

Abstract
Acute kidney injury (AKI) is associated with high morbidity and mortality, and no drugs are available clinically. Metabolic reprogramming resulting from the deletion of S-nitroso-Coenzyme A reductase 2 (SCoR2; AKR1A1) protects mice against AKI, identifying SCoR2 as a potential drug target. Of the few known inhibitors of SCoR2, none are selective versus the related oxidoreductase AKR1B1, limiting therapeutic utility. To identify SCoR2 (AKR1A1) inhibitors with selectivity versus AKR1B1, analogs of the non-selective (dual 1A1/1B1) inhibitor imirestat were designed, synthesized, and evaluated. Among 57 compounds, JSD26 has ten-fold selectivity for SCoR2 versus AKR1B1 and inhibits SCoR2 potently through an uncompetitive mechanism. When dosed orally to mice, JSD26 inhibited SNO-CoA metabolic activity in multiple organs. Notably, intraperitoneal injection of JSD26 in mice protected against AKI through S-nitrosylation of pyruvate kinase M2 (PKM2), whereas imirestat was not protective. Thus, selective inhibition of SCoR2 has therapeutic potential to treat acute kidney injury.
Graphical Abstract
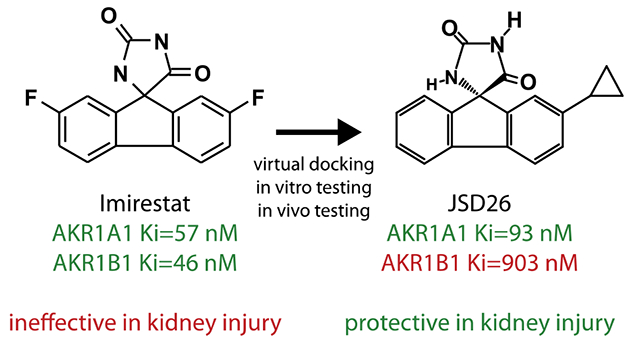
Introduction
Acute kidney injury (AKI) is a syndrome characterized by the rapid loss of kidney excretory function and is associated with high morbidity and mortality.1, 2 AKI can be caused by renal ischemia, direct damage to the kidneys, or blockage of the urinary tract. Low blood pressure, vasoconstrictive drugs, radiocontrast agents, blood loss, severe allergic reactions and trauma may often contribute.3, 4 When delivery of oxygen and metabolic substrates becomes inadequate during ischemia, sublethal injury of renal proximal tubular epithelium occurs.5 This in turn leads to the initiation of acute tubular necrosis and a rapid decline in glomerular filtration rate. After a period of temporary ischemia, acute restoration of the blood supply (reperfusion) results in oxidative damage and robust inflammation.6 AKI is thus a complicated illness with manifold causes and contributing mechanisms, and no drugs are available for clinical treatment.7, 8
We have recently discovered that a novel protein S-nitrosylation system, including S-nitroso-Coenzyme A (SNO-CoA) and its cognate denitrosylase SNO-CoA reductase (SCoR1 in yeast; SCoR2 in mammals), plays an important role in protecting kidneys from AKI.9 SNO-CoA, formed by coupling of NO to CoA, serves as an endogenous S-nitrosylating agent and regulates multiple metabolic pathways via S-nitrosylation of specific substrates.10, 11 The cellular level of SNO-CoA is strictly regulated by SCoR2, which eliminates SNO-CoA.12 Deletion of the SCoR2 thus leads to elevated SNO-CoA and increased S-nitrosylation of proteins. Knockout of SCoR2 in mice promotes renal protection through S-nitrosylation of pyruvate kinase M2 (PKM2).9 S-nitrosylation of PKM2 causes metabolic shunting of glycolytic intermediates into both one-carbon anabolic precursors and into the pentose phosphate pathway to generate NADPH thereby ameliorating oxidative kidney injury.9, 13 Thus, inhibition of SCoR2 provides a novel therapeutic opportunity in AKI.
SCoR2 is encoded by the aldo-keto reductase family member A1 (AKR1A1) gene. Among the AKR family, AKR1B1 is the closest isoform to AKR1A1, sharing around 50% protein sequence identity. Both AKR1B1 and AKR1A1 are characterized by a well-known (β/α)8-barrel motif.14 The C-terminal region of the (β/α)8-barrel delineates the active site, which is optimized for high-affinity interaction with NADPH. The active site possesses two contacting domains potentially involved in inhibitor binding. The first is a hydrophilic region, which contains a recognition sequence for hydrogen bond acceptors. The second is a hydrophobic pocket lining the active site cleft. The active sites of AKR1B1 and AKR1A1 differ in the C-terminal loop region, providing opportunity to design specific inhibitors. The C-terminal loop of AKR1B1 exhibits a higher level of plasticity, while AKR1A1 possesses an insertion of 10 amino acids between Met301 and Arg311 that likely affects its dynamic properties.15, 16 Because of its important role in hyperglycemic injury and diabetic complications, AKR1B1 is by far the most studied of the aldo-keto reductase isoforms, and several AKR1B1-specific inhibitors have been developed.17 Most AKR1B1 inhibitors are either carboxylic acids (tolrestat and zopolrestat) or spirohydantoins (sorbinil, imirestat and ranirestat), and several have entered clinical trials. By contrast, the primary function of AKR1A1 in humans had remained a mystery until we identified AKR1A1 as a SNO-CoA reductase.12 Thus, AKR1A1 (SCoR2) has not previously been viewed as a desirable drug target.18
Here, we report that AKR1B1 co-inhibition using a non-selective drug is detrimental to renal protection expected by inhibition of AKR1A1. To develop specific inhibitors of SCoR2 (AKR1A1), we employed in silico docking to aid in the design of compounds that bind the active site of SCoR2 versus AKR1B1. Thirty compounds were identified that inhibit SCoR2 activity with an IC50 below 100 nM. One of these, JSD26, selectively inhibits SCoR2 versus AKR1B1, and we used JSD26 in vivo to demonstrate the therapeutic potential of a specific SCoR2/AKR1A1 inhibitor in renoprotection.
Results
Imirestat inhibits both AKR1B1 and SCoR2 activity.
Our previous study indicated that inhibition of SCoR2 may be therapeutic in AKI.9 AKR1B1 is the closest isoform to SCoR2 in the aldo-keto reductase family. Thus, to discover potential SCoR2 inhibitors, we tested the well-established AKR1B1 inhibitors (imirestat, sorbinil, epalrestat and tolrestat) to determine whether they also inhibit SCoR2 activity. All four compounds inhibit the activity of SCoR2 to metabolize DL-glyceraldehyde (a common substrate of the aldo-keto reductase family) in an in vitro assay (Figure 1A–1D). Of these, imirestat was the most potent inhibitor of the DL-glyceraldehyde-metabolizing activity of SCoR2, with an IC50 of 45 nM (Figure 1A). Imirestat also efficiently inhibited the SNO-CoA-metabolizing activity of SCoR2, with an IC50 of 57 nM (Figure 1E).
Figure 1. Imirestat inhibits SCoR2 activity but fails to protect against AKI.
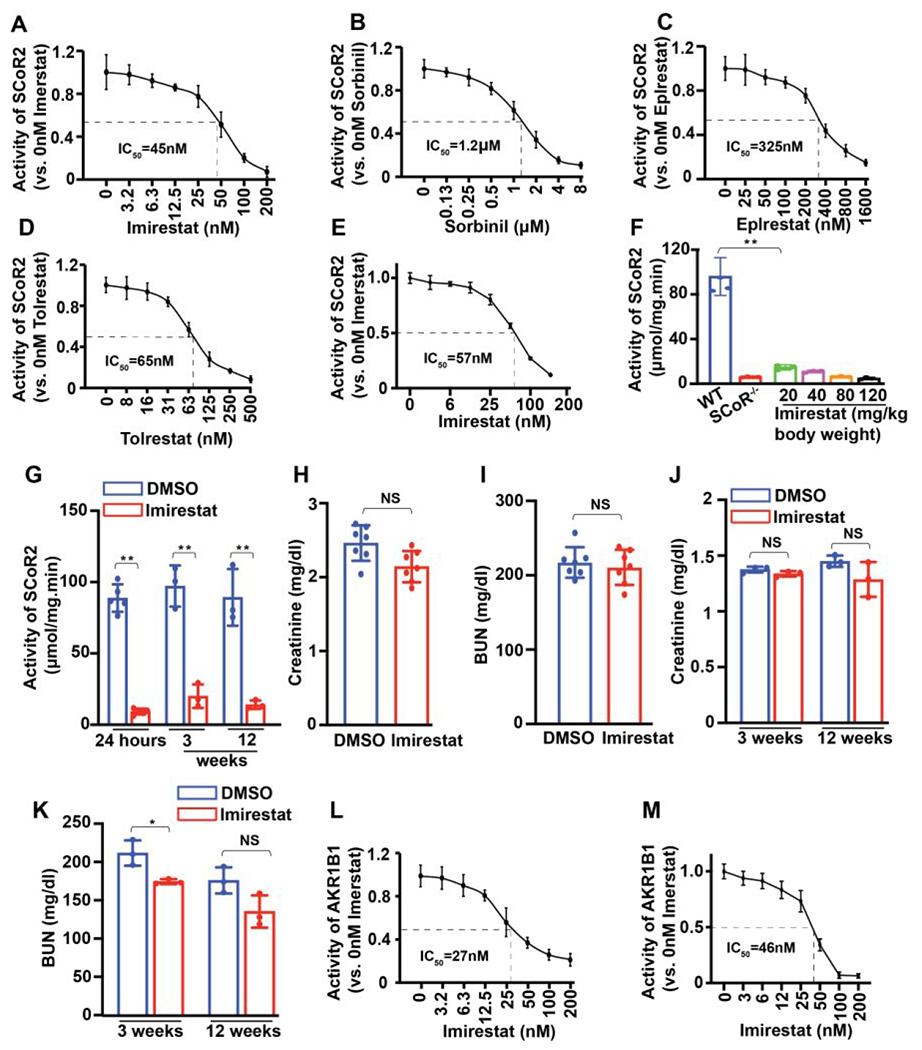
(A-D) IC50 values of four known AKR1B1 inhibitor compounds (imirestat, sorbinil, epalrestat and tolrestat) against purified SCoR2 using DL-glyceraldehyde as substrate (n=3). (E) IC50 value of imirestat to inhibit SCoR2 using SNO-CoA as substrate (n=3). (F) SNO-CoA metabolizing activity of kidneys from imirestat-treated mice (i.p. at the indicated dose) vs vehicle (DMSO)-treated or SCoR2-knockout (SCoR2−/−) mice (n=3). (G) SNO-CoA-metabolizing activity in AKI-injured kidneys from mice on normal diet (n=5) and imirestat diet-fed mice (for the indicated times) (n=3). (H-I) Serum creatinine and blood urea nitrogen (BUN) levels in serum from mice injected i.p. with imirestat 18h prior to I/R-induced AKI (n=7). (J-K) Serum creatinine and BUN in imirestat diet-fed mice (for the indicated times) subjected to I/R-induced AKI (n=3). (L) IC50 value of imirestat against AKR1B1 using DL-glyceraldehyde as substrate (n=3). (M) IC50 value of imirestat against AKR1B1 using methylglyoxylate as substrate (n=3). All results are presented as mean ± SD. Two-tailed Student’s t-test was used to detect significance. *, p<0.05; **, p<0.01; NS, not significant.
