

Abstract
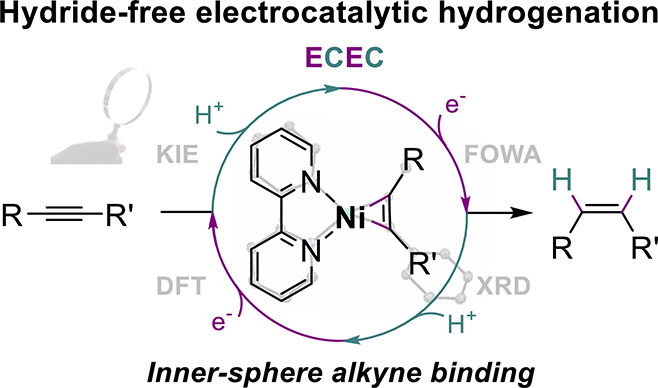
Hydrogenation reactions of carbon–carbon unsaturated bonds are central in synthetic chemistry. Efficient catalysis of these reactions classically recourses to heterogeneous or homogeneous transition-metal species. Whether thermal or electrochemical, C–C multiple bond catalytic hydrogenations commonly involve metal hydrides as key intermediates. Here, we report that the electrocatalytic alkyne semihydrogenation by molecular Ni bipyridine complexes proceeds without the mediation of a hydride intermediate. Through a combined experimental and theoretical investigation, we disclose a mechanism that primarily involves a nickelacyclopropene resting state upon alkyne binding to a low-valent Ni(0) species. A following sequence of protonation and electron transfer steps via Ni(II) and Ni(I) vinyl intermediates then leads to olefin release in an overall ECEC-type pattern as the most favored pathway. Our results also evidence that pathways involving hydride intermediates are strongly disfavored, which in turn promotes high semihydrogenation selectivity by avoiding competing hydrogen evolution. While bypassing catalytically competent hydrides, this type of mechanism still retains inner-metal-sphere characteristics with the formation of organometallic intermediates, often essential to control regio- or stereoselectivity. We think that this approach to electrocatalytic reductions of unsaturated organic groups can open new paradigms for hydrogenation or hydroelementation reactions.
Introduction
The electrification of chemical processes is a major challenge to be met in the transition from petrochemical to defossilized production.1−4 Redox reactions are extensively present in chemical synthesis and provide a particularly relevant entry for electrons produced from renewables into the chemical value chains.5−7 In the specific case of hydrogenation reactions, which are conceptually simple but widely applied in bulk chemical processing as in fine synthesis,8−10 electrons can be directly used as reducing agents in combination with protons. Developing electrosynthetic strategies for the efficient and selective hydrogenation of organic unsaturated compounds would thus represent a major step forward. The electrochemical nature of these reactions may also lead to innovative reactivity patterns, which can be controlled or triggered using adequate electrocatalysts. In that aim, the tunability offered by transition-metal complexes place these species as ideal candidates in the exploration of the electrocatalytic space.
Most of the hydrogenation reactions of C–C unsaturated bonds are assumed or even proven to involve hydrides as catalytically relevant species.10−15 In the organometallic formalism, the reaction of a metal hydride with an unsaturated fragment can occur during four elementary key steps: migratory insertion (MI), hydride transfer (HT), hydrogen atom transfer (HAT), and reductive elimination (RE) (Scheme 1).16,17 Interestingly, these intermediates and steps are observed regardless whether the hydrogen source is molecular H2, hydride reagents, or protons in conjunction with electrons obtained from sacrificial reductants or an electrode. In the prototypical hydrogenations using H2 gas, metal hydrides are classically encountered both in the MI and the RE steps. Recent reports of C–C bond hydrogenations by transition-metal-based photocatalytic systems18−20 also propose metal hydride intermediates, except for one study suggesting a mechanism exempt of hydride.19
Scheme 1. Main Mechanistic Routes for the Molecular (Electro)catalytic Hydrogenation of C–C Multiple Bonds.
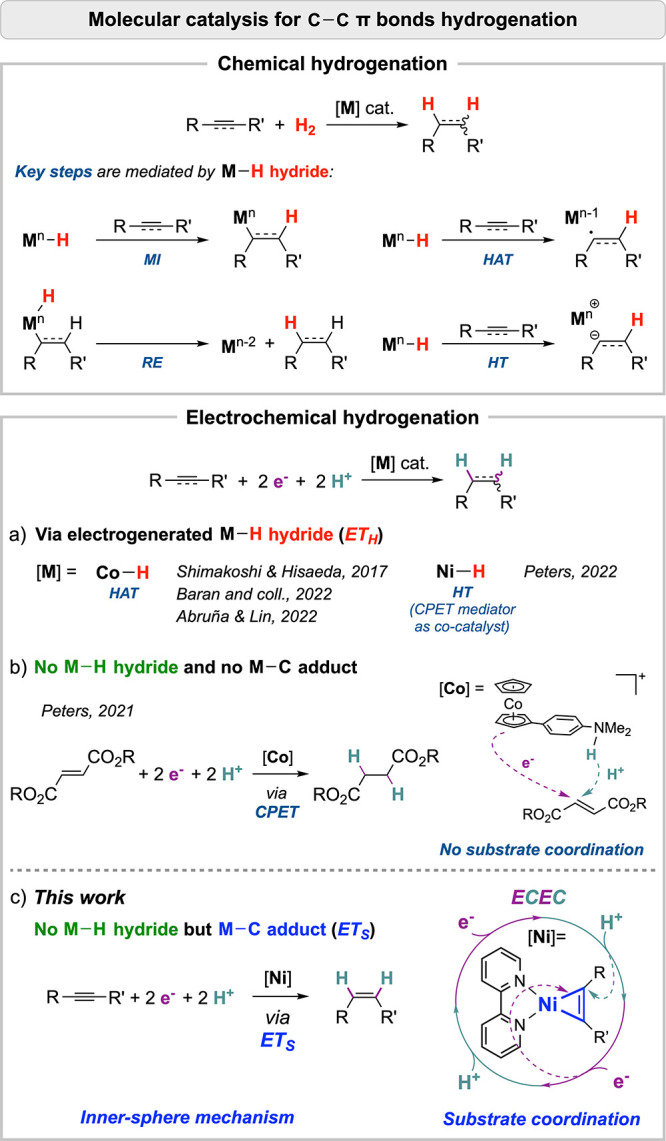
Molecular electrocatalysis has also been recently disclosed to achieve C–C unsaturated bond hydrogenation (Scheme 1), with metal hydrides highlighted as key intermediates.21−23 This point stands, for instance, in the electrochemical hydrogenation/deuteration of alkenes and alkynes using catalytic systems relying on a vitamin B12 model complex,24 a cobalt bipyridine complex,25 or salen complexes25,26 or the combination of a proton-coupled electron transfer (PCET) mediator with a nickel bis-diphosphine catalyst (Scheme 1a).27 These examples all share as common feature the generation of a M–H intermediate that further reacts with the C–C bond by hydride or hydrogen atom transfer.24−27 In such steps, electrons and protons are delivered together as H– or H•, subscribing to pathways coined as electron transfer through hydride (ETH).28 At variance, the electrochemical hydrogenation of α,β-unsaturated esters directly catalyzed by a concerted proton–electron transfer (CPET) mediator (Scheme 1b) reported by Peters and co-workers29 is, to our knowledge, the only supported example of molecularly electrocatalyzed C–C hydrogenation where no hydride is involved. By ensuring a separate delivery of electron and proton, that strategy bypasses the ETH route and thus doing outcompetes the undesired hydrogen evolution reaction (HER). In that case, however, the molecular construct of the CPET mediator precludes the formation of a metal–substrate adduct, often associated with improved selectivity and concertedness.30,31
Within that frame, our group has recently disclosed that [Ni(bpy)3]2+ is an efficient electrocatalyst for alkyne semihydrogenation providing the corresponding (Z)-olefins in good to high yields and faradaic efficiencies (FE).32 In the present work, we document that our system operates by an original pathway exempt of hydride intermediates. This mechanism instead involves a sequence of proton transfer (PT) and electron transfer (ET) steps from a nickelacyclopropene species as a resting state, as supported by organometallic, electrochemical, and kinetic studies (including isotope effect) as well as density functional theory (DFT) calculations. A most salient feature is that, while excluding the recourse to a catalytically competent hydride, the mechanism does proceed in the inner sphere of the metal via Ni-alkyne adduct formation and further PTs and ETs to the bound substrate. The system thus subscribes to the original frame of an ETS mechanism (electron transfer to the substrate)28 in which electrons are delivered via the metal–substrate adduct and that remained so far elusive for electrocatalytic C–C hydrogenation (Scheme 1c). In addition, our findings also rationalize that the observed (Z)-stereoselectivity most likely ensues from a barrierless isomerization of two nickel vinyl isomers.
