

Abstract
Purpose:
To describe the clinical impact of commercial laboratories issuing conflicting classifications of genetic variants.
Methods:
Results from 2,000 patients undergoing a multi-gene hereditary cancer panel by a single laboratory were analyzed. Clinically significant discrepancies between the lab provided test reports and other major commercial laboratories were identified, including differences between pathogenic/likely pathogenic (P/LP) and variant of uncertain significance (VUS) classifications, via review of ClinVar archives. For patients carrying a VUS, clinical documentation was assessed for evidence of provider awareness of the conflict.
Results:
50/975 (5.1%) patients with non-negative results carried a variant with a clinically significant conflict, 19 with a P/LP variant reported in APC or MUTYH, and 31 with a VUS reported in CDKN2A, CHEK2, MLH1, MSH2, MUTYH, RAD51C, or TP53. Only 10/28 (36%) patients with a VUS with a clinically significant conflict had a documented discussion by a provider about the conflict. Discrepant counseling strategies were utilized for different patients with the same variant. Among patients with a CDKN2A variant or a monoallelic MUTYH variant, providers were significantly more likely to make recommendations based on the laboratory-reported classification.
Conclusion:
Our findings highlight the frequency of variant interpretation discrepancies and importance of clinician awareness. Guidance is needed on managing patients with discrepant variants to support accurate risk assessment.
Keywords: Cancer genetics, variant interpretation, variant interpretation discrepancy, conflicting variant interpretation, variant of unknown significance, variant of uncertain significance, VUS, ClinVar, clinical cancer genetics, genetic counseling, germline variant, risk assessment
Introduction
In cancer genetics practice, multiple commercial germline genetic testing laboratories may be used each providing their own categorization of genetic variants, which can lead to clinicians receiving discrepant classification of genetic variants1. Variant classification often determines whether the variant has implications for a patient’s clinical care, which may include type and frequency of cancer surveillance strategies, prophylactic surgeries, surgical or medical treatment decisions, and recommendation for a cancer genetics evaluation for a patient’s family members2,3. Individuals with pathogenic or likely pathogenic variants in hereditary cancer predisposition genes are often provided management recommendations based on the variant, while individuals with benign or likely benign variants or variants of uncertain significance (VUS) are managed based on their personal or family history of cancer2–7. Therefore, discrepancies in variant classification by different genetics laboratories can have significant clinical implications.
ClinVar is a publicly available online database of variant interpretations, created in 2013 with the goal of sharing evidence about variant pathogenicity and establishing consensus interpretations8. Numerous major commercial and research laboratories, expert panels, and other organizations contribute to ClinVar, and clinicians use it as a tool to evaluate variant classifications.
Genetic testing laboratories largely adhere to the joint American College of Medical Genetics (ACMG) and Association for Molecular Pathology (AMP) guidelines for classifying variants, which incorporate evidence such as clinical, population, computational, functional, and segregation data1. Nevertheless, discrepancies in variant classification have been identified in 12–83% of variants9–15, with the rate depending on year of the published study, date the variants were last evaluated, specific genes evaluated, and types of laboratories and ClinVar submissions included in the study14,16. Clinically significant discrepancies between the lab report and ClinVar, i.e. discrepancies between P/LP and VUS, have been reported in 11% of patients with germline findings in cancer susceptibility genes10.
Despite the adoption of the ACMG/AMP guidelines, discrepant classifications among laboratories remains an issue due to factors including the subjectivity of determining when ACMG criteria are met, laboratory-specific classification schemes, and differences in each laboratory’s internal clinical data from patients tested at that particular laboratory9,10,13–15,17,18. Rarer variants and those in lower penetrance genes are predicted to take a longer time to correctly classify, as larger sample sizes are needed19. For example, well-studied high penetrance genes such as BRCA1 and BRCA2 have a lower reported frequency of variant interpretation discrepancies than other high and moderate penetrance hereditary cancer genes20. Additionally, very few (0.1%) discrepancies in BRCA1 and BRCA2 involve opposite classifications (pathogenic/likely pathogenic and benign/likely benign).15
Ninety-six percent of cancer genetic counselors report encountering a variant classification discrepancy, and 99% have concerns about counseling patients with these variants21. While the presence of clinically significant conflicting variant classification has been established, limited research has assessed the frequency of conflict solely among major commercial clinical laboratories and the impact on patient care. Additionally, the prevalence of conflicting interpretations has not been studied in a defined population tested through a single laboratory. This study aims to describe the clinical impact of conflicting variant interpretations by quantifying the proportion of patients tested through a single commercial laboratory that were found to have a variant with a clinically significant discrepancy, assessing genetics providers’ awareness of conflicts, and comparing management recommendations provided to patients with discrepant classifications of the same variant within the same clinical practice.
It is likely that the broad community of cancer genetics practitioners has difficulty interpreting, integrating, and incorporating discordant results into clinical counseling, and this practice is likely to lead to discrepant clinical recommendations given to patients with the same variant. We hypothesize that recommendations for cancer surveillance and genetic testing of family members are likely to correspond with the classification of the variant on the report even when conflicts exist between laboratories.
Methods
Ascertainment of Conflicting Variant Interpretation Prevalence
All reported research was approved by institutional review boards of the participating centers. A cohort of 2,000 patients was recruited as part of a multicenter, prospective cohort study of germline panel testing, which has been described previously in detail22. Individuals were invited to enroll between July 2014 and November 2016 during their genetic counseling appointment at three academic centers, USC Norris Comprehensive Cancer Center and Hospital (USC Norris), the Los Angeles County + USC Medical Center (LAC+USC), and Stanford University Cancer Institute. Written informed consent was obtained. All individuals underwent pre-test counseling with a board-certified genetic counselor (CGC) or an advanced practice genetics nurse practitioner (APNG), and 688 patients (34%) also met with a medical oncologist or gastroenterologist specializing in cancer genetics. Testing was performed with a 25- or 28-gene panel (Myriad Genetic Laboratories, Inc) that included the following: APC, ATM, BARD1, BMPR1A, BRCA1, BRCA2, BRIP1, CDH1, CDK4, CDKN2A (p14ARF and p16INK4a), CHEK2, EPCAM, MLH1, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, PTEN, RAD51C, RAD51D, SMAD4, STK11, and TP53. In July 2016, GREM1, POLD1, and POLE were added. After genetic test results were disclosed, medical management recommendations and recommendations for family member testing were recorded in a case report form (CRF). Many variants were reclassified over time; however, the aim of our study was to evaluate interpretation discrepancies at the time of the original test result disclosure. Data was stored in a Progeny database23.