Imirestat has been tested clinically as an AKR1B1 inhibitor and is efficiently absorbed into blood after intraperitoneal injection.19 We injected imirestat i.p. into the lower right quadrant of the mouse abdomen at a single dose of 20, 40, 80 or 120 mg/kg body weight. After 18 hours, renal SNO-CoA metabolic activity was reduced by 80% in kidneys from imirestat-treated mice, even at the lowest dose (20 mg/kg), compared with DMSO control (Figure 1F). This degree of inhibition was essentially equal to that seen in the kidneys of SCoR2-knockout mice (SCoR2−/−), indicating that imirestat is a highly effective inhibitor of SCoR2 in vivo.
Next, we investigated whether pretreatment with imirestat would provide protection against AKI, as seen with knockout of SCoR2 in mice.9 AKI surgery was performed 18 hours after administration of imirestat (20 mg/kg i.p.). Blood and kidneys were harvested 24 hours post-surgery. Although SNO-CoA-metabolic activity in kidneys following AKI was strongly inhibited and almost equivalent to inhibition in the kidneys of SCoR2−/− mice (Figure 1G), we found that both serum creatinine and blood urea nitrogen (BUN) level—indicators of kidney dysfunction—were the same in imirestat-treated mice as in DMSO-treated mice, indicating that imirestat treatment does not protect against AKI (Figure 1H and 1I). To investigate if longer-term inhibition of SCoR2 activity is required for protection against AKI, we fed mice a diet containing imirestat (AIN-93 Rodent diet, with 125 mg imirestat/kg diet) for 3 weeks or 12 weeks. SCoR2 activity in the kidney was potently inhibited on the imirestat diet (Figure 1G) confirming its oral bioavailability, but the mice still did not show protection against AKI (Figure 1J and 1K). Since imirestat inhibits the activity of AKR1B1 (Figure 1L and 1M), we inferred that the AKR1B1 inhibition mediated by imirestat might counteract renal protection.
JSD26 is a selective SCoR2 inhibitor.
Based on the high SCoR2 inhibitory potency of imirestat, we elected to develop SAR around its tricyclic hydantoin core using an enzymatic assay with DL-glyceraldehyde as the substrate (Figure 2A). Beginning with imirestat, we explored the effects of substituents at C2, C3, C4 and C7 (Figure 2B and Table S1). Synthesis of the hydantoin ring was accomplished using the Bucherer-Berg reaction starting from substituted fluorenones.20 For compound 5-1 (JSD26) and 5-2, a modified synthesis from the imine was carried out (Supplemental Figure 1). Of the 57 imirestat analogs synthesized, 23 inhibited DL-glyceraldehyde-metabolizing activity with an IC50 below 150 nM (Figure 2C). SAR studies indicate: 1) hydantoin analog 2 which lacks the C2 and C7 fluorine atoms of imirestat is less potent; 2) introduction of substituents at C2, with or without a C7 fluoro substituent, gave loss of potency, with chloro, bromo, cyclopropyl and cyclobutyl being the only exceptions; 3) substitution at C3 and C4 was better tolerated with several larger and more polar substituents giving potency equal to or better than that of imirestat; 4) all analogs with substituents larger than fluorine at either C2 and C7 were less potent than imirestat (Table 1).
Figure 2. Screening for selective SCoR2 inhibitors.
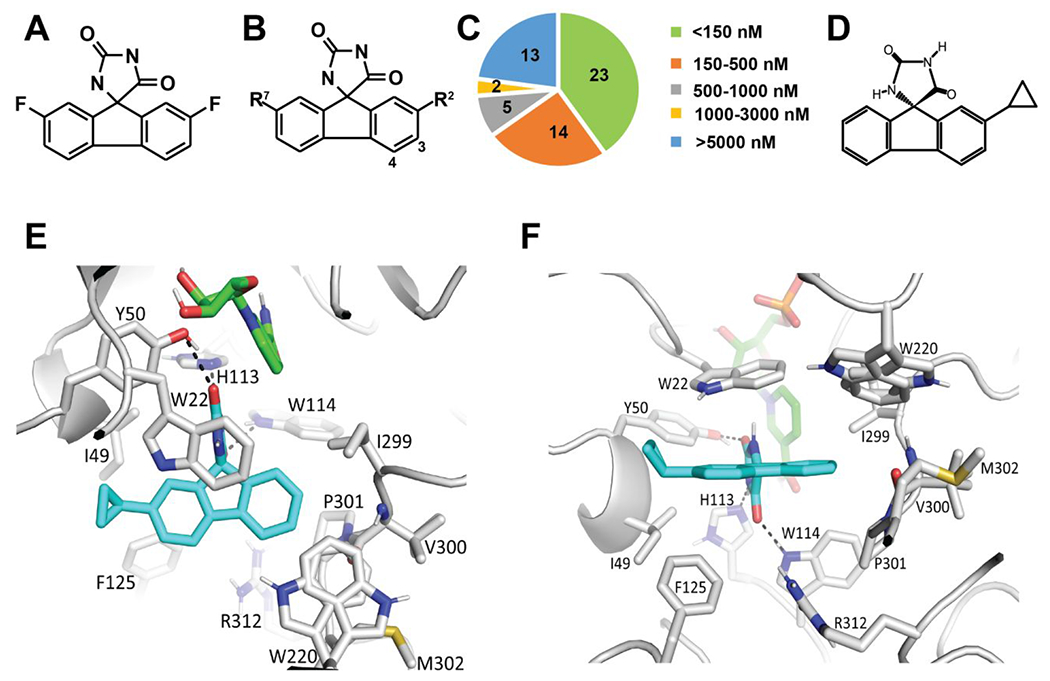
(A) Chemical structure of imirestat. (B) Generic structure of imirestat scaffold showing positions (C2, C3, C4, C7) modified in design of new compounds. (C) IC50 value of 57 compounds for inhibition of the DL-glyceraldehyde-metabolizing activity of SCoR2. (D) Chemical structure of JSD26. (E-F) Docked binding model of JSD26 bound to SCoR2. SCoR2 (grey carbons), NADPH (green carbons) and JSD26 (blue carbons). Predicted hydrogen bonds are depicted as dashed lines.
