

Abstract
Introduction
Primary erythromelalgia (EM) is a rare clinical syndrome characterized by recurrent erythema, burning pain and warmth of the extremities. The symptoms greatly compromise the patients’ quality of life leading to severe disability. SCN9A mutations can be the cause of the disease. Dermatologists are often the specialists these patients turn to for assistance.
Objectives
To describe the demographic and clinical characteristics of patients with primary EM, to assess the presence and mutation types in the SCN9A gene, to evaluate the effectiveness of several therapeutic approaches, and to propose a diagnostic algorithm with therapeutic implications.
Methods
A monocentric retrospective study using the database of patients with a discharge diagnosis of primary EM of our Center. Demographic, clinical, instrumental and laboratory data of patients were reviewed.
Results
Eleven female patients (age range 16 to 57) were selected. All patients were affected in both the lower and upper extremities. Follow-up ranged from 2 to 9 years. Four patients had four different heterozygous variants of the SCN9A gene. Two patients, although genetically negative, had a suggestive family history. A variety of medications were tried in all our patients to alleviate symptoms, but their efficacy was variable, partial and/or transitory. The most effective therapies were antihistamines, venlafaxine, and mexiletine.
Conclusions
The diagnosis and treatment of EM remain challenging. Patients with this condition display a wide spectrum of clinical manifestations and severity, as well as a paucity of resources and structures to support them. Mutations in the SCN9A gene are not always detected.
Keywords: erythromelalgia, SCN9A protein, NAV1.7 Voltage-Gated Sodium Channel, mexiletine, treatments
Introduction
Erythromelalgia (EM) is a rare clinical syndrome characterized by the triad of recurrent erythema, burning pain and warmth of the extremities. The burning sensation is the most predominant symptom and is usually triggered and exacerbated by heat, ambulation, physical activity, emotional stress, fever, sitting, leg dependence, and covering of extremities [1]. The symptoms may occur intermittently or, in rare cases, continuously [2]. Rest, cold temperatures and elevation of the affected limbs alleviate symptoms. Indeed, pain is often relieved by putting affected parts in ice cold water, which results in immersion injury of the affected parts, uncovering their feet during sleep or walking barefoot in winter [1–3]. The lower limbs are more commonly affected than the upper limbs, usually with bilateral and symmetrical involvement [2,3]. Occasionally, the face, nose, ears, neck, and the genital area may be affected [2,4–6]. Despite not being always considered of dermatologic competence, EM patients are often referred to a dermatologist due to these symptoms.
EM may be primary or secondary. As a primary disease, EM can be inherited (familial) or non-inherited (sporadic) [3,4]. As a secondary disease, EM is usually associated with myeloproliferative disorders such as polycythemia vera and thrombocytopenia, however paraneoplastic, drug-induced, connective-tissue diseases, hypertension, poxviruses and vaccinations associated cases have also been described [2,7–19].
The primary form has an estimated annual incidence of 1.1 cases per 100,000, with a female predominance [2,5]. The discovery of a gain-of-function missense mutations in SCN9A, a gene encoding the voltage-gated sodium channel alpha subunit Na(v)1.7, in primary familial EM has provided insights into this disorder [20]. Yet, the pathogenesis remains complex, involving both neurological and vascular mechanisms, and resulting in a challenging patient management.
As a consequence of the disease, patients’ quality of life is greatly compromised, with patients avoiding school, working and limiting social activities. Disability is not uncommon as a result of intolerable pain, secondary tissue damage, or self-mutilation, leading in some cases to suicide [2,4]. As symptoms and signs of EM are subtle, especially in early stage, and/or mimicking other diseases, diagnosis is often delayed [21]. Thus, early recognition of the affection is crucial in order to establish an effective treatment. Further, despite the significant clinical impact, no clinical guidelines are currently available for EM management or treatment.
Objectives
The aims of this study were i) to describe the demographic and clinical characteristics of a series of patients with primary EM in our Center, ii) to assess the presence and mutation types in the SCN9A gene, iii) to evaluate the effectiveness of several therapeutic approaches, and iiii) to propose a diagnostic algorithm with therapeutic implications.
Methods
We designed a monocentric retrospective study using the database of patients with a discharge diagnosis of EM visited between January 2009 and August 2022 at the Dermatology Clinic in collaboration with the Center for Inherited Diseases of our third-level referral Center.
Clinical criteria for the diagnosis of EM included: occurrence of erythema, heat (subjectively and/or objectively observed), and associated discomfort (pain, burning, or tingling) in the extremities [22]. When no plausible underlying disease could be identified through anamnesis, examinations, or follow-up, EM was considered primary.
