acetylation of non‐histone proteins as a target effect of class I HDACis in modulating transcription of ER‐mitochondria cross‐talk in AD pathogenesis. Alternatively, striking findings of a non‐canonical mechanism linking transcription repression to histone hyperacetylation have been reported. These authors demonstrated that HDACis inhibit transcription by redistributing elongation factors to newly acetylated sites, as gene bodies, resulting in reduced occupancy at promoters and enhancers as well as enhancer activity (Cheng et al., 2018; Greer et al., 2015; Marié et al., 2018; Pinz et al., 2015). Collectively, these findings suggest that HDACis‐induced transcription regulation of MAM function may not be limited to a single pathway but rather a broad effect of both histone and non‐histone acetylation, which requires further investigation.
A previous study reported that hippocampal neurons from McGill‐R‐Thy1‐APP transgenic rats exhibited ER swelling and fragmented mitochondrial cristae under conditions of significant Aβ secretion (Martino Adami et al., 2019). Here, we found that extracellular added AβO disrupted mitochondrial morphology through a reduction in mitochondrial surface area, along with increased fission and cristae fragmentation. Our data also indicate that Tac partially restores mitochondrial morphology by increasing mitochondrial elongation and cristae area, maintaining mitochondrial size and number of cristae. The formation of mitochondrial contact site and cristae organizing system (MICOS) in proximity to MERCS was recently found in yeast (Tirrell et al., 2020), whereas Miro‐1 and ‐2, two proteins required for normal mitochondrial cristae architecture, were shown to regulate MERCS structure and function, namely ER‐mitochondrial Ca2+ signaling (Modi et al., 2019). These observations provide new insights into the mechanism that regulate ER‐mitochondria cross‐talk, suggesting that transcriptional modulation of MAM function exerted by Tac in cells exposed to AβO may directly contribute to improved mitochondrial cristae organization.
To further investigate the mechanisms underlying the epigenetic modulation exerted by Tac on ER‐mitochondria dysfunction elicited by AβO, we focused on the effect of nuclear HDAC2 and HDAC3. Our results indicate that HDAC2 silencing abrogated the excessive ER‐mitochondrial Ca2+ transfer and decreased GRP75 expression in response to AβO exposure. In contrast, HDAC3 knockdown downregulated the expression of Ca2+‐handling associated proteins at MAM, namely InsP3R1, Sigma1R, GRP75, and VDAC1, and reduced the elevated ER‐Ca2+ levels in AβO‐treated cells. Interestingly, selective knockdown of HDAC2 or HDAC3 exhibited a similar effect on mitochondrial function by preventing mitochondrial depolarization induced by AβO. More recently, HDAC2 shRNA was reported to enhance mitochondrial respiration and elongation, and reduce the levels of secreted Aβ1–42 and Aβ1–40 in induced pluripotent stem cell‐derived neurons obtained from AD patients fibroblasts (Frankowski et al., 2021).
In conclusion, we show increased levels of HDAC2 and HDAC3 in PBMCs from MCI individuals, AD human brain cortex (Braak staging III–IV) as well as in the hippocampus of APP/PS1 mice and AβO‐treated mouse hippocampal cell line. Data highlight that pharmacologically inhibiting the activity of class I HDACs (including HDAC2 and HDAC3) with Tac and complementary effects of silencing HDAC2 or HDAC3 alleviates mitochondrial dysfunction and the impairment in ER‐mitochondria cross‐talk in AD hippocampal neural cells. Tac modified expression of MAM‐related proteins involved in ER‐mitochondria Ca2+‐transfer, affecting MAM connectivity and ameliorating mitochondrial Ca2+ handling, as well as partially restoring mitochondrial morphology. These findings reveal MAM transcriptional regulation as a promising target for innovative therapeutics in AD early stages. In this respect, targeting class I HDACs is expected to impact on epigenetic modulation, potentially having a protective role in AD pathogenesis.
4. MATERIALS AND METHODS
4.1. Materials
Neurobasal medium, B27 supplement, gentamicin, trypsin, and penicillin–streptomycin were purchased from Gibco, Life Technologies. The synthetic Aβ1–42 peptide was from Bachem. Tacedinaline (CI994) was purchased from Sigma‐Aldrich or from MedChemExpress. Fluorescent probes Fura2‐AM, Rhod2‐AM, tetramethylrhodamine methyl ester (TMRM+), Hoechst 33342 and propidium iodide (PI), and Pluronic acid F‐127 were obtained from Molecular Probes, Invitrogen. Antibodies against acetyl‐H3 (#06‐599), β‐actin (A5316) and β‐tubulin (T7815) were obtained from Sigma‐Aldrich, as well as RPMI‐1640 Medium (R4130), minimum essential medium (MEM M0268), Dulbecco's modified Eagle's medium (DMEM D5648), DMEM D5030, SB, glutamate, glutamine, 3‐(4,5dimethylthiazol‐2‐yl)‐2,5diphenyltetrazolium bromide (MTT), NMDA, ifenprodil, MK‐801, oligomycin, trifluoromethoxy carbonylcyanide phenylhydrazone (FCCP), ionomycin, antimycin, rotenone, phenylmethylsulfonyl fluoride (PMSF), chymostatin, pepstatin A, leupeptin, antipain, paraformaldehyde, and Duolink in situ Red Starter Kit (DUO92101). SAHA was from Biovision. Primary antibodies anti‐GRP75 (#2816), anti‐H3 (#9715), anti‐HDAC2 (#2540), and anti‐HDAC3 (#3949) were from Cell Signaling; anti‐InsP3R1 (19962‐1‐AP) from Proteintech; anti‐VDAC1 (ab14734) and anti‐APP (ab32136) and Coomassie Blue G‐250 were from Abcam and anti‐Aβ (NBP2‐13075) from Novus Biologicals. Thapsigargin was purchased from Tocris Bioscience. Anti‐mouse (#31320) and anti‐rabbit (#31340) IgG secondary antibodies were from Invitrogen. Enhanced chemifluorescence (ECF) substrate and Ficoll‐Paque were obtained from GE Healthcare. Dithiothreitol, NZYol reagent and NZY first‐strand cDNA synthesis kit were from NZYTech. Bradford protein assay reagent and iQ SYBR Green supermix were purchased from BioRad. Pierce BCA protein assay kit was from Thermo Scientific. Control‐, HDAC2‐ and HDAC3‐siRNA were from Santa Cruz Biotechnology. NFAT luciferase reporter was obtained from Addgene. Polyethylene glycol (PEG) 400 was from LabChem Inc. OCT compound embedding medium was from Bio Optica. All other reagents were from analytical grade.
4.2. Aβ1–42 oligomers preparation
Aβ1–42 preparation containing a high percentage of Aβ oligomers and monomers (about 60% and 40%, respectively; Figure S1A) was obtained from synthetic Aβ1–42 as previously described (Klein, 2002). In brief, lyophilized Aβ peptide was dissolved to 1 mM in 1,1,1,3,3,3‐hexafluoro‐2‐propanol (HFIP) in pyrogen/endotoxin‐free tubes and incubated at room temperature for 1 h. HFIP was then evaporated overnight in a chemical hood and completely removed under speed vacuum. Dried monomeric peptide film was further resuspended in anhydrous DMSO to a 5 mM final concentration. The peptide was diluted into Ham's F12 medium to a final concentration of 100 μM and incubated at 4°C for 24 h. The preparation was then centrifuged at 14,000 g for 10 min at 4°C to remove insoluble aggregates, and the supernatant (corresponding to the oligomeric peptide) collected. Peptide concentration was determined using the BioRad protein dye assay reagent. To confirm oligomer formation and the presence of different Aβ peptide forms, the preparation was evaluated by 6%–15% Tris–Tricine SDS‐PAGE gel electrophoresis and further staining with Coomassie Blue G‐250.