Table 1:
Selectivity of top 23 compounds inhibiting SNO-CoA-metabolizing activity of SCoR2 versus inhibiting methylglyoxylate-metabolizing activity of AKR1B1.
| Number | C2 | C3 | C4 | C7 | SCoR2 IC50 (nM) | AKR1B1 IC50 nM | AKR1B1/SCoR |
|---|---|---|---|---|---|---|---|
| Imirestat (1) | F | H | H | F | 57 | 46 | 0.81 |
| 5-1* (JSD26) | Cyclopropyl | H | H | H | 93 | 903 | 9.7 |
| 9 | Cl | H | H | H | 53 | 16 | 0.30 |
| 16 | Cyclopropyl | H | H | F | 122 | 212 | 1.7 |
| 17-1 * | Cyclobutyl | H | H | F | 33 | 55 | 1.7 |
| 17-2 * | Cyclobutyl | H | H | F | 67 | 44 | 0.66 |
| 20 | Cl | H | H | Cl | 127 | 56 | 0.44 |
| 33 | F | Cyclopropyl | H | F | 72 | 15 | 0.21 |
| 34 | H | OCH3 | H | F | 68 | 16 | 0.24 |
| 38 | H | H | F | F | 67 | 16 | 0.24 |
| 45 | H | OCH2CO2H | H | F | 42 | 53 | 1.3 |
| 46 | H | OCH2CO2H | H | F | 74 | 13 | 0.18 |
| 47-1 * | F | OH | CH2CO2H | F | 25 | 42 | 1.7 |
| 47-2 * | F | OH | CH2CO2H | F | 48 | 32 | 0.67 |
| 48-1 * | F | OCH2CO2H | H | F | 63 | 27 | 0.43 |
| 48-2 * | F | OCH2CO2H | H | F | 82 | 22 | 0.27 |
| 49-1 * | F | H | CH2CO2H | F | 57 | 30 | 0.53 |
| 49-2 * | F | H | CH2CO2H | F | 57 | 15 | 0.26 |
| 50 | F | 2-Azetidin-1-yl-2-oxoethoxy | H | F | 80 | 19 | 0.24 |
| 51 | F | H | 2-(Azetidin-1-yl)-2-oxoethoxy | F | 59 | 18 | 0.31 |
| 52 | F | H | 1-Cyclopentyl-1H-1,2,3-triazol-4-yl | F | 66 | 11 | 0.17 |
| 53 | F | (Oxetan-3-yl)oxy | H | F | 29 | 14 | 0.48 |
| 30 | H | H | F | H | 82 | 52 | 0.63 |
| 19 | Br | H | H | F | 51 | 22 | 0.43 |
individual enantiomers
Active compounds from the SAR studies were docked to the published crystal structure of Sus scrofa AKR1A1 (SCoR2, PDBid 3H4G).21 There are three reasons why this PDB structure was chosen: 1) the human AKR1A1 structure does not have the NADPH/NADP+ cofactor bound (PDBid 2ALR);22 2) Sus scrofa AKR1A1 has a high 94.2% sequence identity with human AKR1A1; and 3) the Sus scrofa AKR1A1 structure had fidarestat bound, which shares the same key imidazolidine-2,4-dione moiety with imirestat.21 To probe the selectivity of the designed compounds, we additionally docked the compounds against AKR1B1 (PDBid 4JIR, complexed with epalrestat)23 and AKR1B1 (PDBid 4GAB, complexed with fidarestat).24 Our docking studies suggested that introduction of a hydroxyl group at C3 would allow a hydrogen-bond to be made with the backbone oxygen of Val300 present in SCoR2 and that carboxymethyl substituent at C4 could make an electrostatic interaction with Arg312, present in SCoR2 but not in AKR1B1, and a hydrogen bond with the backbone nitrogen of M302. These docking studies led to the synthesis of inhibitors in Table S2, all of which retained good potency at SCoR2.
To further identify selective SCoR2 inhibitors, we picked the top 23 compounds that inhibited DL-glyceraldehyde-metabolizing activity with an IC50 below 150 nM (Figure 2C). We determined the IC50 of these 23 compounds to inhibit the SNO-CoA (specific SCoR2 substrate)-metabolizing activity of SCoR2 and the methylglyoxylate (specific AKR1B1 substrate)-metabolizing activity of AKR1B1 (Table 1). Seventeen imirestat analogs more efficiently inhibited AKR1B1 activity than the parent imirestat (IC50 below 46 nM), while seven imirestat analogs more effectively inhibited SCoR2 than the parent imirestat (IC50 below 57 nM) (Table 1).
One of these 23 imirestat analogs, compound 5-1 (henceforth called JSD26), inhibited SCoR2 with an IC50 of 93 nM and AKR1B1 with IC50 of 903 nM (Table 1, Figure 2D, 3A and 3B). Thus, JSD26 is a relatively selective compound (9.7 times more potent at SCoR2 vs AKR1B1 compared to imirestat, which is 1.2 times more potent at AKR1B1). JSD26 is the C2-cyclopropyl and C7-H-substituted imirestat analog. SAR studies indicated that 1) analogs with C3 or C4 can tolerate larger polar or non-polar groups substituents and inhibit SCoR2 activity with better efficiency than imirestat, but none improved the selectivity for SCoR2 versus AKR1B1; 2) C2 and C7 positions in imirestat can tolerate only small substituents such as –H, –F, –Cl, –cyclopropyl, –cyclobutyl, –OH or –OCH3; and 3) only compound 5-1 (JSD26) selectively inhibited SCoR2 activity (Table 1).
Figure 3. JSD26 is a selective SCoR2 inhibitor.
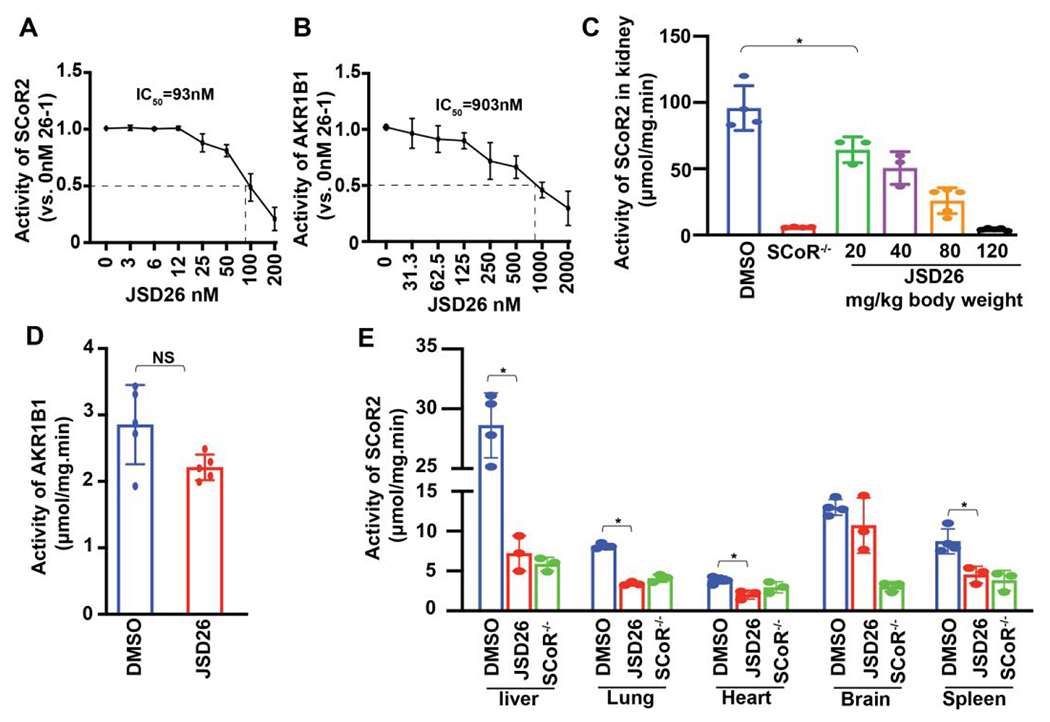
(A) IC50 value of JSD26 against the SNO-CoA-metabolizing activity of SCoR2 (n=3). (B) IC50 value of JSD26 against the methylglyoxylate-metabolizing activity of AKR1B1 (n=3). (C) SNO-CoA-metabolic activity in kidneys from JSD26-treated mice (i.p. at indicated dose, 18h) vs vehicle-treated WT and SCoR−/− mice (n≥3). (D) Methylglyoxylate-metabolizing (AKR1B1) activity in kidneys from JSD26-treated mice (i.p. 120 mg/kg, 18h) (n≥5). (E) SNO-CoA-metabolizing activity in liver, lung, heart, brain and spleen from JSD26-treated mice (i.p. 120 mg/kg, 18h) (n≥3). All results are presented as mean ± SD. Two-tailed Student’s t-test was used to detect significance. *, p<0.05; NS, no significant.
Molecular modeling showed binding of JSD26 to the active site of SCoR2 (with the NADPH cofactor bound and His113 singly protonated at ND1) with a docking score of −11.4 kcal/mol (Figure 2E). The imidazolidine-2,4-dione moiety of JSD26 in the docked binding mode is in the same position as the crystallographically-determined binding mode of fidarestat. The docking suggests that this JSD26 moiety makes hydrogen bonds with Tyr50, His113, and Trp114 of SCoR2 (Figure 2E and 2F). One of the oxygens in the imidazolidine-2,4-dione moiety is situated within 3.2 Å of NADPH. The tricyclic ring of JSD26 makes hydrophobic interactions with Trp22, Ile49, Phe125, Ile299, Trp220, Pro301, and the nonpolar face of the backbone chain of Val300. The cyclopropyl moiety of JSD26 is situated at the entrance of the active site and is predicted to interact with Ile49 (both side chain and main chain) and Trp22. The enantiomer of JSD26 (JSD26-2) did not dock well to SCoR2, yielding a docking score of −7.1 kcal/mol, and did not inhibit SNO-CoA reductase activity. This is likely due to inability to accommodate the cyclopropyl moiety sterically on the other end of the tricyclic ring, since that end of JSD26 is making van der Waals interactions with the nonpolar face of the main chain of Val300.
