

Abstract
Hydrogen atom transfer (HAT) processes are among the most useful approaches for the selective construction of C(sp3)-C(sp3) bonds. 1,5-HAT with heteroatom-centered radicals (O•, N•) have been well established and are favored relative to other 1,n-HAT processes. In comparison, net 1,2-HAT processes have been scarcely observed. Reported is the first amidyl radicals that preferentially undergo a net 1,2-HAT over 1,5-HAT. Beginning with single electron transfer (SET) from 2-azaallyl anions to N-alkyl N-aryloxy amides, the latter generate amidyl radicals. The amidyl radical undergoes a net-1,2-HAT to generate a C-centered radical that participates in an intermolecular radical-radical coupling with the 2-azaallyl radical to generate 1,2-diamine derivatives. Mechanistic and EPR experiments point to radical intermediates. Density functional theory (DFT) calculations provide support for a base-assisted, stepwise-1,2-HAT process. It is proposed that generation of amidyl radicals under basic conditions can be greatly expanded to access α-amino C-centered radicals that will serve as valuable synthetic intermediates.
Graphical Abstract
1. INTRODUCTION
The significance and impact of selective C(sp3)–C(sp3) bond constructions have gained widespread appreciation in recent years.1–6 Hydrogen atom transfer (HAT) processes that enable regioselective functionalization of C–H bonds are strategically valuable, because they can enable the formation of bonds between two C(sp3) carbons.7–11 In particular, heteroatom-centered radicals (O•, N•), which often undergo intramolecular 1,5-HAT reactions to generate translocated carbon-centered radicals (Scheme 1a), have been well established and widely applied in modern organic synthesis.7, 10, 12–14 In comparison with 1,5-HAT processes, net-1,2-HAT reactions are rare, having been observed with a few oxygen-centered radicals (X = O, Scheme 1a).8, 15 These 1,2-HAT processes are generally initiated by transition-metal catalysts, including Ir, Cu and Ag catalysts, or undergo photocatalytic reactions (Scheme 1b).16–20 Another proposed net-1,2-HAT is with aminyl radicals. Aminyl radicals are intermediates in the reaction of amino acid systems with hypochlorite.21–24 In such a process, 1,2-HAT reactions to form captodative C-centered radicals have been proposed, as have both intra- and intermolecular pathways.25 Evidence for carbon-centered radical products in these systems has been presented in spin trapping experiments by EPR. Other studies26, 27 into aminyl radical 1,2-HAT rearrangements found them to be higher energy processes or did not observe products derived from net-1,2-HAT.
Scheme 1.

Reactions of heteroatom-centered radicals. a. 1,5- vs. 1,2-HAT processes with heteroatom centered radicals (X). b. Transition-metal-catalyzed 1,2-hydrogen atom transfer (1,2-HAT) of heteroatom-centered radicals. c. Net-1,2-HAT of aminyl radicals in biological systems.
The 1,n-HAT reaction of heteroatom radicals to carbon-centered radicals are thermodynamically down hill. The transition states for O- and N-centered radicals in intramolecular 1,2-HAT processes have a high barrier due to the constrained nature of 3-centered transition states (Scheme 1a). In contrast, the transition state for 1,5-HAT can approach a linear geometry, which has been calculated to have a much lower activation energy.28 Thus, 1,5-HAT rearrangements generally occur to the exclusion of 1,2-HAT processes.
Since Murphy’s groundbreaking research on organic super-electron-donors (SEDs),29–31 SEDs have found useful applications in the construction of C–C bonds. Our team,32–35 and other groups,36–38 have demonstrated that the 2-azaallyl anions can be used as effective SEDs for transition-metal-free C–C bond-formations. We developed a series of radical-radical coupling approaches enabled by single electron transfer (SET) from 2-azaallyl anions (Scheme 2a).32, 33, 39 Specifically, 2-azaallyl anions undergo SET with various aryl/alkyl halides or allyl ethers to generate 2-azaallyl radicals and aryl, alkyl or allyl radicals followed by radical–radical coupling processes to form new C–C bonds. This strategy has proven useful in tandem reactions to prepare heterocycles of medicinal chemistry interest.40–42
Scheme 2.

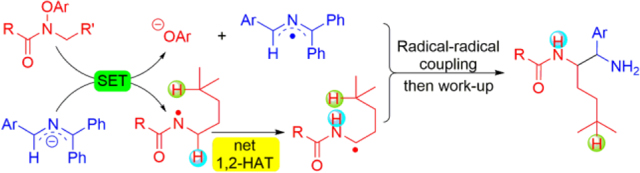
Reaction development. a. SET of 2-azaallyl anions with various electrophiles followed by radical-radical coupling. b. Generation of amidyl radicals, net-1,2-HAT and radical-radical coupling to generate 1,2-diamine derivatives (This work).
Recently, amidyl radicals, which are usually generated under thermal, photochemical or photoredox conditions, have proven to be useful hydrogen abstractors in organic synthesis.8, 43, 44 Knowles45 and Alexanian46 demonstrated synthetically valuable intramolecular HAT processes of amidyl radicals. Wang and Flechsig developed a unique visible-light-mediated allylation of C(sp3)–H bonds via an amidyl radical 1,5-HAT process.47 Inspired by these elegant works, we desired to explore the HAT process of amidyl radicals for radical coupling enabled by 2-azaallyl anions as SEDs (Scheme 2b). We hypothesized that the 2-azaallyl anions and pre-functionalized amides would undergo an SET process to generate 2-azaallyl radicals and amidyl radicals. Surprisingly, the amidyl radical intermediates preferentially underwent net-1,2-HAT rather than the expected 1,5-HAT pathway. The newly formed α-amino C-centered radical then underwent radical-radical coupling with the 2-azaallyl radical to provide 1,2-diamine derivatives.
Herein, we report a strategy to prepare 1,2-diamine derivatives (38 examples, up to 85% yield) without the assistance of added transition metals. 1,2-Diamine derivatives are valuable in synthetic chemistry and pharmaceutical sciences, but their synthesis remains a significant and important challenge.48–50 Notably, this selective net-1,2-HAT protocol of nitrogen-centered radicals (N•) is rare and remains underdeveloped in the synthetic community. After demonstrating the utility of the new net-1,2-HAT rearrangement, selectivity studies with substrates that could undergo 1,5- and 1,6-HAT reaction manifolds are performed, as are radical trapping and cross-over experiments. DFT calculations and mechanistic studies provide support for a base-assisted, stepwise-1,2-HAT process.
2. RESULT AND DISCUSSION
Generation of amidyl radicals and net-1,2-HAT; reaction discovery and optimization.