4.3. Animals
3xTg‐AD mice on a C57BL6/129S background harboring APPswe, PS1/M146V and tauP301L transgenes (genetically engineered as previously described by Oddo et al., 2003) were gifted from Dr. Frank LaFerla, University of California, Irvine. APP/PS1 mice harboring APPswe and PS1dE9 transgenes were acquired from the Jackson Laboratory (MMRRC stock #34829) and fully backcrossed onto a B6C3F1/J background. Offspring were genotyped by PCR amplification of genomic DNA extracted from ear clippings. Mice were bred and maintained under a constant temperature, humidity and 12 h light/dark cycle at CNC‐Faculty of Medicine animal house. All procedures were performed in order to minimize exposure to stress and animal suffering, in accordance with EU Directive 2010/63/UE. The study was approved by the Food and Veterinary Directorate under reference 003927_0421/000/000/2018.
4.4. Cell cultures
4.4.1. Hippocampal neuronal culture
Primary cultures of hippocampal neurons were prepared from 17 days embryos derived from 3xTg‐AD or WT strain mice. The hippocampi were dissected and collected into pre‐cooled Ca2+, Mg2+‐free Hank's balanced salt solution (HBSS containing: 137 mM NaCl, 5.36 mM KCl, 0.44 mM KH2PO4, 4.16 mM NaHCO3, 0.34 mM NaH2PO4.H2O, 5 mM glucose, 5.36 mM Hepes, 1 mM pyruvate, pH 7.2) and then digested in 0.003% (w/v) trypsin for 3 min at 37°C. HBSS containing 5% heat‐inactivated FBS was used to halt enzymatic digestion. Following two washing steps with HBSS, the cells were dissociated mechanically in plating medium (MEM supplemented with 10% horse serum, 0.6% (w/v) glucose and 1 mM pyruvate) and plated onto poly‐d‐lysine‐coated plates, at a final density of 5 × 104 cells/cm2. After 2 h, the plating medium was replaced by Neurobasal medium supplemented with 2% (v/v) B27, 0.5 mM glutamine, 50 μg/mL gentamicin and 25 μM glutamate. Neuronal cultures were maintained at 37°C in a humidified incubator chamber with 95% air and 5% CO2 for 10–11 DIV. Every 3 days, the medium was changed with supplemented Neurobasal medium without glutamate.
4.4.2. HT22 cell culture
Mouse hippocampal‐derived HT22 cell line, a neuronal cell line lacking NMDAR (Maher & Davis, 1996), was generously acquired from Dr. Dave Schubert (Salk Institute). Cells were cultured in high‐glucose DMEM D5648 supplemented with 10% heat‐inactivated FBS, 12 mM NaHCO3, 5 mM Hepes and 100 μg/mL penicillin–streptomycin, pH 7.3 in T‐75 culture flasks in a humidified incubator with 5% CO2 and 95% air, at 37°C. Sub‐cultures were obtained after a 2 min dissociation step using a Ca2+/Mg2+‐free dissociation medium containing 140 mM NaCl, 1.47 mM KCl, 8.1 mM Na2HPO4, 1.47 mM KH2PO4, 0.55 mM EDTA, pH 7.3 at 37°C. Cells were then centrifuged at 800 rpm for 5 min and the pellet resuspended in fresh culture medium. Cells were cultured at a density of 9 × 103 cells/cm2 for 48 h or 3 × 103 cells/cm2 for 72 h in 6, 12, or 96‐well plates or 18 mm coverslips (in 12‐well plates).
4.5. Cell treatment
Hippocampal neurons at 10–13 DIV were treated for 24 h with HDACis, namely SB, SAHA, and Tac. WT neurons were also treated for 23 h with 1 μM AβO following 1 h pre‐incubation with Tac. On the day after plating, HT22 cells were pre‐exposed for 1 h with HDACis and then co‐treated with 1 μM AβO for the remaining 23 h of incubation. To induce selective HDACs knockdown, siRNA‐calcium phosphate precipitates containing 80 pmol of the indicated siRNA duplex were added to each well containing culture medium on the day following plating. Scrambled siRNA was used as a control. After 5 h incubation, culture medium was changed to fresh medium. On the third day after plating, cells were treated with 1 μM AβO and incubated for additional 24 h. For NFAT transcriptional activity studies, hippocampal neurons or HT22 cells were transfected with 0.75 μg of NFAT luciferase reporter for 48 h prior to analyses and treated with Tac plus AβO for 24 h.
4.6. Human samples
Human temporal cortex was obtained from the Neurological Tissue Bank, Biobanc Hospital Clinic‐IDIBAPS, as previously described (Esteves & Cardoso, 2020), from AD patients at Braak stages III–IV (n = 5; age range 74–89) and age‐matched cognitively unimpaired controls (n = 6; age range 63–76). The study was approved by the local regional ethical committee (Law 14/2007 on Biomedical Research).
Peripheral blood mononuclear cells (PBMCs) from 26 participants were obtained as part of a previous study (Mota et al., 2015). Briefly, patients in the clinical continuum of AD and healthy age‐matched controls were selected from the ageing‐cohort of the Memory Clinic (Neurology Unit) of the Centro Hospitalar e Universitário de Coimbra (CHUC) where they are routinely and prospectively assessed with comprehensive neuropsychological, full biomarker characterization and ApoE genotyping. Patients included individuals (n = 5, age range 59–76) fulfilling the established criteria for MCI due to AD according to standard criteria (Albert et al., 2011) and patients with dementia due to AD (McKhann et al., 2011) in the various stages of severity: mild AD (n = 5, age range 58–83), moderate AD (n = 5, age range 64–85), and severe AD (n = 4, age range 62–89). Age‐matched cognitively‐healthy volunteers (n = 7, age range 55–78) confirmed by formal neuropsychological assessment were also recruited as controls; exclusion criteria for all these participants are history of neurologic or psychiatric diseases, including major depressive disorder in the last 6 months, history of severe/chronic unstable disease or substance abuse or medication that interferes with the biochemistry. Samples were previously obtained from peripheral venous blood and stored in liquid N2. The study was approved by the Ethics Committee of CHUC and written informed consent was obtained from all subjects prior to inclusion in the study.
4.7. Cell metabolic activity, proliferation, and viability assays
For cell metabolic activity assay, hippocampal neurons and HT22 cells were incubated with 0.5 mg/mL MTT in sodium‐based medium (Na+ medium containing: 140 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 10 mM glucose, 10 mM Hepes, pH 7.4) or Krebs medium (135 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 0.4 mM KH2PO4, 1 mM MgSO4, 5.5 mM glucose, 20 mM Hepes, pH 7.4), respectively, for 2 h at 37°C. The resulting formazan crystals were dissolved in acidic‐isopropanol (0.04 M HCI in isopropanol). Absorbance was measured at 570 nm using a SpectraMAX Plus microplate reader (Molecular Devices). MTT reduction extent for each condition was normalized to the corresponding control.
To evaluate HT22 cells proliferation, cells were detached from 12‐well plates at each timepoint, resuspended in fresh culture medium and then diluted in 0.4% Trypan blue solution. Viable cells (unstained cells) were counted under an inverted microscope.
For viability assay, cells were subjected to double staining detection method by using Hoechst/PI to distinguish normal, apoptotic and necrotic cells. Cells were incubated with 2 μg/mL Hoechst 33342, a blue‐fluorescence dye (excitation/emission ~350/461 nm when bound to DNA) and 2 μg/mL PI, a red‐fluorescence dye (excitation/emission ~535/617 nm when bound to DNA), in the dark for 3 min at room temperature, then washed with Krebs medium and further examined and scored using the Axioscope 2 Plus upright microscope (Zeiss). Nuclear condensation was used to distinguish apoptotic cells. PI‐positive cells were identified as necrotic cells.
