

Abstract
The activation and utilization of substrates mediated by Frustrated Lewis Pairs (FLPs) was initially believed to occur solely via a two-electron, cooperative mechanism. More recently, the occurrence of a single-electron transfer (SET) from the Lewis base to the Lewis acid was observed, indicating that mechanisms that proceed via one-electron-transfer processes are also feasible. As such, SET in FLP systems leads to the formation of radical ion pairs, which have recently been more frequently observed. In this review, we aim to discuss the seminal findings regarding the recently established insights into the SET processes in FLP chemistry as well as highlight examples of this radical formation process. In addition, applications of reported main group radicals will also be reviewed and discussed in the context of the understanding of SET processes in FLP systems.
1. Introduction
In 1942, Brown and co-workers were the first to observe the influence of steric hindrance on the interaction between Lewis acids and Lewis bases.1 They showed that the combination of trimethyl borane and lutidine are unable to form a classic Lewis acid–base adduct, even at temperatures as low as −80 °C (Scheme 1A). The authors explained these findings by suggesting that steric hindrance prevents the formation of the Lewis adduct. Decades later, in 2006, the group of Stephan et al. showed for the first time that a combination of sterically encumbered Lewis acids and bases can also induce intriguing reactivity.2 They demonstrated that phosphine borane 1 can bind dihydrogen and release it again upon heating (Scheme 1B). A year later, Stephan et al. introduced the term Frustrated Lewis Pairs (FLPs) to highlight cases in which the Lewis acid and base are unable to form a classical Lewis adduct.3 Since these seminal reports, the use of FLPs has been widely explored and rejuvenated the field of main group chemistry and catalysis.4−7
Scheme 1. First Report on (A) Prevention of Adduct Formation and (B) Reactivity Induced by a Sterically Encumbered Lewis Acid and Base.

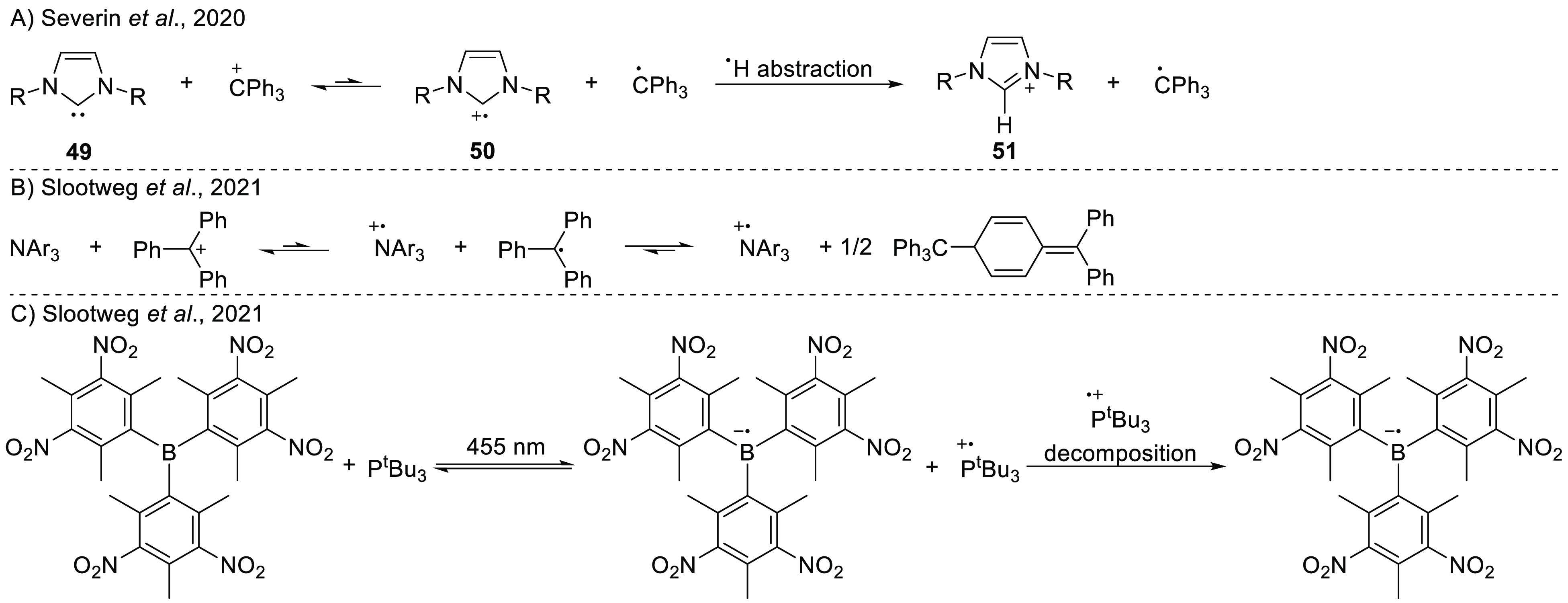
Typically, the reactivity of FLP systems is described by two-electron chemistry, where the Lewis acid and base act cooperatively.8,9 In this mechanism, it is now recognized that the Lewis acid and base first form a so-called encounter complex EDA, as shown in Scheme 2A.10,11 Next, the substrate is heterolytically activated by simultaneous donation of electron density from the Lewis base lone pair into an antibonding orbital of the substate (e.g., the H–H σ*-orbital of H2) and the acceptance of electron density from a bonding orbital of the substrate (e.g., the H–H σ-orbital of H2) by the Lewis acid. In addition to these early reports, Piers et al. postulated in 2011 that FLPs can also react via one-electron pathways.12 For the activation of dihydrogen by the PtBu3/B(C6F5)3 FLP, the authors envisioned that one-electron oxidation of the phosphine by the borane via a single-electron transfer (SET) event occurs as the first reaction step, yielding the radical ion pair PtBu3•+/B(C6F5)3•–2 (Scheme 2B). Subsequently, dihydrogen is activated homolytically to yield the same phosphonium borohydride salt [tBu3PH][HB(C6F5)3] as obtained via the heterolytic cleavage of the H–H bond. Piers et al. presumed that reaction progress through this radical mechanism was unlikely to be the major contribution due to the large disparity in redox potentials between the PtBu3 (0.90 V vs Fc/Fc+ in MeCN13) and B(C6F5)3 (−1.79 V vs Fc/Fc+ in DCM14). This difference results in only a small amount of the intermediate radical ion pair 2 being present in solution, which does not correlate with the observation that PtBu3/B(C6F5)3 promptly reacts with dihydrogen.15 Later, Slootweg and co-workers showed with ultrafast transient absorption spectroscopy that the lifetime of the radical ion pair PtBu3•+/B(C6F5)3•–2 is very short (6 ps) due to rapid back electron transfer to the ground-state electron donor–acceptor (EDA) complex [PtBu3, B(C6F5)3], which prevents subsequent radical reactivity.16 Therefore, in this case, dihydrogen is solely activated heterolytically.17
Scheme 2. (A) Two-Electron Reactivity of an FLP System via the Formation of an Encounter Complex, EDA; (B) First Postulation of Radical Formation in an FLP System; (C) Single-Electron Oxidation of 3 by B(C6F5)3 Leads to the Formation of Radical Cation 4.

Two years after the proposal of radical ion pairs in FLP chemistry by Piers et al., the Wang group provided the first direct experimental evidence of SET between a Lewis base and Lewis acid.18 Namely, upon mixing arylamine 3 and B(C6F5)3 and stirring for 3 days at room temperature, the authors obtained a deep blue solution due to the formation of the amine radical cation 4, which they proved using electron paramagnetic resonance (EPR) spectroscopy (Scheme 2C; Figure 1; see Table 1 for parameters). The corresponding borane radical anion B(C6F5)3•– was however not observed, which is associated with its facile decomposition. Yet, the EPR spectrum of the B(C6F5)3•– radical anion has been previously reported at low temperatures and is characterized by a rich hyperfine structure originating from the nuclear spin-active 10,11B isotopes and fluorine substituents at all positions on the phenyl ring (Figure 1; see Table 1 for parameters).14,19−21
Figure 1.

Isotropic CW X-band EPR spectra of radicals 4 and B(C6F5)3•–; simulated using data reported in Table 1.
Table 1. Spin Hamiltonian Parameters for Radical Species Generated during FLP Reactions.
| radical | giso | aiso/mTa | reference |
|---|---|---|---|
| Phosphorus | |||
| PMes3•+ | 2.009; g|| = 2.010; g⊥ = 2.013 | 23.8; A|| = 40.30; A⊥ = 15.98 | (33) |
| PtBu3•+ | 2.005; g|| = 2.0012; g⊥ = 2.0065 | 30.00; A|| = 48.65; A⊥ = 20.67 | (16) |
| PTipp•+ | 2.007; g|| = 2.002; g⊥ = 2.009 | 22.55; A|| = 41.58; A⊥ = 13.03 | (20) |
| (PEt3)2•+ | 2.008; g|| = 2.00; g⊥ = 2.012 | 45.44; A|| = 53.76; A⊥ = 41.27 | (83) |
| (PtBu3)2•+ | 2.008; g|| = 2.00; g⊥ = 2.012 | 46.18; A|| = 54.80; A⊥ = 41.88 | (83) |
| (PMes3)2•+ | 2.008; g|| = 2.003; g⊥ = 2.011 | 16.54; A|| = 26.90; A⊥ = 11.39 | (57) |
| [(p-ClC6H4)N2(PtBu3)]•(37) | 2.005 | P = 0.88; N1 = 0.42; N2 = 0.36; Ho = 0.36; Cl = 0.28 | (55) |
| Nitrogen | |||
| (C(CH3)2C6H3)3N (4) | 2.002 | 14N: 0.94; 1Hp(3): 0.304; 1Hm(6): 0.071 | (18) |
| tBu3P–NO• | 2.0071 | 14N = 1.05; 31P = 1.21 | (13) |
| C6H4(NMe2)2(70•+) | 2.0034 | 14N(2) = 0.7; 1H(4) = 0.195; 1H(12) = 0.655 | (75) |
| (Me2PhN–C6H5)2 (72) | 2.0033 | 14N(2) = 0.45 | (75) |
| Br–C6H4–N(Me)–CH2–TMS (85) | 2.0033 | 14N(1) = 0.824; 1H(3) = 0.738 ;1H(2) = 0.992; 1H(2) = 0.345; 1H(2) = 0.133; Si(1) = 0.313 | (81) |
| Br–C6H4–N(Me)2(88) | 2.0029 | 14N(1) = 1.174; 1H(3) = 0.773; 1H(2) = 0.414; 1H(2) = 0.205 | (81) |
| Boron | |||
| B(C6F5)3•– | 2.011 | B: 1.10; Fo(6): 0.46; Fm(6): 0.13; Fp(3): 0.53 | (19) |
| Germanium | |||
| [BCHGe]•+(47′) | 1.988 | 177,179Hf: 8.5 | (59) |
| Carbon | |||
| CPh3• | 1.999 | Ho(6): 0.26; Hm(6): 0.11; Hp(3): 0.28 | (20) |
| C(C6F5)3• | 2.003 | n.r. | (52) |
| C(C6H3O)3•(27) | 2.005 | Hm(6) = 0.091; Hp(3) = 0.328 | (53) |
| (C6H4)2CH2•(42) | 2.006 | H1 = 2.28; H3(2) = 0.33; H6(2) = 0.27 | (56) |
| (B(C6F5)3)2-9,10-anthraquinone (70•–) | 2.004 | n.r. | (75) |
aiso/MHz = 10–9 (gμB/h) aiso/mT.
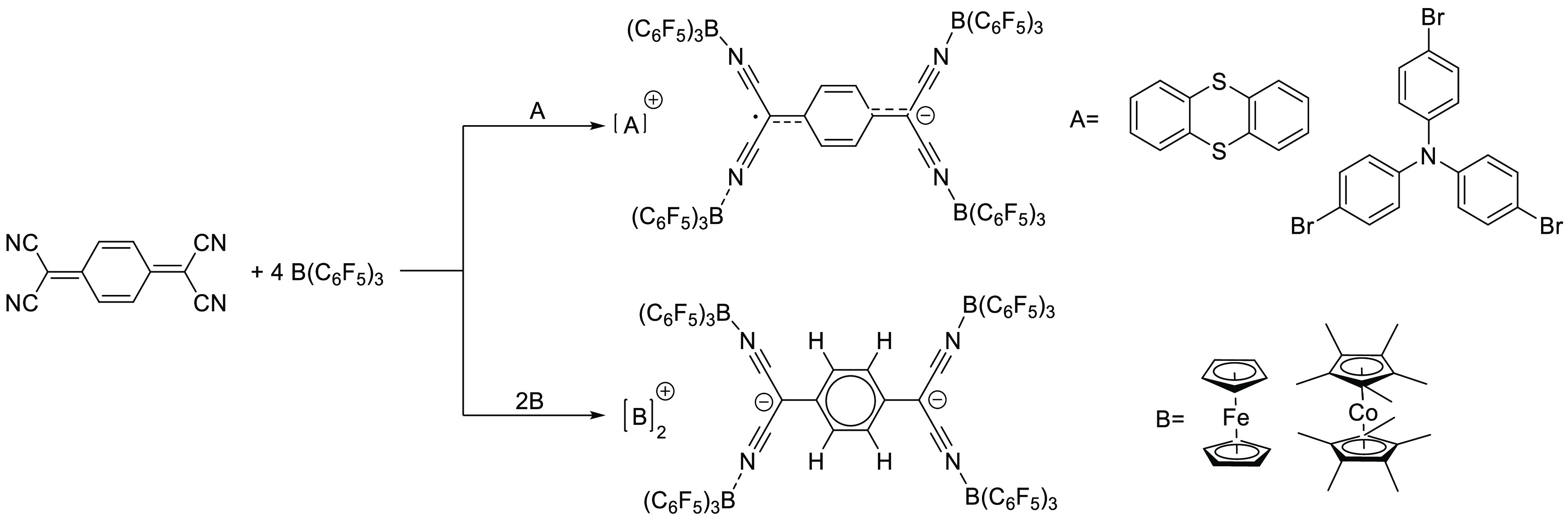
To date, three different approaches for the formation of radicals in FLP chemistry have been identified.22−25 First, the FLP can react with a radical substrate X, such as nitric oxide, to yield a “trimer” radical of the form LA–X–LB 5 (Scheme 3A).26−31 The second route involves the use of a transition-metal-based electron donor, such as ferrocene or cobaltocene, to reduce the Lewis acid to the corresponding radical anion (Scheme 3B).23 Finally, the generation of radical ion pairs (RIPs) can proceed via direct SET between a Lewis base and Lewis acid that function as an electron donor and electron acceptor, respectively (Scheme 3C). These RIPs were coined Frustrated Radical Pairs (FRPs) by Stephan et al. Notably, the radical ion pair can be converted back to the closed-shell FLP system via back electron transfer (BET),32 depending on the relative energies of the two states. In this review, to support discussion of the three alternative approaches to radical formation in FLP chemistry, we shall highlight selected main-group chemistry examples where we believe SET mechanisms are prevalent and focus on the mechanistic details of the SET process and the corresponding spectroscopic findings.
Scheme 3. Three Methods to Obtain Radicals in FLP Chemistry: (A) Incorporation of a Radical Substrate into an FLP to Form a Radical “Trimer”; (B) One-Electron Reduction of a Lewis Acid by a Transition-Metal-Based Single-Electron Donor; (C) SET between a LB and LA to Form an RIP.

LA = Lewis Acid, LB = Lewis base, ED = electron donor, X = radical substrate.
2. Photoinduced Single-Electron Transfer
In 2020, Slootweg et al. reported on key insights into the mechanism of SET in FLP chemistry.16 Prior to this contribution, Stephan et al. had observed only the phosphine radical cation for the PMes3/B(C6F5)3 combination in chlorobenzene.33 In-line with the findings of Piers et al., Slootweg et al. concluded that thermally activated SET for this combination of Lewis acid and base is not feasible. Namely, DFT calculations at the SCRF (toluene)/ωB97X-D/6-311+G(d,p) level of theory showed that the electron affinity of B(C6F5)3 is 3.03 eV (conversion factor: 1.00 eV = 23.0621 kcal/mol), while the ionization energy of PMes3 is 5.54 eV, resulting in an energy difference between the closed shell PMes3/B(C6F5)3 and the radical ion pair PMes3•+/B(C6F5)3•– of 2.51 eV (57.8 kcal/mol; Figure 2). A similar result was computed for PtBu3/B(C6F5)3, for which an even larger energy difference of 2.92 eV (67.2 kcal/mol) for the PtBu3•+/B(C6F5)3•– RIP was found due to the reduced stabilization of the phosphorus radical cation.20,21,32 These energy differences indicate that whereas SET is unlikely to be induced by a thermal reaction, interaction with visible light (λ = 400–800 nm, ΔE = 71.4–35.7 kcal/mol) is however feasible.
Figure 2.