Results and Discussion
Initiation from [Ni(bpy)3]2+
Previous work of our group established that the complex [Ni(bpy)3](BF4)2 (1;2 BF4–) is a selective electrocatalyst for alkynes semihydrogenation into the corresponding (Z)-olefins.32 Electrochemical studies showed that the two-electron reduction of 1 is coupled with the release of a bpy (bpy = 2,2′-bipyridine) ligand evolving [Ni(bpy)2] (2) (Scheme 2a).33 Subsequent coordination of an alkyne at 2 was then suggested on the basis of cyclic voltammetry (CV) analysis and postulated to lead to a [Ni(bpy)(alkyne)] species upon displacement of a second bpy.32,34
Scheme 2. Generation, Structure and Protonation of Nickelacyclopropene 3.

(a) Steps Involved in the Initiation Pathway and in the Synthesis of the Nickelacyclopropene 3; (b) Molecular Structure of 3 Obtained by XRD (Front and Side Views; ORTEP; 50% Probability; H Atoms Omitted for Clarity; Color Code: Gray: C; Purple: N; Green: Ni); (c) Computed Electron Density of the HOMO of 3 (H Atoms Are Omitted for Clarity); (d) Protonation Reactions Starting from the Nickelacyclopropene 3; and (e) 1H NMR Spectrum of the Vinyl Region of the Crude Mixture of 3 and BzOH (1 equiv) in DMF-d7 with nBu4NPF6 (0.1 M).
To confirm the hypothesis and the nature of the generated complex, synthetically isolated 2 (obtained from [Ni(COD)2]; COD = 1,5-cyclooctadiene; see SI Section 3.1) was reacted with the model alkyne 1-phenyl-1-propyne (S1), upon inspiration from the literature.35−38 Addition of 1 equiv of S1 to 2 in THF-d8 at room temperature (Scheme 2a) shows the formation of a dark red species. The spectroscopic signatures and molecular structure obtained by single-crystal X-ray diffraction (XRD) (Scheme 2b) identify the resulting isolated compound as the heteroleptic nickelacyclopropene complex [Ni(bpy)(PhCCMe)] (3), formed in an overall 65% yield from [Ni(COD)2] (see SI Section 3.2).39 In parallel of the synthetic isolation, the electrosynthetic formation of 3 was addressed. The passage of 2 electrons per 1 during bulk reductive electrolysis of a mixture of 1 and alkyne S1 (1/S1 1:10 ratio, in 0.1 M nBu4NPF6 DMF electrolyte) produces spectroscopic features revealing the formation of 3, as in particular the two characteristic 1H NMR signals of the 6/6′ positions of the ligated bpy at 10.14 and 10.01 ppm (see SI Section 3.4.6). These results unambiguously demonstrate that the nickelacyclopropene complex 3 is evolved upon 2-electron reduction of 1 in the presence of the alkyne S1.
Having identified 3 as a plausible catalytically relevant species, we aimed at engaging the complex in electrocatalytic assays of alkyne semihydrogenation. However, we noted that the addition of benzoic acid (BzOH), a suitable proton source for our system,32 to the pre-electrolysis medium containing 3 readily results in the fading of the dark red solution. This observation suggests that 3 is converted in the presence of acid. This result was confirmed by an independent experiment, in which contacting 2 equiv of BzOH acid with 3 in THF-d8 produces a pale blue precipitate (Scheme 2d). 1H NMR analyses of the isolated solid dissolved in DMSO-d6 (low solubility) reveal signals with paramagnetic behavior and that can be attributed to [Ni(bpy)(BzO)2] (4) (see SI Sections 3.3 and 3.4.3). As a conclusion, 3 readily converts to 4 when BzOH is used as the proton source.
We found that 4 is electrocatalytically active for the selective semihydrogenation of S1 into (Z)-β-methylstyrene ((Z)-S1H2) and interestingly provides a large increase in faradaic efficiency (>99 vs 64%) and experimental turnover frequency (TOFexp; 9.9 vs 6.8 10–3 s–1) toward S1H2 compared to 1 (Scheme 3A(a)). In addition, when controlled potential electrolysis of S1 using 4 is performed in the presence of excess bipyridine (2 equiv/4), TOFexp (7.0 10–3 s–1) and FE (68%) decrease to levels consistent with that obtained using 1. These results collectively suggest that excess free bipyridine in solution released upon reduction of [Ni(bpy)3]2+ or purposely added to 4 hinders the electrocatalytic semihydrogenation of interest. Thus, complex 1 was discarded in the rest of the study, and we instead focused on complexes 3 and 4 as these compounds derive from a more competent catalysis (see SI Section 2.2.2 for CV comparison of precatalysts 1 and 4). We also note that the generation of heterogeneous deposits responsible for the electrocatalytic semihydrogenation could be discarded (see SI Section 2.3.2), in line with our previous report using 1.32 With complexes 3 and 4 at hands, we further analyzed the framework of underlying electrochemical (E) and chemical (C) steps to withdraw mechanistic information.
Scheme 3. Electrochemical and Electrocatalytic Results.

