

Abstract
Objectives:
Anti-synthetase syndrome (ASSD) is a heterogeneous autoimmune disease characterized by multi-system involvement with a wide variety of manifestations. Validated classification criteria are necessary to improve recognition and prevent misclassification, especially given the lack of reliable and standardized autoantibody testing. We systematically reviewed the literature to analyze proposed ASSD criteria, characteristics, and diagnostic performance.
Methods:
We searched PubMed and Embase databases (01/01/1984 to 06/11/2018) and the ACR and EULAR meeting abstracts (2017–2018). Sensitivities, specificities, positive, negative likelihood ratios and risk of bias were calculated for ASSD criteria and key variables reported in the literature. We performed meta-analysis when appropriate.
Results:
We retrieved 4,358 studies. We found 85 proposed ASSD criteria from a total of 82 studies. All but one study included anti-synthetase autoantibody (ARS) positivity in the ASSD criteria. Most studies required only one ASSD feature plus anti-ARS to define ASSD (n=64, 78%), whereas 16 studies required more than one ASSD variable plus anti-ARS. The only criteria not including anti-ARS positivity required 5 ASSD clinical features. We found limited data and wide variability in the diagnostic performance of each variable and definition proposed in the literature. Given these limitations we only meta-analyzed the performance of individual muscle biopsy and clinical variables in diagnosing ASSD which performed poorly.
Conclusions
The current ASSD criteria include a variety of serological, clinical, and histological features with wide variability amongst proposed definitions and the performance of these definitions has not been tested. This systematic literature review suggests the need for additional data and consensus-driven classification criteria for ASSD.
Keywords: Anti-synthetase syndrome, Systematic Literature Review, Diagnosis
INTRODUCTION
Anti-synthetase syndrome (ASSD) is a rare and heterogeneous systemic autoimmune disease (SAID) characterized by the positivity of anti-aminoacyl-tRNA synthetase (anti-ARS) autoantibodies, including anti-Jo-1, anti-PL-7, anti-PL1–2, anti-EJ, anti-OJ, anti-KS, anti-Zo, anti-YRS/HA and by the occurrence of a broad spectrum of clinical features involving many organs, including the muscle, lung, joints and skin (1–3).
ASSD was defined as a distinct clinical entity for the first time in 1990 by Marguerie et al, as the presence of idiopathic inflammatory myositis, interstitial lung disease (ILD) and other clinical manifestations such as arthritis, Raynaud phenomenon, keratoconjunctivitis sicca, and subcutaneous calcinosis in patients with anti-ARS autoantibodies (4). Since then, ASSD has been considered a subtype of idiopathic inflammatory myopathy (IIM) due to being closely associated with myositis and myositis associated ILD, arthritis, and other clinical features (5). The first criteria sets for the diagnosis of ASSD, based on expert opinion, were proposed in 2010 by Connors et al. (2). A subsequent definition was proposed in 2011 by Solomon et al. (6), and in 2015 by Lega et al. (7) (Table 1). The presence of anti-ARS autoantibodies was mandatory for an ASSD diagnosis in all these criteria sets. In recent years, several studies analyzed the clinical and prognostic heterogeneity among cohorts of patients with different anti-ARS autoantibodies with highly variable results (8–15).
Table 1.
Summary of available criteria for anti-synthetase syndrome. ILD: interstitial lung disease; ATS: American thoracic society
| Connors (2) | Lega (7) | Solomon (6) |
|---|---|---|
| Anti-aminoacyl-tRNA synthetase autoantibody positivity plus one among: • Myositis (Bohan and Peter’s criteria) • ILD by ATS criteria • Arthritis (clinic, X-rays, self-report) • Unexplained fever • Raynaud’s phenomenon • Mechanic’s hands |
Anti-aminoacyl-tRNA synthetase autoantibody positivity plus one among: • Myositis (overt or hypomyopathic) • ILD by ATS criteria • Arthritis or arthralgia Or two among: • Unexplained fever • Raynaud’s phenomenon • Mechanic’s hands |
Anti-aminoacyl-tRNA synthetase autoantibody positivity plus (2 major criteria or 1 major plus 2 minor criteria) Major criteria: • Myositis (Bohan and Peter’s criteria) • ILD by ATS criteria Minor criteria: • Arthritis • Raynaud’s phenomenon • Mechanic’s hands |
Despite the quite unique clinical phenotypes associated with anti-ARS autoantibodies (16–22) and few proposed definitions, ASSD is not universally recognized as an autonomous and established disease entity. Moreover, anti-ARS antibodies are not universally considered to be a diagnostic marker of this syndrome, as demonstrated by the classification criteria for both IIM (23) and interstitial pneumonia with autoimmune features (IPAF) (24), that may lead to misclassification of patients positive for anti-ARS antibodies, as polymyositis or IPAF, and not ASSD specifically. Moreover, the results of many commercial assays used to detect anti-ARS autoantibodies are limited by a high rate of false positive or negative results, particularly in cases of weak positivity or low pre-test likelihood (25–27). Similar to most classification criteria in rheumatic disease, ASSD classification criteria should also require some combination of characteristic clinical features and/or serological markers with definite and probable thresholds.
The lack of a data driven and validated set of classification criteria for ASSD may lead to under-recognition and misclassification of this syndrome which is associated with high mortality and morbidity. An effort to overcome these limitations has led to the ACR/EULAR Classification of Anti-Synthetase Syndrome (CLASS) project with the goal to develop and validate a data and consensus-driven classification criteria set for ASSD using clinical and/or serological features. In this study, we performed a systematic literature review of the available definitions and unique variables of ASSD which could be evaluated as candidate definitions and variables for the development of classification criteria.
MATERIAL AND METHODS
Two research questions were identified (Q1: How is ASSD defined in the literature?; Q2: What is the diagnostic performance of the definition?). The population, intervention, comparison, outcomes and study type (PICOs) for inclusion were predetermined (supplementary Table S1), following the Preferred Reporting Items for Systematic Literature Reviews (SLR) and Meta-analyses (PRISMA) guidelines (28). A protocol for the SLR was circulated among authors), although not registered.
Literature search
PubMed and Embase were searched from January 1, 1984 (when anti-ARS autoantibodies were first described) to November 6, 2018, following the strategy derived from PICOs (supplementary Table S2). Abstracts from the EULAR and ACR 2017 and 2018 meetings, as well as references within the included studies, were hand-searched. Articles in English, French, Spanish and Italian were included. No restriction based on quality was applied. All references were transferred into a reference manager software and duplicates were eliminated.
Study selections
Two independent reviewers (S.F.K. and G.Z.) screened the titles and abstracts following the criteria defined by the PICOs. Papers were considered for full-text review when the two reviewers agreed that patients with ASSD were included. Subsequently, the full-text review selected only studies in which an explicit definition of ASSD was provided. Reasons for exclusion were standardized, prioritized, and specified for every excluded study. Disagreements about inclusion/exclusion or reason for exclusion were resolved by a third reviewer (G.S.).
Data extraction and quality assessment
We extracted the variables used to define ASSD (clinical, serological, and histological features) into pre-specified forms, and information about study design, sample number, and follow-up duration. We collected the number of the studies reporting each variable used to define ASSD, alone or in combination with others. For studies answering both Q1 and Q2, we considered the variables used to identify ASSD as the definition given by the author (answering Q1), while the diagnostic performances of other variables were tested against this definition. To assess the diagnostic accuracy, 2×2 tables were made when possible and sensitivity, specificity, likelihood ratios with 95% CIs were calculated.
For Q2, when the accuracy of a variable was evaluated by at least by 2 studies, sensitivities and specificities were presented as forest plots and sensitivity/specificity graphs. When the performance of a variable was assessed in at least four homogeneous studies, meta-analysis for data synthesis was conducted, using STATA (Stata Statistical Software: Release 11. College Station, TX: StataCorp LP). The results of each study were presented in the summary of findings tables.
The methodological quality and the risk of bias were estimated with different tools depending on study design: ROBINS (29) for systematic literature reviews, QUADAS-2 (30) for diagnostic accuracy studies, and Newcastle-Ottawa scale (31) for observational studies.
