

Significance
Genetic variants that influence body fat distribution have been reported to impact the risk of type 2 diabetes. Loss-of-function variants in INHBE (activin E) are associated with reduced abdominal obesity and protection from metabolic disease in humans, but how activin E regulates energy metabolism is unclear. This study reveals that liver-derived activin E controls energy storage by suppressing adipose lipid mobilization. Conversely, loss of activin E in mice enhances adipose lipolysis and lowers fat mass. Mechanistically, activin E elicits adipose ACVR1C signaling to suppress the activity of PPARG, a key transcriptional regulator of adipocyte function. Our findings establish a mechanistic framework for a hepatokine that drives interorgan communication to determine fat storage and metabolic health.
Keywords: INHBE (activin E), ACVR1C, body fat distribution, lipolysis, diabetes
Abstract
Body fat distribution is a heritable risk factor for cardiovascular and metabolic disease. In humans, rare Inhibin beta E (INHBE, activin E) loss-of-function variants are associated with a lower waist-to-hip ratio and protection from type 2 diabetes. Hepatic fatty acid sensing promotes INHBE expression during fasting and in obese individuals, yet it is unclear how the hepatokine activin E governs body shape and energy metabolism. Here, we uncover activin E as a regulator of adipose energy storage. By suppressing β-agonist-induced lipolysis, activin E promotes fat accumulation and adipocyte hypertrophy and contributes to adipose dysfunction in mice. Mechanistically, we demonstrate that activin E elicits its effect on adipose tissue through ACVR1C, activating SMAD2/3 signaling and suppressing PPARG target genes. Conversely, loss of activin E or ACVR1C in mice increases fat utilization, lowers adiposity, and drives PPARG-regulated gene signatures indicative of healthy adipose function. Our studies identify activin E–ACVR1C as a metabolic rheostat promoting liver–adipose cross talk to restrain excessive fat breakdown and preserve fat mass during prolonged fasting, a mechanism that is maladaptive in obese individuals.
Obesity or excess of body fat results from an imbalance in the intake and expenditure of calories, conferring increased risk for cardiovascular disease, type 2 diabetes, osteoarthritis, and cancer (1–3). Individuals with a given level of body fat mass can differ in the amount and distribution of adipose tissue, which also has an impact on cardiometabolic disease risk. Epidemiologic and genetic studies examining the impact of the waist-to-hip circumference ratio (WHR), a surrogate for abdominal versus gluteofemoral adiposity, on cardiometabolic disease and mortality revealed that body fat distribution plays a major role in determining disease risk independently of total fat mass (4–8). A larger WHR or waist circumference is strongly associated with higher incidence of cardiovascular disease and type 2 diabetes, while a lower WHR is protective against these disorders, even in individuals with a high BMI (9, 10). Thus, efforts to discover genetic variants associated with beneficial fat distribution are of considerable interest, as they might help to uncover therapeutically amenable pathways for obesity-related disorders.
Large multiancestry exome sequencing efforts have been instrumental in identifying naturally occurring predicted loss-of-function (pLOF) alleles affecting body shape and metabolic disease risk. Recently uncovered INHBE rare pLOF variants are the first example of a liver-specific gene associated with a lower WHR (11, 12) and protection from type 2 diabetes (11), yet a higher BMI. INHBE encodes for the circulating protein activin E, whose expression is increased by fatty acid-sensor PPAR-alpha, e.g., in response to adipose-derived fatty acids (13). Accordingly, INHBE is up-regulated during fasting and permanently increased in obese individuals with insulin resistance or NASH (11, 14–16), in which elevated basal lipolysis or hepatic lipogenesis increases liver fatty acid levels. Activin E belongs to the TGFβ family (17) but unlike other activin/TGFβ ligands, activin E’s receptor is unknown. Besides, the mechanistic insights into activin E function are scarce, and the currently published findings are conflicting. Thus, Inhbe−/− mice were previously reported to be unremarkable with respect to body weight, adiposity, and glycemic control on chow diet (18, 19). At the same time, short-term treatment of obese and diabetic db/db mice with Inhbe siRNA was associated with lower fat mass and enhanced lipid utilization (14). However, Inhbe overexpression (OE) (activin E transgenic mice) was reported to reduce fat mass as well and promoted energy expenditure (19). While both OE and knockdown studies point to white adipose tissue (WAT) as a potential target organ for activin E, the comparable phenotypes obtained with gain- and loss-of-function approaches are difficult to reconcile and raise questions about the mechanistic basis for the observed effects. Thus, it is unclear whether liver-derived activin E acts directly on adipose tissue, how it impacts fat distribution, and whether timing and levels of activin E exposure might differentially affect lipid homeostasis.
Human genetic analyses provided some hints to unravel these questions. pLOF variants in ACVR1C, an activin receptor enriched in WAT, have similarly strong associations with fat distribution as INHBE (11, 12, 20). ACVR1C enables canonical activin-SMAD2/3 signaling, resulting in SMAD-mediated transcriptional suppression of Pparg and Cebpa, and subsequent blockade of adipose lipolysis (21–23). While several activin/TGFβ ligands are known to engage ACVR1C, the phenotypes of mice lacking these ligands do not support their roles in modulating fat distribution through ACVR1C (18, 24–27). Thus, the human genetic associations led us to hypothesize that liver-secreted activin E may represent a bona fide ACVR1C ligand, allowing for biological communication between the liver and adipose tissue. If and how such an interorgan cross talk governs fat distribution is not known, but in vivo analyses of activin E signaling may elucidate the mechanisms that promote healthy fat storage.
Here, by employing gain- and loss-of-function studies, we uncover activin E as a regulator of energy storage in adipose tissue. Specifically, we demonstrate that hepatokine activin E promotes adipose SMAD2/3 signaling and suppresses metabolic genes involved in energy homeostasis. This, in turn, reduces lipid mobilization, promotes inflammation, and contributes to adipocyte hypertrophy. Importantly, these observed effects were dependent on ACVR1C, providing an important link to the liver–adipose communication. Conversely, activin E deletion results in induction of PPARG, increased rates of adipose lipolysis, and improved adipocyte capacity for nutrient uptake. Thus, activin E blockade in humans has a potential to improve adipose energy metabolism and might represent an attractive antidiabetes strategy. Given the targeted impact on adipose, activin E blockade is expected to have phenotypes similar to PPARG agonists but is unlikely to be accompanied by the adverse effects resulting from systemic PPARG activation with thiazolidinedione treatment (28). In conclusion, our findings shed light on two key players of liver–adipose cross talk that define fat distribution and provide a mechanistic link between human genetic associations for INHBE pLOF variants and protection from metabolic disease.
Results
Activin E Suppresses Lipolysis and Increases Adiposity in Mice.