(A) (a) Electrolysis with 1 (from ref (32)) or 4 as a Precatalyst. Yields, F.E., and Z/E Ratios Are Reported at Full Conversion; TOFexp Values Are Estimated Based on Time to Full Conversion; (b) CVs of 3 Alone (Dotted Blue, Oxidation First), 4 Alone (Black), with S1 (10 equiv) (Blue), or with S1 and BzOH (50 equiv) (red); and (c) Experimental and Computed Redox Potentials (V vs Fc+/0) for Compounds 3 and 4; (B) CVs of Mixtures of 4, S1, and BzOH; (a) Increasing [S1] from 5 to 50 mM (Light to Dark Green) and Plot of log(TOFMAX) with Respect to log([S1]); (b) Increasing [BzOH] from 5 to 50 mM (Light to Dark Blue) and Plot of log(TOFMAX) with Respect to log([BzOH]); (c) Increasing [4] from 0.5 to 4 mM (Light to Dark Red) and Plot of log(TOFMAX) with Respect to log([4]); and (d) with BzOD (Purple) or BzOH (Blue); Conditions for Electrolysis: [Catalyst] = 1 mM, [S1] = 10 mM, [BzOH] = 100 mM, Eapp = −1.72 ± 0.02 VFc; Conditions for CV: Unless Otherwise Stated [4] = 1 mM, [S1] = 10 mM, [BzOH] = 50 mM, 0.1 V·s–1 as Scan Rate (ν); Supporting Electrolyte: DMF 0.1 M nBu4NPF6. bEp,a. cE1/2. dEp,c. eE°. fQuadruplet Configuration (See SI Section 5.2 for Other Spin Configurations). gAfter the Release of One Benzoate Ligand (See Scheme S1).
Initial Electron Transfer Steps
CV studies were conducted to understand the behavior of complex 4 under electrocatalytic conditions. The CV of 4 shows a pseudoreversible reduction characterized by a broad cathodic wave at Ep,c = −1.76 V vs Fc+/0 (abbreviated VFc) featuring a shoulder at ca. −1.65 VFc and associated with a reoxidation wave at Ep,a = −1.64 VFc (Scheme 3A(b,c)).40 We tentatively attribute the broad cathodic wave of 4 to one-electron reduction events from Ni(II) to Ni(I) species and further to Ni(0) ones and that are closely located in potentials and underpinned by BzO– ligand dissociation/association equilibria (see SI Section 2.2.4 for more details). Adding 10 equiv of the alkyne S1 (Scheme 3A(b)) positively shifts both the shoulder of the reduction wave of 4 and in a drastic manner the reoxidation wave by 440 mV up to Ep,a = −1.20 VFc. This latter value matches the oxidation potential of the native nickelacyclopropene 3 (Ep,a = −1.19 VFc), which confirms the in situ formation of that species when 4 is doubly reduced in the presence of S1. A similar reoxidation event is observed when 1 is used instead (see SI Section 2.2.2). Adding BzOH to the previous mixture, a catalytic wave develops from the shoulder of the cathodic wave of 4 and reaches a first pseudoplateau at ca. −1.78 VFc and a second one at ca. −1.93 VFc (Schemes 3A(b) and Figure S6b). Interestingly, the reoxidation wave corresponding to the nickelacyclopropene species 3 can still be traced but is largely decreased (ip,a/ip,a0 = 0.13). Observing the nickelacyclopropene 3 under conditions of electrocatalytic turnover suggest that this species is the resting state in our conditions. Therefore, we posited that a protonation from 3 would be rate-determining. Pathways involving a stepwise electron transfer (ET) to 3 can be discarded under our electrocatalytic conditions since the applied potential is strongly positive to the reduction of 3 (Eapp = −1.7 VFc vs E1/2(30/–I) = −2.18 VFc). We thus turned to the more detailed study of pathways where a proton transfer (PT) proceeds from 3.
Protonation Steps
The protonolysis of 3 with 2 equiv of BzOH in DMF-d7 at room temperature quantitatively evolves, along with the precipitation of 4 (vide supra), the olefinic hydrogenation product S1H2 in a Z/E ratio of 8:2 (see SI Section 3.4.3). This observation clearly indicates that a two-fold protonation is accessible at 3. The same experiment in the presence of the supporting electrolyte nBu4NPF6 (0.1 M) induces solubilization of the paramagnetic species 4 which prevents a sensible 1H NMR analysis. To tentatively trap the protonation sequence after the first PT, we contacted 3 with only 1 equiv of BzOH and at low temperature (−30 °C) in DMF-d7 with 0.1 M nBu4NPF6 (Scheme 2d). The 1H NMR spectrum of the reaction mixture shows the appearance of two quadruplets at 5.00 and 5.30 ppm, which we attribute to α-methyl vinylic protons of putative nickel(II) vinyl species (I2 and I2E, Scheme 2e), based on the literature41 and the shifts expected for the corresponding free olefins. We attribute the build-up of two vinylic signals to the presence of the (Z)- and the (E)-vinyl complexes, respectively (see SI Section 3.4.2), found in a Z/E 8:2 ratio very close to the one of alkenes generated under stoichiometric (using 2 equiv of BzOH) and electrocatalytic conditions. The evolution of a Ni-vinyl species was also observed when using diphenylacetylene as the alkyne substrate (see SI Section 3.4.5). The facile protonation at the nickelacyclic carbons in 3 is further rationalized by the strong electron density located at these carbons in the highest occupied molecular orbital (HOMO) of this complex (Scheme 2c). However, only the vinyl protons in α-methyl position could be observed, which strongly support that the protonation is regioselective for that position. In the absence of nBu4NPF6, the vinylic signals were not obtained and the benzoic acid (1 equiv) was fully consumed toward the formation of alkenes. We surmise that, without nBu4NPF6, precipitation of 4 constitutes a driving force toward the formation of alkenes preventing the observation of the nickel vinyl intermediates. We also found that a similar body of results is obtained when 3 is electrogenerated from 1, followed by addition of BzOH in the mixture (see SI Section 3.4.6).
The observation of a Ni(II) vinyl intermediate indicates that the lifetime of such species may be long enough to afford for ET, in the case of electrocatalytic turnover. As such, this vinyl intermediate can constitute a bifurcating point between two mechanisms, namely, an EECC-type one where the two PTs are successive and an ECEC-type one, where the second PT is preceded by ET.42
Electrocatalytic Cycle
To gain further information on the mechanism, kinetic data were extracted from CV experiments. As we could not identify suitable conditions leading to a canonical S-shaped CV, we resorted to foot-of-wave analysis (FOWA).43 Regardless of the mechanism considered (EECC or ECEC), FOWA can be used to provide an estimate of the maximal turnover frequency (TOFMAX; see SI Section 4 for details).43 Following this methodology, we have found an average TOFMAX of 1.37 ± 0.09 105 s–1, leading to a reaction span of 10.5 ± 0.4 kcal mol–1 (see SI Section 4). Applying the same methodology while varying the concentrations in reaction partners, we obtained that the reaction is first order in the catalyst 4, zeroth order in alkyne, and first order in BzOH (Scheme 3B). Moreover, a kinetic isotope effect (KIE) of 6.3 ± 0.6 derives from FOWA analyses of CVs recorded using either BzOH or BzOD. Taken together, these results support that a protonation of a nickel-based intermediate after alkyne binding is rate-determining. Capitalizing on the kinetic data and reactivities highlighted in the previous sections, we discuss now possible mechanistic pathways by putting DFT calculations (Scheme 4) in perspective with experimental results.