RESULTS
Study selection
After the exclusion of duplicates, 4358 studies were retrieved, and the full text of 375 was assessed to define eligibility. Among them, 77 met the criteria for inclusion. Three additional studies were included from the 2017 and 2018 EULAR abstracts and two from the hand search. The selection process is shown in Figure 1. Seventy-two studies were included only in Q1, and ten in both Q1 and Q2.
Figure 1. Flow-chart showing the study selection process and numbers of publications identified.
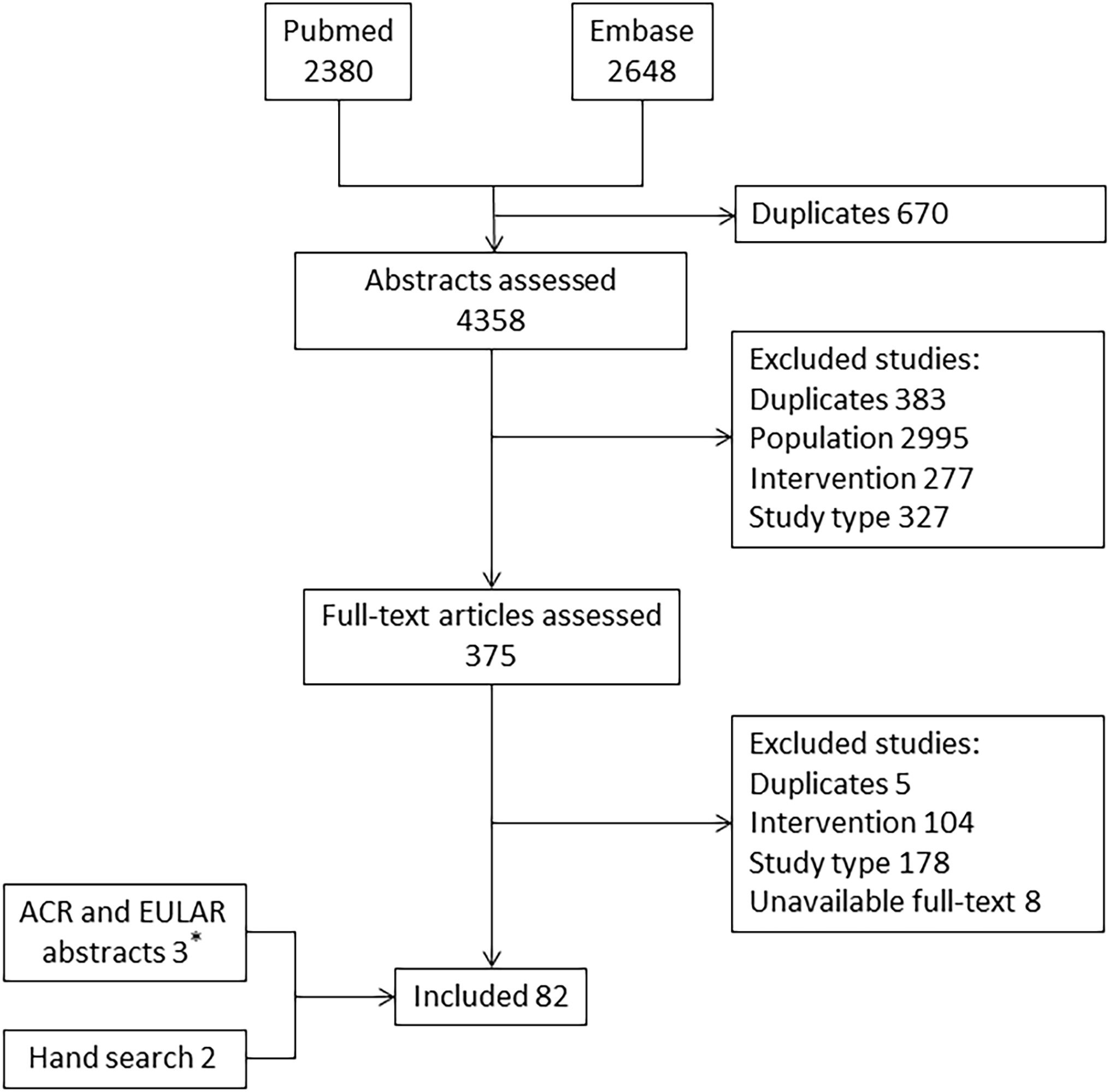
* All 3 abstracts were included from 2017 and 2018 EULAR congress.
Q1: How has ASSD been defined in the published literature?
A total of 82 studies were included. Most studies (79 out of 82) provided a single definition of ASSD. Three studies provided two definitions (3,12,32) for a total of 85 definitions. The complete list of variables retrieved is displayed in Figure 2.
Figure 2. List of variables used to define ASSD.
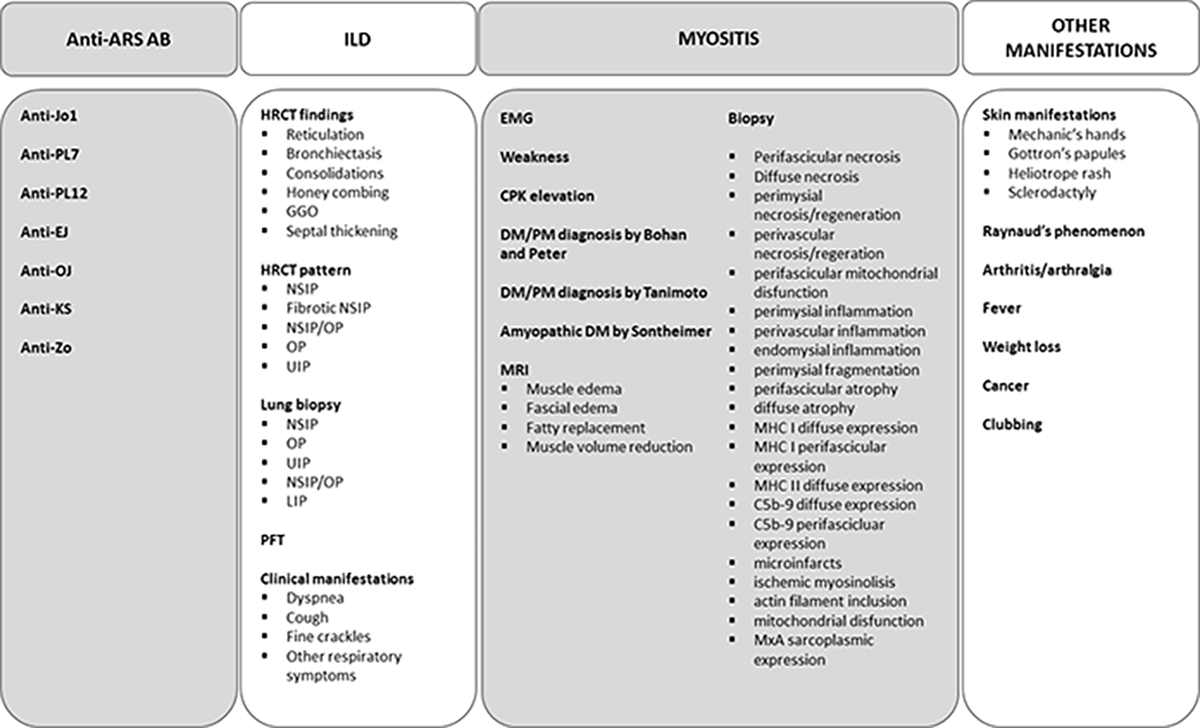
ARS-Ab: anti aminoacyl-RNA-synthetase autoantibodies; ILD: interstitial lung disease; HRCT: high resolution computed tomography; NSIP: non-specific idiopathic pneumonia; OP: organizing pneumonia; UIP: usual interstitial pneumonia; GGO: ground glass opacities; LIP: lymphoid interstitial pneumonia; PFT: pulmonary function tests; EMG: electromyography; CPK: creatine phosphokinase; DM: dermatomyositis; PM: polymyositis; MRI: magnetic resonance imaging; MHC: major histocompatibility complex.
In every included study, the presence of anti-ARS autoantibodies was used to define ASSD. Almost half of the studies used more than one (median of only three) anti-ARS autoantibody in their analysis (44 studies, 54%), while 37 only one anti-ARS autoantibody (25 anti-Jo-1, four anti-PL-12, four anti-PL-7, three anti-EJ, and one anti-KS). In only two studies, the presence of anti-ARS autoantibodies was sufficient to define ASSD (33,34), whereas only one study defined ASSD without including anti-ARS positivity. (32).