Energy required for the formation of a radical ion pair can be estimated from the electron donor’s ionization energy (IE) and the electron acceptor’s electron affinity (EA).16 Calculations at the SCRF/ωB97X-D/6-311+G(d,p) (solvent = toluene) level of theory.
Inspired by the pioneering work of Mulliken and Kochi et al.,34,35 Slootweg et al. realized that the Mulliken theory can be used to explain the observation of these radical ion pairs.16 Namely, an electron donor and an electron acceptor (classified as Lewis base and Lewis acid in Frustrated Lewis Pair chemistry) can combine to form an electron donor–acceptor (EDA) complex, which is visualized in Scheme 2A as the encounter complex EDA.34,35 This EDA complex can be characterized by UV–vis spectroscopy36 and shows a new absorption band at longer wavelengths compared to the absorptions of the individual FLP components. Irradiation of the FLP system using selective wavelengths that align with the absorption band of the EDA leads to a photoinduced SET from the electron donor to the acceptor.
The reported UV–vis spectra of the PMes3/B(C6F5)3 (violet) and PtBu3/B(C6F5)3 (pale yellow) FLPs in toluene showed a new absorption band to be present for both (at λmax = 534 nm and λmax ≈ 400 nm, respectively; see Figure 3).16 Time-dependent DFT (TD-DFT) calculations also predicted the presence of these new absorption bands [PMes3/B(C6F5)3: 439 nm (fosc = 0.0184); and PtBu3/B(C6F5)3: 400 nm (fosc = 0.0719)]. Analysis of the frontier molecular orbitals showed that the donor orbital (HOMO) contains the phosphine lone pair, while the acceptor is the empty orbital (LUMO) located on B(C6F5)3, underlining that FLP systems are ideally suited for single-electron-transfer processes.
Figure 3.
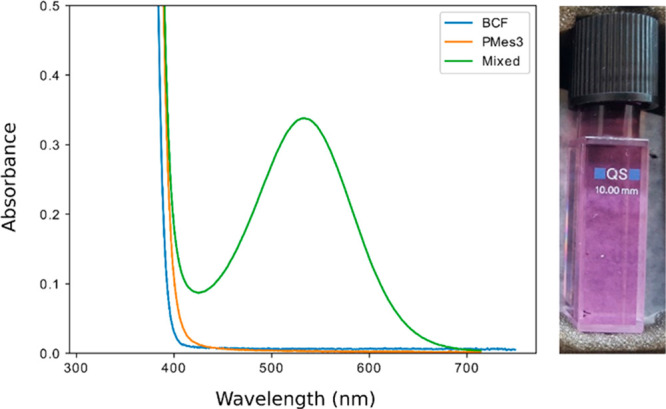
UV–vis absorption spectrum of a 1.5 × 10–2 M mixture of B(C6F5)3 and PMes3 in toluene, indicating the formation of a new EDA absorption band.
To confirm that the SET in these phosphine/borane systems is photoinduced, EPR spectroscopy was performed in toluene at 30 K.16 When measuring the samples in the dark, no signals were observed in the EPR spectra, highlighting that the color of the solution stems from the closed-shell EDA complex and not from the presence of radicals. On the other hand, upon irradiation with visible light (390–500 nm), SET was promoted to form the corresponding RIP, as evidenced by the observation of two separate signals in the EPR spectra (Figure 4), which could be assigned for the first time to both the phosphine radical cation and the borane radical anion. Subsequently, by using transient absorption spectroscopy, the lifetime of the radical ion pair state in toluene at room temperature could be determined to be 237 and 6 ps for PMes3/B(C6F5)3 and PtBu3/B(C6F5)3, respectively. These results indicate that BET occurs quickly after SET, leading to an equilibrium between the ground state and the radical state, which lies heavily on the side of the EDA complex, as shown in Scheme 4. These results prove that the color of these solutions (in the dark) is due to the existence of a charge-transfer band, in contrast to earlier reports where typically the color was assigned to the presence of radicals.33
Figure 4.
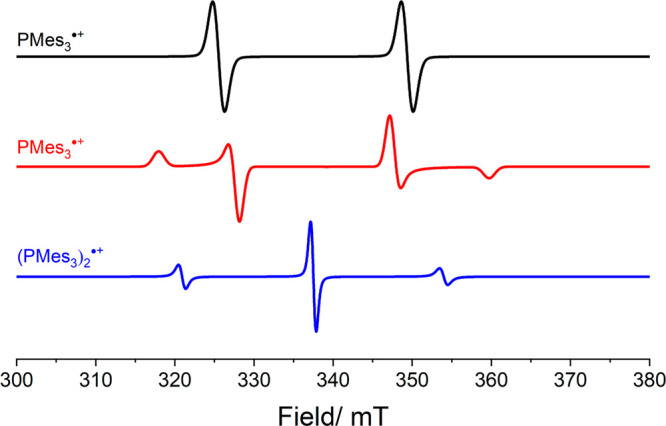
Isotropic (top) and anisotropic (middle) CW X-band EPR spectra of PMes3•+, and (PMes3)2•+ (bottom); simulated using data reported in Table 1.
Scheme 4. Photolytic Single-Electron Transfer between a Lewis Acid (LA) and a Lewis Base (LB) Following Mulliken Theory As Postulated by Slootweg et al.16.

Similarly, an equimolar mixture of Mes3P and Al(C6F5)3 in dry toluene or chlorobenzene was reported by Stephan et al. to yield, without exclusion of light, a distinctly purple-colored solution.33 The room-temperature EPR spectrum of the reaction mixture showed a doublet resonance with a hyperfine coupling constant of 23.8 mT centered at giso = 2.0089, assigned to PMes3•+ of the corresponding radical ion pair PMes3•+/Al(C6F5)3•– (Figure 4).17 No evidence of the radical anion Al(C6F5)3•– could be observed via EPR, analogous to the boron-based radical anion.33 Based on the findings of the photoinduced SET, we expect the formation of PMes3•+ in this case is due to irradiation of the charge-transfer band by visible light and that the charge-transfer band causes the purple color. Similar to B(C6F5)3•–, the absence of Al(C6F5)3•– can be explained by facile decomposition.
Subsequently, Marques and Ando investigated photoinduced SET in FLP chemistry using resonance Raman spectroscopy aided by supporting DFT calculations.37 Resonance Raman spectroscopy of the archetypal PMes3/B(C6F5)3 system employing 457 nm excitation to overlap with the charge-transfer band showed the increase in signal intensity of specific signals compared to the normal Raman spectra (excitation at 1064 nm). The excitation in resonance with the charge-transfer band causes greater changes in the polarizability tensor of the vibrational modes associated with the EDA complex [PMes3, B(C6F5)3] and hence selective enhancement of Raman bands that were specifically attributed to the radical ion pair PMes3•+/B(C6F5)3•–. The author’s experimental results were supported by complementary DFT calculations, leading to the important conclusion that the enhancement of intensity in Raman signals originated from vibrations in both B(C6F5)3 and PMes3. The involvement of both components of the FLP system further confirms the occurrence of SET between Lewis base and acid upon irradiation of the charge-transfer band in the EDA.
As the concept of photoinduced SET in FLP chemistry is now well recognized within the literature as demonstrated here, the following sections highlight examples to showcase the potential applications accessible via these remarkable one-electron processes.
2.1. Application of Photoinduced Single-Electron Transfer: Utilization in Materials Science
In search of multicomponent polymers with new properties, Meijer et al. reported on the photophysical properties of a copolymer consisting of stacked boranes and amines as monomers (Figure 5).38,39 For a combination of 6 and 7, the authors observed a new absorption band in the UV–vis spectrum upon mixing the two components in decaline (λabs ∼500 nm).38 As neither of the individual LA or LB components absorb above 500 nm, this absorption band was assigned to the charge-transfer band of the encounter complex [6, 7]. To explore the emission properties of the corresponding radical ion pair 7•+/6•– formed upon excitation, photoluminescence spectroscopy was employed. Excitation (λex = 387 nm) of the FLP resulted in an additional emission band at 550 nm, which was not observed in corresponding measurements of the individual components even though both components have absorption bands at the selected excitation wavelength. The authors assigned this long wavelength emission to BET from the borane to the amine. The emission decay was characterized by two different lifetimes of ∼96 ns and ∼6 μs, which the authors assigned to decay of the singlet and triplet (caused by two electrons with the same spin in close proximity) states. Similar emission spectroscopy results were also reported by the authors for the combination 6 and 8, although it should be noted that no UV–vis spectrum of the combination was reported to confirm the presence of a charge-transfer band.
Figure 5.

Copolymer formation resulting from stacking of amines and borane components. These copolymers were shown to undergo photoinduced single-electron transfer from the amine to the borane.
In a subsequent study, the authors used EPR spectroscopy to characterize the combination of arylamine 8 and B(C6F5)3 in a stacked copolymer.39 Complementary UV–vis spectroscopy showed no evidence of a charge-transfer band for this combination, although this could be due to the relatively low concentration of the components used in this study (both 25 μM). Despite this, two signals were observed in the low-temperature EPR spectra upon 320–390 nm irradiation of the copolymer, at g = 2.0 and g = 4.2 (Figure 6). The signal at g = 2.0 was weakly observed in the dark and grew in intensity upon irradiation, therefore indicating the formation of a low population of doublet-state isolated radical ions upon low-temperature excitation. The signal at g = 4.2 is characteristic of a triplet state (S = 1), arising from two coupled electrons of the same spin being localized in close proximity, likely arising from an N-to-B SET process. However, the individual components do also absorb the 320–390 nm light, therefore making it possible that these EPR results are due to absorption by one of the individual components.
Figure 6.

CW X-band EPR spectra of two independent EPR signals originating from low- and high-spin systems of a copolymer radical.
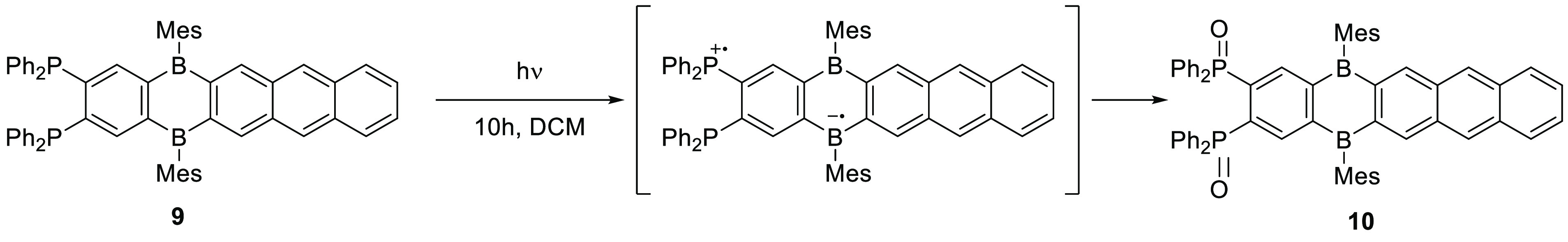
In search of photoluminescent aryl boranes as novel optoelectronic materials, the Wagner group synthesized a doubly Ph2P-substituted dihydrodiborapentacene 9.40 This compound is an air-stable solid in the dark, but when a DCM solution was exposed to ambient light, quantitative yield of the oxidized product 10 was obtained within 10 h (Scheme 5). The authors related this to a photoinduced, intramolecular charge transfer from the phosphine toward the borane backbone, yielding the corresponding radical ion pair. Phosphorus radical cations are known to react readily with oxygen to yield the corresponding phosphine oxide, just as the authors observed.40 The photoinduced SET is further supported by the calculation of the IE (5.50 eV) and EA (−2.79 eV) with DFT (SCF/B3LYP-D4/def2TZVP(-f) with SMD treatment of DCM).40 This results in the radical ion pair state calculated to be 2.71 eV (458 nm) higher in energy, which is comparable to the longest wavelength absorption band reported in benzene solution (470 nm, ε = 20800 mol–1 dm3 cm–1). TD-DFT also predicted the possibility of a photoinduced SET where electron density is moved from the phosphorus centers to the boranes (2.86 eV, 433 nm, fosc = 0.186). It is important to note that the TD-DFT calculations (at the OT-LRC-ωPBEh/def2-TZVP level of theory with pt(SS+LR)-PCM treatment of toluene) also predicts other charge-transfer states in close proximity. For these excitations, the electron density is transferred from the anthracene toward the borane centers.
Scheme 5. Intramolecular, Photoinduced SET from the Phosphines toward Boranes, Facilitating Oxidation toward Phosphine Oxides.
2.2. Photoinduced Single-Electron Transfer: Utilization in Synthesis
As previously noted, dihydrogen activation was one of the first examples of an FLP-induced chemical modification.2 In this reaction, as clearly evidenced by Piers and co-workers, both radical (SET) and two-electron-transfer mechanisms can be envisioned (Scheme 6).12 In order to elucidate the prevalent reaction pathway, Slootweg et al. further investigated the PMes3/B(C6F5)3 combination.17 As shown above, the formation of the RIP PMes3•+/B(C6F5)3•– via SET for this FLP pair occurs only under the influence of light. However, irradiation (534 nm, 2.2 W) was reported to have no significant influence on the reaction kinetics of H2 activation. Therefore, it was concluded that the radical pathway plays an insignificant role due to the transient nature of the RIP PMes3•+/B(C6F5)3•– and rather the major contribution to this reaction chemistry is via a two-electron-transfer mechanism.
Scheme 6. Activation of Dihydrogen by the FLP PtBu3/B(C6F5)3 via Two-Electron (Top Reaction) or Single-Electron (Bottom Reaction) Mechanisms.