4.8. Mitochondrial membrane potential measurement
Hippocampal neurons and HT22 cells were incubated with 100 nM TMRM+ dye (quench mode) plus 0.2% (v/v) pluronic acid in Na+ medium or Krebs medium, respectively, for 30 min at 37°C. To directly assess mitochondrial function, glycolysis was inhibited by replacing glucose with 2 mM 2‐deoxy‐d‐glucose and by adding 10 mM pyruvate, in order to directly feed mitochondria (“pyruvate‐based medium”). Basal fluorescence (540 nm excitation and 590 nm emission) was measured using Spectrofluorometer Gemini EM (Molecular Devices) or SpectraMax iD3 (Molecular Devices). Maximal mitochondrial depolarization was also performed in every individual experiment by adding oligomycin (2 μg/ml) to prevent ATP synthase reversal and a protonophore (2 μM FCCP). Results were expressed as the difference between the increase in TMRM+ fluorescence upon addition of FCCP plus oligomycin and basal fluorescence values. All plotted values were normalized for initial baseline value.
4.9. Seahorse analysis
O2 consumption rate (OCR) was measured by using the Seahorse XF24 flux analyzer (Agilent) as previously described (e.g., Ferreira et al., 2018). In brief, neurons were rinsed and equilibrated for 1 h at 37°C in DMEM D5030 supplemented with 2 g/L glucose, 0.2 g/L glutamine, and 0.1 g/L pyruvate, pH 7.4. Then, oligomycin (1 μg/mL), FCCP (1 μM), and antimycin A (1 μM) plus rotenone (1 μM) were sequentially added to evaluate basal respiration, maximal respiration induced by FCCP, ATP synthesis, spare respiratory capacity, H+ leak, coupling efficiency, and non‐mitochondrial respiration. Results are expressed in pmol O2/min.
4.10. Analysis of intracellular free Ca2+ levels
Hippocampal neurons and HT22 cells were incubated with 5 μM Fura2‐AM or 1 μM Rhod2‐AM plus 0.2% (v/v) pluronic acid in Na+ medium or Krebs medium, respectively, for 30–40 min at 37°C. Neuronal experiments were performed in the absence of Mg2+ to maximize NMDAR activation in a pyruvate‐based medium containing 20 μM glycine. Following stimulation with 100 μM NMDA, the neurons were submitted to 10 μM MK‐801, a non‐competitive NMDAR antagonist. Maximal mitochondrial depolarization was achieved by the addition of protonophore FCCP (2 μM) in the presence of 2 μg/mL oligomycin to prevent ATP hydrolysis. In HT22 cells, mitochondrial Ca2+ content was also evaluated under maximal mitochondrial depolarization. Calibration of intracellular free Ca2+ (Ca2+ i) responses was performed at the end of every individual experiment upon addition of 2 μM ionomycin. ER Ca2+ content in both neurons and HT22 cells was estimated following exposure to 1 μM thapsigargin in Ca2+‐free medium. Fura2 fluorescence was measured at 340/380 nm excitation and 510 nm emission, and Rhod2 fluorescence at 552 nm excitation and 581 nm emission using Spectrofluorometer Gemini EM (Molecular Devices) or SpectraMax iD3 (Molecular Devices). All plotted values were normalized for initial baseline value.
4.11. Animal treatment
APP/PS1 versus WT females aged 9 months old were treated daily with intraperitoneal injections of Tac (30 mg/kg/day; n = 7/group) or vehicle (DMSO: PEG: Saline [5:30:65]; n = 9/group) for 30 days. The body weight of all mice was measured and recorded once a week.
4.12. Brain tissue preparation
On 30th day approximately 30–40 min after Tac/vehicle administration, mice were anesthetized intraperitoneally with 80 mg/kg pentobarbital and then perfused transcardially with 20 mL saline solution (0.9% NaCl). For protein/RNA samples, hippocampi were quickly dissected, frozen in liquid nitrogen, and stored at −80°C until further extraction. For the immunohistochemical analysis, the hemisphere was removed, fixed with 4% paraformaldehyde at 4°C for 24 h, cryoprotected in 30% sucrose at 4°C until it sank, frozen in liquid nitrogen and stored at −80°C until use. Brain blocks embedded in OCT medium were sectioned coronally on a cryostat at a thickness of 40 μm and collected onto SuperFrost adhesion slides.
4.13. Protein extraction and Western blotting
Human PBMCs, hippocampal neurons, and HT22 cells were lysed in ice cold RIPA buffer (150 mM NaCl, 50 mM Tris–HCl, 5 mM EGTA, 1% Triton X‐100, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with 1 mM dithiothreitol, 1 mM PMSF, and 1 μg/mL protease inhibitor cocktail containing chymostatin, pepstatin A, leupeptin, and antipain. Following a freeze–thaw cycle, cellular extracts were centrifuged at 14,000 rpm using an Eppendorf Centrifuge 5417R for 10 min at 4°C and protein quantification performed by the BioRad protein assay. Human cortex and mouse hippocampi were homogenized in pre‐boiled 1% SDS and boiled at 100°C for 10 min. After centrifugation, protein content was quantified using BCA protein assay. Equivalent amounts of protein samples (15–30 μg) were denatured with sample buffer (containing 50 mM Tris–HCl pH 6.8, 2% SDS, 5% glycerol, 100 mM dithiothreitol, 0.01% bromophenol blue) at 95°C for 5 min and separated on a 7.5%–15% SDS‐PAGE. Proteins were then transferred onto PVDF membranes and blocked with 5% BSA or non‐fat dry milk in Tris‐buffered saline (25 mM Tris–HCl, 150 mM NaCl, pH 7.6) plus 0.1% Tween‐20 (TBS‐T) according to the manufacturer's recommendation for 1 h at room temperature. Membranes were incubated with primary antibodies against acetyl‐H3 (0.1 μg/mL; 17 kDa), H3 (1:1000; 17 kDa), InsP3R1 (1:300; 290 kDa), GRP75 (1:1000; 75 kDa), VDAC1 (1 μg/mL; 39 kDa), HDAC2 (1:1000; 60 kDa), and HDAC3 (1:1000; 49 kDa) overnight at 4°C or β‐actin (1:20,000; 42 kDa) and β‐tubulin (1:20,000; 55 kDa) for 1 h at room temperature. After a washing step with TBS‐T, membranes were incubated with the respective secondary antibodies (1:10,000) for 1 h at room temperature. Membranes were washed in TBS‐T, further incubated with ECF substrate and immunoreactive bands visualized on a BioRad ChemiDoc Touch Imaging System and further quantified using Image Lab analysis software (BioRad).