For all variants identified in individual lab provided reports in the cohort (1326 total variant calls in 975 patients) ClinVar data were analyzed to identify whether there was a clinically significant discrepancy between the classification on the original report and the overall classification in ClinVar. For each variant, the overall ClinVar classification and all submissions to ClinVar prior to the patient’s test report date were recorded. We used application programming interfaces (API) E-utilities and Entrez Direct to access and retrieve ClinVar data on November 19, 2019. ClinVar records were manually retrieved for variants that could not be accessed with the application programming interfaces. A clinically significant discrepancy (“discrepancy”) was defined as a discrepancy between a clinically non-actionable classification (benign, likely benign, or VUS) and a clinically actionable classification (P/LP) between the patient’s test report and ClinVar. If the overall ClinVar classification was “conflicting interpretations of pathogenicity,” the breakdown of classifications was reviewed to determine whether at least one entry had a clinically significant discrepancy from the report classification.
We manually reviewed the available ClinVar entries as well as ClinVar archives from the month of the patient’s report. Each ClinVar archive XML file was searched to identify all records for the particular variant. The record submissions were then evaluated to determine whether there was a discrepancy at that time by a major commercial laboratory, defined as a commercial laboratory in the United States that is Clinical Laboratory Improvement Amendments certified and College of American Pathologists accredited and has at least 1000 submissions to ClinVar. Examples of major commercial laboratories include Ambry Genetics, Color, Counsyl, Fulgent Genetics, GeneDx, Invitae, Prevention Genetics, Quest Diagnostics, and University of Washington. Additionally, submissions from ClinVar-determined expert panels were included. Submissions from research laboratories, GeneReviews, Online Mendelian Inheritance in Man (OMIM), and other laboratories were not included.
If there was no conflict in ClinVar during the month of the patient’s report, ClinVar archives for 12 months following the date of the lab provided report were reviewed to search for any discrepancies pending ClinVar submissions. All patients found to have a conflict at the time of their report date through the methods described were combined into a single dataset.
Evaluation of Records for Clinical Suspicion of VUS Pathogenicity
Of the patients with discrepant variants, those in which the variant was classified as a VUS on the report were evaluated to determine if their genetics provider(s) had knowledge of other laboratories’ classification of P/LP or suspicion of pathogenicity. This was assessed by reviewing the CRF and patient medical records. For three patients, the CRF was missing and the clinic note was unavailable; these patients were excluded. Provider interpretations and recommendations were evaluated in the context of patient and family history to determine if there was evidence of a provider’s suspicion of pathogenicity. Criteria (Supplemental Table 1) were created based on standard NCCN guidelines2,3 and well-established cancer risks associated with specific genes. For example, evidence of a provider’s suspicion of pathogenicity included written documentation in the medical record or CRF of another laboratory’s classification as pathogenic/likely pathogenic (P/LP), use of the word “suspicious,” or providing screening or risk reduction recommendations according to NCCN Guidelines for P/LP variants in the respective gene in the absence of a significant family history that would warrant such recommendations. Examples that were considered to not demonstrate provider suspicion of pathogenicity included not recommending screening beyond general population guidelines when there were established cancer risks and NCCN guidelines for P/LP variants in the respective gene and not recommending genetic testing of family members for the variant. Complete list of general and gene-specific criteria utilized is detailed in Supplemental Table 1.
Comparison of Counseling Strategy in Discrepant Interpretations of the Same Variants
To compare patients with discrepant classifications of variants between different laboratories, we queried the Cancer Genetics Registry at USC Norris and LAC+USC, where participants underwent multigene panel testing through a variety of commercial laboratories between April 2013 and September 2019. All unique variants within the original cohort of 2,000 patients that were identified to have a discrepancy per the methods described above were queried in the Registry database. Four variants in CDKN2A, CHEK2, and MUTYH were identified with discrepancies such that another laboratory categorized the variant differently. All patients with these four variants who were seen either at the USC Norris or LAC+USC were combined into a single dataset that included 57 patients. For USC Norris and LAC+USC patients, recommendations are likely to be consistent among different providers due to a weekly case conference attended by three genetic counselors, a genetics nurse practitioner, and three genetics physicians for clinical practice discussions. Recommendations provided for medical management and genetic testing of family members were assessed through review of CRF and clinical documentation of the results disclosure. There were five patients for which no clinical or research documentation of results disclosure was available.
Data Analysis
Descriptive statistics and Fisher’s exact tests were performed using IBM SPSS Statistics software version 26. A p-value of less than 0.05 was considered statistically significant. All Fisher’s exact tests were two-sided.
Results
Demographic Characteristics of the Study Population
A total of 2,000 participants were recruited for hereditary cancer panel testing. The most frequently self-reported races and ethnicities were non-Hispanic White (40.6%), Hispanic (39.0%), and Asian (11.7%); 6.8% of participants reported Ashkenazi Jewish ancestry. Most participants (72.1%) were affected with at least one primary cancer, and 8.3% had multiple primaries. Genetic test results included 243 (12.2%) positive (with or without an additional VUS), 732 (36.6%) VUS, and 1,025 (51%) negative (Table 1).
Table 1.
Characteristics of 2,000 study participants
| N | % | |
|---|---|---|
| Gender | ||
| Female | 1614 | 80.7% |
| Male | 386 | 19.3% |
| Agea | ||
| <30 | 100 | 5.0% |
| 30–39 | 283 | 14.2% |
| 40–49 | 533 | 26.7% |
| 50–59 | 503 | 25.2% |
| 60–69 | 411 | 20.5% |
| 70–79 | 139 | 7.0% |
| >79 | 31 | 1.6% |
| Race/Ethnicity | ||
| Non-Hispanic White | 811 | 40.6% |
| Hispanic White | 779 | 39.0% |
| Asian | 234 | 11.7% |
| Unknown/More than one | 91 | 4.6% |
| Black or African Americanb | 75 | 3.8% |
| American Indian/Alaska Native | 5 | 0.3% |
| Native Hawaiian/Pacific Islander | 5 | 0.3% |
| Cancer Status | ||
| Affected | 1442 | 72.1% |
| Unaffected | 558 | 27.9% |
| Original Overall Result | ||
| Positive (with or without a VUS) | 243 | 12.2% |
| VUS | 732 | 36.6% |
| Negative | 1025 | 51.2% |
| Any VUS Identified | ||
| Yes | 810 | 40.5% |
| No | 1190 | 59.5% |
Mean = 51.5. SW. Dev. = 13.378.