Demographic and clinical data of patients were reviewed. Data as to gender, age at onset of symptoms, age at diagnosis, family history, comorbidities, symptom description, triggers and relieving factors, complications, treatment and outcome were obtained for all cases. Laboratory investigations, including complete blood count, coagulation panel, autoimmunity panel, basic metabolic panel, hormone panel, and urinalysis, were reviewed as well. In all cases, laboratory studies such as neurophysiological, neuroimaging, and vascular studies were available. In 3 cases, skin biopsy specimens were available and reviewed by one of us (CT). In all patients, genetic consultation with SCN9A gene sequencing and post-test genetic counseling have been provided. Sequence variants were classified according to the American College of Medical Genetics and Genomics (ACMG) standards [23].
Results
A total of 17 patients met the clinical criteria for EM (14 females, 3 males). Fourteen (13 females, 1 male) were classified as primary EM. Three patients were excluded because they had less than 2 accesses in our hospital, resulting in 11 enrolled patients. All the included patients were females, and their average age at the time of the first visit was 36 years (range: 16 to 57). The clinical data are summarized in Table 1. Acral erythrosis, edema, and dysesthesias were experienced by all patients (Figure 1). In 5 cases, the condition developed during infancy, in 2 cases during adolescence and in 4 during adulthood. All patients were affected in both the lower and upper extremities. Five patients developed EM symptoms first in the lower distal limbs (#2, #3, #4, #10, #11), whereas 5 patients developed symptoms in both the upper and lower distal limbs simultaneously (#1, #5, #6, #7, #8). Only patient 9 presented with hand involvement before feet involvement. The EM episodes affected the face in 2 patients (#5, #8), the ears in 2 patients (#6, #8), the knees in 3 patients (#5, #6, #8). Patient 9 complained of a heat sensation throughout her body during EM attacks.
Table 1.
Clinical data of the patients.
| Patient | Age at lower extremities involvement (y) | Age at upper extremities involvement (y) | Age at visit (y) | Other involved sites | Persistent pain | Seasonality | Triggering factors | SCNA9 mutation | Erythromelalgia crisis frequency | Disease control |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 7 | 7 | 18 | yes | heat, humidity, emotional stress | c.2572C>T(e) (p.Leu858Phe) (pathogenetic); c.58C>T(e) (p.Leu20Phe) (VUS) | every day | not sufficient | ||
| 2 | 8 | 12 | 43 | yes, summer | sun exposure | c.3734A>G (p.Asn1245Ser) (benign) | once or twice weekly during summer months | good | ||
| 3 | 10 | 43 | 43 | heat, evening, alcohol | c.1999G<A (p.Asp678Asn) (VUS) | every day in the evening | good | |||
| 4 | 15 | 21 | 21 | yes | heat, humidity, physical activity | none | every day | good | ||
| 5 | 4 | 4 | 42 | knees, face | yes, summer | sun exposure | none | once or twice weekly in summer months | discrete | |
| 6 | 42 | 42 | 44 | knees, ears, diffuse heat over the entire body | no, but persistent erythrosis | heat, humidity, emotional stress | none | once or twice weekly | discrete | |
| 7 | 24 | 24 | 37 | no, but persistent erythrosis | yes, winter | heat | none | every day in winter months | poor | |
| 8 | 12 | 12 | 16 | face, ears, knees | emotional stress, evening | none | every day | poor | ||
| 9 | 39 | 37 | 39 | occasionally burning sensation in other body sites | yes, summer | heat, evening | none | once or twice weekly in summer months | discrete | |
| 10 | 7 | 14 | 57 | yes, summer | heat, sun exposure | none | once or twice weekly, every day in summer months | discrete | ||
| 11 | 37 | 39 | 39 | yes, with persistent edema | heat | c.4067G>A (p.Arg1356His) (benign) | every day | poor |
VUS = variant of uncertain significance.
Figure 1.

Erythromelalgia. Red, hot, and painful right (A) and left (B) leg in patient 1. The left leg presents ulceration due to repeated cold-water immersions. (C) Characteristic redness and edema in patient 8 with small bilateral ulcerations. (D) Extreme measures to improve their symptoms, such as going barefoot or cooling affected areas with cold water, are common in patients with erythromelalgia. (E) Persistent erythrosis and pain of the hands in patient 11.
Symptoms were persistent in 3 cases (#1, #4, and #11) and intermittent in 8 cases. The frequency of EM attacks in these patients varied from one episode a day to 1 or 2 episodes per week. In patients 6 and 7, erythrosis persisted between attacks.
A history of sweating disorders was present in 3 patients before the onset of EM: 2 patients had respectively palmoplantar and axillary hyperhidrosis (#1, #6), and 1 had hypohidrosis (#7). In one case (#1) the sweeting disorder presented in infancy, whilst in 2 cases (#6, #7) in early adulthood.
The triggering factors included heat exposure (8 patients), sun exposure (3 patients), humidity (three patients), emotional stress (3 patients), evening time (3 patients), physical activity (1 patient), and alcohol intake (1 patient). In 4 patients (#2, #5, #9, #10), EM crises were more frequent in summer, while 1 patient (#7) reported a winter seasonality. Seven patients reported that cold was a reliving factor (#1, #2, #3, #4, #5, #6, #8), with one of them (#1) walking barefoot in winter in the attempt to alleviate symptoms.
