

Significance
COVID-19 remains a global challenge necessitating continuing research that provides insights into factors that predispose to the diversity of outcomes—from mild disease to death. Preclinical models are the cornerstone of such research, but they are often based on the use of transgenic animals that express human angiotensin-converting enzyme 2, or genetically modified viruses that present significant caveats and complicate interpretation of data. Here, we introduce a mouse-adapted strain of SARS-CoV-2 (P21) that causes severe disease in young and lethality in aged mice. P21 infection causes weight loss, lung inflammation, and cytokine storm, triggered by a transcriptomic response that correlates with COVID-19 disease. Our preclinical model provides a much-needed tool to study host responses to COVID-19 and test drug candidates.
Keywords: SARS-CoV-2, COVID-19, mouse adapted, cytokines, inflammation
Abstract
The diversity of COVID-19 disease in otherwise healthy people, from seemingly asymptomatic infection to severe life-threatening disease, is not clearly understood. We passaged a naturally occurring near-ancestral SARS-CoV-2 variant, capable of infecting wild-type mice, and identified viral genomic mutations coinciding with the acquisition of severe disease in young adult mice and lethality in aged animals. Transcriptomic analysis of lung tissues from mice with severe disease elucidated a host antiviral response dominated mainly by interferon and IL-6 pathway activation in young mice, while in aged animals, a fatal outcome was dominated by TNF and TGF-β signaling. Congruent with our pathway analysis, we showed that young TNF-deficient mice had mild disease compared to controls and aged TNF-deficient animals were more likely to survive infection. Emerging clinical correlates of disease are consistent with our preclinical studies, and our model may provide value in defining aberrant host responses that are causative of severe COVID-19.
COVID-19 usually manifests as a self-limiting respiratory illness, but around 15% of people experience severe disease, with age and comorbidities increasing the likelihood of adverse clinical outcomes (reviewed in refs. 1–3). The elderly and immunocompromised are highly vulnerable to COVID-19, with reports indicating a case-fatality rate 60 times higher in people aged 65 y or older compared to younger individuals (4, 5). Although certain demographic factors increase the risk of severe COVID-19, a proportion of people unpredictably succumb to the disease. COVID-19 severity and mortality are associated with systemic inflammatory responses and acute respiratory distress syndrome. Multiple proinflammatory markers have been suggested as predictors of severe disease, including ferritin, interleukin-1β (IL-1β), IL-6, IL-8, D-dimer, and Tumor Necrosis Factor (TNF) (6–14).
Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) infection requires interaction between the viral spike protein and the human host cell surface receptor angiotensin-converting enzyme 2 (ACE2) (15–17), while numerous other host cell proteins promote establishment of infection (15, 18–21). The human tropism of early SARS-CoV-2 strains has restricted mouse preclinical studies to the use of transgenic animals that express human ACE2 (hACE2), or of genetically modified ancestral SARS-CoV-2 with introduced adaptive mutations for mouse ACE2 (mACE2), defined by molecular modeling (SARS-CoV-2 MA) (22). While SARS-CoV-2 MA only causes moderate disease (22), neuroinvasiveness, systemic viral spread (23), and encephalitis (24) contributed to death in hACE2 models, which do not match the majority of COVID-19 clinical observations. Given the data stemming from published mouse adaptive mutations of the original SARS-CoV, and given the need for animal models that mimic human disease to understand COVID-19 pathogenesis, we monitored the spike protein sequence in public databases for evolution of key sites of SARS-CoV-2 during transmission among the Australian population. We found a naturally occurring N501Y variant (VIC2089) that was capable of infecting wild-type C57BL/6 mice. We passaged VIC2089 in 8-wk-old mice and recovered a virulent isolate that caused severe disease in young adult and lethality in aged animals. We linked viral adaptation to mutations in Orf1AB. We infected young adult and aged mice with the original and mouse-adapted SARS-CoV-2 to define host factors that contribute to disease. Transcriptomic analyses identified several overrepresented cytokine pathways linked to severe disease in mice, including interferon (IFN), IL-6, TNF, and Transforming Growth Factor β (TGF-β) pathways, that may have relevance to severe COVID-19. Congruent with our pathway analysis, we showed that TNF-deficient mice had mild disease compared to controls and were more likely to survive lethal infection. Our two viruses provided an opportunity to understand how host immune responses contribute to the spectrum of disease severity and test the efficacy of therapeutics in mitigating severe COVID-19.
Results
Generation of a Mouse Pathogenic Variant of SARS-CoV-2.
In June 2020, during a period of strict border control and quarantine of SARS-CoV-2 in Australian communities, genomic surveillance identified a contained 35-person outbreak with a Spike N501Y mutant (25). The N501Y mutation has been correlated with viral adaptation to mACE2 (26) and viral isolates carrying the substitution have been successfully used in mouse models (27, 28). To select for a variant that causes more disease in mice, we intranasally infected young C57BL/6 mice with a SARS-CoV-2 N501Y clinical isolate (VIC2089) and used homogenized infected lung filtrate as intranasal inoculum to infect a further cohort of animals, generating a virus referred to as P2. We repeated this process 19 times to generate virus passages P2 to P21 (Fig. 1A). The first 9 passages of the virus did not significantly alter viral burdens or the manifestations of illness in mice. At 3 days postinfection (dpi), virus derived from the 14th (P14) animal passage resulted in significant loss in body weight compared to P2, while further passages to generate P21 did not significantly increase weight loss or viral burdens in mice (Fig. 1 B and C). Compared to P2, P21 infection caused significantly increased viral burdens in the lungs 2 to 4 d dpi, but clearance kinetics were indistinguishable between passages, with all mice clearing infection by 7 to 10 dpi. While viral burdens peak between days 2 and 4, weight loss of P21-infected mice is more pronounced at days 3 to 4 postinfection (Fig. 1 D and E). We examined lung viral burdens upon P2 or P21 infections at 3 dpi and found that while P21-infected mice tended to have worse disease (greater weight loss) when presented with higher viral loads, this phenomenon was not observed in mice infected with P2 virus (SI Appendix, Fig. S1 A and B). Interestingly, when mice with similar lung viral burdens were compared, animals infected with P21 showed increased weight loss compared to mice infected with P2 (SI Appendix, Fig. S1 C and D).
Fig. 1.