Most studies required only one variable in association with anti-ARS autoantibodies to define ASSD (64 studies, 78%), thus fitting Connor’s criteria for ASSD. The variable was myositis in 30 studies (10,32,35–61), ILD in 14 (3,62–74), and arthritis in one (75). The remaining 19 studies required one of the following features: myositis, ILD, arthritis, fever, skin rash or mechanic’s hands, and Raynaud phenomenon (12,76–93). Among the remaining definitions, 16 studies defined ASSD as the presence of more than one variable associated with anti-ARS positivity (11,12,94–107). In two studies, ASSD was defined as having a connective tissue disease (CTD) diagnosis and simultaneous positive anti-ARS (3,108). The only definition that did not encompass anti-ARS positivity required all five clinical features (myositis, ILD, arthritis, Raynaud’s phenomenon and mechanic’s hands) to define ASSD (32). The frequency of use for each variable, alone or in combination with others, is reported in Figure 3 and Table 2.
Figure 3. Available definitions of anti-synthetase syndrome, based on the combination of autoantibodies and other variables.
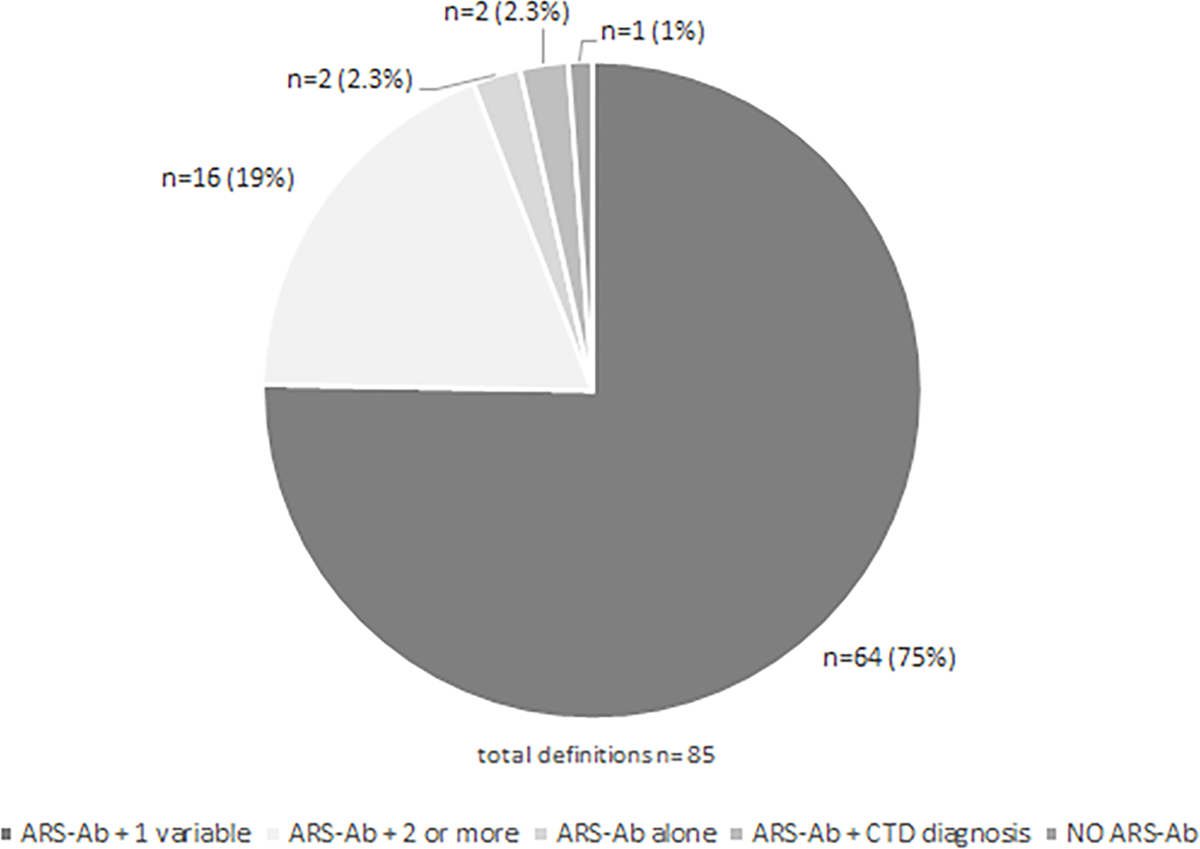
ARS-Ab: anti-synthetase autoantibodies; CTD: connective tissue disease.
Table 2.
Frequency of variables used to define anti-synthetase syndrome.
| VARIABLE | VARIABLE ALONE (*EXCLUSIVE) | VARIABLE ALONE (**NOT EXCLUSIVE) | VARIABLE IN ASSOCIATION WITH OTHERS | TOTAL (Total definitions N=85) | |
|---|---|---|---|---|---|
|
Anti-synthetase
autoantibodies |
Anti-Jo1 | 0 | 0 | 0 | 67 |
| Anti-PL12 | 0 | 0 | 0 | 46 | |
| Anti-PL7 | 0 | 0 | 0 | 40 | |
| Anti-EJ | 0 | 0 | 0 | 23 | |
| Anti-OJ | 0 | 0 | 0 | 16 | |
| Anti-KS | 0 | 0 | 0 | 10 | |
| Anti-Zo | 0 | 0 | 0 | 1 | |
| Myositis | Overall definitions | 30 | 19 | 16 | 65 |
| DM/PM Bohan and Peter | 31 | ||||
| CPK | 10 | ||||
| EMG | 9 | ||||
| Biopsy | 9 | ||||
| Weakness | 8 | ||||
| Aldolase | 2 | ||||
| MRI | 2 | ||||
| Not defined/Other definitions | 21 | ||||
| ILD | Overall definitions | 14 | 19 | 16 | 49 |
| HRCT | 39 | ||||
| PFT | 23 | ||||
| Biopsy | 7 | ||||
| Clinical | 6 | ||||
| Other clinical manifestations | Arthritis/arthralgia | 1 | 14 | 11 | 26 |
| Raynaud’s phenomenon | 0 | 3 | 10 | 13 | |
| Skin manifestations | 0 | 4 | 10 | 14 | |
| Fever | 0 | 2 | 5 | 7 | |
| Mechanic’s hands | 0 | 3 | 8 | 11 | |
| Sclerodactyly | 0 | 0 | 2 | 2 |
EXCLUSIVE: the variable was considered sufficient and necessary for ASSD diagnosis
NOT EXCLUSIVE: the variable was sufficient but not necessary to make the diagnosis.
EMG: electromyography; CPK: creatine phosphokinase; DM: dermatomyositis; PM: polymyositis; MRI: magnetic resonance imaging; HRCT: high resolution computed tomography; PFT: pulmonary function tests.
Among the features of the clinical triad of ASSD, myositis was the most frequently used (64 studies) (10–12,32,35–61,76–107,109), followed by ILD (49 studies) (3,11,12,32,62–74,76–107) and arthritis (26 studies) (11,12,32,75–77,79,80,83–90,93–101,104). Interestingly, the presence of myositis was defined predominantly as a combination of features, often fulfilling Bohan and Peter’s criteria (31 studies).
However, 21 studies did not report how myositis was defined. The remaining 13 definitions were obtained by a combination of muscle enzyme elevation (10 studies), electromyographic alterations (9 studies), muscle weakness (8 studies), and MRI alterations (2 studies). One study defined myositis using a single variable, muscle biopsy (61).
Nine studies used muscle biopsy findings for ASSD definition (41,55,61,76,83,86–88,90). Details of histological findings were available only in four papers. In particular, perifascicular atrophy and diffuse necrosis were the most used variables. Table S3 includes the list of all muscle biopsy variables.
ILD was mainly defined by high resolution computed tomography (HRCT) (39/49 studies), alone (15 studies), or in combination with pulmonary function tests (PFTs) (21 studies) or symptoms (three studies). ILD was not clearly defined in five studies. In four studies, ILD was defined by chest x-ray abnormalities and PFTs or by the presence of dyspnea and acute respiratory failure (two studies each). Lung biopsy, mostly in combination with other variables, was performed in seven studies. Among the 39 studies using HRCT, seven did not report any details about radiographic features or patterns. Figure S1 demonstrates the list of HRCT findings and patterns used to define ILD.
