

Abstract
Background and Objectives
Epstein-Barr virus (EBV) is a ubiquitous herpesvirus that establishes lifelong latency in memory B cells and has been identified as a major risk factor of multiple sclerosis (MS). B cell depletion therapies have disease-modifying benefit in MS. However, it is unclear whether this benefit is partly attributable to the elimination of EBV+ B cells. Currently, there are no EBV-specific antiviral therapies available for targeting EBV latent infection in MS and limited experimental models to study EBV in MS.
Methods
In this study, we describe the establishment of spontaneous lymphoblastoid cell lines (SLCLs) generated ex vivo with the endogenous EBV of patients with MS and controls and treated with either an Epstein-Barr virus nuclear antigen 1 (EBNA1) inhibitor (VK-1727) or cladribine, a nucleoside analog that eliminates B cells.
Results
We showed that a small molecule inhibitor of EBNA1, a critical regulator of the EBV life cycle, blocks the proliferation and metabolic activity of these SLCLs. In contrast to cladribine, a highly cytotoxic B cell depleting therapy currently used in MS, the EBNA1 inhibitor VK-1727 was cytostatic rather than cytotoxic and selective for EBV+ cells, while having no discernible effects on EBV− cells. We validate that VK-1727 reduces EBNA1 DNA binding at known viral and cellular sites by ChIP-qPCR.
Discussion
This study shows that patient-derived SLCLs provide a useful tool for interrogating the role of EBV+ B cells in MS and suggests that a clinical trial testing the effect of EBNA1 inhibitors in MS may be warranted.
Introduction
Multiple sclerosis (MS) is a chronic, heterogenous, and incurable demyelinating disease that significantly detracts from an individual's quality of life.1,2 The etiology of MS is complex, involving a convergence of known genetic susceptibilities, heightened inflammatory responses, and suspected environmental triggers that ultimately lead to the development of focal inflammatory lesions in the CNS and subsequent axonal damage.3 The armamentarium of disease-modifying treatment modalities for patients with MS has expanded greatly in the past 20 years, particularly with the development and widespread use of B cell depletion therapies.4-6 Nevertheless, a definitive cure for this common neuroinflammatory disorder remains elusive; this may partly stem from the paucity of disease-modifying treatments that specifically target environmental agents that are believed to serve as both triggers and drivers of disease pathogenesis.
For more than a century, infectious agents have been implicated in the pathogenesis of MS. Conclusive support for these infectious agents was, historically, insufficient to gain universal acceptance. However, evidence for a role of Epstein-Barr virus (EBV) in MS has mounted over the past 30 years.7,8 EBV is a ubiquitous γ-herpesvirus and the first human tumor virus described; it is estimated to be involved in approximately 1% of human cancers, including Burkitt lymphoma, Hodgkin lymphoma, 10% of gastric cancers (EBVaGC), and nasopharyngeal carcinoma (NPC).9 EBV establishes a latent infection in long-lived memory B lymphocytes, which are believed to be the reservoir for lifelong infection, and epithelial cells of the oropharynx and gut mucosa.10,11 EBV is capable of transforming resting B cells into immortalized lymphoblastoid cell lines (LCLs) through a stepwise process dependent on several viral oncogenes. Although the role of EBV in cancers and infectious mononucleosis (IM) is well-established, the role of EBV in MS is less clear,12,13 and other risk factors including vitamin D deficiency, cigarette smoking, obesity, and other viruses have been investigated as etiologic factors.14 The association between EBV and MS is supported by numerous epidemiologic studies demonstrating that most patients with MS have higher EBV-specific antibody titers 15–20 years before the onset of neurologic symptoms; a history of IM is more common in patients with MS and in areas where MS is prevalent; and there is a reduced risk of MS among individuals who are EBV seronegative, which increases sharply if the same individuals seroconvert.15 In a recent landmark study following up 10 million young adults in the US Army over a time span of 20 years, it was demonstrated that seroconversion to EBV+ increased the risk of developing MS 32-fold, suggesting that EBV is likely a prerequisite for developing MS.16
Although EBV is a leading candidate infectious agent for MS, the mechanism by which EBV may either cause or contribute to the pathogenesis of MS is unclear. However, many studies have described aberrant immune responses, including cross-reactive autoimmune responses, targeting the EBV nuclear antigen 1 (EBNA1) in patients with MS.17-21 EBNA1 is the viral-encoded origin-binding protein required to maintain the viral genome during latent infection and the only EBV latency protein present in all EBV-mediated cancers. Moreover, EBNA1 is known to promote host cell transformation, survival, and proliferation, encouraging the development of cancer and putatively contribute to its function as a trigger and driver of autoimmunity.22
Currently, there are no vaccines or antiviral drugs available to specifically target latent EBV infection.23 Several therapeutic approaches, including anti-EBV T-cell therapies and anti-EBV vaccines, are in development for potential use in MS.24-26 We and others have developed small molecule inhibitors that target EBNA1 DNA binding function, for use in EBV-associated cancers.27-30 Previous studies have determined that these EBNA1 inhibitors inhibit the proliferation of EBV+ tumor cells in vitro and in murine xenograft models of NPC, B cell lymphoma, and EBVaGC.27,28 EBNA1 inhibitors show both potency and specificity for a variety of EBV+ cells,27 but their selectivity for EBV+ B cells derived from patients with MS has not yet been investigated.
In this study, we characterized EBV-DNA load in peripheral blood mononuclear cells (PBMCs) and CD19+ B cells from patients with MS in relation to healthy controls (HCs) and described the generation of a collection of spontaneous LCLs (SLCLs) immortalized with endogenous EBV from these individuals. We characterized these SLCLs for EBV DNA load and for their response to treatment with the EBNA1 inhibitor, VK-1727. We found that the EBV copy numbers were higher in the peripheral blood mononuclear cells (PBMCs) and sorted B cells of patients with active MS (AMS) in comparison with PBMCs and B cells of patients with stable MS (SMS) and of HCs. Moreover, we demonstrate that an EBNA1 inhibitor, VK-1727, blocks cell proliferation for both MS-derived and HC-derived SLCLs. Our studies suggest that EBV latency may be deregulated in patients with MS and their derived SLCLs. These findings also indicate that EBNA1 inhibitors can selectively block SLCL proliferation with no discernible toxicity in EBV− B cells. These studies provide a new opportunity to study endogenous EBV-transformed B cells from patients with MS and explore therapies that selectively target these EBV-driven B cells.
Methods
Patient and Donor Information
Twenty-five patients with MS with stable disease (SMS), 25 patients with MS with active disease (AMS), and 26 HC donors were enrolled in this study. Patients with MS with active disease had ≥1 cerebral-enhancing lesion at the sample date. The blood from HCs was collected from volunteers at the Transfusion Medicine Blood Bank of the NIH.
Cell Isolation
PBMCs were isolated from whole blood by ficoll-hypaque density centrifugation. PBMCs were cryopreserved in freezing media containing 20% DMSO. CD19+ cells were isolated from 2.5 × 107 frozen PBMCs of HCs and patients with MS using a flow cytometer (FACSAriaII SORP; BD Biosciences, NJ) and magnetic beads (Milteny, Gaithersburg, MD). Cells isolated by flow cytometry were identified as lymphocytes by side scatter area and forward scatter area and as singlets by doublet discrimination (forward scatter height by forward scatter width). CD19+ cells were then sorted by positive selection using the CD3−/CD45+ and CD19+/CD45+ gates.31 For the magnetic beads isolation, PBMCs were labeled with a cocktail of non–B-cell biotinylated antibodies (CD2, CD14, CD16, CD36, CD43, CD235a). Subsequently, those cells were magnetically labeled with antibiotin microbeads for depletion. Non–B cells were retained in a Macs column, while B cells passed through the column and were collected as the enriched, unlabeled cell fraction. The percentage of purity (frequency of CD19+ cells) ranged from 92% to 99%, and it was evaluated by flow cytometry after sorting and magnetic isolation.
Generation of Spontaneous Lymphoblastoid Cell Lines and Tissue Culture Conditions
SLCLs were generated by spontaneous expansion of PBMCs from HC donors and patients with MS in the presence of cyclosporin A.32 In brief, PBMCs were thawed in growth medium comprising RPMI 1640 (Gibco, Gaithersburg, MD) with 10% fetal bovine serum (FBS; Gibco), 1% gentamicin (50 mg/mL; Quality Biological, Gaithersburg, MD), and 1% l-Glutamine (200 mM; Quality Biological). PBMCs were rested overnight and then were plated into 96-well plates (round bottom) at a density of 1 ×x 106/well in a 200-µL total volume. A minimum of 10 wells to a maximum of 30 wells were prepared for each sample according to the available PBMCs of controls and patients. Once a week, 100 µL of medium was removed and replaced with fresh growth medium. After 21 days, cyclosporin A (2 µg/mL; Sigma-Aldrich, Inc., St. Louis, MO) was added to the wells. Three to 6 weeks later, clusters of B-lymphoblastoid cells started to develop. Those clusters were then collected in bigger wells until further expanded in T25 flasks and maintained in growth medium at 37°C under a 5% CO2-humidified atmosphere. PCR for a known deletion in EBV laboratory strain B95.8 was used to confirm that the lines were transformed with endogenous EBV, and ddPCR was used to confirm the EBV infection in the newly generated SLCLs.