Three individuals identified as Stack Hispanic.
Prevalence of Clinically Significant Discrepancies
Among the 975 participants with a positive or VUS result, there were a total of 1,326 variant calls reported, representing 943 unique variants. Data were retrieved from ClinVar for 1,133 variant calls; 81 were found through a manual search; 112 were not reported in ClinVar. A total of 50/1,326 (3.8%) variant calls were found to have a discrepancy at the time of the patient’s original genetic test report, representing 50/2,000 (2.5%) patients who underwent the panel, and 50/975 (5.1%) unique patients who had at least one variant identified (Figure 1). Classifications were captured for each variant call by each major laboratory and expert panel during the month that the patient’s original report was issued (Supplemental Table 2).
Figure 1. Variant evaluation flowchart.
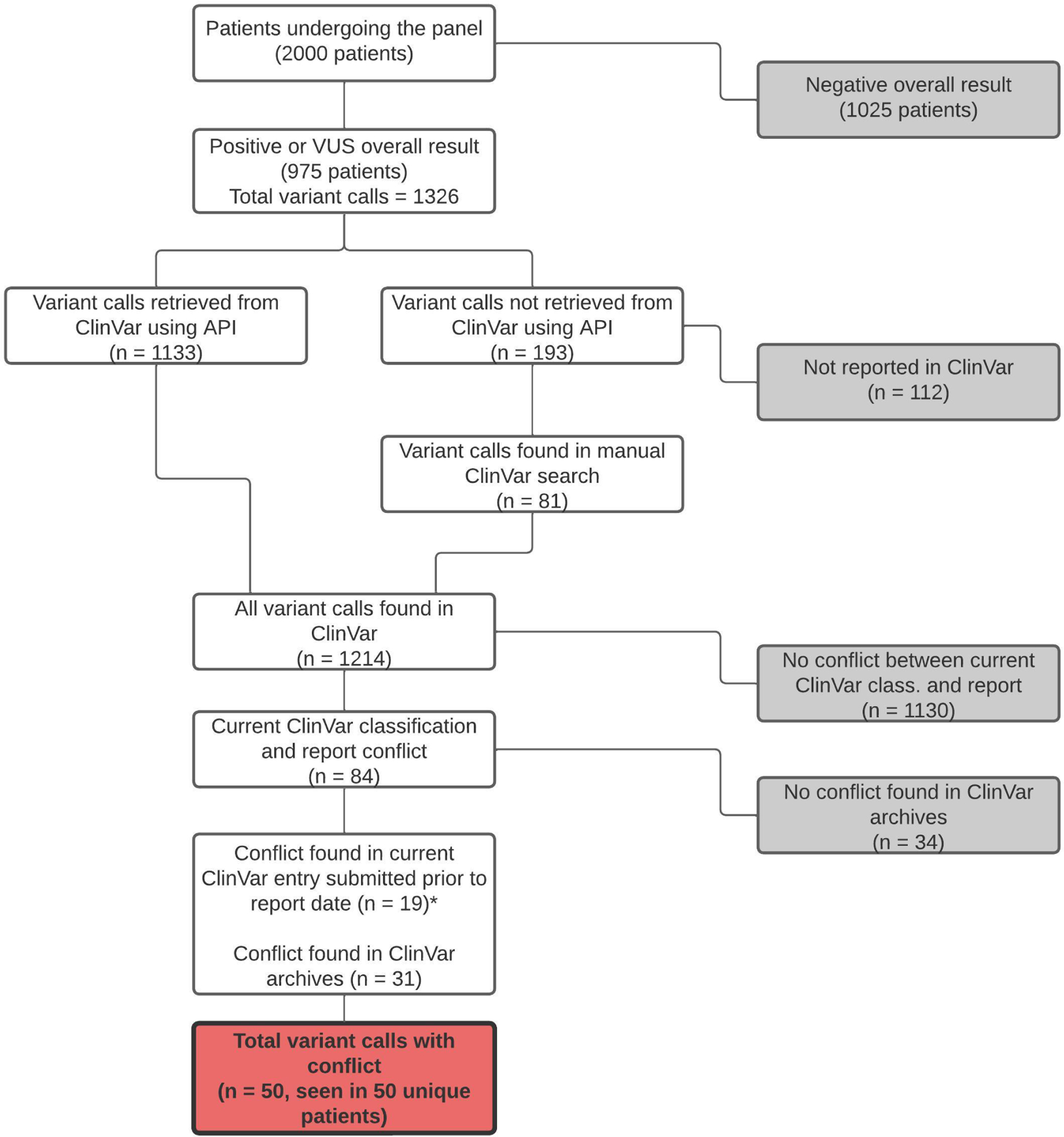
The 1,326 variant calls were evaluated individually to determine whether there was a clinically significant conflict among major commercial laboratories at the time of the specific patient’s report. Final conflicts are shown in the red box and represent conflicts among major laboratories as outlined by criteria in the Methods. *ClinVar archive search required for the other 65 variants.
There were 14 unique variants in which a conflict was identified. CHEK2 was the most frequently identified gene with a discrepancy (5 unique variants among 17 patients). Discrepancies were also seen in APC, CDKN2A, MLH1, MSH2, MUTYH, RAD51C, and TP53 (Figure 2). Complete HGVS nomenclature for each variant is in Supplemental Table 3 and was validated by VariantValidator24. Of note, the testing laboratory utilized transcript NM_001128425.2 for MUTYH. The variants we describe as c.857G>A and c.934–2A>G are also known as c.773G>A and c.850–2A>G, respectively, when using MANE (Matched Annotation from NCBI and EMBL-EBI) transcript NM_001048174.2.
Figure 2. Distribution of variants with clinically significant conflicts.