Three patients (#2, #8, #10) were misdiagnosed with solar urticaria, rosacea, venous insufficiency, and connective tissue disorders. One patient (#1) was diagnosed with restless legs syndrome and dermatitis factitia secondary to obsessive-compulsive disorder.
In 2 cases (#1, #8), foot ulcers, intertrigo, and maceration were present as a result of prolonged immersion in cold water or ice (Figure 1 ABC). In all cases there was no evidence of rheumatological disease. In two patients anti-nuclear antibodies (ANA) were positive (1:160). In all cases, extractable nuclear antigen (ENA) panel and other autoimmune serology tests were negative. Follow-up ranged from 2 to 9 years (median 5 years). In all patients, the coagulation parameters were within the normal range.
Neuroimaging and vascular studies were normal in all patients. Despite needle electromyography being normal in all patients, nerve conduction studies and quantitative sudomotor axon reflex revealed small fiber neuropathy in patients 1, 6 and 7.
Four patients with primary EM had 4 different heterozygous variants of the SCN9A gene (Nav1.7 channel). According the American College of Medical Genetics and Genomics (ACMG) criteria [23], Patient 1 was found to have a pathogenic variant, c.2572C>T(e) (p.Leu858Phe), and a variant of uncertain significance (VUS), c.58C>T(e) (p.Leu20Phe). Patient 2 had a benign variant, c.3734A>G (p.Asn1245Ser), patient 3 presented a VUS, c.1999G<A (p.Asp678Asn), and patient 11 presented a benign variant, c.4067G>A (p.Arg1356His).
Two patients (#4, #10), although genetically negative, had a family history of EM-like disorder, but their relatives were not examined. The father of patient 4 suffered from of paroxystic pain in the hands and feet and the patient’s grandmother was diagnosed with palmar-plantar erythrodysesthesia. Patient 10 described EM-like symptoms in her paternal aunt. It is likely that both these patients had familial EM harboring an unknown gene mutation.
The histopathology examination from skin biopsies performed in 3 patients (#1, #2, #6) showed substantially similar changes consisting of increased numbers of small vessels throughout the dermis, arranged in clusters in the superficial dermis (Figure 2A-B-C) and around the periadnexal and neurovascular plexuses in the mid and deep reticular dermis (Figure 2D). The vessel lumen appeared narrowed with wall thickening by PAS positive, hyaline material (Figure 2E). In one case, endothelial cell hyperplasia was prominent with nuclear atypia and mitoses suggesting a malignant vascular process (Figure 2F). No intraluminal thrombi or inflammatory infiltrate were found. The adnexal structures were preserved, and no quantitative or qualitative changes were observed in the small dermal nerves (Figure 2D). Extravasated erythrocytes were a common finding in all cases (Figure 2F).
Figure 2.
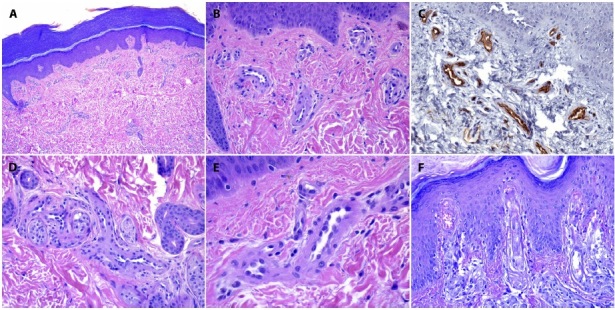
(A,B) The biopsy specimen of patient 1 shows an increased number of small vessels in the papillary and mid dermis, mainly clustered in small groups. The epidermis with a cornified layer is typical for the region (H&E ×20, H&E ×100). (C) CD34 positivity in endothelial cells (CD34, ×100). (D) Neurovascular plexus with increased numbers of capillaries with narrowed lumen and wall thickening. Note the close relation of small vessels with small nerve endings ((H&E ×40). (E) Groups of capillaries with narrowed lumen, endothelial swelling and wall thickening by hyaline material (H&E ×400). (F) Endothelial cell hyperplasia was prominent with nuclear atypia, mitoses and extravasated erythrocytes (H&E ×100).
A variety of medications were tried in all our patients in order to alleviate symptoms, but their efficacy was variable, partial and/or transitory (Table 2). Three patients (#2, #3, #4) reported a good reduction in pain intensity and episodes, while four patients (#5, #6, #9, #10) reported a sufficient improvement with the therapy. Four patients (#1, #7, #8, #11) had a poor response to the multiple treatments and one of them committed suicide as a result of the disability and depression caused by the disease (#1). As a result of the persistent edema and pain, patient 11 was unable to maintain an upright position and had severe limitations in her walking ability, even for short distances, needing a walker.
Table 2.
