

CONSPECTUS.
DNA-encoded library technology (DELT) is a new screening modality that allows efficient, cost-effective, and rapid identification of small molecules with potential biological activity. This emerging technique represents an enormous advancement that, in combination with other technologies such as high-throughput screening (HTS), fragment-based lead generation, and structure-based drug design, has the potential to transform how drug discovery is carried out. DELT is a hybrid technique in which chemically synthesized compounds are linked to unique genetic tags (or “bar codes”) that contain readable information. In this way, millions to billions of building blocks (BBs) attached on-DNA via split-and-pool synthesis can be evaluated against a biological target in a single experiment. Polymerase chain reaction (PCR) amplification and next-generation sequencing (NGS) analysis of the unique sequence of oligonucleotides in the DNA-tag are used to identify those ligands with high affinity for the target. This innovative fusion of genetic and chemical technologies was conceived in 1992 by Brenner and Lerner (Proc. Natl. Acad. Sci. 1992, 89, 5381–5383), and is under accelerated development with the implementation of new synthetic techniques and protocols that are compatible with DNA. In fact, reaction compatibility is a key parameter to increasing the chances of identification of a drug target ligand, and a central focus has been the development of new transformations and the transition to robust protocols for on-DNA synthesis. Because the sole use of the DNA tag is as an amplifiable identification barcode, its structural integrity during a new chemical process is mandatory. As such, the use of these sensitive, polyfunctional biological molecules as substrates typically requires aqueous solutions within defined pH and temperature ranges, which is considered a notable challenge in DEL synthesis.
Using low energy visible light as the driving force to promote chemical transformations represents an attractive alternative to classical synthetic methods, and it is an important and well-established synthetic tool for forging chemical bonds in a unique way via radical intermediates. Recent advances in the field of photocatalysis are extraordinary, and this powerful research arena is still under continuous development. Several applications taking advantage of the mild reaction conditions of photo-induced transformations have been directed toward DEL synthesis, allowing the expansion of chemical space available for the evaluation of new building blocks on-DNA. There are no doubts that visible light-driven reactions have become one of the most powerful approaches for DELT, given the easy way they provide to construct new bonds and the challenges to achieve equal success via classical protocols.
Key characteristics of photocatalytic synthesis include the short reaction times and efficiency, which translate into retention of DNA integrity. In this Account, we describe recent advances in the photo-induced diversification of building blocks prepared on-DNA, highlighting the amenability of the techniques employed for preserving the genetic structure of the molecules. We demonstrate with recent research from our group the applicability of photocatalysis to the field and include in the summary a table containing all the photo-induced methods reported to date for DELT, demonstrating their key aspects such as scope, applications, and DNA-compatibilities. With this information, practitioners are provided with compelling reasons for developing/choosing photocatalytic methods for DELT applications.
Graphical Abstract

1. Introduction
DNA-encoded library technology (DELT) is a strategy used extensively in both academic and industrial realms. This technology is a powerful method employed to accelerate the discovery of novel leads in medicinal and agricultural chemistry. Therefore, it represents a fundamental approach that is still in its infancy but has tremendous promise for the discovery of biologically active small molecules. In fact, this approach has already been incorporated into an integrated lead generation strategy, along with others such as high-throughput screening (HTS), fragment-based lead generation, and structure-based drug design, for example.1
Twenty years ago, the extraordinary contributions of Brenner and Lerner led to the establishment of this new screening modality, defined by them as a “retrogenetic” method for specifying each building block in a library of compounds.2 The method brought together the advantages of genetic and synthetic approaches to discovering the structure of biologically active molecules. The application of DELT consists of chemically binding a synthetic building block (BB) to a genetic tag. During the process of constructing a library by pooling and division (“split and pool”), each chemical unit attached to the polymeric structure is encoded by an oligonucleotide sequence, which serves as a “barcode” (Figure 1a). After being incubated with a biological target immobilized on a resin or bead, the chemical units are selected by their binding affinities, and those with low affinity are washed away. For the hit identification step, polymerase chain reaction (PCR) amplified copies of the unique genetic tags are obtained and analyzed via next-generation sequencing (NGS), and the potential drug target ligands are thereby decoded (Figure 1b).
Figure 1.

DELT as a new screening modality for the interrogation of billions of clinical candidates in a single experiment.
In that seminal work, the authors pointed out the importance of introducing chemical diversity to the library to increase the chances of hit identification by exploring amide-linkage formation, locating the amino- and carboxylic acid moieties on different compounds, and installing important heterocycles in the chains of non-natural amino acids. In fact, this is a crucial parameter for the effectiveness of a DEL synthesis because a large variety of compounds containing different functional groups can be screened against a target in a single experiment. As a numerical comparison, this number varies from billions of compounds in DELT versus some millions in a HTS modality and thousands or hundreds for fragment-based screening.3,4
As a demonstration of the potential cost-effectiveness of DELT, classical combinatorial synthesis has a total cost of ~$1.10 per candidate compound, while the use of DELT would allow a reduction of the costs to around $0.0002 per candidate, considering a library of only 800 compounds!5 The rapid scanning of new chemical space brings the promise of a faster development of pharmaceuticals, accelerating their availability, and has the potential to make enormous contributions to human health.
An early example from GSK reveals the potential value of the method. Thus, using a DELT approach targeting R1P1 inhibitors,6 GSK researchers identified a potent and selective benzoxazepinone,7 which later resulted in the discovery of the clinical candidate GSK2982772, associated with immune-mediated inflammatory diseases.8 Optimization of the initial benzoxazepinone candidate revealed that replacing the isozaxole unit with a triazole ring significantly improved solubility, lipophilicity, and rat oral exposure. This became the first-in-class RIPK1 (receptor-interacting protein kinase-1) inhibitor to enter clinical trials.
To maximize the efficiency of a DEL, it is crucial to have synthetic methods that are compatible with the sensitive nature of DNA molecules.9 An ideal reaction to be performed on-DNA should be water-compatible (≥20% water as co-solvent) to avoid precipitation, tolerate protic solvents under dilute conditions (1 mM), and avoid the use of harsh conditions, such as high temperatures (ideally maintaining temperatures between 25−90 °C), extremally acidic conditions (ideal pH range between 4−14), and strong oxidants and reductants. It is of extreme importance to preserve the integrity of the genetic tag because it contains the readable information for the building block identification. Additionally, the chemical protocols for the introduction of new building blocks must preserve the integrity of the previously installed building blocks.
As a combined genetic and synthetic technique, an important part of the development of DELT is the design of the libraries and the search for amenable transformations on-DNA. A productive DEL must incorporate structural diversity by introducing building blocks with pharmacophore features and biologically relevant motifs. In terms of design, Pfizer created an algorithm for monomer-based selections and the definition of representative sublibraries.10 Eli Lilly researchers also made a significant contribution by developing a system that generates library designs by considering all available building blocks and on-DNA compatible transformations.11 All these advances, along with broader synthetic protocols, expand the practical application of DELT and diffuse this powerful technology in the pharmaceutical arena. At this point, there are several well-established on-DNA protocols that have adapted and applied classical organic reactions to diversify these libraries. Regarding classical approaches, at least thirty different reactions are known to be DNA-compatible, including acylation, SNAr, Sonogashira, Suzuki, Heck, 1,3-dipolar cycloaddition, click, Henry, and Fmoc/Boc removal reactions.12,13,14,15
The fast development of this screening technology has been accompanied by advances in the organic synthesis field. The renaissance of visible light-promoted chemistry thus became a landmark in extending the toolkit of on-DNA compatible transformations.14–17 Photocatalysis has already proven its importance and unique manner of accessing C-C and C-X bonds that would be challenging to achieve under traditional methods, and it is still demonstrating its rich applications through continuous development.
Photocatalytic approaches make use of mild reaction conditions, being performed mostly at room temperature and using the potential energy of visible-light irradiation to access the excited state of organic molecules, which can subsequently engage in unique reaction pathways. An absorbing species, the photocatalyst, can reach its triplet excited state from the absorption of energy at a determined wavelength. The excited state species is prone to undergo a series of electron transfer events, leading to the donation/acceptance of electrons to generate open-shell radical intermediates. Instead of transferring electrons, the photocatalyst excited state can also activate substrates via energy transfer.18 Blue LED irradiation has promoted such efficient transformations, which can proceed through well-established mechanisms. The aim of this Account is not to provide a deep understanding of photoredox chemistry, because remarkable reviews have already expertly covered the activation modes of this catalysis field,19–27 but rather to outline the ways in which this technology has been adapted to DEL synthesis.
This Account is organized by the mechanism of the photoinduced transformations on-DNA, including a) classical photoredox reactions, b) synergistic protocols in the presence of transition metal (TM) catalysts, and c) via the formation of visible light-absorbing electron-donor/electon-acceptor (EDA) complexes. The principal mechanistic pathway in visible light-mediated photoredox catalysis is via single-electron transfer (SET) events with other substrates, leading to the generation of reactive neutral, cationic, or anionic radical intermediates that can engage in successive chemical transformations. In a representative mechanism (Figure 2a), after light excitation of the photocatalyst (PC), the reaction proceeds via an oxidative or reductive quenching of the excited state photocatalyst (PC*) via SET with electron-acceptor (EA) or electron-donor (ED) species, consecutively, leading to the generation of the reactive radical intermediates. In redox-neutral protocols, the oxidation and reduction steps occur without the need for sacrificial oxidants or reductants. This type of mechanism can be combined with other catalysts, such as organo- and transition-metal (TM) species. In dual (or synergistic) catalysis (Figure 2b), the TM catalyst traps and stabilizes the radical intermediate generated in the photoredox cycle. Further steps follow the traditional mechanisms in TM cycles, which include an oxidative addition in the presence of aryl halides followed by the reductive elimination of the product. The oxidized M(II) species formed can then be reduced in the photoredox cycle to recycle the M(0) catalyst. This type of dual mechanism opened a new avenue for the development of stereoselective strategies and the construction of new C-C bonds. Finally, another mode of light-driven mechanism proceeds via EDA (or charge-transfer) complexes in the absence of any exogenous photocatalyst. As the name indicates, these complexes are formed by the ground-state association between an electron-donor (ED) and an electron-acceptor (EA) in a diffusion event, leading to the formation of a radical ion pair [D•+/A•-] upon irradiation that engages in further one-electron reactions (Figure 2c).28
Figure 2.

