

ABSTRACT
Understanding how applied voltage drives the electrocatalytic reaction at the nanoscale is a fundamental scientific problem, particularly in non-metallic electrocatalysts, due to their low intrinsic carrier concentration. Herein, using monolayer molybdenum disulfide (MoS2) as a model system of non-metallic catalyst, the potential drops across the basal plane of MoS2 (ΔVsem) and the electric double layer (ΔVedl) are decoupled quantitatively as a function of applied voltage through in-situ surface potential microscopy. We visualize the evolution of the band structure under liquid conditions and clarify the process of EF keeping moving deep into Ec, revealing the formation process of the electrolyte gating effect. Additionally, electron transfer (ET) imaging reveals that the basal plane exhibits high ET activity, consistent with the results of surface potential measurements. The potential-dependent behavior of kf and ns in the ET reaction are further decoupled based on the measurements of ΔVsem and ΔVedl. Comparing the ET and hydrogen evolution reaction imaging results suggests that the low electrocatalytic activity of the basal plane is mainly due to the absence of active sites, rather than its electron transfer ability. This study fills an experimental gap in exploring driving forces for electrocatalysis at the nanoscale and addresses the long-standing issue of the inability to decouple charge transfer from catalytic processes.
Keywords: non-metal electrocatalyst, surface potential, electron transfer, hydrogen evolution reaction, mapping
By measuring the MoS2's surface potential under various applied potentials, we observe the insulator-to-conductor transition in the basal plane and establish a significant connection to spatially mapped electrocatalytic activity.
INTRODUCTION
Electrochemical reactions are a key strategy for realizing carbon neutrality and advancing the utilization of clean energy through the production of green hydrogen via water electrolysis with renewable electricity. Unraveling the electrocatalytic mechanism is of paramount importance to achieve a comprehensive understanding of the process and develop high-performance catalysts. Therefore, investigating the electrocatalytic mechanism holds significant promise for promoting a sustainable future. Applying voltage is an effective method for changing the electrochemical potential of an electrocatalyst [1,2], thereby determining the direction and rate of the reaction. However, the applied voltage (Vappl) regulates the electrochemical process differently for metal and non-metal catalysts. For metal catalysts with a huge density of states (DOS) near the Fermi levels (EF), the potential mainly drops within the electric double layer (EDL), modulating the electrostatic potential (φ) and changing the reaction coordinates directly [1]. While for non-metallic catalysts, due to the low electrical conductivity and complex changes in chemical composition, the potential distribution across the solid–liquid interface becomes complicated [3–8]. A part of the potential drop may drop across the non-metallic catalysts, while another part of the potential drops across the EDL [6–8]. In this case, both the chemical potential (μe) and the electrostatic potential (φ) are changed, resulting in the complex electrocatalytic mechanism. Due to the wide variety of non-metal electrocatalysts, finding the universal rules is a great challenge. For example, it was reported that electrocatalytic oxygen evolution reaction (OER) is driven by the chemical potential (μe) in an IrOx catalyst, rather than its electrostatic potential [2,9]. While for M–N-doped carbon catalysts, the electrocatalytic reaction is driven predominantly by the change in electrostatic potential across the EDL [2,10,11]. If the potential distribution across the solid–liquid interface can be directly measured, it will be very convenient for studying the electrocatalytic mechanism of non-metallic catalysts.
Up until now, distinguishing the potential distribution between the non-metallic catalyst and the EDL has still relied on complex theoretical calculations [3,4,12]. By using empirical formulas of capacitance, Bediako et al. showed that the potential distribution between twisted bilayer graphene and EDL is a function of Vappl [8]. In the field of conventional electrochemical technology, Mott-Schottky analysis is a common method for calculating the flat band potential of semiconductors. However, it cannot provide information on how the band structure moves, which is related to the interfacial potential distribution. Through I-V curve, the onset potential of a low-DOS semiconductor at which the EF reaches the band edge can be obtained [13]. However, the potential drop across the semiconductor–electrolyte interface still remains unknown, and hence it is not possible to obtain the respective shift of Ec and EF. The value of the total interface capacitance (Ci) can be obtained by electrochemical impedance spectroscopy (EIS). However, the specific capacitance of the non-metallic electrode and EDL cannot be directly measured by EIS, which requires calculations based on the non-metallic structure model and the double layer (Gouy–Chapman-Stern) model, respectively [6]. In summary, there is a lack of in-situ techniques for directly measuring the potential distribution between the non-metallic catalyst and the EDL. Moreover, conventional electrochemical characterization only provides the ensemble information for electrode materials, neglecting the spatial heterogeneity in the electronic structures of catalysts [14–20]. Therefore, a spatially resolved in-situ characterization technique is needed.
Scanning electrochemical potential microscopy (SECPM), invented by Bard's group, has been used to measure EDL profiles on electrode surfaces, demonstrating the feasibility of scanning probe techniques for potential measurements in liquid [21]. Boettcher et al. developed a contact-based potential-sensing electrochemical atomic force microscopy technique and successfully mapped the photovoltage at nanoscopic semiconductor–catalyst interfaces [22,23]. This shows that changes in surface potential of catalysts can be sensed through contact mode. Based on these pioneering works, we focus on the key issues in electrocatalysis, to reveal the working principle of electrocatalysts through in-situ surface potential microscopy. Recently, the spatial heterogeneity of local electrostatic potential at Au nanoplate–solution interfaces was revealed under open circuit potential (OCP) conditions [24]. However, it is still a challenge to figure out how the electronic structure of non-metallic electrocatalysts changes under applied voltage.