Medications used for erythromelalgia symptoms by the study patients and their reported efficacy.
| Patient | Desloratadine | Rupatadine | Magnesium supplement | Cardioaspirin | Amlodipine | Pregabalin | Venlafaxine | Duloxetin | Escitalopram | Propranolol | Metoprolol | Mexiletine | Clonazepam | Brotizolam | Opioids |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | |||||||||||||||
| 2 | |||||||||||||||
| 3 | |||||||||||||||
| 4 | |||||||||||||||
| 5 | At need | ||||||||||||||
| 6 | |||||||||||||||
| 7 | |||||||||||||||
| 8 | |||||||||||||||
| 9 | |||||||||||||||
| 10 | At need | ||||||||||||||
| 11 | At need |
| Not helpful | |
| Somewhat helpful | |
| Helpful |
The association of 2 second-generation non-sedating antihistamines, desloratadine 4 mg/day and rupatadine 10 mg/day, was the most frequently prescribed treatment (10 patients) and was associated with magnesium pidolate supplementation in 5 cases. Serotonin-norepinephrine reuptake inhibitors (SNRIs) such as venlafaxine 75 mg to 150 mg/day or duloxetine were prescribed in 6 cases. Five cases were treated with mexiletine a sodium channel blocker, at the dosage of 600–1200 mg/day, with good results in 3 of them. Other treatments included caardioaspirin, amlodipine, escitalopram, beta-blockers and pregabalin. An attempt was made to treat patient 1 with a spinal cord stimulator, but it was unsuccessful.
Conclusions
The dermatologist faces two challenges with EM patients: determining the correct path for a definite diagnosis and engaging other specialists in a multidisciplinary integrated approach. To assist clinicians, we propose here a diagnostic-therapeutic flowchart for EM (Figure 3).
Figure 3.

Diagnostic-therapeutic flow-chart proposal or erythromelalgia patients.
Anamnesis
The diagnosis of EM depends on clinical history and physical examination. When EM is suspected, a detailed review of patients’ medical history should be collected to provide information about possible factors that might lead to secondary EM. A positive or negative family history can further help to confirm familial/sporadic form. Due to the intermittent nature of this disorder, many Authors consider an EM diagnosis even if the typical findings are absent at the time of examination but the history of recurrent episodes of redness, heat and discomfort of the extremities is suggestive for the disease [2–4,24,25]. According to the patients, symptoms have been variably described as “hot”, “burning” or “numbness” feeling, or “tingling”, “prickling”, “throbbing” [2,24,26].
The onset age and the family history can assist in the determination of a primary or secondary form of EM, but this is not always conclusive [3,4,24]. Most cases of primary EM are not inherited [4]. Episode frequency, characteristics, and seasonality, as well as presence of triggering and relieving factors, should be investigated. Asymmetric or unilateral involvement should raise the suspicion of a secondary form [27,28]. An accurate drug anamnesis is essential, since erythromelalgia-like symptoms have been reported in association with a variety of drugs (iodine contrast, calcium channel blockers, bromocriptine, ticlopidine, vaccines) and even intoxicants [2,9,11,28–30].
Diagnostics
To rule out myeloproliferative disorders and paraneoplastic cases, the laboratory examination should include a complete blood count, peripheral blood smear, and serum tumor markers. Cancer screening exams, based on gender and age, should be performed. Inflammatory markers, serum protein electrophoresis, cryoglobulins, anti-nuclear antibody (ANA), extractable nuclear antigen (ENA) panel, autoantibody panel, complement levels should be assessed to rule out connective tissue diseases, including systemic lupus erythematosus, rheumatoid arthritis, and systemic sclerosis [2,4,12,31]. If a coexisting connective tissue disease or Reynaud’s phenomenon is suspected, nailfold capillaroscopy should also be performed. Other neuropathies, such as those caused by diabetes or other neurological diseases, should be excluded [32].
EM can be confused with complex regional pain syndrome (CRPS), which also has similar symptoms but is often unilateral, proximal and less commonly triggered by warming [33]. EM should also be distinguished from Fabry disease, a rare genetic disorder that presents with burning pain, acroparesthesias, angiokeratomas, proteinuria, hypohidrosis/anhidrosis, and cardiac symptoms. Fabry disease can be diagnosed by testing for a deficiency of alpha-galactosidase activity [34].
Instrumental investigations may provide diagnostic complementary information. Infrared thermography, laser Doppler flow and transcutaneous oxygenation measurements usually document an increase in skin temperature and blood flow and a paradoxical decrease in oxygenation in affected areas during EM attacks [35].
Nerve conduction studies, needle electromyography, and quantitative sudomotor axon reflex test (QSART, or sweat test) are useful and sensitive tools to demonstrate a small-fiber distal neuropathy, which is most prevalent in EM patients [2,22,35]. Quantitative sudomotor axon reflex test (QSART, or sweat test) may provide evidence of a sudomotor abnormality, which was clinically evident in three of our patients even before the onset of EM symptoms [35].