HRCT findings were reported in 21 studies. Ground-glass opacities (GGO) were included in all studies. Reticulation was reported in 17 studies, parenchymal consolidation in 14, honeycombing in 11, traction bronchiectasis in 10, and septal thickening in seven. Only 16 studies reported the ILD pattern. Among these, 16 reported non-specific interstitial pneumonia (NSIP), 14 organizing pneumonia (OP), 11 usual interstitial pneumonia (UIP), and six overlaps between NSIP and OP.
Among the 26 studies that included the articular involvement, ten provided no details on how this was defined, whereas six studies defined joint symptoms as the presence of either arthritis or arthralgia, and nine studies required joint swelling. Morning stiffness was considered a sufficient criterion in one study. Polyarticular involvement was required in four studies.
Other clinical findings were used to define ASSD in only a smaller number of studies. Only 14 studies used skin manifestations (mechanic’s hands, Gottron papules, heliotrope rash) as part of the definition (11,12,32,84,85,88,94–98,100,101,104). We noted similar observations for Raynaud phenomenon (13 studies) (11,12,32,84,85,88,94,95,97–100,104) and fever (7 studies) (12,85,88,94–96,98). Table 2 summarizes the frequencies of use of each variable.
In summary, the most frequent definition for ASSD included the presence of anti-ARS autoantibodies plus one clinical feature of ASSD (64/85 definitions, 75%). The most commonly used single variable, was myositis (65/85 definitions, 76%), primarily defined according to Bohan and Peter’s criteria (31 cases/65, 48%). Table S4 shows the complete report, including the summary of findings of each study.
The assessment of the risk of bias (Newcastle-Ottawa scale) was extremely variable due to different study designs and long temporal intervals for inclusion. The risk of bias of the systematic literature review by Lega et al.(3) was defined as unclear (ROBINS). The complete report of risk of bias for studies included in Q1 is available in Table S4.
Q2: What is the accuracy of the different definitions for diagnosing ASSD?
Ten studies were included for Q2. As a pre-specified inclusion criterion, all studies tested accuracy against the reference standard of a clinical diagnosis of ASSD, based on the presence of anti-ARS plus one or more typical clinical manifestations. In one study, anti-ARS was defined as the presence of anti-ARS autoantibodies and CTD diagnosis, characterized as IIM, SSc, SLE, RA, UCTD or MCTD (3). Table S5 includes results of these studies. The majority were cohort studies (49,54–56,58,59,62,92), except for one systematic review with meta-analysis (3) and one diagnostic accuracy study (57).
The tested variables mostly derived from muscle biopsy (7/10 studies). The only two variables assessed in at least four studies were perifascicular necrosis/atrophy and perimysial connective tissue fragmentation. Meta-analysis was performed only for the perifascicular necrosis/atrophy(six studies) (49,54,56–59), showing a pooled sensitivity of 0.54 (95%CI 0.3–0.76) and a pooled specificity of 0.55 (95%CI 0.43– 0.66). The pooled positive likelihood ratio (LR+) was 1.21 (95%CI 0.68–2.14) and the pooled negative likelihood ratio (LR−) was 0.83 (95%CI 0.44–1.55) (Figure S3). It was not possible to perform a metanalysis for perimysial connective tissue fragmentation due to model instability.
We were only able to test the diagnostic accuracy of individual variables rather than the accuracy of the provided definitions of ASSD due to the inherent nature of these studies. We extracted data on accuracy for muscle biopsy variables. Perifascicular atrophy/necrosis demonstrated a highly variable sensitivity, from 0.2 to 1, and a similar specificity, from 0.27 to 0.71 (49,54,56–59). Perimysial fragmentation showed a relatively high sensitivity (0.74–1) and a variable specificity (0.18–0.73) (49,54,57,59). Diffuse myofiber necrosis also showed highly variable sensitivity (0.1–0.89) and a specificity from 0.17 to 0.83 (49,56,57). Perimysial inflammation showed a sensitivity from 0.55 to 1 and a specificity from 0.14 to 0.46 (49,57,59). Sarcolemmal C5b-9 deposition on non-necrotic fibers was analyzed in two studies (54,57) and showed a sensitivity from 0.53 to 0.84 and a specificity from 0.25 to 0.81. The perifascicular distribution of C5b-9 was assessed in two studies (49,57) and displayed a sensitivity from 0.26 to 0.72 and a specificity from 0.14 to 0.19. Mitochondrial dysfunction showed a sensitivity from 0 to 0.2 and a specificity from 0 to 0.71 (49,56,59). HLA-ABC expression had a sensitivity from 0.79 to 0.94 and a specificity from 0.37 to 0.71 (54,57). Results are summarized in Figure 4. The biopsy variable with the best performance was the presence of nuclear actin inclusion, which discriminated ASSD from other myositis subsets with good sensitivity and specificity (0.8 and 1, respectively) (55). Also HLA-DR deposition, was able to discriminate ASSD from DM in one study, showing a sensitivity of 0.84 and a specificity of 0.72 (54). Only one study analyzed the performance of a composite definition of muscle biopsy parameters, combining the presence of perifascicular necrosis, atrophic fibers, and perimysial fragmentation. This definition was tested against a population of other myositis subsets and provided an area under the curve of 0.91 (57).
Figure 4: Studies included in Q2, assessing the performance of muscle biopsy variables to diagnose ASSD.

95% CI: 95% confidence interval; TP: true positive; FP: false positive; FN: false negative; TN: true negative.
The diagnostic accuracy for clinical variables was calculated only from two studies that defined ASSD as anti-ARS+CTD diagnosis (3) and anti-ARS+ILD (62). These variables were tested against very different control populations; IIM patients with MSA positivity other than anti-ARS and IPF patients, respectively. The sensitivity of arthritis/arthralgia ranged from 0.15 to 0.62 with a specificity of 0.64 to 0.98. The sensitivity of fever ranged from 0.15 to 0.43, with a specificity of 0.64 to 0.95. The sensitivity of Raynaud phenomenon ranged from 0.08 to 0.47, with a specificity of 0.55 to 0.98. The sensitivity of cutaneous symptoms, including Gottron sign and heliotrope rash, was around 0.31 with a specificity of 0.46 to 0.98. Mechanic’s hands’ performance was retrievable in only one study (3), with a sensitivity of 0.27 and a specificity of 0.98. The accuracy of ILD in differentiating ASSD from other IIMs was tested in the first study (3) and displayed a sensitivity of 0.7 and a specificity of 0.85.
Only one study analyzed the performance of muscle MRI (defined as anti-ARS autoantibodies plus ILD or myositis) against healthy controls. The sensitivity and specificity of muscle edema was 0.38 and 0.87, fascial edema 0.29 and 0.93, fatty replacement 0.41 and 0.96, and muscle mass reduction 0.14 and 0.93 (92).
The assessment of the risk of bias for the studies included in Q2, performed by QUADAS-2, revealed a high risk of bias, especially for patient selection. In particular, most were case-control studies, including patients with an established diagnosis and not incident cases. The risk of bias and applicability concerns are summarized in Figure S2.
DISCUSSION
This systematic literature review highlights that proposed definitions of ASSD in the literature are highly variable, purely eminence-based and lack any supporting data or consensus. ASSD appears to be a very heterogeneous condition with a variety of phenotypic and prognostic features, mimicking many other conditions clinically such as DM, RA and IPF. However, recent findings suggest that ASSD is a unique entity with a different immunopathogenic pathway, risk profile and prognostic factors compared to other forms of myositis. Therefore, it may be appropriate to consider ASSD a distinct syndrome from its mimickers, since individual manifestations of ASSD, such as mechanic’s hands, may be indistinguishable from other diseases (110). Unfortunately, the result of this systematic literature review confirms the lack of adequate data and/or a consensus-driven validated criterion for ASSD. The most widely used definitions of ASSD, fulfilling Connor’s criteria (2), consisted of anti-ARS autoantibodies plus one feature of the classical clinical triad (arthritis, myositis, or ILD). Although useful in clinical practice, this approach comes from expert opinion and lacks validation or performance assessment. This systematic review was also unable to retrieve or confirm any measure of diagnostic performance for these expert proposed definitions. This relates to the nature of the published ASSD studies that are primarily observational describing the clinical phenotype of anti-ARS positive patients. Moreover, anti-ARS autoantibodies appear necessary for ASSD diagnosis, since every study except one (32) used them to define the disease.