Semiconductors have been predicted to be non-ideal catalysts due to their low intrinsic carrier concentration [25]. For example, it is widely believed that the basal plane of MoS2 impedes charge transfer in electrocatalytic reactions [26–31]. According to classical electron transfer theories, the Schottky-analogue junction is broken and becomes ohmic once the EF is tuned into the bands, resulting in the low conductivity of the semiconductor in liquid [13,32]. However, it is difficult to explain the high electrocatalytic activities of some semiconductor catalysts [13,33,34]. Liu et al. found that the ultrathin MoS2 can be modulated to be highly conductive (‘on’) or insulating (‘off’), strongly correlating with the hydrogen evolution reaction (HER) [13]. Morpurgo's group also observed liquid-gating-induced superconductivity in thin MoS2 crystals [35]. The Y1.75Co0.25Ru2O7−δ electrocatalyst was found to enhance the charge transfer step in the oxygen evolution reaction (OER) [36], while the β-Co(OH)2 electrocatalyst exhibited gradually increasing conductivity after the onset potential of the OER [17]. The above results showed that a charge transport pathway can be open under solution conditions, reflecting the dramatic change of the energy band structure of the semiconductor. However, the underlying reason for the enhanced conductance is not clear.
MoS2 is a representative material in both the fields of electrocatalysis and semiconductor devices. Studying how the voltage affects the band structure and conductivity of MoS2 under solution conditions is of great research significance. Herein, using MoS2 as a non-metallic model system, the potential distribution between the basal plane of monolayer (ML) MoS2 (ΔVsem) and the electric double layer (ΔVedl) were quantificationally decoupled as a function of the applied voltage. We visualized how EF moves into Ec of the basal plane of MoS2, thereby realizing the transition of conductivity, which is the origin of the enhanced conductance in liquid. We provide a method for studying electrochemical driving force at the nanoscale and demonstrate that only when the electron transfer site and the chemical site are spatially coincident can the energy conversion efficiency be maximized. This work has significant importance in advancing our understanding of the electrocatalytic mechanism, and provides theoretical guidance for the conversion of clean energy and the achievement of carbon neutrality.
RESULTS AND DISCUSSION
Decoupling the potential drops across the semiconductor (ΔVsem) and the electric double layer (ΔVedl)
Schematic illustrations of the home-built in-situ surface potential microscope are shown in Fig. 1A and Fig. S1. By combining a high impedance amplifier (1 TΩ) to the atomic force microscopy (AFM) positioning, the local surface potential Vs of the electrode can be obtained. This is achieved by reading the potential of the tip (Vtip) in direct contact with the electrode surface relative to the Ag/AgCl reference based on an assumption: the EF of the tip is controlled by the solution and not by the semiconductor in the ranges of applied voltage (the tip and the semiconductor are difficult to equilibrate, so the EF of the tip does not follow the EF of the semiconductor, and the potential of the tip (Vtip) reflects the EDL potential at the surface of the semiconductor (Vs)).
Figure 1.

Local surface potential measurement on ML MoS2. (A) Schematic of in-situ surface potential measurement set-up and potential drops across the electrode–electrolyte interface for metal, semi-metal and semiconductor respectively. The potential of the tip (Vtip) with respect to an Ag/AgCl reference electrode can be recorded through a high impedance amplifier with negligible leakage current. The potential distribution between catalyst (ΔVsem) and electrolyte (ΔVedl) is determined by the series of quantum capacitor (Cq) and EDL capacitor (Cedl), 1/Ctot = 1/Cq + 1/Cedl. (B) Surface potential values of the basal plane of ML MoS2, Au and solution were measured with a tip in 0.1 M K2SO4. LSV was performed on the substrate (v = 10 mV/s, sample interval = 0.05 V), while the tip recorded the surface potential relative to the Ag/AgCl reference electrode.
We take Vs (Vappl = 0) as a reference. The change of potential drop over the EDL (ΔVedl) can be read directly by Vs (Vappl ≠ 0)−Vs (Vappl = 0). By combining with the change of applied voltage (ΔVappl), the change of potential drop across the electrocatalyst (ΔVsem) can also be obtained by ΔVsem = ΔVappl−ΔVedl. To eliminate the effect of redox reactions on the tip surface, which affect the potential measurements, we used a solution that contains only the supporting electrolytes (0.1 M K2SO4) and studied the intrinsic properties of ML MoS2 at different Vappl.
Due to the small Debye screening length, the distance between the tip and the sample determines whether an accurate surface potential can be measured. To demonstrate the feasibility of surface potential measurement, a metal (Au) substrate was used as a standard sample. The tip was rested on the surface with an applied force of ∼29 nN, slightly larger than that reported by Boettcher et al. (∼25 nN) [21] but sufficient to prevent damage to the sample. As shown in Fig. 1B, Vs faithfully tracked Vappl, reflecting that the surface potential Vs is equal to the applied voltage. This is consistent with the common view for bulk metal electrodes [1,37] that there is no potential drop inside the metal. In this situation, the surface electron concentration ns can be considered a constant value and ΔVappl is applied to increase the electric field intensity of EDL (ΔVappl = ΔVedl), as indicated by curve III of Fig. 1A. With the same applied force (29 nN), the tip can be considered to be able to contact the surface of other samples. Then, the tip was lifted off ∼1 mm from the surface and the potential of the solution (background) was measured (Fig. 1B). It shows that the change in solution potential (∼12 mV) is negligible because the thickness of the EDL is less than 1 nm in a 0.1 M K2SO4 solution [38]. Therefore, the solution can be considered a blank background and does not affect the surface potential measurement.
Next, the surface potential of the basal plane of ML MoS2 at different Vappl was monitored, as shown in Fig. 1B. Under OCP conditions (0.14 V), there was no apparent Vs change when the tip was transferred from the solution to the surface of MoS2. The result reflects that the solution has equilibrated the EF of the tip and MoS2 before contact and the electron transfer between the tip and MoS2 can be ignored. The result demonstrates that the introduction of the nanotip did not change the initial structure of MoS2. When Vappl > −0.15 V (vs. Ag/AgCl), there is no change in Vs. Vs starts to track Vappl when Vappl < −0.15 V. The change in EDL potential (ΔVedl) as a function of applied potential can be obtained by Vs(Vappl ≠ 0) −Vs (Vappl = 0), as shown in Fig. 2A (blue circuit). Since a part of the potential drop over EDL is obtained, another part of the potential drop across the basal plane of MoS2 can also be given by ΔVsem = ΔVappl−ΔVedl (red circuit, Fig. 2A). The results clearly show that when Vappl > −0.15 V, the total potential is dropped across the ML MoS2 (ΔVappl = ΔVsem), as shown in Fig. 1A, potential curve, type I, while the surface potential remains constant. When Vappl is below −0.15 V, the surface potential begins to change, resulting in a partial drop of the potential across the solution (ΔVappl = ΔVsem + ΔVedl, Fig. 1A, potential curve, type II). ΔVedl increases as Vappl becomes more negative. After decoupling the potential drops in the basal plane (ΔVsem) and the EDL (ΔVedl), the evolution of the band structure under electrode conditions can be further revealed.