Skin biopsy is considered of little help in EM due to the lack of specific pathologic changes [24]. Davis et al suggested that the presence of thrombi within vessels would distinguish primary from secondary EM [24,36]. In our study, capillary proliferation, narrowing of the lumina, endothelial swelling and hyperplasia were common histopathologic findings, whilst neither luminal thrombi or reduction of small nerve fibers were observed. Intermittent skin hypoxia may well be considered a stimulus for capillary proliferation in the affected skin, as also previously suggested [37].
Once a primary EM diagnosis is established, the patient should be referred to the center where a SCN9A gene sequencing can be performed. Unfortunately, even in the presence of a family history, only approximately 10% of families have mutations in SCN9A [38]. In our case series, among the 11 patients, seven presented with a classic early onset primary EM, but only four had SCN9A mutations and only in one of them the mutation was pathogenetic. In Patients 4 and 10 the genetic SCN9A gene mutation analysis was negative, although the family history was highly suggestive for hereditary EM. This underlies the limitation of SCN9A sequencing, as the same mutation can result in phenotypic variability [39] and patients may also display protein-modifying mutations in other genes [39,40]. Consequently, if the sequencing result is negative for mutations, the primary EM diagnosis should not be questioned. On the other side, if any mutation is found, the Literature should be reviewed to determine if the genotype is associated with sensitivity to specific treatments.
Treatment
As a matter of fact, EM is resistant to many treatments and our findings confirm its refractory nature [1,2,4,15]. Various pharmacologic trials have been conducted with varied results, and no single medication or drug combination has been found to be universally helpful in relieving patients symptoms.
In our study, the most effective therapies were antihistamines, venlafaxine, and mexiletine. With regard to antihistamines, therapeutic efficacy in our EM patients might be attributed to their action against neurogenic inflammation [41]. Although these drugs are safe, their use in EM is off-label. Five patients were given magnesium pidolate in combination with antihistamines, which would act as a calcium antagonist, a sympatholytic and a muscle-relaxant [42].
In 3 patients, venlafaxine has provided some relief from symptoms of by acting both on sympathetic fibers by inhibiting the reuptake of noradrenaline, as well as on hormonal vascular control by inhibiting the reuptake of serotonin [43–45]. As this medication is approved to treat major depression, which is a common comorbidity among these patients, it is likely to prove useful for both diseases.
Mexiletine is a class IB orally active sodium channel blocker structurally similar to lidocaine, which appears to be particularly effective in neuropathic pain by shortening the action potential duration [46]. This drug was very effective in 3 patients and moderately effective in one, whilst in another was totally ineffective. Pharmacodynamic factors and genetic polymorphisms of the SCN9A gene, encoding the sodium channel Nav1.7, may for the variable efficacy of mexiletine in EM. Based on the available literature, a therapeutic attempt with mexiletine may be recommended for patients with V872G, L858F, and I848T mutant Nav1.7 channels, while the same treatment may not be appropriate for patients with I136V and V1316A mutations [47–49]. Furthermore, a clinically favorable response to intravenous lidocaine may predict a favorable response to oral mexiletine [50]. Patients should undergo periodic cardiological examinations, especially during drug titration, and be informed about common side effects involving the gastrointestinal and central nervous systems [51,52]. The use of mexiletine for EM is off-label.
A literature review may assist in providing other treatment options for EM. These include use of tricyclic antidepressants, gabapentin, anticonvulsants, and others. Carbamazepine, for instance, has been shown to be effective in EM patients harboring V400M and S241T mutations in the Nav1.7 sequence but ineffective in patients with F1449V mutations [53,54].
Finally, a correct and early diagnosis of EM is crucial as it may provide psychological and emotional relief, relieving the burden of a possible malingering or other psychiatric disease diagnosis. Chronic pain may lead to sleeping disorders, school difficulties and poor participation in physical activity in children. Unusual or peculiar behaviors that patients adopt to relieve their pain, such as repeated immersions in water or walking barefoot in the snow can be misinterpreted by the clinician as obsessive-compulsive behaviors or signs of another mental illness. In our case series, Patient 1 had an EM patient typical history: symptoms misinterpretation and misdiagnoses over a period of many years, poor response to many therapies, progressive worsening, social isolation and severe psychiatric complications culminating in suicide. Extreme self-harm and even suicide as in our patient are a rare but well-recognized consequence of EM, with high social costs and great impact on the patients and their families [2,4]. Information about online patient communities, which may offer users a valuable educational resource and a source of psychological and social support, should be offered (https://erythromelalgia.org/).
To conclude, EM involves several medical fields, and the path from diagnosis to treatment, involving all the necessary specialists, may be frustrating, both for patients and clinicians. In our experience, patients with this condition display a wide spectrum of clinical manifestations and severity, as well as a paucity of resources and structures to support them.
Footnotes
Funding: None.
Competing Interests: None.
Authorship: All authors have contributed significantly to this publication.