4.14. RNA isolation and quantitative real time PCR (qRT‐PCR)
Total RNA was extracted from HT22 cells or mouse hippocampi using NZYol (1 mL per 10 cm2 growth area or 10 mg tissue), according to the instructions of the supplier. Briefly, lysates were incubated for 5–10 min at room temperature and chloroform (200 μL per 1 mL NZYol used) was added and mixed vigorously before incubating for another 3 min. After centrifugation at 12,000 g for 15 min at 4°C, the clear upper aqueous layer was transferred to a new tube, isopropanol (500 μL per 1 mL NZYol) was added and the samples incubated for 10 min at −20°C before centrifugation at 12,000 g for 10 min, at 4°C. The RNA precipitate was washed with 75% ethanol and the samples were centrifugated at 12,000 g for 10 min, at 4°C. The pellet was dried at room temperature for 10 min and then solubilized in DEPC‐treated water. RNA concentration was determined with NanoDrop 2000c spectrophotometer (Thermo Scientific). cDNA was synthesized from 500 ng of total extracted RNA using the NZY First‐Strand cDNA Synthesis Kit, following the manufacturer instructions. PCR reactions were performed in 10 μL volumes containing 5 μL of iQ SYBR Green Supermix, 300 nM of each primer and 50 ng of cDNA template in a BioRad CFX96 Real‐Time PCR Detection System using the following cycling conditions: initial denaturation at 95°C for 3 min, followed by 40 cycles of denaturation at 95°C for 15 s and annealing at 60°C for 45 s. At the end, samples were subjected to a melting curve analysis in order to confirm the absence of unspecific amplification products and primers dimers. Samples containing no template were included as negative controls in all experiments. Reactions were run in duplicates. Analysis of gene expression was performed using the ΔΔCT method. Actin was used as an internal control for all samples. PCR primer sequences used were as follows (forward primer/reverse primer):
Insp3r: 5′‐GTATGCGGAGGGATCTACGA/5′‐AACACAACGGTCATCAACCA
Sigmar1: 5′‐ACCAATGGAAAGAGGGCAC/5′‐AACAGGGTAGACGGAATAACAC
Grp75: 5′‐CAGGCAGCATCTTCCCTACAG/5‐CCAGTGCCAGAACTTCCAGAA
Vdac1: 5′‐AAGTGAACAACTCCAGCCTGA/5′‐CACCAGCATTGACGTTCTTG
Mcu: 5′‐CGCCAGGAATATGTTTATCCA/5′‐CTTGTAATGGGTCTCTCAGTCTCTT
Pdzd8: 5′‐TCAACTGATGGGTATGCTGG/5′‐ATAGCAATGAGCCGATCTCC
Vapb: 5′‐GAAGGTGATGGAAGAGTGCAG/5′‐CCCGAAGTCCGTCTTCTTC
Rmdn3: 5′‐GGTCCGCTCTCATATAGAAGAGAAC/5′‐CCTCTCTCTGGCAAAAGGGAAC
Mfn2: 5′‐TCCTCTGTTCCAGTTGTA/5′‐TCGCTTATCCTTCTTGAC
Mfn1: 5′‐GCAGACAGCACATGGAGAGA/5′‐GATCCGATTCCGAGCTTCCG
Actin: 5′‐GTGACGTTGACATCCGTAAAGA/5′‐GCCGGACTCATCGTACTCC
4.15. Proximity ligation assay (PLA)
Cells were fixed with 4% paraformaldehyde at room temperature for 15 min and permeabilized with 0.5% Triton X‐100 for 5 min and in situ Duolink PLA was then performed using 40 μL of solution per 1 cm2 sample on a slide according to manufacturer's instruction. In brief, the samples were incubated with blocking buffer at 37°C for 1 h in a humidified chamber, followed by incubation with primary antibodies against InsP3R1 (1:50) and VDAC1 (1:50) overnight at 4°C. The slides were rinsed in wash buffer, the oligo‐linked secondary probes (1:5) anti‐mouse MINUS and anti‐rabbit PLUS were added and the samples were incubated at 37°C for 1 h in a humidified chamber. After a washing step, samples were incubated with ligase (1:40) for 30 min at 37°C, further rinsed in wash buffer and then incubated with polymerase (1:80) in amplification buffer for 100 min at 37°C. Finally, the slides were mounted in mounting media containing DAPI. Fluorescence signal amplification was recorded as a Z‐stack using the Zeiss LSM 710 confocal laser scanning microscopy under a 63x/1.4 objective and analyzed using the IMARIS analysis software.
4.16. Transmission electron microscopy (TEM)
HT22 cells were collected and centrifuged at 2500 rpm for 5 min and the pellets fixed in 2.5% (v/v) glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) for 2 h at room temperature. Following rinsing in the same buffer, postfixation was performed using 1% (w/v) osmium tetroxide for 1 h. After rinsing in phosphate buffer and distilled water, 1% (w/v) aqueous uranyl acetate was added to the cells during 1 h for contrast enhancement. Following embedding in 2% (w/v) molten agar, samples were dehydrated in a graded ethanol series (30%–100%), impregnated and embedded in Epoxy resin. Ultrathin sections (70 nm) were mounted on copper grids and stained with 0.2% (w/v) lead citrate for 7 min. Observations of 10 independent cells per condition were carried out on a FEI Tecnai G2 Spirit BioTwin electron microscope at 100 kV. Mitochondria‐ER contacts sites (MERCS) were considered when the distance between ER and mitochondria was less than 25 nm. MERCS length, MERCS distance, number of MERCS per mitochondria, mitochondrial area, perimeter, Feret's diameter (defined as longest distance between any two points in the mitochondria), and aspect ratio (defined as the ratio between major and minor mitochondrial axis) were quantified using image analysis software ImageJ (Fiji) according to Lam et al., 2021.
4.17. Immunohistochemistry
Tissue sections were heated in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween‐20, pH 6.0) for 10 min for antigen retrieval. After cooling, the slides were rinsed with PBS and blocked with 0.3% Triton X‐100/3% BSA for 1 h at room temperature. Sections were incubated with primary antibodies against Aβ1–42 (1:200) and APP (1:500) overnight at 4°C, then washed with PBS and incubated with the respective secondary antibodies (1:1000) for 1 h at room temperature. Finally, nuclei were counterstained with 1 μg/mL Hoechst 33342 for 5 min. Fluorescent photomicrographs of whole cross‐sectional hippocampal regions were serial scanned under a 20× objective using a Zeiss Axio Scan Z1 microscope slide scanner and fluorescence levels quantified by using image analysis software ImageJ (Fiji).
4.18. Luciferase reporter assay
Cells were collected in ice‐cold lysis buffer (1.15 M Tris, 1 mM EDTA, 8 mM MgCl2, 15% glycerol, 1 mM DTT, 1% Triton X‐100, pH 7.4) after a freeze–thaw cycle and then samples were transferred to a 96‐well white opaque plaque. Luminescence was measured with 10 s integration time after addition of reading buffer (1.15 M Tris, 1 mM EDTA, 8 mM MgCl2, 15% glycerol, 1 mM DTT, 2 mM ATP‐Mg2+, pH 7.4) and luciferin (50 mg/mL; pH 8) in a LMAX II 384 Luminometer (Molecular Devices). Data were normalized to protein content.
4.19. Statistical analysis
Data were analyzed by GraphPad Prism 8 software and expressed as the mean of at least three independent experiments ± SEM. Statistical significance was assessed using the Mann–Whitney test, unpaired Student's t‐test, Kruskal–Wallis followed by uncorrected Dunn's multiple comparison test or one‐way ANOVA followed by uncorrected Fisher's LSD multiple comparison test. Sample normality was tested using Shapiro–Wilk test. Statistical significance was defined as *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
AUTHOR CONTRIBUTIONS
DM acquired and analyzed the data. ILF and ACR conceptualized and designed the study. DM, ILF and ACR interpreted the results. DM wrote the paper with critical edits from ILF and ACR. RL carried out mice treatment and sacrifice. SMC provided the postmortem human brain tissue. IS contributed in participant's examination and recruitment for blood donation. All authors read, edited, and approved the final version of the manuscript.
CONFLICT OF INTEREST STATEMENT
The authors confirm that there are no financial or non‐financial competing interests to report.
CONSENT FOR PUBLICATION
All authors read the manuscript and consent with its publication.