Generation of Lymphoblastoid Cell Lines With B95.8 Strain EBV
B95-8 cells (ATCC # CRL 1612; 13) were seeded at 3 × 105 cells/mL in a 75-cm2 tissue culture flask in complete RPMI 1640 containing 10% heat-inactivated fetal bovine serum (FBS), penicillin/streptomycin at 100U/mL and 100 μg/mL. After 2 days, cells were resuspended in fresh complete RPMI 1640 at 1 × 106 cells/mL and stimulated with 20 ng/mL of tetradecanoyl phorbol acetate (TPA; Sigma-Aldrich, Inc.) for 1 hour to induce virus production and then resuspended in complete RPMI for 96 hours before centrifugation at 600×g for 10 minutes at 4°C. Cell supernatant was then filtered through a 0.45-μm filter and stored at −80°C.33
To generate LCLs, 500 µL of EBV supernatant was added to 2 × 106 cells in 5 mL of complete RPMI medium in a 25-cm2 tissue culture flask and placed upright in a CO2 incubator at 37°C for 2–4 hours. Subsequently, 10 mL of additional medium and cyclosporin A (1 µg/mL; Sigma-Aldrich, Inc.) were added. On days 3, 6, 9, and 12, half of the medium was replaced with fresh complete RPMI with cyclosporin A. After 2 weeks, stable EBV + LCLs were formed, and anti-CD19 staining was used to confirm that the culture was 100% B cells by flow cytometry.
Digital Droplet PCR (ddPCR)
DNA was extracted from PBMCs, isolated CD19+ cells, and SLCL using the QIAamp MiniElute Virus Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. All DNA samples were eluted in 25 µL of elution buffer. DNA concentration was calculated using NanoDrop 2000 Spectrophotometer (ThermoFisher, Waltham, MA).
EBV-BamHI W primers and FAM/MGB probe (Fwd:CTTCTCAGTCCAGCGCGTTT; Rvs: CAGTGGTCCCCCTCCCTAGA; probe: 6FAM CGTAAGCCAGACAGCAGCCAATTGTCAG MGBNFQ) were designed for ddPCR using NCBI Primer Blast and Primer3Plus. For the duplex ddPCR, the housekeeping gene, Ribonuclease P protein subunit 30 (RPP30) primers (VIC/MGB probe),34,35 was used to determine the cellular quantities.
The restriction enzyme HindIII (New England Biolabs, Ipswich, MA) was used to digest DNA for 30 minutes at 37°C. DNA was then diluted 1:2.5 with nuclease-free water, added to 2× Supermix and EBV-BamHI and RPP30 primers and probes, and emulsified with droplet generator oil (Bio-Rad, Hercules, CA) using a QX-100 droplet generator. After emulsification, PCR amplification was performed within each droplet using a GeneAmp 9700 thermocycler (Applied Biosystems, Grand Island, NY) with the cycling conditions as previously described.36,37 After PCR amplification, a QX200 droplet reader (Bio-Rad, Hercules, CA) was used to quantify droplets. Thresholds were established manually for each experiment, based on negative controls, which included a no template control and a negative sample (EBV− cell lines, Ramos or BJAB). The experiment was considered valid only if no positive droplets were detected in the negative controls. Droplet positivity was established by fluorescence intensity >10,000 droplet sets; the average total number of droplets generated was approximately 30,000. The experiment was replicated if only 2 or fewer positive droplets were visualized in each well. All samples were run in duplicate. The lower limit of detection using 0.5 µg of DNA corresponds to approximately 40 copies/1 × 106 cells with a marginal error of <10%.
PCR for Discrimination of Wild-Type vs Laboratory Strain (B95.8) EBV
DNA was isolated from 5 × 106 cells from each SLCL (HC1, HC2, SMS 1–3, and AMS1-4) and additional EBV+ B cell lines (B95.8 LCLs, Mutu1 cells, Mutu1 LCLs) with a DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany). PCR for the B95.8 deletion (∆139724-151554) was performed using a BioRad C1000 Thermocycler and primers spanning BART Ex 1/2 and Ex 5. Primers used include BART Ex 1//2 F (139422; 5′-ATGTCGCCTTACCTCCCCTA-3′), BART Ex 5 R Primer 1 R (140490; 5′- TATGTGCTGCGTTGGGAACT’3-), and BART Ex 5 R Primer 2 R (151794, 5′- CAGTCACCGCCAGATACTTCA).
Compound Synthesis and Formulation
Methods of synthesis for VK-1727 and structurally related inhibitors of EBNA1 have been described.27,38 VK-1727 was weighed and resuspended in DMSO at a concentration of 50 mM, before dilution to desired concentration while maintaining a final concentration of DMSO at 0.4%.
[3H] Thymidine Incorporation Assay
Cells from HCs, AMS, and SMS SLCLs were cultured in triplicate in 96-well round-bottom microplates at 5 × 103 and 1 × 104 cells/well with defined concentrations of VK-1727 (0, 2, 5, 10, 25, and 50 μM). DMSO was used as control at 0.3% final concentration. The cells were pulsed after 3 and 7 days of culture at 37°C. In brief, 50 μL of 1 μCi [3H] thymidine solution (1:50) (PerkinElmer, Waltham, MA) was added to each well. Cells were incubated for 4 hours at 37°C and subsequently washed and harvested onto a film mat. Five milliliters of scintillation fluid were added to the film mat, and thymidine incorporation was measured using a β plate counter (Microbeta TriLux 1450, PerkinElmer, Waltham MA). The average counts per minute was determined for each well.
Resazurin Cell Respiration Assay
Cells were plated at 8 × 103 cells (in 100 μL) per well into a 96-well plate. VK-1727 or cladribine (2-chloro-2′-deoxyadenosine [2-CdA; Selleckchem, Radnor, PA) was added 24 hours later over a 10-point concentration range with 2-fold dilutions (0.1953–100 μM) in quadruplicate wells. As positive and negative controls, DMSO alone (0.4%) and puromycin (20 μg/mL)–treated wells, respectively, were plated in quadruplicate. After 72 hours, resazurin (20 μL, final 50 μM; Sigma-Aldrich) was added to each well. After 6 hours, plates were read on a CLARIOstarPlus (BMG Labtech; Ortenberg, Germany) multilabel plate reader (excitation, 560 nm; emission, 590 nm). The % cell viability for each drug was calculated relative to complete inhibition of cell viability with puromycin (0%) and treatment with DMSO (100%).
Cell Cycle Analysis
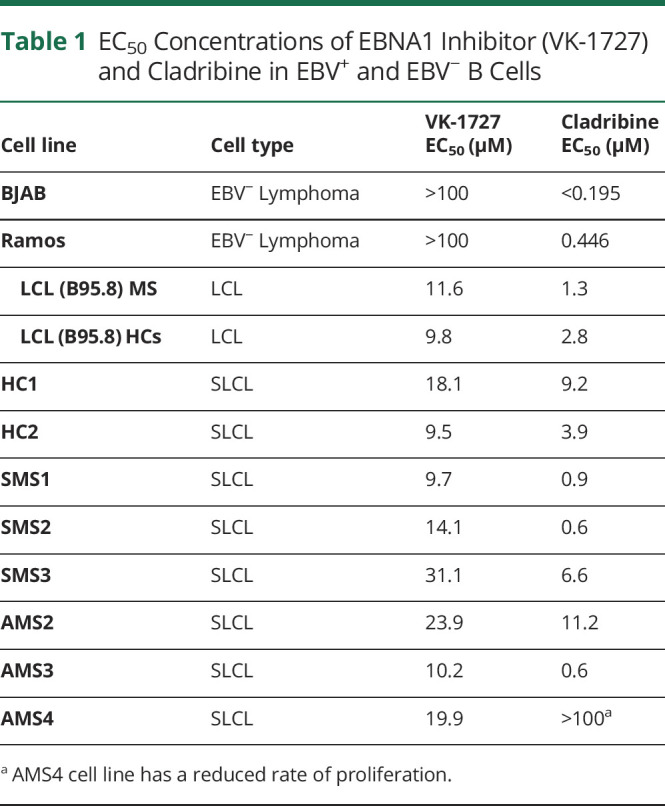
EBV− B cell lines (BJAB and Ramos) and EBV+ SLCLs were seeded at 2.4 × 105 cells/well in 6-well plates and exposed to VK-1727 (25 μM) or cladribine (2.5 μM) in biological triplicates per each condition. Cladribine is highly toxic and has a much lower EC50 than VK-1727. Therefore, Cladribine was used at much lower concentrations than VK-1727 for these experiments. (Table 1) and must be used at lower concentrations than VK-1727, which is not toxic or as effective at lower concentrations. After 72 hours, cells were permeabilized with cold, 70% ethanol and resuspended in PBS containing PI (10 mg/mL; Sigma Aldrich, St. Lois, MO) and RNAse A solution (100 μg/mL; Roche, Branchburg, NJ28). Flow cytometry was performed on a BD-LSR II (BD Biosciences; Bedford, MA), and data were analyzed using FloJo software (Ashland, OR).