Statement of Informed Consent: Written informed consent was obtained from the patients for their anonymized information to be published in this article.
References
- 1.Tang Z, Chen Z, Tang B, Jiang H. Primary erythromelalgia: a review. Orphanet J Rare Dis. 2015;10:127. doi: 10.1186/s13023-015-0347-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davis MD, O’Fallon WM, Rogers RS, 3rd, Rooke TW. Natural history of erythromelalgia: presentation and outcome in 168 patients. Arch Dermatol. 2000;136(3):330–336. doi: 10.1001/archderm.136.3.330. [DOI] [PubMed] [Google Scholar]
- 3.Kalgaard OM, Seem E, Kvernebo K. Erythromelalgia: a clinical study of 87 cases. J Intern Med. 1997;242(3):191–197. doi: 10.1046/j.1365-2796.1997.00185.x. [DOI] [PubMed] [Google Scholar]
- 4.Cook-Norris RH, Tollefson MM, Cruz-Inigo AE, Sandroni P, Davis MD, Davis DM. Pediatric erythromelalgia: a retrospective review of 32 cases evaluated at Mayo Clinic over a 37-year period. J Am Acad Dermatol. 2012;66(3):416–423. doi: 10.1016/j.jaad.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 5.Mantyh WG, Dyck PJB, Dyck PJ, et al. Epidermal Nerve Fiber Quantification in Patients With Erythromelalgia. JAMA Dermatol. 2017;153(2):162–167. doi: 10.1001/jamadermatol.2016.4404. [DOI] [PubMed] [Google Scholar]
- 6.Prevost N, English JC., 3rd Case reports: red scrotal syndrome: a localized phenotypical expression of erythromelalgia. J Drugs Dermatol. 2007;6(9):935–936. [PubMed] [Google Scholar]
- 7.Mørk C, Kalgaard OM, Kvernebo K. Erythromelalgia as a paraneoplastic syndrome in a patient with abdominal cancer. Acta Derm Venereol. 1999;79(5):394. doi: 10.1080/000155599750010409. [DOI] [PubMed] [Google Scholar]
- 8.Han JH, Lee JB, Kim SJ, Lee SC, Won YH, Yun SJ. Paraneoplastic erythromelalgia associated with breast carcinoma. Int J Dermatol. 2012;51(7):878–880. doi: 10.1111/j.1365-4632.2010.04643.x. [DOI] [PubMed] [Google Scholar]
- 9.Eisler T, Hall RP, Kalavar KA, Calne DB. Erythromelalgia-like eruption in parkinsonian patients treated with bromocriptine. Neurology. 1981;31(10):1368–1370. doi: 10.1212/wnl.31.10.1368. [DOI] [PubMed] [Google Scholar]
- 10.Thami GP, Bhalla M. Erythromelalgia induced by possible calcium channel blockade by ciclosporin. BMJ. 2003;326(7395):910. doi: 10.1136/bmj.326.7395.910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michelerio A, Derlino F, Brazzelli V, Vassallo C. Secondary erythromelalgia: a tryptophan dietary supplement-induced case associated with elevated 5-hydroxyindoleacetic acid (5HIAA) urinary levels. Int J Dermatol. 2018;57(1):83–85. doi: 10.1111/ijd.13760. [DOI] [PubMed] [Google Scholar]
- 12.Parker LK, Ponte C, Howell KJ, Ong VH, Denton CP, Schreiber BE. Clinical features and management of erythromelalgia: long term follow-up of 46 cases. Clin Exp Rheumatol. 2017;35(1):80–84. [PubMed] [Google Scholar]
- 13.Grinnell M, Keyes E, Wat M, et al. Erythromelalgia associated with dermatomyositis: A case series. JAAD Case Rep. 2021;16:37–40. doi: 10.1016/j.jdcr.2021.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drenth JP, Michiels JJ, Ozsoylu S. Acute secondary erythermalgia and hypertension in children. Erythermalgia Multidisciplinary Study Group. Eur J Pediatr. 1995;154(11):882–885. doi: 10.1007/BF01957497. [DOI] [PubMed] [Google Scholar]
- 15.Mann N, King T, Murphy R. Review of primary and secondary erythromelalgia. Clin Exp Dermatol. 2019;44(5):477–482. doi: 10.1111/ced.13891. [DOI] [PubMed] [Google Scholar]
- 16.Zheng ZM, Zhang JH, Hu JM, Liu SF, Zhu WP. Poxviruses isolated from epidemic erythromelalgia in China. Lancet. 1988;1(8580):296. doi: 10.1016/s0140-6736(88)90372-8. [DOI] [PubMed] [Google Scholar]
- 17.Confino I, Passwell JH, Padeh S. Erythromelalgia following influenza vaccine in a child. Clin Exp Rheumatol. 1997;15(1):111–3. [PubMed] [Google Scholar]
- 18.Rabaud C, Barbaud A, Trechot P. First case of erythermalgia related to hepatitis B vaccination. J Rheumatol. 1999;26(1):233–234. [PubMed] [Google Scholar]