The over reliance on autoantibody results without establishing pre-test probability of the syndrome based on comprehensive set of clinical feature poses significant challenges. That is, the diagnosis of ASSD is not feasible if the clinician has no access to a reliable anti-ARS assay or lacks the expertise to interpret the results correctly as well as create significant dilemma in cases of false positive and negative results, which itself a significant challenge due to lack of standardized and reliable autoantibody testing (25–27,110). In addition, there are significant delays in receiving timely anti-ARS test results when done by validated assays using reliable laboratories, or by the gold standard method (immunoprecipitation) which is rarely available in clinical practice. Moreover, the search for these antibodies is often neglected especially when other diagnoses are possible, as recently reported in a cohort of ASSD patients misdiagnosed as having rheumatoid arthritis for 3–20 years (111).
Furthermore, the different ASSD manifestations, when taken singularly, may not be specific for ASSD, even in the case of anti-ARS autoantibody positivity as highlighted in the two studies that allowed the assessment of the diagnostic performance of various clinical variables (3,62). These variables were tested singularly and against different control populations, thus making their performance poorly comparable to each other and, in any case, far from being satisfactory. The performance of muscle MRI was also evaluated (92), revealing a high specificity but poor sensitivity in detecting ASSD-related myositis compared to healthy controls. Most muscle variables were not able to distinguish ASSD from other types of myositis with optimum sensitivity and specificity (49,54–59) with the exception of nuclear action aggregation, which, however, has only been examined in one study (55). Moreover, all studies included in Q2 displayed a high risk of bias related to patient selection and a case-control design that tends to overestimate diagnostic accuracy. It should also be noted that a number of ASSD patients do not undergo muscle biopsy or never develop clinical myositis. Therefore, the information emerging from these studies might not reflect a typical clinical setting.
This systematic literature review has highlighted how ASSD, since its first description, has been defined by a pleiotropic combination of clinical, laboratory, histological, and imaging measures. However, there have been absolutely no data or consensus driven attempt to develop a formal validated and widely accepted definition for ASSD, despite recent findings suggest that ASSD is a unique entity with a different immunopathogenic pathway, risk profile and prognostic factors compared to other forms of myositis or ILD (16,17,22), although individual manifestations of ASSD, such as mechanic’s hands, may be indistinguishable from other diseases (112). Meanwhile, there is a lack of performance data of the most used eminence-based definitions proposed for ASSD.
To the best of our knowledge, this is the first systematic literature review regarding ASSD definition. However, there are several limitations to the present work, mostly related to the limitations of the primary studies. In particular, the scarcity of studies reporting the diagnostic performance has limited quantitative synthesis of the results, resulting in limited evidence. More importantly, it was not possible to test a definition of ASSD based on multiple variables. This is of utmost importance since the lack of a data-driven definition of ASSD reflects the impossibility of establishing a confident and early diagnosis. The absence of a universal definition hinders the design of interventional trials in ASSD, with a major consequence on the availability of effective treatments for this condition. The present work, therefore, underpins the need to develop a data-driven and consensus led definition of ASSD as the first step of a long process with an ideal positive impact on the early diagnosis and prognosis improvement of this disease. In our view, it is necessary to develop classification criteria of probable and definite ASSD (even in the absence of serologic data), a syndrome encompassing a variety of clinical manifestations, with subsequent serological confirmation. This is crucial, especially given that the complete syndrome is not always fully expressed at the time of presentation, and the symptoms may evolve over time.
Supplementary Material
Acknowledgments
The Authors would like to thank Dr. Chiara Rebuffi, librarian at the IRCCS Policlinico San Matteo Foundation of Pavia, for developing the search strategies.
Funding
This research was supported in part by the Intramural Research Program of the NIH, National Institute of Arthritis and Musculoskeletal and Skin Diseases and National Institute of Environmental Health Sciences.
References
- 1.Mirrakhimov AE. Antisynthetase syndrome: a review of etiopathogenesis, diagnosis and management. Curr Med Chem 2015;22:1963–75. [PubMed] [Google Scholar]
- 2.Connors GR, Christopher-Stine L, Oddis CV, Danoff SK. Interstitial lung disease associated with the idiopathic inflammatory myopathies: what progress has been made in the past 35 years? Chest. 2010;138:1464–74. [DOI] [PubMed] [Google Scholar]
- 3.Lega J-C, Fabien N, Reynaud Q, et al. The clinical phenotype associated with myositis-specific and associated autoantibodies: a meta-analysis revisiting the so-called antisynthetase syndrome. Autoimmun Rev 2014;13:883–91. [DOI] [PubMed] [Google Scholar]
- 4.Marguerie C, Bunn CC, Beynon HL, et al. Polymyositis, pulmonary fibrosis and autoantibodies to aminoacyl-tRNA synthetase enzymes. Q J Med 1990;77:1019–38. [DOI] [PubMed] [Google Scholar]
- 5.Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet 2003;362:971–82. [DOI] [PubMed] [Google Scholar]
- 6.Solomon J, Swigris JJ, Brown KK. Myositis-related interstitial lung disease and antisynthetase syndrome. J Bras Pneumol 2011;37:100–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lega J-C, Reynaud Q, Belot A, Fabien N, Durieu I, Cottin V. Idiopathic inflammatory myopathies and the lung. Eur Respir Rev 2015;24:216–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monti S, Montecucco C, Cavagna L. Clinical spectrum of anti-Jo-1-associated disease. Curr Opin Rheumatol 2017;29:612–7. [DOI] [PubMed] [Google Scholar]
- 9.Trallero Araguas E, Selva O’Callaghan A, Scire CA, et al. Clinical spectrum time course comparison between pl-7, pl-12 and ej positive antisynthetase syndrome. Ann Rheum Dis 2017;76:1272–3. [Google Scholar]
- 10.Labirua-Iturburu A, Selva-O’Callaghan A, Vincze M, et al. Anti-PL-7 (anti-threonyl-tRNA synthetase) antisynthetase syndrome: clinical manifestations in a series of patients from a European multicenter study (EUMYONET) and review of the literature. Medicine (Baltimore) 2012;91:206–11. [DOI] [PubMed] [Google Scholar]
- 11.Kalluri M, Sahn SA, Oddis CV, et al. Clinical profile of anti-PL-12 autoantibody. Cohort study and review of the literature. Chest 2009;135:1550–6. [DOI] [PubMed] [Google Scholar]
- 12.Sasano H, Hagiwara E, Kitamura H, et al. Long-term clinical course of anti-glycyl tRNA synthetase (anti-EJ) antibody-related interstitial lung disease pathologically proven by surgical lung biopsy. BMC Pulm Med 2016;16:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sato S, Kuwana M, Hirakata M. Clinical characteristics of Japanese patients with anti-OJ (anti-isoleucyl-tRNA synthetase) autoantibodies. Rheumatol 2007;46:842–5. [DOI] [PubMed] [Google Scholar]