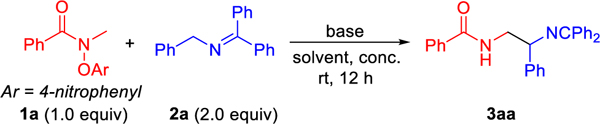
Initially, the focus will be on net-1,2-HAT reactions as outlined above, with the exclusion of possible 1,5- and 1,6-HAT pathways. Thus, we selected pre-functionalized amide 1a as the model substrate, which was readily prepared using the method of Leonori.51–54 We initiated our reaction development and optimization using the amide 1a and ketimine 2a with LiN(SiMe3)2 in DMSO at room temperature for 12 h. The net-1,2–HAT amidyl α-C(sp3)–H coupling product 3aa was generated in 38% assay yield (AY, as determined by 1H NMR integration against an internal standard, Table 1, entry 1). Next, a careful survey of various bases, such as NaN(SiMe3)2, KN(SiMe3)2, LiOtBu, NaOtBu and KOtBu, was carried out (entries 2–6). NaOtBu served as the top base with 64% AY (entry 5). We then examined the effect of solvents [DMF, THF, dioxane, MTBE (methyl tert-butyl ether) and DME (dimethoxyethane)] (entries 7–11). Only DMF delivered the product in 37% AY, while others led to no reaction. Concentration is also a crucial factor in radical coupling reactions of 2-azaallyl radicals.40 Varying the concentration from 0.05 M to 0.03 M, 0.1 M or 0.2 M (entries 12–14) demonstrated that 0.2 M was the optimal concentration, with 74% AY and 71% isolated yield (entry 14). When 1.0 equiv or 3.0 equiv of NaOtBu were employed, the AY decreased to 18% or 66% (entries 15 and 16). Finally, increasing the temperature to 40 °C led to a decrease of AY (60%, entry 17). Based on these results, the best conditions for the radical coupling in Table 1 are those in entry 14.
Table 1.
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Base (equiv) | Solvent | Conc. [M] | Assay yield (%) |
| 1 | LiN(SiMe3)2 (2.0) | DMSO | 0.05 | 38 |
| 2 | NaN(SiMe3)2(2.0) | DMSO | 0.05 | 36 |
| 3 | KN(SiMe3)2 (2.0) | DMSO | 0.05 | 23 |
| 4 | LiOtBu (2.0) | DMSO | 0.05 | 50 |
| 5 | NaOtBu (2.0) | DMSO | 0.05 | 64 |
| 6 | KOtBu (2.0) | DMSO | 0.05 | 43 |
| 7 | NaOtBu (2.0) | DMF | 0.05 | 37 |
| 8 | NaOtBu (2.0) | THF | 0.05 | 0 |
| 9 | NaOtBu (2.0) | Dioxane | 0.05 | 0 |
| 10 | NaOtBu (2.0) | MTBE | 0.05 | 0 |
| 11 | NaOtBu (2.0) | DME | 0.05 | 0 |
| 12 | NaOtBu (2.0) | DMSO | 0.03 | 55 |
| 13 | NaOtBu (2.0) | DMSO | 0.1 | 65 |
| 14 | NaOtBu (2.0) | DMSO | 0.2 | 74 (71)c |
| 15 | NaOtBu (1.0) | DMSO | 0.2 | 18 |
| 16 | NaOtBu (3.0) | DMSO | 0.2 | 66 |
| 17d | NaOtBu (2.0) | DMSO | 0.2 | 60 |
Reaction conditions: 1a (0.1 mmol, 1.0 equiv), 2a (0.2 mmol, 2.0 equiv), base, rt., 12 h.
Assay yields (AY) determined by 1H NMR spectroscopy of the crude reaction mixtures using CH2Br2 as an internal standard.
Isolated yield after chromatographic purification.
40 °C.
Reaction scope of N-benzyl ketimines.
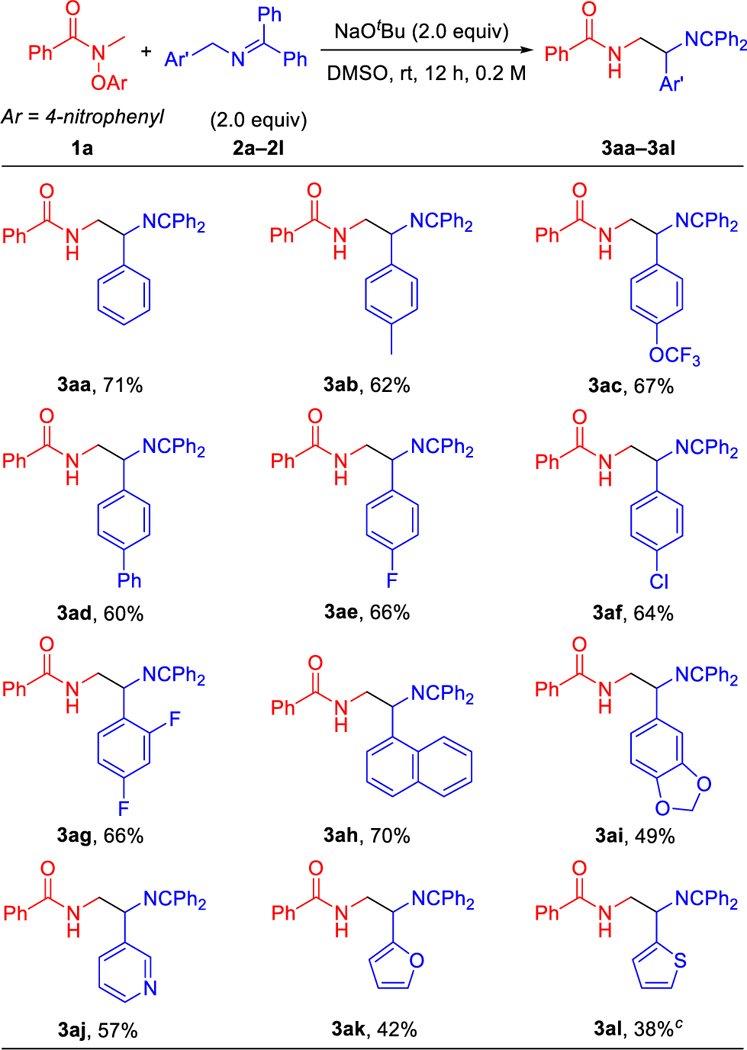
With the optimized conditions in hand (Table 1, entry 14), we initiated exploration of the scope of ketimines 2. As presented in Table 2, generally, ketimines with various substituted Ar’ groups delivered products in moderate to good yields under the optimized conditions. N-Benzyl ketimines bearing electron donating groups, such as 4-Me (2b), 4-OCF3 (2c) and 4-Ph (2d), gave coupling products 3ab, 3ac and 3ad in 62%, 67% and 60% yields, respectively. N-Benzyl ketimines containing electronegative and electron-withdrawing groups (4-F, 2e; 4-Cl, 2f; and 2,4-di-F, 2g) generated the coupling products 3ae, 3af and 3ag in 64–66% yields. The sterically hindered 1-naphthyl (2h) derivative led to coupling product 3ah in 70% yield. Interestingly, medicinally relevant heterocyclic N-benzyl ketimines containing piperonyl (2i), 3-pyridyl (2j), 2-furyl (2k) and 2-thienyl (2l) were also capable coupling partners, providing products3ai, 3aj, 3ak and 3al in 38–57% yields.
