

Abstract
We showed previously that a single intranasal vaccination of mice with a recombinant vesicular stomatitis virus (VSV) expressing an influenza virus hemagglutinin (HA) protein provided complete protection from lethal challenge with influenza virus (A. Roberts, E. Kretzschmar, A. S. Perkins, J. Forman, R. Price, L. Buonocore, Y. Kawaoka, and J. K. Rose, J. Virol. 72:4704–4711, 1998). Because some pathogenesis was associated with the vector itself, in the present study we generated new VSV vectors expressing HA which are completely attenuated for pathogenesis in the mouse model. The first vector has a truncation of the cytoplasmic domain of the VSV G protein and expresses influenza virus HA (CT1-HA). This nonpathogenic vector provides complete protection from lethal influenza virus challenge after intranasal administration. A second vector with VSV G deleted and expressing HA (ΔG-HA) is also protective and nonpathogenic and has the advantage of not inducing neutralizing antibodies to the vector itself.
Vesicular stomatitis virus (VSV) is the prototype of the family Rhabdoviridae and infects a broad range of animals, including cattle, horses, and swine (12). VSV infection of livestock is associated with significant disease, including vesicular lesions around the mouth, hoofs, and teats, and with loss of beef and milk production (3, 38). Although rarely fatal, VSV infection mimics early symptoms caused by a notorious veterinary pathogen of horses and cattle, foot-and-mouth disease virus (38). VSV infection of livestock occurs periodically within the United States, is associated with one of two serotypes of VSV (Indiana, VSVI, or New Jersey, VSVNJ), and is usually not widespread. However, major VSV epizootics occur approximately every 10 to 15 years within the United States, with the last two having occurred in 1982–1983 and 1966 (29, 39). In 1997, 183 confirmed cases of VSVI were reported in four states (36). Arthropod vectors, such as phlebomite sandflies (8, 27) and Aedes mosquitoes (34), have been found to harbor VSVI in nature and likely participate in the spread of the virus from animal to animal and possibly from animals to humans.
Naturally occurring human infections with VSV are rare. However several cases of VSV infection have been reported for individuals directly exposed to infected livestock (6) and for researchers directly exposed within laboratory environments (12, 13, 25). Most VSV infections are thought to be asymptomatic in humans; however, febrile illness, including chills, myalgia, and nausea, has been reported in infected individuals (5, 10, 12, 13). A single case of encephalitis associated with VSVI infection in a 3-year-old boy was reported in 1988 (28).
The seroprevalence of VSV antibodies within the general human population is extremely low except in limited regions of enzooticity in Georgia (VSVNJ) (11) and Central America (VSVI and VSVNJ) (1). Seroprevalence is also moderately high in individuals with a high risk for exposure (i.e., laboratory workers who work with the virus directly and veterinarians and ranchers exposed to infected livestock). An example of high seroprevalence of VSV antibodies within a human population (94% seropositive) was reported for a single rural Panamanian community where VSV infection was enzootic in the surrounding wildlife (35). The surrounding rural Panamanian communities had seroprevalence rates ranging from 3 to 54% in adult lifetime residents, and urban Panamanian populations had no seroprevalence of VSV antibodies (35). The seroprevalence of VSV antibodies has also been examined in high-risk groups for VSV exposure during epizootics. A report on human infection during the 1965 epizootic included a study of 41 individuals at high risk for exposure to VSV (6). Eight of the 41 individuals (20%) had serological evidence of VSV infection. This and other studies (11, 29, 39) indicate limited seroprevalence of antibodies to VSV in the categories with the highest risk of exposure and demonstrate an absence of antibodies to VSV in the general population. The low VSV seropositivity in the general population and the lack of serious pathogenicity in humans are advantages in the potential use of recombinant VSV-vectored vaccines in humans.
In addition to the low seroprevalence of VSV antibodies in the general population, other characteristics of VSV suggest that recombinant VSVs expressing foreign viral glycoproteins would be very good vaccine candidates. VSV elicits strong humoral and cellular immune responses in vivo (3, 37, 41) and naturally infects at mucosal surfaces. Mucosal immunization is a less invasive route of immunization and has been shown to elicit both mucosal and systemic immunity (17, 21, 22). Additionally, recombinant VSVs are able to accommodate large inserts and multiple genes in their genomes. This ability to incorporate large gene inserts in replication-competent viruses offers advantages over other RNA virus vectors, such as those based on alphaviruses (2, 20, 40) and poliovirus (23). Furthermore, like alphaviruses, VSV grows to very high titers in vitro, and VSV infection shuts down host cell protein synthesis. This inhibition of host cell protein synthesis allows rapid identification of viral proteins in infected cell lysates and facilitates rapid purification of large amounts of virus and viral proteins.
VSV recombinants would have advantages over other live recombinant vaccine vectors as well. Compared to large, complex genomes of viruses from the Poxviridae family, the VSV genome is relatively simple, more fully understood, and easier to manipulate. VSV’s single-stranded RNA genome does not undergo reassortment and therefore lacks the potential of reassorting with wild-type viruses in vivo. Furthermore, VSV replicates within the cytoplasm of infected cells and does not undergo genetic recombination.
Previously we reported that intranasal vaccination of mice with recombinant VSV expressing influenza virus hemagglutinin (HA) protein (VSV-HA) was able to protect mice from lethal intranasal influenza virus challenge (30). Prior to challenge, vaccinated mice had high levels of serum neutralizing antibodies to influenza virus A/WSN. After challenge, the mice were completely protected from death, weight loss, pneumonia-associated pathology, and influenza virus replication in the lungs. Although the mice were completely protected from influenza virus challenge, there was pathogenesis associated with the initial VSV-HA vaccination. Initial pathogenesis was indicated by inactivity, ruffled coats, weight loss, and VSV-HA replication in the lungs (assayed 48 h after inoculation). The vector-associated pathogenesis remained a significant concern in the development of a nonpathogenic VSV-vectored vaccine. In addressing this concern, we reported the further attenuation of recombinant VSVs containing truncations within the cytoplasmic tail of the glycoprotein (VSV-CT1 and VSV-CT9) (30). Here we report on the complete attenuation of a VSV-CT1 recombinant expressing influenza virus HA (CT1-HA), its immunogenicity, and its efficacy in protecting mice from a lethal influenza virus challenge. Additionally, we report the construction, recovery, and characterization of a recombinant VSV containing a complete substitution of the VSV G protein with influenza virus HA (ΔG-HA). We also examine and report the absence of vector-associated pathogenesis and the immunogenicity and protective efficacy of recombinant ΔG-HA in the mouse model system.
MATERIALS AND METHODS
Construction of plasmids and recovery of recombinant viruses. (i) pBSIIKS(−)HA.
The HA gene (influenza virus A/WSN HA protein) was PCR amplified from pVSV-HA (16) with the following primers (restriction sites are underlined): XmaI HA(+), 5′-GAGCCCGGGAAAATGAAGGCAAAAC-3′ (containing an XmaI restriction enzyme sequence followed by 5′ sequence from the HA gene), and XhoI ssHA(−), 5′-CCACTCGAGATCGATCTCTG TTAGTTTTTTTCATACCTCAGATGCATATTCTGC-3′ (containing an XhoI restriction enzyme site followed by reverse complements of the VSV transcriptional START/STOP sequence and the 3′ HA gene sequence). The ∼1.7-kb HA PCR fragment was digested with restriction enzymes XmaI and XhoI, agarose gel purified, electroeluted, and ligated with T4 DNA ligase into pBSIIKS(−) vector (product no. 212208; Stratagene, La Jolla, Calif.) which had been digested with XmaI and XhoI. C600 competent bacteria were transformed with ligation reactions and plated on Luria-Bertani medium agar plates containing ampicillin at 100 μg/ml. Colonies were screened by PCR for the 1.7-kb HA insert. Positive colonies were expanded in 250- to 500-ml Luria-Bertani medium-ampicillin cultures, and plasmids (pBSIIKS(−)HA) were purified from these cultures with a Plasmid Maxi kit (product no. 12163; Qiagen Inc., Santa Clarita, Calif.). Expression of HA was confirmed by indirect-immunofluorescence screening of BHK cells transfected with pBSIIKS(−)HA and infected with vTF7-3, recombinant vaccinia virus expressing T7 polymerase (7).