Using photoinduced single-electron transfer employing B(C6F5)3 as the electron acceptor and catalyst, Tang et al. reported the sulfenylation of 2-phenyl indoles (Scheme 7A).41 UV–vis spectroscopy showed the formation of a charge-transfer band around 430 nm for the EDA [2-phenyl indole, B(C6F5)3]. Irradiation of this band with 455–460 nm light resulted in single-electron transfer from the indole to the borane, yielding the radical ion pair 2-phenyl indole•+/B(C6F5)3•- (11), as confirmed by the observation of a signal in the EPR spectrum, characterized by giso = 2.00296 with no resolved hyperfine structure. The narrow spectral width of the signal (∼1.7 mT) precludes the assignment of this signal to the B(C6F5)3•– anion (ΔB = 8 mT); therefore, this signal likely originates from the indole radical cation. The 2.4 V (517 nm) energy difference between indole oxidation (0.6 V vs Fc/Fc+ in acetonitrile42) and borane reduction (−1.79 V vs Fc/Fc+ in DCM14) confirms the feasibility of a photoinduced single-electron transfer. The authors proposed that following generation of the indole radical cation, addition of a thiyl radical and loss of a proton occurred, leading to the formation of the final product 12. The formation of the thiyl radical is suggested to occur concomitantly during oxidation of the borane radical anion using oxygen as an oxidant. Furthermore, this process closes the borane catalytic cycle with selective oxidation of the reactive borane radical anion, achieving a high turnover number (TON) of 19 for B(C6F5)3.
Scheme 7. Synthetic Applications of the Photoinduced SET between Lewis Acid and Base: (A) Sulfenylation of 2-Phenyl Indole, Catalyzed by B(C6F5)3 via the Formation of an EDA Complex and Subsequent SET (Ar = p-tBuPh); (B) Hydroboration of Alkenes via the Photoinduced Radical Pair (X = Cl or Br).
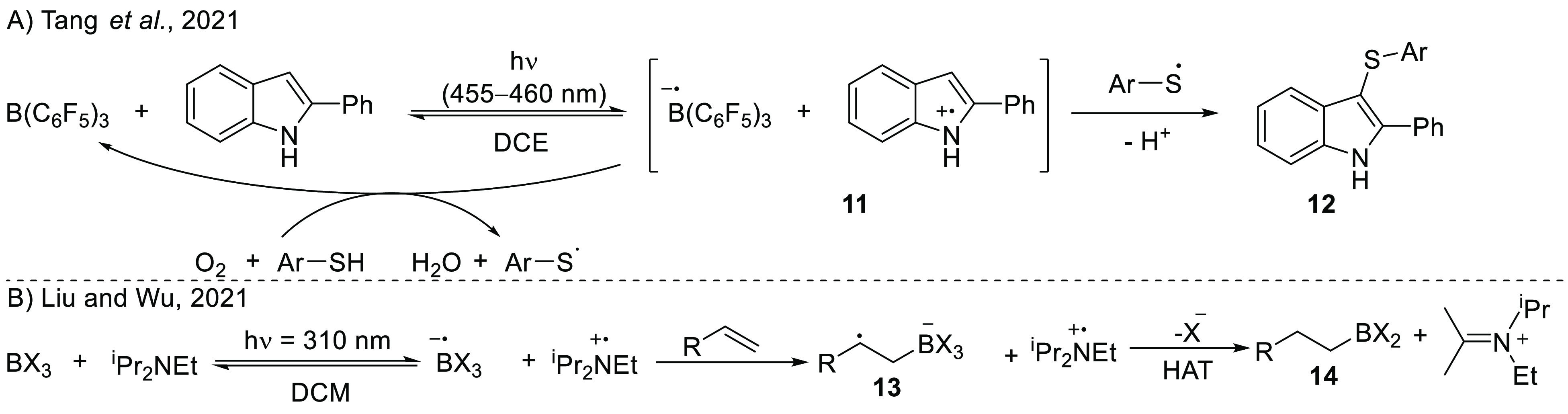
Liu, Wu, and co-workers also used the photoinduced formation of a borane radical anion in the hydroboration of alkenes (Scheme 7B).43 The reaction begins with SET from ethyl di-iso-propyl amine to BX3 (with X = Cl or Br) to form the corresponding radical ion pair iPr2NEt•+/BX3•–, which was calculated via DFT to be 3.40 eV (78.3 kcal/mol) higher in energy than the closed shell pair, hence requiring photoinduced SET for its formation. TD-DFT calculations of Liu et al. also predict a charge-transfer band at 323 nm for SET from the amine to borane. After the formation of the RIP, the authors propose that the borane radical anion adds to the alkene, yielding radical intermediate 13, based on more DFT calculations. After hydrogen atom abstraction from the amine radical cation and halogen loss from 13, the final product 14 is obtained. To determine the proton source, deuterium-labeling experiments were performed using d8-styrene and d2-dichloromethane. For both deuterium sources, no d-incorporation was observed in the β-position of the product. Instead, the presence of an iminium cation was confirmed by observation of a resonance at 152.6 ppm in the 13C NMR spectra and by ESI-MS analysis (128.1435 (observed) vs 128.1434 (calculated)), indicating the amine radical cation to be the hydrogen atom source. Further mechanistic studies regarding halogen loss indicated the formation of BBr4– by the observation of a peak at −24.32 ppm in the 11B-NMR spectrum. An inhibition of the reaction was observed in the presence of radical scavengers like 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) or 2,6-ditert-butyl-4-[(3,5-ditert-butyl-4-λ-1-oxidanylphenyl)methylidene]cyclohexa-2,5-dien-1-one (galvinoxyl), once more proving a radical mechanism.
Besides the typical borane Lewis acids as electron acceptors, there are also reports of using sulfonium salts as the acceptor in combination with, among others, amines or sulfur-based electron donors.44 A prime example is the C–H functionalization of arenes using triarylsulfonium salt 15, as recently reported by Bednar et al. and Procter et al. (Scheme 8).45,46 Mixing 15 with the arene gave the adduct 16, which together with the amine 17 formed EDA complex [17, 16], which is characterized by a small red-shifted absorption in the UV–vis spectrum. The subsequent photoinduced SET (λem = 456 nm) led to the formation of both the amine radical cation 18 and the sulfur radical 19, of which the latter undergoes homolytic bond cleavage to yield the sp2-arene radical 20. Next, this radical is trapped by the nucleophile tBu-isocyanide (route a) or an enol silane (route b) to yield the functionalized arene radical 21. Oxidation of this radical by the amine radical cation 18 or sulfonium cation 15 yields the corresponding arene cation 22. The final product is obtained after elimination of the tBu cation (route a) or silyl cation (route b). With this one-pot procedure, the authors were able to obtain a wide range of α-arylated carbonyl compounds using route a47,48 and cyanated arenes with route b,49,50 which are hard to prepare in the absence of transition-metal catalysts, organometallic reagents, and toxic cyanides.
Scheme 8. Photoinitiated Donor–Acceptor Pairs Catalyzed Site-Selective C–H Cyanation and Alkylation of Arenes.

X = O or single bond. Ar = 1-Naphthyl, phenyl, para-bromo phenyl or para-chloro phenyl.
3. Thermal Single-Electron Transfer
As shown in section 2, for SET to be induced with visible light, the energy gap between the ionization energy of the Lewis base and the electron affinity of the Lewis acid should be in the range 1.5–3.1 eV. It is also possible to thermally induce SET by reducing the energy difference between IE and EA, and as such reducing the energy difference between the ground state EDA complex and the corresponding radical ion pair. This will lead to an equilibrium between the closed shell and radical states as shown in Scheme 9. Following the Boltzmann equation, for 0.06 M solutions (or 0.03 M of both donor and acceptor) an energy gap of ±0.4 eV (±9 kcal/mol) can already lead to detectable concentrations of the radical pair in EPR experiments16 since concentrations as low as the 10–10 M region can be detected.51 In the following sections several FLP and main group systems will be discussed to highlight the feasibility of a thermal SET.
Scheme 9. For Combinations of Lewis Acids (LA) and Bases (LB) with a Sum of the Electron Affinity and Ionization Energy, Respectively, Smaller than 0.4 eV, a Thermal SET Can Potentially Be Observed.

3.1. Evidence of Thermal Single-Electron Transfer
Riedel et al. recently reported thermal SET using the fluorinated trityl cation [C(C6F5)3][Al(OTeF5)4] (23).52 This substituted carbon-based Lewis acid was determined to have an increased electron affinity compared to the nonfluorinated trityl cation (CPh3+) (−7.33 eV vs −5.86 eV, respectively). This is in agreement with trends in the oxidation potential, which increases from −0.11 V vs Fc/Fc+ in MeCN for CPh3•/CPh3+ to 1.11 V vs Fc/Fc+ in o-difluorobenzene for C(C6F5)3•/C(C6F5)3+. Addition of tris(p-bromophenyl)amine (0.74 V vs Fc/Fc+ in DCM) to the fluorinated trityl cation 23 led to the formation of the radical pair (pBr-Ph)3N•+/C(C6F5)3• (24), as directly evidenced by the observation of both radical ions in the EPR spectrum as broad, featureless overlapping singlets characterized by giso ∼ 2.0031 and giso ∼ 2.012 for the perfluorinated trityl and arylamine radical cation, respectively (Scheme 10A). As the redox potential of the fluorinated trityl cation is 0.4 V higher than the redox potential of the amine, thermal SET is indeed possible for this combination of electron donor and acceptor.
Scheme 10. Selected Examples of Thermal SET in FLP Chemistry: (A) Perfluorinated Trityl Cation as an Electron Acceptor with a Triarylamine; (B) Thermal SET between TOTA+ and ItBu and the Decomposition of Both Radicals; (C) SET Proposed to Occur upon Mixing a Boron-Substituted Pyridine and the Trityl Cation.
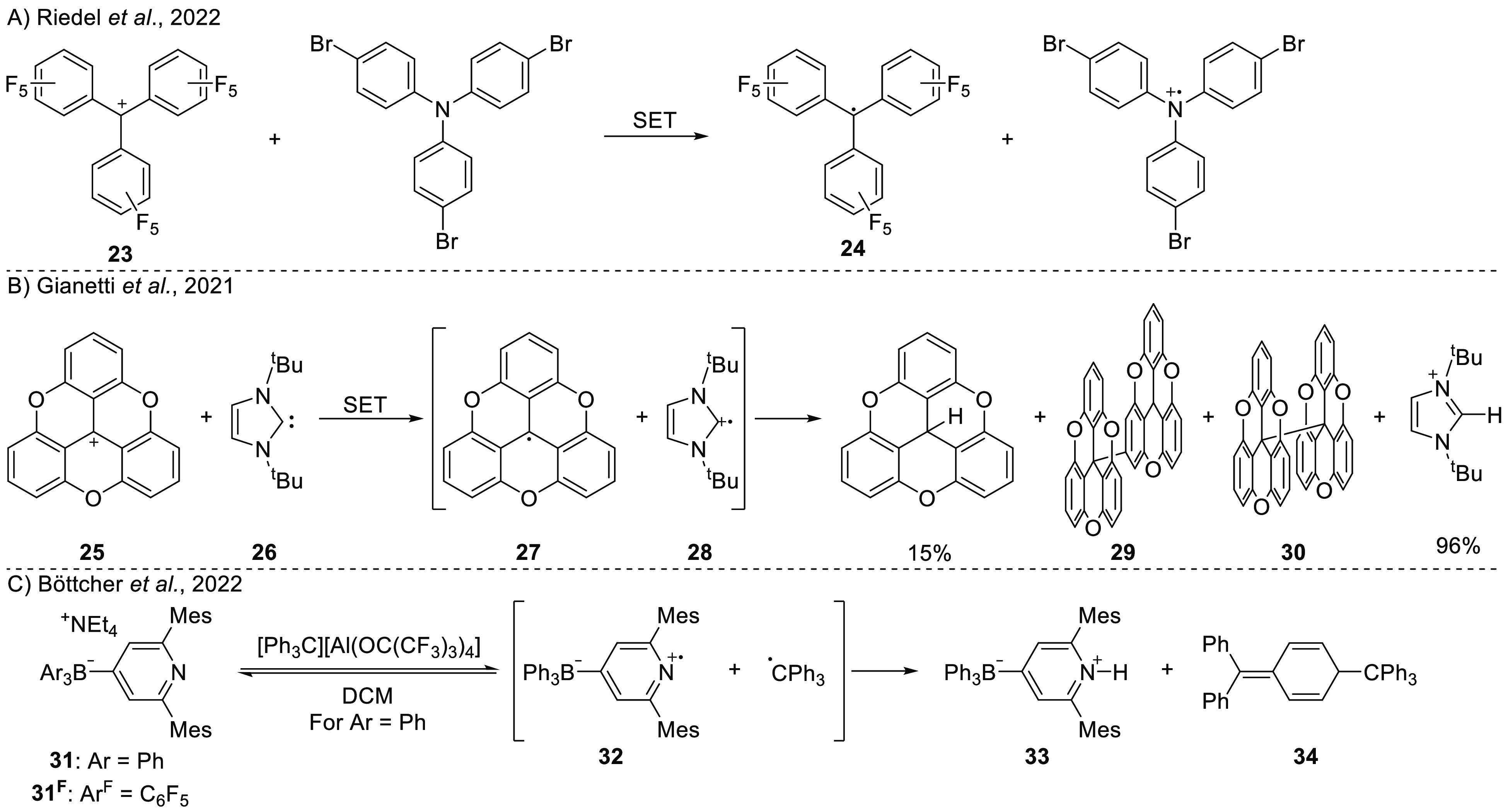
For the fluorinated pyridine 31F no reaction occurred.
The group of Gianetti studied the reactivity between trioxatriangulenium (TOTA+, 25) with 1,3-di-tert-butylimidazol-2-ylidene (ItBu, 26; Scheme 10B).53 Mixing the two compounds in toluene or acetonitrile gave directly a green solution, which is an indication of possibly a SET event. The corresponding radicals TOTA•27 and ItBu•+28 were not observed via EPR spectroscopy, either directly or via trapping experiments with TEMPO and benzoyl peroxide. However, the formation of TOTA-dimers 29 and 30 as components of the product mixture suggested the presence of radical intermediates. The calculated electron affinity of TOTA+ (−7.41 eV, PCM/CAM-B3LYP/6-311G(d,p) in acetonitrile) and the ionization energy of ItBu (5.59 eV) support the premise that a thermal SET mechanism is possible. As a final proof for the occurrence of a thermal SET, the authors studied the properties of the TOTA dimer. Upon measuring a solution of the dimer in m-xylene, the TOTA monomer radical could be observed at temperatures above 340 K with EPR spectroscopy and was identified by a multiline EPR signal characterized by giso = 2.005, aiso(1Hn=6) = 0.091 mT and aiso(1Hn=3) = 0.328 mT (Figure 7).
Figure 7.
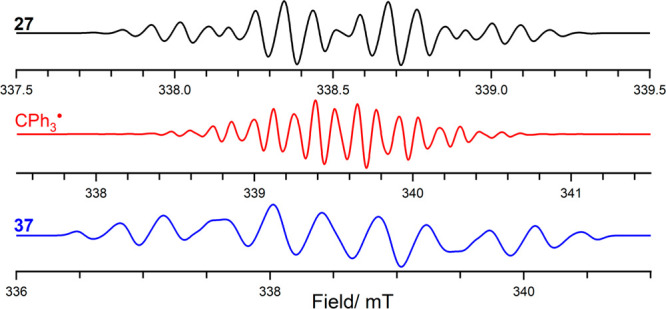
CW EPR spectrum of 27, trityl CPh3• radical, and 37; simulated using values listed in Table 1.
The reduction of metals by pyridine scaffolds was observed by Böttcher et al. during their attempts of coordinating para-borane-substituted pyridines 31 and 31F (Scheme 10C) to metal centers.54 To study the SET process in more detail, they mixed 31 with the trityl cation (CPh3+) in DCM. The obtained products were the protonated pyridine 33 and Gomberg’s dimer, 34. Detection of the Gomberg dimer indicates the occurrence of SET, and indeed the redox potentials (0.56 V vs Fc+/Fc for 31, and 0.66 V vs Fc+/Fc for the trityl cation) show that a thermal SET is feasible. The authors proposed that hydrogen atom abstraction from the solvent by the pyridine radical cation 32 proceeds following the SET event. No evidence for SET was observed using the fluorine-substituted 31F analogue, which is in-line with the higher oxidation potential (1.32 V vs Fc+/Fc) of this borate.
Although the broad field of FLP chemistry is mainly based on the use of B(C6F5)3 and related electrophilic boranes, in addition to a small selection of Al-based examples, Stephan et al. have recently reported an FLP system featuring a Lewis acidic nitrogen center to uncover the use of new Lewis acids in FLP chemistry.55 Treatment of PR3 (R = Ph, tBu, or Mes) with a diazonium salt afforded the diazonium cation 35,16,17 featuring a Lewis acidic nitrogen center (Scheme 11). The PPh3-containing diazonium salt directly undergoes a second phosphine addition to give the stable bis-phosphine [(p-ClC6H4)N(PPh3)N(PPh3)]+ species 36. The use of the more bulky phosphines PtBu3 and PMes3 prevents a second addition, making the observation of 35 feasible.
Scheme 11. Reactivity of FLPs Involving a Nitrogen-Centered Lewis Acid.

