

Abstract
Purpose
Aniridia is a rare congenital eye disease, characterized by a constellation of symptoms including hypoplastic irides, foveal hypoplasia, early cataract, corneal stem cell deficiency, and glaucoma. Large chromosomal deletions spanning the PAX6 gene cause WAGR syndrome (Wilms tumor, aniridia, genitourinary anomalies, and intellectual disability [formerly called mental retardation]). We describe clinical and genetic studies of a three-generation pedigree with aniridia along with additional systemic conditions (morbid obesity, diabetes) suggesting the possibility of a contiguous-gene syndrome like WAGR.
Methods
Clinical records were obtained and DNA was prepared from blood samples from three of the four patients and tested for mutations in the coding sequences of the PAX6 gene. The index patient also had cardiomyopathy and was tested for known cardiomyopathy genetic mutations using a next-generation DNA sequencing assay.
Results
We discovered a novel intragenic PAX6 mutation, a 16 bp heterozygous deletion c.203delCCAGGGCAATCGGTGG, with Sanger sequencing that is the likely cause of autosomal dominant aniridia in this pedigree. This PAX6 deletion causes a frameshift in predicted protein translation and a subsequent premature termination, p.Pro68Leufs*6. The PAX6 deletion was detected in all three available family members with aniridia, the index patient, his mother, and his maternal aunt but was not observed in the Exome Aggregation Consortium (ExAC) database. Targeted sequencing of known cardiomyopathy genes in the index patient identified a second mutation, a 1.7 Mp deletion that spans the MYBPC3 gene.
Conclusions
We report a pedigree with aniridia and other systemic abnormalities that were initially suspicious for a contiguous-gene syndrome like WAGR. However, genetic analysis of the pedigree revealed two independent genetic abnormalities on chromosome 11p: 1) a novel PAX6 mutation, and 2) a large chromosome deletion spanning MYBPC3, a known cardiomyopathy gene. It is unclear if morbid obesity and type II diabetes mellitus have a related genetic cause.
Keywords: Aniridia, PAX6, glaucoma, obesity, diabetes
INTRODUCTION
Aniridia is a rare congenital eye disease with a prevalence of 1/64,000 to 1/96,000 that may have an autosomal dominant inheritance or may occur as an isolated case due to a de novo mutation.(1) Key features of aniridia are hypoplastic irides, foveal hypoplasia, cataract, and pannus secondary to corneal stem cell deficiency. Although the name suggests a complete absence of iris, patients with aniridia have some iris tissue that may range from a small, rudimentary stump of iris tissue to a normal-appearing iris.(2,3) The underdeveloped iris may prolapse against the trabecular meshwork and obstruct the outflow of aqueous humor from the eye leading to high intraocular pressure and secondary glaucoma. (4)
Mutations in the paired box 6 (PAX6 OMIM #607108) gene have been associated with aniridia.(5–7) PAX6 encodes a transcription factor that has a key role in promoting the development of neural and ocular tissues.(8) PAX6 has two functional domains, the homeobox domain (amino acids 211–260) that has DNA binding activity and the paired domain (amino acids 26–88) that may activate or repress local transcription.(5)
Missense, nonsense, and intragenic deletion PAX6 mutations cause familial aniridia that has an autosomal dominant inheritance.(5–7,9–11) Mutations in downstream regulatory sequences have also been shown to cause aniridia.(3,7,12–13) Large chromosome 11p13 deletions that span PAX6 and neighboring genes cause a contiguous-gene syndrome with the acronym WAGR (OMIM #194072), which comes from key features of the syndrome: Wilms tumor, Aniridia, Genito-urinary abnormalities, and intellectual disability (previously described as mental Retardation).(14) Cases of WAGR are associated with reduced fertility and are sporadic.(15)
In this report, we describe clinical and genetic studies of a three-generation pedigree with aniridia as well as several additional systemic conditions (morbid obesity, diabetes mellitus, and hypertrophic cardiomyopathy) suggesting the possibility of a chromosome 11 contiguous-gene syndrome like WAGR. Instead, we discovered a novel intragenic PAX6 mutation that appears to be an isolated cause of aniridia in this pedigree.
METHODS
Clinical analysis
Research protocols adhered to guidelines of the Tenets of Helsinki. Patients provided informed consent and research was conducted with the approval of the University of Iowa’s Institution Review Board (IRB) for human research. Patients had a complete ophthalmological examination including tonometry, gonioscopy, slit-lamp examination, dilated ophthalmoscopy, and Goldmann visual field testing.