- 14.Hamaguchi Y, Fujimoto M, Matsushita T, et al. Common and distinct clinical features in adult patients with anti-aminoacyl-tRNA synthetase antibodies: heterogeneity within the syndrome. PloS One 2013;8:e60442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cavagna L, Trallero-Araguás E, Meloni F, et al. Influence of Antisynthetase Antibodies Specificities on Antisynthetase Syndrome Clinical Spectrum Time Course. J Clin Med 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pinal-Fernandez I, Casal-Dominguez M, Derfoul A, et al. Machine learning algorithms reveal unique gene expression profiles in muscle biopsies from patients with different types of myositis. Ann Rheum Dis 2020;79:1234–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.López-Mejías R, Remuzgo-Martínez S, Genre F, et al. Influence of MUC5B gene on antisynthetase syndrome. Sci Rep 2020;10:1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ponce-Gallegos MA, Ramos-Martínez E, García-Carmona A, et al. Genetic Susceptibility to Antisynthetase Syndrome Associated With Single-Nucleotide Variants in the IL1B Gene That Lead Variation in IL-1β Serum Levels. Front Med 2020;7:547186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Remuzgo-Martínez S, Atienza-Mateo B, Ocejo-Vinyals JG, et al. HLA association with the susceptibility to anti-synthetase syndrome. Joint Bone Spine 2021;88:105115. [DOI] [PubMed] [Google Scholar]
- 20.Barbasso Helmers S, Englund P, Engström M, et al. Sera from anti-Jo-1-positive patients with polymyositis and interstitial lung disease induce expression of intercellular adhesion molecule 1 in human lung endothelial cells. Arthritis Rheum 2009;60:2524–30. [DOI] [PubMed] [Google Scholar]
- 21.Adams RA, Fernandes-Cerqueira C, Notarnicola A, et al. Serum-circulating His-tRNA synthetase inhibits organ-targeted immune responses. Cell Mol Immunol 2019;1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galindo-Feria AS, Albrecht I, Fernandes-Cerqueira C, et al. Proinflammatory Histidyl-Transfer RNA Synthetase-Specific CD4+ T Cells in the Blood and Lungs of Patients With Idiopathic Inflammatory Myopathies. Arthritis Rheumatol 2020;72:179–91. [DOI] [PubMed] [Google Scholar]
- 23.Lundberg IE, Miller FW, Tjarnlund A, Bottai M. Diagnosis and classification of idiopathic inflammatory myopathies. J Intern Med 2016;280:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fischer A, Antoniou KM, Brown KK, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J 2015;46:976–87. [DOI] [PubMed] [Google Scholar]
- 25.To F, Ventín-Rodríguez C, Elkhalifa S, Lilleker JB, Chinoy H. Line blot immunoassays in idiopathic inflammatory myopathies: retrospective review of diagnostic accuracy and factors predicting true positive results. BMC Rheumatol 2020;4:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lackner A, Tiefenthaler V, Mirzayeva J, et al. The use and diagnostic value of testing myositis-specific and myositis-associated autoantibodies by line immuno-assay: a retrospective study. Ther Adv Musculoskelet Dis 2020;12:1759720X20975907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tansley SL, Li D, Betteridge ZE, McHugh NJ. The reliability of immunoassays to detect autoantibodies in patients with myositis is dependent on autoantibody specificity. Rheumatol 2020;59:2109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ 2021;372:n71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whiting P, Savović J, Higgins JPT, et al. ROBIS: A new tool to assess risk of bias in systematic reviews was developed. J Clin Epidemiol 2016;69:225–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bristol U of. Team | Bristol Medical School: Population Health Sciences | University of Bristol [Internet]. Available on: https://www.bristol.ac.uk/population-health-sciences/projects/quadas/quadas-2/team.html
- 31.Ottawa Hospital Research Institute [Internet]. Available on: http://www.ohri.ca/programs/clinical_epidemiology/oxford.asp
- 32.Braillard Poccard AS, Gomez R, Pino M, Garcia Carrasco M, Dubinsky D. Comparison of a single-centre idiopathic inflammatory myopathy cohort from argentina with the euromyositis international registry. Ann Rheum Dis 2018;77:1504. [Google Scholar]
- 33.Marie I, Josse S, Decaux O, et al. Outcome of anti-PL12 positive patients with antisynthetase syndrome. Presse Medicale 2013;42:e153–158. [DOI] [PubMed] [Google Scholar]
- 34.Stanciu R, Guiguet M, Musset L, et al. Antisynthetase syndrome with anti-Jo1 antibodies in 48 patients: pulmonary involvement predicts disease-modifying antirheumatic drug use. J Rheumatol 2012;39:1835–9. [DOI] [PubMed] [Google Scholar]
- 35.Cobo-Ibanez T, Lopez-Longo F-J, Joven B, et al. Long-term pulmonary outcomes and mortality in idiopathic inflammatory myopathies associated with interstitial lung disease. Clin Rheumatol 2019; 38(3):803–815. [DOI] [PubMed] [Google Scholar]
- 36.Schneider F, Yousem SA, Bi D, Gibson KF, Oddis CV, Aggarwal R. Pulmonary pathologic manifestations of anti-glycyl-tRNA synthetase (anti-EJ)-related inflammatory myopathy. J Clin Pathol 2014;67:678–83. [DOI] [PubMed] [Google Scholar]
- 37.Schneider F, Yousem SA, Oddis CV, Aggarwal R. Pulmonary Pathologic Manifestations of Anti-Alanyl-tRNA Synthetase (Anti-PL-12)-Related Inflammatory Myopathy. Arch Pathol Lab Med 2018;142:191–7. [DOI] [PubMed] [Google Scholar]
- 38.Bauhammer J, Blank N, Max R, et al. Rituximab in the Treatment of Jo1 Antibody-associated Antisynthetase Syndrome: Anti-Ro52 Positivity as a Marker for Severity and Treatment Response. J Rheumatol 2016;43(8):1566–74. [DOI] [PubMed] [Google Scholar]
- 39.Marie I, Dominique S, Janvresse A, Levesque H, Menard J-F. Rituximab therapy for refractory interstitial lung disease related to antisynthetase syndrome. Respir Med 2012;106:581–7. [DOI] [PubMed] [Google Scholar]
- 40.Marie I, Hatron PY, Dominique S, et al. Short-term and long-term outcome of anti-Jo1-positive patients with anti-Ro52 antibody. Semin Arthritis Rheum 2012;41:890–9. [DOI] [PubMed] [Google Scholar]
- 41.Noguchi E, Uruha A, Suzuki S, et al. Skeletal Muscle Involvement in Antisynthetase Syndrome. JAMA Neurol 2017;74(8):992–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spath M, Schroder M, Schlotter-Weigel B, et al. The long-term outcome of anti-Jo-1-positive inflammatory myopathies. J Neurol 2004;251:859–64. [DOI] [PubMed] [Google Scholar]
- 43.Casal-Dominguez M, Pinal-Fernandez I, Mego M, et al. High-resolution manometry in patients with idiopathic inflammatory myopathy: Elevated prevalence of esophageal involvement and differences according to autoantibody status and clinical subset. Muscle Nerve 2017;56:386–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marie I, Hatron P-Y, Cherin P, et al. Functional outcome and prognostic factors in anti-Jo1 patients with antisynthetase syndrome. Arthritis Res The 2013;15:R149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Allenbach Y, Guiguet M, Rigolet A, et al. Efficacy of Rituximab in Refractory Inflammatory Myopathies Associated with Anti- Synthetase Auto-Antibodies: An Open-Label, Phase II Trial. PloS One 2015;10:e0133702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marie I, Josse S, Decaux O, et al. Comparison of long-term outcome between anti-Jo1- and anti-PL7/PL12 positive patients with antisynthetase syndrome. Autoimmun Rev 2012;11:739–45. [DOI] [PubMed] [Google Scholar]