(ii) Recombinant CT1-HA.
Plasmid pVSVCT-1XMN (31) (containing the VSV antigenome with an additional transcriptional STOP/START sequence and an XmaI restriction enzyme site downstream of the CT1-G) was digested with restriction enzymes XmaI and NotI and purified. The 1.7-kb HA fragment was PCR amplified from pBSIIKS(−)HA plasmid with primer XmaI HA(+) and primer NotI HA(−), 5′-CTCGCGGCCGCTCAGATGCATATTCTGC-3′ (containing a NotI restriction enzyme site and reverse complement sequence of the 3′ HA gene). The HA-PCR fragment was digested with restriction enzymes XmaI and NotI, purified, and ligated to the XmaI and NotI sites of the pVSVCT-1XMN vector. Plasmid pVSVCT1-HA was isolated and used in the standard VSV recovery system (19) to generate the recombinant CT1-HA virus.
(iii) Recombinant VSVΔG-HA.
Plasmid pVSVMXA2XN2 was digested with restriction enzymes XmaI and XhoI to generate a VSVΔG vector fragment which was purified from the excised G fragment. The HA gene, trailed by the VSV transcriptional STOP/START sequence, was excised from pBSIIKS(−)HA by digestion with restriction enzymes XmaI and XhoI, purified, and ligated to the VSVΔG vector fragment. A plasmid confirmed for the HA insert and VSV G deletion (pVSVΔG-HA) was used in a modified VSV recovery system previously described by Schnell, et al. (33) to generate the recombinant VSVΔG-HA virus.
(iv) Recombinant VSVΔG.
Plasmid pVSV-XMN was digested with restriction enzymes MluI and NheI to generate a VSVΔG vector fragment. The VSVΔG fragment was made blunt with T4 DNA polymerase in the presence of dATP, dCTP, dGTP, and dTTP and ligated with T4 DNA ligase. Transformations, PCR screenings, and expansions of positive colonies (those lacking VSV G) were performed as described above. A plasmid confirmed for the VSV G deletion (pVSVΔG) was used in a modified VSV recovery system previously described by Schnell et al. (33) to generate the recombinant VSVΔG virus.
Indirect immunofluorescence assays (IFA).
Immunofluorescence assays were performed on baby hamster kidney cells (BHK-21; American Type Culture Collection) or BHK-G cells (expressing VSV G [33]) as previously described (32). The cells were either transfected with pBSIIKS(−)HA plasmid (10 μg/plate) and infected with vTF7-3 (multiplicity of infection [MOI] = 10) or infected with supernatants from VSV recovery assays. The cells were fixed in 3% paraformaldehyde, washed in phosphate-buffered saline (PBS) and incubated with primary antibody (mouse monoclonal antibody 523/6 directed to influenza virus A/WSN HA protein; from Robert Webster) at 1:100 dilutions in PBS-glycine with 5 mg of bovine serum albumin/ml added. A second incubation was done in the presence of secondary antibody (fluorescein isothiocyanate-conjugated goat anti-mouse antibody) at 1:50 in PBS-glycine-bovine serum albumin.
Metabolic labeling of cells and recombinant viruses.
BHK or BHK-G cells (in 3.5-cm-diameter dishes) were infected with recombinant viruses (MOI = 1 to 10) and incubated at 37°C in Dulbecco’s modified Eagle medium (with l-glutamine, sodium pyruvate, high glucose, and high bicarbonate [3.7 g/liter]) (product no. 56 499; JRH Biosciences, Lenexa, Kans.) (DMEM) containing 5% fetal bovine serum (FBS) and 100 U of penicillin-streptomycin (PS)/ml for 1 h. The medium was aspirated and replaced with 1 ml of DMEM–5% FBS–PS, and the plates were incubated for an additional 3 h. The cells were washed twice with PBS, and 1 ml of labeling medium (10% DMEM, 90% DMEM minus methionine and cysteine, 100 μCi of 35S translabel [product no. NEG772, Easy Tag EXPRESS protein labeling mix; NEN Life Sciences, Boston, Mass.]) was added to each plate. For radiolabeled cell extracts, the cells were incubated for 1 h, washed twice in PBS, and lysed in 500 μl of ice-cold detergent lysis buffer (1% Nonidet P-40, 0.4% deoxycholate, 66 mM EDTA, and 10 mM Tris-Cl, pH 7.4) for 30 min on ice. The lysates were transferred to cold 1.5-ml Eppendorf tubes and centrifuged at 16,000 × g for 2 min at 4°C. The supernatants were transferred to fresh, cold 1.5-ml Eppendorf tubes and stored at −20°C. For radiolabeled viruses, the cells were incubated at 37°C overnight. The supernatants, containing radiolabeled virus, were collected in 15-ml conical tubes and clarified by spinning at 1,250 × g for 10 min at room temperature. The clarified supernatants were layered onto 4 ml of 20% sucrose in Beckman polyallomer centrifuge tubes (13 by 51 mm) and centrifuged at 38,000 rpm for 1 h at 4°C in a Beckman SW50.1 rotor in a Beckman L8-M ultracentrifuge. The pellets were resuspended in 250 to 500 μl of Tris-EDTA, pH 7.6, and stored at −20°C until analyzed. Sample buffer containing 2-mercaptoethanol was added to the purified viruses to a 1× final concentration, and samples were boiled for 3 to 5 min before being loaded onto sodium dodecyl sulfate (SDS)-polyacrylamide gels (10% acrylamide) for analysis.
Radiolabeled immunoprecipitation assays.
Primary antibody was added to 200 to 300 μl of radiolabeled cell extracts at 1:100 dilutions, and SDS was added to 0.2% final concentration. The antibody-extract solutions were incubated at 37°C for 45 min. Samples were precipitated by adding 30 μl of fixed Staphylococcus aureus (Pansorbin; Calbiochem) and incubating the mixture at 37°C for 30 min. Precipitates were pelleted by centrifugation, washed in 1.5 ml of RIPA buffer (1% Nonidet P-40, 1% deoxycholate, 0.1% SDS, 150 mM NaCl, and 10 mM Tris-Cl, pH 7.4) three times, and pelleted again. The pellets were resuspended in 30 to 40 μl of sample buffer containing 2-mercaptoethanol, boiled for 3 to 5 min, and pelleted again. The supernatants were carefully removed and loaded onto SDS–10% polyacrylamide gels.
Viruses and inoculum.
Recombinant viruses were grown and titers were determined as previously described for VSV-HA (30). The plaque-purified recombinant CT1-HA was grown and its titers were determined on BHK cells, and the plaque-purified recombinants VSVΔG-HA and VSVΔG were grown on BHK-G cells (33) and passaged once through BHK cells, and titers were determined on BHK-G cells. All recombinants were thawed and diluted with DMEM to appropriate titers immediately prior to inoculation. Influenza virus A/WSN/33 (H1N1) (1.4 × 106 PFU/ml), used for challenges and neutralization assays, was grown in MDBK cells in DMEM–10% FBS–PS, and titers were determined by standard plaque assay on MDBK cells with 1% agarose–1× DMEM (serum free)–2 μg · ml−1 acetylated trypsin (product no. T-6763; Sigma, St. Louis, Mo.) overlays. The WSN virus was thawed immediately prior to challenge, diluted 1:10 in DMEM, and administered in a 50-μl total volume (∼7 × 104 PFU/mouse). Viruses or DMEM were administered intranasally as described below.