Genetic analysis
DNA was prepared from blood samples from Patient II-4, II-6, and Patient III-1 and tested for mutations in the coding sequences of the PAX6 gene (RefSeqGene NG_008679.1) with Sanger sequencing as we have previously described.(16) DNA samples were unavailable from patient I-2 who is deceased and patient III-2 who could not be reached for this study. Briefly, each of the exons of the PAX6 gene was PCR amplified using standard reactions. Amplified PCR product was either directly sequenced on an ABI 3730 automated sequencer with BigDye Direct Sequencing kits (ThermoFisher, Waltham, MA) or was cloned into the vector pCR4-TOPO (ThermoFisher) using the manufacturer’s protocol prior to sequencing.
Genetic analyses were conducted to investigate the cause of the proband’s cardiomyopathy including a cytogenetics analysis at the University of Iowa and two commercial assays for mutations: a next-generation DNA sequencing assay to test for coding sequence defects, deletions, and duplications in a panel of 50 known cardiomyopathy genes: ABCC9, ACTC1, ACTN2, AGL, BAG3, CACNA1C, CAV3, CRYAB, CSRP3, DES, DMD, DOLK, DSC2, DSG2, DSP, EMD, EYA4, FHL1, FKRP, FKTN, FLNC, GAA, GLA, HCN4, JUP, LAMP2, LMNA, MYBPC3, MYH7, MYL2, MYL3, PKP2, PLN, PRKAG2, RAF1, RBM20, RYR2, SCN5A, SGCD, SLC22A5, TAZ, TCAP, TMEM43, TNNC1, TNNI3, TNNT2, TPM1, TTN, TTR, VCL (Invitae, San Francisco, CA) and Whole-Genome Oligonucleotide Array Comparative Genome Hybridization (CGH) assay to precisely map deletions and insertions (GeneDx, Gaithersburg, MD). The GRCh37 human genome build was used for the CGH analysis.
Computer modeling of PAX6 mutations
We investigated the likely effects of the p.Pro68Leufs*6 mutation on the protein structure of PAX6 based on a wild-type structure determined by X-ray crystallography.(17) The analysis began by replacing 6 amino acids p.68–73 with the residues LeuValAspArgGlu and truncating the protein after these residues, followed by repacking of nearby residues using a rotamer optimization algorithm(18) and a potential energy function defined by the polarizable atomic multipole AMOEBA force field (19) in the program Force Field X.(20)
RESULTS
Clinical analysis
Ophthalmological features of aniridia
We conducted a complete ophthalmological examination of four members of a three-generation pedigree that have aniridia in the Department of Ophthalmology and Visual Sciences at the University of Iowa (Figure 1). All affected members seen at the University (I-2, II-4, II-6, and III-1) had classic signs of aniridia, including corneal pannus, under-developed iris, nystagmus, foveal hypoplasia, and secondary glaucoma (Figure 2). Visual acuity at their last-recorded visits ranged from 20/60 to count fingers (). In three of the four members (I-2, II-4, II-6), the corneal stem cell deficiency and pannus were severe enough to warrant corneal transplantation (penetrating keratoplasty in I-2, Boston Keratoprosthesis in II-4 and II-6). All four members had secondary glaucoma and had either a trabeculectomy (I-2), or more commonly a glaucoma drainage device (Ahmed or Baerveldt). All patients had early onset posterior capsular cataracts, requiring surgeries at young ages ranging from 30 years (III-1) to 50 years (II-6). Two of the members of our pedigree (I-2, and II-6) deferred surgical treatment of one of their eyes after II-6 experienced a complicated course and episode of treated endophthalmitis in her operated eye ().
Figure 1.