The presence of the cyclopropyl group in JSD26 enlarges the large planar hydrophobic portion of the inhibitor, allowing JSD26 to make hydrophobic and van der Waals interactions from both ends of the molecule, which potentially leads to JSD26 selectively binding to SCoR2/AKR1A1. First, the cyclopropyl group in JSD26 is predicted to interact with I49 and F125 in SCoR2 (Fig. 2F and 2G), but residue I49 is changed to alanine in AKR1B1, leading to a smaller hydrophobic residue, and F125 in AKR1B1 adopts more variable conformations. Second, at the other end of the planar hydrophobic tricyclic ring of JSD26, the phenyl ring is predicted to make tightly packed hydrophobic/van der Waals interactions with SCoR2 I299, W220, and the non-polar face of the main chain atoms of P301, including the main chain nitrogen that is part of that proline ring (Fig.2F and 2G). In AKR1B1, P301 is changed to an isoleucine that adopts a different conformation with its main chain nitrogen pointing towards the binding pocket. This residue difference and conformational main chain difference makes this end of the active site more polar in AKR1B1 than in SCoR2. This more polar region in AKR1B1 would, therefore, likely be less prone to interact with the larger, hydrophobic JSD26.
To investigate whether JSD26 can be effectively absorbed into the blood following intraperitoneal injection and distributed throughout the body, JSD26 was injected i.p. into mouse abdomen at single doses (20, 40, 80 or 120 mg/kg body weight), and renal SNO-CoA-metabolic activity was measured after 18h. SNO-CoA-metabolic activity in kidney was reduced by 80% in JSD26-injected mice at the highest dose (120 mg/kg body weight) compared with DMSO control, almost equal to the activity reduction in SCoR2-knockout mice (SCoR2−/−). Notably, however, injection of JSD26 did not inhibit the activity of kidney AKR1B1 (Figure 3C and 3D). JSD26 treatment also inhibited the activity of SCoR2 in liver, lung, heart and spleen, but not in brain (Figure 3E). We measured JSD26 in serum at 24 hours post-injection using HPLC/mass spectrometry. As shown in Figure S4, JSD26 is readily detectable in serum 24 hours after i.p. injection, verifying bioabsorption and indicating sustained bioavailability. Thus, JSD26 is an absorbable and selective inhibitor of SCoR2, but appears unable to cross the blood-brain barrier.
Characterization of inhibitory mechanism
To determine the inhibitory mode of action of JSD26 on SCoR2 activity, we compared steady-state enzyme kinetic parameters of SCoR2 in the presence of varying concentrations (0 nM, 100 nM or 400 nM) of JSD26. JSD26 robustly reduced both Vmax and Km (Figure 4A and 4B), indicating that JSD26 is an uncompetitive inhibitor with substrate, and suggesting that JSD26 does not compete for substrate binding, but instead binds only to the complex formed between SCoR2 and its substrates (NADPH or SNO-CoA).
Figure 4. JSD26 is an uncompetitive inhibitor.
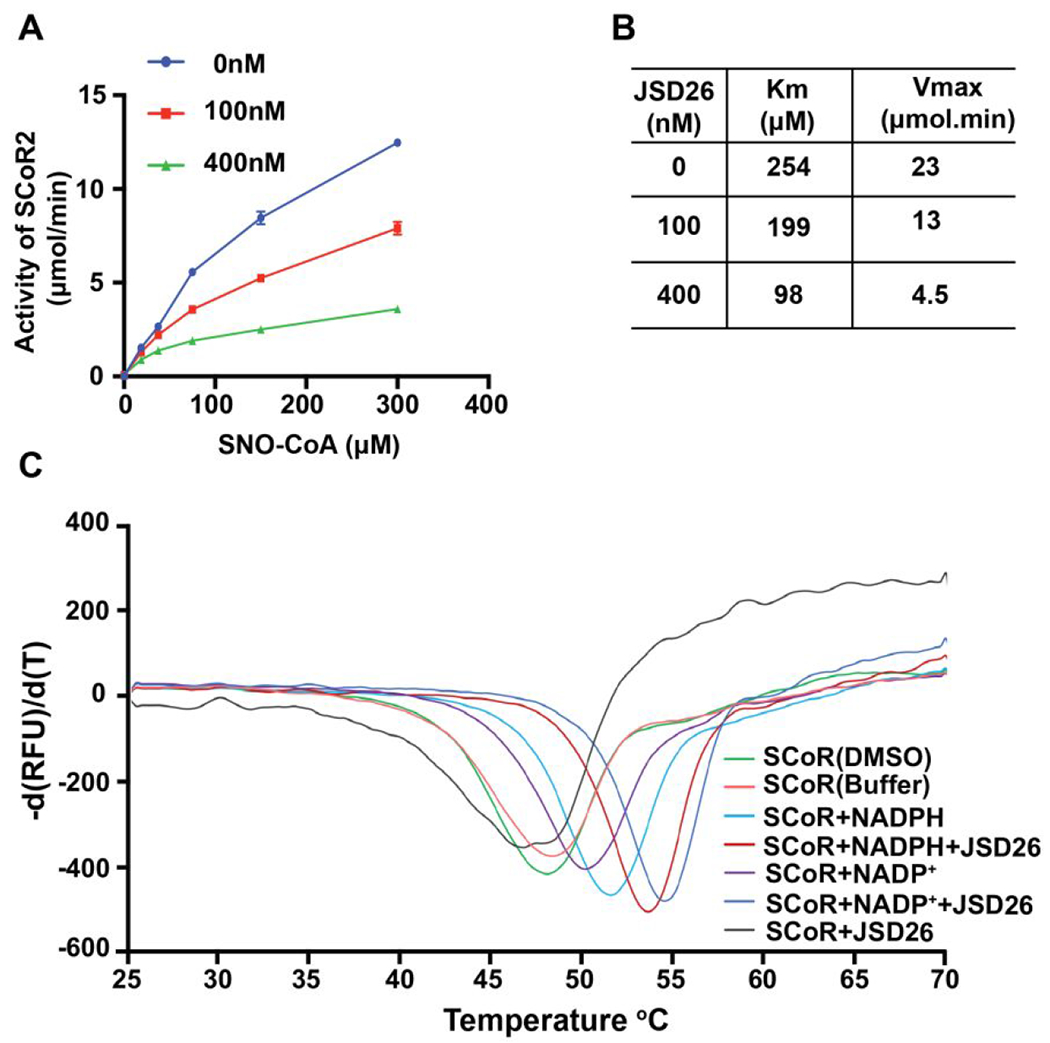
(A) SNO-CoA-metabolizing activity of purified SCoR2 was assayed in the presence of the indicated concentrations of JSD26 and SNO-CoA (n=3). (B) The enzymatic data in (A) were fitted to Michaelis-Menten kinetics. Vmax and Km were calculated for different JSD26 concentrations (n=3). (C) Differential scanning fluorimetry (DSF)/thermal shift assay probing protein stabilization of purified SCoR2 by JSD26 and NADPH (substrate). The rate of change in SYPRO orange fluorescence is plotted versus temperature for SCoR2 in the absence and presence of combinations of NADPH, NADP+, and JSD26. Experiments were done in duplicate.
To study the effects of JSD26 and NAPDH (co-factor) on SCoR2 thermal stability, we perform differential scanning fluorimetry (DSF)/thermal shift assays. SCoR2 alone in either buffer or buffer with 1% DMSO yielded melting temperatures (Tm) of 48.3°C and 48.0°C, respectively (Figure 4C). The presence of NADPH alone significantly increased the Tm of SCoR2 to 51.4°C, while the presence of JSD26 alone reduced the Tm of SCoR2 to 46.6°C, indicating that JSD26 destabilized SCoR2. Interestingly, JSD26 plus NADPH increased the Tm to 53.4°C, while JSD26 plus NADP+ increased the Tm even further, to 54.4°C (Figure 4C), suggesting that JSD26 binds and stabilizes the complexes of SCoR2•NADPH and SCoR2•NADP+. These DSF studies show that JSD26 stabilizes SCoR2 more strongly when complexed to NADP+ than when complexed to NADPH, which could indicate a higher affinity for the NADP+-containing complex. If JSD26 preferentially binds the product-bound SCoR2•NADP+ complex compared to the SCoR2•NADPH complex, then inhibitor binding is likely to be uncompetitive, as was found for aldose reductase.25 Overall, these DSF results support our previous conclusion that JSD26 is an uncompetitive inhibitor.
Of note, docking of JSD26 suggests that His113 is singly protonated at Nδ2 allowing the Nε2 atom to be a hydrogen bond acceptor for the hydantoin ring nitrogen of JSD26. Residue His113 in AKR enzymes adopts different protonation states during catalysis;26 thus the NADP+ oxidation state may promote the nearby singly protonated His113 state over the doubly protonated His113 state thus allowing His113 to form a hydrogen bond with JSD26 (Figure 2F).
JSD26 treatment protects against AKI
We investigated if JSD26 treatment can protect against acute kidney injury in mice. JSD26 was intraperitoneally injected into mice at 120 mg/kg body weight. AKI surgery was performed 18 hours after administration of JSD26 or DMSO, and blood and kidneys harvested 24 hours post-surgery. First, to confirm whether SNO-CoA metabolizing activity was inhibited by JSD26 after surgery, we measured the activity and expression level of SCoR2. At 24 hours post-surgery (42 hours after drug injection), the SNO-CoA-metabolizing activity in kidney from JSD26-treated mice was reduced by 80% compared with DMSO-treated control mice (Figure 5A). The expression level of SCoR2 in kidneys from JSD26-treated mice was the same as the DMSO-treated mice (Figure 5B and 5C). Thus, JSD26 inhibited the activity of SCoR2 through at least 1 day after I/R surgery. Next, we measured serum creatinine and BUN level. Both creatinine and BUN level were significantly lower in JSD26-treated mice than DMSO-treated mice, indicating that administration of JSD26 protected against AKI (Figure 5D and 5E). To histologically confirm JSD26-mediated renal protection, we analyzed tubular injury in the kidneys using H&E staining. The anatomy of cortex and medulla of healthy kidneys was unaffected by JSD26 under basal conditions (Figure 5F). However, after acute kidney injury, tubular damage, including severe tubular lysis, loss of brush borders, and sloughed debris in the tubular lumen, was mitigated in JSD26-treated mice compared with DMSO-treated mice (Figure 5G and 5H). Thus, treatment of mice with the selective SCoR2 inhibitor JSD26 protects against AKI, recapitulating SCoR2 deletion.
