

Abstract
Cell-free DNA (cfDNA) in blood, viewed as a surrogate for tumor biopsy, has many clinical applications, including diagnosing cancer, guiding cancer treatment and monitoring treatment response. All these applications depend on an indispensable, yet underdeveloped task: detecting somatic mutations from cfDNA. The task is challenging because of the low tumor fraction in cfDNA. Recently, we developed the computational method cfSNV, the first method that comprehensively considers the properties of cfDNA for the sensitive detection of mutations from cfDNA. cfSNV vastly outperformed the conventional methods that were developed primarily for calling mutations from solid tumor tissues. cfSNV can accurately detect mutations in cfDNA even with medium-coverage (e.g., ≥200×) sequencing, which makes whole-exome sequencing (WES) of cfDNA a viable option for various clinical utilities. Here, we present a user-friendly cfSNV package that exhibits fast computation and convenient user options. We also built a Docker image of it, which is designed to enable researchers and clinicians with a limited computational background to easily carry out analyses on both high-performance computing platforms and local computers. Mutation calling from a standard preprocessed WES dataset (~250× and ~70 million base pair target size) can be carried out in 3 h on a server with eight virtual CPUs and 32 GB of random access memory.
Introduction
Cell-free DNA (cfDNA) in the blood has received growing interest because of its clinical utility as a surrogate for tumor biopsy, especially in cases when tumor biopsy is unavailable or insufficient1. Compared to a tissue biopsy, cfDNA in the blood can be obtained noninvasively and can provide more comprehensive coverage of heterogeneous genetic alterations of tumors in a patient1,2. Hence, various applications have emerged on the basis of tumor-derived somatic mutations in cfDNA, including detecting cancer3–6, guiding cancer treatment7–9 and monitoring cancer10–14.
However, although it has great research and clinical potential, cfDNA contains only a minor fraction of tumor-derived cfDNA in an overwhelming amount of normal cfDNA1,15. Despite the low fraction, tumor-derived cfDNA shows high heterogeneity, because it comes from the entire volume of a tumor and every tumor present in a patient1,2. As a result, tumor-derived somatic mutations in cfDNA, including clonal mutations and subclonal mutations, usually have a very low variant allele frequency (VAF). This poses a great challenge to conventional mutation callers, which are primarily developed for genomic DNA of solid tumors. Without consideration of the cfDNA-specific properties, these methods are not well equipped to handle the complicated scenarios in cfDNA. Especially in medium-coverage sequencing data (e.g., whole-exome sequencing (WES) data), tumor signals can be represented by only a few tumor reads; therefore, a powerful tool to distinguish signals from noise is needed. Lacking a cfDNA-specific mutation caller, most current applications focus on deep sequencing of a small panel of genes3,8,10,11. The profiling of the small genomic range, however, severely limits the applications of cfDNA. Computational methods for sensitive and accurate mutation calling are paramount for exploiting the great potential of cfDNA and can enable a broad range of downstream applications.
Recently, we developed the computational method cfSNV9 as the first method that comprehensively addressed the aforementioned challenge. By statistically incorporating the biological properties of cfDNA, cfSNV achieves high sensitivity and accuracy in the detection of low-frequency mutations (<5% VAF) in cfDNA9. As a result, it not only enhances the existing clinical applications of cfDNA (e.g., deep sequencing of gene panels) but also enables new applications by making cfDNA WES a viable clinical option. Here, we present a comprehensive workflow and a Docker image16 to detect tumor-derived somatic mutations and estimate the tumor fraction by using cfSNV. Specifically, we implement the cfSNV method in C++ and Python for high computing performance and include all the dependencies in the Docker image for facilitating use of the tool. Therefore, this package exhibits much faster computation and much easier usage than the original prototype9. The package covers the entire workflow from raw paired-end sequencing data of cfDNA and matched white blood cells (WBCs) to a biologically interpretable output of somatic mutations. In addition, the package includes a parameter recommendation function that provides users with parameter-setting options for high-quality mutation detection from the input data of different experimental protocols, sequencing coverages and tumor fractions. Overall, the package is designed to enable researchers and clinicians with a minimal computational background to easily carry out analyses on both high-performance computing platforms and local computers.
Development of the protocol
Unlike genomic DNA, cfDNA has unique biological properties, including low tumor fraction1,15, high heterogeneity in clonal composition1,2, short fragment size17 and nonrandom fragmentation18. These properties impair tumor-derived mutation detection in three ways. First, the low tumor fraction and high heterogeneity result in the low VAF of tumor-derived mutations in cfDNA1,2,15,19. Second, the short fragment size (~166 base pairs (bp)) of cfDNA17, which arises from the enzymatic digestion of cfDNA, leads to a large fraction (>50%9) of overlapping read mates from the standard paired-end sequencing (usually 100 or 150 bp per read). It also causes double-counting in DNA fragments and, therefore, biases estimation of allele frequencies. Third, cfDNA is nonrandomly fragmented with preferred end sites18. Therefore, true mutations in cfDNA could cluster at certain positions on the supporting reads. Conventional methods, which assume random fragmentation20, tend to eliminate true mutation candidates in cfDNA as misalignment artefacts. Therefore, to achieve sensitive and accurate detection of tumor-derived mutations in cfDNA, these cfDNA-specific properties need to be addressed. We developed the cfSNV protocol to statistically take these cfDNA properties into consideration in the mutation calling in cfDNA9. cfSNV introduces five novel techniques (Fig. 1) to the conventional methods, focused on the three major stages schematically outlined in Fig. 1. These stages are briefly outlined here and are detailed in our previous work9:
Fig. 1 |.
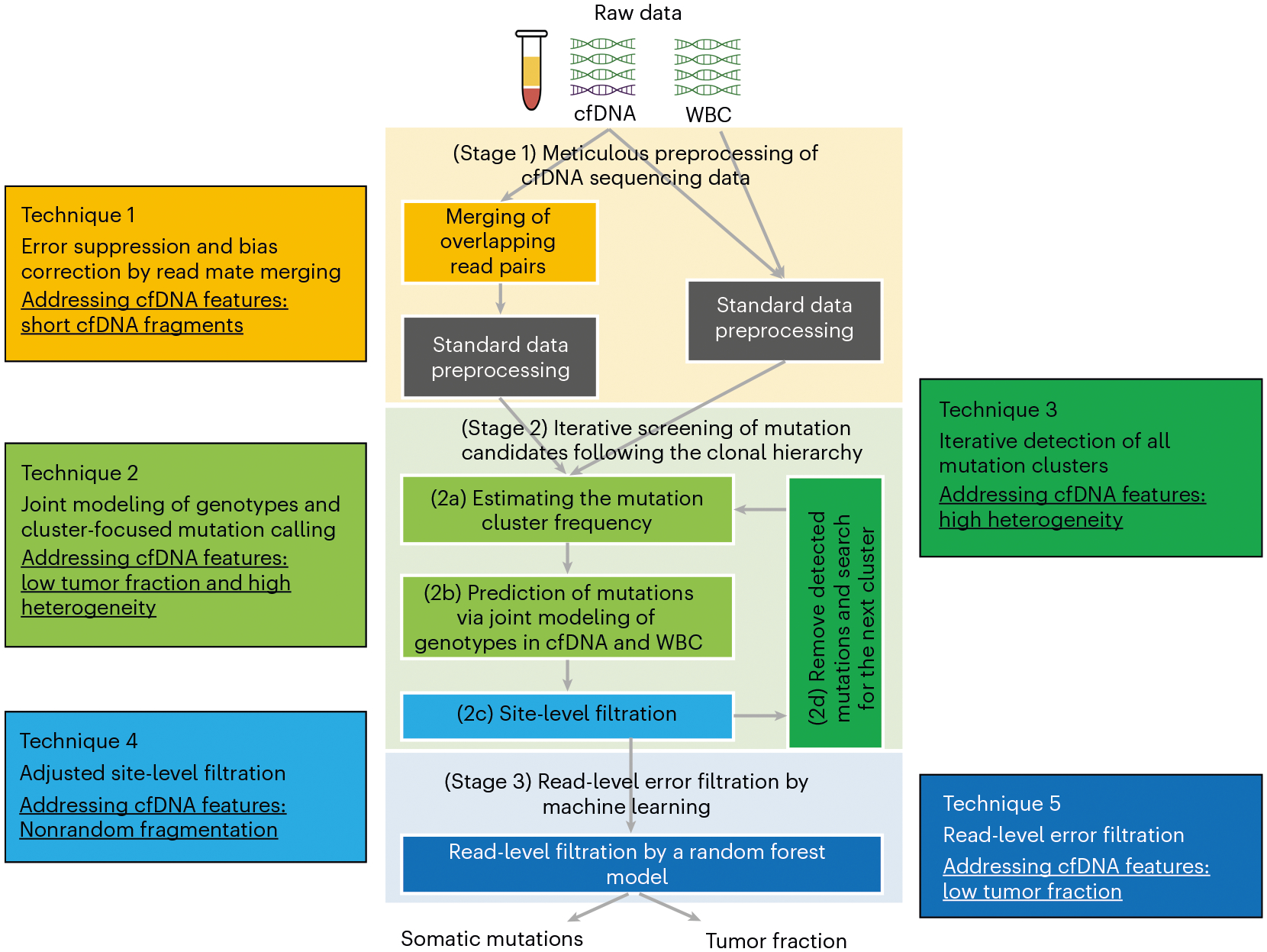
The cfSNV workflow and its techniques. cfSNV takes cfDNA and the matched WBC sequencing data as inputs; no tumor samples are needed. Stage 1 and Technique 1: overlapping read pairs in cfDNA sequencing data are first merged, and then standard data preprocessing tools are used. Stage 2 and Techniques 2–4: an iterative procedure (Stage 2d and Technique 3) then detects mutation clusters and estimates their frequencies on the basis of multiple, automatically selected potential mutation loci (Stage 2a and Technique 2). Each iteration predicts somatic single-nucleotide variant candidates by jointly modeling the genotypes in the cfDNA and the matched WBC (Stage 2b and Technique 2) and masks the mutation candidates before proceeding (Stage 2c and Technique 4). Stage 3 and Technique 5: after all clusters and mutation candidates have been detected, a random forest classifier identifies raw read pairs with sequencing errors. Finally, somatic single-nucleotide variants are reported and detected only if enough variant-supporting read pairs pass the read-level filtration. The background colors in different stage boxes correspond to different techniques; specific features of cfDNA, which are addressed by different techniques, are underlined. Adapted with permission from ref.9 under a Creative Commons licence CC BY 4.0.