Figure 2.

Decoupling of potential drops across MoS2 and EDL, and evolution of MoS2 band structure. (A) The decoupled contribution of each potential drop (ΔVsem and ΔVedl) across the ML MoS2 basal plane–electrolyte interface as a function of Vappl. ΔVedl is obtained by Vs (Vappl ≠ 0)−Vs (Vappl = 0), while ΔVsem can be obtained by ΔVappl−ΔVedl. A cartoon of the DOS of the ML MoS2 basal plane is shown in the top panel of (A). The EF is tuned into the band edge of the conduction band (Ec) at −0.15 V, leading to an increase in local density of states at the EF. (B) The change of applied voltage (ΔVappl) shifts the EF of the basal plane by −eΔVappl relative to its initial position. The potential change in EDL (ΔVedl) shifts the full band structure (including the conduction band (Ec) and the valence band (Ev)) by −eΔVedl relative to its initial position. The potential change in the semiconductor (ΔVsem) corresponds to the relative movement rate between EF and Ec, shifting the EF by −eΔVsem with respect to the Ec, which contributes to the change of surface conductance. (C) Energy diagrams for the basal plane of ML MoS2 at different Vappl. (D) The ns-Vappl curve shows the switching effect of ns: the surface is turned ‘on’ with a high electron concentration under cathodic potential with an onset potential of approximately −0.15 V, while remaining insulated under anodic potential. Inset of (D), in-situ electric conductivity measurement on the basal plane of ML MoS2 in 0.1 M K2SO4. The AFM-SECM tip directly contacted the surface of the basal plane and recorded the conductivity current I (SG/TC mode, lift = 0, ETip = 0 V (vs. Ag/AgCl), LSV was performed on the substrate at v = 10 mV/s).
The evolution of the band structure under electrode conditions
The evolution of the potential curve type I to type II reflects a dramatic change in band structure of the basal plane. As shown in Fig. 2B, the change in applied voltage (ΔVappl) shifts EF of the basal plane by −eΔVappl relative to its initial position [4,8], which is considered as the movement rate of EF in the energy coordinates. The potential change in EDL (ΔVedl) shifts the full band structure (including the conduction band (Ec) and the valence band (Ev)) by −eΔVedl relative to its initial position [1], which can be seen as the movement rate of Ec. The potential change in the semiconductor (ΔVsem) corresponds to the relative rate difference between EF and Ec, shifting EF by −eΔVsem with respect to Ec, which contributes to the change in surface conductance or ns [4,8]. The potential distribution between the catalyst (ΔVsem) and the electrolyte (ΔVedl) is determined by the series of semiconductor capacitors (Csem) and EDL capacitors (Cedl) as shown in Fig. 1 A (bottom). Additionally, due to the smaller DOS of 2D semiconductors compared to bulk semiconductors, 2D semiconductor capacitors function as quantum capacitors (Cq), where 1/Ctot = 1/Cq + 1/Cedl [56,39].
When Vappl > −0.15 V, ΔVappl = −ΔEF/e and ΔVedl = −ΔEc/e = 0. ΔVsem = ΔVappl, indicating that Cq ≪ Cedl (that is, the EF is located in the band gap with zero DOS near the EF). In this case, Ec is pinned at a fixed value in the energy coordinates, and EF moves to the band edge of Ec at the maximum rate (Fig. 2C, iv, iii and ⅱ). However, the transition from type I to type II potential drop occurs at an onset potential of approximately −0.15 V, indicating that the EF is tuned into the bottom of the Ec at Vappl = −0.15 V (Fig. 2C, ⅱ). This results in an increase in Cq in the basal plane of ML MoS2 due to the large DOS near EF, as shown in the top panel of Fig. 2B. When Vappl < −0.15 V, ΔVappl = −ΔEF/e, ΔVedl = −ΔEc/e and ΔVsem = ΔVappl−ΔVedl. The Ec starts to move. As Vappl becomes more negative, Ec moves faster. In this situation, the relative rate between EF and Ec gradually decreases, as shown in Fig. 2C, i. However, the difference in the movement rate between EF and Ec still exists, which means that the Fermi level keeps moving into the conduction band. These results are inconsistent with the classical theory that the Schottky-analogue junction is broken once the EF is tuned into the conduction band, and it cannot be further tuned deep in bands [32]. The bandgap Egap = 1.85 eV of ML MoS2 can be determined from photoluminescence spectroscopy (PL), depicted in Fig. S10C. Since the energy difference between EF and Ec determines the surface charge concentration (ns), for 2D semiconductor materials, ns can be calculated by (see Supplementary Data for derivation details):
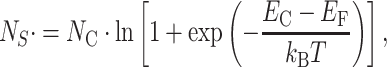 |
(1) |
where Nc ∼8.6 × 1012 cm−2 is the effective density of states in the conduction band [40]. The conductivity of the basal plane is constantly changing since the relative positions of EF and Ec in the energy coordinates change with the Vappl. Figure 2D plots the relationship between ns and Vappl. The ns-Vappl curve shows the switching effect of ns: the surface is turned ‘on’ with high electron concentration (over 1 × 1013 e cm−2 in the basal plane of ML MoS2, consistent with the ‘liquid gating’ effect [13,35,41,42]) at Vappl below −0.15 V, and is turned ‘off’ with low electron concentration (insulating) Vappl higher than −0.15 V. The results show that the surface electron concentration can be effectively tuned under electrolyte conditions. Through the in-situ surface potential measurement, we visualized the evolution of the band structure under electrode conditions and how the high conductivity on the semiconductor surface occurs.
