

Abstract
Glycolysis provides building blocks for the proinflammatory activation of macrophages and simultaneously generates pyruvate. In this issue of Nature Metabolism, Ran et al. provide evidence that the transport of pyruvate to fuel the Krebs cycle in the mitochondria is not required in the inflammatory response.
Macrophages have long been dismissed as merely the ‘garbage collectors’ of the immune system — that is, before the reevaluation of their notable functional plasticity during both steady state and inflammation. Macrophages can be classically activated by lipopolysaccharide (LPS) and interferon-γ with a proinflammatory profile or, alternatively, by interleukin-4 (IL-4) with a proresolving phenotype. These states exist as two extremes within a spectrum of varying shades of activation. The metabolic processes that regulate immune cell responses (‘immunometabolism’) have rapidly gained a growing interest by the scientific community, especially in chronic inflammatory diseases. In macrophages, glycolysis is a hallmark of classical proinflammatory activation. Glucose catabolism fuels the necessary building blocks for the rapid mobilization of the innate immune response, such as after infection1. An end product of glycolysis is pyruvate, which in inflammatory macrophages is often metabolized to lactate through high rates of aerobic glycolysis (known as Warburg metabolism). Alternatively, pyruvate can be transported into the mitochondria by the action of mitochondrial pyruvate carrier (MPC), after which pyruvate enters the tricarboxylic acid cycle (also known as the Krebs cycle). In this issue of Nature Metabolism, Ran et al.2 show that, counter to previous experimental evidence, the Mpc1 gene is not required for proinflammatory macrophage activation, both in vitro and in vivo during experimental endotoxaemia.
Debate continues surrounding the origin of Warburg and inflammatory metabolism in classically activated macrophages. Several scenarios have been proposed that are not mutually exclusive. For example, the accumulation of succinate may stabilize hypoxia-inducible factor 1α (HIF1α) through the induction of reactive oxygen species (ROS), which in turn could upregulate glycolytic genes such as Slc2a1 (which encodes glucose transporter 1) or Ldha (which encodes lactate dehydrogenase A), all while inhibiting pyruvate dehydrogenase (PDH)-dependent glucose oxidation3. In this scenario, limiting PDH-dependent generation of acetyl-coenzyme A (CoA) for the Krebs cycle would have an effect not only in generating NADH (which fuels the electron transport chain for oxidative phosphorylation) but also in lipogenesis. That is, unless this pathway is compensated by carbon flux through pyruvate carboxylase (PC)-dependent generation of oxaloacetate (OAA)4 (Fig. 1a). Classical macrophage activation is also characterized by the accumulation of citrate and itaconate; this is considered the result of a broken Krebs cycle, due to a decline in isocitrate dehydrogenase (Idh1) and an increase in aconitate decarboxylase (Acod1 (also known as Irg1)) mRNA, and decreased succinate dehydrogenase (SDH) activity1,3. This promotes citrate accumulation for lipogenesis, succinate accumulation for ROS regulation and modulation of shared metabolite intermediates of the urea cycle, a process that eliminates nitrogen that arises from amino acid catabolism (Fig. 1a). Nitric oxide production generated through the urea cycle competes with oxygen to inhibit complex II of the electron transport chain, and reduced malic-enzyme-dependent NADPH production perturbs the balance between NADPH oxidase-dependent ROS generation and the regeneration of antioxidant defences to orchestrate classical macrophage activation. Consistently, targeting the shared malate–aspartate shuttle nested close to the aspartate–argininosuccinate shunt through glutamic-oxaloacetic-transaminase-dependent transamination prevented classical macrophage activation5.
Fig. 1 |. Metabolic rewiring upon macrophage activation.