Representative mechanisms via visible-light photocatalysis. RP = radical precursor.
The Pfizer and Molander laboratories were pioneers in translating these conditions to increase the arsenal of available tools for the exploration of chemical space on-DNA. The Baran group had previously disclosed a radical-based protocol for the Giese addition of alkyl radicals from redox-active esters to acrylates on-DNA.29 The seminal work from Pfizer appeared in 2018, demonstrating the utility of photocatalytically-generated alkyl radicals from a-amino acids to promote new C(sp3)-C(sp3) bonds with DNA-tagged Michael-acceptors.30 This contribution was built on a previous off-DNA report from the MacMillan group,31 revealing the potential of transitioning other powerful photocatalytic approaches to DELT. Subsequently, the Molander group developed a C(sp2)−C(sp3) aryl−alkyl cross-coupling reaction by merging Ni/photoredox dual catalysis and radical/polar crossover reactions, which enabled a rapid and efficient incorporation of Csp3 on-DNA, demonstrating applicability in the enhancement of solubility and specificity in druggable molecules.32 These seminal contributions opened a new arena in the visible-light-mediated catalysis field, and since then many important advances have enriched the library of compounds available for DELT. In this Account, we focus on the potential of these recent transformations, highlighting the mildness of the protocols to retain the integrity of the DNA genetic tag, which is critical to the development of this emerging technology.
2. Assembling the DNA-headpieces (DNA-HP) and experimental set-up
As mentioned, DELT is a unique technique, and assembly of the functionalized DNA-HP is a basic step. The adopted tag can be comprised of either single-stranded (ssDNA) or double-stranded DNA (dsDNA); however, the duplex structure is often employed because it preserves the DNA structure from potential damage to a greater degree, and, additionally, the dsDNA has less potential to interfere with enzymatic targets.6 Another important factor explaining why dsDNAs are the most commonly used format for encoding DELs is that ssDNAs are less abundant and frequently require special encoding methods.33–36 Still, ssDNAs have important applications as well, because they are compatible with selection methods suitable for non-immobilized proteins, and a recent study showed that ssDEL provides approximately two-fold higher enrichment factors and comparable fingerprints to the double-strand format.37 Given that, it has become of interest to convert them to dsDELs, as recently reported by Li and colleagues, where this conversion was demonstrated without the need of library redesign and resynthesis using nuclease digestion.38 This possibility broadens the applications and possibilities for the DELT scenario.
The DNA-HP-NH2 tags, such as 5′-/5Phos/GAGTCA/iSp9/iUniAmM/iSp9/TGACTCCC-3′ (shown in Figure 3) can be commercially acquired. These DNA-HPs are attached to a linker (spacer); for example, Fmoc-15-amino-4,7,10,13-tetraoxapentadecanoic acid (AOP), which separates the genetic tag from the synthetic BBs. The small molecules can be attached to the free amino group (obtained after AOP deprotection) via classical protocols, as in substitution and amidation reactions.
Figure 3.

Chemical structure of the 5′-/5Phos/GAGTCA/iSp9/iUniAmM/iSp9/TGACTCCC-3′ DNA-headpiece. The DNA starting material containing a free amino group is named here as DNA-HP-NH2.
For the amidation protocol, for example, different carboxylic acids containing reactive functional groups can be explored for attachment to the genetic tag and used in sequence for the photoredox transformation. For this first step of assembling the DNA-HP starting material, it is common to employ well-recognized methods using bases and coupling reagents. As demonstrated and represented in Figure 4a, a common amidation strategy makes use of excess DIPEA, HATU, and carboxylic acid. The experimental procedure is simple and proceeds by mixing a cool solution of the base, coupling agent, and carboxylic acid in DMA with a solution of the DNA-HP-NH2 in 250 mM sodium borate buffer at pH 9.5. After 1 hour at room temperature and vortexing the sample several times during this period, the crude material must be purified by the ethanol precipitation method. To this end, it is treated with a cold solution of 5 M NaCl (~10% by volume) and cold ethanol (~2.5 volumes), followed by precipitation at −20 °C and centrifugation at 3.000–14.000 rpm depending on the sample and equipment used.13 After completion, the material is lyophilized and dissolved in biological-grade water to the desired concentration. The integrity of the material is confirmed by LC-MS analysis, and it can be purified by reverse-phase HPLC or directly used when obtained with high purity. Usually, the final functionalized DNA-HP is stable when stored at low temperatures in water and can be used several times during the study’s development.
Figure 4.