Inoculation of mice.
Five- to 6-week-old female BALB/c mice from Charles River Laboratories were housed in filter-isolette cages upon arrival. The mice were inoculated no earlier than 4 days after arrival. Prior to inoculation (day 0), the mice were lightly anesthetized with Methoxyflurane (Mallinckrodt Veterinary, Inc., Mundelein, Ill.) and marked by ear punch. Twenty-five microliters of inoculum were delivered intranasally by 200-μl pipette to the anesthetized mice, and the mice were weighed in a plastic beaker to ±0.02 g. Boosts were administered in an identical fashion with viruses of equal kind, titer, and volume on day 21, unless otherwise indicated. Challenge was also administered as described but in a total volume of 50 μl per mouse on day 35, unless otherwise indicated. The mice were observed and weighed unanesthetized on a daily basis.
Neutralization assays.
Blood samples were collected from anesthetized mice by retro-orbital bleeds and allowed to clot at room temperature. The clots were removed, and samples were centrifuged in a TOMY MTX-150 centrifuge (TMA-11 fixed-angle rotor) at 4°C for 10 to 15 min at 5,500 rpm. The clarified sera were transferred to sterile Eppendorf tubes and heat inactivated at 56°C for 45 to 60 min. The heat-inactivated sera were diluted with PBS in serial twofold dilutions in 96-well plates. Generally, 50 μl of serum was mixed with 50 μl of PBS and 50 μl was transferred for serial dilutions. For analysis of titers of neutralizing antibodies to influenza virus, equal volumes (50 μl) of influenza virus A/WSN (∼200 PFU) were then added to the diluted sera and mixed. The 96-well plates containing virus and sera were incubated at 37°C for 30 to 45 min. Approximately 2,000 MDBK cells in DMEM–20% FBS–PS (100-μl total volume) were added to each well. The plates were incubated at 37°C, 5% CO2, for 2 to 3 days. In each assay, the sera were diluted and analyzed in duplicate, and each assay was repeated at least once. Neutralization titers are those dilutions which correspond to complete inhibition of virus-associated cytopathic effect (CPE). Neutralization titers are reported as averages from all assays. For neutralization to VSV, the assays were identical except ∼200 PFU of recovered wild-type VSV (VSVrwt) was added to each well instead of influenza virus A/WSN and ∼200 to 500 BHK cells in DMEM–10% FBS–PS were added to each well instead of MDBK cells.
Recovery of viruses from tissue.
Two days after inoculation or 2 days after challenge, two mice from each group were heavily anesthetized. Blood was collected via cardiac puncture and transferred into K2EDTA-treated 2-ml Microcontainer tubes (product no. 365974; Becton Dickinson, Franklin Lakes, N.J.) and stored at 4°C. The lungs were excised bilaterally, rinsed externally in sterile PBS, and placed in sterile, preweighed 2-ml Nunc cryotube vials. The cryotube vials were immediately dropped in liquid nitrogen and later transferred to −80°C storage. Plasma samples were prepared by centrifugation as described earlier for serum samples. The lungs were thawed, suspended in 9 volumes (wt/vol) of DMEM–2.5% FBS–PS, and homogenized in Dounce homogenizers. To assay for recoverable virus from initial infections with recombinant viruses, BHK or BHK-G cells ∼80% confluent in 6-well plates were infected with 40 μl of plasma or 200 μl of the 10% lung suspensions per well, incubated for 1 h at room temperature, and washed in PBS. Two milliliters of DMEM–5% FBS–PS was added to each well, and the plates were incubated at 37°C and observed daily for 3 days for virus-associated CPE. The procedure was the same for assaying for recoverable virus from challenge except MDBK cells were used instead of BHK or BHK-G cells and DMEM–10% FBS–PS was added after the PBS wash. The remainder of the 10% lung suspensions were stored at −80°C for further analysis.
The lung suspensions were thawed and serially diluted in DMEM, and viral titers were determined by standard plaque assays. Plaque assays were done in duplicate on BHK cells with a 1% methyl cellulose–1× DMEM (5% FBS–PS) overlay or on MDBK cells with 1% agarose–1× DMEM (serum free)–2 μg · ml−1 acetylated trypsin overlays. The plates were incubated at 37°C, and plaque formation was observed and counted 2 to 3 days later.
RESULTS
To determine if attenuated VSV vectors could be employed for vaccine applications, we generated three new VSV recombinants. The first, designated VSVCT1-HA (CT1-HA), encodes a VSV G protein with 28 of its 29 cytoplasmic amino acids deleted (31) and expresses an additional protein, the influenza virus A/WSN HA (WSN HA), between the truncated G gene and the L gene (Fig. 1). Because the VSV-CT1 mutant is greatly attenuated (30, 31), we anticipated attenuation of the CT1-HA recombinant also. A second recombinant, designated VSVΔG-HA (ΔG-HA), has a complete deletion of the VSV G gene and expresses the WSN HA gene from the site of the G deletion between the VSV M and L genes (Fig. 1). We anticipated that this virus would not be able to propagate in culture without VSV G supplied in trans. A VSVΔG (ΔG) virus with VSV G deleted was prepared as a control. Our standard recovery system was used to make the first recombinant (19), and a modified recovery system in which VSV G protein is provided in trans was used to recover the two recombinants with VSV G deleted (33).
FIG. 1.

Recombinant VSV-influenza virus A/WSN constructs. A schematic representation of recombinant viruses indicating gene order is shown 3′ to 5′ on the negative-strand genomic RNA. Each intergenic region contains a transcriptional STOP/START signal recognized by the VSV polymerase (32). Restriction enzyme sequences used for constructing cDNAs of the recombinants are also shown.
Growth characteristics of the CT1 and ΔG recombinants.
Upon recovery of recombinant CT1-HA, ΔG-HA, and ΔG viruses, we examined the growth of each virus in culture. CT1-HA, containing a truncated VSV G protein, could be grown and passaged on BHK cells, but both ΔG-HA and ΔG viruses had to be maintained on BHK-G cells induced for G expression (33). Infectivity of ΔG-HA and ΔG viruses was lost after two low-multiplicity passages through BHK cells. However, it is possible to passage ΔG-HA and ΔG recombinants through BHK cells once and recover a low titer of infectious virus from the supernatant. Although the levels of G protein in the first-passage virus were below the limits of detection in IFA, radiolabeled immunoprecipitation assays, and SDS-polyacrylamide gel electrophoresis (PAGE) analysis of radiolabeled cell extracts and purified viruses, these recombinants must contain a residual amount of G protein, since neutralization of the infectivity was accomplished with antibody to VSV G protein.
Recombinant CT1-HA, like VSV-CT1 (31), grew to titers of 107 PFU/ml on BHK cells. In contrast, VSV-HA grew to titers of 109 PFU/ml on BHK cells, comparable to the titers obtained with VSVrwt. Recombinant CT1-HA made plaques on BHK cells much smaller than those of VSV-HA and VSVrwt. ΔG-HA and ΔG recombinants that were grown and had their titers determined on BHK-G cells reached titers comparable to CT1-HA (107 PFU/ml). These ΔG recombinants did not form plaques on BHK cells; however, they did form large plaques on BHK-G cells. Single-cycle growth kinetics were examined for all three recombinants (CT1-HA, ΔG-HA, and ΔG), and all three showed similar growth kinetics, reaching peak titers at ∼8 h after infection. Recombinant ΔG-HA and ΔG, passaged once on BHK cells, were also assayed on BHK-G cells; the growth kinetics remained the same, but the peak titers were reduced 1,000-fold.
CT1-HA and ΔG-HA recombinants express HA.