Scheme 4. Computed ECEC-Type Mechanism of the Electrocatalytic Semihydrogenation of Alkyne S1 with 3.

ΔG values are given in kcal mol–1 and reaction coordinates are depicted in the middle of the cycle. Conditions: PBE-D3/6-311+G(d,p) level of theory and CPCM model to account for solvent effect (DMF). Geometries of 3, TS3-I1, I4, and I4E are given with H atoms omitted for clarity (except vinylic).
Electrochemical studies support that 3 is the resting state in the catalytic cycle, which was thus taken as the entry point and as origin of our thermodynamic scale for calculations. While the reduction of 3 was ruled out (vide supra), protonolysis of this species observed above indicates that protonation is feasible at this stage. A first outer-sphere protonation of 3 by BzOH was indeed found accessible (ΔG‡(TS3-I1) = +10.2 kcal mol–1) and leads to a cationic Ni(II) vinyl species I1 at +8.6 kcal mol–1, which undergoes barrierless coordination of a benzoate ligand to give I2 (ΔG = −5.5 kcal mol–1). This thermodynamically favored protonation yielding I2 is in good agreement with the NMR investigation (vide supra).
This first protonation then leads to a bifurcation point between the EECC- and ECEC-type pathways. In the case of EECC routes, the first protonation is followed by a second PT, before ET occurs. The most favorable second PT (outer sphere) was found to proceed via a TS at +15.6 kcal mol–1 (see SI Section 5.4.1). The span associated with this reaction path would then be 21.1 kcal mol–1, which is substantially higher than the experimental estimate (10.5 ± 0.4 kcal mol–1). Therefore, EECC-type pathways have been discarded (see SI Section 5.4.1 for details), and we thus turned to scrutinize in more detail possible pathways of the type ECEC.
In this case, the reductions of cationic or neutral vinyl species I1 or I2 are computed at E0 = −1.56 and −1.71 VFc, respectively, which are more anodic than or close to our applied potential and thus possible under our conditions. Regardless of the relevant Ni(II) vinyl intermediate, subsequent ET is computed to lead in turn to a nickel(I) vinyl species I4 (ΔG = −4.3 kcal mol–1). From I4, the different transition states obtained for inner-sphere protonation with BzOH coordinated were all found to be very high in energy (>24 kcal mol–1, see SI Section 5.4.3, Scheme S5).
In contrast, outer-sphere protonation by BzOH is barrierless and thus the most plausible next step, giving the nickel alkene complex I5 at −16.1 kcal mol–1. At this stage, coordination of a benzoate ligand is easy viaTSI5-I6 at −14.4 kcal mol–1 and readily triggers the release of the alkene S1H2 from the resulting alkene benzoate adduct intermediate I6 (ΔG = −29.0 kcal mol–1). The decoordination of the alkene leads to the Ni(I) benzoate complex I7 at −26.7 kcal mol–1. The favored binding of alkyne S1 to I7 displaces the BzO– ligand and generates I8, the cationic analogue of 3 (ΔG = −22.5 kcal mol–1). Electron transfer on this intermediate (E0 = −1.41 VFc) yields 3 and thus closes the catalytic cycle. A pathway including the reduction of I7 prior to alkyne coordination is also accessible, although at a slightly more negative potential (E0 computed at −1.76 VFc) and is further discussed in SI Section 2.2.5.
In the ECEC frame, whether I1 undergoes BzO– coordination to I2 or a direct reduction to I4 is arguable. The first pathway defines an energetic span scaling to 16.1 kcal mol–1 between I2 as a TOF-determining intermediate (TDI) and BzO– decoordination TSI3-I4 as TOF-determining TS (TDTS). The second one displays a span of 10.2 kcal mol–1 between 3 and TS3-I1, which corresponds to the first PT. In the first case yet, a dependence of the apparent reaction rate on [H+] is not to be expected, contradicting experimental findings (vide supra). For that reason and because of the lower span offered by the second pathway, we favor the hypothesis of the direct reduction of I1 to I4, with rate-determining protonation of 3.
Considering this pathway, the computed ECEC-type mechanism agrees with kinetic results obtained from experiments. In particular, the calculated span (10.2 kcal mol–1) is in good match with the expected span of 10.5 ± 0.4 kcal mol–1 derived from experimental TOFmax. In addition, the rate-limiting first protonation of 3 is corroborated by experimental orders in reaction partners (vide supra) and good agreement is found between experimentally observed and computed KIEs for that first protonation (6.3 ± 0.6 vs 6.8, respectively; see SI Section 5.3). An alternative and close pathway involving a concerted proton–electron transfer from 3 to I4 could also be considered. The BDE of I4 was calculated to be 65.2 kcal mol–1. This value is thermodynamically suitable for such a pathway.44,45 The latter can therefore not be fully excluded compared to the stepwise PT-ET sequence from 3.
We stress that the ECEC-type mechanism proposed was identified as the most favored one under our conditions of the applied potential (Eapp ≈ −1.7 VFc), although we recognize that initiation processes and evolution of BzO– may limit the direct quantitative comparison between experimental and computed kinetic values. Especially, the build-up of BzO– concentration at advanced alkyne conversion is likely to shift the mechanism into a pathway operating at a slightly more negative potential, via the reduction of I7 preceding alkyne binding into 3 and that also gives good agreement between electrochemical and computational results. At more negative applied potentials (Eapp ≤ −1.8 VFc), we note a strong degradation of F.E. (Figure S8), which is suggestive that other, less selective mechanistic pathways come into play. These points are further discussed in SI Section 2.2.5.
It is interesting to mention here that the ECEC-type mechanism formally consists in the hydrogenation of a ligand—in that case, an alkyne—which is well-known as a degradation pathway of molecular electrocatalysts46−55 or sometimes as an initiation to access more active catalytic species.56−58 In the present work, this phenomenon is desired and fully exploited as it is part of the catalytic cycle. The absence of substantial decomposition may be here due to the stabilization of the nickel catalyst by benzoate ligands. We also note that the protonation of metallacyclopropenes into vinyl intermediates and further leading to olefin release is not an unprecedented reactivity. Such a pattern was, for instance, explored with early transition-metal complexes, typically [M(Cp)2(alkyne)] metallocenes with M = Ti, Zr, Hf.59−62 However, these systems have, to our knowledge, not been exploited under catalytic conditions. The generation of oxygenated bases upon protonation of the metallacyclopropene by R–OH acids usually results in the formation of strong M–O bonds that irreversibly poison the catalyst and thus preclude turnover. In our case, we surmise that the comparatively lower oxophilicity of Ni combined with the electroreductive conditions enable to displace the bound benzoate ligand and thus entry into a catalytic cycle.