a) A classical method for coupling with carboxylic acids; b) representative experimental set-up.
One aspect of the library’s preparation is that the reactions are performed on a very small scale, in the range of μmol to nmol. This factor allows the use of excess reactants and guarantees the efficiency of the transformations. Here, the principle of atom economy is of no concern, because even a large excess of reactants still represents an exceedingly small quantity. In a representative example where a functionalized DNA-HP is used as the limiting starting material in a 25 nmol scale photoredox reaction, the use of 0.50 equivalent of a costly catalyst in the synthesis, for example the iridium catalyst [Ir[dF(CF3)ppy]2(dtbpy))PF6], would represent only 14 μg. In comparison, approximately 400 on-DNA reactions would use the same amount of this iridium catalyst as a single 0.5 mmol scale off-DNA reaction using 1 mol % of photocatalyst! In terms of excess reactants, in a transformation using 250 equivalents of a radical precursor, for example, the amino acid (tert-butoxycarbonyl)proline for a C(sp2)−C(sp3) cross-coupling on-DNA, only 6.25 μmol of this material would be necessary, which corresponds to just 1.3 mg!32
The use of functionalized DNA-HPs in photocatalytic protocols has demonstrated excellent robustness regarding the sensitive nature of these molecules. Despite the misconceptions about radical chemistry,39 the generation of these reactive one-electron intermediates can be carried out in a controlled and chemoselective way, making use of mild reaction conditions that guarantee the DNA’s integrity. The practicality of the protocols is another key advantage of translating these photocatalytic transformations to DELT.
For the experimental set-up, stock solutions of the starting materials are prepared in water or a water-compatible solvent, mostly DMSO, given its biocompatibility. Other solvents have also been used, such as acetonitrile and DMA. Given the small scale of the transformations, PCR Eppendorf tubes can be used as reaction vessels, which are charged with the synthetic components and the solution of the functionalized DNA-HP in water. In some cases, degassed conditions are required to avoid the quenching of the photocatalyst excited-state in the presence of oxygen, a powerful quencher of almost all fluorophores.40 Irradiation of the solutions with blue LED lamps to promote the organic transformations is usually fast, requiring only a few minutes! Subsequently, the final product on-DNA can be purified by ethanol precipitation. The reaction is analyzed by preparing the sample in water, and the conversions are calculated by examination of the UV and total ion count (TIC) traces of the LCMS chromatograms (Figure 4b). Even in the crude reactions, the DNA bioconjugate materials can be easily analyzed and differentiated from the other organic starting materials used in excess via mass spectrometry, which facilitates the characterization step of these transformations.
Building a functionalized DNA-HP requires the rationalization of which BB’s would be interesting, not only from the point of view of a DEL preparation but also regarding the compatibility of their reactive functional groups with the DNA-conjugated starting material. In addition to side reactions on the DNA tag molecule, reactive and electrophilic functional groups can interfere with the resin used for the immobilization of the biological target in the screening studies or with the protein target itself. Additionally, the nature of the pharmacophore should also be taken into consideration because BB’s with potential mutagenic and toxic effects have to be excluded from the library’s design.13 In terms of availability, there is an immense number of diverse, commercially available BB’s with reactive functional groups that can be easily accessed, especially amines, carboxylic acids, aldehydes, halides, alcohols, amino acids, alkenes, and alkynes.
For the photoredox transformations using a functionalized DNA-HP (containing aryl halides, alkenes, or alkynes, for example), several reaction partners can be commercially acquired or easily prepared from well-established synthetic routes. N-(Acyloxy)phthalimides (redox-active esters), for example, which are broadly used for the generation of alkyl radicals, are quickly prepared from the coupling of inexpensive and abundant carboxylic acids with N-hydroxyphthalimide.41,42
The chemical processes employed during the DNA-HP assembly or functionalization reactions can cause damage to the DNA molecule, and these protocols cannot be applied to DELT because the DNA barcode is lost. Examples of degradation of the biological tag involve acid- or metal-promoted depurination, deamination, purine oxidation, nucleophile addition, and phosphate hydrolysis.43 Some chemical protocols, such as SNAr reactions, are well-known for their compatibility on-DNA; others, such as Cu(I) catalyzed azide-alkyne cycloadditions, should be used with caution because they can cause mutations because of changes in nucleobases and the incorporation of incorrect bases.44 Damaging radical pathways can also be operative, especially by the formation of reactive oxygen-centered species, such as hydroxyl radicals, or metal-bound oxy radicals, depending on the reaction conditions. The reactive and electrophilic hydroxyl radicals can add to the π-bonds of DNA bases and promote hydrogen abstraction from the deoxyribose sugar units. These species are mostly found in the water radiolysis process using ionizing radiation (γ-rays or X-rays).45
In DELT, the quantification of the DNA damage is of extreme importance for the hit elucidation step, and the Sanger sequencing and qPCR analysis are methods routinely used to this end. In a traditional method, a post-transformation of enzymatic ligation introduces the templates for PCR amplification. The the amount of amplifiable DNA is then analyzed via qPCR after the determination of the ligation efficiency via gel electrophoresis. Paegel and colleagues recently published a quantitative assay protocol that generates information on chemical and encoding fidelity as well as DNA compatibility, laying the groundwork for the identification of compatible on-DNA reactions.46 Deep sequencing is recommended in addition to the qPCR technique for analyzing new DNA transformations, and it would be extremely useful to have an assessment of DNA damage regarding different classes of reaction for future developments in this field.44
Gratifyingly, most of the reported visible light-promoted methods guarantee the structural integrity of the DNA molecule, and thus this catalytic mode is one of the most powerful synthetic methods to be employed in combination with DELT.
In this Account, we focus on highlighting the DNA integrity, given that it is of basic importance when developing an on-DNA protocol and should be among the first parameters evaluated. Herein, landmark contributions to the field that have been made in combination with photocatalysis are covered, and a summary of the reported methods regarding their applicability, scope, and compatibility with the on-DNA conditions is discussed. With this material, the reader is provided with a guide containing the well-established visible light-induced protocols for choosing the ideal reaction conditions for on-DNA studies, as well as an incentive for the development of the new transformations for the valuable and promising field of DELT.
3. On-DNA Catalyzed Reactions in the Presence of Photocatalyst
As pointed out, the first proof-of-concept showing the potential of photoredox catalysis as a powerful tool for the preparation of DELs was reported by Flanagan and co-workers.30 They developed a photoinduced conjugate radical addition to DNA-tagged styrenes and Michael acceptors via reductive quenching (Figure 2a) using carboxylic acids as radical progenitors and an iridium photocatalyst (Scheme 1). The scope of this transformation included primary- (1), secondary- (2), and tertiary (3) N-Boc protected amino acids, and suitable radical acceptors for this transformation were comprised of on-DNA α-substituted styrenes, acrylamides, and vinyl benzamides.
Scheme 1.

Protocol developed by Pfizer for the on-DNA decarboxylative alkylation using α-amino acids
One year later, Molander’s group pioneered the on-DNA synthesis of gem-difluoroalkenes32 from trifluoromethylated alkenes by exploring a radical/polar crossover defluorinative alkylation pathway (Scheme 2).47 Importantly, the exploration of diverse radical precursors, such as silicates, 4-alkyl-1,4-dihydropyridines (DHPs), and carboxylic acids was successfully demonstrated. The organophotocatalyst 4CzIPN [1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene] worked well when silicates or DHPs were used as radical precursors, and Ir[dF(CF3)ppy]2(bpy)PF6 provided the best reactivity for the carboxylic acids. Remarkably, the reactions were accomplished within 10 minutes. The scope using silicates (10–13) included fluorinated aliphatic motifs, alkenes, epoxides, esters, and protected amines. Secondary- and primary alkyl DHPs (14–16) readily provided the on-DNA gem-difluoroalkene with good functional group tolerance, including a saccharide scaffold. A wide array of amino acids (17–21) as well as carboxylic acids reacted efficiently, tolerating free alcohols or pyridyl units. Of note, the study was pioneering in demonstrating that photoredox conditions are amenable to the DNA barcode. A 4-cycle tagged DNA successfully underwent PCR amplification and quantitation, and no difference was found in the frequency of misreads between the product formed under the reaction conditions versus the control experiments after sequencing. Further, α-silylamines were used as radical precursors (22–23), which resulted in on-DNA gem-difluoroalkenes with an amine functional group.48 The installation of amino acid-based organosilanes, including leucine or tryptophan, was also successful using Ir[dF(CF3)ppy]2(bpy)PF6.
Scheme 2.

Defluorinative alkylation of on-DNA alkenes.
We also reported a general intermolecular hydroarylation protocol of electronically unbiased olefins through selective C–X bond activation in DNA-tagged organohalides (Scheme 3). Remarkably, this photochemical paradigm delivers reactive (hetero)aryl radical species in a controlled manner without jeopardizing DNA integrity. Notably, this open-to-air process is chemoselective, operates under mild and dilute reaction conditions, and can be completed within a few minutes, making it suitable for late-stage functionalization and high-throughput settings in the pharmaceutical industry. These features make the protocol highly attractive for the acceleration of drug discovery research and triggered new developments in radical-mediated DEL synthesis.
Scheme 3.

Alkylation of on-DNA aryl halides with alkenes.
Pursuing our focus on the exploration of methods for DEL, a direct C–H carbofunctionalization of medicinally relevant heteroarenes via photoreduction of DNA-bound halogenated (hetero)aryl subunits was reported (Scheme 4).49 This heteroarylation method proceeds without the need for pre-functionalization of the heteroarene moiety or the presence of acidic additives. Furthermore, it is also amenable to both electron-rich as well as electron-deficient medicinally relevant heterocyclic building blocks, expanding the chemical space available for DEL synthesis. Additionally, free alcohol and amine handles were tolerated, presenting opportunities for rapid library diversification. In this area, a C–H carbofunctionalization of electron-deficient isoquinolines and quinolines and a palette of electron-rich pyrroles as well as indoles were investigated. Of note, successful arylations were observed in the absence of nitrogen protecting groups (28–31). This method also demonstrated the easy incorporation of multifunctional handles including building blocks with ester, nitrile, ketone, aldehyde, and amide functional groups. Given that peptides and their corresponding macrocycles are important in drug discovery, effective C–H arylations of electron-rich indole heterocycles in monomeric tryptophan and tryptophan-proline dipeptide were then carried out.
Scheme 4.