- 47.Marie I, Josse S, Decaux O, et al. Clinical manifestations and outcome of anti-PL7 positive patients with antisynthetase syndrome. Eur J Intern Med 2013;24:474–9. [DOI] [PubMed] [Google Scholar]
- 48.Pinal-Fernandez I, Casal-Dominguez M, Huapaya JA, et al. A longitudinal cohort study of the anti-synthetase syndrome: increased severity of interstitial lung disease in black patients and patients with anti-PL7 and anti-PL12 autoantibodies. Rheumatol Oxf 2017;56:999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cerbelli B, Pisano A, Colafrancesco S, et al. Anti-aminoacyl-tRNA synthetase-related myositis and dermatomyositis: clues for differential diagnosis on muscle biopsy. Virchows Arch 2018;472:477–87. [DOI] [PubMed] [Google Scholar]
- 50.Shinjo SK, Levy-Neto M. Anti-Jo-1 antisynthetase syndrome. Rev Bras Reumatol 2010;50:492–500. [DOI] [PubMed] [Google Scholar]
- 51.Vancsa A, Csipo I, Nemeth J, Devenyi K, Gergely L, Danko K. Characteristics of interstitial lung disease in SS-A positive/Jo-1 positive inflammatory myopathy patients. Rheumatol Int 2009;29:989–94. [DOI] [PubMed] [Google Scholar]
- 52.Karadimitrakis S, Plastiras SC, Zormpala A, et al. Chest CT findings in patients with inflammatory myopathy and Jo1 antibodies. Eur J Radiol 2008;66:27–30. [DOI] [PubMed] [Google Scholar]
- 53.Lecouffe-Desprets M, Hemont C, Neel A, et al. Clinical contribution of myositis-related antibodies detected by immunoblot to idiopathic inflammatory myositis: A one-year retrospective study. Autoimmunity 2018;51:89–95. [DOI] [PubMed] [Google Scholar]
- 54.Aouizerate J, De Antonio M, Baba Amer Y, et al. Myofiber HLA-DR expression: A distinctive biomarker for antisynthetase myositis. Eur J Neurol 2014;21:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stenzel W, Preusse C, Allenbach Y, et al. Nuclear actin aggregation is a hallmark of anti-synthetase syndrome-induced dysimmune myopathy. Neurology 2015;84:1346–54. [DOI] [PubMed] [Google Scholar]
- 56.Pinal-Fernandez I, Casciola-Rosen LA, Christopher-Stine L, Corse AM, Mammen AL. The Prevalence of Individual Histopathologic Features Varies according to Autoantibody Status in Muscle Biopsies from Patients with Dermatomyositis. J Rheumatol 2015;42:1448–54. [PMC free article] [PubMed] [Google Scholar]
- 57.Mescam-Mancini L, Allenbach Y, Hervier B, et al. Anti-Jo-1 antibody-positive patients show a characteristic necrotizing perifascicular myositis. Brain 2015;138:2485–92. [DOI] [PubMed] [Google Scholar]
- 58.Uruha A, Allenbach Y, Charuel J-L, et al. Diagnostic potential of sarcoplasmic MxA expression in subsets of dermatomyositis. Neuropathol Appl Neurobiol 2019; 45:513–522. [DOI] [PubMed] [Google Scholar]
- 59.Mozaffar T, Pestronk A. Myopathy with anti-Jo-1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry 2000;68:472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.La Corte R, Lo Mo Naco A, Locaputo A, Dolzani F, Trotta F. In patients with antisynthetase syndrome the occurrence of anti-Ro/SSA antibodies causes a more severe interstitial lung disease. Autoimmunity 2006;39:249–53. [DOI] [PubMed] [Google Scholar]
- 61.Uruha A, Suzuki S, Suzuki N, Nishino I Perifascicular necrosis in anti-synthetase syndrome beyond anti-Jo-1. Brain 2016;139:e50. [DOI] [PubMed] [Google Scholar]
- 62.Watanabe K, Handa T, Tanizawa K, et al. Detection of antisynthetase syndrome in patients with idiopathic interstitial pneumonias. Respir Med 2011;105:1238–47. [DOI] [PubMed] [Google Scholar]
- 63.Fischer A, Swigris JJ, du Bois RM, et al. Anti-synthetase syndrome in ANA and anti-Jo-1 negative patients presenting with idiopathic interstitial pneumonia. Respir Med;103:1719–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Debray M-P, Borie R, Revel M-P, et al. Interstitial lung disease in anti-synthetase syndrome: initial and follow-up CT findings. Eur J Radiol 2015;84:516–23. [DOI] [PubMed] [Google Scholar]
- 65.Doyle TJ, Dhillon N, Madan R, et al. Rituximab in the Treatment of Interstitial Lung Disease Associated with Antisynthetase Syndrome: A Multicenter Retrospective Case Review. J Rheumatol 2018;45:841–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sem M, Molber Ø, Lund MB, Gran JT Rituximab treatment of the anti-synthetase syndrome - A retrospective case series. Rheumatology 2009;48:968–71. [DOI] [PubMed] [Google Scholar]
- 67.Lepri G, Avouac J, Airò P, et al. Effects of rituximab in connective tissue disorders related interstitial lung disease. Clin Exp Rheumatol 2016;34:181–5. [PubMed] [Google Scholar]
- 68.Chartrand S, Swigris JJ, Peykova L, Chung J, Fischer A. A Multidisciplinary Evaluation Helps Identify the Antisynthetase Syndrome in Patients Presenting as Idiopathic Interstitial Pneumonia. J Rheumatol 2016;43:887–92. [DOI] [PubMed] [Google Scholar]
- 69.Zamarron-de Lucas Ester, Gomez Carrera L, Bonilla G, Petit D, Mangas A, Alvarez-Sala R. Antisynthetase syndrome: Analysis of 11 cases. Med Clin 2017;148:166–9. [DOI] [PubMed] [Google Scholar]
- 70.Waseda Y, Johkoh T, Egashira R, et al. Antisynthetase syndrome: Pulmonary computed tomography findings of adult patients with antibodies to aminoacyl-tRNA synthetases. Eur J Radiol 2016;85:1421–6. [DOI] [PubMed] [Google Scholar]
- 71.Koreeda Y, Higashimoto I, Yamamoto M, et al. Clinical and pathological findings of interstitial lung disease patients with anti-aminoacyl-tRNA synthetase autoantibodies. Intern Med 2010;49:361–9. [DOI] [PubMed] [Google Scholar]
- 72.Yura H, Sakamoto N, Satoh M, et al. Clinical characteristics of patients with anti-aminoacyl-tRNA synthetase antibody positive idiopathic interstitial pneumonia. Respir Med 2017;132:189–94. [DOI] [PubMed] [Google Scholar]
- 73.Vuillard C, Pineton de Chambrun M, de Prost N, et al. Clinical features and outcome of patients with acute respiratory failure revealing anti-synthetase or anti-MDA-5 dermato-pulmonary syndrome: a French multicenter retrospective study. Ann Intensive Care 2018;8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ben Salem T, Abdelkafi C, Lamloum M, Ben Ghorbel I, Houman MH. Pulmonary manifestations in antisynthetase syndrome. Tunis Med 2018;96:101–6. [PubMed] [Google Scholar]
- 75.Mumm GE, McKown KM, Bell CL Antisynthetase syndrome presenting as rheumatoid-like polyarthritis. J Clin Rheumatol 2010;16:307–12. [DOI] [PubMed] [Google Scholar]
- 76.Maturu VN, Lakshman A, Bal A, et al. Antisynthetase syndrome: An under-recognized cause of interstitial lung disease. Lung India 2016;33:20–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hervier B, Perez M, Allenbach Y, et al. Involvement of NK cells and NKp30 pathway in antisynthetase syndrome. J Immunol 2016;197:1621–30. [DOI] [PubMed] [Google Scholar]
- 78.Andersson H, Sem M, Lund MB, et al. Long-term experience with rituximab in anti-synthetase syndrome-related interstitial lung disease. Rheumatol 2015;54:1420–8. [DOI] [PubMed] [Google Scholar]
- 79.Bachmeyer C, Tillie-Leblond I, Lacert A, Cadranel J, Aractingi S «Mechanic’s hands»: A misleading cutaneous sign of the antisynthetase syndrome. Br J Dermatol 2007;156:192–4. [DOI] [PubMed] [Google Scholar]
- 80.Hervier B, Meyer A, Dieval C, et al. Pulmonary hypertension in antisynthetase syndrome: prevalence, aetiology and survival. Eur Respir J 2013;42:1271–82. [DOI] [PubMed] [Google Scholar]
- 81.Andersson H, Aalokken TM, Gunther A, et al. Pulmonary Involvement in the Antisynthetase Syndrome: A Comparative Cross-sectional Study. J Rheumatol 2016;43:1107–13. [DOI] [PubMed] [Google Scholar]