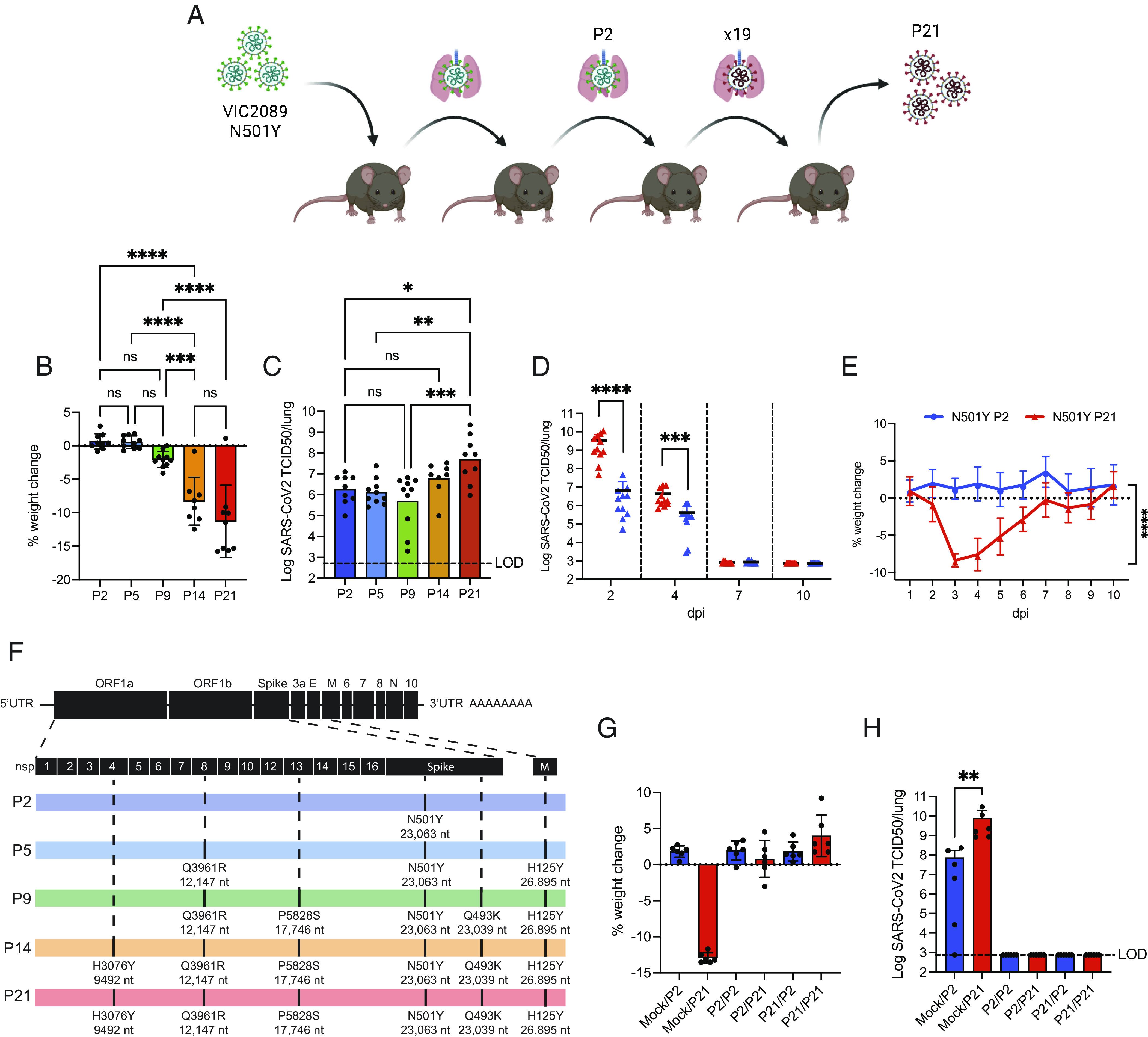
Passage of N501Y VIC2089 in lungs of adult C57BL/6 results in adaptive virulence mutations without affecting response to rechallenge. (A) Graphical representation of the serial passage method to generate SARS-CoV-2 P21 virus. The VIC2089 SARS-CoV-2 N501Y clinical isolate was used to infect C57BL/6 mice (n = 3). Animals were euthanized at 3 dpi, and lungs were homogenized, pooled, and used to infect a new cohort of animals (n = 3) intranasally. This process was repeated 21 times (the image was created with BioRender.com). (B and C) Mice were infected with 104 TCID50 of passage 2, 5, 9, 14, or 21 (n = 8 to 10) and monitored at 3 dpi for (B) percent weight change compared to initial weight and (C) lung viral burden measured by TCID50 assay. (D and E) Mice were challenged intranasally with 104 TCID50 of either the SARS-CoV-2 early passage P2 strain or the mouse adapted, P21 strain (n = 10 to 11) (D) Animals were euthanized at defined time points after infection, and lungs were collected for viral quantification by the TCID50 assay. (E) Daily percent weight change of infected animals compared to initial weight. (F) Virus passages were sequenced using next-generation sequencing, and strains showing nonsynonymous mutations were compared to the original clinical isolate (P2). In black (Top), a schematic of the SARS-CoV-2 mRNA is shown. P2, P5, P9, P14, and P21 mRNA representations are depicted with the acquired mutations marked on their respective locations on the mRNA. (G and H) Mock, P2-, or P21-infected mice were rechallenged 28 d later with P2 or P21 and analyzed 3 d after rechallenge for (G) percent weight change, compared to weight before second infection and (H) lung viral load (TCID50). Data were pooled from (B–E) or are representative of (G and H) 2 to 3 independent experiments. One-way ANOVA with multiple comparisons (B and C), 2-way ANOVA (D and E), and unpaired two-tailed Student’s t test after log10 transformation (H) were performed. Mean ± SD is shown. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
We next performed whole-genome sequencing of the passaged viruses to identify potential nonsynonymous evolutionary mutations that contributed to weight loss and enhanced lung viral loads (SI Appendix, Table S1). We found a small number of point mutations in P21 within regions encoding nonstructural proteins Nsp8 Q3961R (Q19R), which first became prominent in passage 5; Nsp13 P5828S (P504S), which appeared in P9; and H3076Y (H313Y) in Nsp4, which emerged at passages 13 to 14 and became fixed at P21. The substitution in Nsp4 was coincident with a significant increase in weight loss and later fixation in P21 coincided with an increase in lung viral burdens. Only one mutation was observed in the M protein (H125Y), which started to become dominant in P5, and one additional Spike protein (Q493K) mutation emerged in P9 (Fig. 1F).
We next sought to determine whether the Spike mutation acquired at P9 altered the requirement for ACE2 to facilitate infectivity. We administered 104 TCID50 (50%Tissue Culture Infectious Dose) viral stocks intranasally to mice deficient in ACE2 (Ace2−/−) and were unable to detect virus in the lungs 3 dpi (SI Appendix, Fig. S1E). We further questioned whether these mutations allowed P21 virus to escape acquired immunity in mice that had previously recovered from a P2 virus infection. P2-recovered mice were resistant to secondary infection with P21 virus 28 d later, and vice versa, suggesting that viral epitopes from P2 and P21 were not sufficiently different to escape acquired immunity (Fig. 1 G and H). Collectively, these data show that P14 virus caused more severe disease in animals compared to an early passage virus (P2). Increased weight loss was coincident with an H3076Y (H313Y) mutation in Nsp4, which became fixed at later passages (P21).
Lung Histological Features of P21 Infection Reflect Hallmarks of Severe COVID-19.
To define host responses that contribute to mild and severe disease, we compared lung histology in animals infected with P2 and P21 viruses. At 2 dpi, P2-infected lungs showed occasional dropout of bronchiolar epithelial cells and infrequent inflammatory cells along the margins of vessels. In contrast, P21-infected lungs showed moderate multifocal necrotizing bronchiolitis with subpleural inflammation and margination of inflammatory cells with mild vasculitis. By 4 dpi, P2-infected lungs showed clear bronchiolar epithelial necrosis, vascular endothelial cell hyperplasia, and mild interstitial pneumonia. At the same time point, P21-infected lungs displayed vasculitis, endothelial hyperplasia, multifocal alveolitis with necrotic material in the alveoli, subpleural inflammation, and occasional attenuation of the bronchiolar epithelium (Fig. 2A and SI Appendix, Fig. S2A). By 7 dpi, nearly all histological abnormalities had ameliorated in P2-infected lungs except the persistence of some inflammatory infiltrates around the lung vasculature. Conversely, P21-infected lungs at 7 dpi showed severe vasculitis, perivascular inflammation, inflammation of alveolar ducts, and multifocal inflammatory infiltrates (neutrophils, macrophages, and T cells) in the alveoli, vessels, and subpleural space. SARS-CoV-2 nucleocapsid was detected mainly in the bronchiolar epithelium at 2 dpi following infection in both P2- and P21-infected lungs. At 4 dpi, P21 virus localized exclusively to type II pneumocytes, while the localization of P2 virus remained unchanged compared to day 2 (Fig. 2A). Most animals, regardless of virus, had cleared infection by 7 dpi (SI Appendix, Fig S2 B and C). In P21-infected mice, staining for immune cells showed a significant increase in myeloperoxidase (MPO)-positive cells at 2 dpi, while CD3+ and F4/80+ cells were elevated at later time points compared to P2 infection and mock (SI Appendix, Fig. S2 B and C).
Fig. 2.