Stereoselectivity
On the basis of the identified ECEC-type mechanism, we then addressed the experimentally observed (Z)-selectivity (see SI Section 5.4.3 for the different isomerization pathways investigated). We first considered the isomerization of the Ni(II) vinyl complexes I1 or I2. The isomerization of the first intermediate I1 is possible without a transition state and leads to I1E (ΔG = + 10.6 kcal mol–1). In contrast, for the neutral Ni(II) vinyl species I2, the TS associated with such a transformation was found to be very high in energy (>30 kcal mol–1) which makes that transformation very unlikely. Despite this unreachable barrier at I2, the observation of the two isomers I2 and I2E upon stoichiometric protonation of 3 (see Scheme 2d,e) can be rationalized in virtue of the easy (Z)–(E) isomerization at the preceding intermediate I1. Isomerization at the following Ni(I) vinyl species I4 was also considered. The corresponding isomers, I4 and I4E, are both accessible at −4.3 and −3.8 kcal mol–1, respectively (Scheme 4), and also interconvert via a barrierless isomerization. Isomerization at a later stage from a Ni–olefin complex would imply oxidative addition of a C–H olefinic bond into a hydrido-vinyl species, which we rule out (vide infra), or a hydrogenation/dehydrogenation sequence that does not agree with the absence of overhydrogenation products. (Z)–(E) isomerizations at I1 or I4 intermediates are thus the most plausible options under electrocatalytic conditions and the predominance of one versus the other can hardly be assessed. We yet note that I4 follows I1 and that the Gibbs free energy difference for I4 and I4E of 0.5 kcal mol–1 is relatively close to the difference of 1.3 kcal mol–1 expected for a Boltzmann distribution of 9:1 (Z/E), which is the experimental selectivity for the alkene products after electrolysis (vide supra). If stereoselectivity is determined at I4, protonolysis of the nickel–carbon bond by benzoic acid approaching I4 or I4E would lead to the (Z)- or the (E)-olefin, respectively, and in turn generate the Z/E-partitioned product mixture.
Hydride Pathway
While the most plausible mechanistic route identified so far does not involve a hydride, we wanted to evaluate the possible role of such a common intermediate in hydrogenation, participating to migratory insertion, hydride or hydrogen atom transfer, or reductive elimination (vide supra, Scheme 1).11,12,17 Among hydride species conceivable in our system, several could be discarded due to exceedingly endergonic formation (see SI Section 5.4.2), leading to consider a Ni(II) hydride [Ni(H)(bpy)(BzO)] (I10) as the most plausible candidate (Scheme 5).
Scheme 5. Key DFT Calculation Findings on Nickel Hydride Formation and Reactivity.
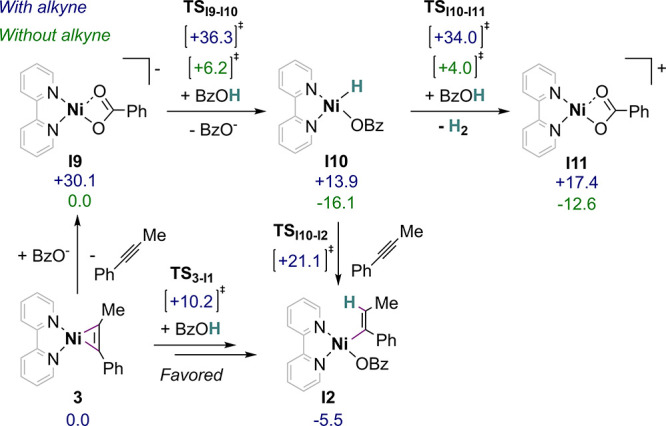
ΔG values are given in kcal mol–1. Conditions: PBE-D3/6-311+G(d,p) level of theory and CPCM model to account for solvent effect (DMF).
Despite an endergonic formation (ΔG = +13.9 kcal mol–1), the hydride species I10 is accessible at room temperature and would proceed by protonation of the anionic Ni(0) benzoate I9 intermediate with BzOH. However, two facts speak against such a pathway. Not only the protonation of I9 displays a high energetic barrier (ΔG‡(TSI9-I10) = +36.3 kcal mol–1) but also, in the presence of an alkyne, the formation of I9 itself is strongly disfavored (ΔG = +30.1 kcal mol–1) due to the high stability of the resting state 3. These results suggest that the presence of the alkyne, and therefore 3, prevents the formation of a hydride and thus hydrogen evolution by protonation of the latter. Such points are well in line with the electrocatalytic selectivity of 4 toward alkyne semihydrogenation, as evidenced by a quantitative faradaic efficiency to the olefin and the absence of detectable amounts of hydrogen in the headspace (see SI Section 2.1.3). We note, however, that, in the absence of the alkyne, a catalytic pathway for the hydrogen evolution can be located by computation. The associated span of 20.1 kcal mol–1 is consistent with a reaction at room temperature. These computational findings are in agreement with the evolution of H2, although poorly effective (turnover number: TON = 0.9, TOFexp = 0.19 10–3 s–1, FE(H2) = 27%), observed under electrocatalytic conditions in the absence of the alkyne (see SI Section 2.2.3 for CV of 4 in the presence of BzOH only).
Conclusions
The electrochemical semihydrogenation of alkynes catalyzed by homogeneous bipyridine-based nickel complexes [Ni(bpy)n(X)m]q has been recently achieved. We have shown here by a combined experimental and theoretical study that, under our conditions, a mechanism of the type ECEC is the most favorable pathway for that reaction. In particular, we found that a reductively induced nickelacyclopropene intermediate is the resting state, from which protonation proceeds as the (genuine) rate-determining step of the catalytic cycle. The selectivity of the reaction was rationalized to be governed by the formation of vinylic intermediate isomers close in energy, at either Ni(II) or Ni(I) stages. Most importantly, our results strongly support that no metal hydride species is involved in the catalytic activity of semihydrogenation in our conditions. We found that the absence of a competent hydride intermediate ensues from the presence and coordination of the alkyne substrate (ETS pathway). The bypass of a hydride is actually possible due to a sequence of electron and proton transfers directly at the Ni-coordinated unsaturated fragment. In addition, triggering a pathway exempt of hydride intermediates also eliminates the hydrogen evolution reaction as a side-reaction. While established systems for the catalytic hydrogenation of C–C unsaturations are so far proven to involve hydride species in migratory insertion, reductive elimination, hydride or hydrogen atom transfer steps (ETH), we document here an alternative ETS mechanism exempt of such hydride intermediates, but that maintains catalysis at the metal center. Such inner-sphere character is a feature of organometallic catalysis that is often integral to the control of regio- or stereoselectivity and brought here to electrocatalytic hydrogenation of C–C multiple bonds. We believe that this original approach to the reduction of organic unsaturations will open new paradigms for hydrogenation or hydroelementation reactions, for instance, with systems able to decouple proton and electron deliveries such as redox, photocatalytic, or electrocatalytic approaches.
Acknowledgments
The authors gratefully acknowledge basic support from the Max Planck Society and the RWTH Aachen University. They thank Annika Gurowski, Alina Jakubowski, and Justus Werkmeister for their help with analytical measurements.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c03340.
