

Significance
The voltage-gated sodium channel isoform 1.7 (NaV1.7) has been widely implicated in chronic pain. We have discovered a unique intracellular region of NaV1.7 that facilitates its regulation by intracellular auxiliary proteins and which can be targeted to ameliorate chronic pain. Targeting this sequence produces robust reversal of mechanical allodynia in preclinical models of nerve injury, as well as a model of chemotherapy-induced peripheral neuropathy. A plasmid coding for this sequence packaged inside a viral capsid reduced NaV1.7 current density and neuropathic pain. The translational utility of our approach is illustrated by the finding that our genetic therapy reduced NaV1.7 function in macaque DRG neurons, which shares complete sequence homology with human NaV1.7.
Keywords: NaV1.7, SUMO, CRMP2, chronic pain, gene therapy
Abstract
Small molecules directly targeting the voltage-gated sodium channel (VGSC) NaV1.7 have not been clinically successful. We reported that preventing the addition of a small ubiquitin-like modifier onto the NaV1.7-interacting cytosolic collapsin response mediator protein 2 (CRMP2) blocked NaV1.7 function and was antinociceptive in rodent models of neuropathic pain. Here, we discovered a CRMP2 regulatory sequence (CRS) unique to NaV1.7 that is essential for this regulatory coupling. CRMP2 preferentially bound to the NaV1.7 CRS over other NaV isoforms. Substitution of the NaV1.7 CRS with the homologous domains from the other eight VGSC isoforms decreased NaV1.7 currents. A cell-penetrant decoy peptide corresponding to the NaV1.7-CRS reduced NaV1.7 currents and trafficking, decreased presynaptic NaV1.7 expression, reduced spinal CGRP release, and reversed nerve injury-induced mechanical allodynia. Importantly, the NaV1.7-CRS peptide did not produce motor impairment, nor did it alter physiological pain sensation, which is essential for survival. As a proof-of-concept for a NaV1.7 -targeted gene therapy, we packaged a plasmid encoding the NaV1.7-CRS in an AAV virus. Treatment with this virus reduced NaV1.7 function in both rodent and rhesus macaque sensory neurons. This gene therapy reversed and prevented mechanical allodynia in a model of nerve injury and reversed mechanical and cold allodynia in a model of chemotherapy-induced peripheral neuropathy. These findings support the conclusion that the CRS domain is a targetable region for the treatment of chronic neuropathic pain.
The generation of action potentials relies in part on the firing threshold set by voltage-gated sodium channels such as NaV1.7. Encoded by the SCN9A gene, NaV1.7 is expressed in nociceptive sensory neurons of the dorsal root ganglion (DRG) (1) and it is a major contributor to human pain signaling (1, 2). Patients carrying activating mutations on SCN9A are burdened with painful heritable syndromes such as inherited erythromelalgia, small-fiber neuropathy, and paroxysmal extreme pain disorder (3). Conversely, SCN9A loss-of-function mutations produce congenital insensitivity to pain (4). NaV1.7 function is increased in preclinical rodent models of chronic neuropathic pain (5–7). While this evidence points to a critical role for NaV1.7 in pain, direct inhibition of this channel failed to reach satisfactory clinical endpoints when evaluated in clinical trials (1, 8–14).
We endeavored to understand how NaV1.7 is trafficked and retained at the presynapse in primary afferents. We identified collapsin response mediator protein 2 (CRMP2) as a critical protein regulating NaV1.7 membrane localization (15–18). When phosphorylated by cyclin-dependent kinase 5 (Cdk5) and SUMOylated, CRMP2 protected NaV1.7 from internalization to maintain its presynaptic localization in chronic neuropathic pain (19, 20). Pharmacological inhibition of CRMP2 phosphorylation or SUMOylation, specifically decreased NaV1.7 membrane localization and function to provide pain relief in rodent models of chronic neuropathic pain (20, 21). However, the mechanism driving the selective regulation of NaV1.7 by CRMP2 has remained elusive.
Using peptide mapping, we identified a unique CRMP2 binding domain in the first intracellular loop of NaV1.7. We further demonstrate that this CRMP2 regulatory sequence (CRS) is necessary for NaV1.7 membrane localization and function. Targeting CRS with a decoy peptide fused to a cell-penetrating motif or a genetically encoded peptide inhibited NaV1.7 in vitro and in vivo to block chronic neuropathic pain, but not physiological pain. Finally, we validated the translational potential of the NaV1.7 CRS domain in sensory neurons and spinal cord from nonhuman primates. Together, our results reveal a unique domain (CRS) amenable to therapeutic targeting to specifically inhibit NaV1.7 and alleviate chronic pain.
Results
Identification of a Unique NaV1.7 Regulatory Domain.
The NaV1.7 intracellular domains mediating channel regulation are not yet well understood (22–24). To identify potential sites of interaction between CRMP2 and NaV1.7, we generated a peptide array spanning the intracellular loops of human NaV1.7 (Fig. 1A). We hybridized the array with spinal cord lysate from rats with chronic neuropathic pain (spared nerve injury, SNI) and found that CRMP2 bound to peptide number 141 (Fig. 1B) (#141; hNaV1.7, 706-SRQKCPPWWYRFAHK-720). CRMP2 bound the same NaV1.7 peptide in lysates from porcine and human DRG or spinal cord (Fig. 1B). Because CRMP2 regulates NaV1.7 exclusively (19, 21), we tested if CRMP2 could bind analogous hNaV1.x-CRS regions (Fig. 1C). CRMP2 bound to hNaV1.7-CRS with a KD six-fold lower than that of any of the other hNaV1.x peptides: KD = 0.99 ± 0.107 µM for hNaV1.7 vs. KD = 6.18 ± 1.16 µM for hNaV1.3 and KD = 6.28 ± 6.83 µM for hNaV1.4 (Fig. 1C and SI Appendix, Fig. S1). Significantly less binding was detected for hNaV1.1 with KD = 223 µM and hNaV1.2 KD = 91.8 µM but no binding was detected for any of the other hNaV1.x-CRS homologous domains (Fig. 1C and SI Appendix, Fig. S1).
Fig. 1.
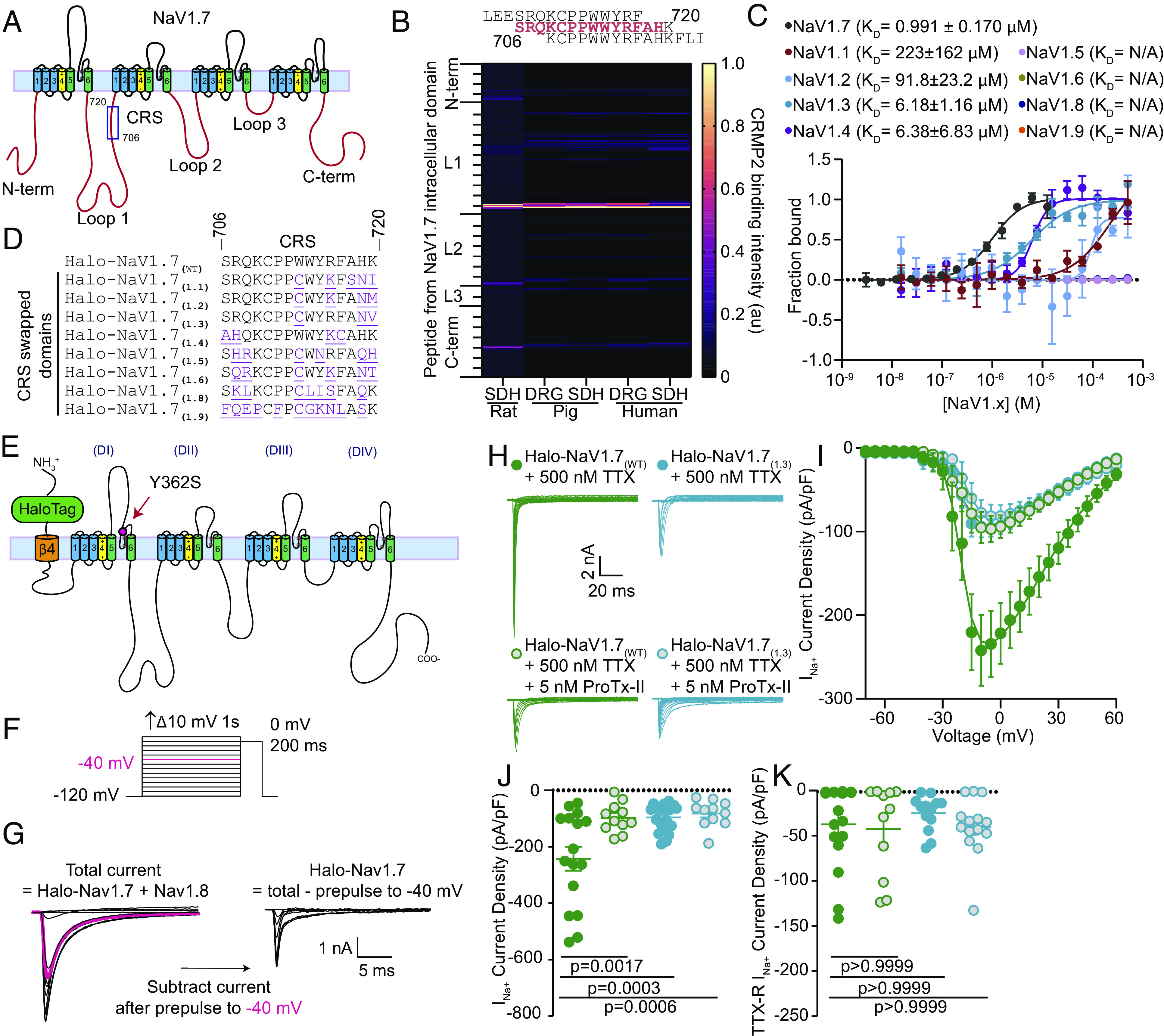