Ar = p-C6H4Cl.
Interestingly, in the presence of a disulfide, the combination of the diazonium Lewis acid–PtBu3 adduct 35 and a second equivalent of PtBu3 is able to cleave the disulfide bond, yielding [PhS–PtBu3][BF4] as evidenced by a chemical shift of 85 ppm in the 31P(1H) NMR spectrum. A color change of the reaction solution from purple to an intense red upon addition of the disulfide indicated the formation of the stable radical species 37 (Scheme 11). This was confirmed upon observation of a paramagnetic species in the EPR spectrum (Figure 7), characterized by multiple hyperfine interactions [aiso(31P) = 0.88 mT, aiso(N1) = 0.42 mT, aiso(N2) = 0.36 mT, aiso(Hortho) = 0.36 mT, and aiso(Cl) = 0.28 mT] (Figure 7).
Independent generation of the radical 37 from the diazonium-phosphine adduct 35 with various reducing agents (Cp2Co, potassium, and PhSNa) suggest that 35 is reduced during the cleavage of the disulfide bond. The reported redox potential of the diazonium-phosphine adduct 35 (Ered = −0.91 V vs Fc/Fc+ in MeCN) is in accordance with a possible thermal SET using PtBu3 as the oxidant (Eox = 0.90 V vs Fc+/Fc in MeCN) yields an energy gap of ΔEox-red = −0.01 V.13
3.2. Reactions with a Thermal Single-Electron-Transfer Step
Melen’s group reported on the use of diaryl esters (38, Scheme 12) for the formation of C–C bonds in the presence of the PMes3/B(C6F5)3 FLP system, providing both experimental and computational details to support proposed reaction mechanisms.56,57 The reaction proceeds initially with the coordination of B(C6F5)3 to the ester 38 (Scheme 12A) to form adduct 39, followed by heterolytic cleavage of the C–O bond, yielding the borane 40 and carbocation 41. The carbocation can undergo a reversible one-electron reduction by PMes3 to the radical 42 (Scheme 12B), as evidenced by room temperature EPR spectroscopy (Figure 9). The authors found that this radical state is only 6.9 kcal/mol (0.30 eV) higher in energy than the ion pair, making a thermally induced SET indeed feasible. The absence of the carbon radical 40 was justified by its quick decomposition. It should be noted that Melen et al. were able to observe a weak signal in the EPR spectrum assigned to a more sterically encumbered carbon radical for a related system, confirming the formation of a radical pair.56
Scheme 12. Proposed Mechanism for the C–C Bond Formation between a Diarylester and Phenylacetylene Using the PMes3/B(C6F5)3 FLP: (A) Formation of the Diaryl Carbocation; (B) Radical Pathway Leading to the Product; (C) Diamagnetic Pathway Leading to the Cross-Coupled Product.
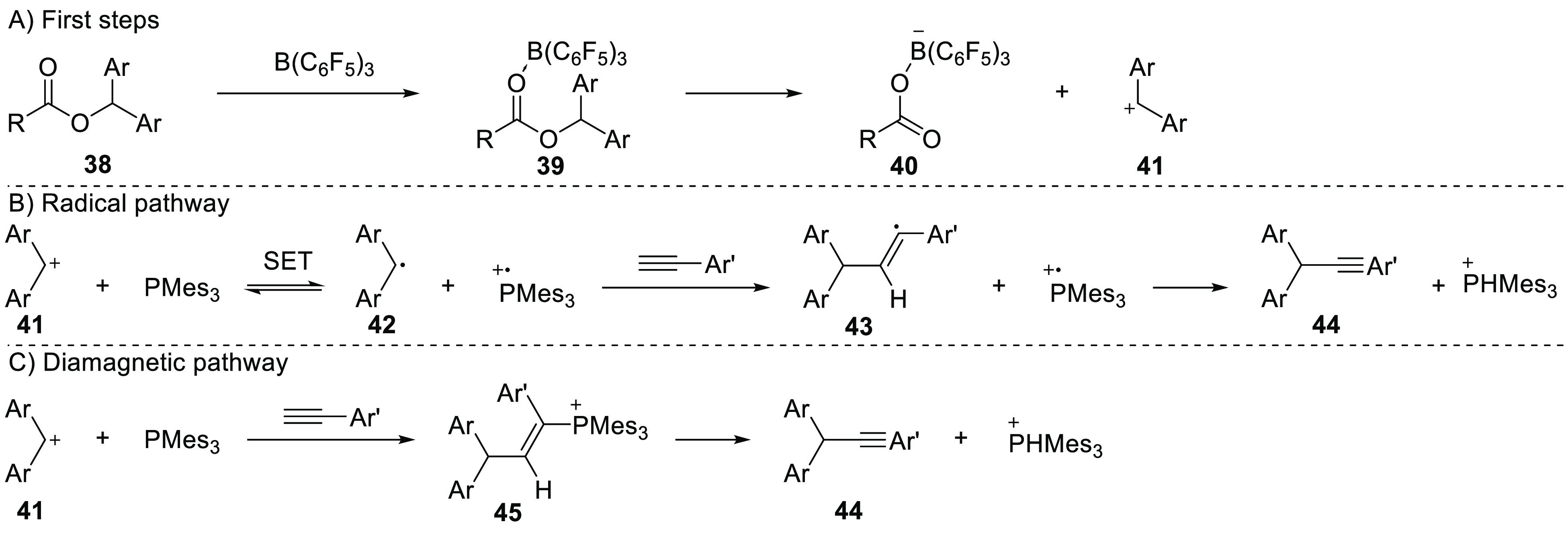
Figure 9.
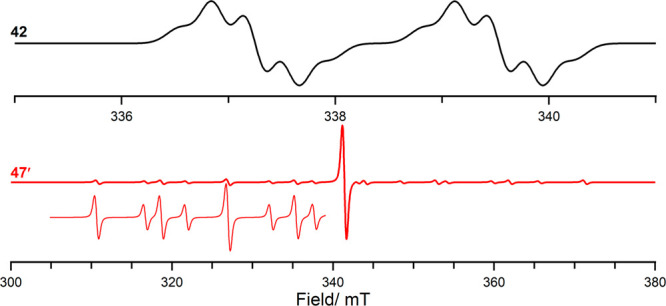
CW EPR spectrum of 42 and 47′; simulated using values listed in Table 1.
Continuing from the equilibrium between cation 41 and radical 42, the reaction can proceed via two alternative pathways.57 The radical mechanism continues from the radical 42 by addition to the alkyne to give 43 as an intermediate, as shown in Scheme 12B. After hydrogen atom abstraction by the phosphine radical cation, the final product 44 is obtained. Alternatively, the diamagnetic pathway proceeds with the addition of both the carbocation 41 and phosphine to phenylacetylene (Scheme 12C). The product 44 is obtained after elimination of PMes3 from the intermediate 45. To probe the operative mechanism, the authors determined the Hammett parameter ρ by doing competition experiments with para-substituted phenyl acetylenes. They found values for ρ of −6.6 ± 1.7 experimentally and −5.7 ± 0.8 computationally, which indicates the buildup of a positive charge during the reaction. These results are consistent with the diamagnetic mechanism. DFT calculations of the energy surface show that for electron-donating para-substitutions on the phenylacetylene, the reaction barrier of the radical mechanism is up to 18.8 kcal/mol (for p-NMe2 phenyl acetylene) higher in energy than the barrier in the diamagnetic pathway. The difference between the radical and diamagnetic mechanism is however smaller when utilizing electron-withdrawing substituents. For example, the difference is only 0.7 kcal/mol for p-NO2 phenyl acetylene, indicating that the radical mechanism can be (partly) operative with electron-poor substrates.
Very recently, Malischewski’s group increased the EA of free TCNQ from experimentally determined 3.38 eV for the first oxidation to 6.04 eV upon addition of four equivalents of B(C6F5)3, as determined by DFT calculations at the B3LYP-D3(BJ)/Def2-SVP level of theory.84 Also the EA of the second oxidation of 3.21 eV was found to be more facile than the first oxidation of free TCNQ. The authors showed that TCNQ-(B(C6F5)3)4 is capable of oxidizing both pBrPh3N and thianthrene in DCM (upper reactions in Scheme 13) by observation of two partly overlapping singlets in either case. In the case of pBrPh3N, the authors assigned the singlet at giso = 2.0143 to the pBrPh3N•+ radical cation and for the TCNQ-(B(C6F5)3)4•– radical anion a signal at giso = 2.0026. In the case of employing thianthrenium, the radical anion was observed at the same position and the thianthrenium radical cation at giso = 2.00788. Independent generation of the monoanion with ferrocene showed a broad singlet at giso = 2.00238, confirming the assignment of the radical anion. Furthermore, for both pBrPh3N and thianthrenium the authors were able to obtain crystal structures in combination with TCNQ-(B(C6F5)3)4, which showed the coordination of the boranes to each of the cyanides of TCNQ.
Scheme 13. Oxidation Reactions of the Combination of TCNQ and B(C6F5)3.
Formation of the dianion [TCNQ-(B(C6F5)3)4]2– was achieved with ferrocene (Fc) and decamethylcobaltocene (Cp*2Co) (bottom reactions in Scheme 13).58 X-ray diffraction confirmed again the coordination of the boranes to the cyanides of TCNQ. The dianion also made it feasible for the authors to measure the oxidation potential of the dianion and monoanion. The first oxidation in DCM of [Cp*2Co]+2[TCNQ-(B(C6F5)3)4]2– was found at −0.065 V vs Fc/Fc+ and the second oxidation at +1.226 V vs Fc/Fc+. Compared to the first reduction potential of −0.30 V vs Fc/Fc+ and −0.88 V vs Fc/Fc+ for the second reduction of TCNQ, this shows a significant increase of oxidation power, in-line with the found EAs.
Slootweg et al. reported the one-electron oxidation of PtBu3 by single-electron transfer (SET) using the strong oxidant nitrosonium salt [NO][BF4] [(NO+/NO•) = 0.87 V vs Fc/Fc+ in MeCN],58 generating [tBu3PH][BF4] as the major product (Scheme 14).13 The reaction was predicted to proceed through the formation of the radical salt intermediate [PtBu3]•+[BF4]− as the oxidation potential of PtBu3 (0.90 V vs Fc/Fc+ in MeCN) is in the range of a thermal SET. The generated phosphorus radical cation readily abstracts a proton from the acetonitrile solvent and subsequent decomposition affords the phosphonium borate product. Using EPR studies, a small side reaction was found to occur as the authors found a six-line pattern characterized by giso = 2.0071, aiso(14N) = 1.05 mT, and aiso(31P) = 1.21 mT (Figure 8). They assigned this to the formation of the nitrosyl–phosphine adduct tBu3P–NO•, which can be established by trapping the in situ generated NO• by residual PtBu3.
Scheme 14. Single-Electron Transfer of PtBu3 with [NO][BF4].

Figure 8.

CW X-band EPR spectra of substituted nitroxyl; simulated using values listed in Table 1.
An exciting example of SET has recently been reported by Müller et al., in which they described the coordination of B(C6F5)3 to the germanium-centered Lewis base 46 (Scheme 15).59 During the reaction, the toluene solution turns into a deep purple color (λmax = 544 nm), eventually turning colorless when the reaction is completed. Room temperature EPR spectra were recorded on reaction aliquots during the course of the reaction, in which the authors observed the formation of two different radical species (Figure 9). The first paramagnetic species was identified as the hafnium-based radical cation 47′ (giso = 1.9881, aiso(Hf) = 8.5 mT), which the authors propose to be in equilibrium with 47. The authors propose that the formation of 47 occurs from 46 by a SET toward B(C6F5)3. Cyclic voltammetry showed indeed an irreversible oxidation ability of compound 46 (−0.51 V vs Fc+/Fc). Furthermore, oxidizing 46 with [Ph3C][B(C6F5)4] showed the formation of the same hafnium radical 47′ that could be detected together with the trityl radical, showing the occurrence of a SET with 46 as the reductant. The radical 47′ was also observed with EPR spectroscopy when a silyl cation was employed as the oxidant.
Scheme 15. Proposed Generation of Radical Ion Pair Species from a Germanium-Centered Lewis Base with B(C6F5)3 and a Control Reaction Using the Trityl Cation as a One-Electron Oxidant for the Lewis Base.

Besides the hafnium radical 47′, the authors observed a second, featureless radical in the EPR spectrum when performing the reaction. They assigned it to the formation of the B(C6F5)3•– radical anion and also proposed that this radical is responsible for the absorption at 544 nm, even though they mention that the B(C6F5)3•– radical anion is highly unstable (t1/2 ≈ 5–10 min at 0 °C in THF).19 Furthermore, based on the reported reduction potential of B(C6F5)3 (−1.79 V vs Fc/Fc+ in DCM), a thermal SET seems unlikely due to the large energy gap (approximately 1.3 V). Therefore, we note here that a photoinduced SET cannot be excluded, where the observed deep purple color of the reaction mixture stems from the EDA complex [46, B(C6F5)3] that features a charge-transfer band in the UV/vis spectrum at λmax = 544 nm. A similar misinterpretation was found for the observed purple color in the case of PMes3/B(C6F5)3 in chlorobenzene.33 For this FLP combination, the purple color was first thought to be indicative of the presence of radicals, but has since been shown to arise from the formation of the EDA complex [PMes3, B(C6F5)3].16
4. Consequences of Radical Decomposition in Radical Pairs Generated via SET
There are cases where a thermally induced SET can be observed even though the energy gap between the closed shell state and the radical (ion) pair is more than 0.4 eV. This can be attributed to the subsequent reaction of one of the radicals in a consecutive step (for example, hydrogen atom abstraction), as illustrated in Scheme 16. Further involvement of the radical in subsequent reactions will shift the SET equilibrium toward the radical products and thus increase the concentration of the more persistent radical, according to Le Chatelier’s principle.60 The consequence of this reactivity is that only one of the two radicals can be directly observed, instead of observing both radicals simultaneously as would be the case if both radicals are persistent. Furthermore, the decomposition of one of the two radicals can also occur after a photoinduced SET. BET is then not feasible anymore, leading to an increasing concentration of the persistent radical upon prolonged irradiation. The next sections contain reports where the decomposition of either of the formed radicals is required to observe radicals or where this is utilized in synthesis (Scheme 16).
Scheme 16. After the Formation of a Lewis Acid (LA) and Lewis Base (LB) Radical Ion Pair, Decomposition of Either Intermediate Will Lead to an Increased Concentration of the Other Radical.

4.1. Observations of Thermal Single-Electron Transfer after Radical Decomposition
Klare et al. and Schilter et al. studied the reactivity of [Ph3C][B(C6F5)4] with different phosphines (Scheme 17A).20,61 Both tris-2,4,6-triiso-propylphenyl phosphine (PTipp3) and PMes3 led to a SET event as both phosphine radical cations were observed by EPR (see Table 1 for details). The sum of the electron affinity and ionization energies were calculated as 0.07 and 0.33 eV (SCRF/M06-2X/6-311+G(d,p) in chlorobenzene), respectively, indicating that a SET thermal process was feasible for these phosphine derivatives. Later, Slootweg and co-workers showed that this is indeed the case as the radicals are formed in the dark.16 For PtBu3, the sum of the electron affinity and ionization energy is 0.67 eV, which is typically too large to observe a thermal SET with EPR.20 However, the authors reported the presence of the trityl radical in the EPR spectrum. The 31P(1H) NMR spectrum showed the formation of tri-tert-butyl phosphonium salt, which was postulated to be formed by hydrogen abstraction by the phosphine radical cation. Therefore, it was postulated that decomposition of the phosphine radical cation leads to an increased concentration of the more persistent trityl radical, and hence increases the likelihood for experimental observation with EPR spectroscopy. The authors also studied the combination [Ph3C][B(C6F5)4] with PoTol3, for which the calculated energy gap (1.04 eV) completely precluded a thermal SET, as confirmed by the absence of detectable radicals in the EPR spectrum; hence, no radicals were available for subsequent reactivity.
Scheme 17. Reactivity of Several Phosphines with Different Electron Acceptors: (A) Trityl Cation As Electron Acceptor; (B) Silyl Cations as Electron Acceptors.