Preprocessing of cfDNA sequencing data (Technique 1 and Stage 1). In our protocol, the preprocessing step not only processes the raw sequencing FASTQ data to analysis-ready BAM files but also removes VAF biases and suppresses sequencing errors from overlapping read mates of short cfDNA fragments. By comparing the context of the read and its mate, we identify the overlapping read mates and combine the two read mates in the overlapping region. If one position has inconsistent bases on the two read mates, we correct it to be the base calls with higher quality; if one position has consistent bases on the two read mates, we increase its base quality. In this way, we avoid double-counting the cfDNA fragments and thereby remove the VAF bias. In addition, taking advantage of the overlapping read mates, we suppress sequencing errors, providing a solid basis for accurate mutation detection (Technique 1 in Fig. 1)9. The bias-corrected and error-suppressed FASTQ files are then aligned to the reference genome, followed by duplicate removal, indel realignment and base quality score calibration by using the standard GATK pipeline21,22.
Iterative screening of mutation candidates following the clonal hierarchy (Techniques 2–4 and Stage 2). Unlike genomic DNA from a solid tumor sample, a blood sample includes DNA fragments from all tumor sites, which covers the full range of clonal and subclonal mutations1,2. However, a mutation caller that is optimized to detect clonal mutations will inevitably sacrifice accuracy to detect subclonal mutations. Therefore, the heterogeneous clonal compositions in cfDNA present a considerable challenge in comprehensive mutation detection. To address this challenge, cfSNV takes advantage of the fact that the mutations associated with the same clone have similar VAF in cfDNA1. The mutations are therefore naturally clustered according to the clonal hierarchy1. This fact allows us to automatically cluster mutations on the basis of their allele frequencies and then detect clusters of mutations from the highest to the lowest frequencies following the clonal hierarchy (Technique 3 in Fig. 1). Specifically, in each iteration, we first focus on the most frequent mutation cluster and estimate the frequency that represents the mutated tumor DNA fraction of this cluster. To obtain a robust estimation of this frequency, we aggregate reads from potential mutation loci, which are identified by directly comparing the cfDNA and the matched WBC data (Technique 2 and Stage 2a in Fig. 1). Given the estimated frequency, we next jointly model the genotypes in the cfDNA and the matched WBC to probabilistically tease apart the low-level tumor-derived signal from the overwhelming normal cfDNA background and identify mutation candidates that can fit into this cluster (Technique 2 and Stage 2b in Fig. 1). This joint model enhances the sensitivity of mutation detection by precisely describing the mixed nature of cfDNA. Then, the mutation candidates are eliminated according to a set of filters that account for the nonrandom fragmentation pattern of cfDNA (Technique 4 and Stage 2c in Fig. 1)18. We stratify the remaining mutation candidates into two groups (high- and medium-quality groups) on the basis of their quality statistics (e.g., base quality, mapping quality and allelic bias). The quality of a mutation candidate determines the number of variant-supporting reads required in Stage 3 to finally report it as a mutation. Note that both the joint genotyping model and the filtration consider the alleles in the matched WBC. In this way, cfSNV effectively removes the non-tumor-related mutations that arise from blood cell expansion (i.e., clonal hematopoiesis of indeterminate potential)23. After the filtration, we remove these detected mutation candidates and repeatedly perform the same operation to identify the next-most-frequent mutation cluster (Technique 3 and Stage 2d in Fig. 1). The process repeats, detecting the next-most-frequent mutation cluster at each iteration until no more mutations are detected with confidence. The iterative process of mutation screening allows comprehensive detection of both clonal and subclonal mutations9.
Read-level error filtration by a random forest classifier (Technique 5 and Stage 3). When the tumor fraction is low, sequencing errors impair the specificity of mutation detection in cfDNA. Traditional mutation detection methods usually use a post-filtration step to improve specificity by using site-level statistics19,24,25 such as averaged base quality of all reads at a mutation. Given the low tumor fraction in cfDNA, the supporting reads for a tumor-derived mutation are limited. Therefore, it is challenging to obtain robust estimates of site-level statistics for reliable error filtration. To address this challenge, machine learning models have been developed on the basis of sequencing context in individual reads26,27. Specifically, we derive a read-level error filtration model (a random forest classifier), which distinguishes true variants from sequencing errors for individual reads. The model incorporates various contextual features (e.g., sequence context and substitution type) and fragment features (e.g., fragment size and alignment concordance) to overcome the low signal-to-noise ratio for low-VAF mutations. After the iterative screening of mutation candidates, all variant-supporting reads at the mutation candidates are filtered by the read-level error filtration model (Technique 5 and Stage 3 in Fig. 1). Only mutation candidates with adequate, true variant-supporting reads are reported. The number of required true variant-supporting reads for a mutation is determined by the quality of a mutation candidate: commonly, we suggest a higher threshold for mutations in the medium-quality mutation group than those in the high-quality mutation group.
cfSNV is implemented in C++ and Python for high computational speed. It is wrapped in R and Docker, so users can simply execute it in high-performance computing environments (e.g., servers, computer clusters or cloud computing platforms), as well as on local computers. To accommodate different sequencing coverages and experimental protocols of the input data and the varied tumor fractions in the cfDNA samples, cfSNV comes with a parameter-recommendation function to help users understand the features of the input sample and the data (e.g., sequencing depth) and provide sample-specific recommendations of parameter settings on the basis of these features for high-quality mutation detection. The software reports a tumor fraction and a list of somatic mutations in the widely acknowledged variant calling format (VCF), which can be easily adopted in the subsequent mutation analysis workflows. The reported tumor fraction can be used to track the tumor burden changes for cancer patients2,11,14, while the somatic mutations contain a wealth of information and can be used to inform cancer treatment decisions6,8,10.
Comparison with other methods
The detection of tumor-derived mutations has been an essential task in cancer research and clinical application, for which many methods have been developed. Because a detailed description and full comparison of these different methods is beyond the scope of this protocol, we refer the readers to recent review articles that outline the features of each tool and discuss their advantages and disadvantages in different use cases28–30. Nearly all of these tools were not designed for cfDNA. Thus, most of them have only been used on genomic DNA sequencing data from solid tumors. Therefore, in our comparison, we focus on the tools that have been applied to cfDNA data in previous studies (i.e., MuTect19 and Strelka225).
In general, mutation detection methods have three stages, as shown in Fig. 1: preprocessing, mutation screening and post-filtration30. cfSNV has unique and specific considerations for cfDNA properties in all three stages9.
Preprocessing
In the preprocessing step, MuTect and Strelka2 directly rely on the standard preprocessing pipeline, including alignment (e.g., bwa31), deduplication (e.g., Picard tools32 or samtools33), optional indel realignment (e.g., GATK21,22) and base quality score recalibration (e.g., GATK21,22 and ABRA34)19,25. In addition to this standard preprocessing, cfSNV explicitly resolves the overlapping read mates in the paired-end sequencing data by identifying and merging the overlapping regions of the read mates. It thereby simultaneously corrects the bias in VAF and suppresses the sequencing errors; other methods do not use the overlapping read mates for this purpose. With a standard read length (100 or 150 bp), the overlapping read mates usually constitute a large fraction (>50%9) of the paired-end sequencing data of cfDNA. Even with a short read length (75 bp), overlapping read mates are expected to be observed according to the cfDNA fragment size distribution16. Therefore, cfSNV exploits the great potential of sequence redundancy naturally in cfDNA data to correct the bias of cfDNA fragment counts and suppress the sequencing errors9. Note that given cfDNA’s short-sized nature, using a shorter read length may be a cost-efficient strategy for cfDNA sequencing, but it may also potentially affect the mappability and the mutation confidence in the repetitive area.