Detailed descriptions of experimental methods, additional electrochemical and electrocatalytic data (e.g., CVs of 1, 3, 4, electrolyses at different applied potentials), additional mechanistic details (e.g., [BzO–] effects), synthesis of compounds and stoichiometric experiments with associated characterizations (e.g., 1H, 13C NMR), and additional computational data (e.g., computed Gibbs energies for an EECC mechanism) (PDF)
The Max Planck Society and the RWTH Aachen University. Open access funded by Max Planck Society.
The authors declare no competing financial interest.
Supplementary Material
References
- Zimmerman J. B.; Anastas P. T.; Erythropel H. C.; Leitner W. Designing for a green chemistry future. Science 2020, 367, 397–400. 10.1126/science.aay3060. [DOI] [PubMed] [Google Scholar]
- Wiebe A.; Gieshoff T.; Mohle S.; Rodrigo E.; Zirbes M.; Waldvogel S. R. Electrifying Organic Synthesis. Angew. Chem., Int. Ed. 2018, 57, 5594–5619. 10.1002/anie.201711060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papanikolaou G.; Centi G.; Perathoner S.; Lanzafame P. Catalysis for e-Chemistry: Need and Gaps for a Future De-Fossilized Chemical Production, with Focus on the Role of Complex (Direct) Syntheses by Electrocatalysis. ACS Catal. 2022, 12, 2861–2876. 10.1021/acscatal.2c00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C.; Zheng Y.; Jaroniec M.; Qiao S.-Z. Electrocatalytic Refinery for Sustainable Production of Fuels and Chemicals. Angew. Chem., Int. Ed. 2021, 60, 19572–19590. 10.1002/anie.202101522. [DOI] [PubMed] [Google Scholar]
- Möhle S.; Zirbes M.; Rodrigo E.; Gieshoff T.; Wiebe A.; Waldvogel S. R. Modern Electrochemical Aspects for the Synthesis of Value-Added Organic Products. Angew. Chem., Int. Ed. 2018, 57, 6018–6041. 10.1002/anie.201712732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta R. C.; Meyer T. H.; Siewert I.; Ackermann L. Renewable resources for sustainable metallaelectro-catalysed C–H activation. Chem. Sci. 2020, 11, 8657–8670. 10.1039/D0SC03578E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Lu L.; Wood D.; Lin S. New Redox Strategies in Organic Synthesis by Means of Electrochemistry and Photochemistry. ACS Cent. Sci. 2020, 6, 1317–1340. 10.1021/acscentsci.0c00549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanfilippo D.; Rylander P. N.. Hydrogenation and Dehydrogenation. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co., 2009; pp 451–461. [Google Scholar]
- Blaser H.-U.; Spindler F.; Thommen M.. Industrial Applications. In The Handbook of Homogeneous Hydrogenation; WILEY-VCH Verlag GmbH & Co., 2006; pp 1279–1324. [Google Scholar]
- Genet J.-P.Reduction of Functionalized Alkenes. In Modern Reduction Methods; Wiley-VCH Verlag GmbH & Co., 2008; pp 1–38. [Google Scholar]
- Kluwer A. M.; Elsevier C. J.. Homogeneous Hydrogenation of Alkynes and Dienes. In The Handbook of Homogeneous Hydrogenation; WILEY-VCH Verlag GmbH & Co., 2006; pp 374–411. [Google Scholar]
- Bouwman E.Nickel. In The Handbook of Homogeneous Hydrogenation; WILEY-VCH Verlag GmbH & Co., 2006; pp 93–109. [Google Scholar]
- Diesen J. S.; Andersson P. G.. Hydrogenation of Unfunctionalized Alkenes. In Modern Reduction Methods; Wiley-VCH Verlag GmbH & Co., 2008; pp 39–64. [Google Scholar]
- Shevick S. L.; Wilson C. V.; Kotesova S.; Kim D.; Holland P. L.; Shenvi R. A. Catalytic hydrogen atom transfer to alkenes: a roadmap for metal hydrides and radicals. Chem. Sci. 2020, 11, 12401–12422. 10.1039/D0SC04112B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swamy K. C. K.; Reddy A. S.; Sandeep K.; Kalyani A. Advances in chemoselective and/or stereoselective semihydrogenation of alkynes. Tetrahedron Lett. 2018, 59, 419–429. 10.1016/j.tetlet.2017.12.057. [DOI] [Google Scholar]
- Crabtree R. H.Alkyls and Hydrides. In The Organometallic Chemistry of the Transition Metals; John Wiley & Sons, Inc., 2014; pp 69–97. [Google Scholar]
- Hartwig J. F.Organotransition Metal Chemistry: From Bonding to Catalysis / John F. Hartwig; University Science Books: Mill Valley, California, 2010, pp 585–602. [Google Scholar]
- Kamei Y.; Seino Y.; Yamaguchi Y.; Yoshino T.; Maeda S.; Kojima M.; Matsunaga S. Silane- and peroxide-free hydrogen atom transfer hydrogenation using ascorbic acid and cobalt-photoredox dual catalysis. Nat. Commun. 2021, 12, 966 10.1038/s41467-020-20872-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcudi F.; Ethordevic L.; Schweitzer N.; Stupp S. I.; Weiss E. A. Selective visible-light photocatalysis of acetylene to ethylene using a cobalt molecular catalyst and water as a proton source. Nat. Chem. 2022, 14, 1007–1012. 10.1038/s41557-022-00966-5. [DOI] [PubMed] [Google Scholar]
- Yuan T.; Huan S.; Ge B.; Lin S.; Zheng M.; Wang X. Semi-Hydrogenation of Alkynes by a Tandem Photoredox System Free of Noble Metal. CCS Chem. 2022, 4, 2597–2603. 10.31635/ccschem.021.202101457. [DOI] [Google Scholar]
- Shi Z.; Li N.; Lu H.-K.; Chen X.; Zheng H.; Yuan Y.; Ye K.-Y. Recent advances in the electrochemical hydrogenation of unsaturated hydrocarbons. Curr. Opin. Electrochem. 2021, 28, 100713 10.1016/j.coelec.2021.100713. [DOI] [Google Scholar]
- Jana S.; Mayerhofer V. J.; Teskey C. Photo- and Electrochemical Cobalt Catalysed Hydrogen Atom Transfer for the Hydrofunctionalisation of Alkenes. Angew. Chem. Int. Ed. 2023, e202304882. 10.1002/anie.202304882. [DOI] [PubMed] [Google Scholar]
- Metal hydrides are also proposed as key intermediates for catalytic hydrogenation using stoichiometric metal reductants in combination with proton sources; see for instance:; van der Puyl V.; McCourt R. O.; Shenvi R. A. Cobalt-catalyzed alkene hydrogenation by reductive turnover. Tetrahedron Lett. 2021, 72, 153047 10.1016/j.tetlet.2021.153047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimakoshi H.; Luo Z.; Tomita K.; Hisaeda Y. Cathodic reductive couplings and hydrogenations of alkenes and alkynes catalyzed by the B12 model complex. J. Organomet. Chem. 2017, 839, 71–77. 10.1016/j.jorganchem.2017.02.002. [DOI] [Google Scholar]