Tipp = 2,4,6-triisopropylphenyl. SiR3 = Si(C6Me5)3, SiiPr3Si, SitBuMe2, or SiEt3.
Klare and Müller et al. have also studied silylium ions as the choice of electron acceptor (Scheme 17B).20 The silylium ions are weaker electron acceptors than the trityl cation, resulting in the IE/EA sum to be larger than 0.4 eV in all cases, which normally would make the observations of radicals with EPR spectroscopy impossible. However, the obtained silyl radicals are known to be highly unstable and can quickly decompose upon formation,62 pushing the equilibrium in favor of the phosphine radical cation. We suggest that this decomposition therefore results in the enhanced ability of the authors to observe in certain cases the partner phosphine radical cation. For example, for the silylium cation [(Me5C6)3Si][B(C6F5)4] in combination with PTipp3 or PMes3, the IE/EA sum is 0.92 and 1.18 eV, respectively, but the phosphine radical cation is clearly visible in the resulting EPR spectrum.20,61 In contrast, no EPR signals were observed for either the PtBu3 and PoTol3 systems, which the authors explained as arising from the energy gap being too large to be thermally overcome for the radical state to be accessed. Furthermore, the phosphine radical cations of PtBu3 and PoTol3 are both thermally unstable, further reducing the possibility of observation of any radicals via EPR spectroscopy as both components, the electron donor and acceptor, are unstable in their corresponding radical state.
Severin et al. reported the use of N-heterocyclic carbenes (NHCs) as electron donors in toluene.63 Upon mixing the NHC IDipp (49 with R = 2,6-di-iso-propylphenyl) and the trityl cation (see Scheme 18A), a short-lived purple solution was obtained. Both the resulting room temperature EPR spectrum and UV–vis spectroscopy measurements (revealing an absorption band at 343 nm) showed evidence of the trityl radical. The IDipp radical cation 50 was not observed by EPR, presumably due to its facile decomposition. On the other hand, the authors were able to assign an absorption band at 591 nm to the IDipp radical cation 50, which was observed during the early stages of the reaction. The authors supported this assignment by independently oxidizing IDipp with [NO][PF6], which showed the same short-lived purple color as observed for the FLP solution. Trapping the IDipp radical cation 50 with hydrogen atom donors (Ph3SnH and THF) led to the formation of the imidazolium salt 51, again supporting an accessible SET pathway. Similar behavior was reported for the ItBu and IMes NHC derivatives (49, where R = tBu or Mes). Calculations by Gianetti et al. determined that the radical pair is 0.94 eV higher in energy for ItBu (49 with R = tBu)/CPh3+ (PCM/CAM-B3LYP/6-311G(d,p) in acetonitrile).53 Typically, this is too high in energy to observe any radical species, but we suggest that the rapid decomposition of the NHC radical cation 50 allows a buildup in concentration of the trityl radical, making its experimental observation with EPR spectroscopy (giso = 2.0025, aiso(o-H) = 0.26 mT, aiso(m-H) = 0.11 mT, aiso(p-H) = 0.28 mT) possible.
Scheme 18. (A) Reduction of the Trityl Cation by NHCs and Subsequent Decomposition of the NHC Radical Cation; (B) Equilibria between a Triarylamine and the Trityl Cation, Leading to Observable Quantities of the Amine Radical Cation; (C) SET between PtBu3 and a Borane, Affording a Thermally Stable Borane Radical Anion.
R = Dipp (2,6-di-iso-propylphenyl), tBu, or Mes (2,4,6-trimethylphenyl). Ar = Ph or para-tolyl (p-Tol).
For a SET between Np-Tol3 or NPh3 and [CPh3][B(C6F5)4] (IE/EA sum = 0.43 and 0.7 eV, respectively, at SCRF/ωB97X-D/6-311+G(d,p) in toluene), the radical pair state is too high in energy to observe any radical formation by EPR.16 However, Slootweg et al. showed that the dimerization of the trityl radical toward Gomberg’s dimer drives the equilibrium toward the radical state (Scheme 18B). This increases the concentration of the amine radical cations, making it possible to observe these radicals in toluene solution at room temperature with EPR spectroscopy.
4.2. Formation of a Single Radical Species after Photoinduced Single-Electron Transfer
It should again be noted that decomposition of one of the radicals can occur after photoinduced SET. Slootweg et al. observed this for the combination PtBu3/B(NO2-Mes)3 in DCM (Scheme 18c), for which the corresponding radical ion pair PtBu3•+/B(NO2-Mes)3•– is 2.91 eV (67.1 kcal/mol) higher in energy than the ground state (SCRF/ωB97X-D/6-311+G(d,p) in toluene).16 Upon irradiation of a mixture of both the donor and acceptor in DCM for 3 h, a red solution was obtained, and the presence of the thermally stable borane radical anion was confirmed by a signal (with some hyperfine features, presumably due to coupling with the boron center) in the EPR spectrum. The red color of the borane radical anion solution was persistent, as BET was prevented by the rapid decomposition of the phosphine radical cation.
4.3. Utilization of Radical Reactivity in Synthesis
Hong’s group reported recently the C–H functionalization of pyridinium salts using alcohols and thiols.64 This reaction was proven to proceed via both a dark reaction and a photoinduced pathway, as evidenced by the results that a 72% yield was obtained in the absence of irradiation, which improved slightly to 84% upon irradiation (λ = 467 nm). The reaction started with an initiation, which the authors proposed for the dark reaction to be a SET between PtBu3 and the pyridinium salt 52 (Scheme 19A). This yielded a radical pair consisting of PtBu3•+ and radical 53. Upon the formation of radical 53, homolytic bond cleavage occurred toward aminyl radical 54 and pyridine. The photoinduced reaction was also proposed to be initiated by the formation of radical 53. The authors showed the presence of a new broad absorption band around 550 nm, which was formed upon mixing pyridinium salt 52 and the in situ formed xanthate anion 55. Irradiation of this band led to SET and via the formation of radical 53 toward aminyl radical 54, which was also obtained in the dark initiation reaction.
Scheme 19. Alkylation of Pyridines via Photoinduced SET between a Pyridinium Salt 52 and Xanthate Anion 55, Starting with (A) Initiation and (B) the Catalytic Cycle.

X = O or S.
The authors proposed the same catalytic cycle for both the dark and light reaction, as shown in Scheme 19B for the formation of the product.64 Initially, the aminyl radical 54 undergoes SET with the xanthate anion 55, to obtain the amide anion 59 and xanthate radical 57. The isolation of Ph3P = S (over 95% yield) showed that the phosphoranyl radical 58 was formed in a reaction between PtBu3 and the xanthate radical in the next step. The phosphoranyl radical undergoes β-scission, yielding the phosphine sulfide and an alkyl radical. In the last step of the catalytic cycle, the alkyl radical reacts with the pyridinium salt, forming the product and regenerating the aminyl radical 54. This example of chemical synthesis illustrates that high conversions of the starting material can be obtained if one of the radicals is reactive, even if the equilibrium does not favor the radical state.
5. Single-Electron Transfer Facilitated by Lewis Acid Coordination
As shown in the previous sections, the electron affinity of the Lewis acid and ionization energy of the Lewis base are of fundamental importance for the possibility of SET events. A possible synthetic strategy to widen the range of electron acceptor affinities is the coordination of Lewis acids. An example is the reduction potential of ferrocyanide as reported by the group of Gray et al.65 The authors showed that the reduction potential could be altered by as much as 2 V by the coordination of a different number of boranes to the metal (Scheme 20). More examples are abound in organometallic chemistry.66,67 Herein, we will present several examples in which the electron affinity of an organic substrate is increased by the coordination of a Lewis acid.
Scheme 20. Coordination of B(C6F5)3 as a Lewis Acid to Ferrocyanide Leads to an Increased Reduction Potential and Thus a Higher Electron Affinity.

PPN+ = bis(triphenylphosphine)iminium cation [Ph3P = N=PPh3]+.
5.1. Observations of Single-Electron Transfer Induced by Lewis Acid Coordination
Stephan and co-workers found that the FLP systems consisting of B(C6F5)3 or Al(C6F5)3 as a Lewis acid and PtBu3 or PMes3 as a Lewis base can readily reduce tetrachloro-p-benzoquinone (Scheme 21A).33 Using equimolar equivalents of PtBu3 in toluene at −78 °C, the adduct 60 was found as the product, in which the phosphine forms a covalent bond with the quinone. On the other hand, when PMes3 was added in excess, the dianionic bis-B(C6F5)3/Al(C6F5)3 adduct 62 resulted, with 2 equiv of the phosphine radical cation as the counterion that was characterized by an absorption at 573 nm in the UV–vis spectrum.
Scheme 21. Reduction of Quinones Facilitated by the Coordination of Lewis Acids Using (A) Phosphines as the Electron Donor and (B) TEMPO, Trityl Radical, or Ferrocene as the Electron Donor.

E = B or Al, R = tBu or Mes (2,4,6-trimethylphenyl).
In 2020, Slootweg et al. set out to explore the mechanistic details of these reductions.17 They found that both the quinone (EA = 4.45 eV, SCRF/ωB97X-D/6-311+G(d,p) in chlorobenzene) and B(C6F5)3 (EA = 3.31 eV) are clearly not strong enough to oxidize PMes3 (5.25 eV) thermally, yet they observed that the reaction proceeds in the dark (ΔE = 0.80 eV for p-benzoquinone and ΔE = 1.94 eV for B(C6F5)3). Encouraged by the report of the groups of Nocera, Jacobsen, and co-workers on the increased electron affinity of quinones by the formation of hydrogen bonds,68 Slootweg et al. calculated the electron affinity of the quinone-B(C6F5)3 adduct 60 (Scheme 21A),17 which increased by 1.12 to 5.57 eV compared to that of the parent quinone. This electron affinity is now large enough to induce a thermal SET using either PtBu3 or PMes3 as the Lewis base. Furthermore, the calculations showed that the coordination of a second equivalent of B(C6F5)3 enhances the electron-accepting ability even further, such that a second reduction event can occur with both PMes3 and PtBu3. In the case of PtBu3, the second reduction is prevented by the barrierless formation of the 61 adduct from the semiquinone fragment.
Slootweg et al. also performed spectroscopic studies on the deep purple reaction mixture of PMes3/B(C6F5)3 (color comes from EDA complex [PMes3, B(C6F5)3]) with 0.5 equiv of tetrachloro-1,4-benzoquinone (TCQ) in the dark.17 For this reaction, Stephan et al. had previously reported the presence of the PMes3 radical cation.33 Slootweg et al. confirmed this by the observation of a doublet signal in the EPR spectrum (giso = 2.0050; aiso= 23.9 mT) and a triplet multiplet pattern assigned to the formation of the (PMes3)2•+ (giso = 2.0060; aiso= 16.6 mT); Figure 4). In addition, an undefined featureless signal (giso = 2.0058) was observed, which was assigned to the TCQ centered radical anion, TCQ–B(C6F5)3•–. These experimental observations were in-line with the computational result that the SET is preceded by the coordination of B(C6F5)3 to the quinone.
Erker et al. showed the reduction of p-benzoquinone in the presence of B(C6F5)3 (Scheme 21B).69 By employing TEMPO and the trityl radical or decamethylferrocene (FeCp*2) as electron donors, they obtained the radical anion bis-B(C6F5)3 adduct 63 (Scheme 21B). Using 2 equiv of the trityl radical, the quinone could be readily reduced to the dianion. However, in the absence of the Lewis acid, none of the electron donors could reduce the quinone moiety. The authors also employed anthraquinone, phenanthrenequinone, and acenaphthenequinone, which all showed similar reactivity to p-benzoquinone, although TEMPO was not always a potent enough reductant for these quinones.
Regarding the mechanism for the formation of 63, the authors found that the reduction of the quinone was reversible, as the addition of THF or DMSO resulted in the backward reaction toward the formation of the quinone.69 The addition of these coordinating solvents led to the decoordination of B(C6F5)3 from the quinone and the formation of a Lewis adduct between B(C6F5)3 and THF or DMSO. Consequently, the quinone became less electron accepting and thus induced a BET to yield the quinone.
The reduction of dioxygen has been reported by Henthorn and Agapie70 and Erker’s group.71 First, Henthorn and Agapie showed that even though ferrocene (FeCp2) is inert to oxygen, in the presence of 2 equiv of B(C6F5)3 in DCM-d2, oxygen (1 atm) can be reduced to obtain [(F5C6)3B–O2–B(C6F5)3]2– (64) in several hours (Scheme 22A). The nature of the product was confirmed by X-ray diffraction, and the control reaction of ferrocene and B(C6F5)3 showed no oxidation of ferrocene, indicating the requirement of both oxygen and B(C6F5)3. In light of the discussed literature, we support the suggestion of the authors that B(C6F5)3 facilities the reduction of oxygen, presumably via coordination in a first step to obtain a strong enough electron donor to be reduced by ferrocene. This is in accordance with the mismatched reduction potentials of ferrocene and O2–/O2 (−1.18 V vs Fc/Fc+ in DMSO).72
Scheme 22. (A) Reduction of Dioxygen Using B(C6F5)3 to Bis(borane)peroxide/Superoxide with Either Ferrocene or TEMPO and BET upon the Addition of THF or DMSO (LB); (B) Pictorial View of the Oxidation States of Quinone and the Lewis Acid Adducts of Each of the Quinone Species.

LA1 = first Lewis acid and LA2 = second Lewis acid.
In the subsequent study of Erker et al., the reduction of oxygen (1.5 bar) in DCM-d2 is achieved using TEMPO as an oxidant in the presence of 2 equiv of B(C6F5)3.71 Instead of obtaining the dianion product, TEMPO can achieve only a single reduction toward the [(F5C6)3B–O2–B(C6F5)3]•– radical anion (65). This radical was characterized by a seven-line signal at giso = 2.01101 due to hyperfine coupling of both borane nuclei (a (11B)iso = 0.323 mT). Trapping the B(C6F5)3 with THF or DMSO leads to BET, as observed by the formation of the TEMPO radical and liberation of presumably oxygen gas. As further proof of the feasibility of SET from TEMPO toward the (F5C6)3B–O2–B(C6F5)3 adduct, the authors recorded the redox potential of the [(F5C6)3B–O2–B(C6F5)3]−/[(F5C6)3B–O2–B(C6F5)3]2– couple to be +0.22 V vs Fc/Fc+ in DCM, which is similar to the TEMPO/TEMPO+ redox potential (+0.24 V vs Fc/Fc+ in DCM).
Thompson and Heiden also studied the influence of Lewis acids on the reduction of quinones (Lewis acid coordination is shown in Scheme 22B).73 Through detailed DFT calculations (SMD/M06-2X/6-311++G(d,p) in acetonitrile), they determined that the reduction potential of quinones can be increased by 0.5–1.5 V upon the coordination of a Lewis acid, dependent on the strength of the Lewis acid. For example, the coordination of B(C6F5)3 increased the computed first redox potential by 1.14 V. A further increase of the reduction potential by 0.7–1.6 V was possible by the coordination of a second Lewis acid. In the case of the coordination of 2 equiv of B(C6F5)3, the computed redox potential is 1.16 V compared to −0.96 V for the parent quinone. These results indicate that the electron affinity of quinones can be tuned over a wide range by the addition of Lewis acids, thus facilitating SET processes.
Interested in obtaining an intermolecular SET induced by Lewis acid coordination, Wang’s group investigated the combination of amines as electron donors for borane-activated quinones.74 Coordination of the borane to the amine quinone 66 yields the donor–acceptor 67 (Scheme 23A). EPR spectroscopy measurements on a powder sample of 67 revealed Δms = ±2 half-field absorption features, characteristic of spin-triplet species. The spectrum was characterized by giso = 2.00427, and the zero-field parameters D = 14.03 MHz, E/D = 0.36. The presence of these signals in the EPR spectrum confirmed the presence of a diradical. As 66 in the absence of the Lewis acid was not EPR-active, this clearly demonstrated that the coordination of the borane activates the quinone such that the SET can occur. This was also proven by DFT calculations, which showed for a model substrate an increased EA of −3.71 eV, in comparison to −1.11 eV before borane coordination at the BP86/6-31G(d) level of theory.
Scheme 23. Quinone-Based RIPs Consisting of an Amine-Centered Radical Cation and Boron-Based Radical Anion: (A) D–A–D Molecule by Connecting Two Triarylborane Donors to 1,4-Dihydroxy-benzoquinone; (B) Formation of Structurally Stable RIP (70) Using Amine (69) and B(C6F5)3 Incorporating Benzoquinone; (C) Formation of RIPs Introducing Benzoquinones as a Link in FLP Reactions via SET Process.