Identification of a unique CRMP2 regulatory domain on NaV1.7. (A) Cartoon of the domain structure of human NaV1.7 with intracellular loops labeled. These loops were divided into 384 15-mer peptides with 12 overlapping amino acids and were printed onto a peptide array. (B) Fluorescent intensity of CRMP2 binding to the peptide array from rat, pig, and human DRG and spinal cord lysate (n = 4). (C) Microscale thermophoresis of NTA-labeled His-CRMP2 (200 nM) with different concentrations of the CRMP2-binding peptide from NaV1.7 or the homologous region of NaV1.X channels, fitted to a one-site binding model (r2= 0.97 for NaV1.7 and 0.76 for NaV1.1; n = 4; Bottom). Peptides with higher sequence similarity to NaV1.7 bound more tightly with CRMP2 in this assay. Those with less sequence similarity bound with little to no detectable affinity. (D) Sequence alignment of the CRMP2 binding domain on NaV1.7 with the CRS analogous domains. (E) Cartoon depiction of the Halo-Nav1.7 construct that was adapted for transfection into rat DRG neurons. (F) Diagram showing the steady-state availability protocol that was used to isolate TTX-R from TTX-S currents using a prepulse to −40 mV to determine the TTX-R current fraction (shown in pink). (G) An electrical subtraction protocol was used to isolate the currents originating from Halo-NaV1.7 channels. This was achieved by subtracting the current obtained during the test pulse (at 0 mV) preceded by a 1-s pulse at −40 mV (pink trace) from the current obtained at the test pulse (at 0 mV) previously subjected to different prepulses ranging from −120 to +10 mV. This approach allowed for the isolation of Halo-NaV1.7 channel currents, as these channels exhibit TTX resistance and possess distinct NaV1.7 inactivation kinetics. (H) Representative traces of sodium currents recorded from DRGs transfected with Halo-NaV1.7(WT) (green) and Halo-NaV1.7(1.3) (teal) in the presence or absence of 5 nM ProTx-II. (I) Current density–voltage relationship for Halo-NaV1.7(WT), Halo-NaV1.7(1.3), and both groups following treatment with 5 nM ProTx-II. Treatment with ProTx-II did not reduce the current density further suggesting maximal reduction was achieved by swapping out the NaV1.7-CRS domain with the analogous domain from NaV1.3. (J) Peak Halo-NaV1.7 current density for Halo-NaV1.7(WT), Halo-NaV1.7(WT) + 5 nM ProTx-II, Halo-NaV1.7(1.3), and Halo-NaV1.7(1.3) + 5 nM ProTx-II. (K) TTX-R peak current density for the conditions listed above. Each group was compared to its own on-plate Halo-NaV1.7(WT) control group and statistical significance was determined using a Mann–Whitney U test. n = 11 to 16 cells; error bars indicate mean ± SEM; P values as indicated (Dataset S1); Kruskal–Wallis test with the Dunn post hoc test. All biophysical parameters are shown in SI Appendix, Table S1. SDH—Spinal Dorsal Horn; DRG—Dorsal Root Ganglia. Panels E–H Green circles—Halo-NaV1.7(WT) + 500 nM TTX; Blue circles Halo-NaV1.7(1.3) + 500 nM TTX; Green circles with gray center - Halo-NaV1.7(WT) + 500 nM TTX + 5 nM ProTx-II; Blue circles with gray center Halo-NaV1.7(1.3) + 500 nM TTX + 5 nM ProTx-II.
To test if NaV1.7-CRS is responsible for the selective regulation of NaV1.7, we used a plasmid encoding a mNaV1.7 channel with resistance to tetrodotoxin (Y362S, TTX-R) (25) and added an N-terminal extracellular HaloTag reporter linked to NaV1.7 via the transmembrane helix from the β4 subunit of VGSC (referred here as Halo-NaV1.7(WT)) (26). We mutated the CRS domain in Halo-NaV1.7(WT) to the homologous CRS sequences from other Nav1.x channels (Fig. 1 D and E). All NaV1.7 mutants expressed well in mouse catecholamine A differentiated (CAD) cells (SI Appendix, Fig. S2A) and could be detected with Halo ligand fluorescence in transfected rat DRG neurons (SI Appendix, Fig. S2B). We recorded sodium currents for each Halo-NaV1.7(WT/1.x) construct expressed in rat DRG neurons where the endogenous TTX-sensitive currents were blocked (500 nM TTX) (Fig. 1F). We chose this concentration of TTX because it will block all endogenous NaV1.7 while leaving the current flowing through our resistant transfected Halo-NaV1.7 channel intact. We then employed a well-validated electrical subtraction methodology (27) to isolate channels based on their inactivation kinetics. This involves applying a conditioning prepulse to −40 mV to inactivate TTX-S channels before applying a test pulse to 0 mV (Fig. 1F). The total current is obtained by applying conditioning pulses over a range of voltages from −120 mV to +10 mV followed by a test pulse to 0 mV. The test pulse current after the conditioning prepulse to −40 mV, which represents the TTX-R current, is then subtracted from the total current. This yields the Halo-Nav1.7 current fraction (Fig. 1G). This allowed us to discriminate between Halo-NaV1.7 and endogenously expressed Nav1.8 channels that are resistant to TTX. We used changes in sodium current density to infer the degree of regulation imposed by substitution of the NaV1.7-CRS. We found that all our mutations in the CRS domain of Halo-NaV1.7 imposed a profound reduction of current density (SI Appendix, Fig. S3). To test whether all the Halo-NaV1.7 current was nonfunctional when the CRS was mutated, we applied the NaV1.7 selective blocker ProTx-II, which did not result in further reduction of current density (Fig. 1 H–K). This experiment was done by comparing Halo-NaV1.7(1.3) to Halo-NaV1.7(WT) to test this idea using the CRS domain with the highest homology with the CRS(1.7) sequence. Importantly, the current density of TTX-R channels–NaV1.8 and NaV1.9–was not affected in these experiments (Fig. 1K and SI Appendix, Fig. S3). The activation and inactivation constants of NaV1.7 remained largely unchanged in our experiments, except for the Halo-NaV1.7(1.1) and Halo-NaV1.7(1.5) constructs, which were shifted to mildly more positive potentials (SI Appendix, Table S1 and Fig. S3). This could suggest the involvement of these substitutions with the NaV1.7 voltage-sensing domain. However, due to the location of these substitutions within intracellular loop 1, it is unlikely that these residues directly impact the function of the voltage sensing domain. Taken together, these results identify a unique regulatory site necessary for NaV1.7 function in DRG sensory neurons.
Competitive Inhibition of CRMP2 Binding to NaV1.7 Decreases Na+ Current.
Having discovered a regulatory domain on NaV1.7, we next tested if this sequence could be targeted to limit NaV1.7 function. This domain (NaV1.7-CRS) was fused to the cell-penetrating trans-activator of transcription (TAT) sequence (YGRKKRRQRRR) (28). A scrambled control peptide was also generated, designated Myr-TAT-SCR (SCR—Scrambled peptide). To increase the juxtamembrane concentration of this decoy peptide, we added a 14-carbon myristate group (Myr) to the N terminus as an anchor to the plasma membrane (29) (Fig. 2A). At 5 µM, Myr-TAT-NaV1.7-CRS disrupted the interaction between CRMP2 and the CRS domain forming peptides on our peptide array (SI Appendix, Fig. S4). Myr-TAT-NaV1.7-CRS also inhibited the CRMP2/NaV1.7 interaction in CAD cells (Fig. 2 B and C). Myr-TAT-NaV1.7-CRS had no effect on CRMP2 SUMOylation, or phosphorylation by either Cdk5 (pS522) or GSK3β (pT514) (SI Appendix, Fig. S5), ensuring that the actions of Myr-TAT-NaV1.7-CRS is not via an effect on CRMP2 posttranslational modifications known to affect its interaction with NaV1.7 (19, 30).
Fig. 2.