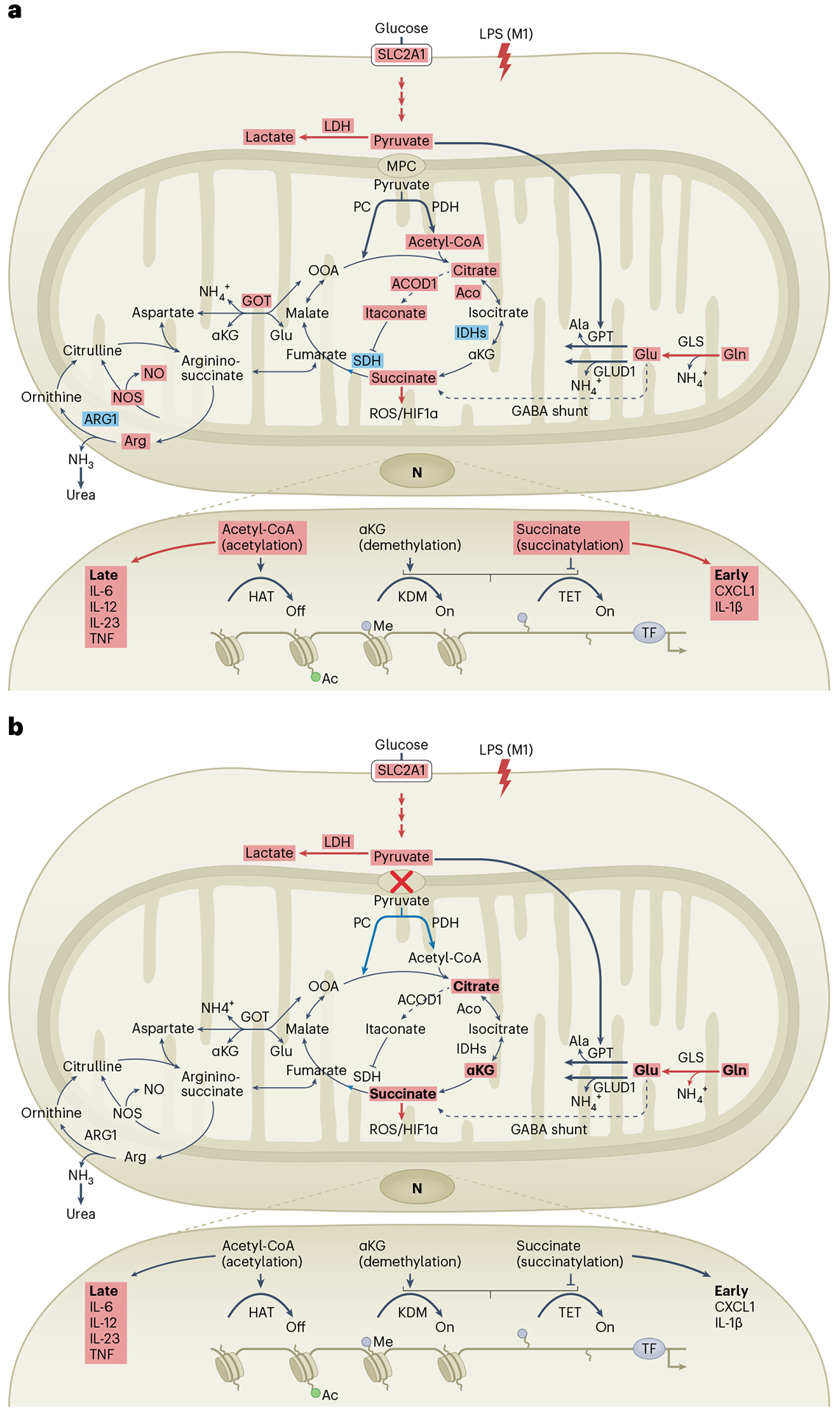
a, A hallmark of classical proinflammatory activation of macrophages is enhanced glycolysis, which provides the necessary building blocks for rapid energy production. This often occurs through Warburg metabolism, which involves aerobic glycolysis with pyruvate (an end product of glycolysis) to produce lactate or, alternatively, pyruvate being transported by the action of MPC into the mitochondria (to fuel the Krebs cycle via anaplerotic reactions). Pyruvate can be converted by PC to produce OAA or by PDH to promote acetyl-CoA production and histone acetylation of inflammatory genes. These mechanisms were proposed to occur within only a few hours of inflammatory triggers such as LPS (‘early response’). The accumulation of citrate, itaconate and succinate are considered to be the result of a broken Krebs cycle, due to a decline in Idh1 and an increase in Acod1 mRNA expression, and decreased SDH activity. Together with enhanced glutaminolysis, this promotes citrate accumulation for lipogenesis and succinate accumulation for ROS regulation and modulation of shared metabolite intermediates of the urea cycle, a process that eliminates nitrogen arising from amino acid catabolism. Nitric oxide (NO) production generated through the urea cycle competes with oxygen to inhibit complex II of the electron transport chain. The glutamine-dependent accumulation of succinate and ROS stabilizes HIF1α and boosts the specific expression of IL-1β that occurs after the first inflammatory wave (‘late response’). Altogether, these mechanisms shape classical macrophage activation. b, The absence of MPC owing to Mpc1 deficiency inhibited respiration driven by pyruvate without affecting glycolysis or lactate production. In these specific Mpc1-deficient macrophages, pyruvate import to the mitochondria was uncoupled from both inflammation and global histone acetylation, although locus specificity remains to be investigated. It is unclear whether this is related to a compensatory metabolic rewiring linked to a raise of carbon flux from glutaminolysis. Ac, acetyl; aco, aconitate; CXCL1, chemokine ligand 1; GABA, γ-aminobutyric acid; GLS, glutaminase; GOT, glutamic oxaloacetic transaminase; HAT, histone acetyltransferase; IDHs, isocitrate dehydrogenases; KDM, histone lysine demethylase; LDH, lactate dehydrogenase; LPS (M1), LPSmediated macrophage M1 activation; me, methyl; NOS, NO synthase; TET, ten-eleven translocation enzymes; TF, transcription factor; TNF, tumour necrosis factor-α.