In all cases, the quinone is made more electron accepting by the coordination of a borane as Lewis acid, thereby facilitating SET processes.
In a follow-up study, Wang et al. aimed to demonstrate intermolecular SET events between amines and borane-activated quinones.75 Upon mixing amine 69 and 9,10-anthraquinone (Scheme 23B) with 2 equiv of B(C6F5)3 in DCM, the authors obtained a dark blue solution from which they could obtain deep purple crystals. UV–vis spectroscopy of 70 in DCM showed two absorption bands at 570 and 620 nm, which were assigned to the amine radical cation. The crystal structure revealed elongation of several quinone bonds, in tandem with the shortening of amine bonds upon addition, attributed to electron transfer to obtain the quinone radical anion and amine radical cation. The presence of the radical cation and anion were confirmed by the observation of two signals in the EPR spectra (Figure 10). The quinone radical anion gave a featureless signal at giso = 2.0040, while the amine radical cation was observed by a multiplet signal centered at giso = 2.0034 with hyperfine splitting originating from coupling to proton and nitrogen nuclei [aiso(1H) = 0.195 and 0.655 mT, and aiso (14N) = 0.700 mT]. Half-field signals were observed for solid samples (both at room temperature and 88 K), indicating magnetic exchange couplings to be present. SQUID measurements confirmed the relatively strong antiferromagnetic exchange coupling.
Figure 10.
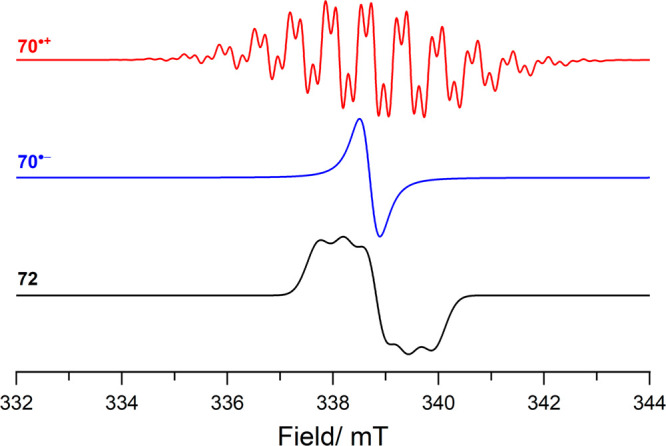
CW X-band EPR spectrum of the individual radical components of 70 (i.e., amine-radical cation 70•+ and quinone radical anion 70•–), and the radical ion pair 72; simulated using values listed in Table 1.
In contrast, for the combination of amine 71 and p-benzoquinone with 2 equiv of B(C6F5)3, the SQUID measurements showed only weak antiferromagnetic interaction (Scheme 23C).75 The authors explained this by the large distance between the cation and anion in the crystal structure. Another difference observed between 69 and 72 was observed in the EPR spectrum. Unlike 69, for 72 only the radical cation could be observed with a characteristic nitrogen hyperfine coupling [giso = 2.0033, aiso(14N) = 0.45 mT] (Figure 10). The presence of the amine radical cation was further confirmed by UV–vis spectroscopy of a DCM solution of 72, in which a broad absorption band around 1500 nm was observed that was earlier already ascribed to the amine radical cation using spectroelectrochemistry.76 The authors suggest that the lack of observation of the quinone radical anion of 72 was due to a low coupling constant or weak signal intensity.75 These results make us on the other hand speculate whether a second reduction of the quinone occurred, yielding a quinone dianion that also could explain the absence of a signal in the EPR spectrum.
Stephan et al. showed the cleavage of the O–O bond of benzoyl peroxides 73 using the FLP system PMes3/B(C6F5)3 (Scheme 24).77 Previously, the authors observed that B(C6F5)3 coordinates readily to peroxides.78 Using Cp*2Fe as reductant, it was possible to cleave the O–O bonds of these peroxides. Upon employment of PMes3/B(C6F5)3 in combination with 0.5 equiv of (ArCOO)2 in DCM, immediately an extremely deep purple solution was obtained containing the salt [PMes3]•+[RCOOB(C6F5)3]− (R = Ph, p-BrC6H5, p-CH3C6H5). The characteristic doublet signal in the EPR spectrum and the absorption maximum at 572 nm further confirmed [PMes3]•+ formation. The authors proposed the mechanism to start with the coordination of B(C6F5)3 to the ketone moiety of benzoyl peroxide 73, forming adduct 74.77 Next, the reduction occurred using PMes3 as the electron donor. We expect that the increase of the electron affinity of the benzoyl peroxide due to B(C6F5)3 coordination is required to enable the use of PMes3 as a one-electron donor.
Scheme 24. Homolytic Cleavage of the O–O Bond of Benzoyl Peroxides by PMes3 in the Presence of B(C6F5)3.

Ar = Ph, p-MePh, or p-BrPh.
Recently, Warren et al. presented the reduction of the nitrite anion using strong Lewis acids, such as B(C6F5)3, without breaking any N–O bonds (Scheme 25). The unsymmetrically capped [Cp*2Co][(C6F5)3B–ONO] (77) was produced by adding 1 equiv of B(C6F5)3 and [Cp*2Co][NO2] (76) in fluorobenzene and showed the B(C6F5)3 bound to one of the O-atoms of nitrite with distinctly different N–O distances of 1.337(10) Å (for capped O atom) and 1.200(10) Å (free O atom), respectively.79 Addition of a second equivalent of B(C6F5)3 in fluorobenzene forms the doubly activated nitrite anion [Cp*2Co][(C6F5)3B–ONO–B(C6F5)3] (78) with symmetric NO distances (1.261(2), 1.225(2)Å), indicating the coordination of the Lewis acid to both O atoms. No reduction wave was observed either for nitrite (76) or monocapped nitrite anion (77) in CV using [PPN] [BArF4] as the electrolyte in fluorobenzene, whereas the free nitrite is proton-dependent, having a range from +0.98 V and −0.48 V vs NHE (normal hydrogen electrode) at pH 0.0 and 14.0, respectively. A quasi-reversible wave was observed for 78, centered at −0.74 V versus NHE, corresponding to the [(C6F5)3B–ONO–B(C6F5)3]−/[(C6F5)3B– ONO–B(C6F5)3]2– couple. Further chemical reduction of 78 with one more equivalent of Cp*2Co in fluorobenzene led to an immediate color change from yellow to gray with the production of borane-capped nitrite radical dianion 79. Although both the capped mono- and dianions 78 and 79 displayed comparable structure features, IR spectroscopy exhibited a distinct lower-energy N–O stretching frequency for dianion 79 (1.010 cm–1), indicating the weakening of the N–O bonds of nitrite upon one-electron reduction, in comparison with monoanion 78 (1.265 cm–1). The EPR spectrum for 79 attained the expected isotropic three-line pattern rising from 14N hyperfine coupling, with an isotropic hyperfine coupling of 1.57 mT at room temperature. This study showed that besides influencing the redox potential of electron acceptors, also the stability can be influenced at the same time.
Scheme 25. Reduction of Nitrite Anion Using Lewis Acid to Produce Capped Nitrite Mono- and Dianions.

5.2. Influence of Lewis Acid Coordination on the Photoinduced SET
Interested in fluorescence properties of organic donor–acceptor diads, Abe’s group studied the influence of B(C6F5)3 coordination.80 The commercial fluorescence dye 80 has an absorption band in DCM at 422 nm (2.94 eV) for the photoinduced SET from the coumarin core toward the pyridyl group. The fluorescence emission after excitation of the absorption band is at 481 nm (2.58 eV). Upon coordination of B(C6F5)3 as illustrated in Scheme 24, the dye 81 is obtained where the B(C6F5)3 is coordinated to the pyridine acceptor unit of 80. The absorption of 81 is shifted to 452 nm (2.74 eV) and the fluorescence to 523 nm (2.37 eV). These red shifts are also observed in TD-DFT calculations (B3LYP/6-31G(d) level of theory) where the absorption changes from 373 nm (fosc = 0.7108) for 80 to 413 nm (fosc = 0.4952) upon B(C6F5)3 coordination.
The authors attributed the observed change in absorption to a smaller HOMO–LUMO gap of 81 (3.30 eV) compared to the 3.57 eV for the parent diad 80 (computed at the B3LYP/6-31G(d) level of theory). The change of orbital energies is also reflected in the change of redox potentials upon the coordination of B(C6F5)3. The oxidation of 80 and 81 showed a small change and are both around 0.5 V vs Fc+/Fc in MeCN, while the reduction in DCM went from about −2.3 V vs Fc+/Fc to −2.0 V vs Fc+/Fc.
These results demonstrate that the coordination of a Lewis acid to the electron acceptor changes the required wavelength in the case of a photoinduced SET. Besides being reflected in orbital energies and redox potentials, also the fluorescence, likely from BET, moves toward a longer wavelength upon coordination of a Lewis acid. Besides dye 80, the authors observed similar results for the coordination of B(C6F5)3 to the dyes 82 and 83 (Scheme 26).
Scheme 26. Abe’s Group80 Studied Donor–Acceptor Dyes and the Influence of Coordination of B(C6F5)3 at the Acceptor Side.