Recent studies suggest that Inhbe (activin E) expression is increased in humans with NASH and insulin resistance (11, 14), as well as in obese mice upon high fat feeding (SI Appendix, Fig. S1A). To test the consequences of elevated activin E, we overexpressed Inhbe in lean adult wild-type (WT) mice by administering AAV8-CAG-mInhbe or AAV8-CAG-eGFP (control). Inhbe mRNA levels in the liver were ~sixfold higher than in control mice 7 wk post-AAV administration, confirming successful OE (Fig. 1A). Activin E OE had little effect on overall body weight or composition (Fig. 1B and SI Appendix, Fig. S1 B and C). However, it led to a significant increase in visceral (epididymal) WAT mass but had no effects on subcutaneous (inguinal) WAT mass (Fig. 1C). Hematoxylin and eosin (H&E) staining and adipocyte size quantification revealed an increase in large adipocytes in epididymal WAT of activin E overexpressing mice (Fig. 1D), with more subtle effects on subcutaneous WAT (SI Appendix, Fig. S1D). To understand the cause of adipose hypertrophy, we performed ex vivo lipolysis assays in which WAT explants from activin E overexpressing mice were stimulated with isoproterenol and glycerol was determined enzymatically in the supernatant. Since activin E is not present in culture media during these measurements, the assay evaluates whether in vivo exposure to elevated activin E had altered the ability of WAT to respond to β-agonists. Indeed, we found that activin E OE led to suppression of stimulated glycerol release from epididymal (Fig. 1E) but not subcutaneous WAT (SI Appendix, Fig. S1E). Plasma nonesterified fatty acids (NEFAs), typically derived from adipose lipolysis, were also lower in activin E overexpressing mice (Fig. 1F). We obtained similar results in hyperlipidemic db/db mice, which upon OE of activin E displayed reduced epididymal adipose lipolysis and lower circulating NEFA levels, while the effect on subcutaneous WAT was again more subtle (SI Appendix, Fig. S1 F–H). Furthermore, activin E OE was associated with reduced expression of lipolytic genes: Beta-adrenergic receptor Adrb3, the lipases Atgl/Pnpla2, Cgi-58, and Hsl, as well as fatty acid transporter Fabp4 were significantly suppressed in epididymal WAT (Fig. 1G). Again, subtle changes were seen in subcutaneous WAT (SI Appendix, Fig. S1I). Thus, our data suggested that activin E suppresses lipolysis in visceral WAT of mice.
Fig. 1.

Activin E lowers fat mobilization and increases adiposity in mice. (A) Liver mRNA levels of Inhbe (n = 10). (B) Body weights in 7- to 14-wk-old male Control AAV and Inhbe AAV-treated WT mice (n = 10). (C) Weights of visceral (epididymal) and subcutaneous (inguinal) white adipose tissue (n = 10). (D) Adipocyte morphology and size distribution measured by imaging software in H&E-stained epididymal WAT sections (n = 534,046 cells for Control AAV and 525,314 cells for Inhbe AAV analyzed from 10 mice/group). (E) Ex vivo lipolysis with epididymal WAT explants from WT mice under basal and isoproterenol-stimulated conditions. Fat mobilization is determined enzymatically by glycerol measurements in supernatant (n = 5). (F) Plasma NEFA levels (n = 10). (G) Epididymal WAT mRNA levels of genes involved in adipose lipolysis, relative to Control AAV (n = 10). Mean ± SEM are shown in all graphs besides (D), where mean is shown. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 relative to Control AAV.
Activin E Lowers Liver Lipid Content and Improves Glycemic Control in Mice.
During fasting, adipose lipolysis provides NEFAs to liver and other peripheral tissues to support energy production (29). Thus, we evaluated functional consequences of activin E–driven suppression of lipolysis on liver homeostasis. We found that activin E OE reduced liver mass during fasting, which was associated with lower hepatic triglyceride content (Fig. 2 A and B), suggesting that activin E–driven suppression of adipose lipolysis deprives hepatocytes of NEFAs during fasting. We also measured liver weight and lipid content of activin E–overexpressing mice in the postprandial state when adipose lipolysis is largely inactive. In this scenario, liver mass and triglyceride levels were similar to those of control animals (Fig. 2 C and D). These findings rule out a general defect in liver fatty acid uptake but rather point to the critical role for activin E in regulating fasting-induced lipolysis and lipid distribution.
Fig. 2.

Activin E regulates glycemic control and liver lipid content in mice. (A–D) Liver weights and lipid content. Fasted liver weight (A) and hepatic triglyceride content (B) in Control AAV or Inhbe AAV-treated mice on HFD (n = 9 to 10). Nonfasted liver weight (C) and hepatic triglyceride content (D) in Control AAV or Inhbe AAV-treated mice on HFD (n = 9 to 10). (E) Fasting plasma insulin levels before (baseline) and 3 wk after Inhbe AAV administration on chow (n = 9). (F and G) Glycemic control in Inhbe overexpressing mice. Oral glucose tolerance (F) and insulin tolerance tests (G) in mice administered with Control AAV or Inhbe AAV and fed chow or high-fat diets (n = 10). Mean ± SEM are shown in all graphs. **P < 0.01, ***P < 0.001.
Hepatic lipid content is associated with systemic insulin sensitivity (30). Besides WHR, INHBE pLOF in humans is associated with lower odds of type 2 diabetes (11, 12) prompting us to evaluate how changes in activin E levels would affect glycemic control in mice. We found that plasma insulin levels were reduced in activin E–overexpressing mice (Fig. 2E). While activin E had no effect on glucose tolerance, it led to improved insulin tolerance on both chow- and HFD-fed mice (Fig. 2 F and G). Thus, OE of activin E in adult mice produced remarkably beneficial effects on glycemic control, consistent with the data from activin E transgenic mice (19) but suggesting a major discrepancy between humans and mice: With regard to diabetes, INHBE loss-of-function is protective in humans (11, 12), while mice appear to benefit from elevated Inhbe expression.
Loss of Activin E Promotes Fat Mobilization.
Since human INHBE pLOF variants are also associated with lower WHR, we sought to evaluate how loss of activin E would affect fat distribution in mice. Activin E–deficient mice (Inhbe−/− or KO) were previously reported to be phenotypically unremarkable (18), besides minor changes in gene expression in BAT (19). However, these mice have not undergone extensive metabolic phenotyping, nor have they been challenged with high-fat diet (HFD). We therefore generated Inhbe−/− mice by deleting the entire coding sequence (Materials and Methods). Hepatic Inhbe mRNA levels were undetectable in KO mice, confirming the gene knockout (SI Appendix, Fig. S2A). Consistent with prior reports, Inhbe−/− mice appeared normal on chow diet. Body weight, composition, and size of adipose depots were similar to WT animals (SI Appendix, Fig. S2 B–E). However, chow-fed Inhbe−/− mice showed a subtle yet significant increase in fasting plasma NEFA levels (SI Appendix, Fig. S2F).
Since Inhbe expression is increased upon high fat feeding (SI Appendix, Fig. S1A), we hypothesized that loss of activin E might reveal a more pronounced phenotype upon HFD challenge. Indeed, Inhbe−/− mice gained significantly less weight during 16 wk of HFD, compared to their WT counterparts (Fig. 3A). While lean mass was comparable between genotypes, Inhbe−/− mice accrued less fat mass on HFD (SI Appendix, Fig. S3 A and B). Both epididymal and subcutaneous WAT were significantly smaller compared to WT (Fig. 3 B and C). Ex vivo lipolysis assays demonstrated that loss of activin E was associated with increased glycerol release from WAT explants, suggesting accelerated rates of lipolysis (Fig. 3D and SI Appendix, Fig. S3C), which was also reflected by the prominent increase in fasting plasma NEFAs and beta-hydroxybutyrate in HFD-fed Inhbe−/− mice (Fig. 3 E and F). Moreover, Inhbe−/− mice had higher adipose expression of lipolytic genes (Fig. 3G and SI Appendix, Fig. S3D), and histological analysis showed a reduced proportion of large adipocytes in WAT (SI Appendix, Fig. S3 F and G), supporting increased fat mobilization from WAT with Inhbe deletion.