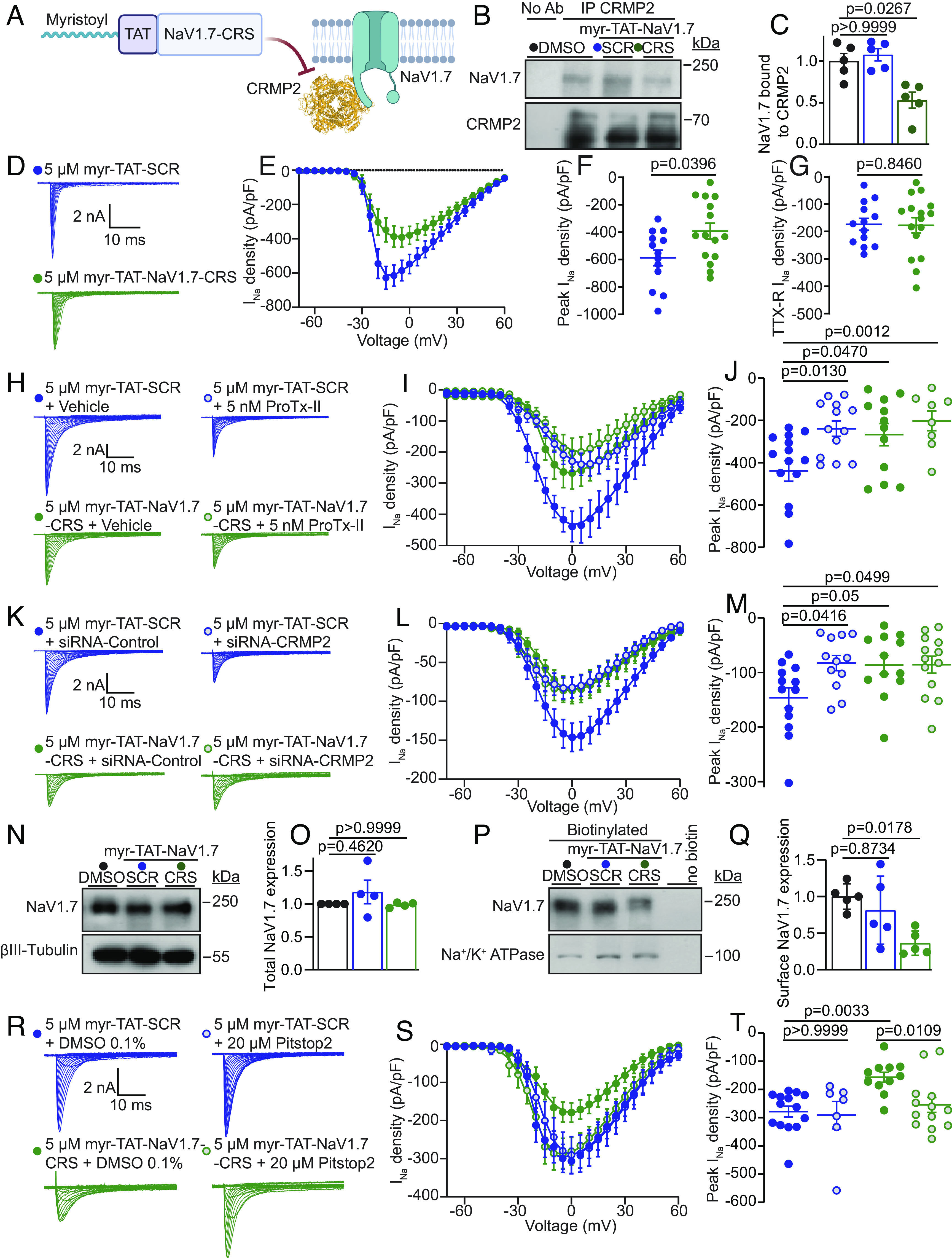
Myr-TAT-NaV1.7-CRS causes CRMP2 dependent reduction in NaV1.7 currents in DRG neurons. (A) The Myr-TAT-NaV1.7-CRS peptide competes for binding to NaV1.7 regulatory proteins. (B) Representative immunoblots and summary (C) of CRMP2 immunoprecipitation (IP) to detect NaV1.7 from CAD cells treated with the indicated peptides (n = 5). (D) Representative current traces recorded from small-sized DRG neurons in the presence of 5 µM Myr-TAT-SCR peptide (blue circles, n = 15) and Myr-TAT-NaV1.7-CRS peptide (green circles, n = 16). (E) Summary of Boltzmann fits for current density–voltage curves, (F) peak current densities, and (G) electrically isolated TTX-R currents. (H) Representative current traces recorded from small-sized DRGs incubated with Myr-TAT-SCR and Myr-TAT-NaV1.7-CRS in the presence and absence of the NaV1.7-specific inhibitor ProTx-II. (I) Boltzmann fits of the current density–voltage curves for each treatment group. (J) Summary of peak current densities. N = 8 to 12 cells; (K) representative current traces recorded from small-diameter DRGs transfected with either siRNA-Control or siRNA-CRMP2 and treated with Myr-TAT-NaV1.7-CRS or Myr-TAT-SCr as indicated. (L) Boltzmann fits for current density–voltage curves and (M) peak current densities. n = 12 to 13 cells; (N) representative immunoblots and (O) summary (Bottom) of NaV1.7 expression in CAD cells treated with the indicated peptides. βIII-Tubulin is used as a loading control (n = 4). (P) Representative immunoblots of streptavidin-enriched surface fractions probed for NaV1.7 and Na+/K+ ATPase as a control. (Q) Bar graph with scatter plot of mean surface localized NaV1.7 in CAD cells treated with the indicated peptides. (R) Representative current traces recorded from small-diameter DRG neurons in the presence of Myr-TAT-SCR or Myr-TAT-NaV1.7-CRS ± 20 µM Pitstop2 as indicated; (S) Boltzmann fits for current density-voltage curves and (T) summary of peak current densities (pA/pF) showing pitsopt2 blocked the current reduction imposed by Myr-TAT-NaV1.7-CRS. n = 7 to 14 cells; error bars indicate mean ± SEM; P values as indicated; Kruskal–Wallis test with Dunnett’s post hoc comparisons (Dataset S1). All biophysical parameters are shown in SI Appendix, Table S2 and statistical comparisons shown in Dataset S1. CRS—CRMP2 regulatory sequence; SCR—Scrambled control peptide (Sequence: KYHPWACFRQWRSPK).
In DRG neurons, acute application of 5 µM Myr-TAT-NaV1.7-CRS reduced sodium current density (Fig. 2 D–F) without affecting TTX-R current density, compared to the Myr-TAT-SCR scrambled control peptide (Fig. 2G). There was a subtle shift in the half-maximal activation voltage and slope of the activation function (SI Appendix, Table S2). To test whether all of the current carried by NaV1.7 is inhibited following treatment with Myr-TAT-NaV1.7-CRS, we added ProTx-II (5 nM) to block any remaining NaV1.7 channels. While ProTx-II decreased sodium current density in DRG neurons treated with the scramble control peptide, coapplication with Myr-TAT-NaV1.7-CRS did not produce any further current reduction (Fig. 2 H–J), thus, demonstrating that Myr-TAT-NaV1.7-CRS achieved maximal NaV1.7 blockade in DRG neurons. We next used our validated siRNA (19) to silence CRMP2 expression in DRG neurons to test whether Myr-TAT-NaV1.7-CRS could reduce NaV1.7 current independently of CRMP2. As before, reducing CRMP2 expression inhibited peak sodium current density by ~43% (Fig. 2 K–M). Cotreatment with Myr-TAT-NaV1.7-CRS failed to elicit a further reduction of current density (Fig. 2 K–M). Furthermore, we found that Myr-TAT-NaV1.7-CRS had no effect on voltage-gated potassium (SI Appendix, Fig. S6 A–C) or calcium channels (SI Appendix, Fig. S6 D–G) in DRGs. The above results demonstrate that the CRS domain can be functionalized to specifically inhibit NaV1.7 in DRG sensory neurons.
Disruption of the CRS Domain Induces NaV1.7 Internalization.
CRMP2 binding to NaV1.7 maintains the membrane localization of the channel (30), so we next examined whether blocking the NaV1.7 CRS domain could induce its internalization. Myr-TAT-NaV1.7-CRS did not change NaV1.7 protein expression (Fig. 2 N-O). In cell surface biotinylation experiments, Myr-TAT-NaV1.7-CRS diminished NaV1.7 surface expression by ~64% (Fig. 2 P-Q). In DRG neurons, Pitstop2 (20 µM)—an inhibitor of clathrin assembly—rescued the decrease of sodium currents imposed by Myr-TAT-NaV1.7-CRS (Fig. 2 R-T). The half-maximal activation voltage was shifted slightly to more positive potential following application of Pitstop2 with the scrambled peptide, but this was not observed for the Myr-TAT-NaV1.7 peptide (SI Appendix, Table S2). These findings show that the reduction of NaV1.7 currents due to the blockade of the CRS domain is elicited via active internalization of the channel.
Inhibition of the NaV1.7 CRS Domain Reduces DRG Excitability and Spinal CGRP Release.
NaV1.7 channels set the threshold for action potential firing in sensory neurons (31). Therefore, inhibition of NaV1.7 currents by Myr-TAT-NaV1.7-CRS may in turn decrease action potential firing in DRG neurons. Compared to neurons treated with the Myr-TAT-SCR control, neurons treated with Myr-TAT-NaV1.7-CRS had decreased excitability (Fig. 3 A and B). Their resting membrane potential was not affected (Fig. 3C) but the rheobase—the minimum current necessary to evoke a single AP—was increased in DRG neurons treated with Myr-TAT-NaV1.7-CRS (Fig. 3 D and E). The input resistance was not different between treatment conditions (Fig. 3F). Analysis of the action potential waveform revealed no alterations of the peak and anti-peak amplitude or the time constants describing the rising and falling phase of the action potential waveforms (rise/decay tau) at any current step tested (SI Appendix, Fig. S7). These findings show that targeting the CRS domain can attenuate the intrinsic excitability of DRG neurons.
Fig. 3.