Table 2.
|
Reactions were conducted on a 0.4 mmol scale using 1.0 equiv 1a, 2.0 equiv 2 and 2.0 equiv NaOtBu at 0.2 M.
Isolated yields after chromatographic purification.
2.5 h.
Reaction scope of amides.
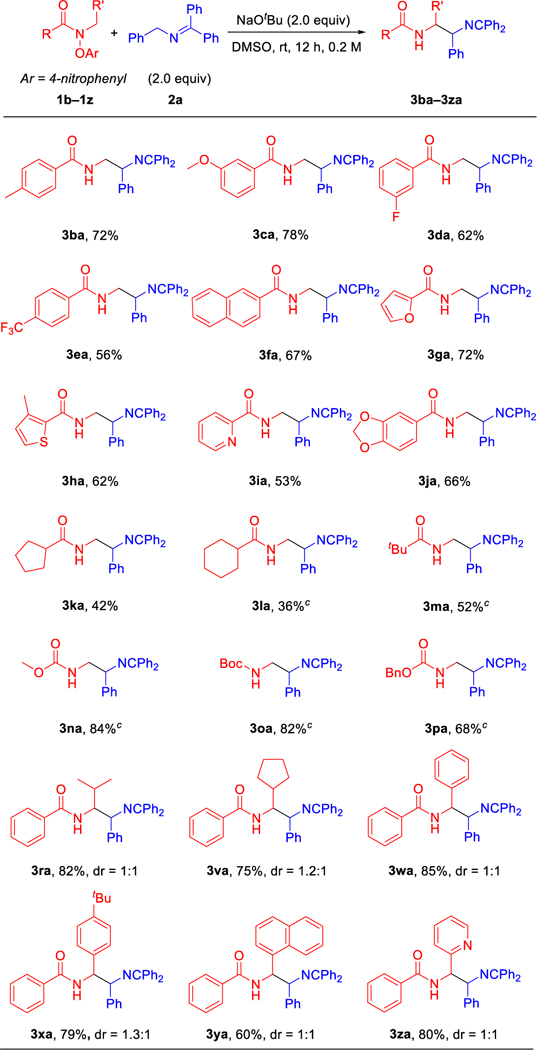
Next, we focused our attention on investigating the scope of substituted amides. As mentioned, N-aryloxy amides were readily synthesized from the corresponding acyl chlorides.52 In general, amides with various substituted aryl, alkyl and alkoxy groups offered moderate to excellent yields under the optimized conditions (Table 3). Aryl substituted amides with electron donating groups, such as 4-Me (1b), or electron-withdrawing groups, such as 3-OMe (1c), 3-F (1d) and 4-CF3 (1e), reacted with N-benzyl ketimine 2a to furnish coupling products 3ba, 3ca, 3da and 3ea in 72%, 78%, 62% and 56% yields, respectively. 2-Naphthyl substituted amide 1f provided coupling product 3fa in 67% yield. Notably, medicinally relevant heterocyclic amides possessing 2-furyl (1g), 3-Me-2-thienyl (1h), 2-pyridyl (1i) and piperonyl (1j) delivered coupling products 3ga, 3ha, 3ia and 3ja in 72%, 62%, 53% and 66% yields, respectively. Furthermore, alkyl substituted amides bearing cyclopentyl (1k), cyclohexyl (1l) and t-butyl (1m) were also suitable coupling partners, offering coupling products 3ka, 3la and 3ma in 36–52% yields. To our delight, alkoxy substituted amides, such as methoxy 1n, t-butyloxy 1o and benzyloxy 1p, performed well, furnishing the desired products 3na, 3oa and 3pa (CCDC 2173282, see SI for details) in 84%, 82%, and 68% yields, respectively. Furthermore, N-aryloxy amides bearing isobutyl (1r), cyclopentanemethyl (1v), benzyl (1w), 4-tBu benzyl (1x), 1-naphthalenemethyl and 2-pyridinemethyl (1z), were also competent coupling partners, leading to the desired products 3ra, 3va, 3wa, 3xa, 3ya and 3za in 82%, 75%, 85%, 79%, 60% and 80% yields, respectively.
Table 3.
|
Reactions were conducted on a 0.4 mmol scale using 1.0 equiv 1, 2.0 equiv 2a and 2.0 equiv NaOtBu at 0.2 M.
Isolated yields after chromatographic purification.
2.5 h.
Gram-scale synthesis and product hydrolysis.
To test the scalability and practicality of this coupling process, the telescoped gram-scale synthesis was carried out. As shown in Scheme 3a, benzylamine and diphenylmethanimine were reacted in THF at 50 °C for 12 h, followed by solvent removal to offer imine 2a. The unpurified 2a was coupled with 5 mmol aryl amide 1d or alkoxy amide 1o under the standard reaction conditions to generate 1.22 g of 3da and 1.52 g of 3oa in 58% and 76% yields, respectively. It is noteworthy that the coupling products 3da and 3oa underwent hydrolysis to deliver the 1,2-diamines 4da and 4oa in 87% and 82% yields (Scheme 3b).
Scheme 3.

Gram-scale synthesis and hydrolysis reactions. a. Gram-scale sequential one-pot synthesis of 3da and 3oa. b. Hydrolysis of the products to access diamines 4da and 4oa.
Probing reaction intermediates and pathways.
Initial experiments were conducted to explore the reaction intermediates and mechanism. As noted in the Introduction, at the outset of this work we imagined that radical intermediates would be involved. To probe this assertation, EPR experiments employing phenyl N-t-butylnitrone (PBN) as the radical spin trap were conducted. Accordingly, treatment of N-phenoxyamide 1a with ketimine 2a under the standard conditions in the presence of PBN resulted in the generation of a PBN-trapped carbon-centred radical, as detected by EPR spectroscopy (Scheme 4). The generated EPR signals (g = 2.0076, AN = 14.5 G, AH = 3.0 G) were in agreement with previously reported literature for trapping C-centered radicals.39, 55, 56 This observation supports our contention that radical intermediates are involved in this transformation.
Scheme 4.

EPR spectrum of the PBN-trapped carbon-centered radical.
Two general mechanisms were initially considered for the generation of the key carbon-centered α-amino radical (Scheme 5a-b). The first is a base promoted elimination to form an N-acyl imine. The N-acyl imine was then envisioned to undergo SET from the azaallyl anion to generate the α-amino radical. The second pathway is initiated by SET to the N-phenoxy amide followed by 1,2-HAT to generate the same α-amino radical. To probe the N-acyl imine formation, N-phenoxyamide 1a was treated with NaOtBu for 12 h in DMSO at RT, but in the absence of ketimine. No elimination product N-acylimine or its hydrolysis product benzamide were observed, although the N-phenoxy N-methyl amide was converted to the N-methyl amide. This result suggests that an elimination pathway is not operative (Scheme 5a).