Fig. 3.

Loss of activin E promotes fat mobilization. (A) Body weights in 7- to 24-wk-old male WT and Inhbe−/− mice on chow and high-fat diet (HFD) (n = 7 to 8). (B and C) Weights of epididymal (B) and subcutaneous (C) white adipose tissue of Inhbe−/− mice following 16 wk of HFD (n = 8). (D) Ex vivo lipolysis with epididymal WAT explants from WT and Inhbe−/− mice under basal and isoproterenol-stimulated conditions (n = 10 to 11). (E and F) Fasted plasma NEFA (E) and beta-hydroxybutyrate (F) levels before (chow) and after 10 or 16 wk of HFD (n = 8). (G) Epididymal WAT mRNA levels of genes involved in adipose lipolysis (n = 6 to 10). (H and I) Fasted liver weight (H) and hepatic triglyceride content (I) in WT and Inhbe−/− mice on HFD (n = 8). (J) Oral glucose tolerance and insulin tolerance tests (K) in WT and Inhbe−/− mice on chow and high-fat diets (n = 7 to 8). (L) Plasma insulin levels before (baseline chow) and 16 wk after HFD (n = 8). (M) Plasma ALT levels in Inhbe−/− mice on chow and HFD for 16 wk. Mean ± SEM are shown in all graphs. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Consequently, fasted Inhbe−/− mice exhibited prominent hepatic fat accumulation and a significant increase in liver weights on both HFD and chow (Fig. 3 H and I and SI Appendix, Fig. S2 G and H, respectively). Thus, with respect to fat handling, Inhbe−/− mice displayed the phenotype opposite to activin E overexpressing mice, demonstrating that activin E has a major impact on fat storage and adipose lipid mobilization. We then assessed glycemic control. Although Inhbe−/− mice on chow behaved similarly to WT in response to oral glucose challenge, HFD-fed KO animals displayed striking glucose intolerance (Fig. 3J). Inhbe−/− mice on HFD were also severely insulin resistant (Fig. 3K). This was accompanied by large increases in fasting insulin in activin E–deficient mice (Fig. 3L and SI Appendix, Fig. S2I). Taken together, the insulin resistance seen in Inhbe−/− mice appears to be—at least in part—due to fat redistribution from adipose to the liver that caused subsequent liver damage, as assessed by liver enzyme elevation (Fig. 3M and SI Appendix, Fig. S3E).
Activin E Signals via ACVR1C to Suppress PPARG in Adipose Tissue.
Our phenotypic characterizations suggested that activin E has a profound impact on adipose tissue and led us to hypothesize that these effects may be driven by activin receptor signaling. In this regard, we were intrigued by recent human genetics analyses suggesting similar associations for INHBE and activin receptor ACVR1C pLOF variants with favorable fat distribution and protection from metabolic disease (11, 12). To determine whether loss of ACVR1C signaling mirrors Inhbe−/− phenotypes, we administered anti-ACVR1C neutralizing antibody (mAb) to adult mice upon HFD challenge. While the timeframe of these studies was limited due to development of anti-human antibodies beyond 4 wk of treatment, the results of short-term ACVR1C blockade were remarkably similar to Inhbe LOF. ACVR1C mAb-treated mice on HFD showed a trend toward reduced body weight (Fig. 4A). Epididymal WAT weight was greatly reduced, while subcutaneous fat mass trended lower (Fig. 4 B and C). Reduced fat mass was associated with increased glycerol release and increased expression of lipolytic genes (SI Appendix, Fig. S4 A–D). Additionally, plasma NEFAs and ketones were significantly higher in ACVR1C mAb-treated mice, which led to increased liver mass, hepatic fat, and ALT levels (Fig. 4D and SI Appendix, Fig. S4 E–H). ACVR1C inhibition was also associated with a rapid increase in insulin levels (Fig. 4E). We then generated Acvr1c−/− mice to allow for longer-term studies and assessed glycemic control. Acvr1c-deficient mice exhibit insulin resistance on HFD, as previously noted (Fig. 4F) (24). Thus, ACVR1C inhibition functionally phenocopied Inhbe−/− mice with respect to adipose lipid mobilization, liver fat content, and glycemic control.
Fig. 4.

Activin E signals via ACVR1C to suppress PPARG in adipose tissue. (A) Body weight increase in 7- to 11-wk-old male WT mice treated with control monoclonal antibody (mAb) or neutralizing ACVR1C mAb while on HFD (n = 8 to 9). (B–D) Fasted weights of epididymal (B) and subcutaneous (C) white adipose tissue and liver (D) (n = 8 to 9). (E) Plasma insulin levels before (baseline) and 2 or 4 wk after Control mAb or ACVR1C neutralizing mAb administration to WT mice (n = 8 to 9). (F) Insulin tolerance test in WT and Acvr1c−/− mice on high-fat diet (n = 10). (G to I) Fasted weights of epididymal (G) and subcutaneous (H) white adipose tissue and liver (I) 6 wk following Control AAV or Inhbe AAV administration to WT or Acvr1c−/− mice on chow diet (n = 11). (J and K) Epididymal WAT mRNA levels of activin target genes Pmepa1 and Serpine1 (J), and adipose transcription factor Pparg (K), relative to Control AAV in WT and Acvr1c−/− mice (n = 10 to 11). Mean ± SEM are shown in all graphs. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To test whether ACVR1C is a putative activin E receptor, we overexpressed activin E by AAV8-CAG-mInhbe delivery to Acvr1c−/− or WT mice using AAV8-CAG-eGFP as a control. We then examined the consequences of activin E over-expression to fat and liver mass on chow diet (Fig. 4 G–I). In WT mice, activin E OE led to increased epididymal WAT and subtle effects on subcutaneous WAT mass, as seen before (Fig. 4 G and H). Importantly, this effect was abrogated in Acvr1c−/− mice, suggesting that ACVR1C facilitates activin E’s impact on adipose tissue (Fig. 4 G and H). This was also reflected on the transcriptional level: Activin E increased epididymal adipose expression of activin target genes Pmepa1, Serpine1, and Fstl3 in WT mice, but had no effect in Acvr1c−/− mice (Fig. 4J and SI Appendix, Fig. S4I). Activin E also suppressed expression of adipose transcription factors Pparg and Cebpa in WT, but not Acvr1c−/− mice (Fig. 4K and SI Appendix, Fig. S4J). Livers were significantly smaller upon activin E OE, but this effect again depended on ACVR1C, as the receptor KO mice showed unchanged liver mass (Fig. 4I). Thus, our data underscore the critical role of activin E–ACVR1C signaling in the regulation of fat storage and distribution.
Activin E Promotes a Gene Signature Related to Adipose Dysfunction.
To gain detailed molecular insights into activin E’s role in adipose energy metabolism, we performed transcriptome analysis on epididymal and subcutaneous WAT from activin E–deficient or overexpressing mice. Comparative analysis of differentially expressed genes revealed numerous adipose genes which were perturbed in response to altered activin E levels (Fig. 5A and SI Appendix, Fig. S5A). Notably, many genes (131 in epididymal WAT and 194 in subcutaneous WAT) were regulated in the opposite direction by Inhbe OE vs. Inhbe deletion (KO) (Dataset S1), confirming their relevance to activin E action. Among the Inhbe-responsive genes obtained by overlapping chow Inhbe−/− and OE signatures, we see similar but amplified gene changes in the HFD-fed Inhbe−/− mice (Fig. 5A and SI Appendix, Fig. S5A).