Table 1.
EC50 Concentrations of EBNA1 Inhibitor (VK-1727) and Cladribine in EBV+ and EBV− B Cells
Annexin V/PI Assay
An Annexin V-FITC apoptosis detection kit (Abcam; Cambridge, UK) was used to confirm the effect of VK-1727 and cladribine on cell viability. Again, EBV− B cell lines (BJAB and Ramos) and EBV+ SLCLs (HC1-2, SMS1-3, AMS1-4) were seeded in 6-well plates and exposed to VK-1727 (25 μM) or cladribine (2.5 μM) in biological triplicates per each condition. After 72 hours, the percentages of live and apoptotic cells were analyzed after double staining with FITC-conjugated Annexin V and propidium iodide (PI). Flow cytometry was performed on a BD-LSR II (BD Biosciences; Bedford, MA), and data were analyzed using FloJo software (Ashland, OR).
EBV Gene Expression Analysis
In brief, SLCLs from patient AMS2 were treated with vehicle control (DMSO, 0.4%) or VK-1727 (10, 25, or 50 μM) with fresh medium (5% FBS) and compound changed every 24 hours for 72 hours. Again, a higher concentration of VK-1727 (25 µM) is used here than in the proliferation and cell respiration assays because in previous studies, this dose is required to observe a cell cycle disturbance in EBV+ cells.28 RNA was isolated using the RNeasy Mini Kit (Qiagen, Hilden, Germany) and treated with deoxyribonuclease (Qiagen, Hilden, Germany). Reverse transcription followed by real-time (or quantitative) PCR (RT-qPCR) was used to measure EBV gene expression levels. Primers used for qPCR of EBV genes were designed not to cross splice junctions and include the following: EBNA1 F (5′-GGTCGTGGACGTGGAGAAAA-3′), EBNA1 R (5′-GGTGGAGAC CCGGATGATG-3′); EBNA2 F (5′-TTGCCCTAGTGGTTTCGGACACA-3′), EBNA2 R (5′-ACTTGCAAATGCTCTAGGCGGGAA-3′); EBER1 F (5′-TTTGCTAGGGAGGAGACGTGTGT-3′), EBER1 R (5′-AAGCAGAGTCTGGGAAGACAACCA-3′); Zta F (5′-TCTGAACTAGAAATAAAGCGATACAAGAA-3′), Zta R (TTGGGCACATCTGCTTCAAC); EA-D F (5′- TTGGGCAGGTGCTGTTGAT-3′), EA-D R (5′- TGCCCACTTCTGCAACGA-3′); LMP1 F (5′-TCCAGAATTGACGGAAGAGGTT-3′), LMP1 R (5′-GCCACCGTCTGTCATCGAA-3′). Glucuronidase beta (GUSB) was used as a cellular control: GUSB (5′-CGCCCTGCCTATCTGTATTC and 5′-TCCCCACAGGGAGTGTGTAG-3′).39
The average cycle threshold (CT) was determined by 3 independent samples. Template-negative (quantitative PCR mixtures without cDNA) and RT-negative (RNA after genomic DNA elimination) conditions were used as controls. All data were normalized to the housekeeping gene GUSB, and quantitative PCR data for the relative quantification were calculated with the ΔΔCt method where the level of EBNA1 transcript in cells treated with vehicle control (DMSO; 0.4%) was set as 1.
Chromatin Immunoprecipitation (ChIP) Assay
ChIP assay to measure EBNA1 protein binding to viral and cellular DNA elements was previously described.27 In brief, SLCLs from patient AMS2 were treated with VK-1727 at 30 μM with fresh medium (5% fetal bovine serum) and compound or vehicle control (0.4% DMSO) changed every 24 hours for 72 hours. After harvesting, cells were cross-linked in 1% formaldehyde for 15 minutes, followed by quenching for 5 minutes with 0.125 M glycine and then lysed in 1 mL of SDS lysis buffer (1% SDS, 10 mM EDTA, and 50 mM Tris-HCl, pH 8.0) containing 1 mM PMSF and protease inhibitor cocktails (Sigma-Aldrich), and kept on ice for 10 minutes. Lysates were sonicated with a Diagenode Bioruptor, cleared by centrifugation to remove insoluble materials, diluted 10-fold into IP buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris pH 8.0, 167 mM NaCl, 1 mM PMSF, and protease inhibitors cocktail), and incubated with rabbit anti-EBNA1 antibody (2 μg/reaction) or IgG control for immunoprecipitation overnight at 4°C before washing 5 times in wash buffer at 4°C and eluting with 150 μL of elution buffer (10 mM Tris, pH 8.0, 5 mM EDTA, and 1% SDS) at 65°C for 30 minutes. The elutes were incubated at 65°C overnight to reverse cross-linking and further treated with Proteinase K in a final concentration of 100 μg/mL at 50°C for 2 hours. ChIP DNA was purified by Quick PCR Purification Kit (Life Technologies) following the manufacturer's instruction. Real-time quantitative PCR (ABI 7900HT Fast Real-Time PCR System; Applied Biosystems) was performed on ChIP DNA to quantitate 2 EBV loci (DS50 and Qp) and 1 cellular locus (CLIC1), which have been reported to associate with EBNA1 (GEO database GSE73887).40 Results were quantified as % input. Primer sets used for ChIP were as follows: DS50 F (5′-ATGTAAATAAAACCGTGACAGCTCAT-3′), DS50 R (5′-TTACCCAACGGGAAGCATATG-3); QP F (5′-AAATTGGGTGACCACTGAGGGAGT-3′), QP R (5′-ATAGCATGTATTACCCGCCATCCG-3′); CLIC1 F (5′-CCTAAGCTGAGGGTGATTCATCTC-3′), CLIC1 R (5′-CCCCACATCCTTGACAGGAA-3′).
Standard Protocol Approvals, Registrations, and Patient Consents
All patients with MS were enrolled in IRB-approved natural history study. Both HCs patients with MS provided informed consent before participation.
Data Availability
All data related to this article is provided in the main text or supplemental data.
Results
Demographic and Clinical Data
Characteristics of patients with MS and HC cohort are summarized in Table 2. Brain MRI was available for all patients with MS at the sample date (data not shown). The cohort was further segregated into 2 major subgroups; those diagnosed with SMS or with AMS, based on clinical and radiologic assessment (defined as patients with 1 or more gadolinium-enhancing lesions) during blood sampling (Table 2). Our MS cohort included 20% (5/25) of naïve never-treated patients while 56% (14/25) of patients with SMS and 52% (13/25) of patients with AMS, respectively, were under disease-modifying therapy (DMT) during sample collection (Table 2). Among the treated patients with SMS, 5 were on high-efficacy drugs; 3 with natalizumab, 1 with alemtuzumab, and 1with daclizumab. None of the 14 treated patients in the SMS group received corticosteroids within 4 weeks before sample collection or were on B cell depleting therapy (Table 2). In the AMS cohort, 3 patients were under high-efficacy drugs; 1 received alemtuzumab, and 2 were on natalizumab, although 1 of those 2 patients received drug for only 12 days (Table 2). In the AMS group, no patients were on B cell depleting therapy, while 3 of the patients with AMS stopped steroid treatment 1 month before sample collection. To determine whether there were any differences in EBV load in patients with MS relative to controls, we quantified EBV-DNA in PBMCs and CD19+ sorted B cells from 26 healthy controls, 25 patients with SMS, and 25 patients with AMS. EBV-DNA was measured by ddPCR assay using primers specific for EBV BamHI W region, and the relative EBV copy number per 106 cells was determined by normalizing to the cellular gene, RPP30. EBV-DNA copy number in PBMCs is known to vary over time and among individuals.41 ddPCR detected EBV-DNA in only a fraction of each blood sample (Figure 1, A and B). As expected, while EBV-DNA was more frequently detected in CD19+ sorted B cells compared with PBMCs in all cohorts consistent with the known B cell tropism of EBV,42 there were some notable differences among cohorts. Of interest, the frequency of EBV detection in CD19+ cells in the AMS cohort was roughly double (20/25; 80%) that of the HC (11/26; 42%) and SMS cohorts (7/25; 28%) (p = 0.0002 AMS vs SMS; 0.006 AMS vs HCs) (Figure 1A). In addition, EBV was also detected most frequently in PBMCs of those with active MS (9/26; 36%), which was significantly more than the detection frequency in those with stable MS (3/25; 12%), but similar to the frequency of detection in PBMCs of HCs (7/26; 27%) (Figure 1A). However, the magnitude of the EBV load was not significantly different between the 3 cohorts as determined by the ANOVA test. Our data are based on the sensitivity of the assay, and zero on the Y axis of Figure 1B represents a value that was below the limits of detection of the assay (Figure 1B). Our data comparing the frequency of detection and viral loads in PBMCs are consistent with other reports describing a similar prevalence of EBV in the PBMCs of patients with RRMS and healthy controls.18,43 Of interest, it is only in the isolated CD19+ cell population where significant increases in frequency of EBV detection are observed in the “active” cohort compared with that in the “stable” cohort and the HCs (Figure 1B). As expected, there is a discordance in the male:female sex ratio in patients with MS relative to healthy controls with a higher percentage of male individuals in the HC cohort and a preponderance of female individuals in the MS cohort, particularly in patients with active disease. This is consistent with many studies demonstrating the increased incidence of MS among female individuals. It is possible that sex-based differences may also influence the increased detection of EBV in CD19+ cells detected in this cohort.44,45
Table 2.