Figure 5. JSD26 treatment protects against acute kidney injury in mice.
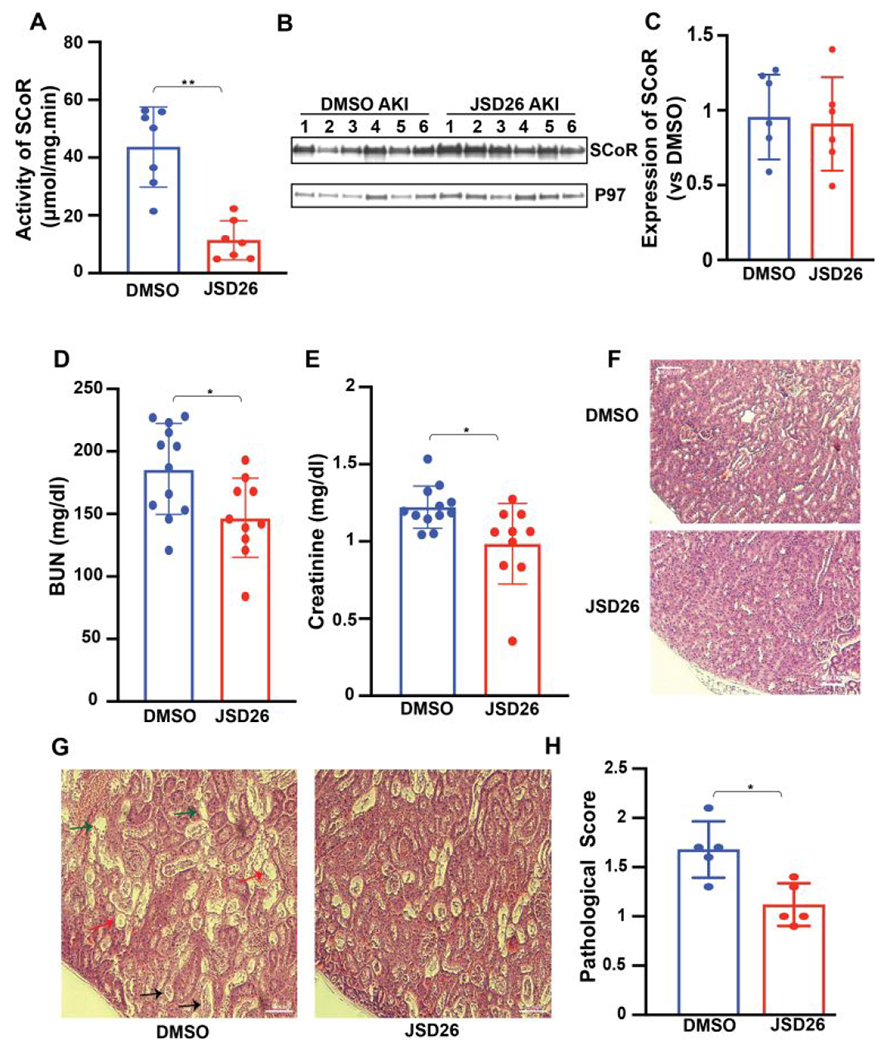
(A) SNO-CoA-metabolizing activity in kidneys from JSD26-treated mice (120 mg/kg i.p. 18h before surgery) assayed 24 hours post AKI (n=7). (B-C) Expression of SCoR2 in kidneys from JSD26-treated mice 24 hours post AKI (B) with quantification (C) (n=6). (D-E) Serum creatinine and BUN in JSD26-treated mice 24h after I/R-induced AKI (n≥10). (F) H&E stain in sham-surgery, and injury surgery vehicle- vs JSD26-treated kidneys. (G) H&E stain in injured kidneys, treated with vehicle or JSD26. AKI induced renal tubular injury includes severe tubular lysis (black arrow), loss of brush borders (green arrow) and sloughed debris in the tubular lumen (red arrow). (H) Pathological scores of tubular injury in vehicle-treated or JSD26-treated mice (n=5 mice per group). All results are presented as mean ± SD. Two-tailed Student’s t-test was used to detect significance. *, p<0.05; **, p<0.01.
JSD26 treatment increases the S-nitrosylation level of PKM2
Next, we investigated the mechanism of renal protection mediated by JSD26 treatment. PKM2 is a major locus of regulation by the SNO-CoA/SCoR2 system in the kidney and responsible for protection against AKI.9, 13 S-nitrosylation of PKM2 in SCoR2−/− mice inhibits the activity of PKM2 and reprograms metabolic pathways in injured kidneys by shunting glycolytic intermediates into the pentose phosphate pathway.9, 13 To verify that PKM2 S-nitrosylation is elevated in renal protection mediated by JSD26, we measured SNO-PKM2 levels in kidneys from JSD26-treated and DMSO-treated mice. We observed higher levels of SNO-PKM2 following sham operation or AKI surgery in JSD26-treated mice than in DMSO-treated mice (Figure 6A and 6B). Further, PKM2 activity was lower in JSD26-treated mice vs. DMSO-treated mice following AKI surgery (Figure 6C). These results indicate that the mechanism of renal protection in SCoR2 knockout mice is also evident in JSD26 treated mice.
Figure 6. JSD26 treatment increases S-nitrosylation of PKM2 and inhibits PKM2 activity.
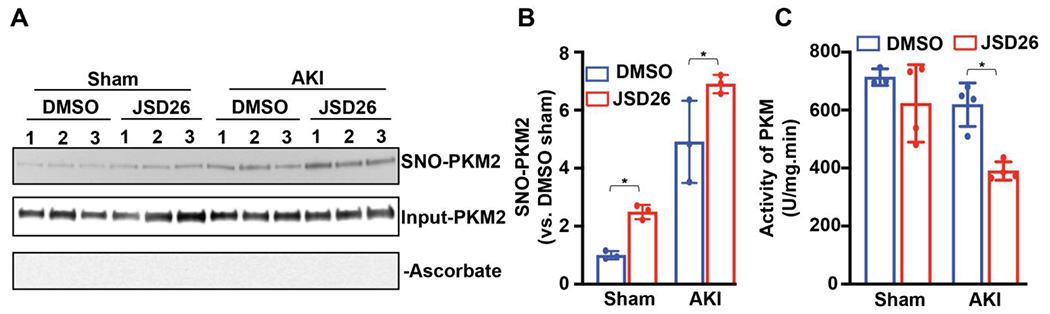
(A) S-nitrosylation of PKM2 in sham-surgery or I/R-injured kidneys from DMSO-treated or JSD26-treated mice. Three mice per group are all shown. Samples processed without ascorbate (−Ascorbate) is a negative control for SNO. Input is used as loading control. (B) Quantification of SNO-PKM2. SNO-PKM2 is normalized to input PKM2 (n=3 per group). (C) Activity of endogenous pyruvate kinase (PK) in sham-surgery or I/R-injured kidneys from DMSO-treated or JSD26-treated mice; (n=4 per group) Uninjured (sham) kidney expresses PKM1 that is insensitive to S-nitrosylation, while AKI kidney expresses mainly PKM2.9 All results are presented as mean ± SD. One-way ANOVA with Tukey post hoc was used to detect significance. *, p<0.05.
Discussion and Conclusions
Imirestat administration can effectively inhibit SCoR2 activity in vivo; however, it fails to protect against AKI. Imirestat is a nonselective drug that inhibits both AKR1A1 and AKR1B1. AKR1B1 serves as an antioxidant defense to metabolize toxic aldehydes,27, 28 which are known to be generated as a consequence of lipid peroxidation following AKI and to cause cell damage through modification of proteins and nucleic acids.29 AKR1B1 is in fact a mediator of ischemic preconditioning by diminishing lipid peroxidation,30 and its abundance in collecting tubule cells and induction by hyperosmolality are consistent with a protective role.31 AKR1B1 also has important physiological roles in glucose metabolism, inflammation and prostaglandin synthesis.32 Mice lacking AKR1B1 exhibit hypercalciuria, hypercalcemia, hypermagnesemia, and reduced ability to concentrate urine and keep cation homeostasis.33 Considering the role of AKR1B1 in antioxidant defense, osmoprotection and cation homeostasis, it is not surprising that the inhibition of AKR1B1 by imirestat aggravates kidney injury and compromises renal protection mediated by inhibition of SCoR2.