Fig. 5.
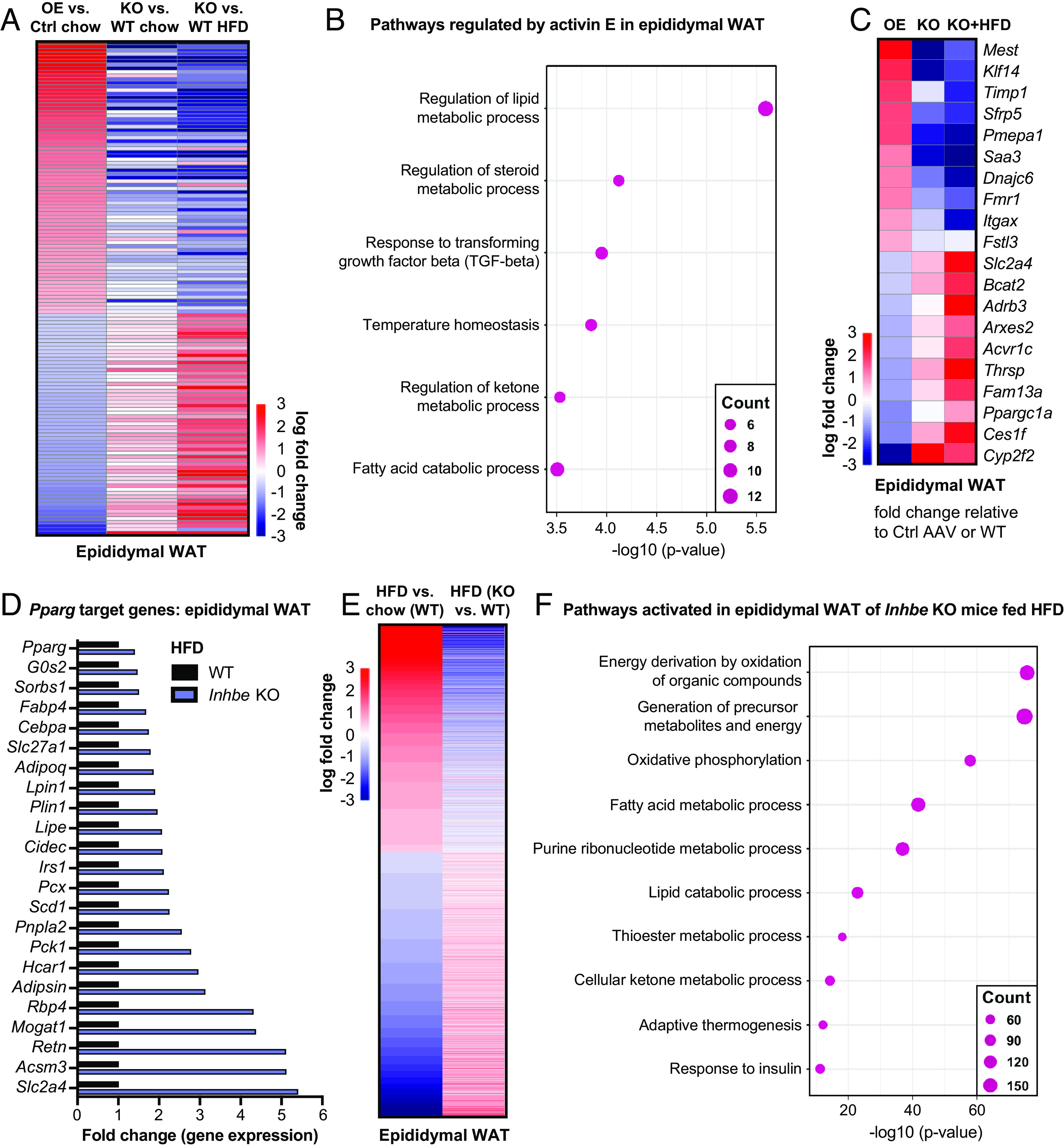
Activin E promotes a gene signature related to adipose dysfunction. (A) Heat map of epididymal adipose transcriptome signatures comparing Inhbe overexpression, Inhbe KO on chow and high-fat diet (HFD) vs. control-treated or WT mice, respectively. (B) Dot plots showing top biological pathways for oppositely regulated genes (Inhbe overexpression vs. KO) in epididymal WAT. (C) Heatmap showing examples of oppositely regulated genes (Inhbe overexpression vs. KO) in epididymal WAT. (D) Relative gene expression levels of PPARG target genes in epididymal WAT, comparing Inhbe KO and WT mice on high-fat diet. (E) Heatmap of epididymal adipose transcriptome signatures comparing WT chow vs. HFD, and Inhbe KO vs. WT on high-fat diet (HFD), demonstrating that Inhbe LOF reverses many of the HFD effects seen in WT mice. (F) Dot plots showing top biological pathways for genes increased in epididymal WAT of Inhbe KO vs. WT on HFD.
Closer inspection of Inhbe-responsive genes revealed activin E’s prominent role in regulating adipose energy metabolism (Fig. 5B and SI Appendix, Fig. S5B). Genes controlled by activin E were involved in lipid- (e.g., Ppargc1a, Adrb3, Ces1f, Thrsp), glucose- (e.g., Slc2a4/Glut4) and amino acid- (e.g., Bcat2) metabolism (Fig. 5 B and C, SI Appendix, Fig. S5 B and C, and Dataset S2). Notably, activin E OE suppressed catabolic pathways, while its loss resulted in stimulation of these processes. Besides metabolic genes, elevated activin E induced markers of adipocyte hypertrophy (e.g., Mest, Sfrp5) (31, 32) and inflammation (e.g., Itgax, Saa3) (Fig. 5C, SI Appendix, Fig. S5 B–D, and Dataset S1). Activation of the innate immune response was particularly evident in subcutaneous WAT, where genes including Ccl2, Arg2, and Tlr7 were prominently up-regulated by activin E OE (SI Appendix, Fig. S5D). These effects appeared to be a consequence of altered activin/TGFβ signaling, as activin E OE and KO correlated with increased or decreased expression of activin-SMAD2/3 target genes Fstl3 and Pmepa1, respectively (Fig. 5 B and C and SI Appendix, Fig. S5C) (33, 34). Taken together, the transcriptome studies corroborated our findings that activin E regulates fat storage and provide a mechanistic basis for how elevated activin E contributes to adipose dysfunction.