Cohort Characteristics and Treatment Information

Figure 1. Detection of EBV and EBV Loads in Patients With MS and HCs.
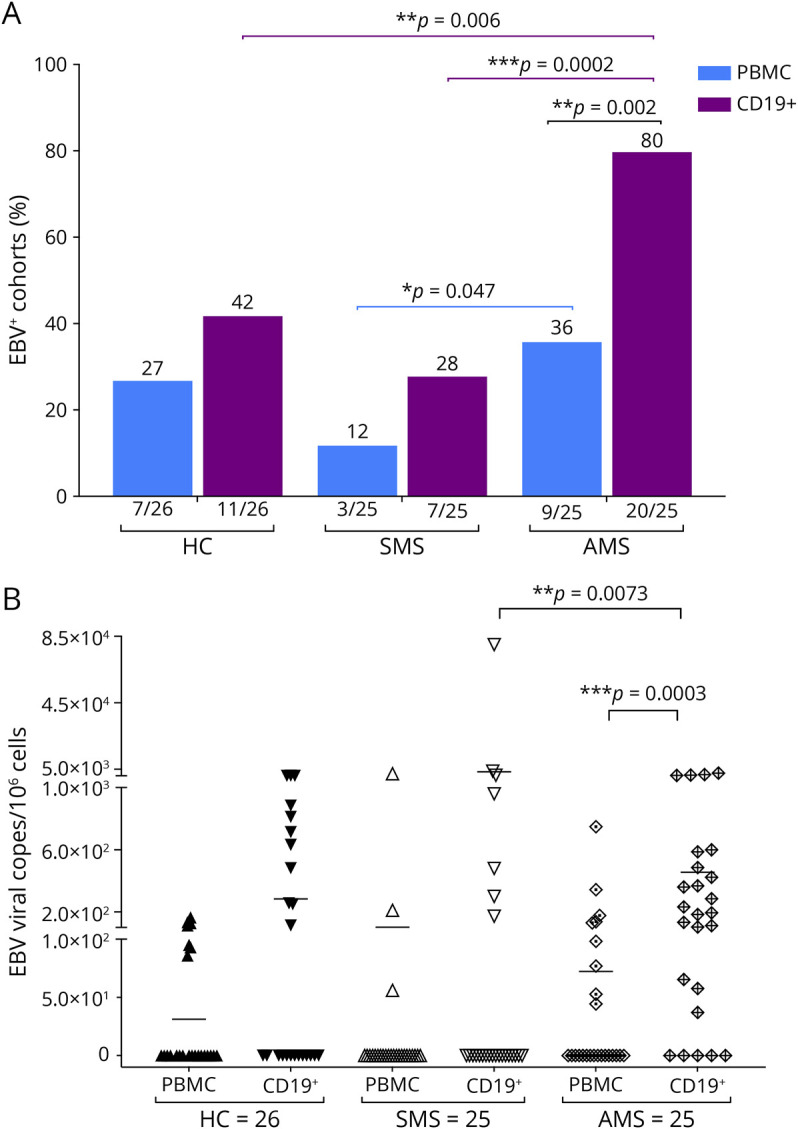
(A) Percent of HCs, SMS, and AMS PBMCs and CD19+ B cells that are positive for EBV by ddPCR (B) EBV-BAMH1 copies/106 cells in PBMCs and CD19+ B cells from HCs, patients with SMS, and patients with AMS by ddPCR (0 on Y-axis represents a value that was below the limits of detection of the assay). EBV = Epstein-Barr virus; HCs = healthy controls; PBMC = peripheral blood mononuclear cell.
Generation and Characterization of Spontaneous Lymphoblastoid Cell Lines From Patients With MS and Healthy Controls
We next attempted to generate SLCLs from 29 individuals from whom sufficient material was available, including: 14 HCs and 15 patients with MS (Table 3). From these individuals, we were able to establish SLCLs from only 2 of 14 (14%) of healthy controls, while SLCLs were successfully generated from 7 of 15 (47%) patients with MS (43% of PBMCs isolated from patients with MS during stable disease, 50% of PBMCs isolated from patients with MS during active disease, as defined by MRI. A schematic representation of the generation of SLCLs is pictured in Figure 2A. Shortly after the addition of cyclosporin A, it was possible to detect an increased number of CD19+ B cells in comparison with CD3+ T cells, as shown by the fluorescence microscopy (data not shown). Three to 6 weeks later, clusters of B-lymphoblastoid cells began to develop. EBV DNA ddPCR on those cultures demonstrated the increased detection of EBV-BamHI (FAM) in the SLCLs that were generated (Figure 2C) in comparison with that in the PBMCs in the initial culture stage (Figure 2B). The increased yield of SLCLs from patients with MS compared with that from healthy controls is consistent with previous reports.46,47 Of interest, 2 of 3 and 2 of 4 patients with SMS and AMS, respectively, were on a DMT during blood collection. Given the small size of the cohort of patients with MS that yielded SLCLs, we are presently unable to determine the influence of DMT on EBV reactivation or the likelihood of generating SLCLs from patients with MS. We also observed that EBV loads, as determined by ddPCR, trended higher in SLCLs derived from AMS than in SLCLs derived from HCs or those with SMS (Table 3).
Table 3.
Generation of SLCLs

Figure 2. Generation of Spontaneous Lymphoblastoid Cell Lines (SLCLs) From the PBMCs of Patients With MS and Healthy Controls.
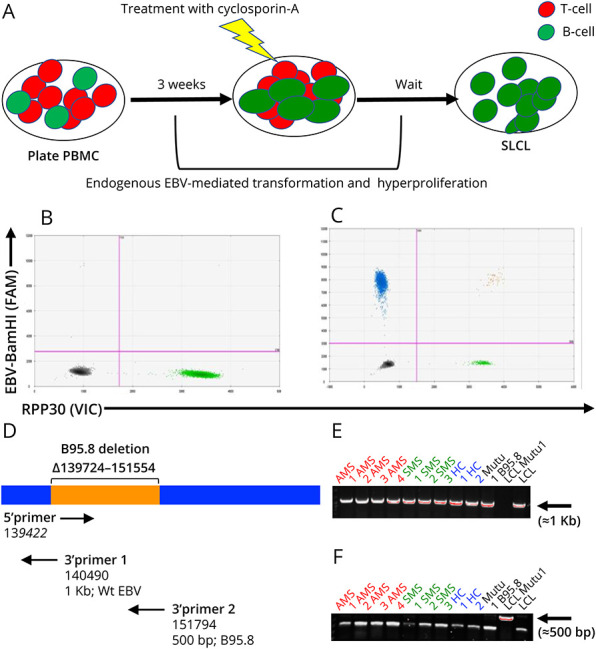
(A) Patient with MS and control PBMC were used to generate SLCLs from endogenous EBV, without adding exogenous/laboratory strain EBV. After 21 days in culture, residual T-cells were eliminated by the addition of cyclosporin A. (B) Digital droplet PCR was performed on newly formed SLCLs. Digital droplet (dd) PCR plot indicates EBV-BamHI W-FAM (Y-axis; upper left quadrant; blue droplets) and the RPP30 cellular housekeeping gene-VIC (X-axis; lower right quadrant; green droplets). (C) Representative 2-dimensional ddPCR plot performed on a generated SLCL culture. Primer sets and fluorescent probes to detect a sequence in the EBV-BamHI W-FAM (Y-axis; upper left quadrant; blue droplets) and RPP30-VIC, housekeeping gene (X-axis; lower right quadrant; green droplets). Populations double positive for EBV-BamHI W and RPP30 are in the upper right quadrant (orange droplets). (D) Diagram of EBV genome. (E) Amplification of EBV WT DNA (≈1 Kb) with 3′ primer 1. (F) Amplification of B95.8 laboratory strain DNA (≈500bp) with 3′ primer 2. EBV = Epstein-Barr virus; PBMC = peripheral blood mononuclear cell.
To confirm that SLCLs were transformed ex vivo from endogenous EBV and not derived from the laboratory strain (B95.8) of EBV present in the laboratory that the SLCLs were generated in, we performed PCR to determine the presence or absence of the known B95.8 deletion ∆139724-151554 that is not present in the WT EBV genome or other laboratory strains (e.g., Mutu) using primers to the miBART region (Figure 2, D–F). In this assay, PCR using 3′ primer 1 yields a 1-Kb product in wild-type EBV, while PCR using 3′ primer 2 yields a 500-bp product when the B95.8 deletion is present (Figure 2D). Our results confirm that the B95.8 deletion is present in only B95.8-transformed LCLs but absent in SLCLs from patients with MS and healthy controls and other B cell lines transformed with Mutu virus (Figure 2, E and F), confirming SLCLs carry only endogenous patient-derived EBV.