Scheme 5.

Possible paths. a. Base-promoted elimination mechanism. b. Amidyl radicals α-C(sp3)–H coupling via net-1,2-HAT.
To gain insight into the coupling process, we performed a series of mechanistic probe experiments. Radical trapping experiments with 2,2,6,6-tetramethyl piperidine-1-oxyl (TEMPO, 5.0 equiv) provided insights into the key radical intermediates. Consistent with the working mechanism, when the standard coupling reaction between N-phenoxyamide 1a and ketimine 2a in the presence of NaOtBu was conducted, the 2-azaallyl radical was engaged by TEMPO and led to the oxidized ketimine 5aa in 70% yield. Interception of the proposed α-amino radical by TEMPO afforded adduct 6aa in 21% yield (Scheme 6a). Of course, trapping of these intermediates resulted in a decrease in the yield of the diamine-derived coupling product 3aa (31%). It is noteworthy that treatment of 1a with NaOtBu in DMSO with 5 equiv TEMPO, but in the absence of ketimine, did not generate the trapping product from α-amino radical (Scheme 6b). Further, use of the aldimine 2a’ derived from 9-amino fluorene (Scheme 6c), which generates a more stabilized and less-reducing 2-azaallyl anion, did not lead to the formation of coupling product. These latter two experiments indicate that the strongly reducing 2-azaallyl anion is needed for the formation of the amidyl radical. In addition, cyclic voltammogram studies (see SI for details) suggested that 2-azaallyl anions (E1/2 = –1.11 V vs. SCE)57 would readily reduce amide substrate 1a (E1/2 = –0.886 V vs. SCE).47
Scheme 6.

Control experiments. a. Radical trapping experiment. b. Reaction in the absence of ketimine. c. Less reducing 2-azaallyl anion.
Additional experiments were performed to probe the formation of the proposed amidyl radical. The cyclopropyl substituted amide 1q was prepared as a radical clock (Scheme 7, see SI for details). The reaction of the cyclopropane radical clock 1q (2.0 mmol) with imine 2a under the standard conditions furnished the ring-opened product 3qa in 67% yield. A possible mechanism is illustrated in Scheme 7. It should be noted that the timing of the double bond migration is unclear. These results suggest that the coupling reaction proceeds through an N-centered radical intermediate.
Scheme 7.

Reaction of radical clock 1q.
It is known that α-amino C–H bonds are relatively weak and provide stabilized radicals after HAT. We were concerned that if a reactive radical were generated under our reaction conditions, it would have a natural tendency to chemoselectively abstract the C–H positioned alpha to the amino group, giving a net-1,2-HAT. With this in mind, we next set out to determine if the net-1,2-HAT reaction was an intra- or intermolecular process by performing cross-over experiments. Thus, treatment of a 1.0 : 1.0 mixture of N-phenoxyamide 1a with N-methyl carbonate 1n’ in the presence of ketimine 2a and NaOtBu resulted in the formation of the coupled product 3aa (55% yield) without the formation of the cross-over product 3na (Scheme 8a). Likewise, use of the Boc protected N-methyl amine with ketimine 1b and base provided 3ba (78% yield) without the formation of detectable 3oa (Scheme 8b). We note that both 3na and 3oa were formed in the net-1,2-HAT in Table 3. The experiments in Scheme 8 support the notion that the formation of the α-amino radical is an intramolecular process.
Scheme 8.

Cross-over experiments.
To understand the unexpected favorability of an apparent 1,2-HAT, the mechanism was probed using density functional theory (DFT) [UM06/6–311+G(d,p)-CPCM(DMSO)//UB3LYP/6–31G(d),58–64 see SI for full computational details]. First, we explored the generation of the azaallyl anion (Figure 1), which occurs via deprotonation of 2a (2a-TS-7, 13.8 kcal/mol) to give 7 which is uphill in energy by 4.9 kcal/mol consistent with the pKa values for tBuOH and azaallyl 2a. Azaallyl anion 7 then undergoes SET to 1a to form azaallyl radical 8, N-centered radical 9a, and phenoxide (downhill in energy by 20.0 kcal/mol), as proposed previously.52, 65, 66
Figure 1.

Generation of azaallyl radical 8 via deprotonation of 2a followed by SET to 1a. Free energies were computed using UM06/6–311+G(d,p)-CPCM(DMSO)//UB3LYP/6–31G(d). See Figure S7 for energetics with UM06–2X/6–311+G(d,p)-CPCM(DMSO)//UB3LYP/6–31G(d).
Next, we explored the formation of the amidyl radical 11a from N-centered radical 9a (Figure 2). DMSO with very low water content in this reaction under the standard conditions caused a more modest decrease in the reaction yield from 71% to 46% compared to other reports16–18 (see Table S9 in the SI). Several pathways were thus explored (for the higher energy pathways considered, including 1,2-HAT assisted by water, see Figure S8 in the SI). For clarity, only the lowest energy pathway and direct 1,2-HAT are shown. As a lower energy pathway involving a discrete water molecule in the proton transfer could not be located, we hypothesize that water aids this process via a more complex hydrogen bonding network.
Figure 2.

Generation of radical-radical coupling product 3aa via base-assisted, stepwise 1,2-HAT of 9a. Free energies were computed using UM06/6–311+G(d,p)-CPCM(DMSO)//UB3LYP/6–31G(d). See Figure S12 for energetics with UM06–2X/6–311+G(d,p)-CPCM(DMSO)//UB3LYP/6–31G(d).
As expected, the direct 1,2-HAT of 9a via 3-centered transition state 9a-TS-11a has a large barrier of 34.2 kcal/mol. Consequently, we explored the formation of amidyl radical 11a by an indirect, base-assisted-1,2-HAT. In this process, NaOtBu first abstracts the C–H proton α to the N-centered radical via 9a-TS-10a (barrier of 2.1 kcal/mol) to form radical anion 10a (see Figure S9 in SI for deprotonation with NaOH`). Notably, the α-C–H’s to the N-centered radical in 9a are more acidic than those on a neutral amide. Formation of radical anion 10a is exergonic by 38.6 kcal/mol, in contrast to the analogous deprotonation of N-methylbenzamide to benzamidomethanide which is endergonic (see Figure S10 in the SI). Resonance structures of 10a are consistent with 1,2-radical shift (1,2-RS).67 We expect that this pathway to form radical anion 10a can be exploited in future reaction design to achieve new modes of reactivity of amidyl radicals compared to documented HAT reactivity of these reactive species (see Figure S11 for structural details on 10a). The α-amino C-centered radical 11a then forms by reprotonation of the nitrogen atom via 10a-TS-11a (barrier of 14.3 kcal/mol). Finally, 2-azaallyl radical 8 and the α-amidyl C-centered radical 11a undergo intermolecular radical coupling (barrier of 0.5 kcal/mol) to generate product 3aa. Thus, the overall mechanism corresponds to that shown in Scheme 9.