Mutation screening
During the mutation screening step, MuTect and Strelka2 use matched tumor-normal samples to detect somatic mutations. The basic idea is to compare the likelihoods of models with somatic mutations, wild-type genotypes or germline mutations. MuTect uses a likelihood model of genotypes in tumor samples to determine mutation candidates19. It selects mutation candidates on the basis of a single log-likelihood ratio cutoff between mutation genotypes and wild-type genotypes19. The cutoff retains the most likely mutation candidates while removing potential artefacts. Strelka2 predicts genotypes in tumor and normal samples25. It considers all positions where a somatic genotype has the highest likelihood to be somatic mutation candidates25. However, without a hard cutoff, the mutation candidates from Strelka2 may include many false positives. cfSNV, like MuTect and Strelka2, uses cfDNA and its matched WBC data for mutation screening, but it has a different statistical model and procedure that are tailored to cfDNA-specific properties. Given the mixed nature of cfDNA and the low tumor fraction, cfSNV jointly models the tumor and the normal genotypes in cfDNA and explicitly incorporates the tumor fraction and mutation cluster frequency into the likelihood model. As a result, it statistically separates the small amount of tumor-derived cfDNA from the pool of normal cfDNA to overcome the low tumor fraction and detect low-frequency mutations9. Furthermore, to address heterogeneity, cfSNV uses an iterative procedure to screen mutation candidates from the highest to the lowest frequency. The iterative procedure explores the full clonal hierarchy, which makes cfSNV sensitive to both clonal and subclonal mutations9. Meanwhile, cfSNV retains all mutation candidates for filtration. Therefore, it may include false-positive candidates at low allele frequencies.
Post-filtration
Sequencing or alignment artefacts may trick the statistical model to pass them as real mutations. Most mutation-detection methods apply a set of filters to identify the artefacts and improve specificity. In the post-filtration step, MuTect and Strelka2 use site-level statistics, including strand bias, clustered read position and averaged mapping quality. MuTect uses hard thresholds for all site-level statistics to filter artefacts19. Strelka2 builds a machine learning model by using these site-level statistics as features to classify mutations and artifacts25. These filters have limited power on the cfDNA data. First, none of these filters consider nonrandom fragmentation of cfDNA, so they may incorrectly remove true mutations. Second, site-level statistics alone may not be robust enough for post-filtration, given that limited variant alleles can be observed for a low-frequency mutation in cfDNA. Instead, cfSNV uses a two-stage post-filtration at the site level and the read level. For site-level filtration, cfSNV adjusts the filters in the existing methods to accommodate the nonrandom fragmentation pattern of cfDNA. These adjusted filters avoid incorrect or aggressive filtration of mutation candidates in cfDNA to keep more true positives. Note that site-level filtration is implemented as a component after identifying a mutation cluster in the iterative mutation-screening procedure9. For read-level filtration, cfSNV builds a random forest model to distinguish individual reads with true mutations from those with sequencing errors, whereby both contextual features and fragmentation features are used in the model. This model can effectively remove artefacts without relying on multiple observations of variant alleles9. Therefore, the read-level filtration can eliminate false negatives and ensure high specificity when the tumor fraction is low9. Note that the read-level filtration is performed as an independent step after the iterative mutation-screening procedure.
Overall, in all three stages, cfSNV has considered cfDNA-specific properties for sensitive and specific somatic mutation detection in cfDNA. As shown in a series of simulation data and real patient data9, cfSNV outperforms MuTect19 and Strelka225 in terms of sensitivity and specificity in detecting somatic mutations in cfDNA.
Applications of the protocol
Because of its noninvasiveness, cfDNA in blood has received enormous attention as a surrogate for solid tumors. Somatic mutations in cfDNA have been widely used as biomarkers in many circumstances. Here, we name a few. (1) Tumor mutation profiling: in many cancer cases (e.g., metastatic tumors), a single tumor biopsy is difficult to access or is insufficient to represent the tumor1,2,35,36. The sensitive detection of mutations in cfDNA can provide a comprehensive somatic mutation profile for the heterogeneous tumor6,9, aiding treatment decisions and personalized therapy selection. (2) Cancer monitoring: to monitor cancer treatment, repeated evaluation of tumor changes is needed, when tumor biopsy is not a viable option. Because cfDNA can be collected noninvasively, the tumor-derived mutations in cfDNA can be analyzed in a timely manner and used for the detection of recurrence5,14, minimal residual diseases13,14,37, multiple primary diseases14,38 and clonal evolution2,14,19. (3) Cancer detection: somatic mutations are stable tumorigenesis markers that have been used for cancer detection3–5. Mutation detection is key for these applications. With the high sensitivity and accuracy of tumor-derived somatic mutation detection in cfDNA, cfSNV can provide a solid basis for these applications. The improved mutation-calling performance of cfSNV expands the clinical utility of cfDNA from small gene panels (at a deep sequencing coverage) to the whole-exome scale (at a medium-coverage sequencing), which further facilitates new applications using exome-wide mutation profiles, such as a blood-based truncal tumor mutation burden for immunotherapy prognosis9. In addition, cfSNV can easily adapt to the analysis of cfDNA data from other organisms (e.g., tumor-bearing mice). Therefore, it will also contribute to general liquid biopsy-related basic and translational cancer research.
Limitations
There are several limitations of cfSNV. First, cfSNV has a focus on tumor-derived single-nucleotide variants (SNVs). To date, cfSNV has not yet supported the detection of somatic indels. Second, cfSNV does not consider haplotypes of somatic mutations. Considering the short size of cfDNA fragments, it might be difficult to resolve haplotypes from cfDNA. Third, the main part of cfSNV (parameter recommendation and mutation detection) can be applied to both paired-end and single-end sequencing data. However, some techniques (e.g., error suppression in the overlapping read mates) at least partly rely on the read mates from the paired-end sequencing data. Fourth, unique molecular identifier (UMI)-tagged ultra-deep sequencing has been used to profile small gene panels in cfDNA, but cfSNV intentionally does not include a module to handle UMI-related preprocessing (e.g., UMI trimming and deduplication), because of the often customized UMI design. Therefore, we recommend that users separately preprocess UMI-tagged data. Then, cfSNV can use the preprocessed data as input and perform mutation detection. Fifth, the error suppression in cfSNV specifically addresses the base call errors of the sequencing-by-synthesis method. Without additional molecular information (e.g., UMI), it cannot handle the PCR errors from the library preparation, but note that the PCR errors are generally not an issue for medium-coverage data without UMI because of the limited sequencing depth.
Experimental design
The procedure outlined in this protocol applies cfSNV to detect somatic mutations for a simulated patient (test demo) and a real breast cancer patient (example data36). In both cases, the input data are the WES data of cfDNA and of its matched WBC. The test demo contains only simulated sequencing reads generated from a prostate cancer patient39 on chromosome 22 for quick testing and troubleshooting. The example data contain raw sequencing reads from a standard WES dataset. We recommend that users who are not familiar with next-generation sequencing data first follow our test demo data and the example data before applying cfSNV to their own datasets.
The input files for cfSNV are described in Box 1; these files are user data files (raw FASTQ files from cfDNA and the matched WBC) and reference files tailored to the users’ experimental protocols. The reference files include a FASTA-format file (genome) of the reference genome; a browser extendable data (BED)-format file (target) that contains targeted genomic regions in the sequencing, such as a targeted panel; and a VCF-format file (database) that contains genomic positions to be blocked from mutation detection. The mutation detection will be performed in the genomic regions in the input BED file target.
Box 1 |. Input files for cfSNV.
cfSNV requires the user to specify data files and reference files as input for analysis. The names used in this box are the same as those used throughout the procedure.
genome: the reference genome sequence in the standard FASTA format. Examples of the reference genome file can be found on the UCSC Genome Browser52 (https://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/hg19.fa.gz).
- target: the genomic coordinate of the targeted regions in BED format. The targeted regions are the genomic regions that are intended to be captured by the sequencing assay (e.g., the target bait of the WES). We recommend that users remove low mappability regions to improve the quality of mutation detection based on their analysis need. The targeted region file is formatted as follows:
- No header
- Column 1: chromosome names (e.g., chr1, chr2, …, chrY; the format must be consistent with the chromosome names in the reference genome)
- Column 2: start coordinate of the target region on the chromosome
- Column 3: end coordinate of the target region on the chromosome
- Other columns are optional.
database: a list of genomic positions in the standard VCF format to be blocked in mutation detection. The possible reasons to block a position from mutation detection include common SNPs, repeated elements and low-complexity sequences. The file should have a header and the same chromosome name format and chromosome order as the reference genome. Examples can be found in the dbSNP database51 (https://ftp.ncbi.nlm.nih.gov/snp/organisms/human_9606_b150_GRCh37p13/VCF/common_all_20170710.vcf.gz).
(cfDNA) fastq1: the raw read 1 data in the standard FASTQ format from the paired-end sequencing of cfDNA.
(cfDNA) fastq2: the raw read 2 data in the standard FASTQ format from the paired-end sequencing of cfDNA.
(WBC) fastq1: the raw read 1 data in the standard FASTQ format from the paired-end sequencing of WBCs.
(WBC) fastq2: the raw read 2 data in the standard FASTQ format from the paired-end sequencing of WBCs.
The cfSNV package consists of four major modules (Fig. 2): (i) index generation, (ii) data preprocessing, (iii) parameter recommendation and (iv) mutation detection. Note that these are not the same as the three stages outlined in Development of the protocol.
Fig. 2 |.