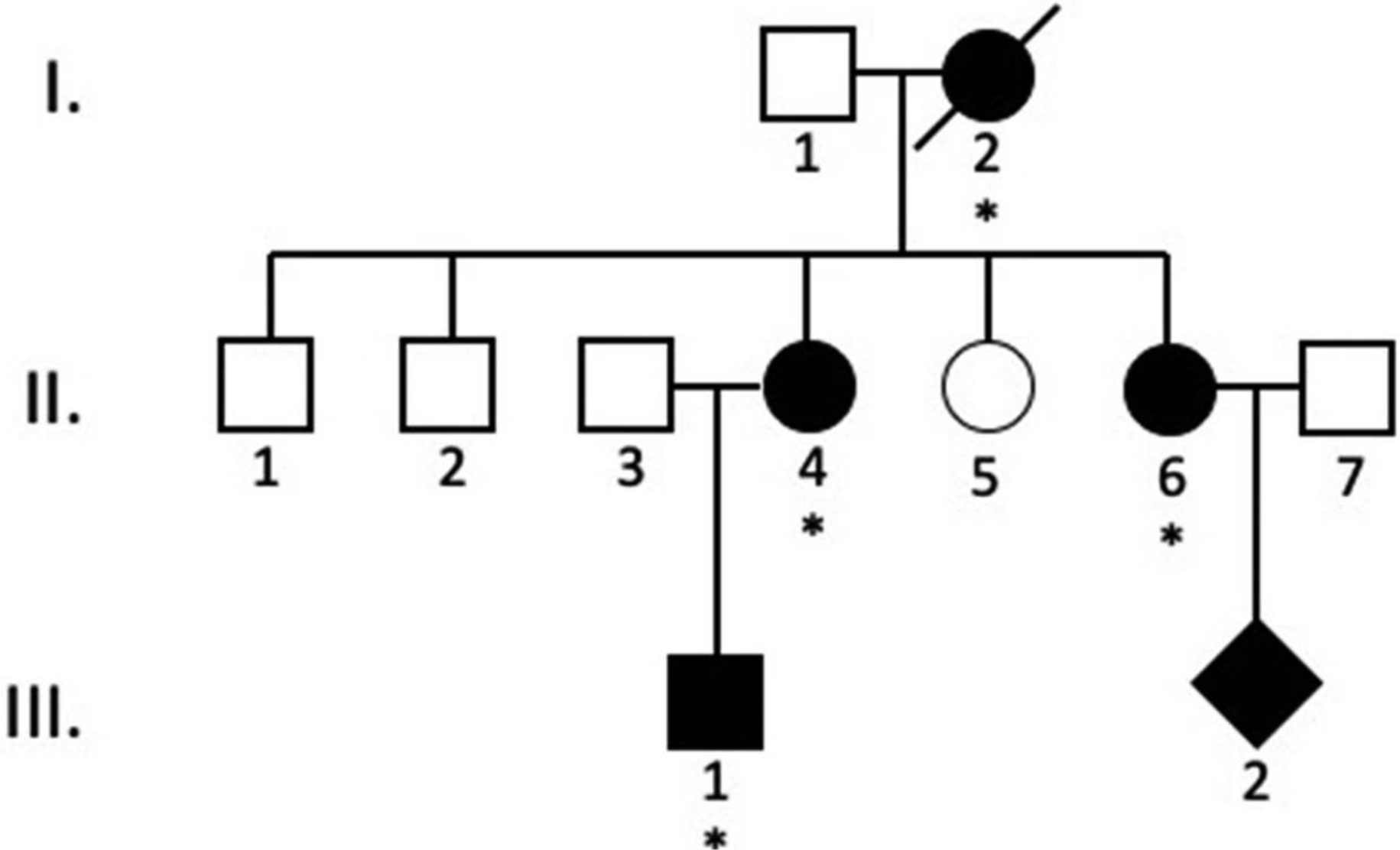
A three-generation aniridia pedigree.
Patients that were either diagnosed with aniridia by one of the authors or that were affected by a report from other family members are indicated by black symbols. Square symbols indicate males, round symbols indicate females, and diamond symbols indicate a family member with the gender that was not reported. The family members that were available for clinical examination are indicated with an asterisk. A slash marking indicates a family member is deceased.
Figure 2.
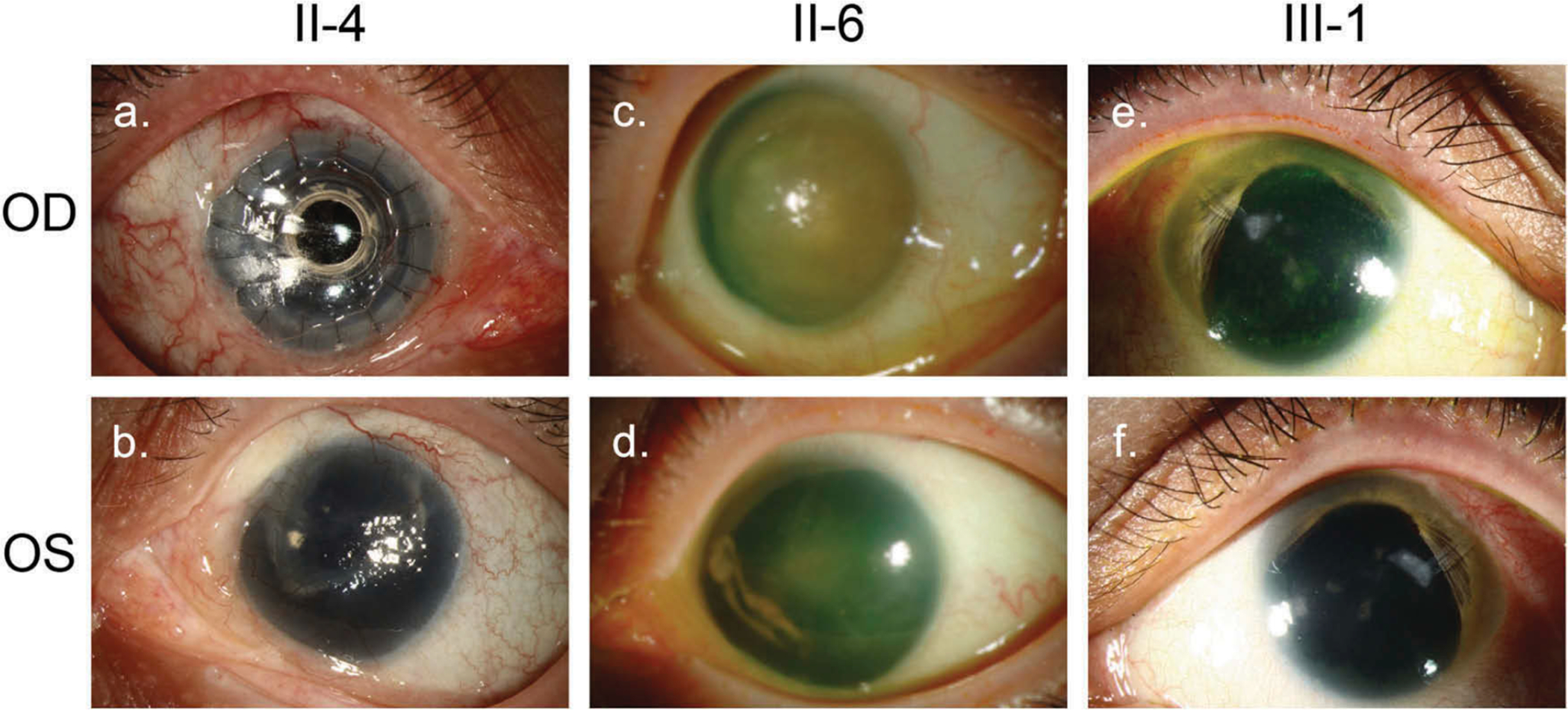
Clinical features of aniridia in the pedigree.
Clinical photos of three members with aniridia, II-4 (A. OD, B. OS), II-6 (B. OD, C. OS), and III-1 (E. OD, F. OS). All have varying degrees of corneal stem cell deficiency and pannus, including one who has undergone a Boston KPro keratoprosthesis (II-4, Frame A). Patients II-4 and II-6 have no clinically visible iris (Frames A, B, C, D). Patient III-1 has symmetric supratemporal crescents of iris remnants (Frames E, F). Tube-shunts are visible in both eyes of patient III-1 (Frames E, F).
Systemic features of disease
Mother (Figure 1, II-4), sister (II-6) and son (III-1) of our pedigree have morbid obesity, while mother (II-4) and son (III-1) had diabetes mellitus in addition to aniridia (Table 1).
Table 1.
Clinical details of four related patients with aniridia.
| Ocular findings | Ocular procedures | Systemic findings | |
|---|---|---|---|
|
| |||
| I-2 | - Aniridia | Right eye | - Hypertension |
| - Foveal hypoplasia | - Kpro/phaco (~ 40) | (No outside records) | |