- Gnaim S.; Bauer A.; Zhang H.-J.; Chen L.; Gannett C.; Malapit C. A.; Hill D. E.; Vogt D.; Tang T.; Daley R. A.; Hao W.; Zeng R.; Quertenmont M.; Beck W. D.; Kandahari E.; Vantourout J. C.; Echeverria P.-G.; Abruna H. D.; Blackmond D. G.; Minteer S. D.; Reisman S. E.; Sigman M. S.; Baran P. S. Cobalt-electrocatalytic HAT for functionalization of unsaturated C–C bonds. Nature 2022, 605, 687–695. 10.1038/s41586-022-04595-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X.; Gannett C. N.; Liu J.; Zeng R.; Novaes L. F. T.; Wang H.; Abruña H. D.; Lin S. Intercepting Hydrogen Evolution with Hydrogen-Atom Transfer: Electron-Initiated Hydrofunctionalization of Alkenes. J. Am. Chem. Soc. 2022, 144, 17783–17791. 10.1021/jacs.2c08278. [DOI] [PubMed] [Google Scholar]
- Derosa J.; Garrido-Barros P.; Li M.; Peters J. C. Use of a PCET Mediator Enables a Ni-HER Electrocatalyst to Act as a Hydride Delivery Agent. J. Am. Chem. Soc. 2022, 144, 20118–20125. 10.1021/jacs.2c09786. [DOI] [PubMed] [Google Scholar]
- Kinzel N. W.; Werlé C.; Leitner W. Transition Metal Complexes as Catalysts for the Electroconversion of CO2: An Organometallic Perspective. Angew. Chem., Int. Ed. 2021, 60, 11628–11686. 10.1002/anie.202006988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derosa J.; Garrido-Barros P.; Peters J. C. Electrocatalytic Reduction of C-C pi-Bonds via a Cobaltocene-Derived Concerted Proton-Electron Transfer Mediator: Fumarate Hydrogenation as a Model Study. J. Am. Chem. Soc. 2021, 143, 9303–9307. 10.1021/jacs.1c03335. [DOI] [PubMed] [Google Scholar]
- Lexa D.; Saveant J. M.; Schaefer H. J.; Su Khac B.; Vering B.; Wang D. L. Outer-sphere and inner-sphere processes in reductive elimination. Direct and indirect electrochemical reduction of vicinal dibromoalkanes. J. Am. Chem. Soc. 1990, 112, 6162–6177. 10.1021/ja00173a002. [DOI] [Google Scholar]
- Kaeffer N.; Leitner W. Electrocatalysis with Molecular Transition-Metal Complexes for Reductive Organic Synthesis. JACS Au 2022, 2, 1266–1289. 10.1021/jacsau.2c00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. Y.; Kahl C.; Kaeffer N.; Leitner W. Electrocatalytic Semihydrogenation of Alkynes with [Ni(bpy)(3)]2+. JACS Au 2022, 2, 573–578. 10.1021/jacsau.1c00574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henne B. J.; Bartak D. E. Metal-vapor synthesis and electrochemistry of bis(bipyridyl)nickel(0). Inorg. Chem. 1984, 23, 369–373. 10.1021/ic00171a020. [DOI] [Google Scholar]
- Derien S.; Dunach E.; Perichon J. From stoichiometry to catalysis: electroreductive coupling of alkynes and carbon dioxide with nickel-bipyridine complexes. Magnesium ions as the key for catalysis. J. Am. Chem. Soc. 1991, 113, 8447–8454. 10.1021/ja00022a037. [DOI] [Google Scholar]
- Rosenthal U.; Nauck C.; Arndt P.; Pulst S.; Baumann W.; Burlakov V. V.; Görls H. Darstellung und eigenschaften des komplexes (dipy)Ni(η2-PhC2SiMe3). Zur korrelation struktureller bindungsparameter mit IR- und NMR-spektroskopischen daten in übergangsmetall-alkin-komplexen. J. Organomet. Chem. 1994, 484, 81–87. 10.1016/0022-328X(94)87189-2. [DOI] [Google Scholar]
- Eisch J. J.; Ma X.; Han; Kyoung I.; Gitua; John N.; Krüger C. Mechanistic Comparison of the Nickel(0)-Catalyzed Homo-Oligomerization and Co-Oligomerization of Alkynes and Nitriles. Eur. J. Inorg. Chem. 2001, 2001, 77–88. . [DOI] [Google Scholar]
- Ritz F. J.; Valentin L.; Henss A.; Würtele C.; Walter O.; Kozhushkov S. I.; Meijere A.; Schindler S. Syntheses, Structural Characterization, and Kinetic Investigations of Metalla[3]triangulanes: Isoelectronic Nickel(0) and Copper(I) Complexes with Bicyclopropylidene (bcp) and Dicyclopropylacetylene (dcpa) as Ligands. Eur. J. Org. Chem. 2021, 2021, 1864–1870. 10.1002/ejoc.202100045. [DOI] [Google Scholar]
- Hoberg H.; Herrera A. Nickelacyclopentene Derivatives from Nickel(0), Carbon Monoxide and Alkyne. Angew. Chem., Int. Ed. 1980, 19, 927. 10.1002/anie.198009271. [DOI] [Google Scholar]
- Based on ref (35), we note that the species 3 is better described as a Ni(II) nickelacyclopropene than as a Ni(0)-alkyne complex. I5 and I6 are better described as Ni(I)-olefin complexes.
- We note that the reduction of Ni(II) mono-polypyridine bis-carboxylate complexes is prone to trigger carboxylate ligand dissociation and disproportionation at Ni(I). These phenomena can produce an ill-defined reduction wave of the initial Ni(II) complex. Yet, the process leads to an overall two-electron reduction to Ni(0) at potentials negative to the wave. The nature of the underpinning events is further discussed in SI Section 2.2.4 and in previous literature:; Day C. S.; Renteria-Gomez A.; Ton S. J.; Gogoi A. R.; Gutierrez O.; Martin R. Elucidating electron-transfer events in polypyridine nickel complexes for reductive coupling reactions. Nat. Catal. 2023, 6, 244–253. 10.1038/s41929-023-00925-4. [DOI] [Google Scholar]
- The difference between the 1H NMR shifts of the olefinic protons in the free alkene and in the vinyl complex is similar to that reported in the case of another Ni(II)-vinyl complex:; Shen R.; Chen T.; Zhao Y.; Qiu R.; Zhou Y.; Yin S.; Wang X.; Goto M.; Han L. B. Facile regio- and stereoselective hydrometalation of alkynes with a combination of carboxylic acids and group 10 transition metal complexes: selective hydrogenation of alkynes with formic acid. J. Am. Chem. Soc. 2011, 133, 17037–44. 10.1021/ja2069246. [DOI] [PubMed] [Google Scholar]
- The primary entry in catalysis is via I7 by initiation from 4. As this initiation does not require a pre-equilibrium to allow for electron transfer at the potential of electrolysis (ca. −1.7 VFc), we classified the mechanism as an ECEC one. We kept this schematic description to describe the mechanism in general as the following cycles also alternate electron and proton transfer steps.