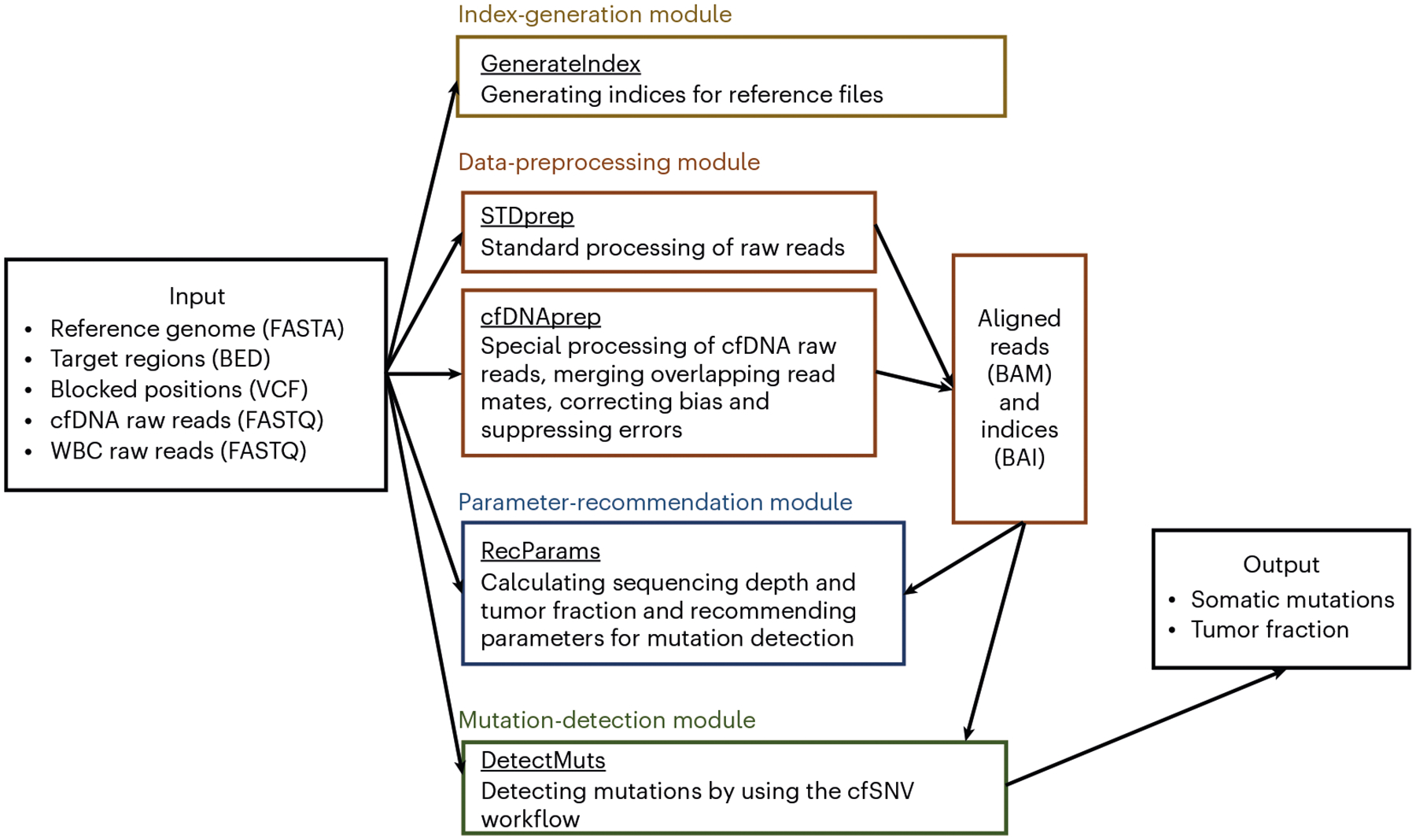
Four modules in the cfSNV Docker package. cfSNV includes four major modules: index generation, data preprocessing, parameter recommendation and mutation detection. The index generation module contains one command, GenerateIndex. It generates indices for the userspecified reference files (reference genome, target regions and blocked positions), which are required for quick search across the genome. We provide two commands, STDprep and cfDNAprep, in the data preprocessing module. STDprep processes the raw sequencing reads (FASTQ files) of cfDNA and WBC to aligned reads and indices (BAM and BAI files). cfDNAprep contains a cfDNA-specific preprocessing pipeline, which accommodates the short cfDNA fragments. It generates aligned reads and indices (BAM and BAI files) from the raw sequencing reads of cfDNA (FASTQ files). The parameter recommendation module contains one command, RecParams. It recommends key parameters for mutation detection on the basis of the estimated sequencing depth and tumor fraction (optional). The mutation detection module has one command, DetectMuts. It takes the aligned reads generated from the data preprocessing module as input, and it outputs a list of somatic mutations and an estimation of tumor fraction.
Index generation
The input reference files need to be indexed before use. Although the index files are not explicitly used in the cfSNV protocol, they are required by bwa31, GATK21,22 and Picard tools32 for efficient random searching of genomic coordinates. cfSNV provides a command (GenerateIndex) to generate all index files required in the subsequent analysis. The users can directly use this command to process their reference files.
Data preprocessing 4
Data preprocessing contains the first major stage of cfSNV (i.e., preprocessing of cfDNA sequencing data (Stage 1 in Fig. 1)). cfSNV provides two commands to process raw sequencing FASTQ files of cfDNA and WBC genomic DNA into analysis-ready BAM files. One command (STDprep) is for the standard preprocessing of both WBC and cfDNA data. The raw sequencing data are checked for data quality and processed with a standard preprocessing pipeline, including alignment (bwa31), deduplication (Picard tools32), indel realignment (GATK21,22) and base quality score recalibration (GATK21,22). The other command (cfDNAprep) is only for cfDNA to deal with the overlapping read mates. The overlapping read mates are first identified and merged by using FLASH240. The merged read pairs are processed as single-ended data by the standard preprocessing pipeline. The read pairs without any overlap remain as paired-end data and are processed by the standard preprocessing pipeline. For a pair of cfDNA and WBC samples, the raw data of cfDNA are processed with both STDprep and cfDNAprep, whereas the raw data of the WBC are processed only with STDprep.
Parameter recommendation
As aforementioned, cfSNV groups the mutation candidates into high- and medium-quality groups. The mutation candidates in different groups require different numbers of true variant-supporting reads to be reported. Thus, there are two key parameters: minPass and minHold (i.e., the minimum number of required true supporting reads for mutation candidates in the high- and medium-quality groups, respectively). These parameters need to be tailored to specific datasets. The parameter-recommendation module inspects the input data, provides users with statistics for their own data and offers options for parameter settings. The parameter recommendation module is wrapped in a single command, RecParams. For a specific input dataset, RecParams checks the sequencing depth and roughly estimates the tumor fraction. The rough estimation of tumor fraction requires a longer running time, so we also provide an option to disable the tumor fraction estimation in RecParams. Theoretically, the expected detection limit is determined by the sequencing depth and the minimum number of required supporting reads for a mutation. For example, given a sequencing depth of 200×, if we report mutations only with ≥10 variant-supporting reads, the detection limit is ~5%. Following this idea, we provide up to three recommendations of parameter settings (i.e., minPass and minHold), given the estimated sequencing depth and different detection limit thresholds (Fig. 3a,b). In other words, the parameter recommendation tells the users that by using a set of recommended minPass and minHold parameters, they can achieve the corresponding detection limit (i.e. the lowest VAF of detectable mutations) from the input data. Then, the users can select parameters that suit their data and meet their expected detection limit. The selection of parameters is not limited to the recommendations.
Fig. 3 |.
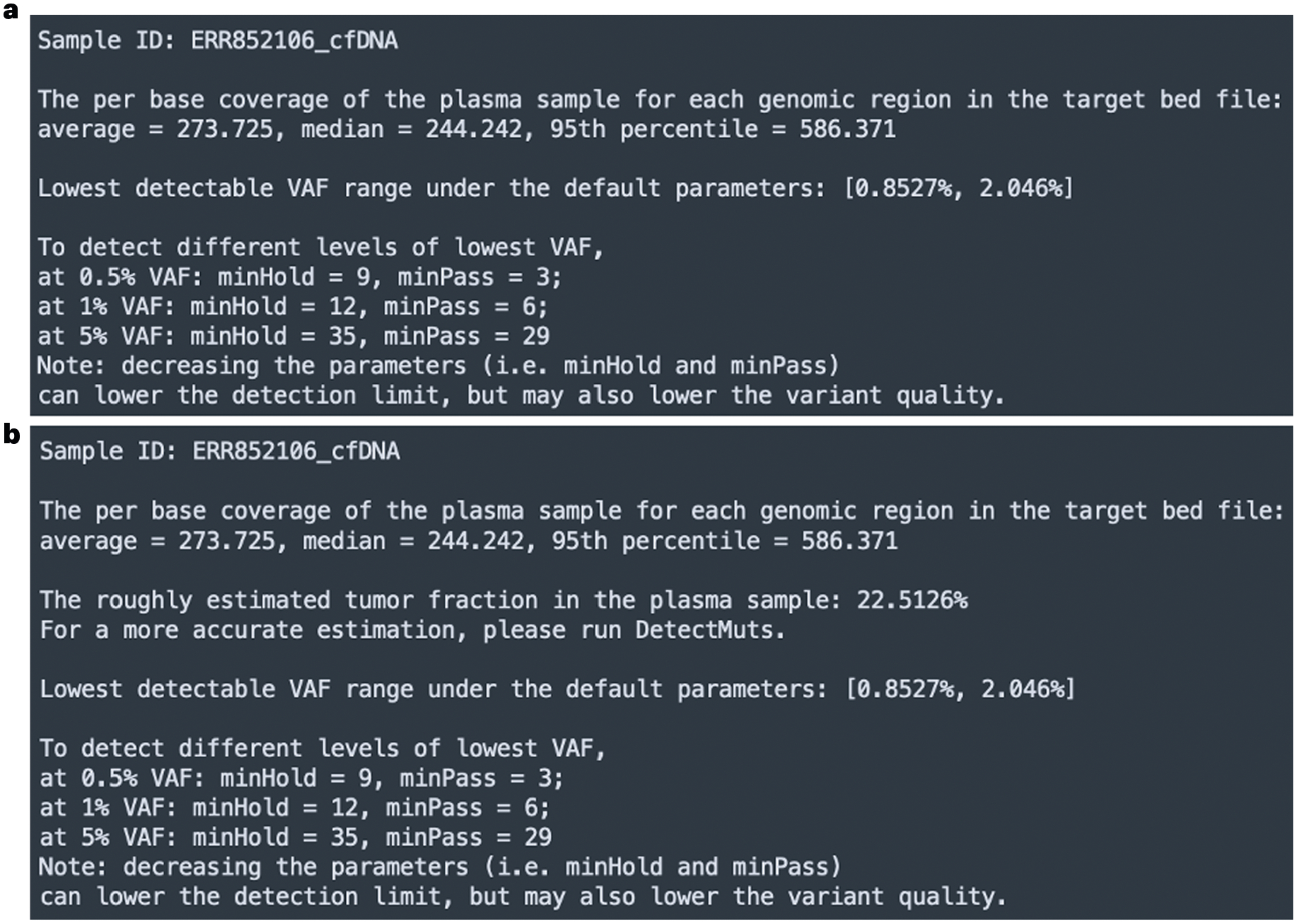
Example outputs of the parameter-recommendation module. a,b, The output is generated by executing the parameter recommendation module (i.e., the command RecParams) on our example data without estimating the tumor fraction (roughEstimate=FALSE) (a) and with estimating the tumor fraction (roughEstimate=TRUE) (b). The command RecParams reports the estimated per-base coverage of the aligned reads (generated by STDprep in Step 3) of the cfDNA sample in the genomic regions included in the target BED file. It also reports a roughly estimated tumor fraction if the option roughEstimate is set to TRUE. On the basis of the estimated per-base coverage, RecParams provides recommended parameters (minHold and minPass) for up to three different detection limits (i.e., the lowest detectable variant allele frequency). The users can determine the parameters for the mutation-detection module on the basis of the recommendations.