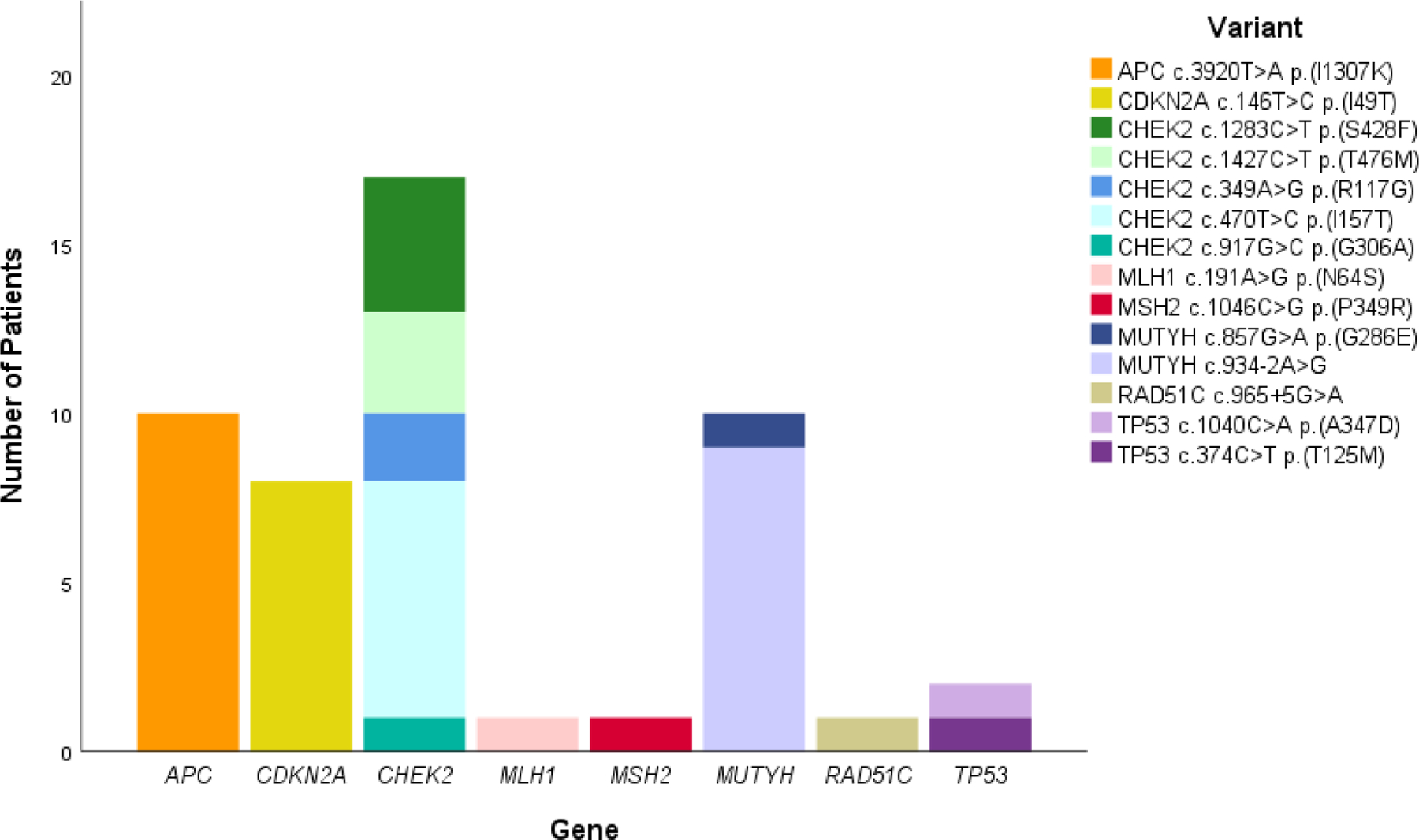
The 50 patients with variants that had clinically significant discrepant classifications are distributed by unique variant and aggregated by gene. The order of variants in the key corresponds to the vertical order of variants in the graph.
Of the 50 patients with conflicting variants, 19 individuals (38%) had a P/LP classification by the lab provided reports (for the variants APC c.3920T>A p.(I1307K) and MUTYH c.934–2A>G), and 31 individuals (62%) had a VUS classification (for the other 12 variants listed in Figure 2). The proportion of the cohort with a discrepancy by race/ethnicity was 4.7% (11/234) for Asians, 1.7% (13/779) for Hispanics, and 3.1% (25/811) for Non-Hispanic Whites. For individuals with Ashkenazi Jewish ancestry, 9.6% (13/136) had a discrepancy; nine were APC c.3920T>A p.(I1307K). When excluding APC c.3920T>A p.(I1307K), Asians had the highest prevalence of discrepancy, largely attributed to MUTYH c.934–2A>G.
For each of the 50 patients with a conflicting variant, the total number of relatives was counted to assess the broader impact of the variant classification, yielding a total of 291 first-degree relatives (215 living), and 790 second-degree relatives.
Provider Suspicion of VUS Pathogenicity
Of the 50 patients with a conflicting variant, 31 (62%) had a variant classified as VUS by the lab provided testing reports; 28 had medical records available. Each patient was seen by one of eight genetic counselors or nurse practitioners, and some patients were also seen by one of five physicians. There was no evidence of provider suspicion of pathogenicity for 64% (18/28). The proportion of patients in which there was provider suspicion varied by specific variant (Figure 3). For high penetrance genes (CDKN2A, MLH1, and TP53), only one out of nine patients received counseling that acknowledged the variant discrepancy.
Figure 3. Suspicion of pathogenicity in patients with variants classified as VUS.