In this context, interest in pyruvate as a modulator of macrophage inflammation has been high, given its central role in bridging glycolysis and mitochondrial metabolic pathways. Previous studies have implicated a role for pyruvate import to the mitochondria in the polarization of proinflammatory macrophages using the pharmacological compound UK5099 (refs. 6,7). One mechanistic explanation for this suggests that glycolysis-derived pyruvate could enter the mitochondria to promote acetyl-CoA production and histone acetylation of inflammatory genes6,8 (Fig. 1a). These mechanisms were proposed to occur within hours of the inflammatory response to LPS, preceding the glutamine-dependent accumulation of succinate that both stabilizes HIF1α and boosts IL-1β (in the context of a broken Krebs cycle)1,3. Indeed, Ran et al. first confirmed that mitochondrial respiration was increased in the acute (3-h) LPS response2, before macrophages shifted to a strong reduction in their oxygen consumption rate at 24 h5,6. In this early phase, U-13C-glucose tracing experiments confirmed the role of glycolysis in feeding citrate metabolites to the Krebs cycle. However, in contrast to the UK5099 compound, genetic deficiency of Mpc1 was dispensable for proinflammatory cytokine production in LPS-stimulated macrophages both for the ‘early’ and ‘late’ LPS-phase response. Thus, the investigators show that pyruvate import to the mitochondria can be uncoupled from both inflammation and global histone acetylation2; however, further in-depth analysis of the epigenetic changes is warranted.
Ran et al. also tested for the contribution of metabolic compensation in Mpc1-deficient macrophages by generating mice with an inducible macrophage-specific deletion of Mpc1, in which they investigated the transcriptome diversity of these cells by RNA sequencing (RNA-seq) and single-cell RNA-seq2. Although Mpc2 (which is located on a separate chromosome to Mpc1) was still expressed in Mpc1-deficient macrophages, the authors reported a loss of MPC2 protein expression as well — most probably because of the requirement for MCP1 and MCP2 to function as a heterodimer within the inner mitochondrial membrane. They found that high concentrations of UK5099 compound were still effective at reducing proinflammatory cytokines in LPS-stimulated Mpc1-deficient macrophages. This contrasted with the use of the MSDC0602 compound (another MPC inhibitor) in infected Mpc2-deficient alveolar macrophages in a previous study9. Thus, it is still possible that germline Mpc1 deficiency could rewire metabolic susceptibility to UK5099. Protein biochemistry approaches provided evidence for UK5099 binding to oxidoreducatase-related proteins. These effects were associated with alterations of a series of cellular respiratory responses in an MPC-independent fashion, including oxidative phosphorylation, mitochondrial membrane potential, stabilization of the glycolysis-inducing transcription factor HIF1α and glutamate oxidation2. In this context, it will be interesting in future studies to elucidate more specifically the MPC-independent mechanism of UK5099 action (particularly given its anti-inflammatory potential), as has been done for other multifunctional compound classes such as metformin.
This is not the first time that the relevance of a metabolite or mitochondrial fuel has been questioned in macrophages owing to off-target pharmacological effects. For example, high-dose etomoxir was used to implicate long-chain fatty-acid oxidation in alternatively activated macrophages, but this was later found to be due to depletion of intracellular free CoA levels10. Separately, and in the context of classical proinflammatory activation, an enhanced inflammatory response has been observed 24 h after the use of 2-deoxyglucose, a nonmetabolizable glucose analogue that blocks the first step in glycolysis7. These findings contrasted with the short-term (3 h) use of 2-deoxyglucose8 or nutritional glucose deprivation6, most probably (and trivially) because long-term energy depletion promotes cellular death. In this context, the MPC-independent effects of UK5099 are yet another reminder of the caveats of conclusions based on high-dose pharmacological inhibition: it will be important to compare UK5099 to alternative MPC-targeting compounds (such as mitoglitazone or MSDC0602) and, to increase rigour, this will be especially important for FDA drugs approved without animal testing.