Mutation detection
Mutation detection consists of two major stages of cfSNV: iterative mutation screening (Stage 2 in Fig. 1) and read-level filtration (Stage 3 in Fig. 1). The details of these two stages have been described in our previous work9 and are briefly discussed in Development of the protocol, Stages 2 and 3. The mutation-detection module is wrapped in a single command, DetectMuts. This command generates the final output of cfSNV, including a list of somatic mutations (Fig. 4a) and a numeric value of the estimated tumor fraction in the cfDNA (Fig. 4b).
Fig. 4 |.
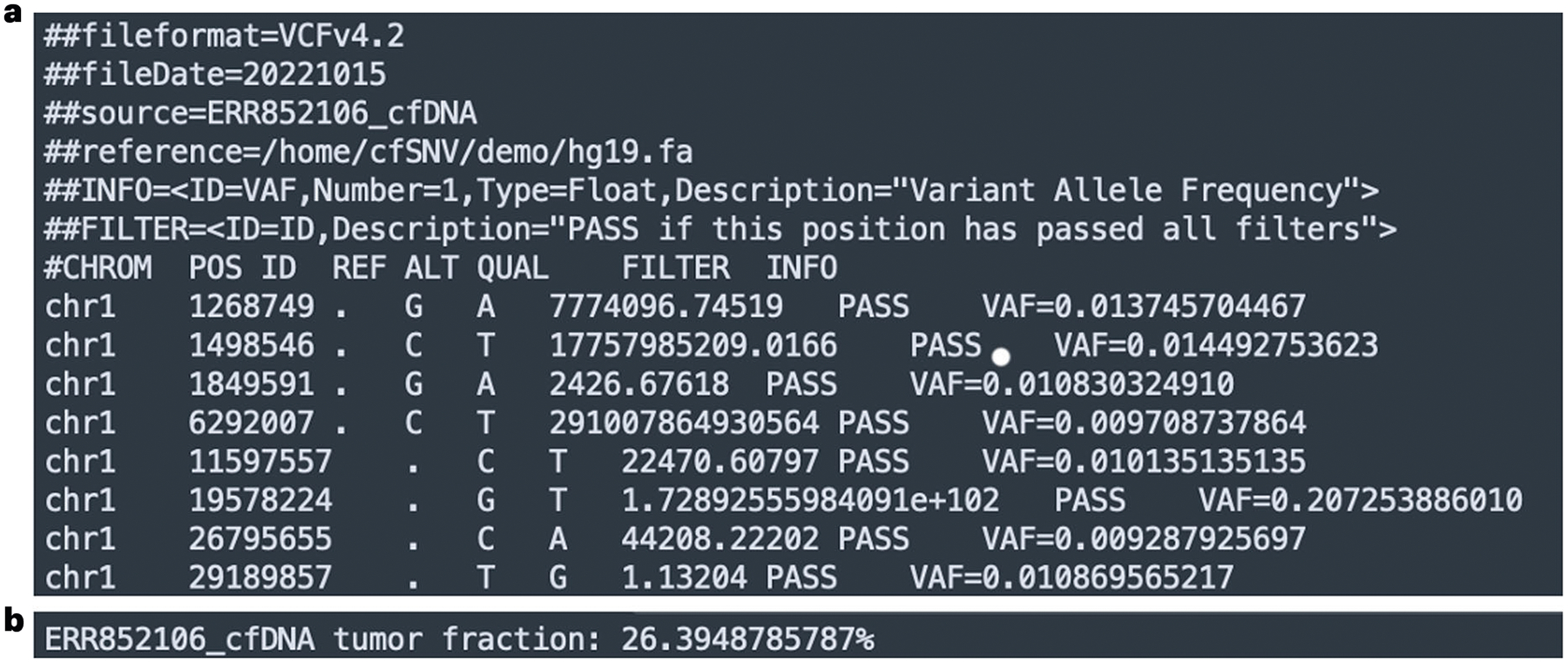
An example output of the variant list and the tumor fraction from cfSNV. a, A screenshot of the output variant list generated from the example data. The variant list is in the standard VCF format. For each mutation, we output the genomic position and the nucleotide change. In addition, we provide a quality score, a flag indicating the filter status and the variant allele frequency of every mutation. b, A screenshot of the output tumor fraction from the example data.
Expertise needed to implement the protocol
We built a Docker image of cfSNV, which contains all dependencies required to run the pipeline. To use the Docker image, only some basic knowledge of the Bash command-line interface is needed. Following the steps of this protocol, users only need to execute a few command lines in the terminal. The parameter recommendation module automatically detects the statistics of the users’ own input dataset and recommends a few parameter settings that are tailored to the specific experimental protocol, sequencing coverage and tumor fraction of the dataset. Therefore, users with limited experience in sequencing data and mutation detection can still execute the pipeline and carry out high-quality analyses. The output format is in the standard VCF and thus requires minimal prior experience to interpret the results, fit into the existing workflow and perform downstream analysis.
Materials
Equipment
Software
Operating system: Windows, MacOS or Linux distributions
Docker41 (http://www.docker.com/). With the cfSNV Docker image as a template, containers can be built and run on most operating systems. ▲ CRITICAL All dependencies of cfSNV are packed into the Docker container, so the user does not need to install any specific tools separately. Versions of software tools and package dependencies are listed as they are packed in the container.
bedtools42 v2.30.0 (https://github.com/arq5x/bedtools2/releases/tag/v2.30.0)
bwa31 v0.7.17 (http://bio-bwa.sourceforge.net/)
FLASH40 v2 (https://github.com/dstreett/FLASH2)
GATK21,22 v3.8.0 (https://github.com/broadgsa/gatk/releases/tag/3.8)
Java43 v1.8 (https://java.com/download)
Picard32 v2.18.4 (https://github.com/broadinstitute/picard/releases/tag/2.18.4)
Python44 v3.6 (https://www.python.org/downloads/)
Python library: numpy45 v1.13.3 (https://numpy.org)
Python library: pandas46 v0.20.3 (https://pandas.pydata.org)
Python library: scikit-learn47 v0.24.1 (https://scikit-learn.org/stable/)
Python library: scipy48 v1.1.0 (https://scipy.org)
R49 v4.0.2 (https://www.r-project.org)
R package: Rcpp50 (https://www.rcpp.org)
samtools33 v1.11 (https://sourceforge.net/projects/samtools/files/samtools/)
Hardware
Computer. cfSNV can be used on laptops, workstations, computer clusters or cloud computing platforms. ▲ CRITICAL We recommend an eight-core processor and 16 GB of random access memory (RAM), but it is not a hard minimum. The required disk storage is determined by the amount of input data. We recommend 20 GB of disk storage for reference files, including the reference genome, the index files and the single-nucleotide polymorphism (SNP) databases. For the WES data of cfDNA (~300 million 100-bp reads, ~250× on target regions of size ~70 million bp) and the matched WBC (~300 million 100-bp reads, ~250× on target regions of size ~70 million bp), ~250 GB of disk storage is needed for temporary files. The complete results of a cfDNA sample use the minimum disk storage (<10 MB).
Equipment setup
In this protocol, we use the test demo data and example data to describe each step in the pipeline. The steps (Fig. 2) are interactively performed inside the Docker container through Bash commands and executable scripts.
Initial setup
Start the Docker software and open the command terminal on the host machine. In the Windows system, the terminal can be found as the Command Prompt from the Start menu. In Linux system, the terminal can be found as a pre-installed application Terminal. In MacOS, the terminal can be found as the Terminal in the Applications list.