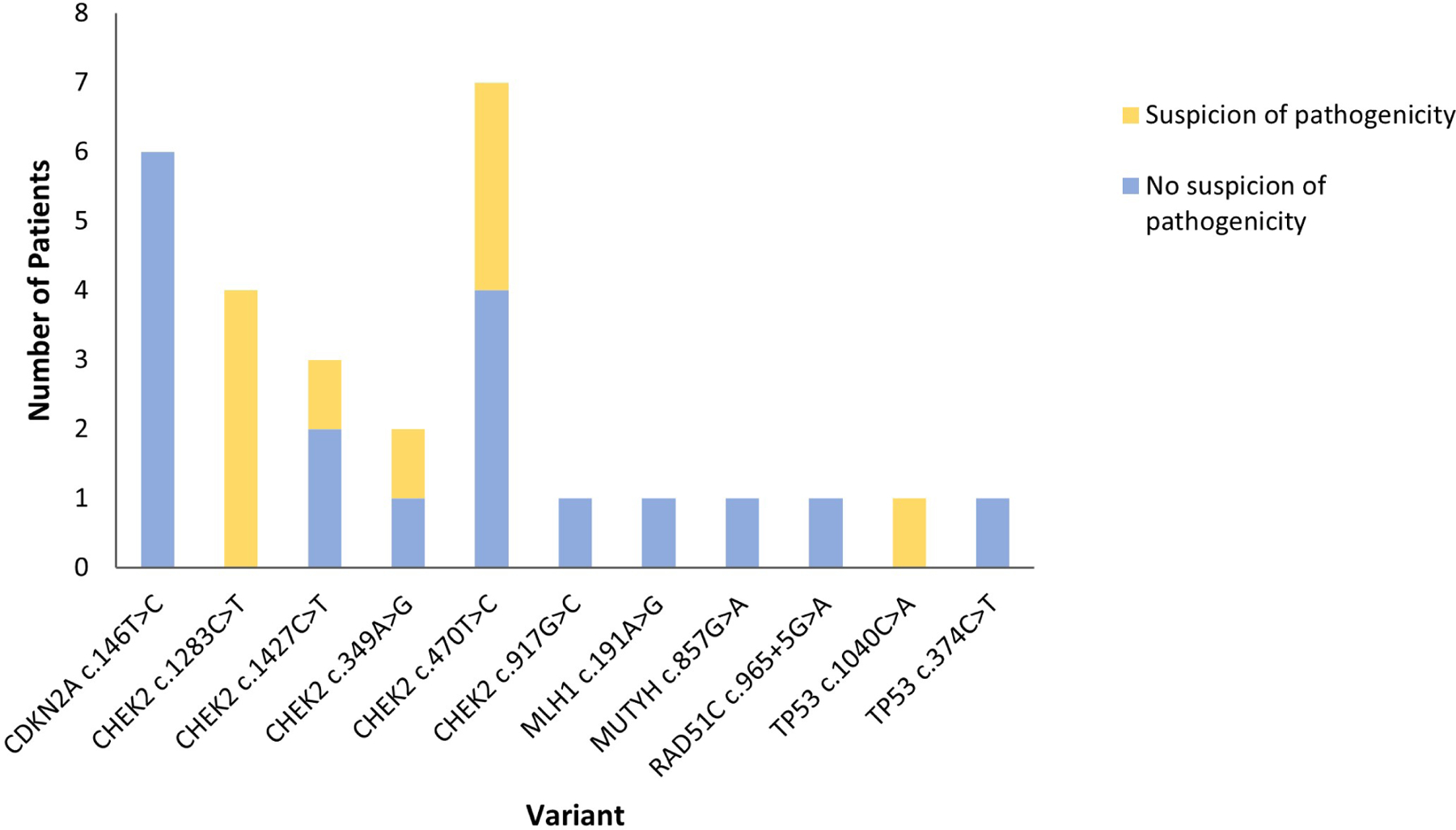
N=28. The number of patients with each variant for which there was and was not evidence of provider suspicion of pathogenicity is shown.
Discrepant Classifications of the Same Variant within a Clinical Practice
The Cancer Genetics Registry at USC Norris and LAC+USC allowed for identification of additional individuals with the same discrepant variants (CDKN2A c.146T>C, CHEK2 c.349A>G, CHEK2 c.470T>C, and MUTYH c.934–2A>G). There were 57 total patients (including 24 from the original cohort and 33 from the Registry) with these four variants. Results for these Registry patients were received between April 2014 and June 2019.
Three of these variants were seen in three or more patients and were analyzed further to compare medical management recommendations between those with a P/LP classification and a VUS classification (Table 2). While the sample is small, it revealed that most patients are counseled according to their test report, and that personal and family history as well as the patient’s current disease status influenced the recommendations. For example, in patients with CDKN2A c.146T>C, two of the 14 patients with a lab reported VUS classification were recommended to undergo a skin exam, but one had a personal history of melanoma, and the other had a family history of melanoma. There was a statistically significant association between report classification of this variant and skin exam recommendation when excluding those with a personal or family history of melanoma (Fisher’s exact p=0.018).
Table 2.
Comparison of medical management recommendations for discrepant variants
| Classification |
Fisher’s exact P-value | ||
|---|---|---|---|
| P/LP |
VUS |
||
| N (%) | N (%) | ||
| CDKN2A c.146T>Ca | |||
| Skin exam recommended | |||
| No | 0 (0%) | 9 (81.8%) | 0.077c |
| Yes | 2 (100%) | 2 (18.2%)b | |
| Total | 2 | 11 | |
| Pancreatic cancer screening recommended | |||
| No | 1 (50.0%)d | 11 (100%) | 0.154f |
| Yes | 1 (50.0%)e | 0 (0%) | |
| Total | 2 | 11 | |
| Targeted variant testing recommended | |||
| No | 1 (50.0%)g | 10 (90.9%) | 0.295 |
| Yes | 1 (50.0%) | 1 (9.1%)h | |
| Total | 2 | 11 | |
| CHEK2 c.470T>C | |||
| Colonoscopy frequencyi | |||
| General population or no recommendation given | 2 (22.2%) | 3 (60.0%) | 0.266 |
| Every 5 years or more frequently | 7 (77.8%) | 2 (40.0%)j | |
| Total | 9 | 5 | |
| Breast MRI recommendedk | |||
| No | 2 (25.0%) | 2 (50.0%) | 0.547 |
| Yes | 6 (75.0%) | 2 (50.0%)l | |
| Total | 8 | 4 | |
| Targeted variant testing recommendedm | |||
| No | 1 (20.0%)n | 5 (83.3%) | 0.08 |
| Yes | 4 (80.0%) | 1 (16.7%)o | |
| Total | 5 | 6 | |
| MUTYH c.934–2A>G | |||
| Colonoscopy frequencyp | |||
| General population or no recommendation given | 1 (6.7%) | 2 (100%) | 0.022* |
| Every 5 years or more frequently | 14 (93.3%) | 0 (0%) | |
| Total | 15 | 2 | |
| Targeted variant testing or MUTYH sequencing recommended | |||
| No | 2 (15.4%) | 2 (100%) | 0.057 |
| Yes | 11 (84.6%) | 0 (0%) | |
| Total | 13 | 2 | |
P/LP represents a classification of pathogenic/likely pathogenic. Cancer screening recommendations are for individual patients, and targeted variant testing refers to testing of family members for the respective variant. MUTYH sequencing refers to sequencing of the gene to assess either for the presence of biallelic P/LP variants, which would result in MUTYH associated polyposis syndrome (MAP), or to assess status of the patient’s partner to ascertain risk of MAP in offspring. Total N for familial testing represents total unique families.
Documentation of results disclosure was unavailable for three individuals with VUS classifications.
One had a personal history of melanoma, and one had a family history of melanoma.
When controlling for no personal or family history of melanoma, Fisher’s exact P = 0.018*.
Patient already affected with this disease. Knowledge of conflicting interpretations was not apparent.