Moreover, it can be seen from the ns-Vappl curve that the basal plane of ML MoS2 is nearly insulated at the open circuit potential (∼0.14 V). It corresponds to the depleted state of ML MoS2 with low electron concentration in solution (the flat band potential is approximately equal to −0.07 V, estimated from a macroscopical Mott-Schottky measurement, Fig. S9).
The switching effect of ns in the basal plane was further proven by conductivity measurements [42–44]. An in-situ local electric conductivity measurement was performed, shown in the inset of Fig. 2D. With the tip directly contacting the surface of the basal plane (lift = 0, substrate generation/tip collection (SG/TC) mode), a linear sweep voltammetry (LSV) curve was collected on the substrate while the potential of the tip remained constant. In this way, the conducting current I is recorded at the solid–liquid interface (containing only the supporting electrolytes to exclude the Faradic reaction). It is shown that the basal plane of ML MoS2 becomes conductive (‘on’) once the Vappl is below the onset potential of approximately −0.15 V. Otherwise, the basal plane maintains an insulating state (‘off’). In previous studies, the onset potential of the conductivity of low DOS semiconductor materials (2D WS2, MoS2, etc.) was regarded as the potential at which EF reaches the position of the band edge of Ec [13,45], proving the accuracy of the surface potential measurement. To date, how the band structure of the semiconductor is transformed under electrolyte conditions has not been clearly studied by conventional electrochemical methods. The difficulty is in distinguishing the potential distribution between semiconductor and EDL, which still depends on theoretical calculations. In our study, the potential drops in the semiconductor (ΔVsem) and the EDL (ΔVedl) are decoupled by direct measurements. Based on the potential values, the evolution of band structure can be revealed under electrolyte conditions.
The role of the applied voltage in electrocatalytic reactions
Electron transfer (ET) and the subsequent formation and rupture of chemical bonds (catalytic reaction) are two fundamental processes in electrocatalytic reactions [9]. However, it is difficult to distinguish their contributions from each other due to their convoluted nature. It becomes even more challenging for non-metal electrocatalysts with spatial heterogeneity in electronic structures and catalytic centers [46,47].
To study how Vappl acts in an electrocatalytic reaction, first we used outer-sphere redox pairs to decouple the ET process from the electrocatalysis process and figure out how the Vappl drives the ET process. Atomic-force-microscope-based scanning electrochemical microscopy (AFM-SECM) [24,48,49] (Fig. S11) was used to map the local electrochemical activity of ML MoS2 electrocatalysts in SG/TC mode [50,51]. A higher iTip reflects a higher local product concentration (that is, higher local electrochemical activity) of the electrocatalysts. The outer-sphere redox pairs with the reaction [Ru(NH3)6]3+ + e− → [Ru(NH3)6]2+ were used to provide information about the ET [20]. The ET image clearly shows that the basal plane of MoS2 has comparable ET activity to that of the edge sites (Fig. 3A). The ET currents in the back of the topographic image are a bit higher than those in the front, due to the change in local concentration of redox pairs near the surface during the imaging process (scanning from back to front in Fig. 3A). Histograms of ET current distributions at different sites are shown in Fig. 3B. The ET currents taken from the single scan line collected in a short time are comparable. Figure 3C gives the topography and ET current values obtained along the same scan line (the dashed line in Fig. 3A). The current values of the basal plane and the edge differ by only 15 pA. The spatially resolved LSV curve of the basal plane largely matches that of the edge (Fig. 3D), indicating no significant difference in the ET rate between basal plane and edges at different biases. ET imaging shows high ET activity of the basal plane, contrary to the traditional consensus under ex-situ conditions [52,53]. It is reasonable to speculate that the high ET activity may derive from the aforementioned ‘liquid gating’ effect with high ns. However, ET currents are jointly decided by both the ns and rate constant kf (i ∝ ns and kf) [1], that is
Figure 3.

ET imaging on the ML MoS2 electrocatalyst. SG/TC mode was used in this experiment. The Pt tip and substrate were used as dual-working electrodes. Semi-ring graphite and Ag/AgCl (saturated KCl) were used as the counter electrode and reference electrode, respectively. Products ([Ru(NH3)6]2+) are generated from the negatively biased substrate and oxidized to reactants ([Ru(NH3)6]3+) by a positively biased tip. (A) ET images of ML MoS2. ET currents are superimposed on a 3D topography map. Inset, 2D topography map of ML MoS2. Fixed height h = 25 nm, ESub = −0.5 V, ETip = 0.3 V (vs. Ag/AgCl). The solution contains 5 mM [Ru(NH3)6]Cl3 and 0.1 M KCl. P++-Si was used as substrate. (B) Histograms of ET current distributions at different sites. (C) Values of topography and ET current obtained along the dashed line in the inset of (A). (D) Spatially resolved LSV curves obtained on the edge and basal plane of ML MoS2 for the [Ru(NH3)6]3+/[Ru(NH3)6]2+ system (h = 25 nm, ETip = 0.3 V (vs. Ag/AgCl), v = 3 mV/s). We set the cathode current to be positive and the anodic current to be negative. (E) The potential-dependent behavior of the rate constant kf and the surface electron concentration ns in the ET reaction are further decoupled based on the measurements of ΔVsem and ΔVedl. (F) Schematic of the potential-dependent behavior of kf and ns in (E).
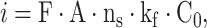 |
(2) |
where F is Faraday constant, A is electrode area, ns is surface electron concentration related to surface electrical conductivity, and C0 is the reactant ([Ru(NH3)6]Cl3) concentration near the surface of the electrode. Here, the reverse reaction is negligible. The rate constant kf can be expressed as k0·exp[−αf(Es–E0')], in which k0 is the standard rate constant, Es is surface potentials of the electrode, α is the transfer coefficient, 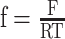 , and E0' is the formal potential. In a solution containing 5 mM [Ru(NH3)6]3+, the formal potential E0′, estimated as E1/2 of the metal substrate, is −0.14 V for [Ru(NH3)6]3+/2+ (Fig. S12B), which is close to the Vappl at which MoS2 becomes semi-metallic. Only when the voltage is applied to change the ns, rather than kf for the basal plane, can the ‘liquid gating’ effect be responsible for the high ET activity. To date, it is still unclear how much the ns contributes to the ET rate in non-metal electrocatalysts.