We took advantage of imirestat’s structure using a docking calculations-guided approach to assist in the identification of a potent and selective SCoR2 inhibitor.34, 35 The compound JSD26, the C2-H and C7-cyclopropyl-substituted analog of imirestat, is a selective inhibitor of SCoR2 (IC50=93 nM for SCoR2/AKR1A1 vs IC50=903 nM for AKR1B1). Although selectivity of JSD26 for SCoR2 vs AKR1B1 is relatively modest, JSD26 is a nanomolar inhibitor of SCoR2, and an effective dose of JSD26 to completely inhibit SCoR2 in vivo does not inhibit AKR1B1 appreciably. Thus, JSD26 provides a new molecular tool to investigate the functions of SCoR2 with very little inhibition of AKR1B1 and provides a potential starting point for designing more selective SCoR2 inhibitors.
Our data provide further validation that S-nitrosylation of PKM2 by SNO-CoA plays a critical role in protection against AKI. After kidney injury, PKM2 is expressed in the proximal tubule, which enables protection through metabolic reprogramming.9 PKM2 expression involves alternative splicing to include a novel S-nitrosylation site and enables the binding of SCoR2.9 SCoR2 activity is inhibited endogenously in injured tissue to increase S-nitrosylation of PKM2. A single intraperitoneal injection of JSD26 increases PKM2 S-nitrosylation and further protects against AKI. Since PKM2 expression is a characteristic feature of injured tissues,9 the SCoR2-specific inhibitor JSD26 may be a useful tool to explore the broader therapeutic potential of SCoR2 inhibition.
In conclusion, the SCoR2/SNO-CoA system plays an important role in kidney injury. Thus inhibition of SCoR2 provides a novel therapeutic opportunity to protect tissues from injury. Current SCoR2 inhibitors, however, have off target effects that mitigate their effectiveness, and none are available clinically. We have identified a novel compound JSD26, the C2-H and C7-cyclopropyl-substituted imirestat analog, and show it to be a fairly selective and potent inhibitor of SCoR2 (IC50=93 nM for SCoR2 vs IC50=903 nM for AKR1B1). By engineering AKR1A1 selectivity, benefit is conferred: JSD26 is absorbed into the blood following i.p. injection, and well distributed throughout the body (but does not appear to cross the blood-brain barrier). Administration of JSD26 into mice by i.p. injection induces the S-nitrosylation and inhibition of PKM2 to protect against AKI. Thus, a newly developed SCoR2 inhibitor JSD26 provides a novel therapeutic approach to AKI.
EXPERIMENTAL SECTION
Design and synthesis of compounds
Analogs of imirestat were designed by varying substituents at four positions (C2, C3, C4 and C7) on its tricyclic ring (Figure 2B, SMILES format molecular formula strings for all compounds are shown in Table S3). Designed compounds were tested for their ability to bind to SCoR2 using docking calculations based on the crystal structure of Sus scrofa AKR1A1 (PDBid 3H4G).21 To probe the selectivity of the designed compounds, we additionally docked the compounds against two AKR1B variants: human ARK1B10 (PDBid 4GAB, complexed with fidarestat) and human AKR1B1 (PDBid 4JIR, complexed with epalrestat).23, 24 AKR1B1 and AKR1B10 share 71% sequence identity, and the amino acid differences do not involve residues interacting with the bound ligands. Docking calculations were carried out using Schrodinger’s GLIDE software as done previously using the Extra Precision XP algorithm.35, 36, 37 Designed imirestat analog compounds were synthesized by TCG Lifesciences (Kolkata, India) and are >95% pure by HPLC analysis.
Purification of recombinant SCoR2 and AKR1B1
For purification of recombinant SCoR2 and AKR1B1, cDNAs encoding human SCoR2 or AKR1B1 were cloned into pET21b (Novagen) to introduce a C-terminal 6xHis tag on the expressed protein. The recombinant SCoR2 and AKR1B1 proteins were purified from BL21-CodonPlus Competent E. coli Cells (Agilent). Overnight E. coli cultures were sub-cultured into 1L of LB medium at 5%. At OD600 of 0.5, cultures were induced with 100 mM IPTG and grown for a further 4 hours at 28°C. Cultures were centrifuged at 4000xg for 10 min to harvest the cells. Cell pellets from 1L cultures were lysed in 10 mL of 1x PBS buffer containing 1 mM PMSF and protease-inhibitor cocktail (Roche) by sonication. After centrifugation at 14500xg for 20 min, the supernatant was collected. The lysate was diluted by addition of 30 ml 1x PBS buffer containing 1 mM PMSF and protease-inhibitor cocktail and incubated with 1 mL of Ni-NTA agarose (Qiagen) at 4°C for 1 hour with rotation. The slurry was then poured into empty PD-10 columns (GE Healthcare). The beads were washed in the column with 100 mL of 50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole (pH 8). Elution was done in 2 mL of 50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole (pH 8). Buffer was exchanged with modified Roeder D [20 mM HEPES (pH 7.9), 20% (v/v) glycerol, 0.1 M KCL, 0.2 mM EDTA] using a Microcon centrifugal filter device (Millipore).
Screening of imirestat analogs
The compounds were dissolved in DMSO to 20 μM, and then diluted serially to generate stock solutions at 10 μM, 5 μM, 2.50 μM, 1.25 μM, 0.625 μM, 0.3125 μM, and 0.0155 μM. For initial screening, cuvettes contained 1 ml of reaction mixture with 20 nM recombinant SCoR2, 3 mM DL-glyceraldehyde as substrate and varying concentrations of inhibitors in 50 mM phosphate buffer (pH 7.0). Reactions were initiated by the addition of NADPH (Cayman) to 100 μM and allowed to proceed for 2 min. The reaction slope was calculated through monitoring the change in NADPH concentration over time using a diode-array spectrophotometer (Cary 4000), and half-maximal inhibitory concentrations (IC50) determined using GraphPad Prism 7. For comparison of selectivity of the compounds, reactions were set up in 96 well plates, with 200 μl of reaction mixture per well, containing 200 nM purifed SCoR2 (or AKR1B1), 100 μM SNO-CoA (or methylglyoxylate for AKR1B1) and varying concentrations of inhibitors in 50 mM phosphate buffer (pH 7.0). Reactions were initiated by the addition of NADPH to 100 μM and allowed to proceed for 2 min. The slope was calculated through monitoring the change in NADPH concentration using a plate reader (Biotech) at 340 nm, and half-maximal inhibitory concentration (IC50) determined by normalizing activity against 20 nM of purified protein.
Kinetic analysis of NADPH-dependent SNO-CoA reductase activity
NADPH-dependent SNO-CoA reductase activity was determined spectrophotometrically as described previously.12 To calculate kinetic parameters, triplicate reactions were performed with 20 nM recombinant SCoR2 (or AKR1B1) and 100 μM NADPH (Sigma) in 50 mM phosphate buffer (pH 7.0) containing 100 μM EDTA and 100 μM DTPA. The reaction was started by addition the substrate 3 mM DL-glyceraldehyde (generic AKR substrate), 100 μM SNO-CoA (SCoR2 substrate) or 100 μM methylglyoxylate (AKR1B1 substrate). The SNO-CoA was prepared by reacting equal volumes of 0.1 μM CoA (Sigma) in 1 M HCl and 0.1 M NaNO2 (Fluka Chemicals) in MilliQ water containing 100 μM EDTA and 100 μM DTPA. SNO-CoA was immediately diluted to 4 mM and used in assays; DL-glyceraldehyde and methylglyoxylate were from Sigma-Aldrich. Initial rates were calculated using absorbance decrease at 340 nm using an extinction coefficient of 7.06 mM−1cm−1 (from both SNO-CoA and NADPH). Inhibition assays were performed in triplicate as above but with a fixed concentration of substrates and varying concentrations of inhibitors dissolved in DMSO. IC50 was determined using GraphPad Prism 7.
To calculate NADPH-dependent SNO-CoA reductase activity in mouse tissues, organs harvested from C57BL/6J mice were mechanically homogenized in lysis buffer (50 mM phosphate buffer, pH 7.0, 150 mM NaCl, 100 μM EDTA and 100 μM DTPA). Extracts were clarified by centrifugation (20,000xg, 4°C, 20 min, ×2), and protein concentration was determined by bicinchoninic acid assay (Pierce). The assays were performed in 50 mM phosphate buffer (pH 7.0), 100 μM EDTA, 100 μM DTPA plus 200 μM SNO-CoA and 100 μM NADPH. Reactions were initiated by the addition of lysate and allowed to proceed for 2 min. All assays were performed in triplicate.
Differential scanning fluorimetry (DSF)/thermal shift assay
His-tagged human SCoR2 protein was expressed and purified using metal affinity chromatography as described. The crude protein was further purified using size-exclusion chromatography (buffer A: 10 mM HEPES, pH 8.0, 50 mM NaCl) prior to DSF experiments. The DSF reaction volume was 30 μl containing 5 μM SCoR2 ± 250 μM NADPH or NADP+ (from 25 mM stock solutions in milliQ water), and ± 250 μM JSD26 (from a 25 mM stock solution in DMSO) in buffer A. Samples were incubated for 45 minutes at room temperature. For samples containing both NADPH/NADP+ and JSD26, NADPH/NADP+ was first added and allowed to incubate for 15 min before JSD26 was added, followed by an additional 30 min incubation. After the 45 min incubation, SYPRO Orange (ThermoFisher) fluorescent dye was added to a final concentration of 10X. Control experiments included protein only, with and without 1% DMSO, to match the concentration of DMSO in the reactions containing JSD26. The temperature was ramped from 25.0–70.0°C with 0.3°C increments and the fluorescence measured using a CFX96 Touch Real-Time PCR Detection System (Bio-Rad).