DNA-encoded C-H arylation of heteroarenes.
Finally, to validate the robustness of this method and to demonstrate that the integrity of the DNA remained intact during the photochemical processes, model DNA conjugates were subjected to the standard reaction conditions under blue light irradiation. These DNA conjugates were then elongated by ligation and then quantified by qPCR. No substantial difference in qPCR amplification was observed across the various reaction samples, indicating DNA compatibility under these developed photochemical heteroarylation conditions.
Lastly, regarding the implementation of methods on-DNA employing simple photocatalysts, the group developed a Giese addition to install highly functionalized bicyclo[1.1.1]pentanes (BCPs) using tricyclo[1.1.1.0]pentane (TCP) as a radical linchpin. Other diverse alkyl groups from the corresponding organohalides as non-stabilized radical precursors were further evaluated (Scheme 5).50 To fill a gap in general methods for introducing diversely substituted arene bioisosteres on DNA, it was hoped that investigating what has previously been viewed as limitations of DEL synthesis (low substrate concentration, excess reagents, etc.) would be advantageous in achieving desired reactivity with incorporation of novel substrates. During the scope of this transformation using a set of 1-halobicyclo[1.1.1]pentanes, a wide range of substrates served as competent partners, including those containing bifunctional handles, free alcohols, N-Boc-protected amines, sugars, and methyl esters, all with moderate to excellent yields. Bicyclopentyl (BCP) iodides containing aryl substituents were also suitable substrates for this transformation. Notably, arenes could be brought in through either functionalization at the benzylic position or through direct substitution of electron-deficient aryl iodides, generating C(sp2)−C(sp3) bonds prior to halo-BCP coupling. These initial results represent the first time that BCP bromides were leveraged for a Giese-type reaction on- or off-DNA. Subsequent exploration of the scope for on-DNA alkene acceptors demonstrated that meta- and para-substituted styrene HPs delivered the desired products in moderate to excellent yields with secondary- and tertiary radicals. In addition, electron-deficient vinyl pyridines have proven to be compatible with a variety of substrates, such as deprotected piperidine-, quinoline- and pyrazole-containing BCPs, and aryl BCPs possessing two useful handles for further post-functionalizations. Additionally, the use of trifluoromethyl-substituted styrene was examined, and the corresponding gem-difluoroalkene adduct was observed instead of the Giese-type addition product. To complete this method, “telescoped” processes for three substrate classes, such as activated esters, carboxylic acids, and alcohols, were pursued, allowing the workflow to be streamlined. In this context, secondary- and tertiary alkyl systems provided products with more than 90% yield. In addition, the present methods (using activated esters and carboxylic acids) allow non-stabilized primary-, secondary-, and tertiary carboxylic acids to be used as precursors for the Giese addition to the less reactive DNA styrene, which had not been previously reported. The last strategy that employed alcohols in the telescoped reaction allowed the introduction of primary- and secondary radicals with >90% yield. Finally, the integrity of the DNA tag was preserved, indicating the method’s compatibility to produce DELs that cover previously inaccessible chemical space.
Scheme 5.
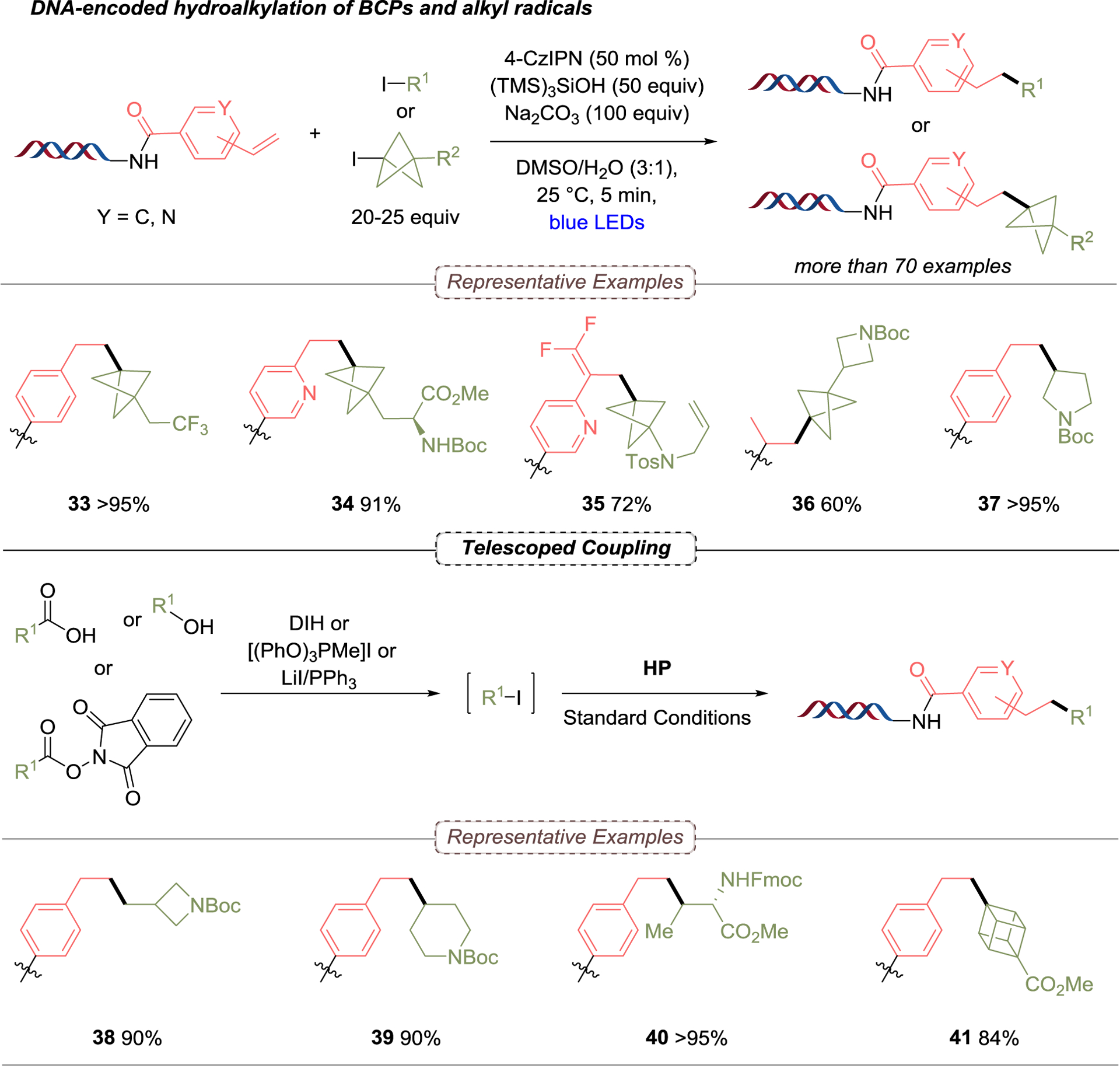
On-DNA hydroalkylation to introduce BCPs and alkyl radicals. DIH = 1,3-diiodo-5,5-dimethylhydantoin.
4. Metallaphotoredox Approaches Applied to DNA-tethered Aryl Halides
In 2014, we first showed that alkyl radicals were suitable partners in Ni-catalyzed cross-coupling reactions, funneling into a cycle to assemble new Csp3–Csp2 bonds.51 Since this disclosure, metallaphotoredox catalysis has emerged as a valuable tool for accelerating the assembly of challenging C–C linkages under very mild reaction conditions.52 Motivated by increasing the Csp3 fraction on DEL platforms and moving away from traditional harsh organometallic or anionic species, a Csp2-Csp3 cross-coupling via photoredox/nickel dual catalysis was translated to on-DNA conditions, promoting the reaction between aryl halides and DHPs or amino acids as radical progenitors (Scheme 6).32 The challenges of applying a photoredox/nickel approach to on-DNA chemistry are to avoid the formation of protodehalogenated and phenolated side products because of the need to use aqueous media. Despite this potential hurdle, suitable conditions were found after screening of different nickel sources as well as equivalences of the radical precursor. The use of the organic dye 4CzIPN and Ir[dF(CF3)ppy]2(dtbbpy)PF6 showed excellent efficiency when using DHPs (42–46) and amino acids (47–50) as radical precursors. The scope of this transformation revealed successful reactions with a variety of substrate combinations, verifying the range of yields that may be experienced in a library setting instead of just those under validation-like conditions. Electron-deficient aryl bromides and electron-neutral aryl iodides efficiently reacted with secondary-, tertiary-, benzyl-, and α-alkoxy DHPs. Both electron-deficient aryl bromides and -iodides allowed the installation of pyrrolidine, piperidine, unprotected indoles, and 5–6 fused rings; the abundant and easily accessible amino acids were also amenable radical precursors for this transformation.
Scheme 6.