, and E0' is the formal potential. In a solution containing 5 mM [Ru(NH3)6]3+, the formal potential E0′, estimated as E1/2 of the metal substrate, is −0.14 V for [Ru(NH3)6]3+/2+ (Fig. S12B), which is close to the Vappl at which MoS2 becomes semi-metallic. Only when the voltage is applied to change the ns, rather than kf for the basal plane, can the ‘liquid gating’ effect be responsible for the high ET activity. To date, it is still unclear how much the ns contributes to the ET rate in non-metal electrocatalysts.
Herein, the electronic structure of the basal plane measured in supporting electrolytes (without Faraday currents) was used to predict the factors affecting the ET activity and give a meaningful reference. According to kf = k0·exp[−αf(Es–E0′)], since k0 is unknown it is hard to obtain the specific value of kf. However, kf at different Vappl can be compared by kf(Es,1)/kf(Es,2) = exp[−αf(Es,1–Es,2)]. Taking kf at Vappl = 0.4 V as the reference point (marked as kf,0), we can obtain kf/kf,0 at different Vappl. Similarly, we can also have ns/ns,0 at different Vappl (the reference point ns,0 is ns at Vappl = 0.4 V). Combining kf/kf,0 and ns/ns,0, the potential-dependent behavior of kf and ns can be obtained (Fig. 3E). As shown in Fig. 3F, when Vappl is more positive than the formal potential E0′ (Vappl > −0.14 V), the electrochemical potential (or EF) of MoS2 is lower than that of the [Ru(NH3)6]3+/2+ redox pair and there is no driving force to transfer electrons from MoS2 to the solution (ET reaction does not occur). In this voltage range, all potential drops within the semiconductor increase the ns rapidly with a |Slope| ≈ 17 for log(ns/ns,0). There are no potential drops within the EDL, so the electric field intensity of EDL remains constant and kf does not change with |Slope| ≈ 0 for log(kf/kf,0). In this voltage range, the effect of the applied voltage can be considered as a ‘preparation’ for the ET reaction, transforming MoS2 from a semiconductor state to a semi-metallic state. When Vappl is more negative than the formal potential E0′ (Vappl < −0.14 V), the electrochemical potential of MoS2 is higher than the [Ru(NH3)6]3+/2+ redox pair, creating a driving force for electrons to transfer from MoS2 to solution, and the ET reaction begins to occur. In this voltage range, the potential drop within the EDL gradually increases. The Vappl begins to act on the electric field intensity of EDL, changing the value of kf. When Vappl < −0.3 V, the Vappl increases kf rapidly with |Slope| ≈ 6 for log(kf/kf,0), while the Vappl increases ns slowly with |Slope| ≈ 1 for log(ns/ns,0). When the ET reaction occurs, the effect of the applied voltage is to a greater extent to change the kf value, prompting electrons to pass through the solid–liquid interface for the ET reaction.
The performance of the ML MoS2 in an electrocatalytic (inner-sphere) reaction was also investigated at the nanoscale. Figure 4A displays HER current mapping superimposed on a 3D topography map. The result shows that the high tip current (iTip) is collected at the edge sites, while the current at the basal plane is weak. Histograms of HER current distribution at different sites are shown in Fig. 4B. Compared with the outer-sphere reaction, the inner-sphere reaction is more sensitive to the chemical properties of the active site. The HER currents at different edge sites vary greatly, ranging from a few pA to hundreds of pA. The differences in HER currents reflect the variation in the chemical properties of edge sites under in-situ conditions. Figure 4C gives four current-topography plots along the dashed lines in Fig. 4A, representing the edge sites with different HER activities. Despite differences in the current, the topography of the edge corresponds exactly to the maximum of the HER current. In addition, the spatially resolved LSV method was also performed for ML MoS2 (Fig. 4D). The basal plane is almost catalytically inert over a wide potential range, while the electrochemical currents at the edge increase exponentially with increasing negative bias. In a previous study, Norskov et al. found that the HER activity of MoS2 is proportional to its edge length, and thus hypothesized that the edges are the active sites of the HER [15]. Our work provides direct evidence for the structure–activity relationship of MoS2 through in-situ HER imaging. With the outer-sphere and the inner-sphere reaction, the spatial mismatch of charge transfer and chemical reaction processes of ML MoS2 was clearly revealed at the nanoscale. The applied voltage plays an essential role in the electron transfer step, increasing the surface electron concentration ns and the rate constant kf of the basal plane. However, the large number of electrons reaching the surface cannot participate in the chemical reactions (Fig. 4E). The large Gibbs free energy of the atomic hydrogen (ΔGH*) adsorbed on the basal plane (1.96 eV) prevents chemical processes at the solid–electrolyte interface (Fig. 4F). Therefore, the low binding energy of H contributes to low HER rates through the effect of high barriers, resulting in low energy-conversion efficiency.
Figure 4.

HER imaging on the ML MoS2 electrocatalyst. Products (H2) are generated from the negatively biased substrate and oxidized to reactants (H+) by a positively biased tip. (A) HER images of ML MoS2. HER currents are superimposed on a 3D topography map. Inset, 2D topography map of ML MoS2. Fixed height h = 25 nm, ESub = −0.5 V, ETip = 0.3 V (vs. Ag/AgCl). The solution contains 10 mM HClO4 and 0.1 M NaClO4. p++-Si were used as substrate. (B) Histograms of HER current distributions at different sites. (C) Values of topography and HER current obtained along the dashed lines in the inset of (A). (D) Spatially resolved LSV curves obtained on the edges and basal plane of ML MoS2 for the H+/H2 system. The tip was biased at 0.3 V (vs. Ag/AgCl) at a fixed height, h = 25 nm, while LSV was performed on the substrate (v = 5 mV/s). We set the cathode current to be positive and the anodic current to be negative. The dashed line in (D) is the fitting results according to the respective current values. (E) A model of ML MoS2 in the HER. (F) Comparison of the Gibbs free energies of the adsorbed H on the basal plane and edge of MoS2.