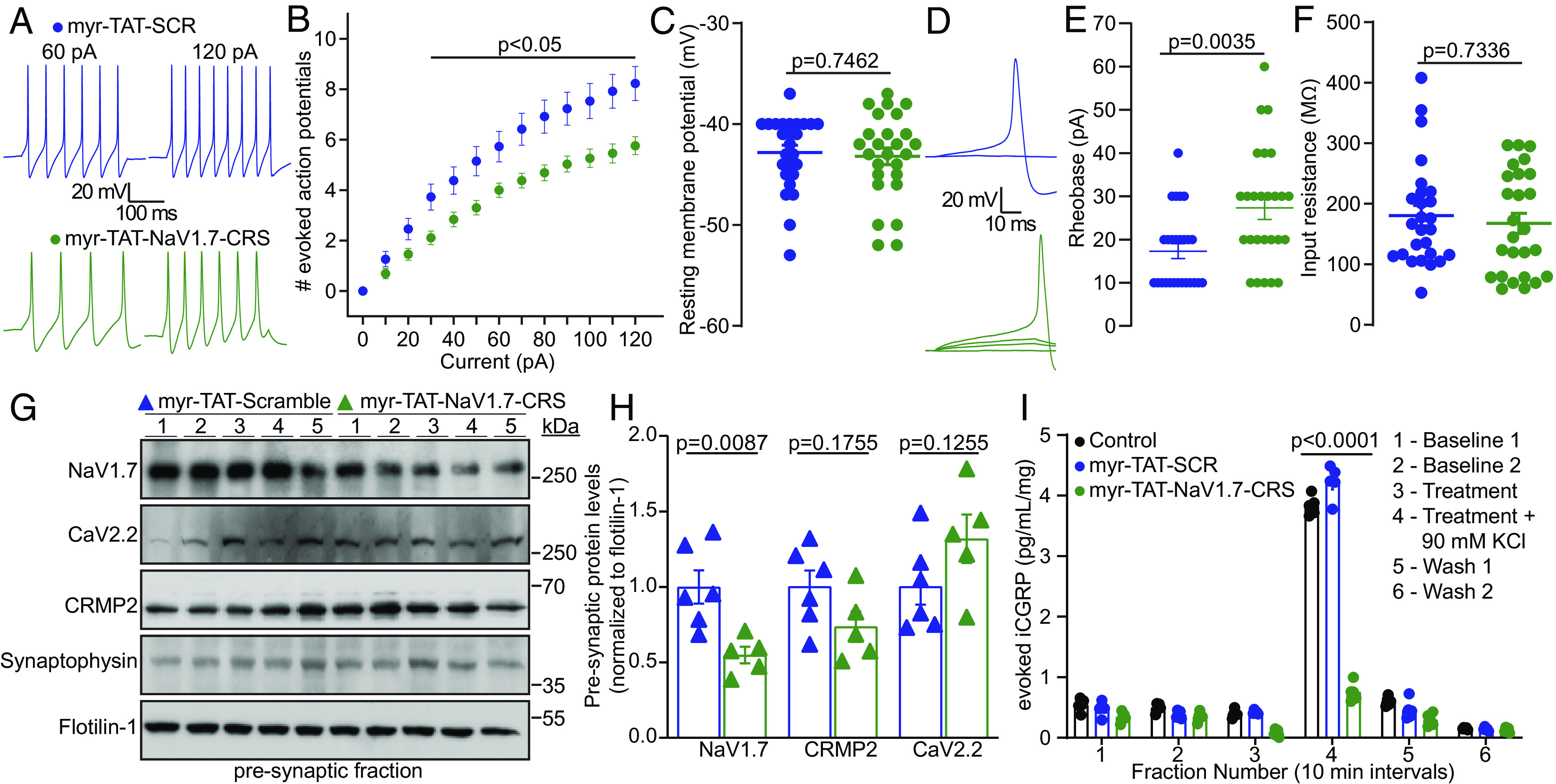
Disruption of the NaV1.7-CRMP2 interaction decreases presynaptic NaV1.7, sensory neuron excitability, and spinal cord neurotransmitter release. (A) Representative action potential recordings from DRGs in response to current injections after adding Myr-TAT-SCR (blue) and Myr-TAT-NaV1.7-CRS peptides (green). (B) Quantification of current-evoked action potentials in response to 0 to 120 pA of injected current. (C) Resting membrane potential of cells recorded in A. (D) Representative traces and (E) quantification indicating an increased rheobase with Myr-TAT-NaV1.7-CRS treatment. n = 26 cells; (F) Summary data for membrane input resistance. (G) Immunoblots of NaV1.7, CRMP2, and CaV2.2 expression in the presynaptic fraction of spinal dorsal horn, 1 h after peptide injection [i.t. injection of 20 µg/5 µL of Myr-TAT-SCR (n = 6) or Myr-TAT-NaV1.7-CRS (n = 5)]. (H) Quantification showing NaV1.7 spinal presynaptic localization of data in G. (I) KCl depolarization-evoked CGRP release measured from isolated spinal cord following incubation with control, Myr-TAT-SCR, or Myr-TAT-NaV1.7-CRS peptides. Histograms show normalized CGRP levels (n = 4 animals); error bars indicate mean ± SEM; data analyzed by Mann–Whitney U test or two-way ANOVA (details in Dataset S1), P values indicated.
To parallel our in vitro data, we used an in vivo synaptic fractionation approach to assess if targeting the CRS domain of NaV1.7 could also reduce NaV1.7 spinal presynaptic localization. Male rats with SNI were injected with Myr-TAT-NaV1.7-CRS (20 µg/5 µL i.t.) or control, then 1 h after intrathecal administration, we collected the lumbar dorsal horn and isolated the pre-synaptic fraction (20). We found that blocking the CRS domain of NaV1.7 with Myr-TAT-NaV1.7-CRS reduced NaV1.7 presynaptic localization without impacting presynaptic CRMP2 or CaV2.2 levels (Fig. 3 F and G). Next, we examined if the Myr-TAT-NaV1.7-CRS induced reduction of presynaptic NaV1.7 would impair evoked release of the nociceptive neurotransmitter calcitonin gene-related peptide (CGRP) (32). Using an ex vivo assay, lumbar spinal cord was stimulated with high concentration KCl to induce depolarization and fractions were collected for analysis of evoked CGRP content. We found an ~80% decrease in evoked CGRP from spinal cords treated with Myr-TAT-NaV1.7-CRS (5 µM) compared to Myr-TAT-SCR or 0.1% DMSO controls (Fig. 3H). Together, these data show that interfering with NaV1.7 CRS domain reduces DRG excitability, presynaptic localization of spinal NaV1.7, and consequently spinal CGRP release.
Disruption of NaV1.7-CRMP2 Binding Confers Antinociception and Spares Physiological Pain without Affecting Motor Control.
Thus far, our results uncover a new intracellular domain on NaV1.7 that can be blocked to limit the function of this channel. Using protein extract from the ipsi- and contra-lateral sides from rats with SNI, we found that the interaction of CRMP2 with the target peptide #141 was specifically increased in tissues from the side experiencing chronic neuropathic pain (Fig. 4A). We next performed an appraisal of nociceptive behaviors that could be mitigated by our blocking peptide Myr-TAT-NaV1.7-CRS (20 µg/5 µL i.t.). We found no effect on behavioral measures of acute and chemical irritant-induced pain (SI Appendix, Fig. S8A), postoperative (SI Appendix, Fig. S8B), visceral (SI Appendix, Fig. S8C), and inflammatory (SI Appendix, Fig. S8D) pain. Next, we administered Myr-TAT-NaV1.7-CRS (20 µg/5 µL i.t.) to male and female rats with SNI and observed a >5 h reversal of the mechanical hypersensitivity induced by nerve injury (Fig. 4 B and C), consistent with a half-life of the ~88 min for the Myr-TAT-NaV1.7-CRS peptide in CSF (SI Appendix, Fig. S9). NaV1.7 loss in mice leads to a profound loss of pain sensation and elevated nociceptive thresholds (33, 34). We found that intrathecal injection of Myr-TAT-NaV1.7-CRS in male and female mice had no effect in the hot plate (Fig. 4D) and tail-flick assays (Fig. 4E). Finally, using the rotarod test, we show that Myr-TAT-NaV1.7-CRS does not impact motor functions, a common unwanted effect of many analgesics (Fig. 4F). Altogether, our results demonstrate that the CRS domain can be targeted to provide pain relief in chronic pain conditions without altering physiological nociception.
Fig. 4.

Disruption of the NaV1.7–CRMP2 interaction alleviates mechanical allodynia without affecting physiological pain. (A) CRMP2-binding intensity to NaV1.7-derived peptide #141 from contralateral (Contra) and ipsilateral (Ipsi) spinal cords of male rats taken two weeks following SNI (n = 4). (B) Time course for male and female rats following administration of Myr-TAT-SCR, or Myr-TAT-NaV1.7-CRS (20 µg in 5 µL, i.t.). (C) Area under the curve for paw withdrawal thresholds showing that Myr-TAT-NaV1.7-CRS reversed mechanical allodynia, n = 6 rats; (D) Cartoon and bar graph with scatter plot of paw withdrawal latency in the hot plate test (52 °C) (n = 10 mice). (E) Cartoon and bar graph with scatter plot of the tail flick (52 °C) test showing no effect of the Myr-TAT-NaV1.7-CRS (n = 10 mice). (F) Cartoon of the rotarod apparatus to assess motor coordination in rodents. Bar graph with scatter plot showing the latency to fall off a rotating rod was not different between the treatments. n = 7 rats; error bars indicate mean ± SEM; Mann–Whitney U test, Kruskal–Wallis, or two-way ANOVA (details are in Dataset S1), P values as indicated. The experiments were conducted by investigators blinded to treatments.
AAV Plasmid Encoding NaV1.7-CRS Reduces NaV1.7 Currents in DRG Neurons, and sEPSC Frequency and Amplitude in Spinal Cord Slices.
Having demonstrated the therapeutic potential of the CRS sequence in control of NaV1.7 function in vivo, we next asked if a gene therapy approach could result in long-term analgesic effects. Employing an adeno-associated virus (AAV) vector, we inserted the CRS sequence at the N terminus of a GFP (35) (AAV-NaV1.7-CRS) to allow tracking of transduced cells. To validate that our construct is functional, we first transfected DRG neurons and recorded whole-cell sodium currents. DRG neurons expressing AAV-NaV1.7-CRS showed a ~70% decrease in peak sodium current density compared to neurons transfected with the scrambled control plasmid (AAV-SCR) (Fig. 5 A–C). The fraction of current carried by TTX-R channels (Fig. 5D) and the biophysical properties of steady-state inactivation and voltage-dependent activation were not affected by AAV-NaV1.7-CRS (SI Appendix, Table S3). We next used ProTx-II to test if the reduction imposed by AAV-NaV1.7-CRS was due to NaV1.7. Indeed, ProTx-II did not further block sodium current in AAV-NaV1.7-CRS transfected cells (Fig. 5 E–G). Finally, we used Pitstop2 to test whether the inhibition of NaV1.7 was elicited through the same mechanism as for the Myr-TAT-NaV1.7-CRS peptide (Fig. 2 R–T). In DRG neurons transfected with AAV-NaV1.7-CRS, Pitstop2 rescued the inhibition of NaV1.7 current compared to the AAV-SCR control (Fig. 5 H–J). These results validate that the genetic expression of the CRS domain recapitulates the inhibition of NaV1.7 in DRG neurons that we observed with the Myr-TAT-NaV1.7-CRS peptide (Fig. 2).
Fig. 5.