| - PSC | - Trab x 2 (75) | ||
| - Esotropia | Left eye | ||
| - Myopia | - None | ||
| Visual acuity: CF 2’, CF 4’ (76) | |||
| II-4 | - Aniridia | Right eye | - Hypertension |
| - Foveal hypoplasia | - Phaco (44) | - Morbid obesity | |
| - Optic nerve hypoplasia | - Ahmed/Kpro (56) | (BMI 62.79, 166 kg) | |
| - Pannus | Left eye | - Type 2 diabetes | |
| - PSC | - Phaco/trab (37) | - Hypothyroidism | |
| - Esotropia | - Baerveldt (40) | ||
| - Nystagmus | |||
| - Myopia | |||
| - Amblyopia, left eye | |||
| Visual acuity: 20/60, 20/800 (63) | |||
| II-6 | - Aniridia | Right eye | - Hypertension |
| - Foveal hypoplasia | - Phaco/Kpro/Ahmed (50) | - Morbid obesity | |
| - Pannus | - Kpro revision (50) | (BMI 38.51, 97 kg) | |
| - PSC | - Kpro replacement (53) | ||
| - Myopia | Left eye | ||
| - Esotropia | - None - deferred | ||
| - Fungal endophthalmitis, right eye Visual acuity: 20/70–1, HM (58) | |||
| III-1 | - Aniridia | Right eye | - Morbid obesity |
| - Foveal hypoplasia | - Ahmed (28) | (BMI 59.2, 169 kg) | |
| - Pannus | - Phaco/Ahmed (30) | - Hypertrophic cardiomyopathy | |
| - PSC cataract | Left eye | - Type 2 diabetes | |
| - Myopia | - Ahmed (28) | ||
| - Esotropia | - Phaco (31) | ||
| Visual acuity: 20/250, 20/250 (33) | |||
Numbers within parentheses are age at the time of procedure or examination. Ahmed = Ahmed glaucoma drainage implant; Baerveldt = Baerveldt glaucoma drainage implant; BMI = body mass index; CF = count fingers; HM = hand motion; kg = kilogram; Kpro = Boston keratoprosthesis; Phaco = phacoemulsification; PSC = posterior subcapsular cataract; Trab = trabeculectomy.