First metallaphotoredox approach on-DNA: cross-coupling between on-DNA attached aryl halides and DHPs/amino acids.
As mentioned in the introduction, the primary goal of DELT is to prepare a large battery of products efficiently with minimal effort in terms of setup complexity and timing. In this context, the use of alkyl bromides as radical feedstocks is considered ideal. Molander’s group presented an efficient cross coupling based on a dual catalytic system between on-DNA aryl halides and alkyl bromides. This strategy relies on the use of Ir(ppy)2(dtbpy)PF6 photocatalyst and NiBr2•bpy in the presence of MgCl2 and NEt3 as base.48 The reaction was applied to a wide range of secondary- (51) and primary (52–53) alkyl bromides (Scheme 7). The functional group tolerance is notable, where alkenes, alkyl chlorides, free alcohols and N-Boc-protected amines were installed on-DNA. In addition to alkyl bromides, the use of unprotected α-silylamines as radical precursors also proved feasible.
Scheme 7.

On-DNA aryl halides cross-coupled with alkyl halides via photoredox/nickel dual catalysis.
The aminomethyl moiety serves as a pivotal linker in bioactive molecules.53 A photoexcited iridium species undergoes effective single-electron oxidation of aminomethylsilanes to yield the corresponding amine radical cations, which induces loss of “Me3Si+”, generating aminomethyl radicals. The aminomethyl radical can then be funneled into a nickel-catalyzed cross-coupling cycle. More than 12 on-DNA aryl halide functionalized DNA-HPs were successfully cross-coupled using aminomethylsilane radical progenitors (Scheme 8).48 Importantly, the PCR amplification, quantification, and sequencing demonstrated that this nickel/photoredox dual manifold can be applied to DEL synthesis without compromising the DNA integrity.
Scheme 8.

The use of aminomethylsilanes as radical precursors on-DNA aryl halides.
5. On-DNA Photoinduced Transformations Circumventing Exogenous Photocatalysts
Although a photoinduced strategy making use of electron donor−acceptor (EDA) complex formation has been widely exploited in small molecule synthesis, it was not until 2020 that this strategy was applied to on-DNA substrates. Mendoza and co-workers demonstrated that dihydronicotinamides and nicotinamide adenine dinucleotide (NADH) promote the coupling of redox-active esters (RAEs) and alkenes upon blue light irradiation (Scheme 9). Given that this reaction could be carried out in aqueous media, they extended the method to on-DNA Michael acceptor substrates.
Scheme 9.

Metal-free alkylation using RAEs and activated on-DNA acrylates/acrylamides.
The exploration of this transformation to Michael acceptor-functionalized DNA-HPs displayed good reactivity with secondary- and tertiary RAEs within 60 minutes. The installation of an α-amino radical was also demonstrated with a single example (59). The aliphatic photocoupling between RAEs and on-DNA electron-poor olefins was achieved without using an exogenous photocatalyst. Although the detailed mechanisms of these transformations remain unclear, one potential scenario involves the initial formation of an EDA complex between the NADH and RAEs. The NADH may thus act as electron-donor in this transformation, with the RAEs acting as electron-acceptors. Through this strategy, on-DNA N-protected amines and acetamides have been prepared in good yields (Scheme 9). Of note, the lipid-lowering agent Gemfibrozil-derived RAE (60) was also used successfully.
In 2021, we reported a decarboxylative-based hydroalkylation of DNA-conjugated trifluoromethyl-substituted alkenes enabled by single-electron transfer (SET) and subsequent hydrogen atom termination through electron donor–acceptor (EDA) complex activation (Scheme 10).54 Thanks to this EDA paradigm, a selective decarboxylative-based hydroalkylation of trifluoromethyl-substituted alkenes through radical addition/hydrogen atom transfer was achieved to access complex benzylic trifluoromethylated scaffolds. This first example unlocked a complementary reactivity outcome to the established carbodefluorinative protocols mediated by an external photoredox catalyst (Scheme 2), again without compromising the DNA integrity. To demonstrate the possibility of incorporating multi-functional building-blocks for subsequent derivatization crucial in DEL settings, a broad array of primary aliphatic systems that lack any radical stabilizing factors were used and exhibited excellent reactivity (61–64). As a further benefit, a broad functional group tolerance, facilitating the introduction of bifunctional handles such as ketones, aryl halides, esters, substituted alkenes, free alcohols, as well as medicinally relevant heteroaromatic scaffolds, was noted. Finally, the scope was extended to the modification of biologically active molecules displaying a high density of pendant functional groups. In short, this method provides a clear advantage in terms of scope over previously reported on-DNA photoinduced decarboxylative alkylation protocols, which are largely limited to α-heteroatom-stabilized radicals or exclusively restricted to secondary- and tertiary radicals.
Scheme 10.