Recombinant viruses were examined initially for protein expression in infected cells by IFA. BHK cells infected with recombinant CT1-HA or ΔG-HA expressed HA at the cell surface as detected by IFA with either polyclonal rabbit serum raised to influenza virus A/WSN (16) or mouse monoclonal antibody raised to WSN HA. BHK cells infected with recombinant ΔG expressed VSV N but lacked HA expression as expected. BHK cells infected with ΔG-HA and ΔG lacked detectable VSV G at the cell surface, while VSV G was readily detected on CT1-HA-infected cells (data not shown).
Protein expression from the recombinant viruses was also examined by SDS-PAGE analysis of radiolabeled, infected cell lysates. BHK cells were infected with VSVrwt, VSV-HA, or CT1-HA, and BHK-G cells were infected with recombinant ΔG-HA or ΔG. The cells were infected at high multiplicities for 1 h and metabolically labeled with [35S]methionine and [35S]cysteine for 3 to 4 h, proteins were immunoprecipitated from cell lysates with mouse monoclonal antibodies to VSV G or WSN HA protein, and the precipitates were analyzed by SDS-PAGE (Fig. 2). When immunoprecipitated with αHA monoclonal antibody, recombinants containing the HA gene showed a characteristic band at ∼85 kDa relative to molecular mass markers which was absent in recombinants lacking HA (Fig. 2A). VSVrwt and VSV-HA, immunoprecipitated with αG monoclonal antibodies, showed a characteristic band at ∼65 kDa for full-length glycoprotein (Fig. 2A, lane 1, and B). A faster-migrating band, which corresponds to the truncated form of the glycoprotein, was detected in the CT1-HA lane (Fig. 2B, lane 7). Precipitation of lysates from ΔG-HA- or ΔG-infected cells with αG monoclonal antibodies illustrated the absence of VSV G (Fig. 2B, lanes 3 to 4).
FIG. 2.

Immunoprecipitations of proteins from 35S-labeled cell extracts. Cells infected with recombinant VSVs were metabolically labeled, lysed, and immunoprecipitated with mouse monoclonal antibodies specific for influenza virus A/WSN HA protein (A, lanes 2 to 8) or for VSV G protein (A, lane 1, and B). VSVΔG and VSVΔG-HA were from BHK-G cell extracts; all other samples were from BHK cell extracts. VSVrwt and VSV-HA recombinants were run on all gels for comparison of immunoprecipitated proteins. Mock, immunoprecipitations of uninfected BHK cell lysates.
Recombinant ΔG-HA incorporates large amounts of HA protein into virions.
Previous studies showed that the influenza virus HA protein, the major influenza virus spike glycoprotein in virions, was incorporated into virions of VSV-HA, although at lower efficiency than with VSV G (16). To determine if the G truncation or the absence of VSV G affected the relative amount of HA incorporation into VSV virions, cells infected with VSV-HA, CT1-HA, ΔG-HA, or ΔG were labeled with [35S]methionine and [35S]cysteine overnight. The cell supernatants were collected when all cells showed signs of virus-associated CPE. Viruses were purified from these supernatants, and the radiolabeled proteins were analyzed by SDS-PAGE. Signal intensities of nucleocapsid and phosphoprotein (N-P) were quantitated for each sample on a phosphorimager after background subtraction, and a second gel was run with volumes of each sample which were normalized for the content of N-P. The phosphorimage of this second SDS-PAGE gel is shown in Fig. 3.
FIG. 3.
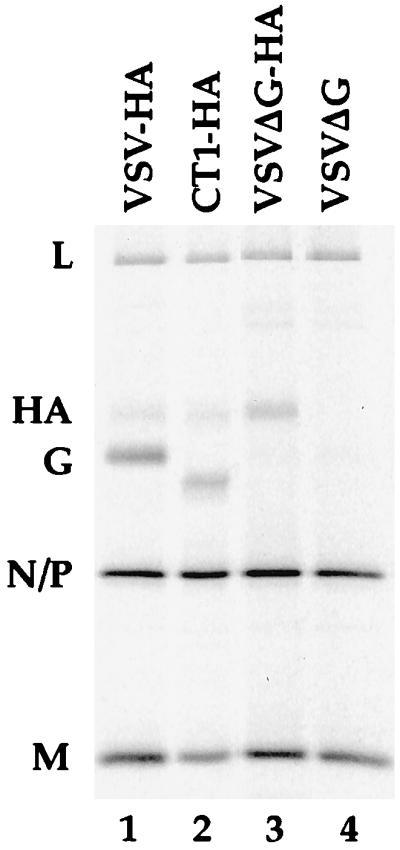
Sucrose-purified recombinant virions. Cells infected with recombinant viruses were metabolically labeled with 35S translabel. Supernatants were collected, and viruses were purified by centrifugation through 20% sucrose. The volumes loaded were normalized for N-P protein content. Lane 1, 1% of total VSV-HA virus from a 3.5-cm-diameter dish of BHK cells; lane 2, 18% of total CT1-HA virus from a 3.5-cm-diameter dish of BHK cells, lane 3, 14% of total VSVΔG-HA virus from a 3.5-cm-diameter dish of BHK-G cells; lane 4, 13% of total VSVΔG virus from a 3.5-cm-diameter dish of BHK-G cells.
The incorporation of HA into virions is seen for all HA recombinants (Fig. 3, lanes 1 to 3), while the HA band is absent from the ΔG recombinant that does not express HA (Fig. 3, lane 4). The faster mobility of the truncated VSV G in the CT1-HA recombinant is also evident (Fig. 3, lane 2), and absence of VSV G is clear for both ΔG-HA and ΔG recombinants (Fig. 3, lanes 3 to 4). Furthermore, it is evident that HA is present in greater amounts relative to N-P in the ΔG-HA recombinant than in either the VSV-HA or CT1-HA recombinant.
The relative amounts of HA and/or G protein incorporated into purified virions was quantified and is shown in Table 1. The protein signals were corrected for methionine and cysteine content, and background radioactivity was subtracted from each sample so that relative molarities could be calculated. As is clear from inspection of the SDS-PAGE gel, the amount of HA incorporated into the ΔG-HA recombinant (relative to N-P) is at least twofold greater than the amount of HA incorporated into either CT1-HA or VSV-HA. The increased incorporation of HA in ΔG-HA virions is obvious even when samples have not been normalized for N-P content. This difference could be due in part to increased synthesis of HA following deletion of G, but it might also result from additional space in the virion membrane in the absence of VSV G protein.
TABLE 1.
Protein content of purified recombinant VSVs
| Recombinant | Relative molarity
|
||
|---|---|---|---|
| N-Pa | HA | G | |
| VSV-HA | 100 | 4.4 | 34 |
| CT1-HA | 100 | 3.5 | 15 |
| ΔG-HA | 100 | 8.8 | 0 |
| ΔG | 100 | 0 | 0 |
N-P values were set at 100.
HA is found in the virion envelope.
To demonstrate that HA was present specifically in the virion envelope and to examine the morphology of the recombinant virions lacking VSV G protein, purified VSVrwt and ΔG-HA were adsorbed onto carbon-coated grids and incubated with αHA monoclonal antibody followed by labeling with gold-conjugated goat anti-mouse immunoglobulin G as described precisely by Schnell et al. (33). The grids were negatively stained with 2% phosphotungstic acid and examined on a Zeiss EM910 electron microscope. Strong labeling of HA proteins on the virion surface of recombinant ΔG-HA was clear in greater than 70% of virions, and all virions had the characteristic bullet shape expected for VSV (Fig. 4B). Although the background labeling is high, it is clear that no label is specifically associated with the surface of VSVrwt in the control (Fig. 4A).
FIG. 4.

Immunogold labeling of sucrose-purified recombinants. Electron micrographs of recombinant VSVrwt (A) and VSVΔG-HA (B) bound to carbon-coated grids, labeled with mouse monoclonal antibody to HA and secondary gold-conjugated antibody, and negatively stained.
Recombinants CT1-HA and ΔG-HA are attenuated in mice.