Supporting information
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Appendix S1
ACKNOWLEDGMENTS
We thank Rodrigo Taveira (CNC) for assisting APP/PS1 mouse treatment. We thank Mónica Zuzarte (Institute for Biomedical Imaging and Life Sciences, University of Coimbra) for assistance with electron microscopy sample preparation and imaging, and Margarida Caldeira, Tatiana Catarino and Luísa Cortes (MICC‐CNC – Microscopy Imaging Center of Coimbra, University of Coimbra) for technical support on confocal microscopy. We are thankful to the IDIBAPS Biobank and to Professor I Ferrer Abizanda, Bellvitge Hospital Universitari, Institut Català de la Salut, Barcelona, Spain for sample and data procurement. This work was supported by the European Regional Development Fund (ERDF) through the Centro 2020 Regional Operational Programme under project CENTRO‐01‐0145‐FEDER‐000012‐HealthyAging2020, and through the COMPETE 2020 – Operational Programme for Competitiveness and Internationalisation and Portuguese national funds via FCT – Fundação para a Ciência e a Tecnologia, I.P., OE FCT/MCTES (PIDDAC) under projects POCI‐01‐0145‐FEDER‐032316, EU Twinning project RESETageing (ref. 952266), UIDB/04539/2020, UIDP/04539/2020 and LA/P/0058/2020. DM was supported by HealthyAging2020/BIM/DPA‐19‐895 and FCT PhD fellowship 2020.07951.BD.
Marinho, D. , Ferreira, I. L. , Lorenzoni, R. , Cardoso, S. M. , Santana, I. , & Rego, A. C. (2023). Reduction of class I histone deacetylases ameliorates ER‐mitochondria cross‐talk in Alzheimer's disease. Aging Cell, 22, e13895. 10.1111/acel.13895
Daniela Marinho and Ildete Luísa Ferreira contributed equally to this work.
Contributor Information
Ildete Luísa Ferreira, Email: ildetelferreira@gmail.com.
A. Cristina Rego, Email: acrego@cnc.uc.pt.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Albert, M. S. , DeKosky, S. T. , Dickson, D. , Dubois, B. , Feldman, H. H. , Fox, N. C. , Gamst, A. , Holtzman, D. M. , Jagust, W. J. , Petersen, R. C. , Snyder, P. J. , Carrillo, M. C. , Thies, B. , & Phelps, C. H. (2011). The diagnosis of mild cognitive impairment due to Alzheimer's disease: Recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's & Dementia, 7(3), 270–279. 10.1016/j.jalz.2011.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Area‐Gomez, E. , Del Carmen Lara Castillo, M. , Tambini, M. D. , Guardia‐Laguarta, C. , de Groof, A. J. , Madra, M. , Ikenouchi, J. , Umeda, M. , Bird, T. D. , Sturley, S. L. , & Schon, E. A. (2012). Upregulated function of mitochondria‐associated ER membranes in Alzheimer disease. The EMBO Journal, 31(21), 4106–4123. 10.1038/emboj.2012.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartok, A. , Weaver, D. , Golenár, T. , Nichtova, Z. , Katona, M. , Bánsághi, S. , Alzayady, K. J. , Thomas, V. K. , Ando, H. , Mikoshiba, K. , Joseph, S. K. , Yule, D. I. , Csordás, G. , & Hajnóczky, G. (2019). IP3 receptor isoforms differently regulate ER‐mitochondrial contacts and local calcium transfer. Nature Communications, 10(1), 3726. 10.1038/s41467-019-11646-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaver, M. , Karisetty, B. C. , Zhang, H. , Bhatnagar, A. , Armour, E. , Parmar, V. , Brown, R. , Xiang, M. , & Elefant, F. (2022). Chromatin and transcriptomic profiling uncover dysregulation of the Tip60 HAT/HDAC2 epigenomic landscape in the neurodegenerative brain. Epigenetics, 17(7), 786–807. 10.1080/15592294.2021.1959742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckers, T. , Burkhardt, C. , Wieland, H. , Gimmnich, P. , Ciossek, T. , Maier, T. , & Sanders, K. (2007). Distinct pharmacological properties of second generation HDAC inhibitors with the benzamide or hydroxamate head group. International Journal of Cancer, 121(5), 1138–1148. 10.1002/ijc.22751 [DOI] [PubMed] [Google Scholar]
- Booth, D. M. , Várnai, P. , Joseph, S. K. , & Hajnóczky, G. (2021). Oxidative bursts of single mitochondria mediate retrograde signaling toward the ER. Molecular Cell, 81(18), 3866–3876.e2. 10.1016/j.molcel.2021.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldeira, G. L. , Ferreira, I. L. , & Rego, A. C. (2013). Impaired transcription in Alzheimer's disease: Key role in mitochondrial dysfunction and oxidative stress. Journal of Alzheimer's Disease, 34(1), 115–131. 10.3233/JAD-121444 [DOI] [PubMed] [Google Scholar]
- Cao, T. , Zhou, X. , Zheng, X. , Cui, Y. , Tsien, J. Z. , Li, C. , & Wang, H. (2018). Histone deacetylase inhibitor alleviates the neurodegenerative phenotypes and histone dysregulation in presenilins‐deficient mice. Frontiers in Aging Neuroscience, 10, 137. 10.3389/fnagi.2018.00137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha, M. Y. , Han, S. H. , Son, S. M. , Hong, H. S. , Choi, Y. J. , Byun, J. , & Mook‐Jung, I. (2012). Mitochondria‐specific accumulation of amyloid β induces mitochondrial dysfunction leading to apoptotic cell death. PLoS One, 7(4), e34929. 10.1371/journal.pone.0034929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, H. , Xuan, H. , Green, C. D. , Han, Y. , Sun, N. , Shen, H. , McDermott, J. , Bennett, D. A. , Lan, F. , & Han, J. J. (2018). Repression of human and mouse brain inflammaging transcriptome by broad gene‐body histone hyperacetylation. Proceedings of the National Academy of Sciences of the United States of America, 115(29), 7611–7616. 10.1073/pnas.1800656115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, H. K. , Choi, Y. , Kang, H. , Lim, E. J. , Park, S. Y. , Lee, H. S. , Park, J. M. , Moon, J. , Kim, Y. J. , Choi, I. , Joe, E. H. , Choi, K. C. , & Yoon, H. G. (2015). PINK1 positively regulates HDAC3 to suppress dopaminergic neuronal cell death. Human Molecular Genetics, 24(4), 1127–1141. 10.1093/hmg/ddu526 [DOI] [PubMed] [Google Scholar]
- Chu, B. , Li, M. , Cao, X. , Li, R. , Jin, S. , Yang, H. , Xu, L. , Wang, P. , & Bi, J. (2021). IRE1α‐XBP1 affects the mitochondrial function of Aβ25‐35‐treated SH‐SY5Y cells by regulating mitochondria‐associated endoplasmic reticulum membranes. Frontiers in Cellular Neuroscience, 15, 614556. 10.3389/fncel.2021.614556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, A. , Butto, T. , Hammer, N. , Jagannath, S. , Fend‐Guella, D. L. , Akhtar, J. , Radyushkin, K. , Lesage, F. , Winter, J. , Strand, S. , Roeper, J. , Zechner, U. , & Schweiger, S. (2020). Inhibition of histone deacetylation rescues phenotype in a mouse model of Birk‐Barel intellectual disability syndrome. Nature Communications, 11(1), 480. 10.1038/s41467-019-13918-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Eletto, M. , Rossin, F. , Occhigrossi, L. , Farrace, M. G. , Faccenda, D. , Desai, R. , Marchi, S. , Refolo, G. , Falasca, L. , Antonioli, M. , Ciccosanti, F. , Fimia, G. M. , Pinton, P. , Campanella, M. , & Piacentini, M. (2018). Transglutaminase type 2 regulates ER‐mitochondria contact sites by interacting with GRP75. Cell Reports, 25(13), 3573–3581.e4. 10.1016/j.celrep.2018.11.09 [DOI] [PubMed] [Google Scholar]