No personal or family history of pancreatic cancer. Results disclosure note discussed other laboratories’ VUS classification, indicating that provider was aware of conflict. Patient was under recommended age to begin screening, and note discussed the possibility for recommendations to change due to the ambiguity of the variant. Patient recommended to return to clinic in two years for updated management recommendations.
When controlling for no personal or family history of pancreatic cancer, Fisher’s exact P = 0.083.
Provider displayed awareness of conflict, and this appeared to play a role in not recommending familial testing.
VUS tracking studies recommended for family history of melanoma.
One deceased patient, one with incomplete data, and three with active metastatic disease were excluded, as cancer screening recommendations were not provided or available.
Both demonstrated provider awareness of conflicting interpretations; no family history of colorectal cancer.
Only females were included.
One also had a pathogenic variant in ATM that appeared to drive this recommendation; the other had demonstrated provider awareness of conflicting interpretations.
There were 13 unique families with this variant; one was excluded because the patient was deceased when the results were received, and no recommendations were provided, and the other was excluded because all at-risk family members had already been tested for the variant prior to presenting to Cancer Genetics.
Unclear why not recommended; not discussed in clinic note. It may be possible that it was recommended but not documented in the note.
Appeared to be driven by knowledge of conflict.
Documentation of results disclosure was available for 18 patients. One patient was added because although there was no CRF or original genetics results disclosure note, there was another provider’s note available which discussed the colonoscopy recommendations from the genetics provider. One deceased patient and one with incomplete data were excluded.
Another example included patients with the MUTYH c.934–2A>G variant, including 18 classified as P/LP and two as VUS (Table 2). Fourteen of those assessed with a P/LP classification of this variant were recommended to undergo colonoscopy every at least every 5 years, which was the clinical group’s recommendation throughout the period of the study for individuals with monoallelic P/LP variants in MUTYH. For both individuals with a VUS classification, no colonoscopy recommendations were given. Among patients with this variant, providers were significantly more likely to make colonoscopy recommendations based on the laboratory-reported classification (Fisher’s exact p=0.022).
Counseling Strategy for Discrepant Classifications by the Same Provider
Three genetic providers were involved in counseling patients with discrepant classifications of the same variant. All three providers displayed differences in counseling strategy when counseling patients with different classifications of the same variant (Supplemental Table 4A-E). For example, one provider saw patients with discrepant classifications of MUTYH c.934–2A>G and disclosed their results one month apart. Although neither patient had any personal or family history of colon cancer or polyps, enhanced colonoscopy screening and targeted variant testing were recommended for the patient with LP classification and not for the patient with VUS classification. The patient with VUS classification was not counseled with knowledge of the conflict. Neither patient had additional P/LP variants identified on testing. Additional case examples are available in Supplemental Table 4A-E.
Discussion
This study describes the clinical impact of variant interpretation discrepancies between laboratories. Clinically significant conflicts were found in 2.5% (50/2000) of the original cohort of patients and 5.1% (50/975) of patients with a non-negative result. Conflicts were found in variants in APC, CDKN2A, CHEK2, MLH1, MSH2, MUTYH, RAD51C, and TP53. Our comparison of recommendations for discrepant variants supports our hypothesis that clinicians are more likely to provide clinical recommendations according to the laboratory-reported classification.
This study builds on prior studies analyzing variant discrepancies. For example, a previous study found that 11% of patients with a variant identified on hereditary cancer panel testing had a clinically significant discrepancy10. However, the study included all ClinVar submissions and was not limited to clinical laboratories. In a critique of the Balmaña et al. study, the variants were re-evaluated to only include submissions from clinical laboratories and ClinVar-determined expert panels (excluding literature and research submissions), and only 5.5% of patients had a clinically significant conflict25. This is consistent with our finding that 5.1% of those with non-negative results had a variant with a clinically significant conflict when their report was issued. By focusing on conflicts that have the potential to impact medical management and only including ClinVar submissions by laboratories that perform a considerable amount of clinical testing, our findings likely reflect the proportion of patients who may actually be impacted by these discrepancies.
The genes and variants identified to have discrepancies are relatively consistent with previously published studies10,14,25. CHEK2 had the greatest number of unique variants with conflicts and affected the greatest number of patients (5 unique variants among 17 patients). In the Balmaña et al. study, 63.2% (36/57) of the variant calls with a clinically significant conflict were in CHEK2. Other genes with conflicts in their study included APC, BRIP1, CDKN2A, FH, MSH6, MUTYH, NBN, PALB2, and RAD51C. We similarly identified discrepancies in APC (specifically p.(I1307K)), CDKN2A, MUTYH, and RAD51C, and additionally in MLH1, MSH2, and TP53. Similarly, in the Harrison et al. study, CHEK2 had the greatest number of conflicts of all cancer-related genes.
Variants in CHEK2, particularly missense variants, are known to be challenging to classify17,26. Perhaps this is because CHEK2 is a moderate penetrance gene, its expected phenotype (breast or colon cancer) is common, and the genetics community’s understanding of its associated cancer types and specific risk estimates is continuing to evolve26. Therefore, data from phenotype studies may not be as useful for determining variant classification. Three variants in CHEK2—c.470T>C p.(I157T), c.1283C>T p.(S428F), and c.1427C>T p.(T476M)—are known founder variants with conflicting data on pathogenicity19,26. Some laboratories and publications describe these missense variants as low penetrance26, which are known to have high rates of discordance16. All conflicting variants in CHEK2 identified in our study were missense, with 14/17 patients having one of the founder variants.
Low penetrance variants are challenging to classify because they do not fall into any of the categories outlined in the ACMG/AMP guidelines1,16,27. Individuals with APC c.3920T>A p.(I1307K) made up 20% (10/50) of conflicts. Previous research and national guidelines have determined that this is a low penetrance founder variant that confers a moderately increased risk of colorectal cancer3. However, this variant is still classified as VUS by several major laboratories. Development of guidelines for classification of low penetrance variants, with criteria similar to the ACMG/AMP guidelines1, may be helpful in resolution of some of these conflicts. Furthermore, gene-specific interpretation guidelines will aid in the interpretation of variants in moderate and low penetrance genes, as the utilization of gene-specific criteria has been shown to decrease the frequency of discordant interpretations11.
Conflicts can also be prevalent in genes that are typically highly penetrant, such as TP53, in which one study showed that 11% of families had a variant with a clinically significant discrepancy28. Both TP53 variants identified in our study were also identified in the Frone et al. study. It is possible that some of the variants with conflicts in high penetrance genes like TP53 may truly be low or moderate penetrance variants and may produce an attenuated phenotype compared to other P/LP variants in the gene.