CONCLUSIONS
Through in-situ surface potential measurements, the potential distribution between the basal plane of ML MoS2 (ΔVch) and electrolyte (ΔVedl) was decoupled quantitatively as a function of the applied voltage. We visualized the evolution of the band structure under electrode conditions and revealed the process of conductivity transformation on the semiconductor surface. To clarify how the applied voltage acts on the electrocatalytic reaction, the ET and catalytic reaction processes on ML MoS2 were identified at the nanoscale through AFM-SECM mapping. We clearly showed that the applied voltage plays an essential role in the ET process of the basal plane of MoS2, increasing the surface electron concentration ns and the rate constant kf to different degrees under different Vappl. However, the applied voltage cannot be precisely applied to the electrocatalytic reaction due to the spatial mismatch of charge transfer and reactant adsorption sites. This work paves the way for the rational design of efficient non-metallic electrocatalysts based on the understanding of how voltage acts on non-metallic catalysts at the nanoscale.
Supplementary Material
Contributor Information
Ziyuan Wang, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), State Key Laboratory of Catalysis, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China; Department of Chemistry, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China.
Jun Chen, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), State Key Laboratory of Catalysis, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China; Energy College, University of Chinese Academy of Sciences, Beijing 100049, China.
Chenwei Ni, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), State Key Laboratory of Catalysis, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China; Energy College, University of Chinese Academy of Sciences, Beijing 100049, China.
Wei Nie, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), State Key Laboratory of Catalysis, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China; Energy College, University of Chinese Academy of Sciences, Beijing 100049, China.
Dongfeng Li, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), State Key Laboratory of Catalysis, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China; Energy College, University of Chinese Academy of Sciences, Beijing 100049, China.
Na Ta, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), State Key Laboratory of Catalysis, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China.
Deyun Zhang, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), State Key Laboratory of Catalysis, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China; Energy College, University of Chinese Academy of Sciences, Beijing 100049, China.
Yimeng Sun, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), State Key Laboratory of Catalysis, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China; Energy College, University of Chinese Academy of Sciences, Beijing 100049, China.
Fusai Sun, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), State Key Laboratory of Catalysis, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China; Energy College, University of Chinese Academy of Sciences, Beijing 100049, China.
Qian Li, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), State Key Laboratory of Catalysis, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China; Energy College, University of Chinese Academy of Sciences, Beijing 100049, China.
Yuran Li, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), State Key Laboratory of Catalysis, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China; Department of Chemistry, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China.
Ruotian Chen, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), State Key Laboratory of Catalysis, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China.
Tiankai Bu, Department of Materials, Imperial College London, London SW7 2AZ, UK.
Fengtao Fan, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), State Key Laboratory of Catalysis, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China.
Can Li, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), State Key Laboratory of Catalysis, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China.
FUNDING
This work was supported by the Fundamental Research Funds for the Central Universities (20720220011), the National Program on Key Basic Research Project of China (2021YFA1500600), the Fundamental Research Center of Artificial Photosynthesis (FReCAP), the National Natural Science Foundation of China (22088102), the Chinese Academy of Sciences Projects for Young Scientists in Basic Research (YSBR-004) and the Dalian Institute of Chemical Physics Innovation Foundation (DICPSZ201801).
AUTHOR CONTRIBUTIONS
Ziyuan Wang performed most of the experiments and wrote the manuscript. Jun Chen and Chenwei Ni performed the theoretical calculations. Wei Nie and Dongfeng Li analyzed the in-situ surface potential data. Na Ta carried out the scanning transmission electron microscopy. Deyun Zhang performed scanning electron microscopy. Yimeng Sun, Ruotian Chen and Tiankai Bu analyzed the in-situ electrochemical imaging data and revised the manuscript. Fusai Sun, Qian Li and Yuran Li helped with the in-situ electrochemical imaging measurement. Fengtao Fan and Can Li proposed and supervised the project.
Conflict of interest statement. None declared.