- Ding, H. , Dolan, P. J. , & Johnson, G. V. (2008). Histone deacetylase 6 interacts with the microtubule‐associated protein tau. Journal of Neurochemistry, 106(5), 2119–2130. 10.1111/j.1471-4159.2008.05564.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteves, A. R. , & Cardoso, S. M. (2020). Differential protein expression in diverse brain areas of Parkinson's and Alzheimer's disease patients. Scientific Reports, 10(1), 13149. 10.1038/s41598-020-70174-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando, W. , Martins, I. J. , Morici, M. , Bharadwaj, P. , Rainey‐Smith, S. R. , Lim, W. , & Martins, R. N. (2020). Sodium butyrate reduces brain amyloid‐β levels and improves cognitive memory performance in an Alzheimer's disease transgenic mouse model at an early disease stage. Journal of Alzheimer's Disease, 74(1), 91–99. 10.3233/JAD-19012 [DOI] [PubMed] [Google Scholar]
- Ferreira, I. L. , Bajouco, L. M. , Mota, S. I. , Auberson, Y. P. , Oliveira, C. R. , & Rego, A. C. (2012). Amyloid beta peptide 1‐42 disturbs intracellular calcium homeostasis through activation of GluN2B‐containing N‐methyl‐d‐aspartate receptors in cortical cultures. Cell Calcium, 51(2), 95–106. 10.1016/j.ceca.2011.11.008 [DOI] [PubMed] [Google Scholar]
- Ferreira, I. L. , Carmo, C. , Naia, L. , Mota, S. I. , & Rego, A. C. (2018). Assessing mitochondrial function in in vitro and ex vivo models of Huntington's disease. Methods in Molecular Biology, 1780, 415–442. 10.1007/978-1-4939-7825-0_19 [DOI] [PubMed] [Google Scholar]
- Ferreira, I. L. , Ferreiro, E. , Schmidt, J. , Cardoso, J. M. , Pereira, C. M. , Carvalho, A. L. , Oliveira, C. R. , & Rego, A. C. (2015). Aβ and NMDAR activation cause mitochondrial dysfunction involving ER calcium release. Neurobiology of Aging, 36(2), 680–692. 10.1016/j.neurobiolaging.2014.09.006 [DOI] [PubMed] [Google Scholar]
- Filadi, R. , Greotti, E. , Turacchio, G. , Luini, A. , Pozzan, T. , & Pizzo, P. (2016). Presenilin 2 modulates endoplasmic reticulum‐mitochondria coupling by tuning the antagonistic effect of mitofusin 2. Cell Reports, 15(10), 2226–2238. 10.1016/j.celrep.2016.05.013 [DOI] [PubMed] [Google Scholar]
- Frankowski, H. , Yeboah, F. , Berry, B. J. , Kinoshita, C. , Lee, M. , Evitts, K. , Davis, J. , Kinoshita, Y. , Morrison, R. S. , & Young, J. E. (2021). Knock‐down of HDAC2 in human induced pluripotent stem cell derived neurons improves neuronal mitochondrial dynamics, neuronal maturation and reduces amyloid beta peptides. International Journal of Molecular Sciences, 22(5), 2526. 10.3390/ijms22052526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaisina, I. N. , Lee, S. H. , Kaidery, N. A. , Ben Aissa, M. , Ahuja, M. , Smirnova, N. N. , Wakade, S. , Gaisin, A. , Bourassa, M. W. , Ratan, R. R. , Nikulin, S. V. , Poloznikov, A. A. , Thomas, B. , Thatcher, G. , & Gazaryan, I. G. (2018). Activation of Nrf2 and hypoxic adaptive response contribute to neuroprotection elicited by phenylhydroxamic acid selective HDAC6 inhibitors. ACS Chemical Neuroscience, 9(5), 894–900. 10.1021/acschemneuro.7b00435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido‐Maraver, J. , Loh, S. , & Martins, L. M. (2020). Forcing contacts between mitochondria and the endoplasmic reticulum extends lifespan in a Drosophila model of Alzheimer's disease. Biology Open, 9(1), bio047530. 10.1242/bio.047530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillmore, T. , Farrell, A. , Alahari, S. , Sallais, J. , Kurt, M. , Park, C. , Ausman, J. , Litvack, M. , Post, M. , & Caniggia, I. (2022). Dichotomy in hypoxia‐induced mitochondrial fission in placental mesenchymal cells during development and preeclampsia: Consequences for trophoblast mitochondrial homeostasis. Cell Death & Disease, 13(2), 191. 10.1038/s41419-022-04641-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindarajan, N. , Rao, P. , Burkhardt, S. , Sananbenesi, F. , Schlüter, O. M. , Bradke, F. , Lu, J. , & Fischer, A. (2013). Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer's disease. EMBO Molecular Medicine, 5(1), 52–63. 10.1002/emmm.201201923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gräff, J. , Rei, D. , Guan, J. S. , Wang, W. Y. , Seo, J. , Hennig, K. M. , Nieland, T. J. , Fass, D. M. , Kao, P. F. , Kahn, M. , Su, S. C. , Samiei, A. , Joseph, N. , Haggarty, S. J. , Delalle, I. , & Tsai, L. H. (2012). An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature, 483(7388), 222–226. 10.1038/nature10849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer, C. B. , Tanaka, Y. , Kim, Y. J. , Xie, P. , Zhang, M. Q. , Park, I. H. , & Kim, T. H. (2015). Histone deacetylases positively regulate transcription through the elongation machinery. Cell Reports, 13(7), 1444–1455. 10.1016/j.celrep.2015.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedskog, L. , Pinho, C. M. , Filadi, R. , Rönnbäck, A. , Hertwig, L. , Wiehager, B. , Larssen, P. , Gellhaar, S. , Sandebring, A. , Westerlund, M. , Graff, C. , Winblad, B. , Galter, D. , Behbahani, H. , Pizzo, P. , Glaser, E. , & Ankarcrona, M. (2013). Modulation of the endoplasmic reticulum‐mitochondria interface in Alzheimer's disease and related models. Proceedings of the National Academy of Sciences of the United States of America, 110(19), 7916–7921. 10.1073/pnas.1300677110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirabayashi, Y. , Kwon, S. K. , Paek, H. , Pernice, W. M. , Paul, M. A. , Lee, J. , Erfani, P. , Raczkowski, A. , Petrey, D. S. , Pon, L. A. , & Polleux, F. (2017). ER‐mitochondria tethering by PDZD8 regulates Ca2+ dynamics in mammalian neurons. Science, 358(6363), 623–630. 10.1126/science.aan6009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honrath, B. , Metz, I. , Bendridi, N. , Rieusset, J. , Culmsee, C. , & Dolga, A. M. (2017). Glucose‐regulated protein 75 determines ER‐mitochondrial coupling and sensitivity to oxidative stress in neuronal cells. Cell Death Discovery, 3, 17076. 10.1038/cddiscovery.2017.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack, C. R., Jr. , Bennett, D. A. , Blennow, K. , Carrillo, M. C. , Dunn, B. , Haeberlein, S. B. , Holtzman, D. M. , Jagust, W. , Jessen, F. , Karlawish, J. , Liu, E. , Molinuevo, J. L. , Montine, T. , Phelps, C. , Rankin, K. P. , Rowe, C. C. , Scheltens, P. , Siemers, E. , Snyder, H. M. , … Contributors . (2018). NIA‐AA Research Framework: Toward a biological definition of Alzheimer's disease. Alzheimer's & Dementia, 14(4), 535–562. 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadiya, P. , Kolmetzky, D. W. , Tomar, D. , Di Meco, A. , Lombardi, A. A. , Lambert, J. P. , Luongo, T. S. , Ludtmann, M. H. , Praticò, D. , & Elrod, J. W. (2019). Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer's disease. Nature Communications, 10(1), 3885. 10.1038/s41467-019-11813-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilgore, M. , Miller, C. A. , Fass, D. M. , Hennig, K. M. , Haggarty, S. J. , Sweatt, J. D. , & Rumbaugh, G. (2010). Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology, 35(4), 870–880. 10.1038/npp.2009.197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, W. L. (2002). Abeta toxicity in Alzheimer's disease: Globular oligomers (ADDLs) as new vaccine and drug targets. Neurochemistry International, 41(5), 345–352. 10.1016/s0197-0186(02)00050-5 [DOI] [PubMed] [Google Scholar]