Scheme 9.

Deprotonation/protonation mechanism to net 1,2-HAT.
Considering the overwhelming prevalence of intramolecular 1,5-HAT of nitrogen-centered amidyl radicals (N•),13 we were curious to explore the selectivity between 1,2-HAT and 1,5- or 1,6-HAT with the amidyl radical generated under our reaction conditions. As shown in Scheme 10a, treatment of the substituted amide 1s, which can undergo either net-1,2-, 1,5- or 1,6-HAT processes, with 2a under basic conditions following the standard procedure provided the net-1,2-HAT product 3sa in 66% yield. Notably, no 1,5- or 1,6-HAT coupling product 12sa or 12sa׳ was detected. Next, a substrate with a weaker benzylic C–H bond positioned to facilitate 1,5-HAT was examined. Thus, the phenylbutyl substituted amide 1s׳ was prepared and employed Scheme 10a. When subjected to the standard conditions 1s׳ gave the net-1,2-HAT product 3sa׳ in 42% yield and the 1,5-product 12 sa׳׳ was obtained in 17% yield.
Scheme 10.

Overview of mechanistic probes. a. 1,2-HAT vs. 1,5-HAT. b. 1,2-HAT against benzylic 1,5-HAT. c. 1,5-HAT.
We wondered if the 1,5-HAT could be favored by employing a substrate with a weaker benzylic C–H bond positioned four atoms away from the site of the amidyl radical. Thus, substrate 1t was prepared and subjected to the standard reaction conditions with ketimine 2a. Here again, the 1,2-HAT product 3ta was obtained (65% yield) and no 1,5-HAT product was found (Scheme 10b).
Finally, we opted to generate an amidyl radical under our conditions that did not have a 1,2-HAT pathway available, leaving only the 1,5-HAT rearrangement as viable. Thus, the N-tBu N-phenoxyamide was prepared and subjected to the coupling conditions with ketimine 2a. In the event, the 1,5-HAT product was obtained in 35% yield (unoptimized). The selectivity experiments in Scheme 10c suggest that net-1,2-HAT takes place with excellent chemoselectivity, which is quite surprising given the overwhelming preference for 1,5-HAT in other systems.47, 68–70
The above selectivity for net 1,2-HAT vs the typically more favorable 1,5-HAT or 1,6-HAT is consistent with the computed mechanism in Figure 2. Formation of the N-centered radical 9s occurs with a similar profile to that of 9a in Figure 1 (see Figure S13 in SI). A comparison of the barriers from the N-centered radical 9s for the direct 1,2-, 1,5-, and 1,6-HAT as well as indirect, base-assisted net-1,2-HAT is particularly instructive (Figure 3, see Figure S14 for the profile for 1s’). Both 1,2-HAT and 1,6-HAT are prohibitively high in energy, with barriers of 28.9 kcal/mol and 51.8 kcal/mol respectively. As expected, 1,5-HAT has a low barrier of 4.8 kcal/mol. However, intermediate 9s undergoes an even lower energy deprotonation of the C–H proton to the nitrogen (barrier of 1.5 kcal/mol) to form radical anion 10s, downhill in energy by 43.6 kcal/mol. Sodium tert-butoxide then transfers a proton to the nitrogen atom of 10s via 10s-TS-11s (barrier of 17.2 kcal/mol) to generate α-amino radical 11s. α-Amino radical 11s undergoes radical-radical coupling with 2-azaallyl radical 8 to form the coupling product 3sa.
Figure 3.

Generation of amidyl radical 11s via indirect, base-assisted 1,2-HAT of 9s. Direct 1,2-, 1,5-, and 1,6-HAT processes are also shown. Free energies were computed using UM06/6–311+G(d,p)-CPCM(DMSO)//UB3LYP/6–31G(d). See Figure S15 for energetics with UM06–2X/6–311+G(d,p)-CPCM(DMSO)//UB3LYP/6–31G(d).
Finally, we note some comparisons between the apparent 1,2-HAT described herein and the chemistry of amine radical cations. It is known that single electron oxidation of tertiary amines generates radical cations,71–75 which have been exploited in photoredox catalysis.76, 77 In these N-centered radicals, the C–H bonds attached to the N-centered radical are dramatically weakened, with estimated bond strengths of 42 kcal/mol.78 The weak C–H’s can then be deprotonated inducing a 1,2-radical shift. In the amidyl radical, a related increase in acidity of the α-C–H’s is proposed. However, this type of reactivity has not, to our knowledge, been documented in amidyl radicals, where 1,5-HAT and intramolecular HAT reactions dominate.
3. CONCLUSION AND OUTLOOK
In summary, we have developed the first synthetically useful amidyl radicals that undergo a net-1,2-HAT to generate α-amino-carbon-centered radicals. This carbon centered radical undergoes C(sp3)-C(sp3) intermolecular radical-radical coupling with 2-azaallyls radicals to generate 1,2-diamine derivatives. Unlike past advances in intramolecular HAT rearrangement processes, this system exhibits excellent chemoselectivity favoring a rare net-1,2-HAT of nitrogen-centered radicals with very high chemoselectivity. A telescoped gram-scale synthesis and product hydrolysis illustrate the potential synthetic utility of this transformation.
Mechanistic, EPR experiments and DFT calculations demonstrate an intramolecular radical net-1,2-HAT pathway. It is noteworthy that this reaction proceeds efficiently at room temperature by combining ketimines, N-phenoxy amides and base to generate diamine derivatives, which are of value in synthetic chemistry and the pharmaceutical industry. Furthermore, this chemistry does not employ transition-metal catalysts, photocatalysts, or organometallic reagents, which increases its attractiveness for applications in the pharmaceutical industry. We are currently investigating net-1,2-HAT processes with amidyl radicals in other applications.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the National Key R&D Program of China (2019YFE0109200), NSFC (21662043), NSF of Yunnan (202207AA110007, 202207AB110002), Ling-Jun Scholars Yunnan Province (202005AB160003), Program for Xingdian Talents (Yun-Ling Scholars) and IRTSTYN. P. J. W. thanks the US National Science Foundation (CHE-2154593) for financial support. M.C.K. thanks the NIH (R35 GM131902) for financial support and XSEDE (TG-CHE120052) for computational support. We thank Prof. Chengfeng Xia for the help with EPR equipments.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c0xxxx.
Experimental procedures, characterization data for all compounds, mechanistic experiments, computational details, and copies of NMR spectra (PDF).
Structure file (XYZ).
Accession Codes
CCDC 2173282 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, U.K.; fax: +44 1223 336033.
The authors declare no competing financial interest.