Once we had demonstrated the incorporation of HA into the recombinant CT1-HA and ΔG-HA virions, we examined their potential as attenuated vaccines for protection from influenza virus in a mouse model system. Six-week-old female BALB/c mice were inoculated intranasally with recombinant viruses at 5 × 104 PFU/mouse. The mice were observed and weighed daily, and weight loss was used as an indication of pathogenesis as previously reported (30). The mice inoculated with recombinant VSV-HA began losing body weight the day after inoculation, as seen previously (30). These mice lost 17% of their initial body weight before they began regaining weight (Fig. 5A). Weight loss corresponded with decreased activity and poor grooming, all of which indicate vector-associated pathogenesis. In contrast, the mice receiving CT1-HA, ΔG-HA, or ΔG recombinants did not lose significant amounts of weight (Fig. 5) compared to mice receiving medium alone. Also, no change in activity or grooming was observed for these mice in comparison to mice receiving medium alone. The lack of significant weight loss and the lack of decreased activity and grooming indicate that the CT1-HA and ΔG-HA vectors are attenuated in the mouse model. The mice were inoculated a second time with identical titers of virus 21 days after the initial inoculation. Weights, activity, and grooming were unaffected in all groups in the days following the second inoculation.
FIG. 5.

Attenuated recombinants provide protection from lethal influenza virus challenge. Average daily weights of mice inoculated with VSV recombinants and challenged with influenza virus A/WSN are shown. (A) Mice were inoculated intranasally with VSV-HA (n = 5) or CT1-HA (n = 5) at 5 × 104 PFU/mouse on day 0. The mice were boosted with an equal amount of inoculum on day 21. The mice were challenged with a lethal dose of influenza virus A/WSN on day 35. (B) Conditions were the same as for panel A, except the mice were inoculated and boosted with VSVΔG (n = 4) or VSVΔG-HA (n = 5) at 5 × 104 PFU/mouse. The error bars indicate ±0.5 × standard deviation.
Vector-associated pathogenesis may be associated with vector replication within the host. Therefore, in addition to monitoring weight loss, we sacrificed two mice from each group at 6, 12, 24, and 48 h after initial inoculation, and their blood and lungs were screened for recombinant virus. Virus was recovered from the lungs of mice inoculated with VSV-HA at all times assayed, whereas viremia (1.9 × 103 PFU/ml) was only detected at 24 h after inoculation. No virus was recovered from mice inoculated with CT1-HA from either the lungs or plasma at any time assayed. However virus was detected in 10% lung suspensions of one ΔG-HA-inoculated mouse at 6 h after inoculation and in 10% lung suspensions of both ΔG-HA-inoculated mice at 12 h after inoculation. No virus was detected in the lungs at 24 and 48 h after inoculation or in any plasma sample from ΔG-HA-inoculated mice. Viral titers recovered from the lungs of VSV-HA- and ΔG-HA-inoculated mice are summarized in Table 2.
TABLE 2.
Recombinant virus recovered from mouse lungs
| Inoculum | Virus (PFU/g) recovered at time postinoculation (h)
|
|||
|---|---|---|---|---|
| 6 | 12 | 24 | 48 | |
| VSV-HA | 1.9 × 104 | 2.3 × 105 | 1.2 × 106 | 3.1 × 105 |
| CT1-HA | 0a | 0a | 0a | 0a |
| ΔG-HA | 7.5 × 101 | 5.3 × 102 | 0a | 0a |
Sensitivity, 50 PFU/g.
Recombinant viruses are immunogenic in mice.
To examine the immunogenicity of the recombinants, mice were bled prior to initial inoculations, second inoculations, and challenges, and surviving mice were bled after challenge. Serum samples were pooled, unless otherwise indicated, and screened for neutralizing antibodies to VSV and influenza virus A/WSN. Serum neutralizing antibody titers correspond to titers which provided complete inhibition of virus-associated CPE when virus was preincubated with serum before the addition of susceptible cells (Table 3). All sera collected before inoculation had no titers of antibody to either VSV or influenza virus. Eighteen days after initial inoculation, mice receiving VSV-HA and CT1-HA had titers of neutralizing antibody to both VSVrwt and influenza virus in the serum. Mice inoculated with ΔG-HA and ΔG had no measurable titers of neutralizing antibody to either VSVrwt or influenza virus. Thirty-two days after the initial inoculation, which was also after a second inoculation (boost), mice receiving the ΔG-HA recombinant had low-level titers of neutralizing antibody to influenza virus. No titers of antibody to VSVrwt were detected in ΔG-HA- or ΔG-inoculated mice. No titers of antibody to influenza virus were detected in ΔG-inoculated mice. Titers of neutralizing antibody to influenza virus in the sera of mice surviving challenge reached 1:4,096.
TABLE 3.
Immunogenicity of recombinant VSVs
| Inoculuma | Serum neutralizing antibody titer to:
|
|||
|---|---|---|---|---|
| influenza virus A/WSN
|
VSV Day 47d
|
|||
| Day 18b | Day 32c | Day 47d | ||
| VSV-HA | 1:512 | 1:2,048 | 1:4,096 | 1:512 |
| CT1-HA | 1:32 | 1:128 | 1:2,048 | 1:512 |
| ΔG-HA | ≤1:8 | 1:16 | 1:1,024 | ≤1:8 |
| ΔG | ≤1:8 | ≤1:8 | ≤1:8c | |
Dose, 5 × 104 PFU.
Prior to boost.
Prior to challenge.
After challenge.
Efficacy of recombinants in generating protective immunity.
In an initial experiment to examine the efficacy of recombinant VSVs expressing influenza virus HA in protecting mice from a lethal influenza virus challenge, mice were inoculated with recombinant virus at 5 × 104 PFU/mouse. The mice were challenged 21 days after this single, initial immunizing inoculation. Fifty microliters of a 1:2 dilution of influenza virus A/WSN in DMEM (∼3.5 × 105 PFU/mouse) was administered for challenge. Mice which had been inoculated with CT1-HA or VSV-HA, which also had titers of neutralizing antibody to influenza virus A/WSN in the serum 18 days after inoculation, were completely protected from lethal challenge as indicated by 100% survival. Mice inoculated with the ΔG-HA recombinant, which lacked titers of neutralizing antibody in the serum at day 18 after initial inoculation, were partially protected from lethal challenge; only two of five mice survived.
In an experiment run simultaneously with the initial experiment, mice were initially inoculated on day zero and boosted on day 21 with recombinant VSVs (5 × 104 PFU/mouse/inoculation). These mice were challenged on day 35 with 50 μl of a 1:10 dilution of influenza virus A/WSN (∼7 × 104 PFU/mouse). The lethal dose of the challenge virus for mice was not determined, but 50-μl doses of 1:50 dilutions (1.4 × 104 PFU/mouse) resulted in 100% lethality in unprotected mice; thus the challenge is at least five 100% lethal doses. Mice which had been inoculated with recombinants containing HA were completely protected from pathogenesis as indicated by negligible weight loss after challenge (Fig. 5), no change in activity or grooming, and 100% survival. Control mice that had been inoculated with ΔG were not protected and died within 7 days after challenge (Fig. 5B). These mice were increasingly inactive and poorly groomed in the days preceding death.
In addition to monitoring weight loss, we sacrificed two mice from each recombinant group in the second set of experiments 2 days after challenge, and their blood and lungs were screened for influenza virus to indicate the extent of protection from influenza virus challenge. Virus was recovered from the lungs of both mice inoculated with ΔG and ΔG-HA at average titers of 1.5 × 106 and 2.5 × 104 PFU/g, respectively, but not from the lungs of CT1-HA- or VSV-HA-inoculated mice. No virus was found in any plasma sample, indicating both a lack of measurable viremia 2 days after challenge and that virus recovered from lung samples was not from blood trapped in pulmonary tissues. Although influenza virus A/WSN was recovered from the lungs of ΔG-HA-inoculated mice after challenge, there was a 2 log unit reduction in virus titers compared to those of unprotected ΔG-inoculated mice. The low level of serum neutralizing antibodies in the ΔG-HA-inoculated mice preceding challenge was obviously not high enough to provide sterilizing immunity to the challenge virus. However, the ΔG-HA-inoculated mice were provided enough protection to avoid death and the external signs of pathogenesis, including weight loss, decreased activity, and poor grooming, preceding death.