- 19.McMahon DE, Amerson E, Rosenbach M, et al. Cutaneous reactions reported after Moderna and Pfizer COVID-19 vaccination: A registry-based study of 414 cases. J Am Acad Dermatol. 2021;85(1):46–55. doi: 10.1016/j.jaad.2021.03.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang Y, Wang Y, Li S, et al. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet. 2004;41(3):171–417. doi: 10.1136/jmg.2003.012153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alhadad A, Wollmer P, Svensson A, Eriksson KF. Erythromelalgia: Incidence and clinical experience in a single centre in Sweden. Vasa. 2012;41(1):43–48. doi: 10.1024/0301-1526/a000162. [DOI] [PubMed] [Google Scholar]
- 22.Davis MD, Sandroni P, Rooke TW, Low PA. Erythromelalgia: vasculopathy, neuropathy, or both? A prospective study of vascular and neurophysiologic studies in erythromelalgia. Arch Dermatol. 2003;139(10):1337–1343. doi: 10.1001/archderm.139.10.1337. [DOI] [PubMed] [Google Scholar]
- 23.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis MD, Weenig RH, Genebriera J, Wendelschafer-Crabb G, Kennedy WR, Sandroni P. Histopathologic findings in primary erythromelalgia are nonspecific: special studies show a decrease in small nerve fiber density. J Am Acad Dermatol. 2006;55(3):519–522. doi: 10.1016/j.jaad.2006.04.067. [DOI] [PubMed] [Google Scholar]
- 25.Reed KB, Davis MD. Incidence of erythromelalgia: a population-based study in Olmsted County, Minnesota. J Eur Acad Dermatol Venereol. 2009;23(1):13–15. doi: 10.1111/j.1468-3083.2008.02938.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurzrock R, Cohen PR. Erythromelalgia: review of clinical characteristics and pathophysiology. Am J Med. 1991;91(4):416–422. doi: 10.1016/0002-9343(91)90160-y. [DOI] [PubMed] [Google Scholar]
- 27.Michiels JJ, Abels J, Steketee J, van Vliet HH, Vuzevski VD. Erythromelalgia caused by platelet-mediated arteriolar inflammation and thrombosis in thrombocythemia. Ann Intern Med. 1985;102(4):466–471. doi: 10.7326/0003-4819-102-4-466. [DOI] [PubMed] [Google Scholar]
- 28.Cohen JS. Erythromelalgia: new theories and new therapies. J Am Acad Dermatol. 2000;43(5 Pt 1):841–847. doi: 10.1067/mjd.2000.109301. [DOI] [PubMed] [Google Scholar]
- 29.Bibb LA, Winter RP, Leicht SS. Cyclosporine-induced Erythromelalgia. Cureus. 2018;10(10):e3506. doi: 10.7759/cureus.3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakajima N, Ueda M, Higashi N, Katayama Y. Erythromelalgia associated with Clitocybe acromelalga intoxication. Clin Toxicol (Phila) 2013;51(5):451–454. doi: 10.3109/15563650.2013.792933. [DOI] [PubMed] [Google Scholar]
- 31.Grinnell M, Keyes E, Wat M, et al. Erythromelalgia associated with dermatomyositis: A case series. JAAD Case Rep. 2021:37–40. doi: 10.1016/j.jdcr.2021.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reach P, Lazareth I, Coudore F, et al. A reappraisal of the presence of small or large fiber neuropathy in patients with erythromelalgia. Neurophysiol Clin. 2021;51(4):349–355. doi: 10.1016/j.neucli.2021.03.008. [DOI] [PubMed] [Google Scholar]
- 33.Shim H, Rose J, Halle S, Shekane P. Complex regional pain syndrome: a narrative review for the practising clinician. Br J Anaesth. 2019;123(2):e424–e433. doi: 10.1016/j.bja.2019.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30. doi: 10.1186/1750-1172-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandroni P, Davis MD, Harper CM, et al. Neurophysiologic and vascular studies in erythromelalgia: a retrospective analysis. J Clin Neuromuscul Dis. 1999;1(2):57–63. doi: 10.1097/00131402-199912000-00001. [DOI] [PubMed] [Google Scholar]
- 36.Michiels JJ, ten Kate FW, Vuzevski VD, Abels J. Histopathology of erythromelalgia in thrombocythaemia. Histopathology. 1984;8(4):669–678. doi: 10.1111/j.1365-2559.1984.tb02379.x. [DOI] [PubMed] [Google Scholar]
- 37.Kalgaard OM, Clausen OP, Mellbye OJ, Hovig T, Kvernebo K. Nonspecific capillary proliferation and vasculopathy indicate skin hypoxia in erythromelalgia. Arch Dermatol. 2011;147(3):309–314. doi: 10.1001/archdermatol.2010.337. [DOI] [PubMed] [Google Scholar]