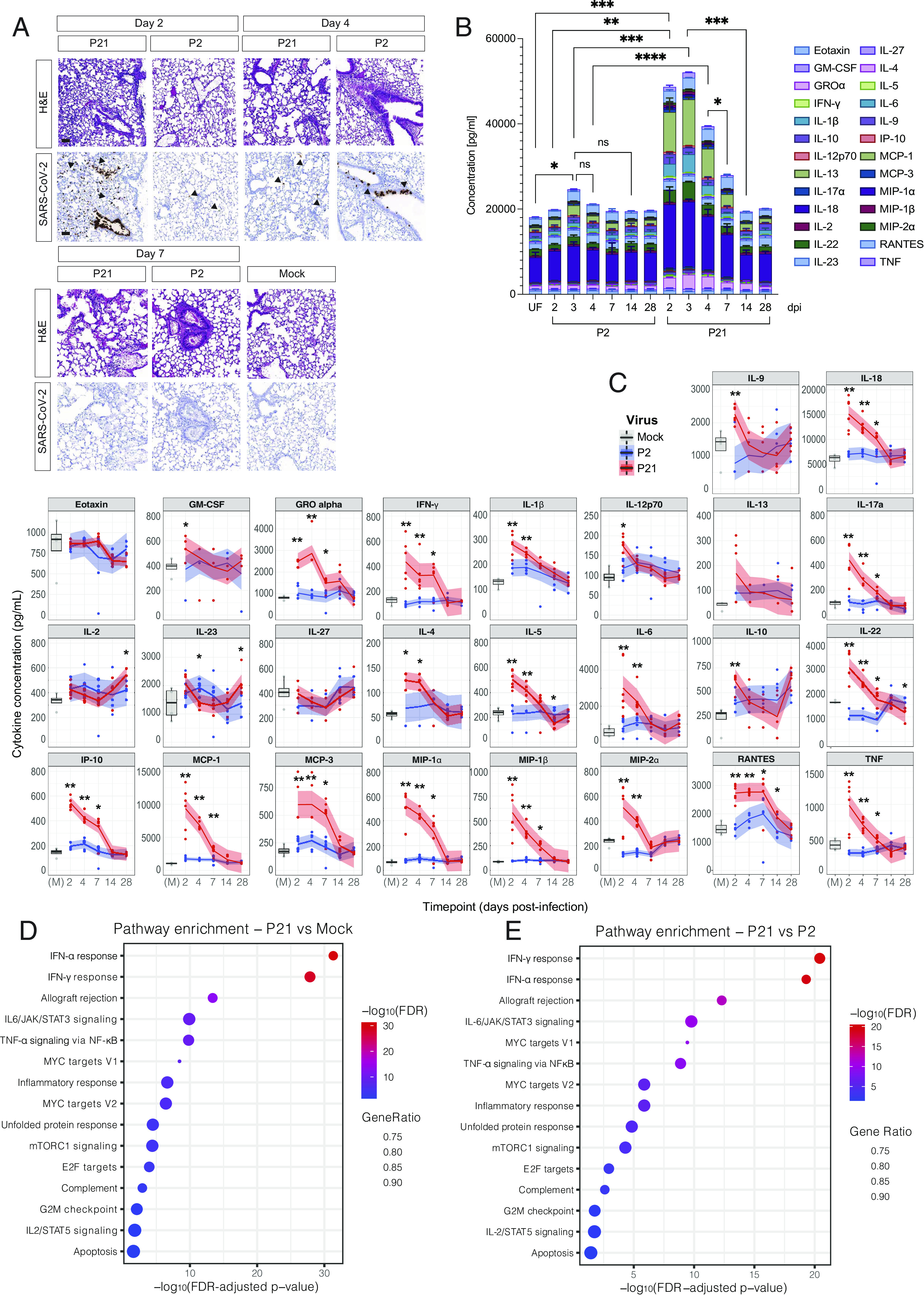
P21 infection induces severe lung disease, cytokine storm, and changes in the host transcriptome. (A) Representative images of hematoxylin and eosin (H&E) and SARS-CoV-2 nucleocapsid stained lungs. Mice were infected intranasally with 104 TCID50 of P2, P21, or mock (media only), and lungs were collected and fixed for histological analysis at days 2, 4, and 7 postinfection. Histological images are representative of at least 3 animals. Black arrows point to exemplary SARS-CoV-2-positive cells. (Scale bars, 50 µm.) (B) Mice were infected intranasally with 104 TCID50 of either P2, P21, or inoculated with vehicle control (M = mock infection). Supernatants of lung homogenates were taken at days 2, 3, 4, 7, 14, and 28 postinfection for analysis of 26 cytokines/chemokines. The concentration of all measured cytokines/chemokines for each animal were summed, and mean ± SD of each cytokine is shown. Colours represent different analytes (n = 4 to 5, 6 to 8-wk old mice per group). (C) Cytokine analysis of infected mice over time. Lungs of animals infected with 104 TCID50 of P2 or P21 were collected at days 2, 4, 7, 14, and 28 postinfection and utilized for ELISA of 26 different cytokines and chemokines. Each panel displays mock- (gray), P2- (blue), and P21- (red) infected animals. Boxplots of mock-infected samples depict the median and interquartile ranges. Loess smoothing was applied to the P2 and P21 infection time course, with the shaded area indicating 95% CIs. (D and E) Pathway enrichment analyses of significantly differentially expressed genes identified from P21- vs. mock-infected mice and P21- vs. P2-infected mice comparisons using Hallmark gene sets. Negative log10 FDR-adjusted P values associated with each pathway are plotted; dot sizes correspond to the proportion of all genes from that pathway that were found to be significantly differentially expressed in a given comparison (Gene Ratio). Two-way ANOVA with multiple comparisons (B) and Wilcoxon rank-sum (C) statistical tests were performed; *P < 0.05, **P < 0.01, and ***P < 0.001.
Cytokine Responses and Transcriptomic Analyses Reveal Greater Activation of Inflammatory Pathways Associated with P21 Virus Infection Compared to P2.
Next, we quantified the levels of 26 cytokines and chemokines in the lungs of uninfected and infected animals (Fig. 2B). P2 infection caused a significant increase in cytokines 3 dpi compared to mice challenged with mock. These cytokine responses were greatly enhanced in mice infected with P21 across a range of proinflammatory cytokines including IFN-γ, IL-6, IL-18, and TNF (Fig. 2C). With the exception of IL-10, all cytokines returned to baseline (mock) levels by day 28 postinfection. To dissect the nature of the enhanced immune response, we performed RNA sequencing (RNA-seq) on bulk lung homogenates. We compared P2 and P21 virus–infected mouse cohorts to uninfected animals to identify significantly differentially expressed genes [DEGs, false discovery rate (FDR)-adjusted P value < 0.05 and absolute log2 fold-change > 1] between the groups. No significant differences were observed between P2-infected lungs and uninfected animals based on these significance criteria (SI Appendix, Fig. S2D). In contrast, P21 virus infection led to the upregulation of 958 and downregulation of 564 DEGs compared to uninfected mice. We identified 516 up-regulated and 314 down-regulated DEGs comparing P21- to P2-infected mice. Consistent with our cytokine data, P21 infection caused an increased expression of genes related to type I and II IFN, TNF, and IL-6 pathways compared to uninfected mice (Fig. 2D). Additionally, genes related to inflammatory responses, mTORC1, complement, and apoptosis were differentially expressed. These DEGs were also noted in a comparison of P21 infection with P2 infection (Fig. 2E). Ingenuity pathway analysis (IPA) showed STAT3 as the top upstream regulator differentiating P2 and P21 virus, through association with genes regulating proinflammatory markers such as MCP-1 (CCL2) and IL-6 (Dataset S1). Plasminogen activator inhibitor-1 (PAI-1) was also shown to be up-regulated in P21-infected mice compared to P2-infected mice, as well as pathways related to T cell responses, accumulation of neutrophils, and IRF1/3 and 7 (Dataset S2 and SI Appendix, Fig. S2E). Collectively, our transcriptomic, cytokine, and histological analyses indicate that P21 SARS-CoV-2 infection, causes substantial upregulation of inflammatory responses particularly across IFN, IL-6/STAT-3, and TNF pathways, similar to severe COVID-19.