Through the terminal, create a working directory called ‘working_directory’, which is used to place the Docker image. Enter the working_directory and download the latest Docker image from https://github.com/jasminezhoulab/cfSNV_docker/releases. For example:
cd <working_directory> && wget
https://github.com/jasminezhoulab/cfSNV_docker/releases/download/v2.0.1/cfsnv_image.tar.gz Load the Docker image:
docker load < cfsnv_image.tar.gz
The Docker image called ‘cfsnv_docker’ will be loaded.
By default, the Docker container cannot access files located on the host machine. Therefore, users need to create a Docker container with a directory on the host machine mounted into the Docker container or copy files from the host machine to the Docker container.
Here, we provide the instructions for mounting a directory inside the container. Specifically, the users need to specify two directory paths for mounting: (i) a local_directory on the host machine, where all input data are located and (ii) a container_directory, through which the data on the host machine can be accessed. ! CAUTION The container_directory cannot be set to /home/cfSNV or /home; otherwise, the executable commands will be removed.
Then, create the Docker container and mount the data directory, with the following command:
docker run -it -d -v <local_directory>:<container_directory> --name <cfsnv_container> cfsnv_docker bash
In this command, please replace local_directory with a local directory in the host machine, replace container_directory with the directory in the Docker (a path name in Linux; e.g., /home/cfSNV/demo) and replace cfsnv_container with a name as the container’s name. ! CAUTION The command in general applies to different operating systems (Windows, Linux and MacOS), but users need to follow the syntax of path names on their host machine. An example path name in Windows is C:\Documents\cfSNV\demo, and an example path name in Linux or MacOS is /home/users/cfSNV/demo.
Run the Docker container:
docker exec -it <cfsnv_container> bash
After the above steps, users will enter the cfSNV Docker container directory “/home/cfSNV”, where the pipeline can be executed by using interactive bash commands. See Troubleshooting section.
Reference files
The pipeline requires several reference files as input (Box 1), including the reference genome sequence in FASTA format, the genomic coordinate of the targeted regions in BED format and a list of genomic positions in VCF format to be blocked in the mutation detection (e.g., dbSNP common SNP database51). The required reference files for the example data and the test demo data are available at https://zenodo.org/record/7191202/files/example_reference_files.tar.gz and https://zenodo.org/record/7191202/files/demo_reference_files.tar.gz. Users can download the reference files after starting the container. ▲ CRITICAL The reference files need to be consistently referred to the same genome build and need to be downloaded into the container_directory before users start the analysis procedures:
Inside the container, users can set the variable container_directory as the container directory path used when building the container:
container_directory=/CONTAINER_PATH/TO/INPUT
cd ${container_directory}\
&& wget https://zenodo.org/record/7191202/files/example_reference_files. tar.gz \
&& tar -xzvf example_reference_files.tar.gz \
&& rm example_reference_files.tar.gz
cd ${container_directory} \
&& wget https://zenodo.org/record/7191202/files/demo_reference_files. tar.gz \
&& tar -xzvf demo_reference_files.tar.gz \
&& rm demo_reference_files.tar.gz
The above commands download the reference files for the example data and the demo data to folders named ‘example_reference_files’ and ‘demo_reference_files’, respectively, in the container_directory. Users can prepare their own reference files based on their needs.
Example data and test demo data
The example data are available at the European Nucleotide Archive (https://www.ebi.ac.uk/ena) by using the run accession numbers ERR850376 (WES WBC) and ERR852106 (WES cfDNA)36.
A command (DownloadEg) for easily downloading the example data (four FASTQ files from two samples that are separated into reverse and forward reads) is available in the container.
Users can also use the command in the Docker container to download data to an example_data folder within the container_directory:
mkdir ${container_directory}/example_data \
&& cd ${container_directory}/example_data \
&& /home/cfSNV/DownloadEg
The demo data are available at https://zenodo.org/record/7191202/files/demo_data.tar.gz. To download the demo data to the container_directory folder:
wget https://zenodo.org/record/7191202/files/demo_data.tar.gz && tar -xzvf demo_data.tar.gz \ && rm demo_data.tar.gz \ && mv demo_data ${container_directory}
▲ CRITICAL The data need to be downloaded and put into the container_directory before users start the analysis procedure.
Procedure
Start the Docker container 25CF ● Timing ~1 s
-
1
1 Start the Docker container by running:
docker exec -it <cfsnv_container> bash
If a cfSNV Docker container is successfully built by following the Equipment setup, Initial setup instructions, users can simply execute the above command to enter the bash environment in the container. The command works for different operating systems on the host machine (Windows, Linux or MacOS). The executable files in the /home/cfSNV folder are used to execute cfSNV pipelines. In the following steps, we assume the working directory is /home/cfSNV and that all codes are executed within the directory.
Generate index files for the reference file ● Timing ~1 h
-
2
Create genome index files by GenerateIndex. To check the usage of the command:.
./GenerateIndex -h
Usage:./GenerateIndex –g ${genome}
For instance, to generate index files for the example reference genome:
./GenerateIndex \
-g ${container_directory}/example_reference_files/hg19.fa
To generate index files for the demo reference genome:
./GenerateIndex \
-g ${container_directory}/demo_reference_files/demo_ref_genome.fa
Preprocess the input data ● Timing ~15 h per command
▲ CRITICAL The two preprocessing commands (STDprep and cfDNAprep) are independent, so users can easily execute them in parallel (e.g., running them simultaneously in the background). For the standard preprocessing, instead of using STDprep, our method also allows users to use other preprocessing pipelines and then directly provide the processed BAM files as the input for the next two steps.
-
3
Standard preprocessing of cfDNA and the matched WBC data. cfSNV provides a standard preprocessing workflow for paired-end genomic sequencing data of cfDNA and the matched WBCs, including sequence alignment, duplicate removal, indel local realignment and base quality score recalibration. The complete workflow is wrapped in a single command. The input of this command includes the raw sequencing reads in the FASTQ format, the reference genome in the FASTA format, the database of blocked positions in the VCF format (Box 1), the specified sample name called ‘id’ and the output directory called ‘output’.
Preprocess the data by STDprep. To check the usage of the command:
./STDprep -h
Usage:./STDprep -f1 ${fastq1} -f2 ${fastq2} -g ${genome} -d ${database} -i ${id} -o ${output}
This command needs to be applied to the raw sequencing data of both the cfDNA and the matched WBC. An analysis-ready BAM file (with suffix .recal.bam) and a corresponding index BAI file (with suffix .recal.bai) will be generated for each sample.
Our example data can be preprocessed by using the following specified commands. For the example cfDNA data:
./STDprep \
-f1 ${container_directory}/example_data/ERR852106_1.fastq.gz \
-f2 ${container_directory}/example_data/ERR852106_2.fastq.gz \
-g ${container_directory}/example_reference_files/hg19.fa \
-d ${container_directory}/example_reference_files/dbSNP.hg19.vcf \
-i ERR852106_cfDNA \
-o ${container_directory}/example_preprocess
For the example WBC data:
./STDprep \
-f1 ${container_directory}/example_data/ERR850376_1.fastq.gz \
-f2 ${container_directory}/example_data/ERR850376_2.fastq.gz \
-g ${container_directory}/example_reference_files/hg19.fa \
-d ${container_directory}/example_reference_files/dbSNP.hg19.vcf \
-i ERR850376_WBC \
-o ${container_directory}/example_preprocess
The demo cfDNA data can be preprocessed by:
./STDprep \
-f1 ${container_directory}/demo_data/cfDNA_1.fastq.gz \
-f2 ${container_directory}/demo_data/cfDNA_2.fastq.gz \
-g ${container_directory}/demo_reference_files/demo_ref_genome.fa \
-d ${container_directory}/demo_reference_files/demo_snp_db.vcf \
-i demo_cfDNA \
-o ${container_directory}/demo_preprocess
For the demo WBC data:
./STDprep \
-f1 ${container_directory}/demo_data/WBC_1.fastq.gz \
-f2 ${container_directory}/demo_data/WBC_2.fastq.gz \
-g ${container_directory}/demo_reference_files/demo_ref_genome.fa \
-d ${container_directory}/demo_reference_files/demo_snp_db.vcf \
-i demo_WBC \
-o ${container_directory}/demo_preprocess
? TROUBLESHOOTING
-
4
cfDNA-specific preprocessing. cfSNV especially considers the short fragment size of cfDNA in the preprocessing step and provides a special preprocessing workflow for cfDNA data. The workflow is wrapped in a single command, which includes read mate merging, error suppression, sequence alignment, duplicate removal, indel local realignment and base score calibration. The input of this command is the same as the standard preprocessing command (Step 3).
Preprocess the data by STDprep. To check the usage of the command:
./cfDNAprep -h
Usage:./cfDNAprep -f1 ${fastq1} -f2 ${fastq2} -g ${genome} -d ${database} -i ${id} -o ${output}
This command needs to be applied only to the raw sequencing data of cfDNA. It generates two analysis-ready BAM files (i.e., extended overlapping reads with suffix .extendedFrags.recal.bam and uncombined nonoverlapping reads with suffix .notCombined.recal.bam) and two corresponding index BAI files (with suffixes .extendedFrags.recal.bai and .notCombined.recal.bai, respectively). The BAM file of the extended overlapping reads contains single-end long reads that are merged from the overlapping paired-end read mates. The BAM file of the uncombined nonoverlapping reads contains paired-end original read mates that do not overlap with each other.