Conversely, loss of activin E had protective effects on adipose tissue. The preponderance of metabolic pathways among Inhbe-responsive genes led us to assess the impact of Inhbe ablation on adipose transcription factor PPARG, which was suppressed by activin E OE (Fig. 4K). PPARG is a master regulator required for adipocyte differentiation, regulation of insulin sensitivity, lipogenesis, and adipocyte survival and function (28). Obesity and high fat feeding are associated with reduced PPARG transcriptional activity (28), as was also evident in our studies in WT mice (SI Appendix, Fig. S5E). Interestingly, employing the same high-fat feeding conditions, Inhbe−/− mice exhibited rescue of adipose PPARG target gene expression. This included genes related to lipolysis, fatty acid uptake and storage, adipokines, and insulin sensitization (Fig. 5D and SI Appendix, Fig. S5F). This finding led us to examine the impact of Inhbe LOF on adipose gene expression profiles more broadly. By comparing HFD gene signatures in WT mice with gene signatures from Inhbe−/− mice, we found that loss of activin E reversed many of the detrimental changes induced by high fat feeding (Fig. 5E and SI Appendix, Fig. S5G). Genes associated with inflammation, tissue remodeling, and angiogenesis were increased by HFD in WT but remained low in WAT of Inhbe−/− mice (Dataset S2). Instead, Inhbe−/− mice had increased expression of genes linked to lipid catabolism, oxidative phosphorylation, and insulin responsiveness (Fig. 5F and Dataset S2). The potent increase in oxidative pathways provided an explanation for the reduced adipose tissue mass in Inhbe−/− mice, despite activation of PPARG which typically drives fat expansion. Thus, in mice, high rates of energy expenditure may favor fat loss over accrual, which appears to be a key difference from human individuals with INHBE pLOF exhibiting higher BMI.
In conclusion, our transcriptomics data are highly consistent with the observed effects of activin E on adipose fat storage and utilization and suggest that activin E may exert its function on adipose, at least in part, through regulation of PPARG and its target genes.
Discussion
Exome-wide analyses of the BMI-adjusted WHR, a surrogate for abdominal fat, have previously uncovered INHBE (activin E) as a genetic determinant of body fat distribution (11, 12). Rare INHBE pLOF variants are associated with a lower WHR and protection from metabolic disease, yet how liver-derived activin E alters adiposity and energy homeostasis had remained unclear. In the current study, we demonstrate that activin E controls fat storage in WAT by suppressing lipolysis in mice. By employing OE and knockout studies, we found that adipocytes are keenly sensitive to activin E levels. Elevated activin E promotes SMAD2/3 signaling and results in suppression of lipolytic and glucose metabolic genes, while loss of activin E enhances this set of genes. Hence, Inhbe−/− mice exhibit reduced fat mass and elevated circulating NEFA levels, reflecting increased lipid mobilization from adipose. Interestingly, hepatic Inhbe expression itself is up-regulated in response to adipose-derived fatty acids via PPAR-alpha (13). This suggests an autoregulatory loop wherein adipose lipolysis promotes hepatic activin E production, which in turn suppresses adipose lipid mobilization to prevent excessive fat breakdown. Such a feedback mechanism may have evolved to preserve fat mass during periods of prolonged fasting. Thus, the hepatokine activin E emerges as a key regulator of energy metabolism, facilitating biological interactions between the liver, a critical organ for energy sensing, and adipose tissue, the body’s specialized energy storage system.
Probing mechanism, we revealed that activin E’s function is mediated by ACVR1C in adipose, for which human pLOF variants are similarly associated with beneficial fat distribution. Prior studies have shed light on the role of ACVR1C: Genetic or pharmacological inhibition in mice results in profound fat loss via lipolysis, yet causes hepatic steatosis and severe insulin resistance (21, 22, 24, 35, 36). It has long been unclear which activin ligands are largely responsible for ACVR1C signaling. While various TGFβ family ligands can engage ACVR1C in vitro, lack of individual ligands produces different outcomes in vivo. Although loss of activin B phenocopied the hyperinsulinemia of Acvr1c−/− mice, Inhbb−/− mutants show no signs of insulin resistance or hepatic steatosis and have no changes in fat mass (24, 25). In contrast, Gdf3−/− mice are resistant to diet-induced obesity and display normal insulinemia and glucose control (26). The role of activin C is poorly defined, but Inhbc−/− mice appear phenotypically normal (18, 27). While partial redundancy or tissue-specific signaling could explain these ligand-selective effects, it was remarkable that loss of activin E alone fully phenocopied the Acvr1c-null phenotype. Importantly, activin E’s effects appeared to depend on ACVR1C, as Inhbe OE in receptor null mice lacks the phenotype observed with Inhbe OE in WT mice. Future biochemical studies will shed more light on the intricate details of activin E receptor binding, affinity, and structural interactions.
Furthermore, we demonstrated in vivo that activin E leads to suppression of genes governing adipose energy metabolism. Thus, activin E OE in mice was associated with impaired lipolysis, adipocyte hypertrophy, and inflammation. We conclude that elevated activin E contributes to adipose dysfunction. This could be particularly relevant for patients with NASH and type II diabetes, where hepatic Inhbe expression is increased (11, 14). Conversely, loss of murine Inhbe increased adipose energy metabolism, promoted fat mobilization, and generated smaller, insulin-sensitive adipocytes. Importantly, our studies revealed that Inhbe LOF led to activation of adipocyte master regulator PPARG, which controls adipose health and metabolism (28). By placing PPARG under negative control of activin E signaling, the liver exerts potent influence on adipose function.
The potential therapeutic implications of this pathway are intriguing. PPARG agonists (thiazolidinediones, TZD) are potent antidiabetic agents which act by promoting adipogenesis and improving adipose storage capacity. Yet, the clinical benefits of TZDs are overshadowed by PPARG activation in nonadipose tissues, posing a risk for cardiovascular events, edema, skeletal fractures, and bladder cancer (28). Activin E inhibition may be a safer alternative to TZDs due to its more targeted impact on adipose tissue. Given the preferential expression of ACVR1C in WAT, activin E blockade might lead to endogenous PPARG activation specifically in adipose but is unlikely to be accompanied by the adverse effects resulting from systemic PPARG activation with TZD treatment. Hence, therapeutic inhibition of activin E may promote healthy adiposity and improve glycemic control via adipose-specific PPARG agonism. That said, our data revealed a fundamental difference between species for this pathway. While human INHBE and ACVR1C pLOF carriers indeed have lower odds of developing diabetes (11, 12), loss of Inhbe and Acvr1c in mice led to strongly impaired glucose homeostasis. Such phenotypic inconsistencies are not without precedence. Blockade of activin receptor type IIB (ActRIIB) in mice similarly causes rapid glucose intolerance, while it improves glycemic control in humans (37–39). Beyond activin signaling, Inhbe genetics is reminiscent of Pde3b, which blocks adipose lipolysis downstream of the insulin receptor. Pde3b-deficient mice have lower fat mass due to enhanced lipolysis but exhibit insulin resistance and hepatic steatosis, akin to Inhbe KOs (40). Yet, human PDE3B pLOF is associated with beneficial fat distribution and protection from type 2 diabetes (11, 12). Similar discrepancies are seen for PLIN1 pLOF in humans and mice (11, 41, 42). Disparities have also been noted in the opposite direction: The PPARG P467L mutation has detrimental effects on glycemic control in humans but not in mice (43), and murine models of partial lipodystrophy do not seem to closely mimic the human disorders (44).
These discrepancies may relate in part to a) the interspecies differences in adipose patterning, as mouse fat depots do not precisely mirror those of humans, b) the apparent lack of sexual dimorphism in fat mass and distribution in mice, or c) differences in energy expenditure and fat utilization between species. The latter may be particularly relevant for the phenotypes seen with Inhbe LOF. Given that relative basal metabolic rates are considerably higher in rodents than humans (45), derepression of lipolysis may have more potent effects on adipose mass reduction in mice. Inhbe deletion in mice results in net fat loss and body weight reduction, whereas humans with heterozygous INHBE pLOF display higher BMI and enlarged gluteofemoral (subcutaneous) adipose tissue (11), similar to the effects of PPARG agonists (28). While PPARG controls genes related to both fasting-induced lipid mobilization and postprandial nutrient uptake and storage (46–48), we hypothesize that a key difference between humans and mice may be the relative balance between these opposing processes. In mice, loss of activin E appears to favor high rates of lipolysis, but further studies will be required to better assess the human INHBE pLOF phenotypes. Whether lipolysis plays a significant role in the regulation of human body fat distribution is currently unclear, but the enrichment of lipolysis-related genes in pLOF variants associated with WHR suggests a potential role for this pathway (11, 49).