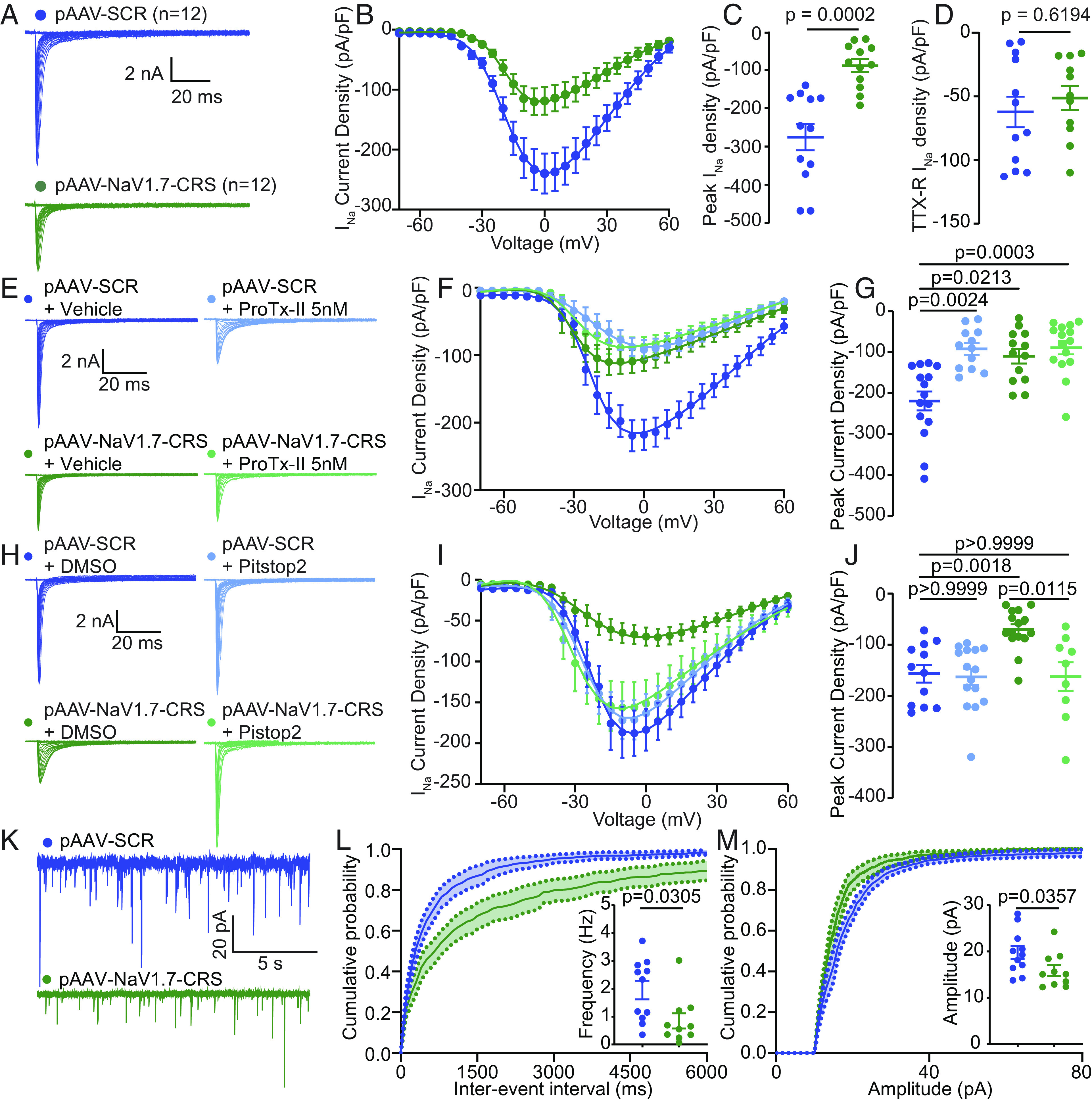
AAV plasmid encoding NaV1.7-CRS reduces NaV1.7 currents through an endocytic mechanism and decreases spontaneous activity in the spinal cord. (A) Current traces recorded from DRGs transfected with pAAV-SCR (blue, n = 12) or pAAV-NaV1.7-CRS (green, n = 12). (B) Summary plots of current density–voltage relationship fitted with Boltzmann curve, (C) peak current density, and (D) electrically isolated TTX-R currents. (E) Current traces from transfected DRG neurons treated with vehicle or 5 nM ProTx-II. (F) Summary plots of current density–voltage relationship fitted with Boltzmann curve and (G) peak current density. (H) Current traces from groups transfected with pAAV-SCR or pAAV-NaV1.7-CRS and treated with Pitstop2 (20 µM). (I) Summary plots of current density–voltage relationship and (J) peak current density. Values for biophysical parameters in SI Appendix, Table S3. n = 9 to 15 cells. (K) Traces of spontaneous excitatory postsynaptic currents (sEPSC) from rat substantia gelatinosa (SG) neurons transduced with AAV-SCR (blue) or AAV-NaV1.7-CRS (green). (L) Cumulative distribution and bar graph of sEPSC interevent intervals from NaV1.7-CRS transduced neurons. (M) Cumulative distribution and bar graph showing decreased sEPSC amplitude in AAV-NaV1.7-CRS-treated rat slices compared to control. Data expressed as means ± SEM. Mann–Whitney U test and Kruskal–Wallis tests (details in Dataset S1).
NaV1.7 regulates spinal nociceptive neurotransmission from sensory neurons that synapse on second-order neurons in outer laminae of the spinal cord dorsal horn. We then tested whether AAV-NaV1.7-CRS could silence spinal neurotransmission similar to NaV1.7 inhibition (21, 36). In spinal cord slices prepared from rats injected with AAV-NaV1.7-CRS (Fig. 5K), we observed a decreased frequency (Fig. 5L) as well as decreased amplitude of spontaneous excitatory postsynaptic potentials (sEPSC) (Fig. 5M), when compared to AAV-SCR control. Collectively, these data show that NaV1.7 inhibition via targeting of the CRS domain recapitulates the spinal phenotype of NaV1.7 loss of function.
AAV9 Delivery of NaV1.7-CRS Reverses and Prevents the Development of Mechanical Allodynia Following SNI.
Our in vitro observations demonstrated that competitive blockade of the CRS domain using either a peptide (Fig. 2) or a genetically encoded sequence (Fig. 5) leads to complete inhibition of NaV1.7 function in DRG sensory neurons. To test the long-term effects of blocking the CRS domain, we packaged the AAV-NaV1.7-CRS plasmid into a functional AAV9 capsid (Fig. 6A). Intrathecal injection (lumbar puncture) of 1 × 1010 viral particles resulted in expression of our construct in both DRG (Fig. 6B) and spinal cord (Fig. 6C). We next leveraged this gene therapy approach to test for the development of potential tolerance and unwanted effects of constant NaV1.7 inhibition by CRS targeting in vivo (SI Appendix, Fig. S10). Male mice with SNI were injected with AAV9-NaV1.7-CRS or control virus. Mice injected with AAV9-NaV1.7-CRS demonstrated a reversal of mechanical allodynia that was maintained for more than 30 d following the injection compared to mice injected with control virus (Fig. 6 D and E). Before termination of the experiment, mice were subjected to an open field test, where they exhibited no impairment of motor capacity (Fig. 6F) or development of anxiety-like behaviors (Fig. 6G). The relief of mechanical allodynia and lack of other off-target effects was recapitulated in female mice injected with AAV9-NaV1.7-CRS (Fig. 6 H–K). These data show that targeting the CRS domain of NaV1.7 can provide sustained relief of painful behaviors in male and female mice with chronic neuropathic pain.
Fig. 6.

AAV9 plasmid encoding NaV1.7-CRS reverses and prevents mechanical allodynia in a mouse model of neuropathic pain. (A) Experimental paradigm for testing AAV9 vector expressing CRS domain in mice. AAV9-NaV1.7-CRS virus was administered intrathecally following establishment of SNI-induced neuropathic pain (SI Appendix, Fig. S10). GFP fluorescence indicates successful injection in DRGs (B) and spinal cord (C). (D) Paw withdrawal thresholds of male mice injected with AAV9-NaV1.7-CRS or control peptide 7 d after SNI. (E) Area under the curve showing pain reversal by AAV-NaV1.7-CRS. (F) Locomotor activity and (G) anxiety-like behavior in open field test. Results replicated in female mice for paw withdrawal threshold (H), area under the curve (I), locomotor activity (J), and anxiety (K), indicating pain reversal in both sexes. Intrathecal injection of AAV9 performed 7 d before SNI surgery (SI Appendix, Fig. S10). Prevention of chronic allodynia in male mice injected with AAV9-NaV1.7-CRS shown by paw withdrawal thresholds (L) and area under the curve (M). No effect on locomotor activity (N) and anxiety-like behavior (O). Data replicated in female mice for paw withdrawal threshold (P), area under the curve (Q), locomotor activity (R), and anxiety (S). AAV9-NaV1.7-CRS prevented chronic pain in both male and female mice. n = 5 to 9 animals; error bars indicate mean ± SEM; detailed statistical analysis in Dataset S1. Experiments were conducted by investigators blinded to treatments.
As other transgenic models of NaV1.7 loss of function (33, 34, 37) were resistant to the development of chronic neuropathic pain, we next asked if targeting the CRS domain could be used to prevent the development of chronic pain following nerve injury. Naive male and female mice were injected with AAV9-NaV1.7-CRS before undergoing spared nerve injury surgery (SI Appendix, Fig. S10). In male mice, no change of the basal mechanical threshold was detected due to the injection of AAV9-NaV1.7-CRS compared to AAV9-SCR control (Fig. 6L). However, mice injected with AAV9-NaV1.7-CRS were resistant to the development of mechanical allodynia after SNI surgery while AAV9-SCR control mice became hypersensitive to mechanical stimuli (Fig. 6 L and M). As above, mice had no impairment of motor or anxiety behaviors (Fig. 6 N and O). We observed no sex difference as female mice showed a similar result (Fig. 6 P–S). Collectively, these findings reveal that the CRS domain can be targeted to prevent the initiation and maintenance of chronic neuropathic pain in both male and female mice. Therefore, the AAV9-NaV1.7-CRS could be used as a genetic therapy to provide long-lasting pain relief.
Intrathecal Injection of AAV9-NaV1.7-CRS Reverses CIPN-Induced Mechanical and Cold Allodynia.