Patient III-1 was also evaluated in the Pediatric Genetics specialty clinic at 18 years of age. He was diagnosed with hypertrophic cardiomyopathy (with preserved ejection fraction) after an evaluation for shortness of breath and edema of the lower extremities. At the time, he had a computed body mass index (BMI) of 44.1 and weighed 127 Kg. He later developed type II diabetes mellitus at age 25 and has since been noted to have an affective schizoid personality disorder. By age 32, he weighed 182 Kg and his BMI was 59.2.
Patient II-4, the mother of III-1, is also morbidly obese. She had a BMI of 62.8 and weight of 166 Kg at her last follow-up examination (12/2018) and she also has type II diabetes mellitus. Patient II-4, however, has not been diagnosed with hypertrophic cardiomyopathy. Patient II-6 is also obese with a BMI of 38.51, but no record of hypertrophic cardiomyopathy or diabetes.
Patient I-2, now deceased, was known to have hypertension (Table 1) from the review of the family medical history of the other members of the pedigree. However, a complete systemic history was not available.
Genetic analysis
DNA samples were collected from three members of the aniridia pedigree, Patient II-4, II-6, and III-1 to facilitate the genetic study of their disease.
Genetic analysis – cytogenetic studies
Given Patient III-1’s health conditions (aniridia, morbid obesity, diabetes mellitus, and hypertrophic cardiomyopathy), he was evaluated for chromosomal abnormalities with a cytogenetics study. He had a normal karyotype (46, XY) and no large deletions, duplications, or rearrangements were detectable by the cytogenetic studies.
Genetic analysis – systemic disease
Patient III-1 was evaluated by a cardiology/genetics specialist to search for a possible genetic cause of his hypertrophic cardiomyopathy. A commercial test for mutations in a panel of 50 known cardiomyopathy genes (Invitae, San Francisco, CA) identified a heterozygous deletion on chromosome 11p11.2 which spanned the entirety of the myosin-binding protein C, cardiac (MYBPC3. OMIM #600958) gene, known to be associated with hypertrophic cardiomyopathy.(21–22) The specific breakpoints of this deletion were subsequently shown to encompass a 1.7 Mbp segment of chromosome 11 (46,842,700–48,588,943 bp) using a comparative genome hybridization (CGH) assay (GeneDx, Gaithersburg, MD). In addition to spanning the MYBPC3 gene, this heterozygous deletion also encompasses an additional 31 genes including 6 genes (LRP4 OMIM #604270; DDB2 OMIM #600811; ACP2 OMIM #171650; SLC39A13 OMIM #608735; RAPSN OMIM #601592; and NDUFS3 OMIM #603846) that have been previously linked with a range of autosomal recessive human diseases (OMIM 614305, 616304, 212780, 178740, 200950, 612350, 208150, 616326, 256000, and 252010) that are not consistent with the patient’s phenotype. Patient II-4 was also tested with the CGH assay and she does not carry the chromosome 11p11.2 deletion.
Genetic analysis – ophthalmic disease
Three members of our aniridia pedigree (Figure 1, II-4, II-6, and III-1) were tested for coding sequence mutations in the PAX6 with Sanger DNA sequencing. A novel 16 bp heterozygous deletion c.203delCCAGGGCAATCGGTGG was detected within exon 6 of the PAX6 gene of both Patient II-4, her sister Patient II-6, and her son Patient III-1. The 16 base pair deletion generates a frameshift and results in a truncated PAX6 protein p.Pro68Leufs*6 that has a profound effect on its structure (Figure 3). This mutation disrupts one of the two key functional domains in the PAX6 protein, a paired box domain, and eliminates the other, a homeobox domain. The p.Pro68Leufs*6 mutation was not detected in the PAX6 DNA sequence of >50,000 control subjects on a public database (exac.broadinstitute.org).
Figure 3.