Molecular modeling
Static protein/flexible ligand modeling of the interaction of JSD26 and other imirestat analogs with SCoR2 was performed using Maestro 9.9 software. The porcine SCoR2 crystal structure (PDB code 3H4G) was prepared for docking by removing H2O and fidarestat from the PDB file. In Maestro, original hydrogens were removed and replaced, bond orders were assigned, and the structure was minimized. A grid was prepared around the active site centered at X 2.0999, Y 24.4072, Z 6.5084. All ligands were prepared for docking in Maestro using the ligand preparation function. Ligands were docked to the active-site grid using XP Glide Docking with post-docking minimization.
NMR spectra
The compound (50mg) was dissolved in 600 uL of DMSO for 13C-NMR. A small aliquot (10mg) was then diluted in another 600 uL of DMSO for 1H-NMR. The spectra were taken on a Bruker AVII 500 MHz NMR instrument. The pulse program was ZG30 on a QNP 5mm 1H/19F/31P/13C with Z gradient probehead. The experiments were performed by the NuMega Resonance Labs (San Diego). Chemical shifts in Parts Per Million (PPM) in 1H-NMR and 13C-NMR spectra of JSD26 were predicted using ChemOffice software.
Mice and AKI injury model
Mouse studies were approved by the Case Western Reserve University Institutional Care and Use Committee (IACUC). Housing and procedures complied with the Guide for the Care and Use of Laboratory Animals and the American Veterinary Medical Association guidelines on euthanasia. AKI surgery was carried out as described previously.9 Mice of similar age (9-11 weeks) and body weight (male, 25-28g) were used for surgery; note that female mice have a very distinct injury profile in this model.9 The mice were anesthetized with isoflurane (1-3%) in oxygen, and anesthesia maintained by adjusting isoflurane (0.75-2.0%) as needed. The fur in the surgical area was removed with clippers and the skin sterilized with 3 alternating washes of betadine (or chlorhexidine) and alcohol. The mouse was placed on a thermostatic station during surgery. The skin and muscle were cut open along the back to expose both right and left kidneys. Gentle blunt dissection revealed the kidney, and a Q-tip was used to mobilize and exteriorize the kidney. A 5-0 silk suture was used to clamp the pedicle to block the blood flow to the kidney to induce renal ischemia for 23 min, then the suture released to allow reperfusion. Identical steps were performed on each kidney. A silk suture was used to close the muscle layer of the incision followed by the closure of the skin wound with vicryl. Immediately after the wound closure, 10-20 ml/kg sterile saline was given intraperitoneally to each mouse. The animal was then kept on a heating pad until it regained full consciousness before being returned to home cage. Mice subjected to surgery without clamping the pedicle were used as sham controls. Mice were euthanized 24 hours after reperfusion, and serum and kidneys collected. Serum creatinine and BUN were measured at University Hospital’s Clinical Laboratories.
SNO-RAC for PKM2 S-nitrosylation
S-nitrosothiol Resin-Assisted Capture (SNO-RAC) to identify S-nitrosylated proteins was carried out as described previously.38 Mouse kidneys were mechanically homogenized in lysis buffer containing 100 mM Hepes, 1 mM EDTA, 100 μM neocuproine (HEN buffer), 50 mM NaCl, 0.1% (vol/vol) Nonidet P-40, 0.2% S-methylmethanethiosulfonate (MMTS) as a thiol-blocking agent, 1 mM PMSF and 1x protease inhibitors (Roche) at 1 mg tissue per 5 μl lysis buffer. After centrifugation (20,000xg, 4°C, 20 min, ×2), SDS and MMTS were added to the supernatants to 2.5% and 0.2% respectively, and the lysate incubated at 50°C for 20 min. Proteins were precipitated with −20°C acetone, and re-dissolved in 1 mL of HEN/1% SDS. Precipitation of proteins was repeated with −20°C acetone, and the final pellets were resuspended in HEN/1% SDS and protein concentrations determined using the Bicinchoninic Acid (BCA) method. Total lysates (2 mg) were incubated with freshly prepared 50 mM ascorbate and 50 μl thiopropyl-Sepharose (50% slurry), and rotated end-over-end in the dark for 4 h. Samples lacking ascorbate were incubated in parallel as controls. The resin-bound SNO-proteins were sequentially washed with HEN/1% SDS and 10% HEN/0.1% SDS; SNO proteins were then eluted with 10% HEN/1% SDS/10% β-meracaptoethanol and analyzed by SDS/PAGE and immunoblotting.
Histological analysis
Kidney samples were fixed with 4% PFA over 24 h, dehydrated and embedded into paraffin blocks. Formalin-fixed, paraffin-embedded blocks were sectioned and stained with H&E by the Tissue Resources core facility at Case Western Reserve University. More than five microscopic fields obtained from each animal were selected for quantitative analysis. Renal histopathologic alterations were evaluated and graded on a 0 to 2 scale as described previously.
Pyruvate kinase (PK) activity
Kidney PKM activity was measured based on generation of pyruvate, which was oxidized by pyruvate oxidase to produce color (λ = 570nm) following the protocol of the Pyruvate Kinase Activity Colorimetric/Fluorometric Assay Kit (Biovision). The kidneys were mechanically homogenized in lysis buffer [50mM phosphate buffer, pH 7.0, 150 mM NaCl, 0.1 mM EDTA, 0.1 mM DTPA, 1 mM PMSF, and protease inhibitor mixture (Roche)]. Extracts were clarified by centrifugation (20,000xg, 4°C, 20 min, ×2), and protein concentration was determined by bicinchoninic acid assay. 10 μl (0.1 μg/μl) lysate was used to measure PKM2 activity.
Western blot analysis
Proteins were extracted from cells or tissues or derived from the SNO-RAC assay and subjected to 4–20% Criterion Precast Midi Protein Gel electrophoresis. Blotted membranes were incubated overnight at 4°C with primary antibodies, and washed with PBS containing 0.1% Tween-20 before incubation with HRP-conjugated secondary antibody (anti-mouse or anti-rabbit IgG (Promega)) for 1 h followed by chemiluminescent detection (ECL (GE Healthcare)). Antibodies employed in western blotting included: rabbit polyclonal Anti-AKR1A1 (15054-1-AP, Proteintech Group), rabbit monoclonal Anti-PKM2 (D78A4, Cell Signaling), and mouse monoclonal p97 (10R-P104A, Fitzgerald). Quantification of western blots was carried out with ImageJ (NIH).
Supplementary Material
Supporting Figure S1, Synthesis of JSD26 (.pdf)
Supporting Figure S2, 1H-NMR of JSD26 (.pdf)
Supporting Figure S3, 13C-NMR of JSD26 (.pdf)
Supporting Figure S4, Purity of JSD26 by mass spectroscopy and HPLC, and detection of JSD26 in mouse serum after i.p. dosing (.pdf)
Supporting Table S1, IC50 of initial compounds to inhibit SCoR2 in vitro (.pdf)
Supporting Table S2, IC50 of secondary compounds to inhibit SCoR2 in vitro (.pdf)
Supporting Table S3. SMILES molecular formula strings for all compounds (.csv)
Supporting Data 1. PDB coordinates for JSD26 docked to SCoR2 (.pdb)
ACKNOWLEDGEMENTS
This work was supported by NIH grant R01 DK119506, R01 HL157151, P01 HL158507 and DK128347 to JSS, as well as R01 DK119506 to HLZ.
Abbreviations
- AKI
acute kidney injury
- BUN
blood urea nitrogen
- I/R
ischemia-reperfusion injury
- PKM
pyruvate kinase muscle isoform
- PKM1
PKM splice variant 1 that is not regulated by S-nitrosylation
- PKM2
PKM splice variant 2 that is inhibited by S-nitrosylation
- SCoR2
S-nitroso-coenzyme A reductase type 2 (encoded by the AKR1A1 gene)
- SNO-CoA
S-nitroso-coenzyme A
- SNO-RAC
S-nitrosothiol Resin Assisted Capture
Footnotes
Supporting Information
Supporting Information for this article includes:
Conflict of Interest
Jonathan Stamler is a founder of and has equity interest in SNO bio, a company developing nitrosylation related therapeutics, and NNOXX, a company developing NO-based device technology. CWRU and UHCMC are aware of these conflicts and appropriate management plans are in place. None of the other authors have relevant conflicts to disclose.