Given the promising finding that AAV9-NaV1.7-CRS alleviates neuropathic pain induced by SNI, we next investigated whether intrathecal injection of AAV9-NaV1.7-CRS could reverse mechanical and cold allodynia induced by paclitaxel administration. Paclitaxel is a chemotherapeutic agent commonly used in the treatment of the most common cancers including those of the breast, ovary, and lung (38). Specifically, paclitaxel causes chemotherapy-induced peripheral neuropathy (CIPN), a result of peripheral nerve fiber dysfunction or degeneration (39). Among the multiple effects of paclitaxel is increased functional expression of NaV1.7 channels in rat and human sensory neurons (40), which enhances their excitability (41). This involvement of NaV1.7 led us to explore whether administration of AAV9-NaV1.7-CRS could significantly reverse the deleterious consequences of CIPN, principally mechanical and cold allodynia.
To answer this question, male and female mice were injected with paclitaxel (8 mg/kg, i.p. for a total of 4 doses) or vehicle and tested at day 7 (mechanical, von Frey) and 8 (cold, acetone test) after the first dose of paclitaxel. Since equivalent effects were observed in females and males in our SNI experiments interrupting the NaV1.7–CRMP2 interaction, we used mice from both sexes. Mice were injected intrathecally (1 × 1010 viral particles in 5 µL) with AAV9-NaV1.7-CRS or AAV9-SCR and tested for mechanical (days 14, 16, and 21) and cold (days 15, 17, and 22) allodynia. Mice treated with paclitaxel and a control plasmid (AAV9-SCR) developed mechanical hypersensitivity (Fig. 7A and Dataset S1). Conversely, mice treated with AAV9-NaV1.7-CRS fully and significantly reversed paclitaxel-induced mechanical allodynia at days 16 and 21 after the first dose of paclitaxel (Fig. 7A). When quantifying area under the curve, we found a significant antinociceptive effect in AAV9-NaV1.7-CRS-paclitaxel-treated mice compared to AAV9-SCR-paclitaxel-treated mice (Fig. 7B). Importantly, we found that AAV9-NaV1.7-CRS significantly reversed paclitaxel-induced cold hypersensitivity 15, 17, and 22 d after the first dose of paclitaxel compared to AAV9-SCR-paclitaxel-treated mice (Fig. 7C and Dataset S1). Analysis of the area under the curve shows that AAV9-NaV1.7-CRS significantly decreased nociceptive behaviors in paclitaxel-treated mice compared to AAV9-SCR-paclitaxel-treated mice (Fig. 7D). Overall, these results show that targeting the CRS domain has significant potential for pain relief in a model of paclitaxel-induced peripheral neuropathy.
Fig. 7.

AAV9-NaV1.7-CRS reverses paclitaxel-induced mechanical and cold allodynia. (A) The paw withdrawal threshold of male and female mice was measured at different time points [Baseline: time 0 (pre injection); and Day 7, 14, 16, and 21] after 4 intraperitoneal injections of paclitaxel (8 mg/kg, i.p.). Mice were treated intrathecally (i.t.) with AAV9-SCR or AAV9-NaV1.7-CRS (1 × 1010 viral particles in 5 µL) as indicated. Results were compared using three-way ANOVA with time, treatment, and virus as factors and post hoc Holm-Šídák test; *P < 0.05 Pac-AAV9-SCR vs. Veh + AAV9-SCR; &P < 0.05 Pac-AAV9-NaV1.7-CRS vs. Veh + AAV9-NaV1.7-CRS; #P < 0.05 Pac-AAV9-NaV1.7-CRS vs. Pac-AAV9-SCR). Detailed statistical analysis is in Dataset S1. (B) Area under the curve in A. Statistical significance as indicated (One-way ANOVA with Tukey’s post hoc test). Taken together, the data show that AAV9-NaV1.7-CRS reversed CIPN-induced mechanical allodynia. (C) Time of aversive responses for male and female mice was measured at different time points [Baseline: time 0 (before any injection); and Day 8, 15, 17, and 22] after 4 intraperitoneal injections of paclitaxel (8 mg/kg, i.p.). Mice were treated intrathecally (i.t.) with AAV9-SCR or AAV9-NaV1.7-CRS (1 × 1010 viral particles in 5 µL) as indicated. Results were compared using three-way ANOVA with time, treatment, and virus as factors and post hoc Holm-Šídák test; *P < 0.05 Pac-AAV9-SCR vs. Veh + AAV9-SCR; &P < 0.05 Pac-AAV9-NaV1.7-CRS vs. Veh + AAV9-NaV1.7-CRS; #P < 0.05 Pac-AAV9-NaV1.7-CRS vs. Pac-AAV9-SCR). Detailed statistical analysis is in Dataset S1. (D) Area under the curve in C. Statistical significance as indicated in the figure (one-way ANOVA with Tukey’s post hoc test). Taken together, the data show that AAV9-NaV1.7-CRS reversed CIPN-induced cold allodynia (acetone test) in both male and female mice. n = 8 animals; error bars indicate mean ± SEM; detailed statistical analysis is in Dataset S1. The experiments were conducted by investigators blinded to treatments.
AAV9-NaV1.7-CRS Reduces Na+ Currents through NaV1.7 Channels in Macaque Sensory Neurons.
To validate our findings in a translationally relevant model organism, we gained access to Rhesus macaque DRG and spinal cord tissues. We hybridized macaque spinal cord lysate on our peptide array and found that macaque CRMP2 bound to the same CRS sequence in the intracellular loop 1 of NaV1.7 (Fig. 8A). Next, using coimmunoprecipitation, we showed that i) the CRMP2/NaV1.7 interaction was conserved from rodents to nonhuman primates and that ii) the Myr-TAT-NaV1.7-CRS peptide could block this interaction in macaque spinal cord lysates (Fig. 8B). We then treated cultured macaque DRG neurons with the AAV9-NaV1.7-CRS and recorded sodium currents (Fig. 8C). Sensory neurons transduced with AAV9-NaV1.7-CRS had reduced total sodium current density (Fig. 8 D and E) compared to the AAV9-SCR treated group. Additionally, treatment with the NaV1.7 inhibitor ProTx-II demonstrated that maximal NaV1.7 inhibition was achieved following transduction with AAV9-NaV1.7-CRS (Fig. 8 D and E). Treatment with AAV9-NaV1.7-CRS had no impact on the half-maximal activation voltage (Fig. 8F and SI Appendix, Table S4). To test if TTX-R currents were impacted by AAV9-NaV1.7-CRS in macaque DRG neurons, we applied TTX. In this condition, AAV9-NaV1.7-CRS and AAV9-SCR control were indistinguishable (Fig. 8 G–J). Taken together, our results show that selective NaV1.7 inhibition can be achieved by targeting the CRS domain in DRG neurons from nonhuman primates.
Fig. 8.
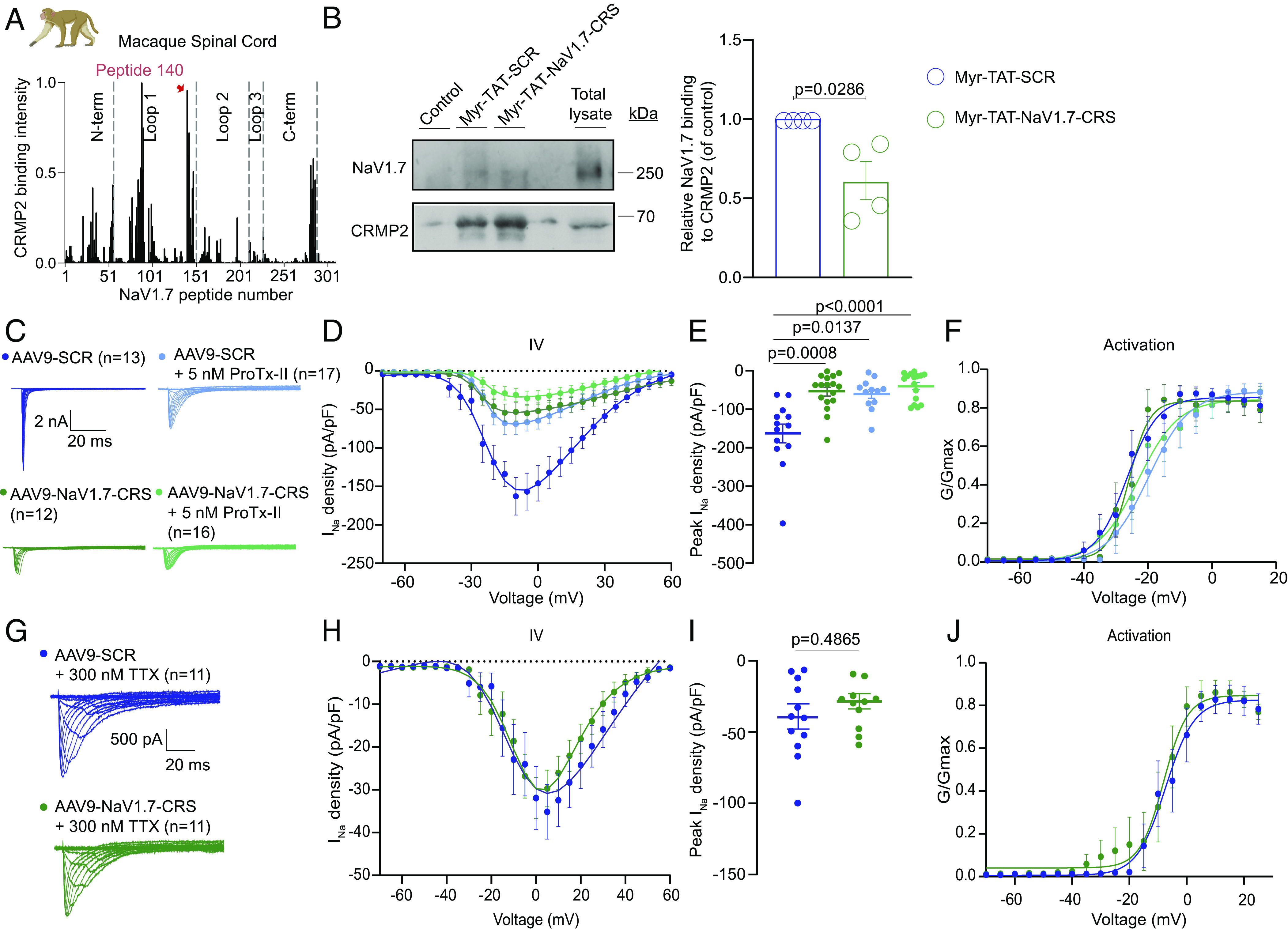
Macaque DRG neurons transduced by AAV9-NaV1.7-CRS show reduced NaV1.7 currents with no effect on TTX-R currents. (A) Fluorescent intensity of CRMP2 binding to peptide array from macaque spinal cord lysate (n = 2). Highlighted in red is the broadest peak with the highest CRMP2 binding, corresponding to peptide #140. (B) Immunoblots (Left) and summary (Right) of mean relative NaV1.7 binding to CRMP2 in spinal cord lysates treated with indicated peptides (n = 4). Error bars show mean ± SEM; P values as indicated; Mann–Whitney U test. (C) Current traces from small-sized DRGs transduced with AAV9-NaV1.7 or AAV9-SCR, coapplying 5 nM ProTx-II, a NaV1.7 inhibitor. (D) Boltzmann fits for current density–voltage curves. (E) Summary of peak current densities (pA/pF). (F) Boltzmann fits of voltage-dependent activation. Half-maximal activation potential and slope values in SI Appendix, Table S4. N = 12 to 17 cells; error bars indicate mean ± SEM; P values as indicated; One-way ANOVA with Tukey’s post hoc test. (G) Current traces from small-sized DRGs transduced with AAV9-NaV1.7 or AAV9-SCR, coapplying 300 nM TTX. The remaining current is exclusively TTX-R (NaV1.8 and NaV1.9) sodium channels. (H) Boltzmann fits for current density–voltage curves. (I) Summary of peak current densities (pA/pF). (J) Boltzmann fits of voltage-dependent activation. Half-maximal activation potential and slope values in SI Appendix, Table S3. N = 11 cells; error bars indicate mean ± SEM; P values as indicated; One-way ANOVA with Tukey’s post hoc test.