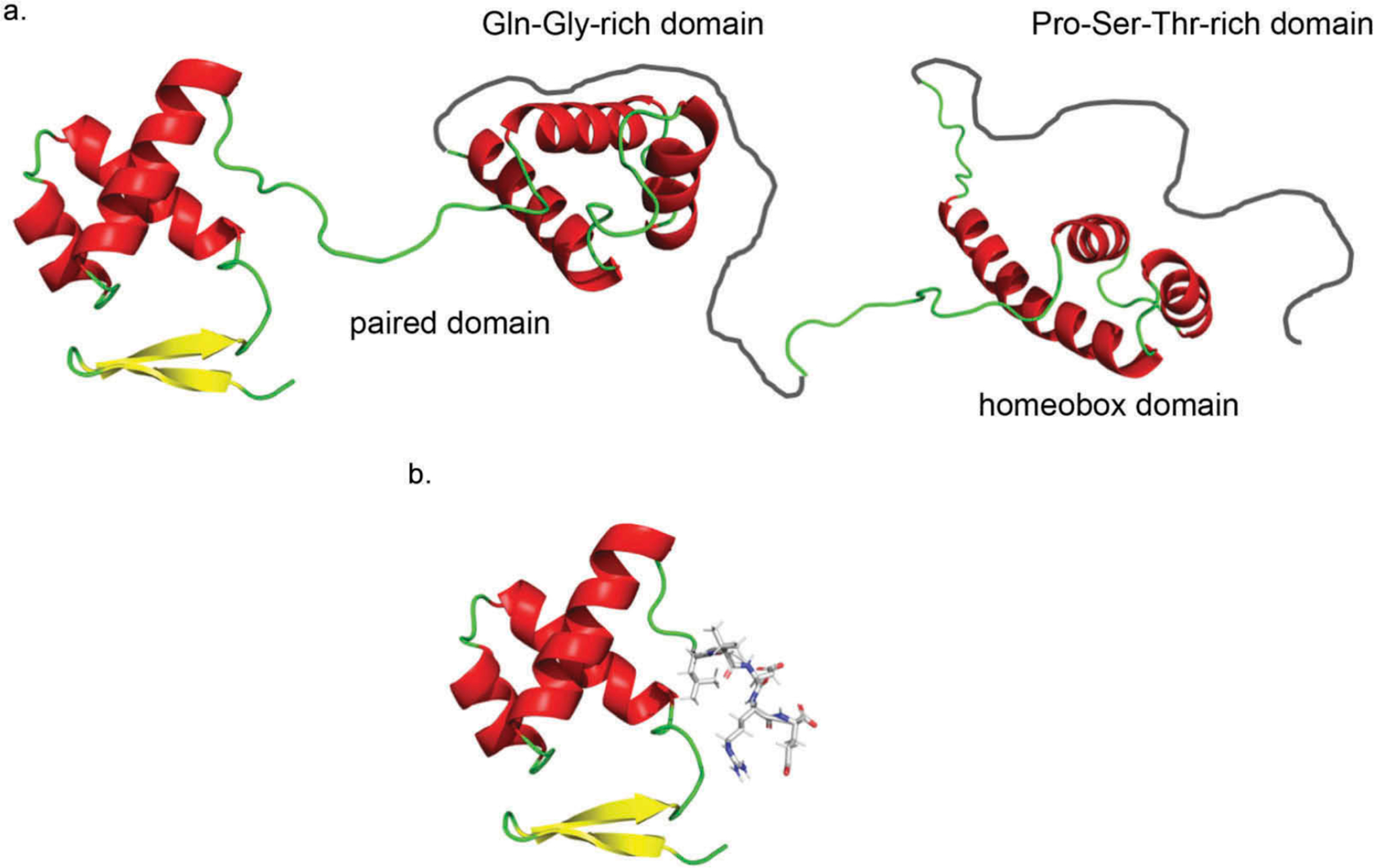
Model of PAX6 protein structure.
A. Wild-type PAX6 protein structure includes a paired box domain sequence (amino adds 4–130), a Gln-Gly-rich domain (amino acids 131–209), a homeobox domain (amino acids 210–269), and a Pro-Ser-Thr-rich domain (amino acids 279–422). B. The Pro68Leufs*6 mutation generates a truncated PAX6 protein with disrupted and missing domains. The Pro68Leufs*6 mutation causes a premature termination within the paired domain produces a truncated protein with an abnormal terminal five amino acids (LeuValAspArgGlu shown with a stick representation) that disrupts the paired domain and eliminates the homeobox domain. These structures were generated using a rotamer optimization algorithm18 and a potential energy function19 in the program Force Field X20 using the known crystal structure of PAX6.