To process the example cfDNA data:
./cfDNAprep \
-f1 ${container_directory}/example_data/ERR852106_1.fastq.gz \
-f2 ${container_directory}/example_data/ERR852106_2.fastq.gz \
-g ${container_directory}/example_reference_files/hg19.fa \
-d ${container_directory}/example_reference_files/dbSNP.hg19.vcf \
-i ERR852106_cfDNA \
-o ${container_directory}/example_preprocess
For the demo cfDNA data,
./cfDNAprep \
-f1 ${container_directory}/demo_data/cfDNA_1.fastq.gz \
-f2 ${container_directory}/demo_data/cfDNA_2.fastq.gz \
-g ${container_directory}/demo_reference_files/demo_ref_genome.fa \
-d ${container_directory}/demo_reference_files/demo_snp_db.vcf \
-i demo_cfDNA \
-o ${container_directory}/demo_preprocess
? TROUBLESHOOTING
(Optional) Recommend parameters based on the input data ● Timing 0.5–3 h
-
5
To accommodate input data from different experimental protocols and different sequencing coverages, cfSNV provides an optional command to help the user better understand the input data and provide the user with recommendations of parameters for mutation calling. Basically, the command runs through the first loop of the mutation screening to provide estimates of the sequencing coverage in the targeted regions, the detection limit range and the roughly estimated tumor fraction (optional).
Execute RecParams to obtain help for parameter recommendation.
./RecParams -h
Usage:./RecParams -p ${plasma} -n ${normal} -e ${extended} -u ${uncombined} -t ${target} -g ${genome} -d ${database} -i ${id} -r ${roughEstimate}
The input of this command includes the standard-preprocessed cfDNA and WBC data in the BAM format (outputs from Step 3, including the .recal.bam file for cfDNA and WBC), the cfDNA-specific-preprocessed cfDNA data in the BAM format (outputs from Step 4, including .extendedFrags.recal.bam and .notCombined.recal.bam files for cfDNA), the targeted regions of the experimental protocol in the BED format, the reference genome in the FASTA format, the database of blocked positions in the VCF format (Box 1), the specified case name id and a Boolean flag roughEstimate indicating whether to estimate the tumor fraction.
This command prints out the estimation of the sequencing coverage, the tumor fraction (optional) and a few recommended parameter settings on the screen. Example outputs are shown in Fig. 3a,b.
For the example data, the recommended parameters can be obtained by executing:
./RecParams \
-p ${container_directory}/example_preprocess/ERR852106_cfDNA.recal. bam \
-n ${container_directory}/example_preprocess/ERR850376_WBC.recal.bam \
-e ${container_directory}/example_preprocess/ERR852106_cfDNA.extendedFrags.recal.bam \
-u ${container_directory}/example_preprocess/ERR852106_cfDNA. notCombined.recal.bam \
-t ${container_directory}/example_reference_files/target.bed \
-g ${container_directory}/example_reference_files/hg19.fa \
-d ${container_directory}/example_reference_files/dbSNP.hg19.vcf \
-i ERR852106_cfDNA \
-r FALSE
For the demo data:
./RecParams \
-p ${container_directory}/demo_preprocess/demo_cfDNA.recal.bam \
-n ${container_directory}/demo_preprocess/demo_WBC.recal.bam \
-e ${container_directory}/demo_preprocess/demo_cfDNA.extendedFrags. recal.bam \
-u ${container_directory}/demo_preprocess/demo_cfDNA.notCombined. recal.bam \
-t ${container_directory}/demo_reference_files/target.bed \
-g ${container_directory}/demo_reference_files/demo_ref_genome.fa \
-d ${container_directory}/demo_reference_files/demo_snp_db.vcf \
-i demo_cfDNA \
-r TRUE
▲ CRITICAL STEP The reference genome, the blocked positions and the targeted regions must be the same as in the preprocessing steps.
Detect mutations with the major functions of cfSNV ● Timing ~3 h
-
6
Call mutations by using DetectMuts. To check the usage of mutation detection:
./DetectMuts -h
Usage:./DetectMuts -p ${plasma} -n ${normal} -e ${extended} -u ${uncombined} -t ${target} -g ${genome} -d ${database} -i ${id} -mh ${minHold} -mp ${minPass} -o ${output}
The input of this command includes the standard-preprocessed cfDNA and the WBC data in the BAM format (outputs from Step 3, including the .recal.bam file for cfDNA and WBC), the cfDNA-specific-preprocessed cfDNA data in the BAM format (outputs from Step 4, including .extendedFrags.recal.bam and .notCombined.recal.bam files for cfDNA), the targeted regions of the experimental protocol in the BED format, the reference genome in the FASTA format, the database of blocked positions in the VCF format (as described in Box 1), the specified case name id, the output directory called ‘output’ and two parameters, minHold and minPass (output from Step 5). These two parameters are the minimum number of supporting read pairs that is required for medium-quality mutation candidates (minHold) and high-quality mutation candidates (minPass).
The command outputs somatic mutations to a VCF file and writes the estimated tumor fraction to a TXT file. The VCF format mutation output can be directly used in many downstream analyses (e.g., mutation filtration from mappability and mutation annotation).
For the example data, detect mutations by executing the following command:
./DetectMuts \
-p ${container_directory}/example_preprocess/ERR852106_cfDNA.recal.bam \
-n ${container_directory}/example_preprocess/ERR850376_WBC.recal.bam \
-e ${container_directory}/example_preprocess/ERR852106_cfDNA.extendedFrags.recal.bam \
-u ${container_directory}/example_preprocess/ERR852106_cfDNA.notCombined.recal.bam \
-t ${container_directory}/example_reference_files/target.bed \
-g ${container_directory}/example_reference_files/hg19.fa \
-d ${container_directory}/example_reference_files/dbSNP.hg19.vcf \
-i ERR852106_cfDNA \
-mh 12 \
-mp 6 \
-o ${container_directory}/example_cfsnv_run
For the demo data:
./DetectMuts \
-p ${container_directory}/demo_preprocess/demo_cfDNA.recal.bam \
-n ${container_directory}/demo_preprocess/demo_WBC.recal.bam \
-e ${container_directory}/demo_preprocess/demo_cfDNA.extendedFrags. recal.bam \
-u ${container_directory}/demo_preprocess/demo_cfDNA.notCombined.recal. bam \
-t ${container_directory}/demo_reference_files/target.bed \
-g ${container_directory}/demo_reference_files/demo_ref_genome.fa \
-d ${container_directory}/demo_reference_files/demo_snp_db.vcf \-i demo_ cfDNA \
-mh 9 \
-mp 3 \
-o ${container_directory}/demo_cfsnv_run
▲ CRITICAL STEP The reference genome, the blocked positions and the targeted regions must be the same as in the preprocessing steps.
? TROUBLESHOOTING
Troubleshooting
Troubleshooting advice can be found in Table 1. In addition, the GitHub Issues page (https://github.com/jasminezhoulab/cfSNV_docker/issues) can be used for unforeseen troubleshooting and discussion.
Table 1 |.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| Equipment setup | The Docker container cannot be created | The specified container name is already in use by an existing container | Remove or rename the existing Docker container so that the name can be reused |
| The mount fails | The link between the local directory and the container directory is incorrect | Rebuild a new container by using the full absolute path of the local directory and the container directory. We recommend that users use /home/cfSNV/${user_specified_name} as the container_directory | |
| 3 and 4 | STDprep or cfDNAprep stops unexpectedly | Step 2 failed to generate index files | Run Step 2 to create genome index files |
| The input files are incorrect | Make sure that the input file content and formats are the same as described in Box 1. Check the error message to see which file is incorrect | ||
| The jobs are killed because the system runs out of memory or space | Enlarge the RAM or the storage | ||
| Previous jobs stop unexpectedly and leave intermediate files | Go to the directory as shown in the error message where the intermediate files are located and remove the files | ||
| 6 | DetectMuts does not output any files to the designated folder | cfSNV has not finished properly | Check if all the input files are correct and if the machine has enough free space |
Timing
The runtime of the pipeline depends on the computing resources and the input data size. Here, we specifically quantified the runtimes for the analysis of a standard WES dataset (i.e., the example data) and a small demonstrative dataset (i.e., the test demo data). The running time is obtained by using eight virtual CPUs, 32 GB of RAM and 500 GB of solid-state drive storage.
Step 1, starting the Docker container: ~1 s
Step 2, the runtime of indexing a genome (GenerateIndex): varies on the basis of the size of the reference genome file. For a standard human genome FASTA file (e.g., hg1952), it takes ~1 h to generate all required index files. The runtime is ~1 h for the example reference file and ~1 min for the demo reference file.
Steps 3 and 4, preprocessing steps (STDprep and cfDNAprep): the runtime can vary widely depending on the available computing resources and data sizes. Our preprocessing steps follow the widely adopted framework21,22, so our runtime is comparable to other mutation-detection tools in general. For example, it takes ~17 h to process ~300 million 100-bp reads but <10 min to process ~2.7 million 100-bp reads by using standard preprocessing (STDprep). For the example data, it takes ~16 h and ~10 h to finish the standard preprocessing of the cfDNA (285.6 million 100-bp reads) and the matched WBC data (181.7 million 100-bp reads), respectively. It takes ~12 h to finish the cfDNA-specific preprocessing (cfDNAprep) of the cfDNA (285.6 million 100-bp reads) example data. For the demo data, there are 2.7 million and 1.1 million 100-bp reads in the cfDNA and the matched WBC data, respectively, which require ~20 min to finish the preprocessing steps. Note that the Docker container allows parallel execution of STDprep and cfDNAprep. By running these commands in parallel, the total runtime can be greatly reduced for the preprocessing of cfDNA and WBC raw data.