REFERENCES
- 1.White MC, Adding Aliphatic C−H Bond Oxidations to Synthesis. Science 2012, 335 (6070), 807–809. [DOI] [PubMed] [Google Scholar]
- 2.He J; Wasa M; Chan KSL; Shao Q; Yu JQ, Palladium-Catalyzed Transformations of Alkyl C-H Bonds. Chem. Rev 2017, 117 (13), 8754–8786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang Z; Lim HN; Mo F; Young MC; Dong G, Transition metal-catalyzed ketone-directed or mediated C-H functionalization. Chem. Soc. Rev 2015, 44 (21), 7764–7786. [DOI] [PubMed] [Google Scholar]
- 4.Newhouse T; Baran PS, If C-H bonds could talk: selective C-H bond oxidation. Angew. Chem. Int. Ed 2011, 50 (15), 3362–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamaguchi J; Yamaguchi AD; Itami K, Funktionalisierung von C-H-Bindungen: neue Synthesemethoden für Naturstoffe und Pharmazeutika. Angew. Chem 2012, 124 (36), 9092–9142. [Google Scholar]
- 6.Yamaguchi J; Yamaguchi AD; Itami K, C-H bond functionalization: emerging synthetic tools for natural products and pharmaceuticals. Angew. Chem. Int. Ed 2012, 51 (36), 8960–9009. [DOI] [PubMed] [Google Scholar]
- 7.Becker P; Duhamel T; Stein CJ; Reiher M; Muñiz K, Kooperative Licht-aktivierte Iod- und Photoredox-Katalyse zur Aminierung von Csp3 -H-Bindungen. Angew. Chem 2017, 129 (27), 8117–8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Capaldo L; Ravelli D; Fagnoni M, Direct Photocatalyzed Hydrogen Atom Transfer (HAT) for Aliphatic C-H Bonds Elaboration. Chem. Rev 2022, 122 (2), 1875–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen MS; White MC, Combined Effects on Selectivity in Fe-Catalyzed Methylene Oxidation. Science 2010, 327, 566–571. [DOI] [PubMed] [Google Scholar]
- 10.Jeffrey JL; Terrett JA; MacMillan DWC, O–H hydrogen bonding promotes H-atom transfer from α C–H bonds for C-alkylation of alcohols. Science 2015, 349 (6255), 1532–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu W; Huang X; Cheng M-J; Nielsen RJ; Goddard WA; Groves JT, Oxidative Aliphatic C-H Fluorination with Fluoride Ion Catalyzed by a Manganese Porphyrin. Science 2012, 337, 1322–1325. [DOI] [PubMed] [Google Scholar]
- 12.Becker P; Duhamel T; Stein CJ; Reiher M; Muniz K, Cooperative Light-Activated Iodine and Photoredox Catalysis for the Amination of Csp3 -H Bonds. Angew. Chem. Int. Ed 2017, 56 (27), 8004–8008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo W; Wang Q; Zhu J, Visible light photoredox-catalysed remote C-H functionalisation enabled by 1,5-hydrogen atom transfer (1,5-HAT). Chem. Soc. Rev 2021, 50 (13), 7359–7377. [DOI] [PubMed] [Google Scholar]
- 14.Kärkäs MD, Photochemical Generation of Nitrogen-Centered Amidyl, Hydrazonyl, and Imidyl Radicals: Methodology Developments and Catalytic Applications. ACS. Catal 2017, 7 (8), 4999–5022. [Google Scholar]
- 15.Nechab M; Mondal S; Bertrand MP, 1,n-Hydrogen-atom transfer (HAT) reactions in which n ≠ 5: an updated inventory. Chem. Eur. J 2014, 20 (49), 16034–16059. [DOI] [PubMed] [Google Scholar]
- 16.Che C; Qian Z; Wu M; Zhao Y; Zhu G, Intermolecular Oxidative Radical Addition to Aromatic Aldehydes: Direct Access to 1,4- and 1,5-Diketones via Silver-Catalyzed Ring-Opening Acylation of Cyclopropanols and Cyclobutanols. J. Org. Chem 2018, 83 (10), 5665–5673. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J; Liu D; Liu S; Ge Y; Lan Y; Chen Y, Visible-Light-Induced Alkoxyl Radicals Enable α-C(sp3)-H Bond Allylation. iScience 2020, 23 (1), 100755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhong LJ; Wang HY; Ouyang XH; Li JH; An DL, Benzylic C-H heteroarylation of N-(benzyloxy)phthalimides with cyanopyridines enabled by photoredox 1,2-hydrogen atom transfer. Chem. Commun 2020, 56 (61), 8671–8674. [DOI] [PubMed] [Google Scholar]
- 19.Che C; Huang Q; Zheng H; Zhu G, Copper-catalyzed cascade annulation of unsaturated alpha-bromocarbonyls with enynals: a facile access to ketones from aldehydes. Chem. Sci 2016, 7 (7), 4134–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Y; Liu D; Zhang J, Investigations on the 1,2-Hydrogen Atom Transfer Reactivity of Alkoxyl Radicals under Visible-Light-Induced Reaction Conditions. Synlett 2021, 32, 356–361. [Google Scholar]
- 21.Schoneich C, Radical rearrangement and transfer reactions in proteins. Essays Biochem 2020, 64 (1), 87–96. [DOI] [PubMed] [Google Scholar]
- 22.Hawkins CL; Davies MJ, Reaction of HOCl with amino acids and peptides: EPR evidence for rapid rearrangement and fragmentation reactions of nitrogen-centred radicals. J. Chem. Soc., Perkin Trans 2, 1998, 1937–1946. [Google Scholar]
- 23.Rees MD; Hawkins CL; Davies MJ, Hypochlorite-Mediated Fragmentation of Hyaluronan, Chondroitin Sulfates, and Related N-Acetyl Glycosamines: Evidence for Chloramide Intermediates, Free Radical Transfer Reactions, and Site-Specific Fragmentation. J. Am. Chem. Soc 2003, 125 (45), 13719–13733. [DOI] [PubMed] [Google Scholar]
- 24.Rees MD; Davies MJ, Heparan Sulfate Degradation via Reductive Homolysis of Its N-Chloro Derivatives. J. Am. Chem. Soc 2006, 128 (9), 3085–3097. [DOI] [PubMed] [Google Scholar]
- 25.Bamatraf MMM; O’Neill P; Rao BSM, OH Radical-Induced Charge Migration in Oligodeoxynucleotides. J. Phys. Chem. B 2000, 104 (3), 636–642. [Google Scholar]
- 26.Pattison DI; Davies MJ; Asmus K-D, Absolute rate constants for the formation of nitrogen-centred radicals from chloramines/amides and their reactions with antioxidants. J. Chem. Soc., Perkin Trans 2, 2002.1461–1467. [Google Scholar]
- 27.Bonifačić M; Armstrong DA; Carmichael I; Asmus K-D, β-Fragmentation and Other Reactions Involving Aminyl Radicals from Amino Acids. J. Phys. Chem. B 2000, 104 (3), 643–649. [Google Scholar]
- 28.Moran D; Jacob R; Wood GPF; Coote ML; Davies MJ; O&Hair RAJ; Easton CJ; Radom L, Rearrangements in Model Peptide-Type Radicals via Intramolecular Hydrogen-Atom Transfer. Helv. Chim. Acta 2006, 89, 2254–2272. [Google Scholar]