Exacerbated P21-Induced SARS-CoV-2 Disease in Aged Mice Resembles Severe COVID-19.
To understand whether age was associated with more severe disease upon SARS-CoV-2 infection, as in humans, we infected 6 to 8-mo-old mice. Disease caused by P2 virus was only marginally exacerbated in older mice, while in contrast, P21 virus infection caused severe weight loss necessitating euthanasia (as per ethical endpoints outlined in the Materials and Methods section) within 5 d of infection (Fig. 3 A and B). Histology showed extensive necrotizing bronchiolitis, vasculitis, and inflammation in aged mice infected with P21 (Fig. 3C), and older P21-infected mice tended to have higher lung viral loads compared to young mice (SI Appendix, Fig. S3 A and B). Cytokine responses, measured by Enzyme-Linked Immunosorbent Assay (ELISA), were exacerbated in P2-infected aged mice compared to P2-infected young mice, but the total lung cytokine responses associated with P21 virus infection did not differ between young and aged mice (Fig. 3D). Comparison of relevant cytokines/chemokines both in terms of transcriptome (RNA-seq) and protein expression (ELISA) showed that Il10 and Il23 gene expression levels were significantly higher in aged P21-infected mice compared to young P21 infected mice, while Ifng and Il27 expression levels were significantly lower. P21 infection in aged mice caused significantly higher expression levels of Tnf, Ifng, Il10, Il6, Ccl3 (MIP-1α), Ccl4 (MIP-1β), and Il23 and reduced Il4 compared to P2 infection in aged animals (Fig. 3E and SI Appendix, Fig. S3B).
Fig. 3.

Inflammation induced by SARS-CoV-2 variants is more pronounced in aged mice, and disease is driven by TNF, TGF-β, and E2F pathways. (A and B) C57BL/6 mice of different ages (young = 6 to 8 wk and aged = 6 to 8 mo) were intranasally inoculated with 104 TCID50 SARS-CoV-2 P2 or P21 and monitored for (A) the proportion of mice that became moribund, reaching humane endpoint and (B) daily percentual weight change over time, relative to the initial weight. (n= 5 to 9 mice per group; results are representative of at least 3 independent experiments) (C) Representative images of hematoxylin and eosin (H&E), SARS-CoV-2 nucleocapsid, MPO (neutrophils), F4/80 (macrophages), or CD3 (T cells) stains of mouse lungs infected with P21 and harvested 3 dpi. Histological images are representative of at least 3 animals. (Scale bars, 50 µm.) (D) Average of sum of 26 cytokines/chemokines expressed in supernatants of lung homogenates of young and aged mice challenged intranasally with 104 TCID50 of P2, P21, or mock at 3 dpi (n = 5 mice per group; mean ± SD of each cytokine is shown; one-way ANOVA with multiple comparisons was performed). (E) Levels of cytokine and chemokines in lung homogenates 3 dpi. Protein levels were measured by ELISA, and corresponding gene expression levels were quantified by bulk RNA-seq. Gene expression data are annotated by protein name rather than the original mouse gene identifier for each of comparison. The Wilcoxon rank-sum test, with Bonferroni adjustment for multiple comparisons, was performed between aged and young mice for each infection group. Boxplots depict the median and interquartile ranges. (F) Pathway enrichment analysis of significantly DE genes identified by directly comparing P21-infected aged and young animals at 3 dpi, using Hallmark gene sets. Negative log10 false discovery rate–adjusted P values associated with each pathway are plotted; dot sizes correspond to the proportion of all genes from that pathway that were found to be significantly DE for a given comparison (Gene Ratio). (G) DE genes attributable to P21 infection in aged and young mice were converted to human homologs and performed a comparative pathway analysis between the two age groups using IPA. Among the top significantly enriched pathways for both age groups was “Role of Hypercytokinemia/Hyperchemokinemia in the Pathogenesis of Influenza” and “Coronavirus Pathogenesis”. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Data are representative of 2 to 3 independent experiments (A–C).
Pathway analysis showed that TNF/Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB), TGFβ, and E2F signaling were differentially regulated in P21-infected aged mice compared to P21-infected young mice (Fig. 3F). To ascertain whether the DEGs linked to severe disease in aged P21-infected mice had correlates in severe COVID-19, we converted DEGs attributable to P21 infection in aged and young mice to human homologs and performed a comparative pathway analysis between the two age groups using IPA. The top significantly enriched pathway for both age groups was “Role of Hypercytokinemia/Hyperchemokinemia in the Pathogenesis of Influenza”, indicating that the murine response to P21 is similar to human viral responses characterized by cytokine storms (Fig. 3G). Additionally, “Coronavirus Pathogenesis” was also among the top commonly significantly enriched pathways, although the pathway z-score was not as high as the hypercytokinemia pathway (Fig. 3G).
TNF Is One Driver of Severe SARS-CoV-2 Disease in Mice.
Our analyses implicated TNF, TGFβ, and E2F signaling pathways in the pathogenesis of severe SARS-CoV-2 disease in aged mice. We utilized TNF gene-targeted animals to examine the role of this cytokine in causing severe SARS-CoV-2 disease upon P21 infection in aged mice. In contrast to the lethality caused by P21 in aged WT animals, aged Tnf −/− P21-infected animals showed a distinct albeit partial survival advantage, as well as reduced weight loss (Fig. 4 A and B), and this was not due to difference in viral burdens (Fig. 4C). Similarly, young adult animals show a reduction in disease, as measured by partial rescue of weight loss 3 dpi (Fig. 4 D and E). The sum of 26 cytokines and chemokines in young adult TNF knockout animals was also significantly reduced compared to WT controls (Fig. 4F). Compared to WT animals, Tnf −/− animals showed decreased levels of IL-22, MCP-1, and MIP-2α, while GM-CSF, IL-4, and IL-5 were increased (SI Appendix, Fig. S4A). To analyze the potential of anti-TNF treatment in the management of severe COVID-19 in our model, we administered the TNF inhibitor etanercept to P21-infected WT mice aged 10 to 12 wk. Treatment initiated at the time of infection did not confer a survival advantage compared to control animals (Fig. 4G). Similarly, prophylactic treatment initiated 2 wk prior to infection did not mitigate mortality in mice infected with P21 compared to controls (Fig. 4H). Although these results do not concur with our genetic studies, several limitations of both the therapeutic and genetic approaches may complicate interpretation of data and contribute to the disparity. Nonetheless, collectively our data implicate a role for TNF in the pathogenesis of severe P21-mediated disease, but the ability to therapeutically target this signaling pathway needs further exploration and may not be simple.
Fig. 4.