EBNA1 Inhibitor Decreases the Proliferation of EBV-Positive SLCLs but Not EBV-Negative B Cells
To investigate the effect of the EBNA1 small molecule inhibitor VK-1727 (Figure 3A) on the proliferation of SLCLs, we treated EBV− B cell lines (BJAB and Ramos) and EBV+ SLCLs (HC1-2 SLCLs, SMS1and 2 SLCLs, and AMS3 and 4 SLCLs) with VK-1727 at a range of doses (0–50 μM) and at 2 cell concentrations (5 × 103 and 1 × 104 cells/well) and evaluated the effect of EBNA1 inhibition on the proliferation of these cell lines over 7 days using a [3H]-thymidine incorporation assay and viable cell counts (Figure 3, B–D). Notably, some of the slower growing lines were not tested in this assay because their rates of proliferation were too slow to assess the magnitude of proliferation or effects on proliferation or drug-mediated effects on proliferation within the time frame of this assay. In EBV− B cell lines (Ramos and BJAB), there was not a significant decrease in cell viability or cell number in cells plated at 2 concentrations (5 × 103 and 1 × 104 cells/well) (Figure 3C). By contrast, VK-1727 inhibited EBV-specific proliferation in EBV+ SLCLs from both patients with MS and healthy controls in a dose-dependent manner (Figure 3D). Inhibition of cell proliferation by VK-1727 correlated with cell viability in the EBV+ cell lines (Figure 3D). These data are consistent with previous studies demonstrating that VK-1727 inhibition of cell proliferation is selective for EBV+ cell lines.27,28 Notably, VK-1727 blocks the proliferation of all EBV+ SLCLs tested and does not distinguish between SLCLs derived from patients with MS and those generated from HCs.
Figure 3. Selective Inhibition of EBV+ SLCL Proliferation From Patients With MS and Controls by EBNA1 Inhibitor, VK-1727.

(A) Structure of EBNA1 inhibitor, VK-1727. (B) Percent inhibition of proliferation as measured by [3H] Thymidine incorporation at day 7 in 2 EBV− B cell lines (BJAB and Ramos) and 6 EBV+ SLCLs treated with VK-1727 (2–10 µM). (C) Cell counts (right Y-axis) and [3H] Thymidine incorporation (left Y-axis) in EBV− B cell lines (BJAB and Ramos). (D) Cell counts (right Y-axis) and [3H] Thymidine incorporation (left Y-axis) EBV+ SLCLs from healthy controls and patients with MS. (C and D) Black bars indicate 3H-CPM when cells were seeded at 1 × 104/well; gray bars indicate 3H-CPM when cells were seeded at 5 × 103/well. Black and gray lines indicate the number of viable cells when cultures were seeded at 1 × 104/well and 5 × 103/well, respectively (*p =< 0.02–0.05; **p =< 0.006; ***p = 0.0006; ****p < 0.0001; Two-way ANOVA). EBV = Epstein-Barr virus; SLCL = spontaneous lymphoblastoid line.
VK-1727 Inhibition Is Selective for EBV+ B Cells Compared With Cladribine
We next compared the inhibitory effects of VK-1727 on EBV+ SLCLs with the effects of cladribine, a nucleoside analog that was first used for its antiproliferative effects in lymphomas and myeloma and is now used as a standard-of-care drug for MS because it depletes B cells. For these studies, we used a resazurin assay to measure changes in metabolic activity of cells treated with VK-1727 (Figure 4A) or cladribine (Figure 4B). To determine the selectivity of VK-1727 and cladribine, we tested these compounds in both EBV+ SLCLs (HC1-2, SMS1-3, AMS1-4), EBV+ B95.8–transformed LCLs (one from a patient with MS and one from an HC), and EBV− B cell lines (BJAB and Ramos) and calculated EC50 values. VK-1727 inhibited metabolic activity in all EBV+ cell lines tested; EC50 values for all EBV+ cell lines tested were between 9.5 and 31.1 μM (15.79 ave ± 6.98 stdev) VK-1727 (Table 1; representative plots in Figure 4A). VK-1727 did not inhibit metabolic activity in the 2 EBV− B cell lines tested where 50% inhibition was not reached at the highest concentration tested, and EC50 values were greater than 100 μM and (Table 1; representative plots in Figure 4A). These results suggest that the metabolic effects of VK-1727 are selective for EBV+ cells. By contrast, cladribine inhibited both EBV+ and EBV− cell lines equally, thus lacking any selectivity for EBV+ cells (Table 1; representative plots in Figure 4B).
Figure 4. Selective Inhibition of Cellular Metabolism by EBNA1 Inhibitor VK-1727 but Not Cladribine.

(A) VK-1727 inhibition of cell metabolism in a resazurin-based assay using EBV+ SLCLs and EBV− control (BJAB). Representative plots shown. (B) Cladribine inhibition of cell metabolism in a resazurin-based assay using EBV+ SLCLs and EBV− control (BJAB). Representative plots shown. EBV = Epstein-Barr virus; SLCL = spontaneous lymphoblastoid line.
EBV Inhibitor Perturbs the Cell Cycle in EBV+ SLCLs but Does Not Induce High Levels of Apoptosis
To determine whether the antiproliferative effect of VK-1727 on EBV+ B cells was cytostatic or cytotoxic, we analyzed cell cycle profiles of EBV− B cell lines and SLCLs treated with VK-1727 (25 μM), cladribine (2.5 μM), or vehicle control. Although cell cycle profiles remained stable in EBV− cells treated with VK-1727 (Figure 5, A and B), EBV− cell cycle populations were greatly affected by the addition of cladribine, which increased the <G1 population and decreased the percentage of cells in G1, S, and G2/M phase. Although there was minimal effect on the cell cycle profile of EBV− cells treated with VK-1727, EBV+ SLCL cell cycle profiles were highly perturbed with a flattening of the G2 peak and an increase in the <G1 and G1 peaks (Figure 5, A and B). Again, these data are similar to data observed for EBV Mutu1-transformed LCL28 and suggest that the antiproliferative effect of VK-1727 is cytostatic rather than cytotoxic in EBV+ SLCLs, while cladribine is highly cytotoxic to all B cells, regardless of their EBV status.
Figure 5. EBNA1 Inhibition Perturbs Cell Cycle Expression While Cladribine Induces Apoptosis in SLCLs.

(A) Cell cycle profiles comparing EBV+ and EBV− B cells treated with DMSO, VK-1727 (25 µM), or cladribine (2.5 µM) measured by flow cytometry analysis of propidium iodide staining (representative graphs for SMS1 and Ramos). (B) Graph of cell cycle kinetics data comparing the effects of VK-1727 with those of cladribine. EBV+ cells (HC1-2, SMS1-3, and AMS1-4) treated with 25 µM of VK-1727 show a significant decrease in the total population of G2 cells that is not observed for EBV− (Ramos and BJAB) B cells. In addition, while cladribine induces apoptosis, marked by an increase in the <G1 population, in all B cells regardless of EBV infection, apoptosis was not increased in EBV− or EBV+ cells treated with VK-1727. Each cell line was treated, stained, and analyzed as biological replicates. Data shown here are batched by group (EBV−, HC SLCL, SMS SLCL, and AMS SLCL) (****p < 0.0001, 2-way ANOVA). (C) Flow cytometry analysis of Annexin V/PI staining comparing EBV− and EBV+ B cells with DMSO and VK-1727 (25 µM) or cladribine (2.5 µM) (representative graphs for SMS1 and Ramos). (D) Graphs summarizing live cell populations (% Q4) observed from the Annexin V/PI experiment. Each cell line was treated, stained, and analyzed as biological replicates. Data shown here are batched by group (EBV−, HC SLCL, SMS SLCL, and AMS SLCL) (****p < 0.0001, 2-way ANOVA). EBV = Epstein-Barr virus; SLCL = spontaneous lymphoblastoid line.
In addition, we performed Annexin V/PI staining on EBV− B cells and EBV+ SLCLs to confirm that VK-1727 does not induce high levels of apoptosis. A small percentage of EBV− or EBV+ VK-1727 (25 μM) or vehicle-treated cells were found to be in early apoptosis (Q3), late apoptosis (Q2), or necrosis (Q1), and we did not observe a significant decrease in the live cell (Q4) population with VK-1727 treatment (Figure 5, C and D). However, cladribine (2.5 μM) was a potent inducer of apoptosis in EBV− and EBV+ cells alike with few live cells remaining (Figure 5, C and D). Collectively, the cell cycle and Annexin V/PI data suggest that VK-1727 specifically inhibits cell cycle progression in EBV+ SLCLs from patients with MS and healthy controls but is cytostatic rather than cytotoxic. This is in contrast to cladribine, which lacks specificity and is highly cytotoxic to B cells.