Efficacy of boost.
Because the preceding experiments did not examine the direct efficacy of a second inoculating dose, a third set of experiments was conducted. Three groups of eight mice each were inoculated with VSV-HA, CT1-HA, or ΔG-HA (104 PFU/mouse). Eighteen days after inoculation sera were collected and assayed for titers of neutralizing antibody to influenza virus A/WSN. On day 21 after initial inoculation four mice from each group were inoculated with DMEM and the other four mice from each group were boosted with an equal titer of initial inoculum. Sera were collected on day 32 and assayed for neutralizing antibodies to influenza virus A/WSN. The serum neutralizing antibody titers are reported in Table 4 and indicate that boosting offers no significant advantage over single inoculations for CT1-HA or VSV-HA immunizations. Antibodies raised to the G protein encoded by these vectors probably prevent infection and could explain the lack of boosting responses. In contrast, mice receiving two inoculations of the ΔG-HA virus had higher concentrations of serum neutralizing antibodies to influenza virus A/WSN than mice receiving a single inoculation at day 32. These data indicate that boosting is efficacious in the instance of ΔG-HA immunization, when the vector does not encode G protein.
TABLE 4.
Effect of a second (boosting) inoculation
| Inoculuma | Mouse no. | Serum neutralizing antibody titer to influenza virus A/WSN
|
||
|---|---|---|---|---|
| Day 18b | Day 32c | Day 32d | ||
| VSV-HA | 1 | 1:1,024 | 1:4,096 | |
| 2 | 1:256 | 1:1,024 | ||
| 3 | 1:512 | 1:2,048 | ||
| 4 | 1:128 | 1:2,048 | ||
| 5 | 1:1,024 | 1:4,096 | ||
| 6 | 1:512 | 1:4,096 | ||
| 7 | 1:512 | 1:1,024 | ||
| 8 | 1:512 | 1:4,096 | ||
| CT1-HA | 1 | 1:32 | 1:128 | |
| 2 | 1:64 | 1:256 | ||
| 3 | 1:128 | 1:256 | ||
| 4 | 1:64 | 1:512 | ||
| 5 | 1:64 | 1:512 | ||
| 6 | 1:64 | 1:256 | ||
| 7 | 1:64 | 1:256 | ||
| 8 | 1:64 | 1:512 | ||
| ΔG-HA | 1 | ≤1:8 | 1:8 | |
| 2 | ≤1:8 | 1:256 | ||
| 3 | ≤1:8 | 1:256 | ||
| 4 | ≤1:8 | 1:32 | ||
| 5 | ≤1:8 | 1:8 | ||
| 6 | ≤1:8 | 1:8 | ||
| 7 | ≤1:8 | ≤1:8 | ||
| 8 | ≤1:8 | 1:16 | ||
| ΔG | 1–8 | ≤1:8 (8 mice) | ≤1:8 (4 mice) | ≤1:8 (4 mice) |
Dose, 104 PFU.
Following single inoculation.
Following single inoculation.
Following boost at day 21.
Attenuation of CT1-HA to confer systemic immunity on mice.
The ability of CT1-HA to provide complete protection from lethal challenge following a single inoculation in the absence of vector-associated pathogenesis led to further characterization of the vector’s immunogenicity. Thirty-six mice were divided into six groups of six mice each and were inoculated with 10, 100, or 1,000 PFU/mouse of either CT1-HA or VSV-HA. Sera were collected and screened 2 and 4 weeks after inoculation. No mice receiving 10 PFU of CT1-HA virus had neutralizing antibody titers to influenza virus A/WSN at 4 weeks postinoculation (Table 5). In contrast, mice receiving 10 PFU of VSV-HA had various levels of serum neutralizing antibodies to influenza virus A/WSN at 4 weeks postinoculation (Table 5). The neutralizing antibody titers of mice receiving 100 PFU of CT1-HA are comparable to those obtained in mice receiving 10 PFU of VSV-HA. This pattern of a 10-fold-greater amount of CT1-HA necessary to elicit a neutralizing antibody response similar to that generated by VSV-HA is consistent throughout the data (Table 5). This demonstrates that CT1-HA is attenuated in immunogenicity in the mouse model. Neutralizing titers at 2 weeks postinoculation were less than those at 4 weeks postinoculation for all mice and therefore are not shown.
TABLE 5.
Dosage of CT1-HA necessary to confer a measurable antibody response
| Inoculum | Serum neutralizing titer to influenza virus A/WSNa at dose of:
|
||
|---|---|---|---|
| 101 PFU/mouse | 102 PFU/mouse | 103 PFU/mouse | |
| CT1-HA | ≤1:8 (6) | ≤1:8 | 1:128 (3) |
| 1:16 | 1:256 (2) | ||
| 1:256 | 1:512 | ||
| 1:512 (2) | |||
| 1:2,048 | |||
| VSV-HA | ≤1:8 (2) | 1:32 | 1:1,024 |
| 1:64 | 1:64 | 1:2,048 (5) | |
| 1:128 (2) | 1:512 (2) | ||
| 1:2,048 | 1:1,024 | ||
| 1:2,048 | |||
Dilutions indicate the lowest titers for which complete inhibition of virus-associated CPE was observed. Sera were collected at 4 weeks postinoculation. The numbers in parentheses indicate the number of mice with a particular titer; no parentheses indicates the titer for a single mouse.
Long-term immunity in CT1-HA- and VSV-HA-inoculated mice.
The longevity of the humoral immune response induced by these vectors has also been examined. Mice inoculated with a single dose of VSV-HA or CT1-HA (104 PFU/mouse) have now been observed for 4 months after an initial intranasal inoculation. These mice still have high levels of serum neutralizing antibodies to influenza virus. CT1-HA-inoculated mice have neutralizing antibody titers ranging from 1:512 to 1:2,048, and VSV-HA-inoculated mice have neutralizing titers ranging from 1:1,024 to 1:4,096. These mice have not yet been challenged with influenza virus, but in all previous experiments these levels of antibody have correlated with 100% protection and little or no weight loss after challenge.
DISCUSSION
We report here the successful attenuation of VSV vectors (VSV-CT1 and VSVΔG) which produce no measurable signs of pathogenesis as shown by weight loss, inactivity, or poor grooming. The attenuated CT1-HA vector was not detected in the lungs or blood of mice examined at 6, 12, 24, and 48 h after inoculation and yet was able to elicit a strong humoral immune response after a single intranasal inoculation. In mice inoculated with the CT1-HA recombinant, humoral immunity is raised to both the VSV G protein and the foreign viral protein (HA). The humoral immune response is also systemic, since serum neutralizing antibody titers are present after a mucosal inoculation. Furthermore, this vector is 100% effective in providing complete protection from a subsequent lethal influenza virus challenge. No significant weight loss is measured in mice after challenge, no change in activity or grooming is observed, 100% of the mice survive challenge, and no influenza virus is recovered from lungs or blood 2 days after challenge.
Although complete neutralization (sterilizing immunity) of the challenge virus was not achieved in mice inoculated with ΔG-HA (low titers of influenza virus were recovered from pulmonary tissues 2 days after challenge), the mice were completely protected from weight loss and lethality associated with more severe pathogenesis when given two immunizing doses of ΔG-HA. A third immunizing inoculation with ΔG-HA virus may further boost the humoral immune response and provide sterilizing immunity to the challenge virus. This possibility will be examined in future assays.