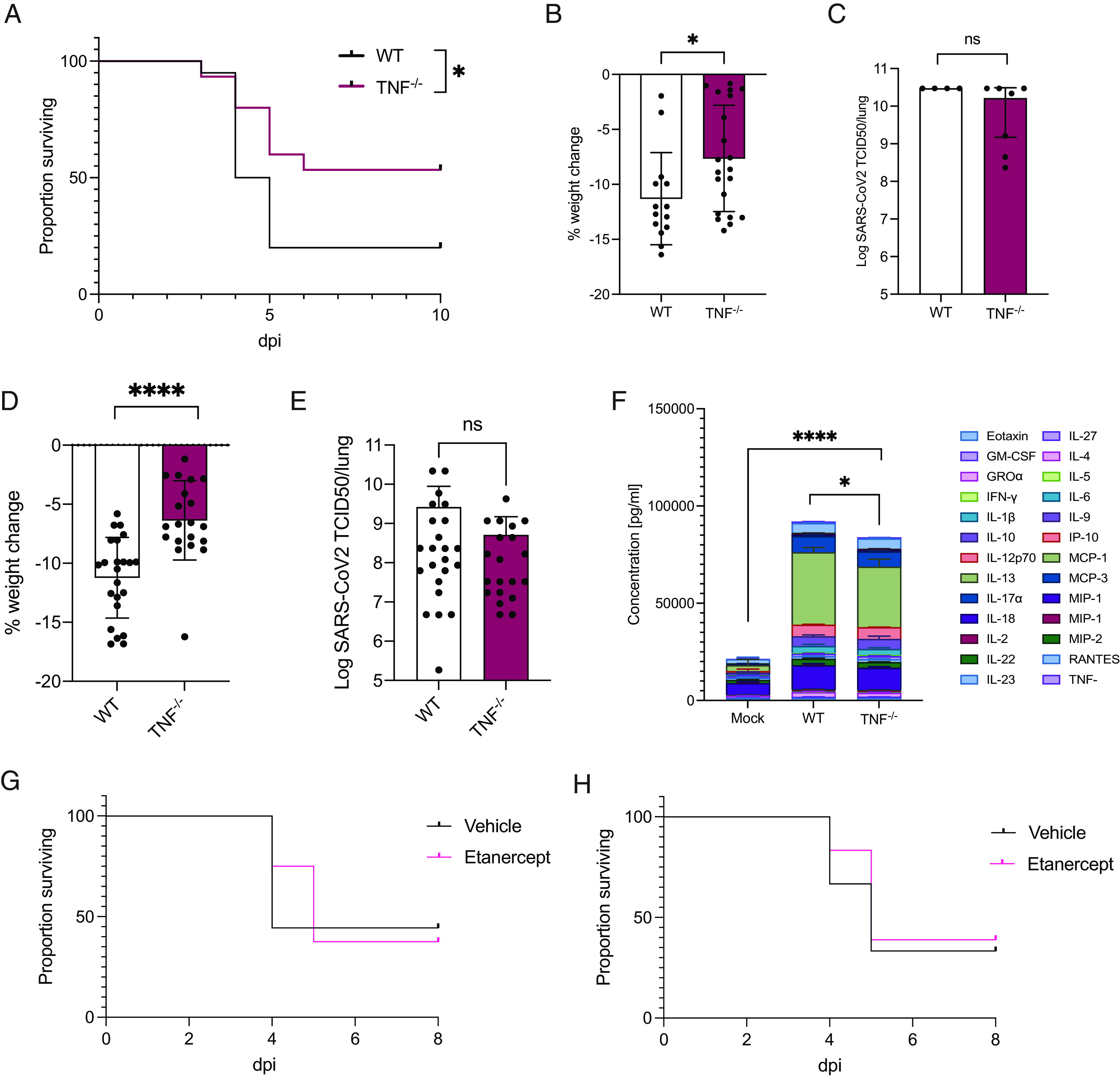
Severe pathology upon SARS-CoV-2 infection is less pronounced in animals lacking the gene encoding the proinflammatory cytokine TNF (A–C) Aged C57BL/6 and Tnf −/− mice (6 to 8 mo) were intranasally inoculated with 104 TCID50 SARS-CoV-2 P21 and monitored for (A) the proportion of mice that became moribund and reached humane endpoint, (B) percent weight change 3 dpi, relative to the initial weight (n = 15 to 20 mice per group), and (C) lung viral loads as measured by TCID50 at 3 dpi (n = 4 to 7 mice per group). (D–F) C57BL/6 and Tnf −/− mice (6 to 8-wk-old) were intranasally inoculated with 104 TCID50 SARS-CoV-2 P21 strain and monitored for (D) percent weight change, relative to the initial weight, (E) lung TCID50 at day 3 postinfection [n = 23 to 24 mice per group (D and E)], and (F) average of sum of 26 cytokines/chemokines expressed in supernatants of lung homogenates (n = 5 mice per group; mean ± SD of each cytokine is shown; mock WT mice were challenged with vehicle only). (G and H) Adult C57BL/6 (10 to 12 wk) were treated with etanercept (anti-TNF) or vehicle and monitored for the proportion of mice that reached humane endpoint (>20% weight loss) after intranasal inoculation with 104 TCID50 SARS-CoV-2 P21. Treatment was commenced either (G) at the time of infection (n = 8 to 9 mice per group) or (H) 2 wk prior to infection (n = 18 mice per group, pooled from 3 independent experiments). Mean ± SD is shown (B–F). Log-rank Mantel–Cox test (A, G, and H), one-way ANOVA (F), and unpaired two-tailed Student’s t test (B and D) after log10 transformation (C and E) were performed; *P < 0.05 and ****P < 0.0001.
Discussion
Our study shows that the mouse-adapted SARS-CoV-2 P21 virus causes severe lethal disease in aged animals with correlates to human severe COVID-19. Several mutations were acquired with passaging virus, including P5828S in Nsp13 and Q493K in Spike and these coincided with mild weight loss, but severe disease coincided with the acquisition of the additional mutation in Nsp4. Nsp4 has been implicated in viral replication/transcription, and it is thought to play a role in ER-derived membrane rearrangement via its interaction with Nsp3. This process is essential for assembly of the replication/transcription complex in coronavirus-infected cells (29, 30). In vivo, we did observe higher viral burdens in the lungs of P21 infected mice compared to P2, but this did not absolutely align with the gain in virulence. This suggests that increased viral replicative capacity in vivo may not be the sole determinant of disease severity in P21 infection.
Compared to P2, P21 virus caused exacerbations in cytokine and inflammatory responses consistent with severe COVID-19. Similar to humans, the severity of disease was associated with age since older mice were more likely to succumb to disease. Transcriptional analysis showed an association between type I and II IFN, IL-6, and TNF responses in P21-induced disease. Although the levels of TNF were not elevated in old P21-infected mice compared to young mice, deficiency in this cytokine ameliorated disease in old mice. This suggests that aging sensitized animals to the pathogenic role of TNF likely in combination with other proinflammatory signals that we observed in P21-infected animals.
We identified differences in the host transcriptional responses between P2 vs. P21 infection in mice of similar ages and also differences between young and old mice. P21 infection in both age groups up-regulated IRF3/7 responses compared to P2 infection, but additionally, young animals activate STAT3 and IRF-1 pathways, while aged animals elicit IL1-β, IL-6, TNF, IFN-γ, and IFN-β responses. The most prominent differences between young and old mice infected with P21 were in the extent of TNF/NF-kB, TGFβ, and E2F pathway activation. We showed using gene-targeted mice that TNF contributed to severe disease, although its deficiency only partially prevented lethality indicating that TNF is not solely responsible for severe disease.
Surprisingly, we were not able to replicate the modest efficacy conferred by loss of TNF in our genetic models when we conducted therapeutic studies. Etanercept, administered prophylactically or at the time of infection, did not protect animals from severe disease. The interpretation of genetic studies is complicated by potential ontological compensatory responses causing confounders that impact outcomes, and equally, therapeutic studies are complicated by pharmacokinetic and pharmacodynamic issues. With regard to the latter, it may be possible that we were not effectively inhibiting TNF activity in the lungs. Indeed, a complete loss of TNF in our genetic studies was required to mitigate severe P21 disease, and anything less would be expected to show substantially reduced efficacy. Nonetheless, our data implicate TNF in the pathogenesis of severe COVID-19 but fall short of supporting a clear therapeutic avenue to target this signaling pathway to mitigate disease. Our data would suggest that TNF is one of potentially several factors that contribute to severe disease.
Clinical studies have found correlations between anti-TNF therapy and lower risk of hospitalization or increased survival (31–33), although none have stratified age with efficacy. Here, we show transcriptomic and genetic evidence that TNF signaling is at least partially responsible for severe SARS-CoV-2 pathology in aged mice. At the transcriptional level, we did not detect differences in TNF gene expression between aged and young animals, but rather differences in genes downstream of TNF signaling, such as Il6, Nfkb1, and serpine-1 (PAI-1). Studies have suggested STAT-1 dysfunction as a potential mechanism of COVID-19 pathogenesis with a compensatory hyperactivation of STAT3 and PAI-1 (34). Interestingly, multiple independent studies on severe COVID-19 corroborate our findings in mice. TNF, IL-8, IL-1β, and IL-6 are consistent predictors of disease severity and mortality (6–13, 35).