Treatment With VK-1727 Reduces Latent Viral Gene Expression and Disrupts EBNA1 DNA Binding
In addition to characterizing the selectivity and cellular action of VK-1727, we were interested in determining the effects of EBNA1 inhibition on EBNA1 function. To confirm that VK-1727 interferes with EBNA1 DNA–binding function in SLCLs, we performed a ChIP assay in an EBV+ SLCL (AMS2) (Figure 6A). Treatment with 30 μM VK-1727 significantly reduced EBNA1 binding to 2 EBV binding sites, including the viral dyad symmetry (DS) and Qp and a known EBNA1 cellular-binding site in the gene for the chloride intracellular channel protein 1 (CLIC1) (Figure 6A). As expected, negligible amplification was shown after immunoprecipitation with the IgG control (Figure 6B). These studies indicate successful target engagement of the EBNA1 inhibitor in cell lines transformed with endogenous virus. We next measured the expression of EBV latency (EBNA1, EBNA2, LMP1, and EBER1) and lytic (Zta and EA-D) genes in the AMS2 SLCL by qRT-PCR. After treatment with 50 μM of VK-1727 for 72 hours, we observed modestly reduced expression of both EBNA1 and EBNA2. Expression of LMP1, EBER1, Zta, and EA-D were unaffected (Figure 6C). These results are similar to what we have observed in EBV (Mutu virus) transformed LCLs, where modest inhibition of EBNA1 expression was also observed after short-term culture in vitro.28 VK-1727 does not seem to induce lytic gene expression (Zta and EA-D) in SLCLs. Of interest, we have shown an increase in lytic gene expression in EBV-associated gastric cancer cell lines treated with VK-1727,28 and other reports have shown lytic gene induction with EBNA1 inhibition.48
Figure 6. VK-1727 Disrupts EBNA1 Binding in SLCLs.
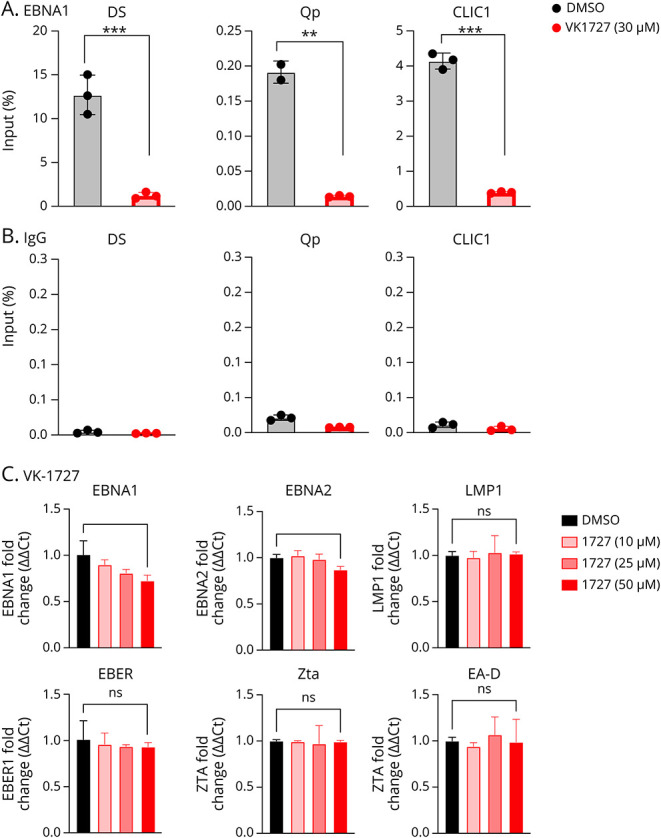
(A) ChIP assay for EBNA1 binding to the DS, Qp, and cellular locus CLIC1 in AMS4 SLCLs treated with DMSO or 30 µM VK-1727 for 72 hours. p values were determined for 3 biological replicates (***p < 0.001, Two-way ANOVA). (B) Immunoprecipitation was performed with IgG as a control bottom panel. (C) Expression of EBV latency (EBNA1, EBNA, EBER, LMP1) and lytic (Zta and EA-D) genes in EBV+ AMS4 SLCL. p values were determined for 3 biological replicates (*p < 0.05, t test). EBV = Epstein-Barr virus; SLCL = spontaneous lymphoblastoid line.
Discussion
Many groups have compared plasma EBV loads in patients with MS and controls and have not found significant differences between the 2 groups.43,49,50 In this study, we report that the frequency of EBV+ cells were statistically higher in the PBMCs and CD19+ populations of patients with AMS in contrast to that of those with SMS. In particular, 80% of patients with active MS were EBV+ in CD19+ purified B cells compared with 42% and 28% of HCs and patients with SMS, respectively.
Notably, there is a discordance in the male:female sex ratio in the healthy controls compared with that in patients with MS. It has been demonstrated that salivary shedding of EBV is increased in female individuals, but not male individuals during periods of psychological distress,44,45 suggesting that there maybe sex-based and hormonal influences that contribute to EBV reactivation. There is a 3.5:1 female-to-male ratio in MS, and it is possible that sex-based differences may also influence the increased detection of EBV in CD19+ cells detected in this cohort.
In addition, the failure to detect EBV in PBMC or CD19+ from most of the HCs or patients with SMS may reflect the lower limit of detection of the assay, which was 40 copies/1 × 106 cells.
We also describe the generation and partial characterization of spontaneous EBV-transformed LCLs derived from a cohort of patients with MS and healthy controls. The higher frequency of EBV detection in CD19+ B cells of patients with AMS and the greater rate of SLCL generation in patients with MS suggest that there may be a fundamental failure of host control of EBV latent infection in AMS. These findings are consistent with known abnormalities in the immune response to EBV in MS, including deficient cytotoxic T-lymphocyte (CTL) control of EBV infection.42 Of interest, early clinical data using adaptive T-cell therapies specific for EBV also indicate that improved CTL control of EBV infection has therapeutic benefit in MS.51 Future studies including serology for EBV coupled with detection of EBV in CD19+ cells in longitudinal samples from patients with MS and healthy controls are necessary to fully elucidate the dynamics of EBV infection in the context of disease activity.
We also show that targeting EBV through EBNA1 inhibitors can block the proliferation of SLCLs from MS and from HCs. EBNA1 inhibitor VK-1727 significantly reduced the proliferation of EBV+ B cells, regardless of whether they are transformed with a laboratory strain (B95.8) or WT virus. These data suggest that VK-1727 targets EBV in cells isolated from individuals with a broad spectrum of disorders, including EBV-mediated cancers and MS and further extends these observations to B cell lines from genetically disparate individuals carrying WT virus. Previous studies have demonstrated that EBNA1 inhibitors, including VK-1727, do not affect the growth of EBV− lung carcinoma cells or EBV− gastric carcinoma cell lines.27,28 Of importance, this study confirms that the proliferation of EBV− B cell lines is not affected by VK-1727, suggesting that there are limited, if any, off-target effects. Currently, an EBNA1 inhibitor is being tested in a phase I/II clinical trial for EBV+ nasopharyngeal carcinoma (ClinicalTrials.gov Identifier: NCT04925544). It is not yet known whether EBNA1 inhibitors will confer benefit to patients with MS or whether additional modifications such as enhanced CNS penetrance may be required to achieve clinical benefit in MS. Other strategies for EBNA1 inhibition, including EBNA1 inhibitors that function as heat shock protein 90 inhibitors, block EBNA1-dependent maintenance or transcription, and block EBNA1 RGG-like linking regions have been developed and validated, but have yet to be tested for their therapeutic potential in MS patient–derived cells or model systems.29
Many effective disease-modifying therapies for MS have their origins as therapeutic agents developed for the treatment of cancer. For example, anti-CD20 chimeric monoclonal antibody therapies, such as rituximab, were developed as B cell depletion therapies for use in non-Hodgkin lymphoma and chronic lymphocytic anemia before their efficacy was established in MS. B cell depletion therapies eliminate EBV− and EBV+ cells. Therefore, it is unclear whether their benefit is related to the elimination of EBV+ B cells. Accordingly, a therapy that specifically targets EBV+ B cells is of interest as an EBV-specific treatment modality in MS that has the potential to improve disease outcomes and allow for further understanding of the specific role of EBV+ B cells in MS. Cladribine is one of many B cell depletion therapies that are now used as standard-of-care therapies in patients with highly active relapsing MS. When we compared the activity of our EBNA1 inhibitor with cladribine in our resazurin-based metabolic assay (Figure 4, A and B; Table 1), we demonstrated that while cladribine decreases the proliferation of EBV− and EBV+ cells, VK-1727 only decreased the proliferation in EBV+ B cells. Moreover, cell cycle analysis (Figure 5A) showed that while both VK-1727 and cladribine perturbed cell cycle kinetics and reduced the percentage of cells in G2/M in EBV+ B cells, cladribine also effected the cell cycle of EBV− cells, while the effect of VK-1727 was EBV specific. In addition, Annexin V/PI staining confirmed cladribine was a potent inducer of apoptosis in EBV+ and EBV− cells, while VK-1727 was not. Again, these data indicate that VK-1727 has a cytostatic rather than cytotoxic mechanism of action and has little off-target activity in human B cells. We have reported similar findings in xenograft models of EBV-associated cancers, where EBNA1 inhibitors were shown to have excellent safety profiles at high doses.27,28 An important caveat here is that VK-1727 and other EBNA1 inhibitors will only target proliferating EBV+ cells and will not affect EBV-infected cells exhibiting a “latency 0” program (no EBNA1 and only noncoding RNAs expressed), making it unlikely that EBNA1 inhibitors will completely eliminate EBV infection.10 However, other compounds with antiproliferative effects in immune cells have been shown to have clinical efficacy in neuroimmune disorders associated with viral infection, including MS and HTLV-I associated myelopathy/tropical spastic paraparesis.52-55
In summary, a role for EBV in MS is supported by observations of significantly increased detection of EBV in CD19+ sorted B cells from patients with MS compared with that from controls. This prompted us to generate a collection of SLCLs from patients with MS and HCs for further studies of EBV latency in MS patient–derived B cells. We show that these SLCLs are more readily generated from patients with MS. We also provide evidence that a small molecule inhibitor of EBNA1 can selectively block cell proliferation and EBNA1 DNA binding and may slightly decrease latent gene expression in EBV+ cells in SLCLs from patients with MS and healthy controls and has the potential to counteract the prosurvival and proinflammatory effects of EBNA1 in different anatomical compartments. Of importance, these EBNA1 inhibitors do not affect EBV− B cells nor induce inflammatory apoptosis as does cladribine, a drug presently used as front-line therapy to treat MS. This study follows several earlier studies showing cross-reactivity between EBV EBNA1 and host CNS antigens17,18,56 and provides compelling evidence that EBV EBNA1 is involved in molecular mechanisms that drive CNS autoimmunity. Collectively, these studies provide justification for further exploring the use of EBNA inhibitors in MS.