DISCUSSION
The index patient of our pedigree (Patient III-1) has a complex set of systemic medical conditions including aniridia, morbid obesity, diabetes mellitus, and cardiomyopathy, which suggested to us that he might have a contiguous-gene syndrome involving a deletion or rearrangement of genes on chromosome 11p that includes PAX6 and neighboring genes to explain some or all of these features. There is evidence to support our hypothesis for a contiguous-gene syndrome in this patient. Several of his clinical features have been previously associated with genes in the region. While some reports suggest that PAX6 mutations may cause glucose intolerance and promote diabetes,(23) there is also evidence that defects in another gene in the region may stimulate the development of diabetes mellitus.(24) Moreover, there are cardiomyopathy genes on chromosome 11.(21,22) Coupled with his aniridia, these observations suggested to us that Patient III-1 might have a novel contiguous-gene syndrome involving PAX6.
The simplest answer, however, is not always the correct answer. In a potential violation of Occam’s razor, at least two separate chromosome 11 abnormalities are responsible for Patient III-1’s clinical features. A novel PAX6 frameshift mutation, p.Pro68Leufs*6, is the likely cause of his aniridia and may also contribute somewhat to his development of diabetes mellitus.(23) His mother (Patient II-4) and aunt (Patient II-6) also have aniridia and this same PAX6 mutation. The detrimental effects of this truncating mutation on the encoded protein, the co-inheritance of this mutation with aniridia in the pedigree, and the absence of this mutation in a large control population strongly suggests the p.Pro68Leufs*6 mutation causes aniridia in the pedigree. Patient III-1 has a second deletion on chromosome 11p11.2 (15 Mbp centromeric to PAX6) that may cause haploinsufficiency of MYBPC3 and is likely responsible for his hypertrophic cardiomyopathy. The causes of his other medical problems are unclear. Patient III-1 may have morbid obesity and diabetes mellitus that are somehow due to his PAX6 mutation, due to the many genes on chromosome 11p11.2 that were deleted along with MYBPC3, or due to one or more other genetic defects that we were unable to discern.
In summary, we report the members of a pedigree that have aniridia and multiple other systemic abnormalities that initially were suspicious for a contiguous-gene syndrome like WAGR. However, genetic analysis of the pedigree revealed two independent genetic abnormalities on chromosome 11p: (1) a novel, intragenic PAX6 mutation, p.Pro68Leufs*6, that is most likely responsible for the autosomal dominant aniridia and (2) a large chromosome 11p11.2 deletion that spans a cardiomyopathy gene, MYBPC3, in Patient III-1 that most likely causes his hypertrophic cardiomyopathy. The precise causes of the other clinical features in this family are unclear.
Acknowledgments
This research was support in part by unrestricted grants from the Research to Prevent Blindness and The Hadley-Carver Chair in Glaucoma.
Footnotes
Declaration of interest
The authors have no conflicts of interest.
REFERENCES
- 1.Shaw MW, Falls HF, Neel JV. Congenital aniridia. Am J Hum Genet. 1960;12:389–415. [PMC free article] [PubMed] [Google Scholar]
- 2.Brauner SC, Walton DS, Chen TC. Aniridia. Int Ophthalmol Clin. 2008;48(2):79–85. [DOI] [PubMed] [Google Scholar]
- 3.Lewis C, Hedberg-Buenz A, AP D, EM S, WLM A, JH F. Primary congenital and developmental glaucomas. Hum Mol Genet. 2017;26(R1):R28–R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grant WM, Walton DS. Progressive changes in the angle in congenital aniridia, with development of glaucoma. Trans Am Ophthalmol Soc. 1974;72:207–28. [PMC free article] [PubMed] [Google Scholar]
- 5.Ton CC, Hirvonen H, Miwa H, Weil MM, Monaghan P, Jordan T, Van Heyningen V, Hastie ND, Meijers-Heijboer H, Drechsler M. Positional cloning and characterization of a paired box- and homeobox-containing gene from the aniridia region. Cell. 1991;67(6):1059–74. [DOI] [PubMed] [Google Scholar]
- 6.Jordan T, Hanson I, Zaletayev D, Hodgson S, Prosser J, Seawright A, Hastie N, Van Heyningen V. The human PAX6 gene is mutated in two patients with aniridia. Nat Genet. 1992;1(5):328–32. [DOI] [PubMed] [Google Scholar]