Taken together, activin E ablation had remarkably protective effects on adipose, particularly during conditions of excess caloric intake. While species differences raise questions for further exploration, our data in mice provide a mechanistic basis for the beneficial human genetic associations and suggest that loss of activin E promotes healthy adiposity.
Materials and Methods
Mouse Models and Procedures.
C57BL/6NTac mice were from Taconic. Db/db mice were from Jackson Laboratories (#000697). The genetically engineered Inhbe−/− (MAID 6634) and Acvr1c−/− (MAID 981) mouse strains were created using Regeneron’s VelociGene® technology (50, 51). Briefly, C57BL/6NTac embryonic stem cells were targeted for ablation of the entire Inhbe or Acvr1c loci, beginning immediately after the endogenous ATG and ending at the respective stop codons. Ablation was achieved using a modified bacterial artificial chromosome (BAC) targeting construct such that BAC Inhbe or Acvr1c sequences were replaced with a self-deleting, floxed lacZ reporter cassette containing a neomycin resistance gene under the control of the human UBC (ubiquitin) promoter. The lacZ reporter was inserted inframe immediately after the endogenous ATG. This construct was electroporated into C57BL/6NTac embryonic stem cells. Following selection with neomycin, correctly targeted clones were identified by TaqMan analysis and microinjected into eight-cell Swiss Webster embryos (Charles River Laboratories), resulting in F0 VelociMouse® fully derived from the injected modified embryonic stem cells (51).
Heterozygous Inhbe+/−/Acvr1c+/− mice were bred to generate age-matched wild-type Inhbe+/+/Acvr1c+/+ and knock-out Inhbe−/−/Acvr1c−/− littermates that were used for experimentation. Male mice were housed on a 12-h light/dark cycle at 22 ± 1 °C, in static cages (≤5/cage) with free access to food and water and fed either control chow diet (PicoLab Rodent Diet 20, LabDiet 5053) or a HFD (Research Diets D12492i) for up to 16 wk. In OE studies, AAV8 particles were administered through intravenous injection at a dose of 2.5 × 10E11 vg/mouse.
All animals were monitored for changes in body weight on a weekly basis. Body mass composition was assessed in awake mice using echoMRI. No adverse reactions or signs of discomfort were observed during the course of the experiment. Mouse experiments were performed on age-matched and strain-matched pairs (littermates). All animal procedures were conducted in compliance with protocols approved by the Regeneron Pharmaceuticals Institutional Animal Care and Use Committee.
Antibody Administration Studies.
The amino acid sequence of anti-ACVR1C monoclonal antibody is identical to “ALK7 mAb J02” as set forth in US Pat. No. 2017/0306028A1. The antibody sequence was cloned, and the antibody was produced and purified at Regeneron for research purposes. Four-hour fasted baseline serum chemistry was established on chow diet, and mice were sorted into treatment groups based on body weight and lipid levels. Mice were then injected with anti-ACVR1C monoclonal antibody or isotype control antibody (10 mg/kg s.c.) once a week for 4 wk and placed on HFD (Research Diets, D12492i; 60% fat by calories). Body weights were measured weekly. Mice were killed 1 wk after the last injection, and WAT and livers were collected and frozen for subsequent lipid content and/or enzymatic measurements or collected in RNAlater (ThermoFisher) for gene expression studies.
Glycemic Control.
To assess glycemic control, oral glucose tolerance tests (oGTT) and insulin tolerance tests (ITT) were performed. For oGTTs, mice were fasted overnight, after which glucose (2 g/kg) was administered to each mouse by oral gavage. The tip of the tail of each mouse was scratched to draw blood. Blood samples were collected at 0, 30, 60, 90, and 120 min, and glucose was measured using the Accu-check blood glucose monitoring system (Roche). For ITT, mice were fasting were 4 h, followed by insulin administration via intraperitoneal injection (HumulinR; Lilly at 0.75U/kg BW). Blood sample collection and time points were similar to oGTTs. For insulin measurements, mice were fasted for 4 h, and blood was collected in capillary tubes containing protease inhibitors. Blood was centrifuged at 10,000 rpm for 10 min to separate the plasma. The mouse insulin ELISA kit (Mercodia, 10-1247-01) or ultrasensitive mouse insulin ELISA kit (Mercodia, 10-1249-01) was used to measure insulin levels.
Liver and Lipid Analysis.
Blood was collected in EDTA tubes, and plasma was obtained by centrifugation at 10,000 rpm for 10 min at 4 °C. Circulating NEFA, ketones (BHOB), alanine aminotransferase (ALT), and aspartate aminotransferase (AST) levels were measured in serum using an ADVIA® Chemistry XPT blood chemistry analyzer (Bayer). To determine liver lipid levels, snap-frozen liver samples were weighed and homogenized in chloroform:methanol (2:1) solution, followed by addition of saline and centrifugation to achieve phase separation. The organic phase (bottom layer) was transferred into a new tube and evaporated with nitrogen gas. The dried lipids were then solubilized with chloroform:Triton X-100 (3:1) solution. Triglyceride content was measured using Infinity Triglycerides reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions and normalized to wet tissue weight, as previously described (52).
Quantitative Real-Time PCR.
Liver and adipose tissue samples were collected in RNAlater (Thermo Fisher Scientific) and frozen. RNA was extracted using TRIzol reagent and Direct-zol RNA miniprep kits following the manufacturer’s instructions (Thermo Fisher Scientific, Zymo Research). Genomic DNA was removed using DNase I provided in the Direct-zol RNA kit (Zymo Research). mRNA (up to 2 μg) was reverse-transcribed into cDNA using SuperScript® VILOTM Master Mix (Thermo Fisher Scientific). cDNA was amplified in duplicate reactions containing 5 μL of 2× TaqMan Gene Expression Master Mix (Thermo Fisher Scientific), 0.5 μL TaqMan assay probes (20×, Thermo Fisher Scientific), 3.5 μL nuclease-free H2O, and 1 μL cDNA using the QuantStudio 6 Flex Real-Time PCR System (Thermo Fisher Scientific). Beta-actin (Actb) housekeeping gene was used as the internal control gene to normalize cDNA input differences. Expression of genes of interest was calculated relative to the Actb housekeeping gene.
RNA Preparation and RNA-Sequencing Read Mapping.