Discussion
We report the discovery and characterization of a previously unknown intracellular domain of NaV1.7, necessary for the membrane localization and function of the channel. We validated this CRMP2 regulatory sequence in NaV1.7 across five different species from rodents (mice and rats) to more translational systems (pig, macaque, and human). We further demonstrated that while the CRS domain is conserved across species, it is divergent among the NaV1.x subtypes. Inhibition of the CRS domain either with a decoy peptide or genetically i) selectively decreased NaV1.7 current density in rat and macaque DRG neurons, ii) dampened DRG neuron excitability via, iii) decreased membrane localization of NaV1.7, iv) reduced spinal neurotransmission, which v) attenuated neurotransmitter release from the spinal cord. Additionally, we demonstrated that competitive inhibition of the CRS domain with a blocking peptide reversed mechanical allodynia in vivo in male and female rats with chronic neuropathic pain. In mice, we used a gene therapy approach to show that CRS inhibition could prevent the initiation and maintenance of chronic neuropathic pain. Furthermore, we demonstrated that this gene therapy was effective at reversing mechanical and cold allodynia in model of paclitaxel-induced peripheral neuropathy. These effects were achieved without any unwanted effects on physiological pain thresholds, motor coordination, or anxiety. Our findings point to the identification of a targetable domain on NaV1.7 that can be leveraged to develop safe and selective pain-relieving drugs with significant clinical potential.
Our previous studies identified CRMP2 as a major regulator of NaV1.7 membrane localization. We reported that NaV1.7 internalization is regulated by formation of a complex between CRMP2, the endocytic adaptor Numb, the E3 ubiquitin ligase Nedd4-2, and the endocytic protein Eps15 (19). We developed a compound, 194, which inhibits CRMP2 SUMOylation to selectively reduce NaV1.7 currents. 194 reversed mechanical allodynia in multiple animal models of neuropathic pain (21, 42, 43). However, the underlying basis of the selective regulation of NaV1.7 by CRMP2 was never elucidated. We now conclude that this regulation is achieved via a specific binding domain for CRMP2 on NaV1.7 that is not found in any other sodium channel. Using the TTX-R Halo-NaV1.7 construct, when combined with an electrical isolation protocol, allowed us to investigate the role of the CRS domain directly in DRG neurons by identifying transfected cells and eliminating the contribution of endogenous sodium channels. We thus found that the CRS domain on NaV1.7 is necessary for the membrane localization and function of the channel. We further show that competitive inhibition of the CRS domain with a peptide, or by genetic means, can selectively inhibit NaV1.7. This is supported by the lack of further blockade of sodium currents in rat or macaque DRG neurons treated with Myr-TAT-NaV1.7-CRS and ProTx-II treatment. Evidence for this conclusion can also be found in our observation that ProTx-II did not induce a rightward shift of half voltage activation in these neurons, thus showing that NaV1.7 was fully blocked and no longer available. Importantly, our data consistently support clathrin-mediated internalization as the mechanism by which NaV1.7 can be down-regulated to provide pain relief. Rather than blocking the pore of the channel directly, indirect modulation of NaV1.7 function by dissociating accessory proteins such as CRMP2, helped us achieve specific inhibition of the channel. This specificity of interaction was further validated in a screen against all orphan human GPCRs (PDSP) where the Myr-TAT-NaV1.7-CRS peptide had no effect.
Targeting the CRS domain specifically addresses chronic neuropathic pain without affecting physiological pain thresholds. CRMP2 extracted from the ipsilateral (painful) spinal dorsal horn showed increased binding to the CRS sequence, consistent with its involvement in chronic pain. Blocking the CRS sequence did not impact acute pain models or physiological pain thresholds. However, it efficiently reversed mechanical allodynia in injury-based and chemotherapy-induced chronic neuropathic pain models. Our peptide-based approach selectively inhibits NaV1.7 function without affecting physiological pain, as other channels can compensate for NaV1.7 absence. Thus, our work supports targeting the CRS domain as it spares protective pain responses while silencing chronic pain.
Small-molecule inhibitors have many advantages but have been notoriously difficult to develop for NaV1.7. A recent report described an alternative approach based on in situ repression of NaV1.7 using AAV-mediated delivery of zinc finger proteins or dCas9, which successfully produced antinociception in multiple mouse models of inflammatory and neuropathic pain (44). Gene therapy is particularly attractive for pain treatment because it would replace other pharmacological options that require daily dosing and equates to treating a chronic disease with a chronic treatment. Here, we leveraged our identification of this unique domain regulating NaV1.7 to form the foundation of a genetic therapy approach. Viral delivery of the NaV1.7-CRS peptide reversed established chronic neuropathic pain, whether from an injury or from a chemotherapeutic agent, but also prevented its development when given before an injury. This suggests that targeting the CRS sequence could be disease modifying and treat established chronic pain. We did not observe any signs of toxicity for the duration of our experiment, which lasted for more than one month after the viral injection. Our additional measures of motor behavior and anxiety suggest a strong safety profile for sustained inhibition of the CRS domain of NaV1.7. These observations are in line with those made with another gene therapy aiming at silencing NaV1.7 expression in the pain pathway (44).
There are a few limitations to our findings. First, we did not investigate other potential binding partners of the CRS domain besides CRMP2. It’s possible that unknown proteins could also interact with NaV1.7 through the CRS domain and regulate its function. Additionally, we didn’t identify the specific protein complex responsible for internalizing NaV1.7 when the CRMP2/NaV1.7 interaction is disrupted. Previous research suggests that endocytic proteins are recruited when CRMP2 SUMOylation is lost and that preventing CRMP2 SUMOylation could be used as a treatment for chronic pain (17, 21, 37), but the exact proteins involved in NaV1.7 internalization without CRMP2 binding remain unknown. Another limitation is that our study primarily relied on rodent pain models, and it has been shown that the pharmacology of NaV1.7 inhibitors can differ between rat and human sensory neurons (27). However, our findings demonstrate that the NaV1.7–CRS interaction is conserved across species, including rat, pig, macaque, and human, suggesting that our observations in rodent tissues may be relevant to human studies. Lastly, it’s important to consider potential drawbacks of gene therapies. Recent reports have raised concerns about neuronal toxicity associated with the AAV9 serotype (45), which we used in this study. Therefore, it’s possible that long-term sensory neuron toxicity could occur beyond the duration of our experiment, as our focus was primarily on assessing the tolerance to sustained inhibition of the NaV1.7 CRS domain.
In summary, we discovered a key intracellular domain on NaV1.7, essential for the membrane localization and function of the channel. We demonstrate that this domain can be targeted to reduce the excitability of sensory neurons, spinal neurotransmission and can ultimately abolish chronic neuropathic pain. We expect that the targeting of the CRS domain can be used as a viable strategy for developing future therapeutics that are highly specific and safe for treating chronic pain.
Materials and Methods
Detailed descriptions of each experiment and any associated references are available in SI Appendix, Materials and Methods. In brief, all electrophysiology, biochemistry, and behavior experiments were performed according to established protocols (21, 37, 46). All animal protocols were approved by the Institutional Animal Care and Use Committee of the College of Medicine at the University of Arizona and Virginia Commonwealth University and conducted in accordance with the Guide for Care and Use of Laboratory Animals published by the NIH. Sample sizes were determined based on our experience with electrophysiological, biochemical, and behavioral experiments in our laboratory. Experimenters were blind to the treatment and the animals were randomly assigned to experimental groups.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (PDF)
Acknowledgments
We thank Drs. Frank Porreca and Amol Patwardhan for helpful discussions on this project. Supported by NIH awards [NINDS (NS098772 to R.K. and NS120663 to R.K.)], and NIDA (DA042852 to R.K.), A.M. is supported by NINDS NS119263.