5.3. Lewis-Acid Catalyzed Single-Electron Transfer: Utilization in Synthesis
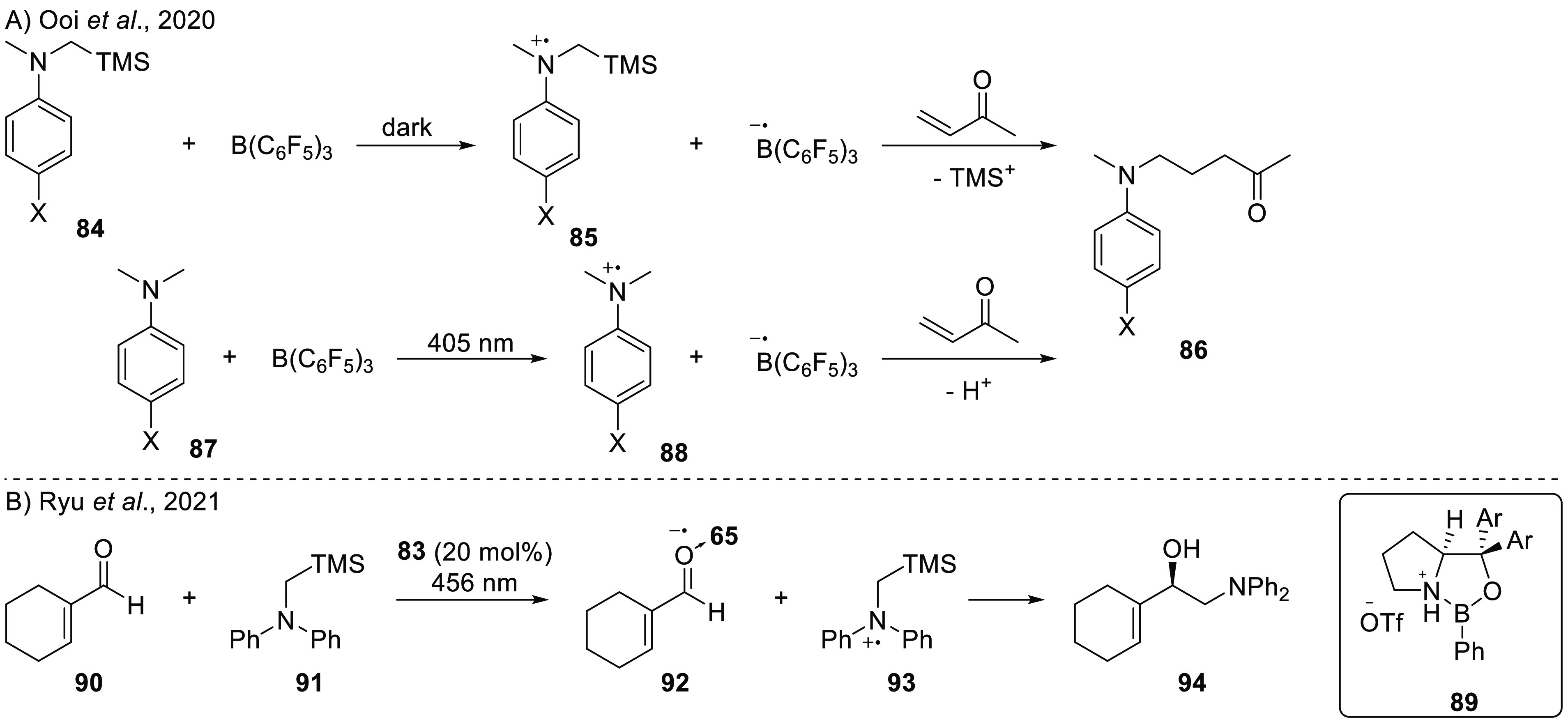
Ooi’s group exploited the use of SET for the formation of C–C bonds between amines and vinyl ketones, catalyzed by B(C6F5)3.81 The authors evidenced that thermal SET occurs with B(C6F5)3 and the TMS amine 84 as the amine radical cation 85 could be observed by EPR spectroscopy both in the presence and in the absence of light (characterized by giso = 2.0033 and hyperfine coupling originating from the amine nitrogen and ring protons Figure 11 and Table 1). Upon addition of methyl vinyl ketone, the formation of the addition product 86 was observed (Scheme 27A). The authors purported that this formed via the loss of the TMS cation before the addition of the ketone. Employing the less electron-rich amine 87 (oxidation potential 0.50 V vs Fc/Fc+, compared to 0.23 V vs Fc/Fc+ for 84, R = Br in both cases), the authors found that light (405 nm) was required for a reversible SET to occur, as the radical 88 was observed only by EPR upon irradiation with 405 nm (Figure 11 and Table 1). TD-DFT calculations for the EDA complex [87, B(C6F5)3] predicted an absorption band at 455 nm (2.74 eV), which allows a photoinduced SET from the amine to B(C6F5)3. The addition of methyl vinyl ketone led to the same coupling product 86, as was obtained by using 84 as the amine. As in this case irradiation (405 nm) is required, the authors conclude the SET is a step in the mechanism for the formation of product 86. Later, Slootweg et al. showed that the SET can be facilitated by the coordination of B(C6F5)3 to the oxygen of the ketone. This results in an increased electron affinity of the ketone from −1.43 eV to −2.73 eV (SCRF/ωB97X-D/6-311+G(d,p), in dichloroethane).17 This reduces the energy required for SET from the amine 87 toward the ketone from 3.68 to 2.38 eV, making a photoinduced SET feasible during the reaction.
Figure 11.
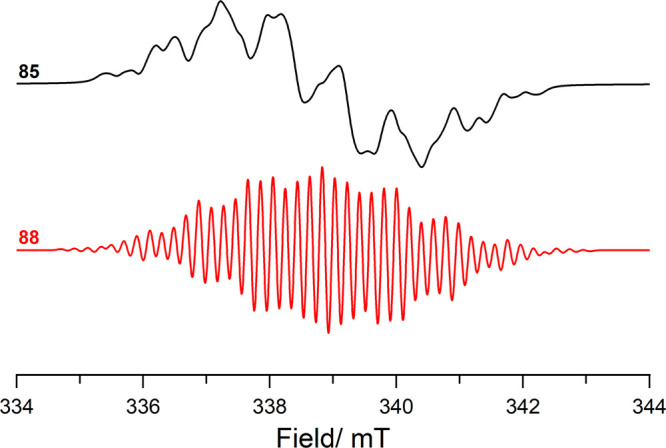
CW X-band EPR spectra of amine radical cations 85 and 88; simulated using data listed in Table 1.
Scheme 27. Synthetic Applications of Using SET Facilitated by the Coordination of a Lewis Acid to the Electron Acceptor: (A) Formation of C–C Bonds between Amines and a Vinyl Ketone via the RIP; (B) Formation of an Enantioselective Addition Product via SET Induced by the Coordination of a Chiral Lewis Acid.
Ar = 2,3-dimethylphenyl, X = Me or Br.
Ryu et al. used the coordination of a chiral Lewis acid 89 to the ketone to obtain an enantioselective addition product (Scheme 27B).82 The Lewis acid first coordinates to the aldehyde 90, which is subsequently able to form an EDA complex with amine 91 (Scheme 27B). The authors proved this using UV–vis spectroscopy, which showed a new absorption peak (a broad peak around 430 nm) in the presence of all three components. Irradiation of this band with 456 nm light promoted photoinduced SET in EDA complex [91, 89] and yielded the RIP consisting of the amine radical cation 93 and the radical anion 92. The amine radical cation 93 subsequently eliminates the TMS cation, yielding an α-alkyl radical, which couples with the radical anion 92 to obtain the product 94 in good yield (up to 94%) (Scheme 27B). The obtained enantioselectivity (up to 95% ee) further proves that Lewis acid coordination to the aldehyde is required to induce SET.
6. Conclusion and Outlook
Since the first report on the simultaneous activation of hydrogen by a Lewis acid and Lewis base, a rise of interest in the chemistry of FLP systems has developed, rejuvenating the field of main group chemistry and catalysis. At first, the reactivity with H2 was solely explained via two-electron-transfer pathways, where simultaneous donation and accepting of electron density in the encounter complex occurs. More recently, reports of radicals in FLP chemistry have been appearing. Here, we set out to review the different mechanisms of SET between Lewis base and acid to understand the underlying fundamental processes of radical formation.
One possible way for a SET to occur is photoinduced. In these cases, the absorption of light by the EDA or encounter complex leads to the formation of the radical pair. Light energy is required as the radical state is significantly higher in energy than the ground state electron donor–acceptor complex, which cannot be reached with common thermal means of heating the reaction mixture. Visible-light energy can be used to accomplish a single-electron shift to form transient radical pairs, which are 1.5–3.1 eV higher in energy than the ground state. If the radical state becomes more stable, the SET could occur thermally. For radical states not more than 0.4 eV higher in energy than the ground state, the radicals can potentially be observed in the reaction mixture with EPR spectroscopy at ambient temperature. Larger energy differences will lead to such small quantities of the radical state following the Boltzmann distribution, which makes their detection with, for example, EPR spectroscopy impossible. Yet, at low temperatures (e.g., 30 K) and upon direct irradiation of the sample in the EPR spectrometer, transient high-energy radical ion pairs like PMes3•+/B(C6F5)3•– have been detected.
Indeed, there are cases where one of the radicals formed following SET is thermally labile and decomposes during the reaction under ambient temperatures, making it only possible to observe the persistent radicals. A consequence of this decomposition is that the equilibrium between the ground state EDA complex and the radical pair shifts toward the radical state. As such, it could be possible to observe radicals even if the radical pairs are too high in energy to be typically observed. In particular, in the case of photoinduced SET, the partial decomposition of the radical pair prevents BET, leading to a buildup of radical concentration. The formation of radicals by SET and subsequent observation can also be facilitated by Lewis acid coordination to the substrate, which acts to increase the electron affinity of the substrate, making the SET event more feasible.
We hope this review leads to more insights and understanding of the SET process in FLP chemistry and related donor–acceptor systems in main group chemistry, which can aid the design of new chemical conversions previously unknown in the two-electron paradigm. During correction of the proofs, the article of Lin et al. was published that highlighted the potential of in-situ generated radical pairs for the regioselective aliphatic C−H functionalization.85 More understanding of the fundamental radical steps should help to stir the development of radical mechanisms, ultimately also leading to more selective pathways or higher reaction rates as typical single-electron-transfer steps and radical mechanisms have low energy barriers. It is especially noteworthy that employing irradiation can potentially lead to increased reaction rates through photoinduced SET events. The occurrence of SET in FLP chemistry could also lead to the activation of thus far unreactive substrates or lead to new synthesis pathways.
Acknowledgments
This research received funding from the Dutch Research Council (NWO) in the framework of the Science PPP Fund for the top sectors via an ENW PPS LIFT grant (ENPPS.LIFT.019.009), and the Leverhulme Trust for research grant RPG-2020-016.
Biographies
Lars van der Zee received his MSc degree in Chemistry cum laude from the University of Amsterdam and the Vrije Universiteit Amsterdam (joint degree). Next, he joined the group of Assoc. Prof. Chris Slootweg at the University of Amsterdam in 2021, first as a researcher before starting his PhD. His current research focus is on the generation of main roup radical ion pairs via single-electron transfer for the activation of unreactive C(sp3)–H bonds.
Dr. Sanjukta Pahar completed her BSc at Asutosh College under the University of Calcutta, and completed her MSc in Chemistry from IIT Madras in 2015. She completed her PhD from CSIR-National Chemical Laboratory, Pune (CSIR-NCL), India, under the supervision of Dr. Sakya S. Sen in 2022, during which she received a “Newton Bhabha internship” to work with Prof. Rebecca Melen for 4 months. After completing her PhD in 2022, she joined the groups of Prof. Rebecca Melen and Dr. Emma Richards as a postdoctoral research associate. Her recent research focuses on the radical-assisted frustrated Lewis pair chemistry.
Emma Richards in a senior lecturer at Cardiff University and coleader of the Electron Paramagnetic Spectroscopy research group. Her research involves utilizing multifrequency CW/pulsed/time-resolved Electron Paramagnetic Resonance, including advanced angular-selective hyperfine techniques to investigate electron-transfer processes in hetero-/homogeneous catalysis. She has a particular interest in the application of first-row complexes and main group systems as sustainable congeners of precious metal alternatives. Emma is coauthor of the popular Oxford University Press textbook “Electron Paramagnetic Resonance”.
Rebecca Melen started her independent career as a lecturer at Cardiff University in 2014 following postdoctoral stays at the University of Toronto and the University of Heidelberg. She was promoted to full professor in 2021. In 2018, she was awarded an EPSRC early career fellowship to investigate main group catalysis. Her research group’s interest lies in developing catalytic systems based upon the p-block elements and exploring their catalytic activity. A key part of her research program involves understanding reaction mechanisms and how ligand design can help tune catalytic activity.
Chris Slootweg started his independent scientific career in 2006 at Vrije Universiteit Amsterdam. He was promoted to Associate Professor in 2014 and moved to the University of Amsterdam in 2016. The mission of his laboratory is to educate students at the intersection of fundamental physical organic chemistry, main group chemistry, and circular chemistry. Chris is cofounder and scientific advisor of SusPhos BV, a pioneering company focused on upcycling of phosphate-rich waste streams to generate high-quality alternatives to replace current fossil-sourced products.
Author Contributions
# L.J.C.Z. and S.P. contributed equally to this work. CRediT: Lars van der Zee writing-original draft, writing-review & editing; Sanjukta Pahar writing-original draft, writing-review & editing; Emma Richards funding acquisition, supervision, writing-review & editing; Rebecca L. Melen funding acquisition, supervision, writing-review & editing; J. Chris Slootweg funding acquisition, supervision, writing-review & editing.
The authors declare no competing financial interest.
Special Issue
Published as part of the Chemical Reviewsvirtual special issue “Persistent and Stable Organic Radicals”.
References
- Brown H. C.; Schlesinger H. I.; Cardon S. Z. Studies in Stereochemistry. I. Steric Strains as a Factor in the Relative Stability of Some Coördination Compounds of Boron. J. Am. Chem. Soc. 1942, 64, 325–329. 10.1021/ja01254a031. [DOI] [Google Scholar]
- Welch G. C.; Juan R. R. S.; Masuda J. D.; Stephan D. W. Reversible, Metal-Free Hydrogen Activation. Science 2006, 314, 1124–1126. 10.1126/science.1134230. [DOI] [PubMed] [Google Scholar]
- McCahill J. S. J.; Welch G. C.; Stephan D. W. Reactivity of “Frustrated Lewis Pairs”: Three-Component Reactions of Phosphines, a Borane, and Olefins. Angew. Chem., Int. Ed. 2007, 46, 4968–4971. 10.1002/anie.200701215. [DOI] [PubMed] [Google Scholar]
- Sarkar P.; Das S.; Pati S. K. Recent Advances in Group 14 and 15 Lewis Acids for Frustrated Lewis Pair Chemistry. Chem. Asian. J. 2022, 17, e202200148 10.1002/asia.202200148. [DOI] [PubMed] [Google Scholar]
- Pal R.; Ghara M.; Chattaraj P. K. Activation of Small Molecules and Hydrogenation of CO2 Catalyzed by Frustrated Lewis Pairs. Catalysts 2022, 12, 201. 10.3390/catal12020201. [DOI] [Google Scholar]
- Tan X.; Wang H. Frustrated Lewis Pair Catalysis: It Takes Two to Make a Thing Go Right. Chin. J. Chem. 2021, 39, 1344–1352. 10.1002/cjoc.202000570. [DOI] [Google Scholar]
- Stephan D. W.; Erker G. Frustrated Lewis Pair Chemistry of Carbon, Nitrogen and Sulfur Oxides. Chem. Sci. 2014, 5, 2625–2641. 10.1039/C4SC00395K. [DOI] [Google Scholar]
- Paradies J. Mechanisms in Frustrated Lewis Pair-Catalyzed Reactions. Eur. J. Org. Chem. 2019, 2019, 283–294. 10.1002/ejoc.201800944. [DOI] [Google Scholar]
- Liu L.; Lukose B.; Jaque P.; Ensing B. Reaction Mechanism of Hydrogen Activation by Frustrated Lewis Pairs. Green Energy & Environment 2019, 4, 20–28. 10.1016/j.gee.2018.06.001. [DOI] [Google Scholar]
- Rokob T. A.; Hamza A.; Stirling A.; Soós T.; Pápai I. Turning Frustration into Bond Activation: A Theoretical Mechanistic Study on Heterolytic Hydrogen Splitting by Frustrated Lewis Pairs. Angew. Chem., Int. Ed. 2008, 47, 2435–2438. 10.1002/anie.200705586. [DOI] [PubMed] [Google Scholar]
- Jupp A. R. Evidence for the Encounter Complex in Frustrated Lewis Pair Chemistry. Dalton Trans. 2022, 51, 10681–10689. 10.1039/D2DT00655C. [DOI] [PubMed] [Google Scholar]
- Piers W. E.; Marwitz A. J. V.; Mercier L. G. Mechanistic Aspects of Bond Activation with Perfluoroarylboranes. Inorg. Chem. 2011, 50, 12252–12262. 10.1021/ic2006474. [DOI] [PubMed] [Google Scholar]
- Habraken E. R. M.; van Leest N. P.; Hooijschuur P.; de Bruin B.; Ehlers A. W.; Lutz M.; Slootweg J. C. Aryldiazonium Salts as Nitrogen-Based Lewis Acids: Facile Synthesis of Tuneable Azophosphonium Salts. Angew. Chem., Int. Ed. 2018, 57, 11929–11933. 10.1002/anie.201806913. [DOI] [PubMed] [Google Scholar]
- Lawrence E. J.; Oganesyan V. S.; Wildgoose G. G.; Ashley A. E. Exploring the Fate of the Tris(Pentafluorophenyl)Borane Radical Anion in Weakly Coordinating Solvents. Dalton Trans. 2013, 42, 782–789. 10.1039/C2DT31622F. [DOI] [PubMed] [Google Scholar]
- Welch G. C.; Stephan D. W. Facile Heterolytic Cleavage of Dihydrogen by Phosphines and Boranes. J. Am. Chem. Soc. 2007, 129, 1880–1881. 10.1021/ja067961j. [DOI] [PubMed] [Google Scholar]
- Holtrop F.; Jupp A. R.; van Leest N. P.; Paradiz Dominguez M.; Williams R. M.; Brouwer A. M.; de Bruin B.; Ehlers A. W.; Slootweg J. C. Photoinduced and Thermal Single-Electron Transfer to Generate Radicals from Frustrated Lewis Pairs. Chem. Eur. J. 2020, 26, 9005–9011. 10.1002/chem.202001494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtrop F.; Jupp A. R.; Kooij B. J.; van Leest N. P.; de Bruin B.; Slootweg J. C. Single-Electron Transfer in Frustrated Lewis Pair Chemistry. Angew. Chem., Int. Ed. 2020, 132, 22394–22400. 10.1002/ange.202009717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X.; Wang X.; Qiu Y.; Li Y.; Zhou C.; Sui Y.; Li Y.; Ma J.; Wang X. One-Electron Oxidation of an Organic Molecule by B(C6F5)3; Isolation and Structures of Stable Non-Para-Substituted Triarylamine Cation Radical and Bis(Triarylamine) Dication Diradicaloid. J. Am. Chem. Soc. 2013, 135, 14912–14915. 10.1021/ja407318h. [DOI] [PubMed] [Google Scholar]
- Kwaan R. J.; Harlan C. J.; Norton J. R. Generation and Characterization of the Tris(Pentafluorophenyl)Borane Radical Anion. Organometallics 2001, 20, 3818–3820. 10.1021/om010272q. [DOI] [Google Scholar]
- Merk A.; Großekappenberg H.; Schmidtmann M.; Luecke M.-P.; Lorent C.; Driess M.; Oestreich M.; Klare H. F. T.; Müller T. Single-Electron Transfer Reactions in Frustrated and Conventional Silylium Ion/Phosphane Lewis Pairs. Angew. Chem., Int. Ed. 2018, 57, 15267–15271. 10.1002/anie.201808922. [DOI] [PubMed] [Google Scholar]
- Murugesan R.; Subramanian S. E.P.R. of Radicals in γ-Irradiated Substituted Phosphines. Mol. Phys. 1979, 38, 1941–1953. 10.1080/00268977900102961. [DOI] [Google Scholar]
- Dasgupta A.; Richards E.; Melen R. L. Frustrated Radical Pairs: Insights from EPR Spectroscopy. Angew. Chem., Int. Ed. 2021, 60, 53–65. 10.1002/anie.202010633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtrop F.; Jupp A.; Slootweg J. C.. Radicals in Frustrated Lewis Pair Chemistry. In Frustrated Lewis Pairs; Jupp A. R., Slootweg J. C., Eds.; Molecular Catalysis (MOLCAT); Springer, Cham, 2021; pp 361–385. [Google Scholar]