Metal-free hydroalkylation of on-DNA trifluoromethylated alkenes and RAEs.
6. Summary and Outlook
Since the first reports on photoinduced approaches for DEL synthesis were published five years ago, many academic and industrial collaborations have contributed significantly to this field (Table 1). The development of photocatalyzed methods, including simple photoredox approaches, photoredox/metal dual catalysis, EDA complex strategies, and energy transfer protocols have demonstrated their powerful and efficient implementation for the development of new synthetic methods on DNA. Importantly, known photoredox approaches as well as new modes of reactivity have been successfully applied to DEL synthesis, given their compatibility with on-DNA conditions, i.e., requiring mild, aqueous, and dilute conditions. This aspect is an important starting point to consider when designing a new transformation for DELT applications, and to help guide the process, a good number of organic reactions under aqueous conditions have been reported off-DNA, including photoredox transformations.
Table 1.
Summary of photo-induced approaches reported for on-DNA transformations.
| Transformation | Employed Method | Substrate Scope | DNA Integrity Studies | Limitations | Ref |
|---|---|---|---|---|---|
| Decarboxylative alkylation of amino acids to on-DNA alkenes | Simple Photoredox | Broad: primary-, secondary-, and tertiary amino acids. | No | Restricted to activated alkenes, styrenes, or Michael acceptors | 30 |
| Csp2–Csp3 Cross-coupling of on-DNA aryl halides and DHPs/amino acids | Metallaphotoredox | Broad for secondary- and tertiary DHPs. Works well with NBoc-protected amino acids | Yes | Absence of non-stabilized primary radicals from DHPs. Restricted to NBoc protected amino acids | 32 |
| on-DNA Decarboxylative arylation | Metallaphotoredox | Works well with primary- and secondary amino acids | Yes | Restricted to NBoc protected AAs. Low yields with tertiary amino acids | 55 |
| Decarboxylative Giese addition of RAEs to on-DNA alkenes | EDA Complex | Secondary- and tertiary RAE | No | Scope limited to a few examples. No primary RAEs tested and only two functionalized DNA-HP used | 56 |
| Csp2–Csp3 Cross-coupling with alkyl bromides | Metallaphotoredox | Secondary- and primary alkyl bromides | Yes | No acyclic secondary examples | 48 |
| Csp2–Csp3 Cross-coupling with alkyl bromides | Metallaphotoredox | Secondary- and primary alkyl bromides | No | No acyclic secondary examples. On-DNA aryl bromides provide low yields | 55 |
| [2+2] Cycloaddition using cinnamate esters | Energy Transfer | Broad | Yes | No limitation | 57 |
| on-DNA Synthesis of 1,2-amino alcohols | Simple Photoredox | Works with on-DNA aldehydes and moderately well with ketones | Yes | Low yield with ketones | 58 |
| Csp2–Csp3 Cross-coupling of on-DNA aryl halides with carboxylic acids | Metallaphotoredox | Works well with secondary cyclic carboxylic acids | Yes | No tertiary carboxylic acid or acyclic secondary examples. In general, moderate yields | 59 |
| C–H Functionalization of N-Boc heterocycles | Photoredox + HAT | 5–6 Membered rings | Yes | Restricted to 4-substituted piperidine; good with EWG but moderate with EDG | 60 |
| Derivatization of quinoxalin-2-ones | Simple Photoredox | Broad, primary-, secondary-, and tertiary amino acids and aliphatic acids | Yes | Scope limited to a few primary amino acids and aliphatic acids. Limited to N-Boc amino acids | 61 |
| C–H Functionalization of I-aryl tertiary amines | Simple Photoredox | Broad, N-aryl tertiary amines | Yes | Only N-arylamines, poor reactivity with N-carbamate or N-alkylamines | 62 |
| on-DNA Sulfonylation and selenation | EDA Complex | Broad scope of sulfinates and diselenides | Yes | Few examples of heterocyclic-based compounds. | 63 |
| Hydroarylation of functionalized olefins | EDA Complex | Broad | Yes | No limitation | 64 |
| C–H Arylation of heteroarenes | Simple Photoredox | Broad | Yes | No limitation | 49 |
| Deaminative alkylation | Simple Photoredox | Secondary- and tertiary examples | Yes | Not very high yield, not very broad scope, no primary | 54 |
| Hydroalkylation of alkenes – BCP | Simple Photoredox | Broad, many examples | Yes | No limitation | 50 |
| C−H sulfonylation of arenes | EDA Complex | electron-rich and electron-poor sulfinates | Yes | Scope limited to few DNA-encoded thianthrenium salts | 65 |
Of note, the DNA integrity (Table 1) of many of the reported new methods have been verified as a critical parameter for valid and useful DEL construction. These studies serve to establish the compatibility of visible-light irradiation and radical species with the DNA-bioconjugated tag. However, in all the reported protocols there is no consensus when performing an experiment to assess the DNA integrity, which means that direct comparisons between the different protocols are not applicable. From our point of view, an analysis showing the % mutated sequences would facilitate the comparison between different methods.
Many important advancements regarding the practical use of DEL in drug discovery programs have been achieved by pharmaceutical industries. However, after almost twenty years from the establishment of the basis of this screening modality, no candidate from a DEL platform has yet made it to the market. This technology has been developed primarily in the private sector, and much of the research has yet to be made public. To find more clinical candidates as quickly as possible, it is critical to continue developing new compatible transformations. Of equal importance would be the advances in bioinformatics and analytical techniques to identify these reactions and quantify the DNA damage associated with the encoded chemistry. This would be a crucial guide for designing the attachment of new BBs on-DNA and understanding the nature of possible mutations in cases where the integrity cannot be guaranteed. The recent progress in translating traditional chemical protocols to explore the milder conditions of photocatalysis has opened a new avenue for the DELT scenario, and we believe that in combination with other suitable methods that can be adapted and designed, this screening modality will soon revolutionize how drug discovery can be performed, and thus it is a promising tool for the present and future developments in the pharmaceutical arena.
ACKNOWLEDGMENT
The authors acknowledge all the current and former Molander group members that have contributed to the development of new synthetic methods for DNA-encoded library synthesis. We would also like to thank our collaborators at GlaxoSmithKline (GSK) and AbbVie for all their assistance in developing projects, useful discussions, providing DNA headpiece-NH2 materials, and in obtaining qPCR and sequencing data.
Funding Sources
The authors are grateful for financial support provided by NIGMS (R35 GM 131680 to G.M.). Financial support for this research was also provided in part by GlaxoSmithKline and AbbVie.
Biographies
Dr. Bianca Matsuo received her B.Sc. (2015) and M.Sc. (2017) degrees in Chemistry from the Federal University of São Carlos, SP, Brazil. She completed her doctoral studies under the supervision of Professor Marcio Paixão at the same university in 2021, working on the development of radical-based protocols by means of organic photoredox catalysis. Currently, she is a postdoctoral researcher in the group of Professor Gary A. Molander at the University of Pennsylvania. Her research interests are focused on the development of new photocatalytic strategies and their application to DEL synthesis.
Dr. Albert Granados was born in Sabadell (Barcelona, Spain). He undertook his undergraduate studies at the Universitat Autònoma de Barcelona (UAB). In 2014, he received his MSc in Electrochemistry, Science and Technology under the supervision of Prof. Iluminada Gallardo. In 2014, he joined the Vallribera group at UAB, where he received his Ph.D. Subsequently, he pursued postdoctoral studies with Prof. Roser Pleixats and Prof. Adelina Vallribera at UAB. In 2021, he began another postdoctoral stint in the Molander group at UPenn. In 2023, he has been appointed as a Lecturer in Organic Chemistry at UAB. His research focuses on organofluorous chemistry, (photo)catalysis, DEL synthesis, and materials science.