Author contributions
M.I.D., A.M., and R.K. designed research; K.G., H.J.S., P.D., S.L.L., C.T., A.C.-R., L.F.-M., M.K., C.L.M., S.L., B.M., E.C., D.R., L.B., S.P.-M., M.I.D., A.M., and R.K. performed research; R.K. contributed new reagents/analytic tools; K.G., H.J.S., P.D., S.L.L., C.T., A.C.-R., L.F.-M., M.K., C.L.M., S.L., B.M., D.R., L.B., A.M., and R.K. analyzed data; R.K. provided funding; and K.G., H.J.S., and R.K. wrote the paper.
Competing interests
R. Khanna, M. Khanna, and V. Gokhale are the co-founders of Regulonix LLC, a company developing non-opioids drugs for chronic pain. R. Khanna, M. Khanna, R. Chawla, and V. Gokhale have patents US10287334 (Non-narcotic CRMP2 peptides targeting sodium channels for chronic pain) and US10441586 (SUMOylation inhibitors and uses thereof) issued to Regulonix LLC.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
All study data are included in the article and/or supporting information.
Supporting Information
References
- 1.Dib-Hajj S. D., Yang Y., Black J. A., Waxman S. G., The Na(V)1.7 sodium channel: From molecule to man. Nat. Rev. Neurosci. 14, 49–62 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Rush A. M., Cummins T. R., Waxman S. G., Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J. Physiol. 579, 1–14 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennett D. L., Woods C. G., Painful and painless channelopathies. Lancet Neurol. 13, 587–599 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Cox J. J., et al. , An SCN9A channelopathy causes congenital inability to experience pain. Nature 444, 894–898 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hong S., Morrow T. J., Paulson P. E., Isom L. L., Wiley J. W., Early painful diabetic neuropathy is associated with differential changes in tetrodotoxin-sensitive and -resistant sodium channels in dorsal root ganglion neurons in the rat. J. Biol. Chem. 279, 29341–29350 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mukai M., et al. , Evaluation of behavior and expression of NaV1.7 in dorsal root ganglia after sciatic nerve compression and application of nucleus pulposus in rats. Eur. Spine J. 23, 463–468 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laedermann C. J., et al. , Dysregulation of voltage-gated sodium channels by ubiquitin ligase NEDD4-2 in neuropathic pain. J. Clin. Invest. 123, 3002–3013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McDonnell A., et al. , Efficacy of the Nav1.7 blocker PF-05089771 in a randomised, placebo-controlled, double-blind clinical study in subjects with painful diabetic peripheral neuropathy. Pain 159, 1465–1476 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Alexandrou A. J., et al. , Subtype-Selective Small Molecule Inhibitors Reveal a Fundamental Role for Nav1.7 in Nociceptor Electrogenesis, Axonal Conduction and Presynaptic Release. PLoS One 11, e0152405 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vetter I., et al. , Na(V)1.7 as a pain target–From gene to pharmacology. Pharmacol. Ther. 172, 73–100 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Siebenga P., et al. , Lack of detection of the analgesic properties of PF-05089771, a selective Na(v) 1.7 inhibitor, using a battery of pain models in healthy subjects. Clin. Transl. Sci. 13, 318–324 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kotecha M., et al. , Design of phase 3 studies evaluating vixotrigine for treatment of trigeminal neuralgia. J. Pain Res. 13, 1601–1609 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Price N., et al. , Safety and efficacy of a topical sodium channel inhibitor (TV-45070) in patients with postherpetic neuralgia (PHN): A randomized, controlled, proof-of-concept, crossover study, with a subgroup analysis of the Nav1.7 R1150W genotype. Clin. J. Pain 33, 310–318 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zakrzewska J. M., et al. , Safety and efficacy of a Nav1.7 selective sodium channel blocker in patients with trigeminal neuralgia: A double-blind, placebo-controlled, randomised withdrawal phase 2a trial. Lancet Neurol. 16, 291–300 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Moutal A., et al. , Blocking CRMP2 SUMOylation reverses neuropathic pain. Mol. Psychiatry 23, 2119–2121 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dustrude E. T., et al. , A single structurally conserved SUMOylation site in CRMP2 controls NaV1.7 function. Channels (Austin) 11, 316–328 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Francois-Moutal L., et al. , Inhibition of the Ubc9 E2 SUMO-conjugating enzyme-CRMP2 interaction decreases NaV1.7 currents and reverses experimental neuropathic pain. Pain 159, 2115–2127 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moutal A., Luo S., Largent-Milnes T. M., Vanderah T. W., Khanna R., Cdk5-mediated CRMP2 phosphorylation is necessary and sufficient for peripheral neuropathic pain. Neurobiol. Pain 5, 100022 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dustrude E. T., et al. , Hierarchical CRMP2 posttranslational modifications control NaV1.7 function. Proc. Natl. Acad. Sci. U.S.A. 113, E8443–E8452 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu J., et al. , Phosphorylated CRMP2 regulates spinal nociceptive neurotransmission. Mol. Neurobiol. 56, 5241–5255 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cai S., et al. , Selective targeting of NaV1.7 via inhibition of the CRMP2-Ubc9 interaction reduces pain in rodents. Sci. Transl. Med. 13, eabh1314 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y., et al. , Nav1.7 is phosphorylated by Fyn tyrosine kinase which modulates channel expression and gating in a cell type-dependent manner. Mol. Pain 14, 1744806918782229 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kerth C. M., Hautvast P., Korner J., Lampert A., Meents J. E., Phosphorylation of a chronic pain mutation in the voltage-gated sodium channel Nav1.7 increases voltage sensitivity. J. Biol. Chem. 296, 100227 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cantrell A. R., Smith R. D., Goldin A. L., Scheuer T., Catterall W. A., Dopaminergic modulation of sodium current in hippocampal neurons via cAMP-dependent phosphorylation of specific sites in the sodium channel alpha subunit. J. Neurosci. 17, 7330–7338 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herzog R. I., Cummins T. R., Ghassemi F., Dib-Hajj S. D., Waxman S. G., Distinct repriming and closed-state inactivation kinetics of Nav1.6 and Nav1.7 sodium channels in mouse spinal sensory neurons. J. Physiol. 551, 741–750 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akin E. J., et al. , Building sensory axons: Delivery and distribution of NaV1.7 channels and effects of inflammatory mediators. Sci. Adv. 5, eaax4755 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang X., Priest B. T., Belfer I., Gold M. S., Voltage-gated Na+ currents in human dorsal root ganglion neurons. ELife 6, e23235 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sarko D., et al. , The pharmacokinetics of cell-penetrating peptides. Mol. Pharm. 7, 2224–2231 (2010). [DOI] [PubMed] [Google Scholar]
- 29.François-Moutal L., et al. , A membrane-delimited N-myristoylated CRMP2 peptide aptamer inhibits CaV2.2 trafficking and reverses inflammatory and postoperative pain behaviors. Pain 156, 1247–1264 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dustrude E. T., Wilson S. M., Ju W., Xiao Y., Khanna R., CRMP2 protein SUMOylation modulates NaV1.7 channel trafficking. J. Biol. Chem. 288, 24316–24331 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bennett D. L., Clark A. J., Huang J., Waxman S. G., Dib-Hajj S. D., The role of voltage-gated sodium channels in pain signaling. Physiol. Rev. 99, 1079–1151 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Lee S. E., Kim J. H., Involvement of substance P and calcitonin gene-related peptide in development and maintenance of neuropathic pain from spinal nerve injury model of rat. Neurosci. Res. 58, 245–249 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Gingras J., et al. , Global Nav1.7 knockout mice recapitulate the phenotype of human congenital indifference to pain. PLoS One 9, e105895 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shields S. D., et al. , Insensitivity to pain upon adult-onset deletion of Nav1.7 or its blockade with selective inhibitors. J. Neurosci. 38, 10180–10201 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu H., et al. , AAV-encoded CaV2.2 peptide aptamer CBD3A6K for primary sensory neuron-targeted treatment of established neuropathic pain. Gene. Ther. 26, 308–323 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alles S. R. A., et al. , Sensory neuron-derived NaV1.7 contributes to dorsal horn neuron excitability. Sci. Adv. 6, eaax4568 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moutal A., et al. , Studies on CRMP2 SUMOylation-deficient transgenic mice identify sex-specific Nav1.7 regulation in the pathogenesis of chronic neuropathic pain. Pain 161, 2629–2651 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hagiwara H., Sunada Y., Mechanism of taxane neurotoxicity. Breast Cancer 11, 82–85 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Beijers A. J., Jongen J. L., Vreugdenhil G., Chemotherapy-induced neurotoxicity: The value of neuroprotective strategies. Neth. J. Med. 70, 18–25 (2012). [PubMed] [Google Scholar]
- 40.Li Y., et al. , DRG voltage-gated sodium channel 1.7 is upregulated in paclitaxel-induced neuropathy in rats and in humans with neuropathic pain. J. Neurosci. 38, 1124–1136 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang H., Dougherty P. M., Enhanced excitability of primary sensory neurons and altered gene expression of neuronal ion channels in dorsal root ganglion in paclitaxel-induced peripheral neuropathy. Anesthesiology 120, 1463–1475 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J., Stratton H. J., Lorca S. A., Grace P. M., Khanna R., Small molecule targeting NaV1.7 via inhibition of the CRMP2-Ubc9 interaction reduces pain in chronic constriction injury (CCI) rats. Channels (Austin) 16, 1–8 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Braden K., Stratton H. J., Salvemini D., Khanna R., Small molecule targeting NaV1.7 via inhibition of the CRMP2-Ubc9 interaction reduces and prevents pain chronification in a mouse model of oxaliplatin-induced neuropathic pain. Neurobiol. Pain 11, 100082 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moreno A. M., et al. , Long-lasting analgesia via targeted in situ repression of Na(V)1.7 in mice. Sci. Transl. Med. 13, eaay9056 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hordeaux J., et al. , MicroRNA-mediated inhibition of transgene expression reduces dorsal root ganglion toxicity by AAV vectors in primates. Sci. Transl. Med. 12, eaba9188 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Moutal A., et al. , SARS-CoV-2 spike protein co-opts VEGF-A/neuropilin-1 receptor signaling to induce analgesia. Pain 162, 243–252 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (PDF)
Data Availability Statement
All study data are included in the article and/or supporting information.