- 29.Barham JP; Coulthard G; Emery KJ; Doni E; Cumine F; Nocera G; John MP; Berlouis LE; McGuire T; Tuttle T; Murphy JA, KOtBu: A Privileged Reagent for Electron Transfer Reactions? J. Am. Chem. Soc 2016, 138 (23), 7402–7410. [DOI] [PubMed] [Google Scholar]
- 30.Barham JP; Dalton SE; Allison M; Nocera G; Young A; John MP; McGuire T; Campos S; Tuttle T; Murphy JA, Dual Roles for Potassium Hydride in Haloarene Reduction: CSNAr and Single Electron Transfer Reduction via Organic Electron Donors Formed in Benzene. J. Am. Chem. Soc 2018, 140 (36), 11510–11518. [DOI] [PubMed] [Google Scholar]
- 31.Murphy JA, Discovery and development of organic super-electron-donors. J. Org. Chem 2014, 79 (9), 3731–3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li M; Berritt S; Matuszewski L; Deng G; Pascual-Escudero A; Panetti GB; Poznik M; Yang X; Chruma JJ; Walsh PJ, Transition-Metal-Free Radical C(sp3)-C(sp2) and C(sp3)-C(sp3) Coupling Enabled by 2-Azaallyls as Super-Electron-Donors and Coupling-Partners. J. Am. Chem. Soc 2017, 139 (45), 16327–16333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li M; Gutierrez O; Berritt S; Pascual-Escudero A; Yesilcimen A; Yang X; Adrio J; Huang G; Nakamaru-Ogiso E; Kozlowski MC; Walsh PJ, Transition-metal-free chemo- and regioselective vinylation of azaallyls. Nat. Chem 2017, 9 (10), 997–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang L; Liu Z; Tian X; Zi Y; Duan S; Fang Y; Chen W; Jing H; Yang L; Yang X, Transition-Metal-Free C(sp3)-H Coupling of Cycloalkanes Enabled by Single-Electron Transfer and Hydrogen Atom Transfer. Org. Lett 2021, 23 (5), 1714–1719. [DOI] [PubMed] [Google Scholar]
- 35.Liu Z; Li M; Deng G; Wei W; Feng P; Zi Q; Li T; Zhang H; Yang X; Walsh PJ, Transition-metal-free C(sp3)-H/C(sp3)-H dehydrogenative coupling of saturated heterocycles with N-benzyl imines. Chem. Sci 2020, 11 (29), 7619–7625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lei Y; Yang J; Qi R; Wang S; Wang R; Xu Z, Arylation of benzyl amines with aromatic nitriles. Chem. Commun 2018, 54 (84), 11881–11884. [DOI] [PubMed] [Google Scholar]
- 37.Tang S; Zhang X; Sun J; Niu D; Chruma JJ, 2-Azaallyl Anions, 2-Azaallyl Cations, 2-Azaallyl Radicals, and Azomethine Ylides. Chem. Rev 2018, 118 (20), 10393–10457. [DOI] [PubMed] [Google Scholar]
- 38.Wang Q; Poznik M; Li M; Walsh PJ; Chruma JJ, 2‐Azaallyl Anions as Light‐Tunable Super‐Electron‐Donors: Coupling with Aryl Fluorides, Chlorides, and Bromides. Adv. Synth. Catal 2018, 360 (15), 2854–2868. [Google Scholar]
- 39.Deng G; Duan S; Wang J; Chen Z; Liu T; Chen W; Zhang H; Yang X; Walsh PJ, Transition-metal-free allylation of 2-azaallyls with allyl ethers through polar and radical mechanisms. Nat. Commun 2021, 12, 3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deng G; Li M; Yu K; Liu C; Liu Z; Duan S; Chen W; Yang X; Zhang H; Walsh PJ, Synthesis of Benzofuran Derivatives through Cascade Radical Cyclization/Intermolecular Coupling of 2-Azaallyls. Angew. Chem. Int. Ed 2019, 58 (9), 2826–2830. [DOI] [PubMed] [Google Scholar]
- 41.Yu K; Li M; Deng G; Liu C; Wang J; Liu Z; Zhang H; Yang X; Walsh PJ, An Efficient Route to Isochromene Derivatives via Cascade Radical Cyclization and Radical‐Radical Coupling. Adv. Synth. Catal 2019, 361 (18), 4354–4359. [Google Scholar]
- 42.Zi Q; Li M; Cong J; Deng G; Duan S; Yin M; Chen W; Jing H; Yang X; Walsh PJ, Super-Electron-Donor 2-Azaallyl Anions Enable Construction of Isoquinolines. Org. Lett 2022, 24 (9), 1786–1790. [DOI] [PubMed] [Google Scholar]
- 43.Czaplyski WL; Na CG; Alexanian EJ, C-H Xanthylation: A Synthetic Platform for Alkane Functionalization. J. Am. Chem. Soc 2016, 138 (42), 13854–13857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quinn RK; Konst ZA; Michalak SE; Schmidt Y; Szklarski AR; Flores AR; Nam S; Horne DA; Vanderwal CD; Alexanian EJ, Site-Selective Aliphatic C-H Chlorination Using N-Chloroamides Enables a Synthesis of Chlorolissoclimide. J. Am. Chem. Soc 2016, 138 (2), 696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choi GJ; Zhu Q; Miller DC; Gu CJ; Knowles RR, Catalytic alkylation of remote C-H bonds enabled by proton-coupled electron transfer. Nature 2016, 539 (7628), 268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmidt VA; Quinn RK; Brusoe AT; Alexanian EJ, Site-selective aliphatic C-H bromination using N-bromoamides and visible light. J. Am. Chem. Soc 2014, 136 (41), 14389–14392. [DOI] [PubMed] [Google Scholar]
- 47.Wu K; Wang L; Colon-Rodriguez S; Flechsig GU; Wang T, Amidyl Radical Directed Remote Allylation of Unactivated sp3 C-H Bonds by Organic Photoredox Catalysis. Angew. Chem. Int. Ed 2019, 58 (6), 1774–1778. [DOI] [PubMed] [Google Scholar]
- 48.Cardona F; Goti A, Metal-catalysed 1,2-diamination reactions. Nat. Chem 2009, 1 (4), 269–275. [DOI] [PubMed] [Google Scholar]
- 49.Kano T; Sakamoto R; Akakura M; Maruoka K, Stereocontrolled synthesis of vicinal diamines by organocatalytic asymmetric Mannich reaction of N-protected aminoacetaldehydes: formal synthesis of (−)-agelastatin A. J. Am. Chem. Soc 2012, 134 (17), 7516–7520. [DOI] [PubMed] [Google Scholar]
- 50.Yu L; Somfai P, Regio- and Enantioselective Formal Hydroamination of Enamines for the Synthesis of 1,2-Diamines. Angew. Chem. Int. Ed 2019, 58 (25), 8551–8555. [DOI] [PubMed] [Google Scholar]
- 51.Davies J; Booth SG; Essafi S; Dryfe RA; Leonori D, Visible-Light-Mediated Generation of Nitrogen-Centered Radicals: Metal-Free Hydroimination and Iminohydroxylation Cyclization Reactions. Angew. Chem. Int. Ed 2015, 54 (47), 14017–14021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davies J; Svejstrup TD; Fernandez Reina D; Sheikh NS; Leonori D, Visible-Light-Mediated Synthesis of Amidyl Radicals: Transition-Metal-Free Hydroamination and N-Arylation Reactions. J. Am. Chem. Soc 2016, 138 (26), 8092–8095. [DOI] [PubMed] [Google Scholar]
- 53.Reina DF; Dauncey EM; Morcillo SP; Svejstrup TD; Popescu MV; Douglas JJ; Sheikh NS; Leonori D, Visible-Light-Mediated 5-exo-dig Cyclizations of Amidyl Radicals. Eur. J. Org. Chem 2017, 2108–2111.