After receiving a two-year technical degree at the UIT of Créteil-Vitry in France (2013), Dr. Guillaume Levitre continued his studies with a B.Sc. in Chemistry-Biology (2014) and then received a M.Sc. in Chemistry (2016) at the Paris-Saclay University (France). From late 2016 to late 2019, he carried out his Ph.D. studies under the supervision of Dr. Géraldine Masson at the Institut de Chimie des Substances Naturelles (ICSN, France), where he developed photocatalytic and enantioselective organocatalytic processes for the synthesis of complex molecules. In 2019, he moved to the University of Geneva (Geneva, Switzerland) as a postdoctoral fellow to study rhodium and ruthenium catalysis for the synthesis and functionalization of 2-vinyloxymalonate enol ethers with Prof. Jérôme Lacour, remaining there until July, 2021. Since September 2021, he has been a postdoctoral research associate in the group of Prof. Gary Molander, investigating functionalizations of phenyl bioisosteres via photoredox processes.
Professor Gary A. Molander completed his undergraduate studies in Chemistry at Iowa State University under the tutelage of Professor Richard C. Larock. He earned his Ph.D. at Purdue University with Professor Herbert C. Brown, where later he was also a postdoctoral research associate. He undertook postdoctoral training with Professor Barry M. Trost at the University of Wisconsin, Madison as an NIH Postdoctoral Fellow. He began his academic career at the University of Colorado, Boulder, moving to the University of Pennsylvania in 1999. Professor Molander has also served as a visiting professor in several countries.
REFERENCES
- (1).Brown DG; Bostrom J Where Do Recent Small Molecule Clinical Development Candidates Come From? J. Med. Chem 2018, 61, 9442–9468. [DOI] [PubMed] [Google Scholar]
- (2).Brenner S; Lerner RA Encoded Combinatorial Chemistry. P. Natl. Acad. Sci. U.S.A 1992, 89, 5381–5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wassermann AM; Camargo LM; Auld DS Composition and applications of focus libraries to phenotypic assays. Front. Pharmacol 2014, 5, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Erlanson DA; Fesik SW; Hubbard RE; Jahnke W; Jhoti H Twenty years on: the impact of fragments on drug discovery. Nat. Rev. Drug Discov 2016, 15, 605–619. [DOI] [PubMed] [Google Scholar]
- (5).Song M; Hwang GT DNA-Encoded Library Screening as Core Platform Technology in Drug Discovery: Its Synthetic Method Development and Applications in DEL Synthesis. J. Med. Chem 2020, 63, 6578–6599. [DOI] [PubMed] [Google Scholar]
- (6).Clark MA; Acharya RA; Arico-Muendel CC; Belyanskaya SL; Benjamin DR; Carlson NR; Centrella PA; Chiu CH; Creaser SP; Cuozzo JW; Davie CP; Ding Y; Franklin GJ; Franzen KD; Gefter ML; Hale SP; Hansen NJV; Israel DI; Jiang JW; Kavarana MJ; Kelley MS; Kollmann CS; Li F; Lind K; Mataruse S; Medeiros PF; Messer JA; Myers P; O’Keefe H; Oliff MC; Rise CE; Satz AL; Skinner SR; Svendsen JL; Tang LJ; van Vloten K; Wagner RW; Yao G; Zhao BG; Morgan BA Design, synthesis and selection of DNA-encoded small-molecule libraries. Nat. Chem. Biol 2009, 5, 772–772. [DOI] [PubMed] [Google Scholar]
- (7).Harris PA; King BW; Bandyopadhyay D; Berger SB; Campobasso N; Capriotti CA; Cox JA; Dare L; Dong XY; Finger JN; Grady LC; Hoffmann SJ; Jeong JU; Kang J; Kasparcova V; Lakdawala AS; Lehr R; McNulty DE; Nagilla R; Ouellette MT; Pao CS; Rendina AR; Schaeffer MC; Summerfield JD; Swift BA; Totoritis RD; Ward P; Zhang AM; Zhang DH; Marquis RW; Bertin J; Gough PJ DNA-Encoded Library Screening Identifies Benzo[b][1,4]oxazepin-4-ones as Highly Potent and Monoselective Receptor Interacting Protein 1 Kinase Inhibitors. J. Med. Chem 2016, 59, 2163–2178. [DOI] [PubMed] [Google Scholar]
- (8).Harris PA; Berger SB; Jeong JU; Nagilla R; Bandyopadhyay D; Campobasso N; Capriotti CA; Cox JA; Dare L; Dong XY; Eidam PM; Finger JN; Hoffman SJ; Kang J; Kasparcova V; King BW; Lehr R; Lan YF; Leister LK; Lich JD; MacDonald TT; Miller NA; Ouellette MT; Pao CS; Rahman A; Reilly MA; Rendina AR; Rivera EJ; Schaeffer MC; Sehon CA; Singhaus RR; Sun HH; Swift BA; Totoritis RD; Vossenkamper A; Ward P; Wisnoski DD; Zhang DH; Marquis RW; Gough PJ; Bertin J Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J. Med. Chem 2017, 60, 1247–1261. [DOI] [PubMed] [Google Scholar]
- (9).Malone ML; Paegel BM What is a “DNA-Compatible” Reaction? ACS Comb. Sci 2016, 18, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zhu HY; Flanagan ME; Stanton RV Designing DNA Encoded Libraries of Diverse Products in a Focused Property Space. J. Chem. Inf. Model 2019, 59, 4645–4653. [DOI] [PubMed] [Google Scholar]
- (11).Martin A; Nicolaou CA; Toledo MA Navigating the DNA encoded libraries chemical space. Commun. Chem 2020, 3, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Nielsen J; Brenner S; Janda KD Synthetic methods for the implementation of encoded combinatorial chemistry. J. Am. Chem. Soc 1993, 115, 9812–9813. [Google Scholar]
- (13).Goodnow RA A handbook for DNA-encoded chemistry : theory and applications for exploring chemical space and drug discovery, Wiley-VCH: Weinheim, 2014. [Google Scholar]
- (14).Shi Y; Wu YR; Yu JQ; Zhang WN; Zhuang CL DNA-encoded libraries (DELs): a review of on-DNA chemistries and their output. RSC Adv. 2021, 11, 2359–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Fitzgerald PR; Paegel BM DNA-Encoded Chemistry: Drug Discovery from a Few Good Reactions. Chem. Rev 2021, 121, 7155–7177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Patel S; Badir SO; Molander GA Developments in Photoredox-Mediated Alkylation for DNA-Encoded Libraries. Trends Chem. 2021, 3, 161–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Fair RJ; Walsh RT; Hupp CD The expanding reaction toolkit for DNA-encoded libraries. Bioorg. Med. Chem. Lett 2021, 51, 1–24. [DOI] [PubMed] [Google Scholar]
- (18).Strieth-Kalthoff F; James MJ; Teders M; Pitzer L; Glorius F Energy transfer catalysis mediated by visible light: principles, applications, directions. Chem. Soc. Rev 2018, 47, 7190–7202. [DOI] [PubMed] [Google Scholar]
- (19).Narayanam JMR; Stephenson CRJ Visible light photoredox catalysis: applications in organic synthesis. Chem. Soc. Rev 2011, 40, 102–113. [DOI] [PubMed] [Google Scholar]
- (20).Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Teegardin K; Day JI; Chan J; Weaver J Advances in Photocatalysis: A Microreview of Visible Light Mediated Ruthenium and Iridium Catalyzed Organic Transformations. Org. Process Res. Dev 2016, 20, 1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Matsui JK; Lang SB; Heitz DR; Molander GA Photoredox-Mediated Routes to Radicals: The Value of Catalytic Radical Generation in Synthetic Methods Development. ACS Catal. 2017, 7, 2563–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Buzzetti L; Crisenza GEM; Melchiorre P Mechanistic Studies in Photocatalysis. Angew. Chem. Int. Edit 2019, 58, 3730–3747. [DOI] [PubMed] [Google Scholar]
- (24).Romero NA; Nicewicz DA Organic Photoredox Catalysis. Chem. Rev 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]
- (25).Marzo L; Pagire SK; Reiser O; Konig B Visible-Light Photocatalysis: Does It Make a Difference in Organic Synthesis? Angew. Chem. Int. Edit 2018, 57, 10034–10072. [DOI] [PubMed] [Google Scholar]
- (26).Tucker JW; Stephenson CRJ Shining Light on Photoredox Catalysis: Theory and Synthetic Applications. J. Org. Chem 2012, 77, 1617–1622. [DOI] [PubMed] [Google Scholar]
- (27).Arias-Rotondo DM; McCusker JK The photophysics of photoredox catalysis: a roadmap for catalyst design. Chem. Soc. Rev 2016, 45, 5803–5820. [DOI] [PubMed] [Google Scholar]
- (28).Lima CGS; Lima TD; Duarte M; Jurberg ID; Paixão MW Organic Synthesis Enabled by Light-Irradiation of EDA Complexes: Theoretical Background and Synthetic Applications. ACS Catal. 2016, 6, 1389–1407. [Google Scholar]
- (29).Wang J; Lundberg H; Asai S; Martin-Acosta P; Chen JS; Brown S; Farrell W; Dushin RG; O’Donnell CJ; Ratnayake AS; Richardson P; Liu Z; Qin T; Blackmond DG; Baran PS Kinetically guided radical-based synthesis of C(sp3)-C(s p3) linkages on DNA. Proc. Natl. Acad. Sci. U.S.A 2018, 115, E6404–E6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kolmel DK; Loach RP; Knauber T; Flanagan ME Employing Photoredox Catalysis for DNA-Encoded Chemistry: Decarboxylative Alkylation of α-Amino Acids. ChemMedChem. 2018, 13, 2159–2165. [DOI] [PubMed] [Google Scholar]