- Knopman, D. S. , Amieva, H. , Petersen, R. C. , Chételat, G. , Holtzman, D. M. , Hyman, B. T. , Nixon, R. A. , & Jones, D. T. (2021). Alzheimer disease. Nature Reviews. Disease Primers, 7(1), 33. 10.1038/s41572-021-00269-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam, J. , Katti, P. , Biete, M. , Mungai, M. , AshShareef, S. , Neikirk, K. , Garza Lopez, E. , Vue, Z. , Christensen, T. A. , Beasley, H. K. , Rodman, T. A. , Murray, S. A. , Salisbury, J. L. , Glancy, B. , Shao, J. , Pereira, R. O. , Abel, E. D. , & Hinton, A., Jr. (2021). A universal approach to analyzing transmission electron microscopy with ImageJ. Cells, 10(9), 2177. 10.3390/cells10092177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, D. , Paillusson, S. , Hartopp, N. , Rupawala, H. , Mórotz, G. M. , Gomez‐Suaga, P. , Greig, J. , Troakes, C. , Noble, W. , & Miller, C. (2020). Disruption of endoplasmic reticulum‐mitochondria tethering proteins in post‐mortem Alzheimer's disease brain. Neurobiology of Disease, 143, 105020. 10.1016/j.nbd.2020.105020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher, P. , & Davis, J. B. (1996). The role of monoamine metabolism in oxidative glutamate toxicity. The Journal of Neuroscience, 16(20), 6394–6401. 10.1523/JNEUROSCI.16-20-06394.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marié, I. J. , Chang, H. M. , & Levy, D. E. (2018). HDAC stimulates gene expression through BRD4 availability in response to IFN and in interferonopathies. The Journal of Experimental Medicine, 215(12), 3194–3212. 10.1084/jem.20180520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks, P. A. , Richon, V. M. , & Rifkind, R. A. (2000). Histone deacetylase inhibitors: Inducers of differentiation or apoptosis of transformed cells. Journal of the National Cancer Institute, 92(15), 1210–1216. 10.1093/jnci/92.15.1210 [DOI] [PubMed] [Google Scholar]
- Martino Adami, P. V. , Nichtová, Z. , Weaver, D. B. , Bartok, A. , Wisniewski, T. , Jones, D. R. , Do Carmo, S. , Castaño, E. M. , Cuello, A. C. , Hajnóczky, G. , & Morelli, L. (2019). Perturbed mitochondria‐ER contacts in live neurons that model the amyloid pathology of Alzheimer's disease. Journal of Cell Science, 132(20), jcs229906. 10.1242/jcs.229906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matilainen, O. , Quirós, P. M. , & Auwerx, J. (2017). Mitochondria and epigenetics – Crosstalk in homeostasis and stress. Trends in Cell Biology, 27(6), 453–463. 10.1016/j.tcb.2017.02.004 [DOI] [PubMed] [Google Scholar]
- McClarty, B. , Rodriguez, G. , & Dong, H. (2021). Dose effects of histone deacetylase inhibitor tacedinaline (CI‐994) on antipsychotic haloperidol‐induced motor and memory side effects in aged mice. Frontiers in Neuroscience, 15, 674745. 10.3389/fnins.2021.674745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann, G. M. , Knopman, D. S. , Chertkow, H. , Hyman, B. T. , Jack, C. R., Jr. , Kawas, C. H. , Klunk, W. E. , Koroshetz, W. J. , Manly, J. J. , Mayeux, R. , Mohs, R. C. , Morris, J. C. , Rossor, M. N. , Scheltens, P. , Carrillo, M. C. , Thies, B. , Weintraub, S. , & Phelps, C. H. (2011). The diagnosis of dementia due to Alzheimer's disease: Recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's & Dementia, 7(3), 263–269. 10.1016/j.jalz.2011.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi, S. , López‐Doménech, G. , Halff, E. F. , Covill‐Cooke, C. , Ivankovic, D. , Melandri, D. , Arancibia‐Cárcamo, I. L. , Burden, J. J. , Lowe, A. R. , & Kittler, J. T. (2019). Miro clusters regulate ER‐mitochondria contact sites and link cristae organization to the mitochondrial transport machinery. Nature Communications, 10(1), 4399. 10.1038/s41467-019-12382-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mota, S. I. , Costa, R. O. , Ferreira, I. L. , Santana, I. , Caldeira, G. L. , Padovano, C. , Fonseca, A. C. , Baldeiras, I. , Cunha, C. , Letra, L. , Oliveira, C. R. , Pereira, C. M. , & Rego, A. C. (2015). Oxidative stress involving changes in Nrf2 and ER stress in early stages of Alzheimer's disease. Biochimica et Biophysica Acta, 1852(7), 1428–1441. 10.1016/j.bbadis.2015.03.015 [DOI] [PubMed] [Google Scholar]
- Naia, L. , Cunha‐Oliveira, T. , Rodrigues, J. , Rosenstock, T. R. , Oliveira, A. , Ribeiro, M. , Carmo, C. , Oliveira‐Sousa, S. I. , Duarte, A. I. , Hayden, M. R. , & Rego, A. C. (2017). Histone deacetylase inhibitors protect against pyruvate dehydrogenase dysfunction in Huntington's disease. The Journal of Neuroscience, 37(10), 2776–2794. 10.1523/JNEUROSCI.2006-14.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo, S. , Caccamo, A. , Shepherd, J. D. , Murphy, M. P. , Golde, T. E. , Kayed, R. , Metherate, R. , Mattson, M. P. , Akbari, Y. , & LaFerla, F. M. (2003). Triple‐transgenic model of Alzheimer's disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron, 39(3), 409–421. 10.1016/s0896-6273(03)00434-3 [DOI] [PubMed] [Google Scholar]
- Oliveira, J. M. , Chen, S. , Almeida, S. , Riley, R. , Gonçalves, J. , Oliveira, C. R. , Hayden, M. R. , Nicholls, D. G. , Ellerby, L. M. , & Rego, A. C. (2006). Mitochondrial‐dependent Ca2+ handling in Huntington's disease striatal cells: Effect of histone deacetylase inhibitors. The Journal of Neuroscience, 26(43), 11174–11186. 10.1523/JNEUROSCI.3004-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao, P. C. , Patnaik, D. , Watson, L. A. , Gao, F. , Pan, L. , Wang, J. , Adaikkan, C. , Penney, J. , Cam, H. P. , Huang, W. C. , Pantano, L. , Lee, A. , Nott, A. , Phan, T. X. , Gjoneska, E. , Elmsaouri, S. , Haggarty, S. J. , & Tsai, L. H. (2020). HDAC1 modulates OGG1‐initiated oxidative DNA damage repair in the aging brain and Alzheimer's disease. Nature Communications, 11(1), 2484. 10.1038/s41467-020-16361-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascoal, T. A. , Chamoun, M. , Lax, E. , Wey, H. Y. , Shin, M. , Ng, K. P. , Kang, M. S. , Mathotaarachchi, S. , Benedet, A. L. , Therriault, J. , Lussier, F. Z. , Schroeder, F. A. , DuBois, J. M. , Hightower, B. G. , Gilbert, T. M. , Zürcher, N. R. , Wang, C. , Hopewell, R. , Chakravarty, M. , … Rosa‐Neto, P. (2022). [11C]Martinostat PET analysis reveals reduced HDAC I availability in Alzheimer's disease. Nature Communications, 13(1), 4171. 10.1038/s41467-022-30653-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinz, S. , Unser, S. , Buob, D. , Fischer, P. , Jobst, B. , & Rascle, A. (2015). Deacetylase inhibitors repress STAT5‐mediated transcription by interfering with bromodomain and extra‐terminal (BET) protein function. Nucleic Acids Research, 43(7), 3524–3545. 10.1093/nar/gkv188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resende, R. , Fernandes, T. , Pereira, A. C. , Marques, A. P. , & Pereira, C. F. (2022). Endoplasmic reticulum‐mitochondria contacts modulate reactive oxygen species‐mediated signaling and oxidative stress in brain disorders: The key role of sigma‐1 receptor. Antioxidants & Redox Signaling, 37(10–12), 758–780. 10.1089/ars.2020.8231 [DOI] [PubMed] [Google Scholar]