Acknowledgment
The authors thank members of the Lieberman laboratory and the Wistar Institute Cancer Center Core Facilities in Flow Cytometry. The authors thank Dr. Dragan Maric in the NINDS Microscope Core Facility and members of the Jacobson laboratory. The authors thank Dr. Maria Gaitan, the Neuroimmunology Clinic at NINDS, and the patients and their families for their contribution to this study.
Glossary
- ChIP
chromatin immunoprecipitation
- CT
cycle threshold
- CTL
cytotoxic T lymphocyte
- ddPCR
digital droplet PCR
- DMT
disease-modifying therapy
- EBNA1
Epstein-Barr Virus Nuclear Antigen 1
- EBV
Epstein-Barr virus
- HC
healthy control
- IM
infectious mononucleosis
- LCLs
lymphoblastoid cell lines
- NPC
nasopharyngeal carcinoma
- PBMC
peripheral blood mononuclear cell
- SLCL
spontaneous lymphoblastoid cell line
Appendix. Authors
Study Funding
This work is supported by NINDS funding and by grants NIH R01-CA093606, R01 AI153508, R01 CA259171-01, SAP# 4100088567 and R01-DE017336 to PML, and the NCI Cancer Center Support Grant P30-CA010815 (Altieri).
Disclosure
The Wistar Institute, on behalf of the authors T.E.M. and P.M.L., has filed patents covering composition of matter and their use on the small molecule disclosed here for the treatment of human cancer and other diseases (patent number WO2015073864, “EBNA1 Inhibitors and Their Method of Use”; WO2016183534, “EBNA1 Inhibitors and Methods using Same”). P.M.L. has an ownership interest in Vironika, LLC. Go to Neurology.org/NN for full disclosures.
References
- 1.McGinley MP, Goldschmidt CH, Rae-Grant AD. Diagnosis and treatment of multiple sclerosis: a review. JAMA. 2021;325(8):765-779. doi: 10.1001/jama.2020.26858 [DOI] [PubMed] [Google Scholar]
- 2.Cotsapas C, Mitrovic M, Hafler D. Multiple sclerosis. Handb Clin Neurol. 2018;148:723-730. doi: 10.1016/B978-0-444-64076-5.00046-6 [DOI] [PubMed] [Google Scholar]
- 3.Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med. 2018;378(2):169-180. doi: 10.1056/NEJMra1401483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gharibi T, Babaloo Z, Hosseini A, et al. The role of B cells in the immunopathogenesis of multiple sclerosis. Immunology. 2020;160(4):325-335. doi: 10.1111/imm.13198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Comi G, Bar-Or A, Lassmann H, et al. Role of B cells in multiple sclerosis and related disorders. Ann Neurol. 2021;89(1):13-23. doi: 10.1002/ana.25927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giovannoni G, Comi G, Rammohan K, et al. Long-term disease stability assessed by the expanded disability status scale in patients treated with cladribine tablets 3.5 mg/kg for relapsing multiple sclerosis: an exploratory post hoc analysis of the CLARITY and CLARITY extension studies. Adv Ther. 2021;38(9):4975-4985. doi: 10.1007/s12325-021-01865-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pender MP. Does Epstein-Barr virus infection in the brain drive the development of multiple sclerosis? Brain. 2009;132(Pt 12):3196-3198. doi: 10.1093/brain/awp312 [DOI] [PubMed] [Google Scholar]
- 8.Bar-Or A, Pender MP, Khanna R, et al. Epstein-Barr virus in multiple sclerosis: theory and emerging immunotherapies: (Trends in Molecular Medicine, 26:3 p:296-310, 2020). Trends Mol Med. 2021;27(4):410-411. doi: 10.1016/j.molmed.2021.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chakravorty S, Afzali B, Kazemian M. EBV-associated diseases: current therapeutics and emerging technologies. Front Immunol. 2022;13:1059133. doi: 10.3389/fimmu.2022.1059133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hochberg D, Middeldorp JM, Catalina M, Sullivan JL, Luzuriaga K, Thorley-Lawson DA. Demonstration of the Burkitt's lymphoma Epstein-Barr virus phenotype in dividing latently infected memory cells in vivo. Proc Natl Acad Sci USA. 2004;101(1):239-244. doi: 10.1073/pnas.2237267100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hochberg D, Souza T, Catalina M, Sullivan JL, Luzuriaga K, Thorley-Lawson DA. Acute infection with Epstein-Barr virus targets and overwhelms the peripheral memory B-cell compartment with resting, latently infected cells. J Virol. 2004;78(10):5194-5204. doi: 10.1128/jvi.78.10.5194-5204.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soldan SS, Lieberman PM. Epstein-Barr virus infection in the development of neurological disorders. Drug Discov Today Dis Models. 2020;32(Pt A):35-52. doi: 10.1016/j.ddmod.2020.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lassmann H, Niedobitek G, Aloisi F, Middeldorp JM, NeuroproMiSe EBV Working Group. Epstein-Barr virus in the multiple sclerosis brain: a controversial issue--report on a focused workshop held in the Centre for Brain Research of the Medical University of Vienna, Austria. Brain. 2011;134(Pt 9):2772-2786. doi: 10.1093/brain/awr197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ascherio A, Munger KL. Epidemiology of multiple sclerosis: from risk factors to prevention-an update. Semin Neurol. 2016;36(2):103-114. doi: 10.1055/s-0036-1579693 [DOI] [PubMed] [Google Scholar]
- 15.Soldan SS, Lieberman PM. Epstein-Barr virus and multiple sclerosis. Nat Rev Microbiol. 2023;21(1):51-64. doi: 10.1038/s41579-022-00770-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bjornevik K, Cortese M, Healy BC, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science. 2022;375(6578):296-301. doi: 10.1126/science.abj8222 [DOI] [PubMed] [Google Scholar]
- 17.Makhani N, Banwell B, Tellier R, et al. Viral exposures and MS outcome in a prospective cohort of children with acquired demyelination. Mult Scler. 2016;22(3):385-388. doi: 10.1177/1352458515595876 [DOI] [PubMed] [Google Scholar]
- 18.Lunemann JD, Edwards N, Muraro PA, et al. Increased frequency and broadened specificity of latent EBV nuclear antigen-1-specific T cells in multiple sclerosis. Brain. 2006;129(Pt 6):1493–1506. doi: 10.1093/brain/awl067 [DOI] [PubMed] [Google Scholar]
- 19.Lunemann JD, Jelcic I, Roberts S, et al. EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-gamma and IL-2. J Exp Med. 2008;205(8):1763-1773. doi: 10.1084/jem.20072397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lang HL, Jacobsen H, Ikemizu S, et al. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat Immunol. 2002;3(10):940-943. doi: 10.1038/ni835 [DOI] [PubMed] [Google Scholar]
- 21.Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. 1995;80(5):695-705. doi: 10.1016/0092-8674(95)90348-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frappier L. EBNA1. Curr Top Microbiol Immunol. 2015;391:3-34. doi: 10.1007/978-3-319-22834-1_1 [DOI] [PubMed] [Google Scholar]
- 23.Soldan SS, Messick TE, Lieberman PM. Therapeutic approaches to Epstein-Barr virus cancers. Curr Opin Virol. 2022;56:101260. doi: 10.1016/j.coviro.2022.101260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ioannides ZA, Csurhes PA, Douglas NL, et al. Sustained clinical improvement in a subset of patients with progressive multiple sclerosis treated with Epstein-Barr virus-specific T cell therapy. Front Neurol. 2021;12:652811. doi: 10.3389/fneur.2021.652811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bar-Or A, Pender MP, Khanna R, et al. Epstein-Barr virus in multiple sclerosis: theory and emerging immunotherapies. Trends Mol Med. 2020;26(3):296-310. doi: 10.1016/j.molmed.2019.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maple PA, Ascherio A, Cohen JI, et al. The potential for EBV vaccines to prevent multiple sclerosis. Front Neurol. 2022;13:887794. doi: 10.3389/fneur.2022.887794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Messick TE, Smith GR, Soldan SS, et al. Structure-based design of small-molecule inhibitors of EBNA1 DNA binding blocks Epstein-Barr virus latent infection and tumor growth. Sci Transl Med. 2019;11:482. doi: 10.1126/scitranslmed.aau5612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soldan SS, Anderson EM, Frase DM, et al. EBNA1 inhibitors have potent and selective antitumor activity in xenograft models of Epstein-Barr virus-associated gastric cancer. Gastric Cancer. 2021;24(5):1076-1088. doi: 10.1007/s10120-021-01193-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang L, Xie C, Lung HL, et al. EBNA1-targeted inhibitors: novel approaches for the treatment of Epstein-Barr virus-associated cancers. Theranostics. 2018;8(19):5307-5319. doi: 10.7150/thno.26823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee EK, Kim SY, Noh KW, et al. Small molecule inhibition of Epstein-Barr virus nuclear antigen-1 DNA binding activity interferes with replication and persistence of the viral genome. Antivir Res. 2014;104:73-83. doi: 10.1016/j.antiviral.2014.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frohman EM, Monaco MC, Remington G, et al. JC virus in CD34+ and CD19+ cells in patients with multiple sclerosis treated with natalizumab. JAMA Neurol. 2014;71(5):596-602. doi: 10.1001/jamaneurol.2014.63 [DOI] [PubMed] [Google Scholar]