In conclusion, we showed that enrichment of a mutation in SARS-CoV-2 Nsp4, generated through passaging the virus in mice, along with other accumulated historical mutations, caused a gain in virulence in animals. Altered host responses contributed to P21-induced disease beyond viral burdens. Responses to P21 were different between young and aged mice, with lethality observed in older mice in part due to aberrant TNF signaling. The tools we have generated may be useful in further dissecting viral virulence factors and aberrant host responses and how host age impacts both.
Materials and Methods
Mice.
Male or female WT C57BL/6J, gene-targeted ACE2 (36) and TNF (37) mice (all on a C57BL/6J background) were bred and maintained in the Specific Pathogen Free Physical Containment Level 2 Bioresources Facility at the Department of Microbiology and Immunology (University of Melbourne) or The Walter and Eliza Hall Institute of Medical Research (WEHI). All procedures involving animals and live SARS-CoV-2 strains were conducted in an OGTR-approved Physical Containment Level 3 (PC3) facility at WEHI (Cert-3621). Mice were transferred to the PC3 laboratory for all SARS-CoV-2 infection experiments at least 4 d prior to the start of experiments. Animals were age and sex matched within experiments (both sexes were used). “Young adult” animal groups were 6 to 8 wk of age at the commencement of experiments, “adults” were 10 to 12 wk, and “aged” cohorts were 6 to 8 mo. Experimental mice were housed in individually ventilated microisolator cages under level 3 biological containment conditions with a 12-h light/dark cycle and provided standard rodent chow and sterile acidified water ad libitum. All mouse strains and procedures were reviewed and approved by The Walter and Eliza Hall Institute of Medical Research Animal Ethics Committee, and were conducted in accordance with the Prevention of Cruelty to Animals Act (1986) and the Australian National Health and Medical Research Council Code of Practice for the Care and Use of Animals for Scientific Purposes (1997).
SARS-CoV-2 Strains.
SARS-CoV-2 VIC2089 clinical isolate (hCoV-19/Australia/VIC2089/2020) was obtained from the Victorian Infectious Disease Reference Laboratory. Viral passages were achieved by serial passage of VIC2089 through successive cohorts of young C5BL/6 mice. Briefly, mice were infected with SARS-CoV-2 clinical isolate intranasally. At 3 dpi, mice were killed, and lungs were harvested and homogenized in a Bullet Blender (Next Advance Inc) in 1 mL Dulbecco’s modified Eagle’s medium (DMEM) media (Gibco/ThermoFisher) containing steel homogenization beads (Next Advance Inc). Samples were clarified by centrifugation at 10,000 g for 5 min before intranasal delivery of 30 µL lung homogenate into a new cohort of naïve C57BL/6 mice. This process was repeated a further 20 times to obtain the SARS-CoV-2 VIC2089 P21 isolate. Lung homogenate from all passages was stored at −80 °C.
SARS-CoV-2 Murine Infection.
Six-to-eight-week-old (young adult), 10-wk-old (adult), or 6 to 8-mo-old (aged) mice were anesthetized with methoxyflurane and inoculated intranasally with 30 μL SARS-CoV-2. Virus stocks were diluted in serum-free DMEM to a final concentration of 104 TCID50/mouse. After infection, animals were visually checked and weighed daily for a minimum of 10 d. Mice were euthanized at the indicated times postinfection by CO2 asphyxiation. For histological analysis, animals were euthanized by cervical dislocation. Lungs were collected and stored at −80 °C in serum-free DMEM until further processing. Infected animals were euthanased immediately if they lost >15% of their starting body weight for 3 consecutive days, or >20% body weight on any day.
Anti-TNF Treatment of Mice.
For depletion of TNF, 10 to 12-wk-old animals were treated intraperitoneally with 4 mg/kg etanercept (Enbrel, Pfizer) starting 2 wk prior, or concomitantly to SARS-CoV-2 infection. Vehicle-treated mice received 4 mg/kg of isotype control antibodies (IgG1 Fc, Bio X Cell, BE0096). Treatment was performed at days 7, 5 3, 2, and 1 preinfection and then every second day after infection. Animals were monitored daily for weight change and euthanized upon reaching the humane endpoint (>20 % weight loss).
Measurement of Viral Loads via TCID50.
TCID50 was performed as previously described in ref. 38. Briefly, African green monkey kidney epithelial Vero cells, purchased from ATCC (clone CCL-81), were seeded in flat bottom 96-well plates (1.75 × 104 cells/well) and left to adhere overnight at 37 °C/5% CO2. Cells were washed twice with PBS and transferred to serum-free DMEM containing TPCK trypsin (0.5 µg/mL working concentration). Infected organs were defrosted, homogenized, and clarified by centrifugation at 10,000 g for 5 min at 4 °C, and the supernatant was added to the first row of cells at a ratio of 1:7, followed by 9 rounds of 1:7 serial dilutions in the other rows. Cells were incubated at 37 °C/5% CO2 for 4 d until virus-induced cytopathic effect was scored. TCID50 was calculated using the Spearman & Kärber algorithm as described in ref. 38.
Sequencing of Viral Passages.
For next-generation sequencing of viral samples, complementary DNA generation and amplification were performed according to the Midnight amplicon protocol (6.0, https://www.protocols.io/view/sars-cov2-genome-sequencing-protocol-1200bp-amplic-bwyppfvn), this representing a 1,200-bp tiled amplicon scheme. Amplicon material was prepared as barcoded sequencing libraries using rapid transposon-based library preparation (RBK-004) and sequenced using an R9.4.1 Flow Cell on a GridION (Oxford Nanopore Technologies Ltd.). Negative controls were introduced at the nucleic acid extraction stage and carried throughout. Sequence data were basecalled using guppy basecaller (5.0.16) and demultiplexed and adapter trimmed using guppy barcoder (5.0.11). Consensus sequences were generated for each sample using the artic network analysis pipeline, with comparison made between sequences and the Wuhan Hu-1 SARS-CoV-2 reference (NC_045512.2) through MUSCLE (3.8.425) alignment. Support for variant sites was assessed further through minimap2 (2.2.4) alignment to the above Wuhan Hu-1 reference genome.
Histological Analysis and Antigen Staining.
Organs were harvested and fixed in 4% paraformaldehyde for 24 h, followed by 70% ethanol dehydration, paraffin embedding, and sectioning. Slides were stained with either hematoxylin and eosin (H&E) or immunohistochemically with anti-CD3 (1:500, Agilent A045201), anti-MPO (1:1,000, Agilent A039829), anti-F4/80 (1:1,000, WEHI in-house antibody), or anti-SARS-CoV-2 nucleocapsid (1:4,000, Abcam ab271180) using the automated Omnis EnVision G2 template (Dako, Glostrup, Denmark). Dewaxing was performed with Clearify Clearing Agent (Dako) and antigen retrieval with EnVision FLEX TRS, High pH (Dako) at 97 °C for 30 min. Primary antibodies were diluted in EnVision Flex Antibody Diluent (Dako). and incubated at 32 °C for 60 min. HRP-labeled secondary antibodies were applied at 32 °C for 30 min. Slides were counterstained with Mayer Hematoxylin, dehydrated, cleared, and mounted with MM24 Mounting Medium (Surgipath-Leica, Buffalo Grove, IL, USA). Slides were scanned with an Aperio ScanScope AT slide scanner (Leica Microsystems, Wetzlar, Germany). Where indicated, a histopathological scoring system (0 to 2) was used by an American board-certified pathologist (Smitha Rose Georgy) to grade histological changes based on H&E staining. The score was based on the average of the following parameters (39): inflammatory cells in the alveolar space (0 = none, 1 = 1 to 5 cells, and 2 = >5 cells), inflammatory cells in the interstitial space/septae (0 = none, 1 = 1 to 5 cells, and 2 = >5 cells), presence of hyaline membranes (0 = none, 1 = 1 membrane, and 2 = > 1 membrane), proteinaceous debris in air space (0 = none, 1 = 1 instance, and 2 = > 1 instance), and alveolar septal thickening (0 > 2× mock thickness, 1 = 2 to 4× mock thickness, and 2 = > 4× mock thickness).