Total RNA was purified from epididymal white adipose and subcutaneous white adipose (n = 8 WT, n = 7 to 8 Inhbe−/−; n = 10 Control AAV, n = 10 mInhbe AAV) using MagMAXTM-96 for the Microarrays Total RNA Isolation kit (Ambion by Life Technologies), according to manufacturer specifications. Genomic DNA was removed using MagMAXTMTurboTMDNase buffer and TURBO DNase from the MagMAX kit listed above (Ambion by Life Technologies). mRNA was purified from total RNA using the Dynabeads® mRNA Purification kit (Invitrogen). Strand-specific RNA sequencing (RNA-seq) libraries were prepared using the KAPA mRNA-Seq Library Preparation Kit (Kapa Biosystems). Twelve-cycle PCR was performed to amplify libraries. Sequencing was performed on Illumina HiSeq®2500 by a multiplexed single-read run with 33 cycles. Raw sequence data (BCL files) were converted to FASTQ format via Illumina bcl2fastq v2.17. Reads were decoded based on their barcodes, and read quality was evaluated with FastQC (www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were mapped to the mouse genome (NCBI GRCm38) using ArrayStudio® software (OmicSoft®, Cary, NC) allowing two mismatches. Reads mapped to the exons of a gene were summed at the gene level. Differential gene expression analysis was performed using the DESeq2 package (53). Signatures were obtained using cutoffs padj<0.05 and shrunken logfc>1.3. Shrinkage of effect size was performed using the ashr package (54). Genes that are highly specific to the liver and epididymis were removed from our signatures as false positives. Pathway enrichment analysis was performed using the clusterProfiler package (55). Top nonredundant significant pathways are shown in the figures. Full lists of significant pathways are provided in Dataset S2.
Histology.
For adipose tissue analysis, mice were fasted for 4 h, and epididymal and subcutaneous (inguinal) adipose tissues were collected. Dissected tissues were fixed in 4% paraformaldehyde, dehydrated in 70% ethanol, and embedded in paraffin. Sections were cut (10 μm in thickness) and affixed to slides, which were then deparaffinized, hydrated, and stained with hematoxylin–eosin. Stained sections were examined with the Leica Aperio AT2 instrument. The cross-sectional area of the adipocytes was analyzed with HALOLink to determine the size distribution of adipocytes (expressed as the frequency of adipocytes with a certain size out of the total number of cells).
Ex Vivo Lipolysis.
Mice were fasted for 4 h prior to tissue collection. Epididymal and subcutaneous fat pads were surgically removed and washed several times with phosphate-buffered saline. Tissue pieces (~20 mg) were incubated in low-glucose (1 g/L glucose) Dulbecco’s modified Eagle’s medium (Invitrogen) containing 2% fatty acid-free bovine serum albumin (Sigma) either in the presence or absence of 10 μM isoproterenol at 37 °C for 60 min. After this preincubation period, the fat pads were transferred into identical, fresh medium and incubated for a further 60 min at 37 °C. Thereafter, aliquots of the medium were removed and analyzed for free glycerol (Sigma) content using enzymatic assays, according to the manufacturer’s instructions. For protein determinations, fat pads were washed extensively with phosphate-buffered saline and then delipidated in chloroform:methanol (2:1) solution for 60 min at 37 °C. Fat pads were then lysed in 0.3 N NaOH, 0.1% SDS overnight at 55 °C. Aliquots of the protein lysates were used to determine protein content using the BCA reagent and bovine serum albumin as standard (Pierce). Lipolysis was performed in triplicate measurements for both basal and stimulated conditions from each tissue sample, and glycerol release was expressed as the mean of the three measurements for each sample.
Statistical Analysis.
Statistical and graphical data analyses were performed using Microsoft Excel and Prism 9 (GraphPad Software, Inc.). Data are expressed as mean ± SEM. Mean values were compared using unpaired two-tailed t-tests, one-way or two-way ANOVA as implemented in the GraphPad Prism 9.0 software (GraphPad Software, Inc.). Grubbs’ test was used to determine and remove significant outliers. A P-value < 0.05 was considered to be significant.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Acknowledgments
We thank Parnian Bigdelou for help with serum chemistry analysis and Lisa Shihanian, Corey Alexa-Braun, and Helen Chen for help with tissue collection. We also thank Zhong Wang and Mary Germino for help with adipocyte size quantification, and Joseph Song, Nicole Negron, Min Ni, and Shuo Li for excellent technical support with RNA sequencing.
Author contributions
R.C.A., G.H., J.M., G.S.A., V.I., A.N.E., and V.G. designed research; R.C.A., D.S.P., J.S.L., Y.Z., I.J.M., S.M., and G.H. performed research; S.A. contributed new reagents/analytic tools; R.C.A., D.S.P., J.S.L., Y.Z., I.J.M., S.M., and G.H. analyzed data; and R.C.A., A.N.E., L.A.L., A.J.M., G.D.Y., M.W.S., and V.G. wrote the paper.
Competing interests
R.C.A., D.S.P., J.S.L., Y.Z., I.J.M., S.M., G.H., J.M., G.S.A., S.A., V.I., A.N.E., L.A.L., A.J.M., G.D.Y., M.W.S., and V.G. are employees and shareholders at Regeneron Pharmaceuticals.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
George D. Yancopoulos, Email: George.Yancopoulos@regeneron.com.
Viktoria Gusarova, Email: Viktoria.Gusarova@regeneron.com.
Data, Materials, and Software Availability
All data associated with this study are present in the paper or supporting information, and all figures have associated raw data. Any materials that can be shared will be released via a material transfer agreement. Transcriptomics datasets have been deposited in Gene Expression Omnibus (GEO): https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?&acc=GSE231444 (56).
Supporting Information
References
- 1.Bluher M., Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 15, 288–298 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Klein S., Gastaldelli A., Yki-Jarvinen H., Scherer P. E., Why does obesity cause diabetes? Cell Metab. 34, 11–20 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Czech M. P., Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 23, 804–814 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Emdin C. A., et al. , Genetic association of waist-to-hip ratio with cardiometabolic traits, type 2 diabetes, and coronary heart disease. JAMA 317, 626–634 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dale C. E., et al. , Causal associations of adiposity and body fat distribution with coronary heart disease, stroke subtypes, and type 2 diabetes mellitus: A mendelian randomization analysis. Circulation 135, 2373–2388 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pischon T., et al. , General and abdominal adiposity and risk of death in Europe. N. Engl J. Med. 359, 2105–2120 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Lotta L. A., et al. , Association of genetic variants related to gluteofemoral vs abdominal fat distribution with type 2 diabetes, coronary disease, and cardiovascular risk factors. JAMA 320, 2553–2563 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lotta L. A., et al. , Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat. Genet 49, 17–26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Emerging Risk Factors Collaboration et al. , Separate and combined associations of body-mass index and abdominal adiposity with cardiovascular disease: Collaborative analysis of 58 prospective studies. Lancet 377, 1085–1095 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.InterAct C., et al. , Long-term risk of incident type 2 diabetes and measures of overall and regional obesity: The EPIC-InterAct case-cohort study. PLoS Med. 9, e1001230 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akbari P., et al. , Multiancestry exome sequencing reveals INHBE mutations associated with favorable fat distribution and protection from diabetes. Nat. Commun. 13, 4844 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deaton A. M., et al. , Rare loss of function variants in the hepatokine gene INHBE protect from abdominal obesity. Nat. Commun. 13, 4319 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fougerat A., et al. , ATGL-dependent white adipose tissue lipolysis controls hepatocyte PPARalpha activity. Cell Rep. 39, 110910 (2022). [DOI] [PubMed] [Google Scholar]