REFERENCES
- 1.Rewa O; Bagshaw SM, Acute kidney injury-epidemiology, outcomes and economics. Nat Rev Nephrol 2014, 10 (4), 193–207. [DOI] [PubMed] [Google Scholar]
- 2.Lameire NH; Bagga A; Cruz D; De Maeseneer J; Endre Z; Kellum JA; Liu KD; Mehta RL; Pannu N; Van Biesen W; Vanholder R, Acute kidney injury: an increasing global concern. Lancet 2013, 382 (9887), 170–9. [DOI] [PubMed] [Google Scholar]
- 3.Zuk A; Bonventre JV, Acute Kidney Injury. Annu Rev Med 2016, 67, 293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kanagasundaram NS, Pathophysiology of ischaemic acute kidney injury. Ann Clin Biochem 2015, 52 (Pt 2), 193–205. [DOI] [PubMed] [Google Scholar]
- 5.Bonventre JV; Yang L, Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest 2011, 121 (11), 4210–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eltzschig HK; Eckle T, Ischemia and reperfusion--from mechanism to translation. Nat Med 2011, 17 (11), 1391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Y; Song M; Liu Y; Liu H; Sun L; Peng Y; Liu F; Venkatachalam MA; Dong Z, Renoprotective approaches and strategies in acute kidney injury. Pharmacol Ther 2016, 163, 58–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jo SK; Rosner MH; Okusa MD, Pharmacologic treatment of acute kidney injury: why drugs haven’t worked and what is on the horizon. Clin J Am Soc Nephrol 2007, 2 (2), 356–65. [DOI] [PubMed] [Google Scholar]
- 9.Zhou HL; Zhang R; Anand P; Stomberski CT; Qian Z; Hausladen A; Wang L; Rhee EP; Parikh SM; Karumanchi SA; Stamler JS, Metabolic reprogramming by the S-nitroso-CoA reductase system protects against kidney injury. Nature 2019, 565 (7737), 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stomberski CT; Zhou HL; Wang L; van den Akker F; Stamler JS, Molecular recognition of S-nitrosothiol substrate by its cognate protein denitrosylase. J Biol Chem 2019, 294 (5), 1568–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li X; Schmohl F; Qi H; Bennewitz K; Tabler CT; Poschet G; Hell R; Volk N; Poth T; Hausser I; Morgenstern J; Fleming T; Nawroth PP; Kroll J, Regulation of Gluconeogenesis by Aldo-keto-reductase 1a1b in Zebrafish. iScience 2020, 23 (12), 101763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anand P; Hausladen A; Wang YJ; Zhang GF; Stomberski C; Brunengraber H; Hess DT; Stamler JS, Identification of S-nitroso-CoA reductases that regulate protein S-nitrosylation. Proc Natl Acad Sci U S A 2014, 111 (52), 18572–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siragusa M; Thole J; Bibli SI; Luck B; Loot AE; de Silva K; Wittig I; Heidler J; Stingl H; Randriamboavonjy V; Kohlstedt K; Brune B; Weigert A; Fisslthaler B; Fleming I, Nitric oxide maintains endothelial redox homeostasis through PKM2 inhibition. EMBO J 2019, 38 (17), e100938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barski OA; Gabbay KH; Bohren KM, The C-terminal loop of aldehyde reductase determines the substrate and inhibitor specificity. Biochemistry 1996, 35 (45), 14276–80. [DOI] [PubMed] [Google Scholar]
- 15.El-Kabbani O; Wilson DK; Petrash M; Quiocho FA, Structural features of the aldose reductase and aldehyde reductase inhibitor-binding sites. Mol Vis 1998, 4, 19. [PubMed] [Google Scholar]
- 16.Wilson DK; Bohren KM; Gabbay KH; Quiocho FA, An unlikely sugar substrate site in the 1.65 A structure of the human aldose reductase holoenzyme implicated in diabetic complications. Science 1992, 257 (5066), 81–4. [DOI] [PubMed] [Google Scholar]
- 17.Ruiz FX; Moro A; Gallego O; Ardevol A; Rovira C; Petrash JM; Pares X; Farres J, Human and rodent aldo-keto reductases from the AKR1B subfamily and their specificity with retinaldehyde. Chem Biol Interact 2011, 191 (1-3), 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barski OA; Tipparaju SM; Bhatnagar A, The aldo-keto reductase superfamily and its role in drug metabolism and detoxification. Drug Metab Rev 2008, 40 (4), 553–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Griffin BW; McNatt LG; Chandler ML; York BM, Effects of two new aldose reductase inhibitors, AL-1567 and AL-1576, in diabetic rats. Metabolism 1987, 36 (5), 486–90. [DOI] [PubMed] [Google Scholar]
- 20.Kalnik M; Gabko P; Bella M; Koos M, The Bucherer-Bergs Multicomponent Synthesis of Hydantoins-Excellence in Simplicity. Molecules 2021, 26 (13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.El-Kabbani O; Carbone V; Darmanin C; Oka M; Mitschler A; Podjarny A; Schulze-Briese C; Chung RP, Structure of aldehyde reductase holoenzyme in complex with the potent aldose reductase inhibitor fidarestat: implications for inhibitor binding and selectivity. J Med Chem 2005, 48 (17), 5536–42. [DOI] [PubMed] [Google Scholar]
- 22.El-Kabbani O; Green NC; Lin G; Carson M; Narayana SV; Moore KM; Flynn TG; DeLucas LJ, Structures of human and porcine aldehyde reductase: an enzyme implicated in diabetic complications. Acta Crystallogr D Biol Crystallogr 1994, 50 (Pt 6), 859–68. [DOI] [PubMed] [Google Scholar]
- 23.Zhang L; Zhang H; Zhao Y; Li Z; Chen S; Zhai J; Chen Y; Xie W; Wang Z; Li Q; Zheng X; Hu X, Inhibitor selectivity between aldo-keto reductase superfamily members AKR1B10 and AKR1B1: role of Trp112 (Trp111). FEBS Lett 2013, 587 (22), 3681–6. [DOI] [PubMed] [Google Scholar]
- 24.Ruiz FX; Cousido-Siah A; Mitschler A; Farres J; Pares X; Podjarny A, X-ray structure of the V301L aldo-keto reductase 1B10 complexed with NADP(+) and the potent aldose reductase inhibitor fidarestat: implications for inhibitor binding and selectivity. Chem Biol Interact 2013, 202 (1-3), 178–85. [DOI] [PubMed] [Google Scholar]
- 25.Nakano T; Petrash JM, Kinetic and spectroscopic evidence for active site inhibition of human aldose reductase. Biochemistry 1996, 35 (34), 11196–202. [DOI] [PubMed] [Google Scholar]
- 26.Penning TM, The aldo-keto reductases (AKRs): Overview. Chem Biol Interact 2015, 234, 236–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rittner HL; Hafner V; Klimiuk PA; Szweda LI; Goronzy JJ; Weyand CM, Aldose reductase functions as a detoxification system for lipid peroxidation products in vasculitis. J Clin Invest 1999, 103 (7), 1007–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spycher SE; Tabataba-Vakili S; O’Donnell VB; Palomba L; Azzi A, Aldose reductase induction: a novel response to oxidative stress of smooth muscle cells. FASEB J 1997, 11 (2), 181–8. [DOI] [PubMed] [Google Scholar]
- 29.Zarkovic K; Jakovcevic A; Zarkovic N, Contribution of the HNE-immunohistochemistry to modern pathological concepts of major human diseases. Free Radic Biol Med 2017, 111, 110–126. [DOI] [PubMed] [Google Scholar]
- 30.Shinmura K; Bolli R; Liu SQ; Tang XL; Kodani E; Xuan YT; Srivastava S; Bhatnagar A, Aldose reductase is an obligatory mediator of the late phase of ischemic preconditioning. Circ Res 2002, 91 (3), 240–6. [DOI] [PubMed] [Google Scholar]
- 31.Aida K; Tawata M; Ikegishi Y; Onaya T, Induction of rat aldose reductase gene transcription is mediated through the cis-element, osmotic response element (ORE): increased synthesis and/or activation by phosphorylation of ORE-binding protein is a key step. Endocrinology 1999, 140 (2), 609–17. [DOI] [PubMed] [Google Scholar]
- 32.Khayami R; Hashemi SR; Kerachian MA, Role of aldo-keto reductase family 1 member B1 (AKR1B1) in the cancer process and its therapeutic potential. J Cell Mol Med 2020, 24 (16), 8890–8902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aida K; Ikegishi Y; Chen J; Tawata M; Ito S; Maeda S; Onaya T, Disruption of aldose reductase gene (Akr1b1) causes defect in urinary concentrating ability and divalent cation homeostasis. Biochem Biophys Res Commun 2000, 277 (2), 281–6. [DOI] [PubMed] [Google Scholar]
- 34.Brazzell RK; Wooldridge CB; Hackett RB; McCue BA, Pharmacokinetics of the aldose reductase inhibitor imirestat following topical ocular administration. Pharm Res 1990, 7 (2), 192–8. [DOI] [PubMed] [Google Scholar]
- 35.Vijayaraghavan J; Kramp K; Harris ME; van den Akker F, Inhibition of soluble guanylyl cyclase by small molecules targeting the catalytic domain. FEBS Lett 2016, 590 (20), 3669–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT, Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. Journal of medicinal chemistry 2006, 49 (21), 6177–6196. [DOI] [PubMed] [Google Scholar]
- 37.Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS, Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J.Med.Chem 2004, 47 (7), 1739–1749. [DOI] [PubMed] [Google Scholar]
- 38.Forrester MT; Thompson JW; Foster MW; Nogueira L; Moseley MA; Stamler JS, Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat Biotechnol 2009, 27 (6), 557–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Figure S1, Synthesis of JSD26 (.pdf)
Supporting Figure S2, 1H-NMR of JSD26 (.pdf)
Supporting Figure S3, 13C-NMR of JSD26 (.pdf)
Supporting Figure S4, Purity of JSD26 by mass spectroscopy and HPLC, and detection of JSD26 in mouse serum after i.p. dosing (.pdf)
Supporting Table S1, IC50 of initial compounds to inhibit SCoR2 in vitro (.pdf)
Supporting Table S2, IC50 of secondary compounds to inhibit SCoR2 in vitro (.pdf)
Supporting Table S3. SMILES molecular formula strings for all compounds (.csv)
Supporting Data 1. PDB coordinates for JSD26 docked to SCoR2 (.pdb)