Quantification of Immunohistochemistry Images.
TIF images were exported from CaseViewer for analysis. Analysis was performed by custom python pipeline. Briefly, cells were detected using a pretrained StarDist network (40) and scored based on signal in the 3,3′-Diaminobenzidine (DAB) channel after color deconvolution (41). Images were split into tiles for memory management purposes and processed on a local high-performance computer.
Lung Cytokine and Chemokine Analysis.
Lungs were thawed, homogenized, and clarified by centrifugation at 10,000 g for 5 min at 4 °C. Supernatants were pretreated for 20 min with 1% Triton-X-100 for viral deactivation, and the Cytokine & Chemokine 26-Plex Mouse ProcartaPlex Panel 1 (EPX260-26088-901) was used as described in the manufacturer’s manual. Twenty-five microliters of clarified lung samples were diluted with 25 µL universal assay buffer, incubated with magnetic capture beads, washed, and incubated with detection antibodies and streptavidin. Cytokines were recorded on a Luminex 200 Analyzer (Luminex) and quantitated via comparison to a standard curve. For analysis of “cytokine storm”, 26 chemokines/cytokines from each animal were summed and compared between relevant groups.
RNA-seq.
Mice were infected, and the left lung lobe was collected for TCID50 assay, while the right lobe was stored in RNAlater™ Stabilization Solution (CAT# AM7020; Thermo Fisher Scientific, Waltham, MA, USA). Lungs were homogenized using pellet pestles, and RNA extraction was performed using the Isolate II RNA mini kit according to the manufacturer’s instruction (Cat# BIO‐52072; Meridian Bioscience, Cincinnati, OH, USA).
An input of 100 ng of total RNA was prepared and indexed for Illumina sequencing using the TruSeq RNA sample Prep Kit (Cat# RS‐122‐2001; Illumina, San Diego, CA, USA) with RiboGlobin depletion as per the manufacturer’s instruction. Each library was quantified using the Agilent Tapestation (using RNA ScreenTape [Cat# 5067‐5576] on a 2200 TapeStation system (Cat# G2964AA; Agilent Technologies, Waldbrunn, Germany)). The indexed libraries were pooled and diluted to 1.5 pM for paired-end sequencing (2 × 81 cycles) on a NextSeq 500 instrument using the v2 150 cycle High Output kit (Illumina) as per the manufacturer’s instructions. The base calling and quality scoring were determined using Real-Time Analysis on board software v2.4.6, while the fastq file generation and demultiplexing utilized bcl2fastq conversion software v2.15.0.4.
Bioinformatic Analysis of RNA-seq.
Quality control analysis of RNA-seq fastq files was performed using FastQC (v0.11.9, https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Individual FastQC files were consolidated into a single report using MultiQC (v1.12, https://multiqc.info/, Dataset S3). A genome index was built using the Mus musculus GRCm39.dna.primary_assembly fasta file (http://ftp.ensembl.org/pub/release-106/fasta/mus_musculus/) using the Rsubread (42) (v4.2) buildindex function. Reads were aligned to the reference genome using the Rsubread align function, and gene counts were quantified from the resultant BAM files using Subread featureCounts (43) (v2.0.3) and the Mus musculus GRCm39.106 reference GTF file (http://ftp.ensembl.org/pub/release-106/gtf/mus_musculus/). The gene count matrix was analyzed in RStudio (44) (v1.4.1743-4, R 4.2.0), and samples were split into one of six experimental groups (Table 1).
Table 1.
RNA-seq sample groups analyzed and their respective sample numbers
| Group (infection/age) | No. of mice/samples |
|---|---|
| Mock-infected young | 4 |
| Mock-infected aged | 4 |
| P2-infected young | 6 |
| P2-infected aged | 6 |
| P21-infected young | 5 |
| P21-infected aged | 6 |
The count matrix was filtered using the edgeR (45) (v3.15) filterByExpr function and including the grouping variable to apply filtration relative to the smallest group size (n = 4). Gene expression distributions were normalized using the trimmed mean of M-values method implemented by the edgeR calcNormFactors function. Sample clustering was assessed by multidimensional scaling analysis (SI Appendix, Fig. S5A). Heteroscedasticity and sample-level variation were removed from the data by applying the limma (46) (v3.15) voomWithQualityWeights function (SI Appendix, Fig. S5 B and C). Linear models were fitted for each of the following group comparisons using the limma lmfit and contrasts.fit functions (Table 2).
Table 2.
Groups compared and fitted through linear models during RNA-seq analysis
| Groups compared | Rationale |
|---|---|
| P21-infected young vs. mock-infected young | To identify genes related to P21 infection in young mice |
| P21-infected aged vs. P21-infected young | To identify differences between P21 infection in young and aged mice |
| P21-infected young vs. P2-infected young | To identify differences between P2 and P21 infection in young mice |
Significance testing was performed using the limma treat function, after empirical Bayes moderation. Significantly differently expressed genes were defined as those with FDR-adjusted P values below 0.05 and an absolute log2 fold-change of greater than 1. Full tables of significantly DE genes are provided in Dataset S2. The Broad Institute’s Mouse Hallmark Gene Sets collection of signatures (https://bioinf.wehi.edu.au/software/MSigDB/) was used to perform gene set testing with the limma camera method. Finally, further pathway enrichment and gene regulatory network analyses were performing using IPA software (v1.20.04, QIAGEN).
Statistical Analysis.
Statistical analyses were performed using Prism v9.3.1 software (GraphPad Software, Inc.). Unpaired two-tailed t tests were used for normally distributed data for comparisons between two independent groups. Data that violated the assumption of normality were transformed by generating log10 prior to statistical analysis. Bars in figures represent either the mean (±SD) or median of normally or non-normally distributed datasets, respectively, and each symbol represents one mouse. Sample sizes (n) replicate numbers, and significance can be found in the figures and figure legends. For all statistical significance indications: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; and ns, not statistically significant (P > 0.05).
Statistical analysis of cytokine data consisted of the Wilcoxon rank-sum test between group medians, with Bonferroni adjustment for multiple comparisons. Boxplots in figures depict the median and interquartile ranges. Loess smoothing was applied to the infection time course data, with the shaded area indicating 95% CIs. For the RNA-seq data, statistical significance was defined based on an FDR-adjusted P value <0.05 and an absolute log2 fold-change > 1.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (PDF)
Acknowledgments
We thank J. Silke for the TNF-deficient mouse strain; Esther Bandala Sanchez for expert advice and help with ELISA experiments; and Stephanie Bound, Lauren Wilkins, and Thomas Kapitelli for excellent animal husbandry support. We would also like to thank The Walter and Eliza Hall histology and imaging departments for their services. We acknowledge the contribution and assistance of Melbourne Health through its Victorian Infectious Diseases Reference Laboratory at the Doherty Institute in providing our laboratory with isolated SARS-CoV-2 material. National Health and Medical Research Council Australia Investigator grant GNT1175011 (M. Pellegrini).
Author contributions
S.M.B., J.P.C., K.C.D., S.W., C.C.A., J.M., D.F.J.P., M. Doerflinger, and M. Pellegrini designed research; S.M.B., J.P.C., L.H., L.M., M. Dayton, C.C.A., J.M., and M. Doerflinger performed research; K.L.R., A.K.C., S.L.G., V.C., M. Pitt, L.C., R.P., M.T., and J.M. contributed new reagents/analytic tools; S.M.B., J.P.C., D.S., G.T., L.W., and S.R.G. analyzed data; S.W., K.L.R., and A.K.C. supervised work; and S.M.B., M. Doerflinger, and M. Pellegrini wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
Viral strains generated in this study are available upon signing of a Materials Transfer Agreement. All analyzed data have been included in this manuscript or in public repositories: RNA-seq data have been deposited at GEO (GSE217556) and are publicly available as of the date of publication. Genomic sequences of SARS-CoV-2 passages (P2 to 21), which were generated in this study, have been deposited to GenBank and are publicly available as of the date of publication. Accession numbers OP848479-98 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA995787). Figures containing full-resolution, original histological images reported in this paper have been deposited at Zenodo (DOI: 10.5281/zenodo.8002877) and are publicly available as of the date of publication. All original code has been deposited at GitHub and is publicly available as of the date of publication (47).