Although the number of patients with discrepancies is relatively small, the clinical impact on these patients can be substantial. NCCN provides guidelines for cancer surveillance and risk-reduction in individuals with P/LP variants in cancer predisposition genes2,3. Patients with potentially pathogenic variants may not be recommended the care associated with the variant. Many insurance companies utilize NCCN guidelines to determine coverage of services29 so services could be denied even if the provider were to recommend the screening based on a known conflict, particularly in individuals who do not meet NCCN criteria for enhanced screening based on family history alone. Additionally, there are now FDA approvals and clinical trials for targeted cancer treatments which utilize germline or tumor variants to inform treatment and are available to individuals with a P/LP variant in specific genes2,3. Variant interpretation discrepancies could lead to discrepancies in treatment options for patients with the exact same cancer type and germline variant. To resolve discrepancies, we encourage collaboration between laboratories and evaluation of variants by ClinGen-determined expert panels.
Our study revealed that only 36% of patients (10/28) with a lab reported VUS were counseled with knowledge of a conflict when their variant was classified as P/LP by another major commercial laboratory. Previous research has shown that most cancer genetic counselors do not evaluate variant evidence beyond the lab report for most of their patients, and the primary barrier is lack of time30. Perhaps another contributing factor is that most VUS are downgraded to benign31–33. Additionally, genetics providers may be less likely to research a VUS in ClinVar when a family history does not fit the respective gene’s phenotype. However, results of this analysis show that cancer genetic counselors cross checking variants in ClinVar could lead to the identification of variant discrepancies in 5% of variants on genetic test reports and may help avoid counselors providing different recommendations to patients with the same variant. Since discrepancies can be critical to patients’ clinical management, it could be helpful for professional organizations such as NCCN to provide guidance to providers about evaluating all variants in ClinVar prior to post-test counseling. This also highlights the importance of clinical laboratories submitting classifications to ClinVar; we encourage professional organizations to consider incorporating this recommendation into practice guidelines.
Awareness of variant conflicts is likely even lower among non-genetics professionals. Non-genetics oncology providers have displayed limited understanding of VUSs32, and are thus more likely to misinterpret results. Non-genetics providers may have lower volume of genetic testing and may be less familiar with recurrent variants or how to handle variant reclassifications. Therefore, the results of our analysis may be even more pronounced among non-genetics providers.
Our review of case examples demonstrated that counseling is challenging even when a provider is aware of a conflict, and recommendations did not always completely align with a VUS or P/LP classification. Clinical genetic counselors are becoming increasingly involved in variant interpretation in determining how to appropriately manage their patients12,30,33. When genetic counselors are aware of a lab conflict or have their own conflicting interpretation based on available evidence, they report discussing this with the laboratory, medical team/colleagues, and patient/family30. Genetic counselors and medical geneticists have reported most often following the laboratory’s classification, but occasionally managing the patient based on their own interpretation of the variant; for VUSs that the clinician suspects is pathogenic, this sometimes includes a recommendation for screening tests but not invasive procedures33. Genetic counselor continuing education and professional organization practice guidelines on how to counsel patients with discrepant variants are desired by genetic counselors21 and would provide awareness and guidance on this issue.
A limitation of our study is that the initial cohort from the longitudinal cohort study was tested in 2014–2016. Patients tested today by an experienced provider may be more likely to receive counseling with knowledge of the conflicts. Additionally, some of the variants we describe have since been reclassified. However, given the increase in identification of VUS over time as multigene panels become increasingly larger and more widely utilized in unselected populations34, conflicts are likely to remain prevalent, and the principles we describe in this paper will continue to impact patient outcomes in clinical practice.
Another limitation of this study is the small chance that variants with clinically significant conflicts were missed by the analysis of ClinVar archives due to variants not being reported at all by a laboratory or laboratories not submitting updates on time. Additionally, when assessing management recommendations and counseling strategy with discrepant classifications, generalizability is limited because the sample size was small and all patients included in this part of the analysis were seen through one institution. This study was not able to identify whether certain racial/ethnic populations are more likely to have variants with discrepancies.
In conclusion, the findings from this study support previously published literature finding that approximately 5% of patients with non-negative results on hereditary cancer panel testing are found to have a variant with clinically significant discrepancies among major commercial laboratories. This study described provider awareness of clinically significant conflicts when counseling patients with a VUS classified as P/LP by other laboratories, and found that a minority of patients appeared to be counseled with provider awareness of the conflict. A detailed case analysis identified discrepant counseling strategies utilized for different patients with the same variant, within the same institution and even by the same genetics provider. Our findings provide evidence that variant interpretation discrepancies can have profound clinical implications, highlighting the importance of clinician awareness and need for guidance on managing patients with discrepant results.
Supplementary Material
Acknowledgments
Supported by Myriad Genetics, National Institutes of Health Grants No. KL2-TR000131.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Ethics Declaration
This work was IRB reviewed and approved by USC, UCI, and Stanford IRBs. Written informed consent was obtained from all patients.
Data Availability
Data are available upon request to corresponding authors.
References
- 1.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine : official journal of the American College of Medical Genetics 2015;17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.NCCN. Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic (Version 1.2022) 2021. (August 11th, 2021). https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf.
- 3.NCCN. NCCN Genetic/Familial High-Risk Assessment: Colorectal 2021.