The study of Ran et al. raises several questions. The complete absence of MPC complex effectively inhibited respiration driven by pyruvate2, which suggests impaired archetypical anaplerotic reactions in mitochondria (that is, entry of intermediates into the Krebs cycle) that support conversion of pyruvate to citrate by PDH or pyruvate to OAA by PC (Fig. 1b). This could affect the global inflammatory response, as classically activated macrophages modulate carbon flux through PC-dependent generation of OAA and subsequent ureagenesis at the expense of PDH-dependent generation of citrate and lipogenesis4. However, maximal respiration was slightly higher at steady state in Mpc1-deficient macrophages before LPS stimulation2. One explanation for this is that pyruvate accumulation can promote the utilization of lactate as a carbon source in the tricarboxylic acid cycle11. By contrast, the authors reported similar U-13C-glucose incorporation into lactate and similar lactate production in Mpc1-deficient macrophages compared to controls2. So, what is the fate of pyruvate? There is an alternative pathway that has not yet been fully considered in macrophages, which converts pyruvate and glutamate to α-ketoglutarate (αKG) and alanine through the cytosolic glutamic-pyruvate transaminase (GPT) (also known, in the liver, as alanine aminotransaminase) (Fig. 1b). Ran et al. observe that MPC1 deficiency exacerbated a boost of carbon flux from glutaminolysis in the early phase of the inflammatory response using U-13C-glutamine tracing experiments2. In another setting of proliferating cancer cells, it has previously been reported that glutamine oxidation maintains the Krebs cycle during impaired mitochondrial pyruvate transport12. Altogether, these findings highlight the versality of glucose and glutamine to compensate for each other and that glutamine supply is sufficient for most of anaplerosis. Apart from GPT-dependent transamination, glutamate can be deaminated by glutamate dehydrogenase 1 (GLUD1) to produce αKG for the Krebs cycle (Fig. 1b). Glud1-deficient macrophages exhibited higher oxidative phosphorylation and energy generation at steady state13, which suggests that glutamate transamination (rather than dehydrogenation) could be the dominant anaplerotic pathway in macrophages. Thus, it remains to be determined whether compensatory pyruvate re-routing through GPT activity could synergize with glutaminolysis to efficiently maintain the Krebs cycle and limit inflammation in Mpc1-deficient macrophages.
Besides classical macrophage activation (which represents the prototype bacterial proinflammatory response), there is a growing interest in the metabolic rewiring of macrophages during other similarly vital macrophage functions such as the clearance and metabolism of apoptotic debris by phagocytic macrophages (efferocytosis). As such, future studies may consider the contribution of these findings to metabolic reprogramming during alternative macrophage functions. Indeed, recent studies have described unique glucose metabolic reprogramming during efferocytosis, relative to classical macrophage activation14,15. Taken together, the present study hints at a therapeutic window to metabolically control inflammation-resolution pathways (such as through efferocytosis in chronic inflammatory disease) without risking the efficient metabolic mobilization of macrophages for bacterial infection, and vice versa.
Footnotes
Competing interests
L.Y.-C. and E.B.T. declare no competing interests.
References
- 1.O’Neill LAJ & Pearce EJ J. Exp. Med 213, 15–23 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ran L et al. Nat. Metab 10.1038/s42255-023-00800-3 (2023). [DOI] [Google Scholar]
- 3.Mehta MM, Weinberg SE & Chandel NS Nat. Rev. Immunol 17, 608–620 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Palmieri EM et al. Nat. Commun 11, 698 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jha AK et al. Immunity 42, 419–430 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Lauterbach MA et al. Immunity 51, 997–1011 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Bae S et al. Cell Rep. 35, 109264 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Langston PK et al. Nat. Immunol 20, 1186–1195 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu B et al. Sci. Immunol 10.1126/sciimmunol.adf0348 (2023). [DOI] [Google Scholar]
- 10.Divakaruni AS et al. Cell Metab. 28, 490–503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Faubert B et al. Cell 171, 358–371 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang C et al. Mol. Cell 56, 414–424 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merlin J et al. Nat. Metab 3, 1313–1326 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morioka S et al. Nature 563, 714–718 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schilperoort M, Ngai D, Katerelos M, Power DA & Tabas I Nat. Metab 5, 431–444 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]