- Rivieccio, M. A. , Brochier, C. , Willis, D. E. , Walker, B. A. , D'Annibale, M. A. , McLaughlin, K. , Siddiq, A. , Kozikowski, A. P. , Jaffrey, S. R. , Twiss, J. L. , Ratan, R. R. , & Langley, B. (2009). HDAC6 is a target for protection and regeneration following injury in the nervous system. Proceedings of the National Academy of Sciences of the United States of America, 106(46), 19599–19604. 10.1073/pnas.0907935106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez‐Fdez, S. , Fernández‐Nevado, L. , Lorenzo‐Martín, L. F. , & Bustelo, X. R. (2020). Lysine acetylation reshapes the downstream signaling landscape of vav1 in lymphocytes. Cell, 9(3), 609. 10.3390/cells9030609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sada, N. , Fujita, Y. , Mizuta, N. , Ueno, M. , Furukawa, T. , & Yamashita, T. (2020). Inhibition of HDAC increases BDNF expression and promotes neuronal rewiring and functional recovery after brain injury. Cell Death & Disease, 11, 655. 10.1038/s41419-020-02897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiner, B. , Hedskog, L. , Wiehager, B. , & Ankarcrona, M. (2015). Amyloid‐β peptides are generated in mitochondria‐associated endoplasmic reticulum membranes. Journal of Alzheimer's Disease, 43(2), 369–374. 10.3233/JAD-132543 [DOI] [PubMed] [Google Scholar]
- Sebollela, A. , Freitas‐Correa, L. , Oliveira, F. F. , Paula‐Lima, A. C. , Saraiva, L. M. , Martins, S. M. , Mota, L. D. , Torres, C. , Alves‐Leon, S. , de Souza, J. M. , Carraro, D. M. , Brentani, H. , De Felice, F. G. , & Ferreira, S. T. (2012). Amyloid‐β oligomers induce differential gene expression in adult human brain slices. The Journal of Biological Chemistry, 287(10), 7436–7445. 10.1074/jbc.M111.298471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao, L. , Zhang, Y. , Hao, Y. , & Ping, Y. (2022). Upregulation of IP3 receptor mediates APP‐induced defects in synaptic downscaling and sleep homeostasis. Cell Reports, 38(13), 110594. 10.1016/j.celrep.2022.110594 [DOI] [PubMed] [Google Scholar]
- Shao, L. W. , Peng, Q. , Dong, M. , Gao, K. , Li, Y. , Li, Y. , Li, C. Y. , & Liu, Y. (2020). Histone deacetylase HDA‐1 modulates mitochondrial stress response and longevity. Nature Communications, 11(1), 4639. 10.1038/s41467-020-18501-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng, B. , Wang, X. , Su, B. , Lee, H. G. , Casadesus, G. , Perry, G. , & Zhu, X. (2012). Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer's disease. Journal of Neurochemistry, 120(3), 419–429. 10.1111/j.1471-4159.2011.07581.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilling, D. , Müller, M. , Takano, H. , Mak, D. O. , Abel, T. , Coulter, D. A. , & Foskett, J. K. (2014). Suppression of InsP3 receptor‐mediated Ca2+ signaling alleviates mutant presenilin‐linked familial Alzheimer's disease pathogenesis. The Journal of Neuroscience, 34(20), 6910–6923. 10.1523/JNEUROSCI.5441-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, J. , Park, S. , Lee, H. , & Kim, Y. (2021). Thioflavin‐positive tau aggregates complicating quantification of amyloid plaques in the brain of 5XFAD transgenic mouse model. Scientific Reports, 11(1), 1617. 10.1038/s41598-021-81304-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therriault, J. , Pascoal, T. A. , Lussier, F. Z. , Tissot, C. , Chamoun, M. , Bezgin, G. , Servaes, S. , Benedet, A. L. , Ashton, N. J. , Karikari, T. K. , Lantero‐Rodriguez, J. , Kunach, P. , Wang, Y. T. , Fernandez‐Arias, J. , Massarweh, G. , Vitali, P. , Soucy, J. P. , Saha‐Chaudhuri, P. , Blennow, K. , … Rosa‐Neto, P. (2022). Biomarker modeling of Alzheimer's disease using PET‐based Braak staging. Nature Aging, 2(6), 526–535. 10.1038/s43587-022-00204-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirrell, P. S. , Nguyen, K. N. , Luby‐Phelps, K. , & Friedman, J. R. (2020). MICOS subcomplexes assemble independently on the mitochondrial inner membrane in proximity to ER contact sites. The Journal of Cell Biology, 219(11), e202003024. 10.1083/jcb.202003024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale, C. , Alonso, E. , Rubiolo, J. A. , Vieytes, M. R. , LaFerla, F. M. , Giménez‐Llort, L. , & Botana, L. M. (2010). Profile for amyloid‐beta and tau expression in primary cortical cultures from 3xTg‐AD mice. Cellular and Molecular Neurobiology, 30(4), 577–590. 10.1007/s10571-009-9482-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Völgyi, K. , Badics, K. , Sialana, F. J. , Gulyássy, P. , Udvari, E. B. , Kis, V. , Drahos, L. , Lubec, G. , Kékesi, K. A. , & Juhász, G. (2018). Early presymptomatic changes in the proteome of mitochondria‐associated membrane in the APP/PS1 mouse model of Alzheimer's disease. Molecular Neurobiology, 55(10), 7839–7857. 10.1007/s12035-018-0955-6 [DOI] [PubMed] [Google Scholar]
- Zhang, F. , Shi, Y. , Wang, L. , & Sriram, S. (2011). Role of HDAC3 on p53 expression and apoptosis in T cells of patients with multiple sclerosis. PLoS One, 6(2), e16795. 10.1371/journal.pone.0016795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H. , Wu, L. , Pchitskaya, E. , Zakharova, O. , Saito, T. , Saido, T. , & Bezprozvanny, I. (2015). Neuronal store‐operated calcium entry and mushroom spine loss in amyloid precursor protein knock‐in mouse model of Alzheimer's disease. The Journal of Neuroscience, 35(39), 13275–13286. 10.1523/JNEUROSCI.1034-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S. , Fujita, Y. , Matsuzaki, R. , & Yamashita, T. (2018). Class I histone deacetylase (HDAC) inhibitor CI‐994 promotes functional recovery following spinal cord injury. Cell Death & Disease, 9(5), 460. 10.1038/s41419-018-0543-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, P. , Chen, Q. , Tian, X. , Qian, N. , Chai, P. , Liu, B. , Hu, J. , Blackstone, C. , Zhu, D. , Teng, J. , & Chen, J. (2018). DNA damage triggers tubular endoplasmic reticulum extension to promote apoptosis by facilitating ER‐mitochondria signaling. Cell Research, 28(8), 833–854. 10.1038/s41422-018-0065-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, X. , Wang, S. , Yu, L. , Jin, J. , Ye, X. , Liu, Y. , & Xu, Y. (2017). HDAC3 negatively regulates spatial memory in a mouse model of Alzheimer's disease. Aging Cell, 16(5), 1073–1082. 10.1111/acel.12642 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Appendix S1
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.