The effectiveness of the VSVΔG-HA vector is presumably due to an ability to infect cells for at least one round of multiplication. We believe the infection is mediated primarily by residual VSV G protein derived from the BHK-G cell line, since neutralization of the ΔG-HA virus is accomplished in vitro with αG antibodies but not with αHA antibodies. We believe that a single round of infection is critical for protective immunity because mice inoculated intranasally with large doses of UV-inactivated VSV-HA did not produce neutralizing antibodies to VSV or to influenza virus and the mice were not protected from subsequent influenza virus challenge. Although the ΔG-HA recombinant was measurable at low levels in the lungs 6 to 12 h after inoculation, it was still highly attenuated, showing no sign of viremia (6 to 48 h after initial inoculation) and causing no measurable weight loss or observable change in activity after initial inoculations or boosts.
The VSVΔG vector also offers a unique attribute not offered by the VSV-CT1 vector. Immunization with VSVΔG recombinants indicates that VSVΔG is a reusable vector, since no neutralizing antibodies are directed to the vector itself while a specific antibody response is elicited to the foreign protein expressed from the vector. We have examined the reusable characteristic of the VSVΔG vector in more detail by taking mice previously inoculated with VSVΔG-HA, which have measurable titers of antibody to influenza virus but not to VSVrwt, and inoculating them with VSVrwt. These mice subsequently develop high titers of neutralizing antibody to VSV in the serum (data not shown) which correspond to production of antibodies to VSV G (14). We have also shown that mice which have first been inoculated with a VSV recombinant expressing VSV G (and have high VSV neutralizing antibody titers) can subsequently be inoculated with a recombinant lacking the VSVI G protein but expressing a G protein of another vesiculovirus (Chandipura virus) and are able to raise high titers of neutralizing antibody to Chandipura virus G (data not shown). Not only is the VSVΔG vector reusable, but an interesting observation is that mice previously exposed to the VSV core proteins but not to G protein do not lose as much weight when subsequently inoculated with constructs containing G protein. This may be due to the time of inoculation, since older mice may be less susceptible to pathogenesis than younger mice (4, 9, 15, 18, 26), or it may be due to partial protection elicited by cell-mediated immunity (24, 41). Cell-mediated immunity to these recombinant VSVs has not yet been examined in the mouse model.
We believe there is significant potential for VSV as a highly attenuated vaccine vector for the delivery of heterologous viral proteins. Vectors such as VSV-CT1 provide strong humoral responses without pathogenesis after single inoculations at doses as low as 103 PFU/mouse, and vectors such as VSVΔG, which are also highly attenuated, have potential as reusable vaccines within an individual for multiple immunizations against heterologous viruses or for viruses, such as influenza virus, which require annual reimmunization. These live attenuated recombinants have the advantage of immunizing with VSV-based vectors without the risks inherent in the heterologous virus(es) themselves. They can be developed rapidly in the native G, CT1, or ΔG backgrounds and can be delivered via the intranasal route, providing systemic immunity. It is likely that cell-mediated immunity and mucosal antibodies (not examined in this report) also provide protection in VSV-immunized mice, since VSV elicits a strong cell-mediated response (41) and since both immunizing inoculations and challenges may be administered through the mucosal, intranasal route. Furthermore, VSV vectors expressing foreign viral glycoproteins are likely to generate long-term immunity characteristic of natural infections. Development of the VSV vector system for vaccine delivery would complement other recombinant systems currently employed and other systems which are being developed.
ACKNOWLEDGMENTS
We thank Karl Haglund for technical assistance, helpful critiques, and editorial comments and Matthias Schnell for technical support. We thank JoAnn Falato for all her assistance. We thank Roxanne Swinsick and other members of the Yale Animal Resource Center, BCMM, as well as Deborah Caruso for their care and assistance with our mice.
This work was supported by NIH grant AI24345. Anjeanette Roberts is supported by a Cancer Research Institute Fellowship. John Forman was supported by a fellowship from Howard Hughes Medical Institute during this work.
REFERENCES
- 1.Cline B L. Ecological associations of vesicular stomatitis virus in rural Central America and Panama. Am J Trop Med Hyg. 1976;25:875–883. doi: 10.4269/ajtmh.1976.25.875. [DOI] [PubMed] [Google Scholar]
- 2.Davis N L, Brown K W, Johnston R E. A viral vaccine vector that expresses foreign genes in lymph nodes. J Virol. 1996;70:3781–3787. doi: 10.1128/jvi.70.6.3781-3787.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dietzschold B, Rupprecht C E, Fu Z F, Koprowski H. Rhabdoviruses. In: Fields B N, Knipe D M, Howley P M, editors. Fields virology. 3rd ed. Vol. 1. Philadelphia, Pa: Lippincott-Raven; 1996. pp. 1137–1159. [Google Scholar]
- 4.Eldadah A H, Nathanson N, Sarsitis R. Pathogenesis of West Nile Virus encephalitis in mice and rats. 1. Influence of age and species on mortality and infection. Am J Epidemiol. 1967;86:765–775. doi: 10.1093/oxfordjournals.aje.a120785. [DOI] [PubMed] [Google Scholar]
- 5.Fellowes O N, Dimopoullos G T, Callis J J. Isolation of vesicular stomatitis virus from an infected laboratory worker. Am J Vet Res. 1955;16:623–626. [PubMed] [Google Scholar]
- 6.Fields B N, Hawkins K. Human infection with the virus of vesicular stomatitis during an epizootic. N Engl J Med. 1967;277:989–994. doi: 10.1056/NEJM196711092771901. [DOI] [PubMed] [Google Scholar]
- 7.Fuerst T R, Niles E G, Studier F W, Moss B. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc Natl Acad Sci USA. 1986;83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galindo P, Srihongse S, de Rodaniche E, Grayson M A. An ecological survey for arboviruses in Almirante, Panama, 1959–1962. Am J Trop Med Hyg. 1966;15:385–400. doi: 10.4269/ajtmh.1966.15.385. [DOI] [PubMed] [Google Scholar]
- 9.Griffin D E, Mullinix J, Narayan O, Johnson R T. Age dependence of viral expression: comparative pathogenesis of two rodent-adapted strains of measles virus in mice. Infect Immun. 1974;9:690–695. doi: 10.1128/iai.9.4.690-695.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanson R P, Brandly C A. Epizootiology of vesicular stomatitis. Am J Public Health. 1957;47:205–209. doi: 10.2105/ajph.47.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanson R P, Karstad L. Further studies on enzootic vesicular stomatitis. Proc US Livestock Sanit Assoc. 1957;61:300–307. [Google Scholar]
- 12.Hanson R P, Rasmussen A F, Jr, Brandly C A, Brown J W. Human infection with the virus of vesicular stomatitis. J Lab Clin Med. 1950;36:754–758. [PubMed] [Google Scholar]
- 13.Johnson K M, Vogel J E, Peralta P H. Clinical and serological response to laboratory-acquired human infection by Indiana type vesicular stomatitis virus (VSV) Am J Trop Med Hyg. 1966;15:244–246. doi: 10.4269/ajtmh.1966.15.244. [DOI] [PubMed] [Google Scholar]