- 54.Svejstrup TD; Ruffoni A; Julia F; Aubert VM; Leonori D, Synthesis of Arylamines via Aminium Radicals. Angew. Chem. Int. Ed 2017, 56 (47), 14948–14952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Das BG; Chirila A; Tromp M; Reek JN; Bruin B, CoIII-Carbene Radical Approach to Substituted 1H-Indenes. J. Am. Chem. Soc 2016, 138 (28), 8968–8975. [DOI] [PubMed] [Google Scholar]
- 56.Buettner GR, Spin trapping: ESR parameters of spin adducts. Free Radic. Bio. Med 1987, 3, 259–303. [DOI] [PubMed] [Google Scholar]
- 57.Panetti GB; Carroll PJ; Gau MR; Manor BC; Schelter EJ; Walsh PJ, Synthesis of an elusive, stable 2-azaallyl radical guided by electrochemical and reactivity studies of 2-azaallyl anions. Chem. Sci 2021, 12 (12), 4405–4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cossi M; Rega N; Scalmani G; Barone V, Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem 2003, 24 (6), 669–681. [DOI] [PubMed] [Google Scholar]
- 59.Krishnan R; Binkley JS; Seeger R; Pople JA, Self‐consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys 1980, 72 (1), 650–654. [Google Scholar]
- 60.Lee C; Yang W; Parr RG, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37 (2), 785–789. [DOI] [PubMed] [Google Scholar]
- 61.McLean AD; Chandler GS, Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys 1980, 72 (10), 5639–5648. [Google Scholar]
- 62.Petersson GA; Al‐Laham MA, A complete basis set model chemistry. II. Open‐shell systems and the total energies of the first‐row atoms. J. Chem. Phys 1991, 94 (9), 6081–6090. [Google Scholar]
- 63.Becke AD, Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 1993, 98 (7), 5648–5652. [Google Scholar]
- 64.Zhao Y; Truhlar DG, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc 2008, 120, 215–241. [Google Scholar]
- 65.Jiang H; Studer A, Chemistry With N-Centered Radicals Generated by Single-Electron Transfer-Oxidation Using Photoredox Catalysis. CCS Chem. 2019, 1, 38–49. [Google Scholar]
- 66.Pratley C; Fenner S; Murphy JA, Nitrogen-Centered Radicals in Functionalization of sp2 Systems: Generation, Reactivity, and Applications in Synthesis. Chem. Rev 2022, 122 (9), 8181–8260. [DOI] [PubMed] [Google Scholar]
- 67.Matsuo B; Granados A; Majhi J; Sharique M; Levitre G; Molander GA, 1,2-Radical Shifts in Photoinduced Synthetic Organic Transformations: A Guide to the Reactivity of Useful Radical Synthons. ACS Org. Inorg. Au 2022, 2 (6), 435–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen H; Fan W; Yuan XA; Yu S, Site-selective remote C(sp3)-H heteroarylation of amides via organic photoredox catalysis. Nat. Commun 2019, 10, 4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim JH; Ruffoni A; Leonori D, Divergent Strain-Release Amino-Functionalization of [1.1.1]Propellane with Electrophilic Nitrogen-Radicals. Angew. Chem. Int. Ed 2020, 59, 8225–8231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morcillo SP; Dauncey EM; Kim JH; Douglas JJ; Sheikh NS; Leonori D, Photoinduced Remote Functionalization of Amides and Amines Using Electrophilic Nitrogen Radicals. Angew. Chem. Int. Ed 2018, 57 (39), 12945–12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lewis FD; Ho T-I, Selectivity of tertiary amine oxidations. J. Am. Chem. Soc 1980, 102 (5), 1751–1752. [Google Scholar]
- 72.Lewis FD; Ho T-I; Simpson JT, Photochemical Addition of Tertiary Amines to Stilbene. Stereoelectronic Control of Tertiary Amine Oxidation. J. Org. Chem 1981, 46 (6), 1077–1082. [Google Scholar]
- 73.Anne A; Hapiot P; Moiroux J; Neta P; Savéant J-M, Dynamics of Proton Transfer from Cation Radicals. Kinetic and Thermodynamic Acidities of Cation Radicals of NADH Analogues. J. Am. Chem. Soc 1992, 114 (12), 4694–4701. [Google Scholar]
- 74.Zhang X; Yeh S-R; Hong S; Freccero M; Albini A; Falvey DE; Mariano PS, Dynamics of α-CH Deprotonation and α-Desilylation Reactions of Tertiary Amine Cation Radicals. J. Am. Chem. Soc 1994, 116 (10), 4211–4220. [Google Scholar]
- 75.Wang ZS; Chen YB; Zhang HW; Sun Z; Zhu C; Ye LW, Ynamide Smiles Rearrangement Triggered by Visible-Light-Mediated Regioselective Ketyl-Ynamide Coupling: Rapid Access to Functionalized Indoles and Isoquinolines. J. Am. Chem. Soc 2020, 142 (7), 3636–3644. [DOI] [PubMed] [Google Scholar]
- 76.Zheng N; Maity S, A Photo Touch on Amines: New Synthetic Adventures of Nitrogen Radical Cations. Synlett 2012, 23 (13), 1851–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nakajima K; Miyake Y; Nishibayashi Y, Synthetic Utilization of α‑Aminoalkyl Radicals and Related Species in Visible Light Photoredox Catalysis. Acc. Chem. Res 2016, 49 (9), 1946–1956. [DOI] [PubMed] [Google Scholar]
- 78.Wayner DM, D; Dannenberg JJ; DavidGriller, Oxidation potentials of α-aminoalkyl radicals: bond dissociation energies for related radical cations. Chem. Phys. Lett 1986, 131 (3), 189–191. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.