- 7.Glaser T, Walton DS, Maas RL. Genomic structure, evolutionary conservation and aniridia mutations in the human PAX6 gene. Nat Genet. 1992;2(3):232–39. [DOI] [PubMed] [Google Scholar]
- 8.Walther C, Gruss P. Pax-6, a murine paired box gene, is expressed in the developing CNS. Development. 1991;113:1435–49. [DOI] [PubMed] [Google Scholar]
- 9.Hanson IM, Seawright A, Hardman K, Hodgson S, Zaletayev D, Fekete G, Van Heyningen V. PAX6 mutations in aniridia. Hum Mol Genet. 1993;2(7):915–20. [DOI] [PubMed] [Google Scholar]
- 10.Davis A, Cowell JK. Mutations in the PAX6 gene in patients with hereditary aniridia. Hum Mol Genet. 1993;2(12):2093–97. [DOI] [PubMed] [Google Scholar]
- 11.Martha A, Ferrell RE, Mintz-Hittner H, Lyons LA, Saunders GF. Paired box mutations in familial and sporadic aniridia predicts truncated aniridia proteins. Am J Hum Genet. 1994;54:801–11. [PMC free article] [PubMed] [Google Scholar]
- 12.Bhatia S, Bengani H, Fish M, Brown A, Divizia MT, de Marco R, Damante G, Grainger R, Van Heyningen V, Kleinjan DA. Disruption of autoregulatory feedback by a mutation in a remote, ultraconserved PAX6 enhancer causes aniridia. Am J Hum Genet. 2013;93(6):1126–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kleinjan DA, Seawright A, Mella S, Carr CB, Tyas DA, Simpson TI, Mason JO, Price DJ, Van Heyningen V. Long-range downstream enhancers are essential for Pax6 expression. Dev Biol. 2006;299(2):563–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.RW M, Fraumeni JF, Manning MD. Association of Wilms’s tumor with aniridia, hemihypertrophy and other congenital malformations. N Engl J Med. 1964;270(18):922–27. [DOI] [PubMed] [Google Scholar]
- 15.Fischbach BV, Trout KL, Lewis J, Luis CA, Sika M. WAGR syndrome: a clinical review of 54 cases. Pediatrics. 2005;116(4):984–88. [DOI] [PubMed] [Google Scholar]
- 16.Scheetz TE, Roos BR, Solivan-Timpe F, Miller K, DeLuca AP, Stone EM, Kwon YH, Alward WLM, Wang K, Fingert JH. SQSTM1 mutations and glaucoma. PLoS ONE. 2016;11(6):e0156001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu HE, Rould MA, Xu W, Epstein JA, Maas RL, Pabo CO. Crystal structure of the human Pax6 paired domain-DNA complex reveals specific roles for the linker region and carboxy-terminal subdomain in DNA binding. Genes Dev. 1999;13(10):1263–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.LuCore SD, Litman JM, Powers KT, Gao S, Lynn AM, Tollefson WTA, Fenn TD, Washington MT, Schnieders MJ. Dead-end elimination with a polarizable force field repacks PCNA structures. Biophys J. 2015;109(4):816–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ponder JW, Wu C, Ren P, Pande VS, Chodera JD, Schnieders MJ, Haque I, Mobley DL, Lambrecht DS, DiStasio RA, et al. Current status of the AMOEBA polarizable force field. J Phys Chem B. 2010;114(8):2549–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fenn TD, Schnieders MJ. Polarizable atomic multipole X-ray refinement: weighting schemes for macromolecular diffraction. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 11):957–65. [DOI] [PubMed] [Google Scholar]
- 21.Watkins H, Conner D, Thierfelder L, Jarcho JA, MacRae C, McKenna WJ, Maron BJ, Seidman JG, Seidman CE. Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat Genet. 1995;11(4):434–37. [DOI] [PubMed] [Google Scholar]
- 22.Bonne G, Carrier L, Bercovici J, Cruaud C, Richard P, Hainque B, Gautel M, Labeit S, James M, Beckmann J, et al. Cardiac myosin binding protein-C gene splice acceptor site mutation is associated with familial hypertrophic cardiomyopathy. Nat Genet. 1995;11(4):438–40. [DOI] [PubMed] [Google Scholar]
- 23.Yasuda T, Kajimoto Y, Fujitani Y, Watada H, Yamamoto S, Watarai T, Umayahara Y, Matsuhisa M, Gorogawa S-I, Kuwayama Y, et al. PAX6 mutation as a genetic factor common to aniridia and glucose intolerance. Diabetes. 2002;51(1):224–30. [DOI] [PubMed] [Google Scholar]
- 24.Macdonald GC, Hesselson SE, Chan JY, Jenkins AB, Laybutt DR, Hesselson D, Campbell LV. Deletion distal to the PAX6 coding region reveals a novel basis for familial cosegregation of aniridia and diabetes mellitus. Diabetes Res Clin Pract. 2018;148:64–71. [DOI] [PubMed] [Google Scholar]