REFERENCES
- 1. Bard AJ, Faulkner LR. Electrochemical Methods: Fundamentals and Applications, 2nd edn.New York: Wiley, 2001. [Google Scholar]
- 2. Boettcher SW, Surendranath Y. Heterogeneous electrocatalysis goes chemical. Nat Catal 2021; 4: 4–5. 10.1038/s41929-020-00570-1 [DOI] [Google Scholar]
- 3. Heller I, Kong J, Hendrik Aet al. Individual single-walled carbon nanotubes as nanoelectrodes for electrochemistry. Nano Lett 2005; 5: 137–42. 10.1021/nl048200m [DOI] [PubMed] [Google Scholar]
- 4. Heller I, Kong J, Williams KAet al. Electrochemistry at single-walled carbon nanotubes: the role of band structure and quantum capacitance. J Am Chem Soc 2006; 128: 7353–9. 10.1021/ja061212k [DOI] [PubMed] [Google Scholar]
- 5. Li J, Burke PJ. Measurement of the combined quantum and electrochemical capacitance of a carbon nanotube. Nat Commun 2019; 10: 3598. 10.1038/s41467-019-11589-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li J, Pham PH, Zhou Wet al. Carbon-nanotube–electrolyte interface: quantum and electric double layer capacitance. ACS Nano 2018; 12: 9763–74. 10.1021/acsnano.8b01427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Waelder J, Vasquez R, Liu Yet al. A description of the faradaic current in cyclic voltammetry of adsorbed redox species on semiconductor electrodes. J Am Chem Soc 2022; 144: 6410–9. 10.1021/jacs.2c00782 [DOI] [PubMed] [Google Scholar]
- 8. Yu Y, Zhang K, Parks Het al. Tunable angle-dependent electrochemistry at twisted bilayer graphene with moiré flat bands. Nat Chem 2022; 14: 267–73. 10.1038/s41557-021-00865-1 [DOI] [PubMed] [Google Scholar]
- 9. Nong HN, Falling LJ, Bergmann Aet al. Key role of chemistry versus bias in electrocatalytic oxygen evolution. Nature 2020; 587: 408–13. 10.1038/s41586-020-2908-2 [DOI] [PubMed] [Google Scholar]
- 10. Warburton RE, Hutchison P, Jackson MNet al. Interfacial field-driven proton-coupled electron transfer at graphite-conjugated organic acids. J Am Chem Soc 2020; 142: 20855–64. 10.1021/jacs.0c10632 [DOI] [PubMed] [Google Scholar]
- 11. Jackson MN, Surendranath Y. Molecular control of heterogeneous electrocatalysis through graphite conjugation. Acc Chem Res 2019; 52: 3432–41. 10.1021/acs.accounts.9b00439 [DOI] [PubMed] [Google Scholar]
- 12. Vasquez R, Waelder J, Liu Yet al. A Gauss's law analysis of redox active adsorbates on semiconductor electrodes: the charging and faradaic currents are not independent. Proc Natl Acad Sci USA 2022; 119: e2202395119. 10.1073/pnas.2202395119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. He Y, He Q, Wang Let al. Self-gating in semiconductor electrocatalysis. Nat Mater 2019; 18: 1098–104. 10.1038/s41563-019-0426-0 [DOI] [PubMed] [Google Scholar]
- 14. Gao X, Chen Y, Sun Tet al. Karst landform-featured monolithic electrode for water electrolysis in neutral media. Energy Environ Sci 2020; 13: 174–82. 10.1039/C9EE02380A [DOI] [Google Scholar]
- 15. Jaramillo TF, Jørgensen KP, Bonde Jet al. Identification of active edge sites for electrochemical H2 evolution from MoS2 nanocatalysts. Science 2007; 317: 100–2. 10.1126/science.1141483 [DOI] [PubMed] [Google Scholar]
- 16. Mariano RG, Kang M, Wahab OJet al. Microstructural origin of locally enhanced CO2 electroreduction activity on gold. Nat Mater 2021; 20: 1000–6. 10.1038/s41563-021-00958-9 [DOI] [PubMed] [Google Scholar]
- 17. Mefford JT, Akbashev AR, Kang Met al. Correlative operando microscopy of oxygen evolution electrocatalysts. Nature 2021; 593: 67–73. 10.1038/s41586-021-03454-x [DOI] [PubMed] [Google Scholar]
- 18. Sun T, Wang D, Mirkin MVet al. Direct high-resolution mapping of electrocatalytic activity of semi-two-dimensional catalysts with single-edge sensitivity. Proc Natl Acad Sci USA 2019; 116: 11618–23. 10.1073/pnas.1821091116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhu J, Wang ZC, Dai Het al. Boundary activated hydrogen evolution reaction on monolayer MoS2. Nat Commun 2019; 10: 1348. 10.1038/s41467-019-09269-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Patten HV, Meadows KE, Hutton LAet al. Electrochemical mapping reveals direct correlation between heterogeneous electron-transfer kinetics and local density of states in diamond electrodes. Angew Chem Int Ed 2012; 51: 7002–6. 10.1002/anie.201203057 [DOI] [PubMed] [Google Scholar]
- 21. Hurth C, Li C, Bard AJet al. Direct probing of electrical double layers by scanning electrochemical potential microscopy. J Phys Chem C 2007; 111: 4620–7. 10.1021/jp0661084 [DOI] [Google Scholar]
- 22. Laskowski FAL, Oener SZ, Nellist MRet al. Nanoscale semiconductor/catalyst interfaces in photoelectrochemistry. Nat Mater 2020; 19: 69–76. 10.1038/s41563-019-0488-z [DOI] [PubMed] [Google Scholar]
- 23. Nellist MR, Laskowski FAL, Qiu Jet al. Potential-sensing electrochemical atomic force microscopy for in operando analysis of water-splitting catalysts and interfaces. Nat Energy 2018; 3: 46–52. 10.1038/s41560-017-0048-1 [DOI] [Google Scholar]
- 24. Nie W, Zhu Q, Gao Yet al. Visualizing the spatial heterogeneity of electron transfer on a metallic nanoplate prism. Nano Lett 2021; 21: 8901–9. 10.1021/acs.nanolett.1c03529 [DOI] [PubMed] [Google Scholar]
- 25. Franklin AD. Electrocatalysis on Non-Metallic Surfaces. Gaithersburg: NBS Special Pub, 1976. [Google Scholar]
- 26. Yu H, Yu X, Chen Yet al. A strategy to synergistically increase the number of active edge sites and the conductivity of MoS2 nanosheets for hydrogen evolution. Nanoscale 2015; 7: 8731–8. 10.1039/C5NR00670H [DOI] [PubMed] [Google Scholar]