Step 5, parameter recommendation (RecParams): the runtime is determined not only by data sizes but also by the ‘-r roughEstimate’ option. If the option is set to TRUE, it takes ~2.8 h and ~1.5 min to finish the parameter recommendation for the example data and the demo data, respectively; if the option is set to FALSE, it takes ~20 min and <1 min to finish the parameter recommendation for the example data and the demo data, respectively.
Step 6, mutation detection (DetectMuts): the runtime relies on the size of targeted regions and the complexity of mutation clonal hierarchies. Because the screening of mutation candidates is iteratively performed along the clonal hierarchy, looping through all mutation clusters consumes hours at a time. It takes ~3 h and ~2 min to finish the mutation detection for the example data and the demo data, respectively.
Anticipated results
The final output of cfSNV is a list of somatic mutations and a numeric value of the estimated tumor fraction in the cfDNA. The results are written into two files in the specified output folder. ${id}.variant_list.vcf contains the genomic coordinates, the substitution types of the somatic mutations and other information in the VCF format (Box 2 and Fig. 4a). ${id}.tumor_fraction.txt contains the estimated tumor fraction in the plain text format (Fig. 4b).
Box 2 |. Description of the output variant list.
The output variant list follows the VCF format, which contains meta-information lines (beginning with ##), a header line (beginning with #) and data lines; each data line describes the information of a gene variant. An example output is provided in Fig. 4a.
CHROM and POS: the genomic position of the mutation.
ID: this field is unused.
REF: the reference allele.
ALT: the alternative allele.
QUAL: the log likelihood ratio of the position to be a somatic mutation.
FILTER: a flag indicating whether the mutation passes the post-filtration. The value is ‘PASS’ if the mutation passes the filters.
VAF: the variant allele frequency of the mutation.
Data availability
No new genomic sequencing data were generated in this study. The datasets used in this protocol included (i) the example data, which are available at the European Nucleotide Archive under the accession numbers ERR850376 and ERR852106; and (ii) the test demo data, which are available at https://zenodo.org/record/7191202/files/demo_data.tar.gz.
Code availability
cfSNV can be obtained at https://github.com/jasminezhoulab/cfSNV_docker. It can be freely used for educational and research purposes by nonprofit institutions and U.S. government agencies only under the UCLA Academic Software License. For information on use for a commercial purpose or by a commercial or for-profit entity, please contact Xianghong Jasmine Zhou (XJZhou@mednet.ucla.edu) and Wenyuan Li (WenyuanLi@mednet.ucla.edu).
Acknowledgements
This work was supported by National Cancer Institute grant nos. U01CA230705 and R01CA264864 to X.J.Z., R01CA246329 to X.J.Z. and W.L. and U01CA237711 to W.L.
Footnotes
Competing interests
X.J.Z. and W.L. are co-founders of EarlyDiagnostics Inc. C.-C.L. and T.-Y.K. are employees of EarlyDiagnostics Inc. S.L. is a former employee of EarlyDiagnostics Inc. The remaining authors declare no competing interests.
References
- 1.VanderLaan PA et al. Success and failure rates of tumor genotyping techniques in routine pathological samples with non-small-cell lung cancer. Lung Cancer 84, 39–44 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murtaza M et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat. Commun 6, 8760 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Phallen J et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med 9, eaan2415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Newman AM et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med 20, 548–554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ueda M et al. Somatic mutations in plasma cell-free DNA are diagnostic markers for esophageal squamous cell carcinoma recurrence. Oncotarget 7, 62280–62291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adalsteinsson VA et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat. Commun 8, 1324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Camus V et al. Digital PCR for quantification of recurrent and potentially actionable somatic mutations in circulating free DNA from patients with diffuse large B-cell lymphoma. Leuk. Lymphoma 57, 2171–2179 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Rothwell DG et al. Utility of ctDNA to support patient selection for early phase clinical trials: the TARGET study. Nat. Med 25, 738–743 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Li S et al. Sensitive detection of tumor mutations from blood and its application to immunotherapy prognosis. Nat. Commun 12, 1–14 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldberg SB et al. Early assessment of lung cancer immunotherapy response via circulating tumor DNA. Clin. Cancer Res 24, 1872–1880 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iwama E et al. Monitoring of somatic mutations in circulating cell-free DNA by digital PCR and next-generation sequencing during afatinib treatment in patients with lung adenocarcinoma positive for EGFR activating mutations. Ann. Oncol 28, 136–141 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Fontanilles M et al. Non-invasive detection of somatic mutations using next-generation sequencing in primary central nervous system lymphoma. Oncotarget 8, 48157–48168 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chaudhuri AA et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov 7, 1394–1403 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li S et al. cfTrack, a method of exome-wide mutation analysis of cell-free DNA to simultaneously monitor the full spectrum of cancer treatment outcomes including MRD, recurrence, and evolution. Clin. Cancer Res 28, 1841–1853 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choudhury AD et al. Tumor fraction in cell-free DNA as a biomarker in prostate cancer. JCI Insight 3, e122109 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li S et al. cfSNV: a software tool for the sensitive detection of somatic mutations from cell-free DNA. Jasminezhoulab/cfSNV_docker: cfSNV docker image. Available at https://github.com/jasminezhoulab/cfSNV_docker (2022). [Google Scholar]
- 17.Jiang P et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc. Natl Acad. Sci. USA 112, E1317–E1325 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang P et al. Preferred end coordinates and somatic variants as signatures of circulating tumor DNA associated with hepatocellular carcinoma. Proc. Natl Acad. Sci. USA 115, E10925–E10933 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abbosh C et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 545, 446–461 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cibulskis K et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol 31, 213–219 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van der Auwera GA et al. From FastQ data to high‐confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinforma 43, 11.10.1–11.10.33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DePristo MA et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet 43, 491–498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pellini B & Chaudhuri AA Circulating tumor DNA minimal residual disease detection of non–small-cell lung cancer treated with curative intent. J. Clin. Oncol 40, 567–575 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roth A et al. JointSNVMix: a probabilistic model for accurate detection of somatic mutations in normal/tumour paired next-generation sequencing data. Bioinformatics 28, 907–913 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim S et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591–594 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Kothen-Hill ST et al. Deep learning mutation prediction enables early stage lung cancer detection in liquid biopsy. Available at https://openreview.net/forum?id=H1DkN7ZCZ (2018).
- 27.Zviran A et al. Genome-wide cell-free DNA mutational integration enables ultra-sensitive cancer monitoring. Nat. Med 26, 1114–1124 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koboldt DC Best practices for variant calling in clinical sequencing. Genome Med 12, 1–13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Z et al. Systematic comparison of somatic variant calling performance among different sequencing depth and mutation frequency. Sci. Rep 10, 3501 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu C et al. A review of somatic single nucleotide variant calling algorithms for next-generation sequencing data. Comput. Struct. Biotechnol. J 16, 15–24 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H & Durbin R Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Broad Institute. Picard tools. Available at https://broadinstitute.github.io/picard/ (2019).
- 33.Li H et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mose LE et al. ABRA: improved coding indel detection via assembly-based realignment. Bioinformatics 30, 2813–2815 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Opasic L et al. How many samples are needed to infer truly clonal mutations from heterogenous tumours? BMC Cancer 19, 1–11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Butler TM et al. Exome sequencing of cell-free DNA from metastatic cancer patients identifies clinically actionable mutations distinct from primary disease. PloS One 10, e0136407 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kurtz DM et al. Enhanced detection of minimal residual disease by targeted sequencing of phased variants in circulating tumor DNA. Nat. Biotechnol 39, 1537–1547 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liebs S et al. Liquid biopsy assessment of synchronous malignancies: a case report and review of the literature. ESMO Open 4, e000528 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramesh N et al. Decoding the evolutionary response to prostate cancer therapy by plasma genome sequencing. Genome Biol 21, 1–22 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Magoč T & Salzberg SL FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Merkel D Docker: lightweight linux containers for consistent development and deployment. Linux J 2014, 2 (2014). [Google Scholar]
- 42.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arnold K, Gosling J & Holmes D The Java Programming Language (Addison Wesley Professional, 2005). [Google Scholar]
- 44.Van Rossum G & Drake FL Python 3 Reference Manual (CreateSpace, Scotts Valley, CA, 2009). [Google Scholar]
- 45.Harris CR et al. Array programming with NumPy. Nature 585, 357–362 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McKinney W Data structures for statistical computing in python. In Proceedings of the 9th Python in Science Conference, Vol. 445 (SCIPY, 2010). [Google Scholar]
- 47.Pedregosa F et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res 12, 2825–2830 (2011). [Google Scholar]
- 48.Virtanen P et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.R Core Team. R: a language and environment for statistical computing. Available at https://www.R-project.org/ (2018).
- 50.Eddelbuettel D & Romain F Rcpp: seamless R and C++ integration. J. Stat. Softw 40, 1–18 (2011). [Google Scholar]
- 51.Sherry ST, Ward M & Sirotkin K dbSNP—database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res 9, 677–679 (1999). [PubMed] [Google Scholar]
- 52.Lander ES et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new genomic sequencing data were generated in this study. The datasets used in this protocol included (i) the example data, which are available at the European Nucleotide Archive under the accession numbers ERR850376 and ERR852106; and (ii) the test demo data, which are available at https://zenodo.org/record/7191202/files/demo_data.tar.gz.