- 38.Goldberg YP, Pimstone SN, Namdari R, et al. Human Mendelian pain disorders: a key to discovery and validation of novel analgesics. Clin Genet. 2012;82(4):367–373. doi: 10.1111/j.1399-0004.2012.01942.x. [DOI] [PubMed] [Google Scholar]
- 39.McDonnell A, Schulman B, Ali Z, et al. Inherited erythromelalgia due to mutations in SCN9A: natural history, clinical phenotype and somatosensory profile. Brain. 2016;139(Pt 4):1052–1065. doi: 10.1093/brain/aww007. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Z, Schmelz M, Segerdahl M, et al. Exonic mutations in SCN9A (NaV1.7) are found in a minority of patients with erythromelalgia. Scand J Pain. 2014;5(4):217–225. doi: 10.1016/j.sjpain.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 41.Rosa AC, Fantozzi R. The role of histamine in neurogenic inflammation. Br J Pharmacol. 2013;170(1):38–45. doi: 10.1111/bph.12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cohen JS. High-dose oral magnesium treatment of chronic, intractable erythromelalgia. Ann Pharmacother. 2002;36(2):255–260. doi: 10.1345/aph.1A186. [DOI] [PubMed] [Google Scholar]
- 43.Moiin A, Yashar SS, Sanchez JE, Yashar B. Treatment of erythromelalgia with a serotonin/noradrenaline reuptake inhibitor. Br J Dermatol. 2002;146(2):336–337. doi: 10.1046/j.1365-2133.2002.4653_6.x. [DOI] [PubMed] [Google Scholar]
- 44.Firmin D, Roguedas AM, Greco M, et al. Treatment of familial erythromelalgia with venlafaxine. J Eur Acad Dermatol Venereol. 2007;21(6):836–837. doi: 10.1111/j.1468-3083.2006.02039.x. [DOI] [PubMed] [Google Scholar]
- 45.DiCaudo DJ, Kelley LA. Alleviation of erythromelalgia with venlafaxine. Arch Dermatol. 2004;140(5):621–623. doi: 10.1001/archderm.140.5.621. [DOI] [PubMed] [Google Scholar]
- 46.Tremont-Lukats IW, Challapalli V, McNicol ED, Lau J, Carr DB. Systemic administration of local anesthetics to relieve neuropathic pain: a systematic review and meta-analysis. Anesth Analg. 2005;101(6):1738–1749. doi: 10.1213/01.ANE.0000186348.86792.38. [DOI] [PubMed] [Google Scholar]
- 47.Choi JS, Zhang L, Dib-Hajj SD, et al. Mexiletine-responsive erythromelalgia due to a new Na(v)1.7 mutation showing use-dependent current fall-off. Exp Neurol. 2009;216(2):383–389. doi: 10.1016/j.expneurol.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 48.Cregg R, Cox JJ, Bennett DL, Wood JN, Werdehausen R. Mexiletine as a treatment for primary erythromelalgia: normalization of biophysical properties of mutant L858F NaV 1.7 sodium channels. Br J Pharmacol. 2014;171(19):4455–4463. doi: 10.1111/bph.12788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu MT, Huang PY, Yen CT, Chen CC, Lee MJ. A novel SCN9A mutation responsible for primary erythromelalgia and is resistant to the treatment of sodium channel blockers. PLoS One. 2013;8(1):e55212. doi: 10.1371/journal.pone.0055212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jang HS, Jung D, Kim S, et al. A case of primary erythromelalgia improved by mexiletine. Br J Dermatol. 2004;151(3):708–710. doi: 10.1111/j.1365-2133.2004.06167.x. [DOI] [PubMed] [Google Scholar]
- 51.Johansson BW, Stavenow L, Hanson A. Long-term clinical experience with mexiletine. Am Heart J. 1984;107(5 Pt 2):1099–1102. doi: 10.1016/0002-8703(84)90181-9. [DOI] [PubMed] [Google Scholar]
- 52.Kerin NZ, Aragon E, Marinescu G, Faitel K, Frumin H, Rubenfire M. Mexiletine. Long-term efficacy and side effects in patients with chronic drug-resistant potentially lethal ventricular arrhythmias. Arch Intern Med. 1990;150(2):381–384. doi: 10.1001/archinte.150.2.381. [DOI] [PubMed] [Google Scholar]
- 53.Fischer TZ, Gilmore ES, Estacion M, Eastman E, Taylor S, Melanson M, Dib-Hajj SD, Waxman SG. A novel Nav1.7 mutation producing carbamazepine-responsive erythromelalgia. Ann Neurol. 2009;65(6):733–741. doi: 10.1002/ana.21678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang Y, Dib-Hajj SD, Zhang J, et al. Structural modelling and mutant cycle analysis predict pharmacoresponsiveness of a Na(V)1.7 mutant channel. Nat Commun. 2012;3:1186. doi: 10.1038/ncomms2184. [DOI] [PMC free article] [PubMed] [Google Scholar]