- 27. Voiry D, Salehi M, Silva Ret al. Conducting MoS2 nanosheets as catalysts for hydrogen evolution reaction. Nano Lett 2013; 13: 6222–7. 10.1021/nl403661s [DOI] [PubMed] [Google Scholar]
- 28. Xie J, Zhang J, Li Set al. Controllable disorder engineering in oxygen-incorporated MoS2 ultrathin nanosheets for efficient hydrogen evolution. J Am Chem Soc 2013; 135: 17881–8. 10.1021/ja408329q [DOI] [PubMed] [Google Scholar]
- 29. Zhang H, Yu L, Chen Tet al. Surface modulation of hierarchical MoS2 nanosheets by Ni single atoms for enhanced electrocatalytic hydrogen evolution. Adv Funct Mater 2018; 28: 1807086. 10.1002/adfm.201807086 [DOI] [Google Scholar]
- 30. Yousaf AB, Imran M, Farooq Met al. Synergistic effect of Co–Ni co-bridging with MoS2 nanosheets for enhanced electrocatalytic hydrogen evolution reactions. RSC Adv 2018; 8: 3374–80. 10.1039/C7RA12692A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Meng X, Yu L, Ma Cet al. Three-dimensionally hierarchical MoS2/graphene architecture for high-performance hydrogen evolution reaction. Nano Energy 2019; 61: 611–6. 10.1016/j.nanoen.2019.04.049 [DOI] [Google Scholar]
- 32. Neamen DA. Semiconductor Physics and Devices: Basic Principles. New Delhi: Tata McGraw Hill Publishing, 1992. [Google Scholar]
- 33. Burke MS, Kast MG, Trotochaud Let al. Cobalt–iron (oxy)hydroxide oxygen evolution electrocatalysts: the role of structure and composition on activity, stability, and mechanism. J Am Chem Soc 2015; 137: 3638–48. 10.1021/jacs.5b00281 [DOI] [PubMed] [Google Scholar]
- 34. Voiry D, Yang J, Chhowalla Met al. Recent strategies for improving the catalytic activity of 2D TMD nanosheets toward the hydrogen evolution reaction. Adv Mater 2016; 28: 6197–206. 10.1002/adma.201505597 [DOI] [PubMed] [Google Scholar]
- 35. Costanzo D, Jo S, Berger Het al. Gate-induced superconductivity in atomically thin MoS2 crystals. Nat Nanotechnol 2016; 11: 339–44. 10.1038/nnano.2015.314 [DOI] [PubMed] [Google Scholar]
- 36. Han N, Feng S, Liang Yet al. Achieving efficient electrocatalytic oxygen evolution in acidic media on yttrium ruthenate pyrochlore through cobalt incorporation. Adv Funct Mater 2023; 33: 2208399. 10.1002/adfm.202208399 [DOI] [Google Scholar]
- 37. Chen S, Liu Y, Chen Jet al. Heterogeneous electron transfer at nanoscopic electrodes: importance of electronic structures and electric double layers. Chem Soc Rev 2014; 43: 5372–86. 10.1039/C4CS00087K [DOI] [PubMed] [Google Scholar]
- 38. Scholz F. Electroanalytical Methods. Berlin: Springer, 2010. [Google Scholar]
- 39. Xia J, Chen F, Tao Net al. Measurement of the quantum capacitance of graphene. Nat Nanotechnol 2009; 4: 505–9. 10.1038/nnano.2009.177 [DOI] [PubMed] [Google Scholar]
- 40. Furchi MM, Polyushkin DK, Pospischil Aet al. Mechanisms of photoconductivity in atomically thin MoS2. Nano Lett 2014; 14: 6165–70. 10.1021/nl502339q [DOI] [PubMed] [Google Scholar]
- 41. Liu X, Li B, Li Xet al. The critical role of electrolyte gating on the hydrogen evolution performance of monolayer MoS2. Nano Lett 2019; 19: 8118–24. 10.1021/acs.nanolett.9b03337 [DOI] [PubMed] [Google Scholar]
- 42. Saito Y, Nakamura Y, Bahramy MSet al. Superconductivity protected by spin–valley locking in ion-gated MoS2. Nat Phys 2016; 12: 144–9. 10.1038/nphys3580 [DOI] [Google Scholar]
- 43. Garrido JA, Härtl A, Dankerl Met al. The surface conductivity at the diamond/aqueous electrolyte interface. J Am Chem Soc 2008; 130: 4177–81. 10.1021/ja078207g [DOI] [PubMed] [Google Scholar]
- 44. Wadhwa P, Seol G, Petterson MKet al. Electrolyte-induced inversion layer schottky junction solar cells. Nano Lett 2011; 11: 2419–23. 10.1021/nl200811z [DOI] [PubMed] [Google Scholar]
- 45. Braga D, Gutiérrez Lezama I, Berger Het al. Quantitative determination of the band gap of WS2 with ambipolar ionic liquid-gated transistors. Nano Lett 2012; 12: 5218–23. 10.1021/nl302389d [DOI] [PubMed] [Google Scholar]
- 46. Li M, Ye KH, Qiu Wet al. Heterogeneity between and within single hematite nanorods as electrocatalysts for oxygen evolution reaction. J Am Chem Soc 2022; 144: 5247–52. 10.1021/jacs.2c00506 [DOI] [PubMed] [Google Scholar]
- 47. Mariano RG, McKelvey K, White HSet al. Selective increase in CO2 electroreduction activity at grain-boundary surface terminations. Science 2017; 358: 1187–92. 10.1126/science.aao3691 [DOI] [PubMed] [Google Scholar]
- 48. Du HY, Huang YF, Wong Det al. Nanoscale redox mapping at the MoS2-liquid interface. Nat Commun 2021; 12: 1321. 10.1038/s41467-021-21660-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jiang J, Huang Z, Xiang Cet al. Nanoelectrical and nanoelectrochemical imaging of Pt/P-Si and Pt/p+-Si electrodes. ChemSusChem 2017; 10: 4657–63. 10.1002/cssc.201700893 [DOI] [PubMed] [Google Scholar]
- 50. Polcari D, Dauphin-Ducharme P, Mauzeroll Jet al. Scanning electrochemical microscopy: a comprehensive review of experimental parameters from 1989 to 2015. Chem Rev 2016; 116: 13234–78. 10.1021/acs.chemrev.6b00067 [DOI] [PubMed] [Google Scholar]
- 51. Bollinger MV, Lauritsen JV, Jacobsen KWet al. One-dimensional metallic edge states in MoS2. Phys Rev Lett 2001; 87: 196803. 10.1103/PhysRevLett.87.196803 [DOI] [PubMed] [Google Scholar]
- 52. Wu D, Li X, Wu Xet al. Uncovering edge states and electrical inhomogeneity in MoS2 field-effect transistors. Proc Natl Acad Sci USA 2016; 113: 8583–8. 10.1073/pnas.1605982113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang C, Johnson A, Hsu C-Let al. Direct imaging of band profile in single layer MoS2 on graphite: quasiparticle energy gap, metallic edge states, and edge band bending. Nano Lett 2014; 14: 2443–7. 10.1021/nl501133c [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.