- 82.Couture P, Brillet P-Y, Varin S, Le Goff B, et al. Sarcoidosis in Patients with Antisynthetase Syndrome: Presentation and Outcome. J Rheumatol 2018;45:1296–300. [DOI] [PubMed] [Google Scholar]
- 83.Lefevre G, Meyer A, Launay D, et al. Seronegative polyarthritis revealing antisynthetase syndrome: a multicentre study of 40 patients. Rheumatol Oxf Engl 2015;54:927–32. [DOI] [PubMed] [Google Scholar]
- 84.Gofrit SG, Yonath H, Lidar M, Shoenfeld Y, Kivity S. The clinical phenotype of patients positive for antibodies to myositis and myositis-related disorders. Clin Rheumatol 2018;37:1257–63. [DOI] [PubMed] [Google Scholar]
- 85.Lilleker JB, Vencovsky J, Wang G, et al. The EuroMyositis registry: an international collaborative tool to facilitate myositis research. Ann Rheum Dis 2018;77:30–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hervier B, Devilliers H, Stanciu R, et al. Hierarchical cluster and survival analyses of antisynthetase syndrome: phenotype and outcome are correlated with anti-tRNA synthetase antibody specificity. Autoimmun Rev 2012;12:210–7. [DOI] [PubMed] [Google Scholar]
- 87.Cavagna L, Nuno L, Scire CA, et al. Clinical Spectrum Time Course in Anti Jo-1 Positive Antisynthetase Syndrome: Results From an International Retrospective Multicenter Study. Medicine (Baltimore) 2015;94:e1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hervier B, Wallaert B, Hachulla E, et al. Clinical manifestations of anti-synthetase syndrome positive for anti-alanyl-tRNA synthetase (anti-PL12) antibodies: a retrospective study of 17 cases. Rheumatol 2010;49:972–6. [DOI] [PubMed] [Google Scholar]
- 89.Trallero-Araguas E, Grau-Junyent JM, Labirua-Iturburu A, et al. Clinical manifestations and long-term outcome of anti-Jo1 antisynthetase patients in a large cohort of Spanish patients from the GEAS-IIM group. Semin Arthritis Rheum 2016;46:225–31. [DOI] [PubMed] [Google Scholar]
- 90.Bartoloni E, Gonzalez-Gay MA, Scire C, et al. Clinical follow-up predictors of disease pattern change in anti-Jo1 positive anti-synthetase syndrome: Results from a multicenter, international and retrospective study. Autoimmun Rev 2017;16:253–7. [DOI] [PubMed] [Google Scholar]
- 91.Hirakata M, Suwa A, Takada T, et al. Clinical and immunogenetic features of patients with autoantibodies to asparaginyl-transfer RNA synthetase. Arthritis Rheum 2007;56:1295–303. [DOI] [PubMed] [Google Scholar]
- 92.Andersson H, Kirkhus E, Garen T, Walle-Hansen R, Merckoll E, Molberg O. Comparative analyses of muscle MRI and muscular function in anti-synthetase syndrome patients and matched controls: a cross-sectional study. Arthritis Res Ther 2017;19:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cavagna L, Fusetti C, Montecucco C, Caporali R. Anticyclic citrullinated peptide antibodies as markers of erosive arthritis in antisynthetase syndrome. J Rheumatol 2010;37:1967. [DOI] [PubMed] [Google Scholar]
- 94.Aguilera Cros C, Ruiz Román A, Méndez Díaz L, et al. Interstitial lung disease in patients with antisynthetase syndrome and anti-RO52 antibodies positive. Ann Rheum Dis 2018;77:505–6. [Google Scholar]
- 95.Pinal-Fernandez I, Pallisa-Nunez E, Selva-O’Callaghan A, et al. Pleural irregularity, a new ultrasound sign for the study of interstitial lung disease in systemic sclerosis and antisynthetase syndrome. Clin Exp Rheumatol 2015;33(4 Suppl 91):S136–141. [PMC free article] [PubMed] [Google Scholar]
- 96.Rojas-Serrano J, Herrera-Bringas D, Mejia M, Rivero H, Mateos-Toledo H, Figueroa JE. Prognostic factors in a cohort of antisynthetase syndrome (ASS): serologic profile is associated with mortality in patients with interstitial lung disease (ILD). Clin Rheumatol 2015;34:1563–9. [DOI] [PubMed] [Google Scholar]
- 97.Carrasco Cubero MC, Rojas Herrera SM, Vargas Pérez ML, Alcalá Peña MI, Espárrago Rodilla M, Chamizo Carmona E Clinical characteristics of a cohort of patients with anti-JO1 antibodies. Ann Rheum Dis 2018;77:1525. [Google Scholar]
- 98.Araujo PAO, Silva MG, Borba EF, Shinjo SK. High prevalence of metabolic syndrome in antisynthetase syndrome. Clin Exp Rheumatol 2018;36:241–7. [PubMed] [Google Scholar]
- 99.Gomard-Mennesson E, Fabien N, Cordier J-F, Ninet J, Tebib J, Rousset H. Clinical significance of anti-histidyl-tRNA synthetase (Jo1) autoantibodies. Ann N Y Acad Sci 2007;1109:414–20. [DOI] [PubMed] [Google Scholar]
- 100.Shi J, Li S, Yang H, et al. Clinical Profiles and Prognosis of Patients with Distinct Antisynthetase Autoantibodies. J Rheumatol 2017;44:1051–7. [DOI] [PubMed] [Google Scholar]
- 101.Cen X, Zuo C, Yang M, Yin G, Xie Q. A clinical analysis of risk factors for interstitial lung disease in patients with idiopathic inflammatory myopathy. Clin Dev Immunol 2013;2013:648570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Targoff IN, Trieu EP, Plotz PH, Miller FW Antibodies to glycyl-transfer RNA synthetase in patients with myositis and interstitial lung disease. Arthritis Rheum 1992;35:821–30. [DOI] [PubMed] [Google Scholar]
- 103.Hervier B, Uzunhan Y, Hachulla E, et al. Antisynthetase syndrome positive for anti-threonyl-tRNA synthetase (anti-PL7) antibodies. Eur Respir J 2011;37:714–7. [DOI] [PubMed] [Google Scholar]
- 104.Dieval C, Ribeiro E, Mercie P, Blanco P, Duffau P, Longy-Boursier M. [Antisynthetase syndrome: a retrospective study of 14 patients]. Rev Med Interne 2012;33:76–9. [DOI] [PubMed] [Google Scholar]
- 105.Labirua-Iturburu A, Selva-O’Callaghan A, Martínez-Gómez X, Trallero-Araguás E, Labrador-Horrillo M, Vilardell-Tarrés M Calcineurin inhibitors in a cohort of patients with antisynthetase-associated interstitial lung disease. Clin Exp Rheumatol 2013;31:436–9. [PubMed] [Google Scholar]
- 106.Johnson C, Connors GR, Oaks J, et al. Clinical and pathologic differences in interstitial lung disease based on antisynthetase antibody type. Respir Med 2014;108:1542–8. [DOI] [PubMed] [Google Scholar]
- 107.Zamora AC, Hoskote SS, Abascal-Bolado B, et al. Clinical features and outcomes of interstitial lung disease in anti-Jo-1 positive antisynthetase syndrome. Respir Med 2016;118:39–45. [DOI] [PubMed] [Google Scholar]
- 108.Yousem SA, Schneider F, Bi D, Oddis CV, Gibson K, Aggarwal R. The pulmonary histopathologic manifestations of the anti-PL7/antithreonyl transfer RNA synthetase syndrome. Hum Pathol 2014;45:1199–204. [DOI] [PubMed] [Google Scholar]
- 109.Marie I, Josse S, Hatron PY, et al. Interstitial lung disease in anti-Jo-1 patients with antisynthetase syndrome. Arthritis Care Res 2013;65:800–8. [DOI] [PubMed] [Google Scholar]
- 110.Piette Y, De Sloovere M, Vandendriessche S, et al. Pitfalls in the detection of myositis specific antibodies by lineblot in clinically suspected idiopathic inflammatory myopathy. Clin Exp Rheumatol 2020;38:212–9. [PubMed] [Google Scholar]
- 111.Kumar RR, Jha S, Dhooria A, Naidu GSRSNK, et al. Anti-Jo-1 Syndrome Often Misdiagnosed as Rheumatoid Arthritis (for Many Years): A Single-Center Experience. J Clin Rheumatol 2021; 27(4):150–5. [DOI] [PubMed] [Google Scholar]
- 112.Evaluating the Cellular Composition of Anti-synthetase Syndrome and Dermatomyositis Skin Lesions Using Image Mass Cytometry. ACR Meeting Abstracts. Available on: https://acrabstracts.org/abstract/evaluating-the-cellular-composition-of-anti-synthetase-syndrome-and-dermatomyositis-skin-lesions-using-image-mass-cytometry/ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.