- Liu L. L.; Stephan D. W. Radicals Derived from Lewis Acid/Base Pairs. Chem. Soc. Rev. 2019, 48, 3454–3463. 10.1039/C8CS00940F. [DOI] [PubMed] [Google Scholar]
- Wass D. F.; Chapman A. M.. Frustrated Lewis Pairs Beyond the Main Group: Transition Metal-Containing Systems. In Frustrated Lewis Pairs II: Expanding the Scope; Erker G., Stephan D. W., Eds.; Topics in Current Chemistry; Springer: Berlin, Heidelberg, 2013; pp 261–280. [DOI] [PubMed] [Google Scholar]
- Cardenas A. J. P.; Culotta B. J.; Warren T. H.; Grimme S.; Stute A.; Fröhlich R.; Kehr G.; Erker G. Capture of NO by a Frustrated Lewis Pair: A New Type of Persistent N-Oxyl Radical. Angew. Chem., Int. Ed. 2011, 50, 7567–7571. 10.1002/anie.201101622. [DOI] [PubMed] [Google Scholar]
- Sajid M.; Kehr G.; Wiegand T.; Eckert H.; Schwickert C.; Pöttgen R.; Cardenas A. J. P.; Warren T. H.; Fröhlich R.; Daniliuc C. G.; Erker G. Noninteracting, Vicinal Frustrated P/B-Lewis Pair at the Norbornane Framework: Synthesis, Characterization, and Reactions. J. Am. Chem. Soc. 2013, 135, 8882–8895. 10.1021/ja400338e. [DOI] [PubMed] [Google Scholar]
- Pereira J. C. M.; Sajid M.; Kehr G.; Wright A. M.; Schirmer B.; Qu Z.-W.; Grimme S.; Erker G.; Ford P. C. Reaction of a Bridged Frustrated Lewis Pair with Nitric Oxide: A Kinetics Study. J. Am. Chem. Soc. 2014, 136, 513–519. 10.1021/ja4118335. [DOI] [PubMed] [Google Scholar]
- Özgün T.; Chen G.-Q.; Daniliuc C. G.; McQuilken A. C.; Warren T. H.; Knitsch R.; Eckert H.; Kehr G.; Erker G. Unsaturated Vicinal Frustrated Lewis Pair Formation by Electrocyclic Ring Closure and Their Reaction with Nitric Oxide. Organometallics 2016, 35, 3667–3680. 10.1021/acs.organomet.6b00627. [DOI] [Google Scholar]
- Liedtke R.; Scheidt F.; Ren J.; Schirmer B.; Cardenas A. J. P.; Daniliuc C. G.; Eckert H.; Warren T. H.; Grimme S.; Kehr G.; Erker G. Frustrated Lewis Pair Modification by 1,1-Carboboration: Disclosure of a Phosphine Oxide Triggered Nitrogen Monoxide Addition to an Intramolecular P/B Frustrated Lewis Pair. J. Am. Chem. Soc. 2014, 136, 9014–9027. 10.1021/ja5028293. [DOI] [PubMed] [Google Scholar]
- Sajid M.; Stute A.; Cardenas A. J. P.; Culotta B. J.; Hepperle J. A. M.; Warren T. H.; Schirmer B.; Grimme S.; Studer A.; Daniliuc C. G.; Fröhlich R.; Petersen J. L.; Kehr G.; Erker G. N, N -Addition of Frustrated Lewis Pairs to Nitric Oxide: An Easy Entry to a Unique Family of Aminoxyl Radicals. J. Am. Chem. Soc. 2012, 134, 10156–10168. 10.1021/ja302652a. [DOI] [PubMed] [Google Scholar]
- Ménard G.; Hatnean J. A.; Cowley H. J.; Lough A. J.; Rawson J. M.; Stephan D. W. C–H Bond Activation by Radical Ion Pairs Derived from R3P/Al(C6F5)3 Frustrated Lewis Pairs and N2O. J. Am. Chem. Soc. 2013, 135, 6446–6449. 10.1021/ja402964h. [DOI] [PubMed] [Google Scholar]
- Liu L. L.; Cao L. L.; Shao Y.; Ménard G.; Stephan D. W. A Radical Mechanism for Frustrated Lewis Pair Reactivity. Chem. 2017, 3, 259–267. 10.1016/j.chempr.2017.05.022. [DOI] [Google Scholar]
- Mulliken R. S. Molecular Compounds and Their Spectra. II. J. Am. Chem. Soc. 1952, 74, 811–824. 10.1021/ja01123a067. [DOI] [Google Scholar]
- Rosokha S. V.; Kochi J. K. Fresh Look at Electron-Transfer Mechanisms via the Donor/Acceptor Bindings in the Critical Encounter Complex. Acc. Chem. Res. 2008, 41, 641–653. 10.1021/ar700256a. [DOI] [PubMed] [Google Scholar]
- Silvi M.; Melchiorre P. Enhancing the Potential of Enantioselective Organocatalysis with Light. Nature 2018, 554, 41–49. 10.1038/nature25175. [DOI] [PubMed] [Google Scholar]
- Marques L. R.; Ando R. A. Probing the Charge Transfer in a Frustrated Lewis Pair by Resonance Raman Spectroscopy and DFT Calculations. ChemPhysChem 2021, 22, 522–525. 10.1002/cphc.202001024. [DOI] [PubMed] [Google Scholar]
- Adelizzi B.; Chidchob P.; Tanaka N.; Lamers B. A. G.; Meskers S. C. J.; Ogi S.; Palmans A. R. A.; Yamaguchi S.; Meijer E. W. Long-Lived Charge-Transfer State from B–N Frustrated Lewis Pairs Enchained in Supramolecular Copolymers. J. Am. Chem. Soc. 2020, 142, 16681–16689. 10.1021/jacs.0c06921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chidchob P.; Jansen S. A. H.; Meskers S. C. J.; Weyandt E.; van Leest N. P.; de Bruin B.; Palmans A. R. A.; Vantomme G.; Meijer E. W. Supramolecular Systems Containing B–N Frustrated Lewis Pairs of Tris(Pentafluorophenyl)Borane and Triphenylamine Derivatives. Organic Materials 2021, 03, 174–183. 10.1055/s-0041-1727235. [DOI] [Google Scholar]
- Jin T.; Bolte M.; Lerner H.-W.; Mewes J.-M.; Wagner M. Charge-Transfer Transitions Govern the Reactivity and Photophysics of Vicinally Diphosphanyl-Substituted Diborapentacenes. Chem. Eur. J. 2022, 28, e202202234 10.1002/chem.202202234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W.; Huang J.; Xu X.; Wang L.; Tang X.-Y. B(C6F5)3-Catalyzed Electron Donor–Acceptor Complex-Mediated Aerobic Sulfenylation of Indoles under Visible-Light Conditions. Org. Lett. 2021, 23, 7139–7143. 10.1021/acs.orglett.1c02553. [DOI] [PubMed] [Google Scholar]
- Ishikawa R.; Iwasawa R.; Takiyama Y.; Yamauchi T.; Iwanaga T.; Takezaki M.; Watanabe M.; Teramoto N.; Shimasaki T.; Shibata M. Synthesis of 1,2-Bis(2-Aryl-1H-Indol-3-Yl)Ethynes via 5-Exo-Digonal Double Cyclization Reactions of 1,4-Bis(2-Isocyanophenyl)Buta-1,3-Diyne with Aryl Grignard Reagents. J. Org. Chem. 2017, 82, 652–663. 10.1021/acs.joc.6b02668. [DOI] [PubMed] [Google Scholar]
- Li S.; Hu C.; Cui X.; Zhang J.; Liu L. L.; Wu L. Site-Fixed Hydroboration of Terminal and Internal Alkenes Using BX3/IPr2NEt. Angew. Chem., Int. Ed. 2021, 60, 26238–26245. 10.1002/anie.202111978. [DOI] [PubMed] [Google Scholar]
- van Dalsen L.; Brown R. E.; Rossi-Ashton J. A.; Procter D. J. Sulfonium Salts as Acceptors in Electron Donor-Acceptor Complexes. Angew. Chem., Int. Ed. 2023, e202303104 10.1002/ange.202303104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednar T. N.; Nagib D. A. Radical Arenes. Nat. Chem. 2023, 15, 3–4. 10.1038/s41557-022-01109-6. [DOI] [PubMed] [Google Scholar]
- Dewanji A.; van Dalsen L.; Rossi-Ashton J. A.; Gasson E.; Crisenza G. E. M.; Procter D. J. A General Arene C–H Functionalization Strategy via Electron Donor–Acceptor Complex Photoactivation. Nat. Chem. 2023, 15, 43–52. 10.1038/s41557-022-01092-y. [DOI] [PubMed] [Google Scholar]
- Hamann B. C.; Hartwig J. F. Palladium-Catalyzed Direct α-Arylation of Ketones. Rate Acceleration by Sterically Hindered Chelating Ligands and Reductive Elimination from a Transition Metal Enolate Complex. J. Am. Chem. Soc. 1997, 119, 12382–12383. 10.1021/ja9727880. [DOI] [Google Scholar]
- Escudero-Casao M.; Licini G.; Orlandi M. Enantioselective α-Arylation of Ketones via a Novel Cu(I)–Bis(Phosphine) Dioxide Catalytic System. J. Am. Chem. Soc. 2021, 143, 3289–3294. 10.1021/jacs.0c13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D.; Xu P.; Ritter T. Palladium-Catalyzed Late-Stage Direct Arene Cyanation. Chem. 2019, 5, 97–107. 10.1016/j.chempr.2018.09.027. [DOI] [Google Scholar]
- McManus J. B.; Nicewicz D. A. Direct C–H Cyanation of Arenes via Organic Photoredox Catalysis. J. Am. Chem. Soc. 2017, 139, 2880–2883. 10.1021/jacs.6b12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möser J.; Lips K.; Tseytlin M.; Eaton G. R.; Eaton S. S.; Schnegg A. Using Rapid-Scan EPR to Improve the Detection Limit of Quantitative EPR by More than One Order of Magnitude. J. Magn. Reson. 2017, 281, 17–25. 10.1016/j.jmr.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann K. F.; Battke D.; Golz P.; Rupf S. M.; Malischewski M.; Riedel S. The Tris(Pentafluorophenyl)Methylium Cation: Isolation and Reactivity. Angew. Chem., Int. Ed. 2022, 61, e202203777 10.1002/anie.202203777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh A. C.; Veleta J. M.; Moutet J.; Gianetti T. L. Trioxatriangulenium (TOTA+) as a Robust Carbon-Based Lewis Acid in Frustrated Lewis Pair Chemistry. Chem. Sci. 2021, 12, 4841–4849. 10.1039/D0SC05893A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidlin N. M. C.; Radtke V.; Schmidt A.; Lõkov M.; Leito I.; Böttcher T. Electronic Modification of a Sterically Demanding Anionic Pyridine Ligand. Z. Anorg. Allg. Chem. 2022, 648, e202200136 10.1002/zaac.202200136. [DOI] [Google Scholar]
- Waked A. E.; Ostadsharif Memar R.; Stephan D. W. Nitrogen-Based Lewis Acids Derived from Phosphonium Diazo Cations. Angew. Chem., Int. Ed. 2018, 57, 11934–11938. 10.1002/anie.201804183. [DOI] [PubMed] [Google Scholar]
- Soltani Y.; Dasgupta A.; Gazis T. A.; Ould D. M. C.; Richards E.; Slater B.; Stefkova K.; Vladimirov V. Y.; Wilkins L. C.; Willcox D.; Melen R. L. Radical Reactivity of Frustrated Lewis Pairs with Diaryl Esters. Cell Rep. Phys. Sci. 2020, 1, 100016. 10.1016/j.xcrp.2020.100016. [DOI] [Google Scholar]
- Dasgupta A.; Stefkova K.; Babaahmadi R.; Yates B. F.; Buurma N. J.; Ariafard A.; Richards E.; Melen R. L. Site-Selective Csp3–Csp/Csp3–Csp2 Cross-Coupling Reactions Using Frustrated Lewis Pairs. J. Am. Chem. Soc. 2021, 143, 4451–4464. 10.1021/jacs.1c01622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht P. A.; Rupf S.; Sellin M.; Schlögl J.; Riedel S.; Malischewski M. Increasing the oxidation power of TCNQ by coordination of B(C6F5)3. Chem. Commun. 2022, 58, 4958–4961. 10.1039/D2CC00314G. [DOI] [PubMed] [Google Scholar]
- Kochi J. K. Inner-Sphere Electron Transfer in Organic Chemistry. Relevance to Electrophilic Aromatic Nitration. Acc. Chem. Res. 1992, 25, 39–47. 10.1021/ar00013a006. [DOI] [Google Scholar]
- Dong Z.; Cramer H. H.; Schmidtmann M.; Paul L. A.; Siewert I.; Müller T. Evidence for a Single Electron Shift in a Lewis Acid–Base Reaction. J. Am. Chem. Soc. 2018, 140, 15419–15424. 10.1021/jacs.8b09214. [DOI] [PubMed] [Google Scholar]
- De Heer J. The Principle of Le Châtelier and Braun. J. Chem. Educ. 1957, 34, 375. 10.1021/ed034p375. [DOI] [Google Scholar]
- Schilter D. Frustration Leads to Radical Behaviour. Nat. Rev. Chem. 2018, 2, 255–255. 10.1038/s41570-018-0047-1. [DOI] [Google Scholar]
- Gynane M. J. S.; Lappert M. F.; Riley P. I.; Rivière P.; Rivière-Baudet M. Triaryl-Silyl, -Germyl, and -Stannyl Radicals. MAr3 (M = Si, Ge, or Sn and Ar = 2,4,6-Me3C6H2) and Ge(2,6-Me2C6H3)3: Synthesis and ESR Studies. J. Organomet. Chem. 1980, 202, 5–12. 10.1016/S0022-328X(00)81378-9. [DOI] [Google Scholar]
- Dong Z.; Pezzato C.; Sienkiewicz A.; Scopelliti R.; Fadaei-Tirani F.; Severin K. SET Processes in Lewis Acid–Base Reactions: The Tritylation of N-Heterocyclic Carbenes. Chem. Sci. 2020, 11, 7615–7618. 10.1039/D0SC01278E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan C.-Y.; Kim M.; Park I.; Kim Y.; Hong S. Site-Selective Pyridine C–H Alkylation with Alcohols and Thiols via Single-Electron Transfer of Frustrated Lewis Pairs. Angew. Chem., Int. Ed. 2022, 61, e202213857 10.1002/anie.202213857. [DOI] [PubMed] [Google Scholar]
- McNicholas B. J.; Grubbs R. H.; Winkler J. R.; Gray H. B.; Despagnet-Ayoub E. Tuning the Formal Potential of Ferrocyanide over a 2.1 V Range. Chem. Sci. 2019, 10, 3623–3626. 10.1039/C8SC04972F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaralingam M.; Lee Y.-M.; Nam W.; Fukuzumi S. Amphoteric Reactivity of Metal–Oxygen Complexes in Oxidation Reactions. Coord. Chem. Rev. 2018, 365, 41–59. 10.1016/j.ccr.2018.03.003. [DOI] [Google Scholar]
- Fukuzumi S.; Ohkubo K.; Lee Y.-M.; Nam W. Lewis Acid Coupled Electron Transfer of Metal–Oxygen Intermediates. Chem. Eur. J. 2015, 21, 17548–17559. 10.1002/chem.201502693. [DOI] [PubMed] [Google Scholar]
- Turek A. K.; Hardee D. J.; Ullman A. M.; Nocera D. G.; Jacobsen E. N. Activation of Electron-Deficient Quinones through Hydrogen-Bond-Donor-Coupled Electron Transfer. Angew. Chem., Int. Ed. 2016, 55, 539–544. 10.1002/anie.201508060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X.; Daniliuc C. G.; Knitsch R.; Hansen M. R.; Eckert H.; Lübbesmeyer M.; Studer A.; Kehr G.; Erker G. The Special Role of B(C6F5)3 in the Single Electron Reduction of Quinones by Radicals. Chem. Sci. 2018, 9, 8011–8018. 10.1039/C8SC03005G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henthorn J. T.; Agapie T. Dioxygen Reactivity with a Ferrocene–Lewis Acid Pairing: Reduction to a Boron Peroxide in the Presence of Tris(Pentafluorophenyl)Borane. Angew. Chem., Int. Ed. 2014, 126, 13107–13110. 10.1002/ange.201408462. [DOI] [PubMed] [Google Scholar]
- Tao X.; Daniliuc C. G.; Janka O.; Pöttgen R.; Knitsch R.; Hansen M. R.; Eckert H.; Lübbesmeyer M.; Studer A.; Kehr G.; Erker G. Reduction of Dioxygen by Radical/B(p-C6F4X)3 Pairs to Give Isolable Bis(Borane)Superoxide Compounds. Angew. Chem., Int. Ed. 2017, 56, 16641–16644. 10.1002/anie.201709309. [DOI] [PubMed] [Google Scholar]
- Maricle D. L.; Hodgson W. G. Reducion of Oxygen to Superoxide Anion in Aprotic Solvents. Anal. Chem. 1965, 37, 1562–1565. 10.1021/ac60231a027. [DOI] [Google Scholar]
- Thompson B. L.; Heiden Z. M. Tuning the Reduction Potentials of Benzoquinone through the Coordination to Lewis Acids. Phys. Chem. Chem. Phys. 2021, 23, 9822–9831. 10.1039/D1CP01266E. [DOI] [PubMed] [Google Scholar]
- Wang J.; Cui H.; Ruan H.; Zhao Y.; Zhao Y.; Zhang L.; Wang X. The Lewis Acid Induced Formation of a Stable Diradical with an Intramolecular Ion Pairing State. J. Am. Chem. Soc. 2022, 144, 7978–7982. 10.1021/jacs.2c02902. [DOI] [PubMed] [Google Scholar]
- Kong S.; Tang S.; Wang T.; Zhao Y.; Sun Q.; Zhao Y.; Wang X. Stable Radical Ion Pairs Induced by Single Electron Transfer: Frustrated Versus Nonfrustrated. CCS Chemistry 2023, 5, 334–340. 10.31635/ccschem.022.202202306. [DOI] [Google Scholar]
- Low P. J.; Paterson M. A. J.; Goeta A. E.; Yufit D. S.; Howard J. A. K.; Cherryman J. C.; Tackley D. R.; Brown B. The Molecular Structures and Electrochemical Response of “Twisted” Tetra(Aryl)Benzidenes. J. Mater. Chem. 2004, 14, 2516–2523. 10.1039/B404731A. [DOI] [Google Scholar]
- Liu L. L.; Cao L. L.; Zhu D.; Zhou J.; Stephan D. W. Homolytic Cleavage of Peroxide Bonds via a Single Electron Transfer of a Frustrated Lewis Pair. Chem. Commun. 2018, 54, 7431–7434. 10.1039/C8CC03522A. [DOI] [PubMed] [Google Scholar]
- Liu L. L.; Cao L. L.; Shao Y.; Stephan D. W. Single Electron Delivery to Lewis Pairs: An Avenue to Anions by Small Molecule Activation. J. Am. Chem. Soc. 2017, 139, 10062–10071. 10.1021/jacs.7b05120. [DOI] [PubMed] [Google Scholar]
- Hosseininasab V.; DiMucci I. M.; Ghosh P.; Bertke J. A.; Chandrasekharan S.; Titus C. J.; Nordlund D.; Freed J. H.; Lancaster K. M.; Warren T. H. Lewis Acid-Assisted Reduction of Nitrite to Nitric and Nitrous Oxides via the Elusive Nitrite Radical Dianion. Nat. Chem. 2022, 14, 1265–1269. 10.1038/s41557-022-01025-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda T.; Tahara K.; Ishimatsu R.; Ono T.; Cui L.; Maeda M.; Ozawa Y.; Abe M. Lewis-Pairing-Induced Electrochemiluminescence Enhancement from Electron Donor-Acceptor Diads Decorated with Tris(Pentafluorophenyl)Borane as an Electrochemical Protector. Angew. Chem., Int. Ed. 2023, 62, e202301109 10.1002/anie.202304251. [DOI] [PubMed] [Google Scholar]
- Aramaki Y.; Imaizumi N.; Hotta M.; Kumagai J.; Ooi T. Exploiting Single-Electron Transfer in Lewis Pairs for Catalytic Bond-Forming Reactions. Chem. Sci. 2020, 11, 4305–4311. 10.1039/D0SC01159B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. Y.; Lee Y. S.; Ryu D. H. Ternary Electron Donor–Acceptor Complex Enabled Enantioselective Radical Additions to α, β-Unsaturated Carbonyl Compounds. ACS Catal. 2021, 11, 14811–14818. 10.1021/acscatal.1c04835. [DOI] [Google Scholar]
- Symons M. C. R.; Tordo P.; Wyatt J. The Structure of Diphosphine Radical Cations. J. Organomet. chem. 1993, 443, C29–C32. 10.1016/0022-328X(93)80310-8. [DOI] [Google Scholar]
- Lu Z.; Ju M.; Wang Y.; Meinhardt J. M.; Alvarado J. I. M.; Villemure E.; Terrett J. A.; Lin S.. Regioselective aliphatic C–H functionalization using frustrated radical pairs. Nature 2023, 10.1038/s41586-023-06131-3. [DOI] [PMC free article] [PubMed] [Google Scholar]