- Costentin C.; Drouet S.; Robert M.; Savéant J.-M. Turnover Numbers, Turnover Frequencies, and Overpotential in Molecular Catalysis of Electrochemical Reactions. Cyclic Voltammetry and Preparative-Scale Electrolysis. J. Am. Chem. Soc. 2012, 134, 11235–11242. 10.1021/ja303560c. [DOI] [PubMed] [Google Scholar]
- Warren J. J.; Tronic T. A.; Mayer J. M. Thermochemistry of proton-coupled electron transfer reagents and its implications. Chem. Rev. 2010, 110, 6961–7001. 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis B. H.; Hammes-Schiffer S. Proton-coupled electron transfer in molecular electrocatalysis: theoretical methods and design principles. Inorg. Chem. 2014, 53, 6427–43. 10.1021/ic5002896. [DOI] [PubMed] [Google Scholar]
- Simándi L.; Szeverényi Z.; Budó-Záhonyi É. Activation of molecular hydrogen by cobaloxime(II) derivatives. Inorg. Nucl. Chem. Lett. 1975, 11, 773–777. 10.1016/0020-1650(75)80098-5. [DOI] [Google Scholar]
- Simándi L.; Budó-Záhonyi É.; Szeverényi Z. Effect of strong base on the activation of molecular hydrogen by pyridinebis(dimethylglyoximato)cobalt(II). Inorg. Nucl. Chem. Lett. 1976, 12, 237–241. 10.1016/0020-1650(76)80158-4. [DOI] [Google Scholar]
- Yamaguchi T.; Miyagawa R. The Effect of Pyridine on the Hydrogen Absorption Process of Bis(dimethylglyoximato)cobalt(II). Chem. Lett. 1978, 7, 89–92. 10.1246/cl.1978.89. [DOI] [Google Scholar]
- Queyriaux N.; Sun D.; Fize J.; Pécaut J.; Field M. J.; Chavarot-Kerlidou M.; Artero V. Electrocatalytic Hydrogen Evolution with a Cobalt Complex Bearing Pendant Proton Relays: Acid Strength and Applied Potential Govern Mechanism and Stability. J. Am. Chem. Soc. 2020, 142, 274–282. 10.1021/jacs.9b10407. [DOI] [PubMed] [Google Scholar]
- El Ghachtouli S.; Fournier M.; Cherdo S.; Guillot R.; Charlot M.-F.; Anxolabéhère-Mallart E.; Robert M.; Aukauloo A. Monometallic Cobalt–Trisglyoximato Complexes as Precatalysts for Catalytic H2 Evolution in Water. J. Phys. Chem. C 2013, 117, 17073–17077. 10.1021/jp405134a. [DOI] [Google Scholar]
- Martin D. J.; McCarthy B. D.; Donley C. L.; Dempsey J. L. Electrochemical hydrogenation of a homogeneous nickel complex to form a surface adsorbed hydrogen-evolving species. Chem. Commun. 2015, 51, 5290–3. 10.1039/C4CC08662G. [DOI] [PubMed] [Google Scholar]
- Kaeffer N.; Morozan A.; Fize J.; Martinez E.; Guetaz L.; Artero V. The Dark Side of Molecular Catalysis: Diimine–Dioxime Cobalt Complexes Are Not the Actual Hydrogen Evolution Electrocatalyst in Acidic Aqueous Solutions. ACS Catal. 2016, 6, 3727–3737. 10.1021/acscatal.6b00378. [DOI] [Google Scholar]
- Lee K. J.; McCarthy B. D.; Dempsey J. L. On decomposition, degradation, and voltammetric deviation: the electrochemist’s field guide to identifying precatalyst transformation. Chem. Soc. Rev. 2019, 48, 2927–2945. 10.1039/C8CS00851E. [DOI] [PubMed] [Google Scholar]
- Queyriaux N. Redox-Active Ligands in Electroassisted Catalytic H+ and CO2 Reductions: Benefits and Risks. ACS Catal. 2021, 11, 4024–4035. 10.1021/acscatal.1c00237. [DOI] [Google Scholar]
- Anferov S. W.; Filatov A. S.; Anderson J. S. Cobalt-Catalyzed Hydrogenation Reactions Enabled by Ligand-Based Storage of Dihydrogen. ACS Catal. 2022, 12, 9933–9943. 10.1021/acscatal.2c02467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R.; Liu M.; Zheng S.-L.; Dogutan D. K.; Costentin C.; Nocera D. G. Proton-coupled electron transfer of macrocyclic ring hydrogenation: The chlorinphlorin. Proc. Natl. Acad. Sci. U.S.A. 2022, 119, e2122063119 10.1073/pnas.2122063119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher A. G.; Liu M.; Nocera D. G. Ligand Noninnocence in Nickel Porphyrins: Nickel Isobacteriochlorin Formation under Hydrogen Evolution Conditions. Inorg. Chem. 2019, 58, 7958–7968. 10.1021/acs.inorgchem.9b00717. [DOI] [PubMed] [Google Scholar]
- Windle C. Electrocatalysis: Reduced ring makes catalyst sing. Nat. Rev. Chem. 2017, 1, 0062 10.1038/s41570-017-0062. [DOI] [Google Scholar]
- Herrmann G. S.; Alt H. G.; Thewalt U. Alkenyl- und Ethylkomplexe des Titanocens. Molekülstruktur von [Cp2Ti(CMe=CHMe)]2μ-O. J. Organomet. Chem. 1990, 393, 83–95. 10.1016/0022-328X(90)87200-W. [DOI] [Google Scholar]
- Beweries T.; Burlakov V. V.; Peitz S.; Arndt P.; Baumann W.; Spannenberg A.; Rosenthal U. Synthesis and Reactions of Cp*2Hf(η2-PhC2SiMe3) with Water and Carbon Dioxide. Organometallics 2008, 27, 3954–3959. 10.1021/om8003064. [DOI] [Google Scholar]
- Pellny P.-M.; Burlakov V. V.; Baumann W.; Spannenberg A.; Rosenthal U. The Influence of the Ligands Cp*(η5-C5Me5) and Cp(η5-C5H5) on the Stability and Reactivity of Titanocene and Zirconocene Complexes: Reactions of the Bis(trimethylsilyl)acetylene Permethylmetallocene Complexes (η5-C5Me5)2M(η2-Me3SiC2SiMe3), M=Ti, Zr, with H2O and CO2. Z. Anorg. Allg. Chem. 1999, 625, 910–918. 10.1002/(SICI)1521-3749(199906)625:63.0.CO;2-6. [DOI] [Google Scholar]
- Rosenthal U. Recent Synthetic and Catalytic Applications of Group 4 Metallocene Bis(trimethylsilyl)acetylene Complexes. Eur. J. Inorg. Chem. 2019, 2019, 895–919. 10.1002/ejic.201801484. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.