- (31).Chu LL; Ohta C; Zuo ZW; MacMillan DWC Carboxylic Acids as A Traceless Activation Group for Conjugate Additions: A Three-Step Synthesis of (+/−)-Pregabalin. J. Am. Chem. Soc 2014, 136, 10886–10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Phelan JP; Lang SB; Sim J; Berritt S; Peat AJ; Billings K; Fan LJ; Molander GA Open-Air Alkylation Reactions in Photoredox-Catalyzed DNA-Encoded Library Synthesis. J. Am. Chem. Soc 2019, 141, 3723–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Debaene F; Mejias L; Harris JL; Winssinger N Synthesis of a PNA-encoded cysteine protease inhibitor library. Tetrahedron. 2004, 60, 8677–8690. [Google Scholar]
- (34).Gartner ZJ; Tse BN; Grubina R; Doyon JB; Snyder TM; Liu DR DNA-templated organic synthesis and selection of a library of macrocycles. Science. 2004, 305, 1601–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Halpin DR; Harbury PB DNA display I. Sequence-encoded routing of DNA populations. PLoS Biol. 2004, 2, 1015–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Vummidi BR; Farrera-Soler L; Daguer JP; Dockerill M; Barluenga S; Winssinger N A mating mechanism to generate diversity for the Darwinian selection of DNA-encoded synthetic molecules. Nat. Chem 2022, 14, 141–152. [DOI] [PubMed] [Google Scholar]
- (37).Bassi G; Favalli N; Oehler S; Martinelli A; Catalano M; Scheuermann J; Neri D Comparative evaluation of DNA-encoded chemical selections performed using DNA in single-stranded or double-stranded format. Biochem. Bioph. Res. Co 2020, 533, 223–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Gui YH; Wong CS; Zhao GX; Xie C; Hou R; Li YZ; Li G; Li XY Converting Double-Stranded DNA-Encoded Libraries (DELs) to Single-Stranded Libraries for More Versatile Selections. ACS Omega. 2022, 7, 11491–11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Yan M; Lo JC; Edwards JT; Baran PS Radicals: Reactive Intermediates with Translational Potential. J. Am. Chem. Soc 2016, 138, 12692–12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Lakowicz JR Principles of fluorescence spectroscopy, 3rd ed.; Springer: New York, 2007. [Google Scholar]
- (41).Murarka S N-(Acyloxy)phthalimides as Redox-Active Esters in Cross-Coupling Reactions. Adv. Synth. Catal 2018, 360, 1735–1753. [Google Scholar]
- (42).Karmakar S; Silamkoti A; Meanwell NA; Mathur A; Gupta AK Utilization of C(sp3)-Carboxylic Acids and Their Redox-Active Esters in Decarboxylative Carbon−Carbon Bond Formation. Adv. Synth. Catal 2021, 363, 3693–3736. [Google Scholar]
- (43).Potowski M; Kunig VBK; Eberlein L; Vakalopoulos A; Kast SM; Brunschweiger A Chemically Stabilized DNA Barcodes for DNA-Encoded Chemistry. Angew. Chem. Int. Edit 2021, 60, 19744–19749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Sauter B; Schneider L; Stress C; Gillingham D An assessment of the mutational load caused by various reactions used in DNA encoded libraries. Bioorgan. Med. Chem 2021, 52. [DOI] [PubMed] [Google Scholar]
- (45).Breen AP; Murphy JA Reactions of Oxyl Radicals with DNA. Free Radic. Biol. Med 1995, 18, 1033–1077. [DOI] [PubMed] [Google Scholar]
- (46).Ratnayake AS; Flanagan ME; Foley TL; Smith JD; Johnson JG; Bellenger J; Montgomery JI; Paegel BM A Solution Phase Platform to Characterize Chemical Reaction Compatibility with DNA-Encoded Chemical Library Synthesis. ACS Comb. Sci 2019, 21, 650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Lang SB; Wiles RJ; Kelly CB; Molander GA Photoredox Generation of Carbon-Centered Radicals Enables the Construction of 1,1-Difluoroalkene Carbonyl Mimics. Angew. Chem. Int. Edit 2017, 56, 15073–15077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Badir SO; Sim J; Billings K; Csakai A; Zhang XG; Dong WZ; Molander GA Multifunctional Building Blocks Compatible with Photoredox-Mediated Alkylation for DNA-Encoded Library Synthesis. Org. Lett 2020, 22, 1046–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Krumb M; Kammer LM; Badir SO; Cabrera-Afonso MJ; Wu VE; Huang MX; Csakai A; Marcaurelle LA; Molander GA Photochemical C-H arylation of heteroarenes for DNA-encoded library synthesis. Chem. Sci 2022, 13, 1023–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Yen-Pon E; Li LB; Levitre G; Majhi J; McClain EJ; Voight EA; Crane EA; Molander GA On-DNA Hydroalkylation to Introduce Diverse Bicyclo[1.1.1]pentanes and Abundant Alkyls via Halogen Atom Transfer. J. Am. Chem. Soc 2022, 144, 12184–12191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Tellis JC; Primer DN; Molander GA Single-electron transmetalation in organoboron cross-coupling by photoredox/nickel dual catalysis. Science. 2014, 345, 433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Chan AY; Perry IB; Bissonnette NB; Buksh BF; Edwards GA; Frye LI; Garry OL; Lavagnino MN; Li BX; Liang Y; Mao E; Millet A; Oakley JV; Reed NL; Sakai HA; Seath CP; MacMillan DWC Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis. Chem. Rev 2022, 122, 1485–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Remeur C; Kelly CB; Patel NR; Molander GA Aminomethylation of Aryl Halides using alpha-Silylamines Enabled by Ni/Photoredox Dual Catalysis. ACS Catal. 2017, 7, 6065–6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Badir SO; Lipp A; Krumb M; Cabrera-Afonso MJ; Kammer LM; Wu VE; Huang MX; Csakai A; Marcaurelle LA; Molander GA Photoredox-mediated hydroalkylation and hydroarylation of functionalized olefins for DNA-encoded library synthesis. Chem. Sci 2021, 12, 12036–12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Kolmel DK; Ratnayake AS; Flanagan ME Photoredox cross-electrophile coupling in DNA-encoded chemistry. Biochem. Biophys. Res. Commun 2020, 533, 201–208. [DOI] [PubMed] [Google Scholar]
- (56).Chowdhury R; Yu Z; Tong ML; Kohlhepp SV; Yin X; Mendoza A Decarboxylative Alkyl Coupling Promoted by NADH and Blue Light. J. Am. Chem. Soc 2020, 142, 20143–20151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Kolmel DK; Ratnayake AS; Flanagan ME; Tsai MH; Duan C; Song C Photocatalytic [2 + 2] Cycloaddition in DNA-Encoded Chemistry. Org. Lett 2020, 22, 2908–2913. [DOI] [PubMed] [Google Scholar]
- (58).Wen H; Ge R; Qu Y; Sun J; Shi X; Cui W; Yan H; Zhang Q; An Y; Su W; Yang H; Kuai L; Satz AL; Peng X Synthesis of 1,2-Amino Alcohols by Photoredox-Mediated Decarboxylative Coupling of alpha-Amino Acids and DNA-Conjugated Carbonyls. Org. Lett 2020, 22, 9484–9489. [DOI] [PubMed] [Google Scholar]
- (59).Ruff Y; Martinez R; Pelle X; Nimsgern P; Fille P; Ratnikov M; Berst F An Amphiphilic Polymer-Supported Strategy Enables Chemical Transformations under Anhydrous Conditions for DNA-Encoded Library Synthesis. ACS Comb. Sci 2020, 22, 120–128. [DOI] [PubMed] [Google Scholar]
- (60).Shan J; Ling X; Liu J; Wang X; Lu X DNA-encoded CH functionality via photoredox-mediated hydrogen atom transformation catalysis. Bioorg. Med. Chem 2021, 42, 116234. [DOI] [PubMed] [Google Scholar]
- (61).Zhang Y; Luo H; Ma H; Wan J; Ji Y; Shaginian A; Li J; Deng Y; Liu G On-DNA Derivatization of Quinoxalin-2-ones by Visible-Light-Triggered Alkylation with Carboxylic Acids. Bioconjug. Chem 2021, 32, 1576–1580. [DOI] [PubMed] [Google Scholar]
- (62).Wu R; Du T; Sun W; Shaginian A; Gao S; Li J; Wan J; Liu G Functionalization of DNA-Tagged Alkenes Enabled by Visible-Light-Induced C-H Activation of N-Aryl Tertiary Amines. Org. Lett 2021, 23, 3486–3490. [DOI] [PubMed] [Google Scholar]
- (63).Lin B; Lu W; Chen ZY; Zhang Y; Duan YZ; Lu X; Yan M; Zhang XJ Enhancing the Potential of Miniature-Scale DNA-Compatible Radical Reactions via an Electron Donor-Acceptor Complex and a Reversible Adsorption to Solid Support Strategy. Org. Lett 2021, 23, 7381–7385. [DOI] [PubMed] [Google Scholar]
- (64).Badir SO; Lipp A; Krumb M; Cabrera-Afonso MJ; Kammer LM; Wu VE; Huang M; Csakai A; Marcaurelle LA; Molander GA Photoredox-mediated hydroalkylation and hydroarylation of functionalized olefins for DNA-encoded library synthesis. Chem. Sci 2021, 12, 12036–12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Zhang Y; Xia S; Shi W.-x.; Lin B; Su X.-c.; Lu W; Wu X; Wang X; Lu X; Yan M; Zhang X.-j. Radical C−H Sulfonation of Arenes: Its Applications on Bioactive and DNA-Encoded Molecules. Org. Lett 2022, 24, 7961–7966. [DOI] [PubMed] [Google Scholar]