- 14.Sugiyama M., et al. , Inhibin betaE (INHBE) is a possible insulin resistance-associated hepatokine identified by comprehensive gene expression analysis in human liver biopsy samples. PLoS One 13, e0194798 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hashimoto O., Sekiyama K., Matsuo T., Hasegawa Y., Implication of activin E in glucose metabolism: Transcriptional regulation of the inhibin/activin betaE subunit gene in the liver. Life Sci. 85, 534–540 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Rodgarkia-Dara C., et al. , The activin axis in liver biology and disease. Mutat. Res. 613, 123–137 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Namwanje M., Brown C. W., Activins and inhibins: Roles in development, physiology, and disease. Cold Spring Harb. Perspect. Biol. 8, a021881 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lau A. L., Kumar T. R., Nishimori K., Bonadio J., Matzuk M. M., Activin betaC and betaE genes are not essential for mouse liver growth, differentiation, and regeneration. Mol. Cell Biol. 20, 6127–6137 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hashimoto O., et al. , Activin E controls energy homeostasis in both brown and white adipose tissues as a hepatokine. Cell Rep. 25, 1193–1203 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Emdin C. A., et al. , DNA sequence variation in ACVR1C encoding the activin receptor-like kinase 7 influences body fat distribution and protects against type 2 diabetes. Diabetes 68, 226–234 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo T., et al. , Adipocyte ALK7 links nutrient overload to catecholamine resistance in obesity. Elife 3, e03245 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ibanez C. F., Regulation of metabolic homeostasis by the TGF-beta superfamily receptor ALK7. FEBS J. 289, 5776–5797 (2022). [DOI] [PubMed] [Google Scholar]
- 23.Yogosawa S., Mizutani S., Ogawa Y., Izumi T., Activin receptor-like kinase 7 suppresses lipolysis to accumulate fat in obesity through downregulation of peroxisome proliferator-activated receptor gamma and C/EBPalpha. Diabetes 62, 115–123 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bertolino P., et al. , Activin B receptor ALK7 is a negative regulator of pancreatic beta-cell function. Proc. Natl. Acad. Sci. U.S.A. 105, 7246–7251 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonomi L., et al. , Activin B regulates islet composition and islet mass but not whole body glucose homeostasis or insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 303, E587–596 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andersson O., Korach-Andre M., Reissmann E., Ibanez C. F., Bertolino P., Growth/differentiation factor 3 signals through ALK7 and regulates accumulation of adipose tissue and diet-induced obesity. Proc. Natl. Acad. Sci. U.S.A. 105, 7252–7256 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee S. J., et al. , Regulation of muscle mass by follistatin and activins. Mol. Endocrinol. 24, 1998–2008 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kahn B. B., McGraw T. E., Rosiglitazone, PPARgamma, and type 2 diabetes. N Engl. J. Med. 363, 2667–2669 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grabner G. F., Xie H., Schweiger M., Zechner R., Lipolysis: Cellular mechanisms for lipid mobilization from fat stores. Nat. Metab 3, 1445–1465 (2021). [DOI] [PubMed] [Google Scholar]
- 30.Shulman G. I., Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N. Engl. J. Med. 371, 1131–1141 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Takahashi M., Kamei Y., Ezaki O., Mest/Peg1 imprinted gene enlarges adipocytes and is a marker of adipocyte size. Am. J. Physiol. Endocrinol. Metab. 288, E117–124 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Anunciado-Koza R. P., Higgins D. C., Koza R. A., Adipose tissue Mest and Sfrp5 are concomitant with variations of adiposity among inbred mouse strains fed a non-obesogenic diet. Biochimie 124, 134–140 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen J. L., et al. , Elevated expression of activins promotes muscle wasting and cachexia. FASEB J. 28, 1711–1723 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Bartholin L., et al. , FLRG, an activin-binding protein, is a new target of TGFbeta transcription activation through Smad proteins. Oncogene 20, 5409–5419 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Zhao M., et al. , Targeting activin receptor-like kinase 7 ameliorates adiposity and associated metabolic disorders. JCI Insight 8, e161229 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Srivastava R. K., Lee E. S., Sim E., Sheng N. C., Ibanez C. F., Sustained anti-obesity effects of life-style change and anti-inflammatory interventions after conditional inactivation of the activin receptor ALK7. FASEB J. 35, e21759 (2021). [DOI] [PubMed] [Google Scholar]
- 37.Wang Q., Guo T., Portas J., McPherron A. C., A soluble activin receptor type IIB does not improve blood glucose in streptozotocin-treated mice. Int. J. Biol. Sci. 11, 199–208 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heymsfield S. B., et al. , Effect of bimagrumab vs placebo on body fat mass among adults with type 2 diabetes and obesity: A phase 2 randomized clinical trial. JAMA Netw. Open 4, e2033457 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garito T., et al. , Bimagrumab improves body composition and insulin sensitivity in insulin-resistant individuals. Diabetes Obes. Metab. 20, 94–102 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Choi Y. H., et al. , Alterations in regulation of energy homeostasis in cyclic nucleotide phosphodiesterase 3B-null mice. J. Clin. Invest. 116, 3240–3251 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tansey J. T., et al. , Perilipin ablation results in a lean mouse with aberrant adipocyte lipolysis, enhanced leptin production, and resistance to diet-induced obesity. Proc. Natl. Acad. Sci. U.S.A. 98, 6494–6499 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wei S., et al. , Spontaneous development of hepatosteatosis in perilipin-1 null mice with adipose tissue dysfunction. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1863, 212–218 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Gray S. L., Dalla Nora E., Vidal-Puig A. J., Mouse models of PPAR-gamma deficiency: Dissecting PPAR-gamma’s role in metabolic homoeostasis. Biochem. Soc. Trans. 33, 1053–1058 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Savage D. B., Mouse models of inherited lipodystrophy. Dis. Model. Mech. 2, 554–562 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Demetrius L., Of mice and men, When it comes to studying ageing and the means to slow it down, mice are not just small humans. EMBO Rep. 6, S39–S44 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodriguez-Cuenca S., et al. , Peroxisome proliferator-activated receptor gamma-dependent regulation of lipolytic nodes and metabolic flexibility. Mol. Cell Biol. 32, 1555–1565 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kershaw E. E., et al. , PPARgamma regulates adipose triglyceride lipase in adipocytes in vitro and in vivo. Am. J. Physiol. Endocrinol. Metab. 293, E1736–1745 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walczak R., Tontonoz P., PPARadigms and PPARadoxes: expanding roles for PPARgamma in the control of lipid metabolism. J. Lipid Res. 43, 177–186 (2002). [PubMed] [Google Scholar]
- 49.Koprulu M., et al. , Identification of rare loss-of-function genetic variation regulating body fat distribution. J. Clin. Endocrinol. Metab. 107, 1065–1077 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Valenzuela D. M., et al. , High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. Nat. Biotechnol. 21, 652–659 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Poueymirou W. T., et al. , F0 generation mice fully derived from gene-targeted embryonic stem cells allowing immediate phenotypic analyses. Nat. Biotechnol. 25, 91–99 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Gusarova V., et al. , ANGPTL3 blockade with a human monoclonal antibody reduces plasma lipids in dyslipidemic mice and monkeys. J. Lipid Res. 56, 1308–1317 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stephens M., False discovery rates: A new deal. Biostatistics 18, 275–294 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu T., et al. , clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb) 2, 100141 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adam R. C., et al. , Transcriptome analysis of adipose tissue of Inhbe knockout and over-expressing mice. NCBI Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE231444. Deposited 1 May 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Data Availability Statement
All data associated with this study are present in the paper or supporting information, and all figures have associated raw data. Any materials that can be shared will be released via a material transfer agreement. Transcriptomics datasets have been deposited in Gene Expression Omnibus (GEO): https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?&acc=GSE231444 (56).