Supporting Information
References
- 1.Chen Y., et al. , Aging in COVID-19: Vulnerability, immunity and intervention. Ageing Res. Rev. 65, 101205 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gasmi A., et al. , Interrelations between COVID-19 and other disorders. Clin. Immunol. 224, 108651 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ejaz H., et al. , COVID-19 and comorbidities: Deleterious impact on infected patients. J. Infect Public Health 13, 1833–1839 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yanez N. D., Weiss N. S., Romand J. A., Treggiari M. M., COVID-19 mortality risk for older men and women. BMC Public Health 20, 1742 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guan W.-J., et al. , Clinical characteristics of Coronavirus disease 2019 in China. N. Engl. J. Med. 382, 1708–1720 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang C., et al. , Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet (London, England) 395, 497–506 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peng R., et al. , ZBP1 induces inflammatory signaling via RIPK3 and promotes SARS-CoV-2-induced cytokine expression. bioRxiv [Preprint] (2021). 10.1101/2021.10.01.462460 (Accessed 30 May 2022). [DOI]
- 8.Blanco-Melo D., et al. , Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181, 1036–1045.e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mehta P., et al. , COVID-19: Consider cytokine storm syndromes and immunosuppression. The Lancet 395, 1033–1034 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li S., et al. , Clinical and pathological investigation of patients with severe COVID-19. JCI Insight 5, e138070 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karki R., et al. , Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell 184, 149–168.e17 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu L., et al. , Single-cell sequencing of peripheral mononuclear cells reveals distinct immune response landscapes of COVID-19 and Influenza patients. Immunity 53, 685–696.e3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liao M., et al. , Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 26, 842–844 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Shah S., et al. , Elevated D-Dimer levels are associated with increased risk of mortality in Coronavirus Disease 2019: A systematic review and meta-analysis. Cardiol. Rev. 28, 295–302 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoffmann M., et al. , SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bestle D., et al. , TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance. 3, e202000786 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cantuti-Castelvetri L., et al. , Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 370, 856–860 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inoue Y., et al. , Clathrin-dependent entry of severe acute respiratory syndrome coronavirus into target cells expressing ACE2 with the cytoplasmic tail deleted. J. Virol. 81, 8722–8729 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bayati A., Kumar R., Francis V., McPherson P. S., SARS-CoV-2 infects cells after viral entry via clathrin-mediated endocytosis. J. Biol. Chem. 296, 100306 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morris G., et al. , The pathophysiology of SARS-CoV-2: A suggested model and therapeutic approach. Life Sci. 258, 118166–118166 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Belouzard S., Chu V. C., Whittaker G. R., Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc. Natl. Acad. Sci. U.S.A. 106, 5871–5876 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dinnon K. H., et al. , A mouse-adapted model of SARS-CoV-2 to test COVID-19 countermeasures. Nature 586, 560–566 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng J., et al. , COVID-19 treatments and pathogenesis including anosmia in K18-hACE2 mice. Nature 589, 603–607 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumari P., et al. , Neuroinvasion and encephalitis following intranasal inoculation of SARS-CoV-2 in K18-hACE2 Mice. Viruses 13, 132 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lane C. R., et al. , Genomics-informed responses in the elimination of COVID-19 in Victoria, Australia: An observational, genomic epidemiological study. The Lancet Public Health 6, e547–e556 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang H., Zhu Y., Niu Z., Zhou L., Sun Q., SARS-CoV-2 N501Y variants of concern and their potential transmission by mouse. Cell Death Differ. 28, 2840–2842 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gu H., et al. , Adaptation of SARS-CoV-2 in BALB/c mice for testing vaccine efficacy. Science 369, 1603–1607 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niu Z., et al. , N501Y mutation imparts cross-species transmission of SARS-CoV-2 to mice by enhancing receptor binding. Signal Transduct. Target. Ther. 6, 284 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Angelini M. M., Akhlaghpour M., Neuman B. W., Buchmeier M. J., Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio 4, e00524-13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hagemeijer M. C., et al. , Membrane rearrangements mediated by coronavirus nonstructural proteins 3 and 4. Virology 458–459, 125–135 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kokkotis G., et al. , Systematic review with meta-analysis: COVID-19 outcomes in patients receiving anti-TNF treatments. Aliment Pharmacol. Ther. 55, 154–167 (2022). [DOI] [PubMed] [Google Scholar]
- 32.Hachem H., et al. , Rapid and sustained decline in CXCL-10 (IP-10) annotates clinical outcomes following TNFα-antagonist therapy in hospitalized patients with severe and critical COVID-19 respiratory failure. J. Clin. Transl. Sci. 5, e146 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson P. C., Richards D., Tanner H. L., Feldmann M., Accumulating evidence suggests anti-TNF therapy needs to be given trial priority in COVID-19 treatment. Lancet Rheumatol. 2, e653–e655 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsuyama T., Kubli S. P., Yoshinaga S. K., Pfeffer K., Mak T. W., An aberrant STAT pathway is central to COVID-19. Cell Death Differ. 27, 3209–3225 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Del Valle D. M., et al. , An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 26, 1636–1643 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crackower M. A., et al. , Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 417, 822–828 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Körner H., et al. , Distinct roles for lymphotoxin-alpha and tumor necrosis factor in organogenesis and spatial organization of lymphoid tissue. Eur. J. Immunol. 27, 2600–2609 (1997). [DOI] [PubMed] [Google Scholar]
- 38.Hierholzer J. C., Killington R. A., Virus isolation and quantitation. Virol. Methods Manual 1996, 25–46 (2007), 10.1016/B978-012465330-6/50003-8. [DOI] [Google Scholar]
- 39.Matute-Bello G., et al. , An official American Thoracic Society workshop report: Features and measurements of experimental acute lung injury in animals. Am. J. Respir. Cell Mol. Biol. 44, 725–738 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmidt U., Weigert M., Broaddus C., Myers G., “Cell detection with star-convex polygons” in Medical Image Computing and Computer Assisted Intervention–MICCAI 2018. MICCAI 2018, A. Frangi, J. Schnabel, C. Davatzikos, C. Alberola-López, G. Fichtinger, Eds. (Springer, Cham, 2018), vol. 11071, pp. 265–273. [Google Scholar]
- 41.Ruifrok A. C., Johnston D. A., Quantification of histochemical staining by color deconvolution. Anal. Quant. Cytol. Histol. 23, 291–299 (2001). [PubMed] [Google Scholar]
- 42.Liao Y., Smyth G. K., Shi W., The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 47, e47 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 44.RStudio Team, RStudio: Integrated Development for R (Version 2023.3.1.446, RStudio, PBC, Boston, MA, 2022). [Google Scholar]
- 45.Robinson M. D., McCarthy D. J., Smyth G. K., edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ritchie M. E., et al. , limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whitehead D. L., HDAB counts, HPC. Github (2022). https://github.com/BioimageAnalysisCoreWEHI/HDAB_counts_HPC. Accessed 17 November 2022.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (PDF)
Data Availability Statement
Viral strains generated in this study are available upon signing of a Materials Transfer Agreement. All analyzed data have been included in this manuscript or in public repositories: RNA-seq data have been deposited at GEO (GSE217556) and are publicly available as of the date of publication. Genomic sequences of SARS-CoV-2 passages (P2 to 21), which were generated in this study, have been deposited to GenBank and are publicly available as of the date of publication. Accession numbers OP848479-98 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA995787). Figures containing full-resolution, original histological images reported in this paper have been deposited at Zenodo (DOI: 10.5281/zenodo.8002877) and are publicly available as of the date of publication. All original code has been deposited at GitHub and is publicly available as of the date of publication (47).