- 32.Kubuschok B, Cochlovius C, Jung W, et al. Gene-modified spontaneous Epstein-Barr virus-transformed lymphoblastoid cell lines as autologous cancer vaccines: mutated p21 ras oncogene as a model. Cancer Gene Ther. 2000;7(9):1231-1240. doi: 10.1038/sj.cgt.7700236 [DOI] [PubMed] [Google Scholar]
- 33.Oh HM, Oh JM, Choi SC, et al. An efficient method for the rapid establishment of Epstein-Barr virus immortalization of human B lymphocytes. Cell Prolif. 2003;36(4):191-197. doi: 10.1046/j.1365-2184.2003.00276.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hindson BJ, Ness KD, Masquelier DA, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83(22):8604-8610. doi: 10.1021/ac202028g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin CT, Leibovitch EC, Almira-Suarez MI, Jacobson S. Human herpesvirus multiplex ddPCR detection in brain tissue from low- and high-grade astrocytoma cases and controls. Infect Agent Cancer. 2016;11:32. doi: 10.1186/s13027-016-0081-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ngouth N, Monaco MC, Walker L, et al. Comparison of qPCR with ddPCR for the quantification of JC polyomavirus in CSF from patients with progressive multifocal leukoencephalopathy. Viruses. 2022;14(6):1246. doi: 10.3390/v14061246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vellucci A, Leibovitch EC, Jacobson S. Using droplet digital PCR to detect coinfection of human herpesviruses 6A and 6B (HHV-6A and HHV-6B) in clinical samples. Methods Mol Biol. 2018;1768:99-109. doi: 10.1007/978-1-4939-7778-9_6 [DOI] [PubMed] [Google Scholar]
- 38.Messick TE, Tolvinski L, Zartler ER, et al. Biophysical screens identify fragments that bind to the viral DNA-binding proteins EBNA1 and LANA. Molecules. 2020;25(7). doi: 10.3390/molecules25071760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lamontagne RJ, Soldan SS, Su C, et al. A multi-omics approach to Epstein-Barr virus immortalization of B-cells reveals EBNA1 chromatin pioneering activities targeting nucleotide metabolism. PLoS Pathog. 2021;17(1):e1009208. doi: 10.1371/journal.ppat.1009208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tempera I, De Leo A, Kossenkov AV, et al. Identification of MEF2B, EBF1, and IL6R as Direct gene targets of Epstein-Barr virus (EBV) nuclear antigen 1 critical for EBV-infected B-lymphocyte survival. J Virol. 2016;90(1):345-355. doi: 10.1128/JVI.02318-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wagner HJ, Fischer L, Jabs WJ, Holbe M, Pethig K, Bucsky P. Longitudinal analysis of Epstein-Barr viral load in plasma and peripheral blood mononuclear cells of transplanted patients by real-time polymerase chain reaction. Transplantation. 2002;74(5):656-664. doi: 10.1097/00007890-200209150-00012 [DOI] [PubMed] [Google Scholar]
- 42.Soldan SS, Lieberman PM. Epstein-Barr virus and multiple sclerosis. Nat Rev Microbiol. 2022. doi: 10.1038/s41579-022-00770-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cocuzza CE, Piazza F, Musumeci R, et al. Quantitative detection of Epstein-Barr virus DNA in cerebrospinal fluid and blood samples of patients with relapsing-remitting multiple sclerosis. PLoS One. 2014;9(4):e94497. doi: 10.1371/journal.pone.0094497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ford JL, Stowe RP. Depressive symptoms are associated with salivary shedding of Epstein-Barr virus in female adolescents: the role of sex differences. Psychoneuroendocrinology. 2017;86:128-133. doi: 10.1016/j.psyneuen.2017.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamanashi H, Akabame S, Miyata J, et al. Association between Epstein-Barr virus serological reactivation and psychological distress: a cross-sectional study of Japanese community-dwelling older adults. Aging (Albany NY). 2022;14(20):8258-8269. doi: 10.18632/aging.204345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Munch M, Moller-Larsen A, Christensen T, Morling N, Hansen HJ, Haahr S. B-lymphoblastoid cell lines from multiple sclerosis patients and a healthy control producing a putative new human retrovirus and Epstein-Barr virus. Mult Scler. 1995;1(2):78-81. doi: 10.1177/135245859500100204 [DOI] [PubMed] [Google Scholar]
- 47.Christensen T, Tonjes RR, zur Megede J, Boller K, Moller-Larsen A. Reverse transcriptase activity and particle production in B lymphoblastoid cell lines established from lymphocytes of patients with multiple sclerosis. AIDS Res Hum Retroviruses. 1999;15(3):285-291. doi: 10.1089/088922299311466 [DOI] [PubMed] [Google Scholar]
- 48.Jiang L, Lung HL, Huang T, et al. Reactivation of Epstein-Barr virus by a dual-responsive fluorescent EBNA1-targeting agent with Zn(2+)-chelating function. Proc Natl Acad Sci USA. 2019;116(52):26614-26624. doi: 10.1073/pnas.1915372116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Farrell RA, Antony D, Wall GR, et al. Humoral immune response to EBV in multiple sclerosis is associated with disease activity on MRI. Neurology. 2009;73(1):32-38. doi: 10.1212/WNL.0b013e3181aa29fe [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Holden DW, Gold J, Hawkes CH, et al. Epstein Barr virus shedding in multiple sclerosis: similar frequencies of EBV in saliva across separate patient cohorts. Mult Scler Relat Disord. 2018;25:197-199. doi: 10.1016/j.msard.2018.07.041 [DOI] [PubMed] [Google Scholar]
- 51.Pender MP, Csurhes PA, Smith C, et al. Epstein-Barr virus-specific T cell therapy for progressive multiple sclerosis. JCI Insight. 2018;3(22). doi: 10.1172/jci.insight.124714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lehky TJ, Levin MC, Kubota R, et al. Reduction in HTLV-I proviral load and spontaneous lymphoproliferation in HTLV-I-associated myelopathy/tropical spastic paraparesis patients treated with humanized anti-Tac. Ann Neurol. 1998;44(6):942-947. doi: 10.1002/ana.410440613 [DOI] [PubMed] [Google Scholar]
- 53.Zhang J, Hutton G, Zang Y. A comparison of the mechanisms of action of interferon beta and glatiramer acetate in the treatment of multiple sclerosis. Clin Ther. 2002;24(12):1998-2021. doi: 10.1016/s0149-2918(02)80094-7 [DOI] [PubMed] [Google Scholar]
- 54.Hauer L, Sellner J. Diroximel fumarate as a novel oral immunomodulating therapy for relapsing forms of multiple sclerosis: a review on the emerging data. Drug Des Devel Ther. 2022;16:3915-3927. doi: 10.2147/DDDT.S236926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li L, Liu J, Delohery T, Zhang D, Arendt C, Jones C. The effects of teriflunomide on lymphocyte subpopulations in human peripheral blood mononuclear cells in vitro. J Neuroimmunol. 2013;265(1-2):82-90. doi: 10.1016/j.jneuroim.2013.10.003 [DOI] [PubMed] [Google Scholar]
- 56.Lanz TV, Brewer RC, Ho PP, et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature 2022;603(7900):321-327. doi: 10.1038/s41586-022-04432-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data related to this article is provided in the main text or supplemental data.