- 14.Kelley J M, Emerson S U, Wagner R R. The glycoprotein of vesicular stomatitis virus is the antigen that gives rise to and reacts with neutralizing antibody. J Virol. 1972;10:1231–1235. doi: 10.1128/jvi.10.6.1231-1235.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim G R, McKee K T., Jr Pathogenesis of Hantaan virus infection in suckling mice: clinical, virologic, and serologic observations. Am J Trop Med Hyg. 1985;34:388–395. doi: 10.4269/ajtmh.1985.34.388. [DOI] [PubMed] [Google Scholar]
- 16.Kretzschmar E, Buonocore L, Schnell M J, Rose J K. High-efficiency incorporation of functional influenza virus glycoproteins into recombinant vesicular stomatitis viruses. J Virol. 1997;71:5982–5989. doi: 10.1128/jvi.71.8.5982-5989.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuno-Sakai H, Kimura M, Ohta K, Shimojima R, Oh Y, Fukumi H. Developments in mucosal influenza virus vaccines. Vaccine. 1994;12:1303–1310. doi: 10.1016/s0264-410x(94)80056-6. [DOI] [PubMed] [Google Scholar]
- 18.Latham P S, Sepelak S B. Effect of macrophage source and activation on susceptibility in an age-dependent model of murine hepatitis caused by a phlebovirus, Punta Toro. Arch Virol. 1992;122:175–185. doi: 10.1007/BF01321126. [DOI] [PubMed] [Google Scholar]
- 19.Lawson N D, Stillman E A, Whitt M A, Rose J K. Recombinant vesicular stomatitis viruses from DNA. Proc Natl Acad Sci USA. 1995;92:4477–4481. doi: 10.1073/pnas.92.10.4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liljestrom P, Garoff H. A new generation of animal cell expression vectors based on the Semliki Forest virus replicon. Bio/Technology. 1991;9:1356–1361. doi: 10.1038/nbt1291-1356. [DOI] [PubMed] [Google Scholar]
- 21.Moldoveanu Z, Clements M L, Prince S J, Murphy B R, Mestecky J. Human immune responses to influenza virus vaccines administered by systemic or mucosal routes. Vaccine. 1995;13:1006–1012. doi: 10.1016/0264-410x(95)00016-t. [DOI] [PubMed] [Google Scholar]
- 22.Moldoveanu Z, Novak M, Huang W Q, Gilley R M, Staas J K, Schafer D, Compans R W, Mestecky J. Oral immunization with influenza virus in biodegradable microspheres. J Infect Dis. 1993;167:84–90. doi: 10.1093/infdis/167.1.84. [DOI] [PubMed] [Google Scholar]
- 23.Morrow C D, Porter D C, Ansardi D C, Moldoveanu Z, Fultz P N. New approaches for mucosal vaccines for AIDS: encapsidation and serial passages of poliovirus replicons that express HIV-1 proteins on infection. AIDS Res Hum Retroviruses. 1994;10:S61–S66. [PubMed] [Google Scholar]
- 24.Murphy B R, Webster R G. Orthomyxoviruses. In: Fields B N, Knipe D M, Howley P M, editors. Fields virology. 3rd ed. Vol. 1. Philadelphia, Pa: Lippincott-Raven; 1996. pp. 1397–1445. [Google Scholar]
- 25.Patterson W C, Mott L O, Jenney E W. A study of vesicular stomatitis in man. J Am Vet Med Assoc. 1958;133:57–62. [PubMed] [Google Scholar]
- 26.Pekosz A, Griot C, Stillmock K, Nathanson N, Gonzalez-Scarano F. Protection from La Crosse virus encephalitis with recombinant glycoproteins: role of neutralizing anti-G1 antibodies. J Virol. 1995;69:3475–3481. doi: 10.1128/jvi.69.6.3475-3481.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peralta P H, Shelokov A. Isolation and characterization of arboviruses from Almirante, Republic of Panama. Am J Trop Med Hyg. 1966;15:369–378. doi: 10.4269/ajtmh.1966.15.369. [DOI] [PubMed] [Google Scholar]
- 28.Quiroz E, Moreno N, Peralta P H, Tesh R B. A human case of encephalitis associated with vesicular stomatitis virus (Indiana serotype) infection. Am J Trop Med Hyg. 1988;39:312–314. doi: 10.4269/ajtmh.1988.39.312. [DOI] [PubMed] [Google Scholar]
- 29.Reif J S, Webb P A, Monath T P, Emerson J K, Poland J D, Kemp G E, Cholas G. Epizootic vesicular stomatitis in Colorado, 1982: infection in occupational risk groups. Am J Trop Med Hyg. 1987;36:177–182. doi: 10.4269/ajtmh.1987.36.177. [DOI] [PubMed] [Google Scholar]
- 30.Roberts A, Kretzschmar E, Perkins A S, Forman J, Price R, Buonocore L, Kawaoka Y, Rose J K. Vaccination with a recombinant vesicular stomatitis virus expressing an influenza virus hemagglutinin provides complete protection from influenza virus challenge. J Virol. 1998;72:4704–4711. doi: 10.1128/jvi.72.6.4704-4711.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schnell M J, Buonocore L, Boritz E, Ghosh H P, Chernish R, Rose J K. Requirement for a non-specific glycoprotein cytoplasmic domain sequence to drive efficient budding of vesicular stomatitis virus. EMBO J. 1998;17:1289–1296. doi: 10.1093/emboj/17.5.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schnell M J, Buonocore L, Whitt M A, Rose J K. The minimal conserved transcription stop-start signal promotes stable expression of a foreign gene in vesicular stomatitis virus. J Virol. 1996;70:2318–2323. doi: 10.1128/jvi.70.4.2318-2323.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schnell M J, Johnson J E, Buonocore L, Rose J K. Construction of a novel virus that targets HIV-1-infected cells and controls HIV-1 infection. Cell. 1997;90:849–857. doi: 10.1016/s0092-8674(00)80350-5. [DOI] [PubMed] [Google Scholar]
- 34.Sudia W D, Fields B N, Calisher C H. The isolation of vesicular stomatitis virus (Indiana strain) and other viruses from mosquitoes in New Mexico, 1965. Am J Epidemiol. 1967;86:598–602. doi: 10.1093/oxfordjournals.aje.a120769. [DOI] [PubMed] [Google Scholar]
- 35.Tesh R B, Peralta P H, Johnson K M. Ecological studies of vesicular stomatitis virus. I. Prevalence of infection among animals and humans living in an area of endemic VSV activity. Am J Epidemiol. 1969;90:255–261. doi: 10.1093/oxfordjournals.aje.a121068. [DOI] [PubMed] [Google Scholar]
- 36.USDA Animal and Plant Health Inspection Service-Veterinary Services, Centers for Epidemiology and Animal Health. 1997 vesicular stomatitis virus (VSV) outbreak in the United States. DxMonitor Anim Health Rep. 1997;Fall 1997:1–2. [Google Scholar]
- 37.Wagner R R, Rose J K. Rhabdoviridae: the viruses and their replication. In: Fields B N, Knipe D M, Howley P M, editors. Fields virology. 3rd ed. Vol. 1. Philadelphia, Pa: Lippincott-Raven; 1996. pp. 1121–1135. [Google Scholar]
- 38.Walton T E, Webb P A, Kramer W L, Smith G C, Davis T, Holbrook F R, Moore C G, Schiefer T J, Jones R H, Janney G C. Epizootic vesicular stomatitis in Colorado, 1982: epidemiologic and entomologic studies. Am J Trop Med Hyg. 1987;36:166–176. doi: 10.4269/ajtmh.1987.36.166. [DOI] [PubMed] [Google Scholar]
- 39.Webb P A, Monath T P, Reif J S, Smith G C, Kemp G E, Lazuick J S, Walton T E. Epizootic vesicular stomatitis in Colorado, 1982: epidemiological studies along the northern Colorado front range. Am J Trop Med Hyg. 1987;36:183–188. doi: 10.4269/ajtmh.1987.36.183. [DOI] [PubMed] [Google Scholar]
- 40.Xiong C, Levis R, Shen P, Schlesinger S, Rice C M, Huang H V. Sindbis virus: an efficient, broad host range vector for gene expression in animal cells. Science. 1989;243:1188–1191. doi: 10.1126/science.2922607. [DOI] [PubMed] [Google Scholar]
- 41.Zinkernagel R M, Adler B, Holland J J. Cell-mediated immunity to vesicular stomatitis virus infections in mice. Exp Cell Biol. 1978;46:53–70. doi: 10.1159/000162882. [DOI] [PubMed] [Google Scholar]