- 4.Domchek SM, Friebel TM, Singer CF, et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA-J Am Med Assoc 2010;304(9):967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindor NM, Petersen GM, Hadley DW, et al. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA-J Am Med Assoc 2006;296(12):1507–1517. [DOI] [PubMed] [Google Scholar]
- 6.Riley BD, Culver JO, Skrzynia C, et al. Essential elements of genetic cancer risk assessment, counseling, and testing: updated recommendations of the National Society of Genetic Counselors. J Genet Couns 2012;21(2):151–161. [DOI] [PubMed] [Google Scholar]
- 7.Welsh JL, Hoskin TL, Day CN, et al. Clinical Decision-Making in Patients with Variant of Uncertain Significance in BRCA1 or BRCA2 Genes. Ann Surg Oncol 2017;24(10):3067–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Landrum MJ, Kattman BL. ClinVar at five years: Delivering on the promise. Hum Mutat 2018;39(11):1623–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amendola LM, Jarvik GP, Leo MC, et al. Performance of ACMG-AMP Variant-Interpretation Guidelines among Nine Laboratories in the Clinical Sequencing Exploratory Research Consortium. Am J Hum Genet 2016;98(6):1067–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balmaña J, Digiovanni L, Gaddam P, et al. Conflicting Interpretation of Genetic Variants and Cancer Risk by Commercial Laboratories as Assessed by the Prospective Registry of Multiplex Testing. J Clin Oncol 2016. [DOI] [PMC free article] [PubMed]
- 11.Amendola LM, Muenzen K, Biesecker LG, et al. Variant Classification Concordance using the ACMG-AMP Variant Interpretation Guidelines across Nine Genomic Implementation Research Studies. Am J Hum Genet 2020;107(5):932–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bland A, Harrington EA, Dunn K, et al. Clinically impactful differences in variant interpretation between clinicians and testing laboratories: a single-center experience. Genetics in medicine : official journal of the American College of Medical Genetics 2018;20(3):369–373. [DOI] [PubMed] [Google Scholar]
- 13.Harrison SM, Dolinsky JS, Knight Johnson AE, et al. Clinical laboratories collaborate to resolve differences in variant interpretations submitted to ClinVar. Genetics in medicine: official journal of the American College of Medical Genetics 2017;19(10):1096–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harrison SM, Dolinksy JS, Chen W, et al. Scaling resolution of variant classification differences in ClinVar between 41 clinical laboratories through an outlier approach. Hum Mutat 2018;39(11):1641–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gradishar W, Johnson K, Brown K, Mundt E, Manley S. Clinical Variant Classification: A Comparison of Public Databases and a Commercial Testing Laboratory. Oncologist 2017;22(7):797–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang S, Lincoln SE, Kobayashi Y, Nykamp K, Nussbaum RL, Topper S. Sources of discordance among germ-line variant classifications in ClinVar. Genetics in medicine : official journal of the American College of Medical Genetics 2017;19(10):1118–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mundt E, Nix P, Brown K, Bowles KR, Manley S. Complexities of Variant Classification in Clinical Hereditary Cancer Genetic Testing. J Clin Oncol 2017;35(34):3796–3799. [DOI] [PubMed] [Google Scholar]
- 18.Pepin MG, Murray ML, Bailey S, Leistritz-Kessler D, Schwarze U, Byers PH. The challenge of comprehensive and consistent sequence variant interpretation between clinical laboratories. Genetics in medicine : official journal of the American College of Medical Genetics 2016;18(1):20–24. [DOI] [PubMed] [Google Scholar]
- 19.Shirts BH, Jacobson A, Jarvik GP, Browning BL. Large numbers of individuals are required to classify and define risk for rare variants in known cancer risk genes. Genetics in medicine : official journal of the American College of Medical Genetics 2014;16(7):529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lincoln SE, Yang S, Cline MS, et al. Consistency of BRCA1 and BRCA2 Variant Classifications Among Clinical Diagnostic Laboratories. JCO Precis Oncol 2017;1. [DOI] [PMC free article] [PubMed]
- 21.Zirkelbach E, Hashmi S, Ramdaney A, et al. Managing Variant Interpretation Discrepancies in Hereditary Cancer: Clinical Practice, Concerns, and Desired Resources. J Genet Couns 2018;27(4):761–769. [DOI] [PubMed] [Google Scholar]
- 22.Idos GE, Kurian AW, Ricker C, et al. Multicenter Prospective Cohort Study of the Diagnostic Yield and Patient Experience of Multiplex Gene Panel Testing For Hereditary Cancer Risk. JCO Precision Oncology 2019(3):1–12. [DOI] [PMC free article] [PubMed]
- 23.Progeny Software, LLC [computer program]. Version 9 Aliso Viejo, CA: 2015. [Google Scholar]
- 24.Freeman PJ, Hart RK, Gretton LJ, Brookes AJ, Dalgleish R. VariantValidator: Accurate validation, mapping, and formatting of sequence variation descriptions. Hum Mutat 2018;39(1):61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nussbaum RL, Yang S, Lincoln SE. Clinical Genetics Testing Laboratories Have a Remarkably Low Rate of Clinically Significant Discordance When Interpreting Variants in Hereditary Cancer Syndrome Genes. J Clin Oncol 2017;35(11):1259–1261. [DOI] [PubMed] [Google Scholar]
- 26.Bychkovsky BL, Agaoglu NB, Horton C, et al. Differences in Cancer Phenotypes Among Frequent CHEK2 Variants and Implications for Clinical Care—Checking CHEK2. JAMA Oncology 2022;8(11):1598–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dolinsky JS, Hruska KS, Pesaran T, et al. Efforts Toward Consensus Variant Interpretation by Commercial Laboratories. J Clin Oncol 2017;35(11):1261–1262. [DOI] [PubMed] [Google Scholar]
- 28.Frone MN, Stewart DR, Savage SA, Khincha PP. Quantification of Discordant Variant Interpretations in a Large Family-Based Study of Li-Fraumeni Syndrome. JCO Precis Oncol 2021;5. [DOI] [PMC free article] [PubMed]
- 29.Rocque GB, Williams CP, Jackson BE, et al. Impact of Nonconcordance With NCCN Guidelines on Resource Utilization, Cost, and Mortality in De Novo Metastatic Breast Cancer. J Natl Compr Canc Netw 2018;16(9):1084–1091. [DOI] [PubMed] [Google Scholar]
- 30.Wain KE, Azzariti DR, Goldstein JL, et al. Variant interpretation is a component of clinical practice among genetic counselors in multiple specialties. Genetics in medicine : official journal of the American College of Medical Genetics 2020;22(4):785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mersch J, Brown N, Pirzadeh-Miller S, et al. Prevalence of Variant Reclassification Following Hereditary Cancer Genetic Testing. JAMA-J Am Med Assoc 2018;320(12):1266–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kurian AW, Li Y, Hamilton AS, et al. Gaps in Incorporating Germline Genetic Testing Into Treatment Decision-Making for Early-Stage Breast Cancer. J Clin Oncol 2017;35(20):2232–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berrios C, Hurley EA, Willig L, et al. Challenges in genetic testing: clinician variant interpretation processes and the impact on clinical care. Genetics in medicine : official journal of the American College of Medical Genetics 2021;23(12):2289–2299. [DOI] [PubMed] [Google Scholar]
- 34.Lucci-Cordisco E, Amenta S, Panfili A, et al. Variants of uncertain significance (VUS) in cancer predisposing genes: What are we learning from multigene panels? European journal of medical genetics 2022;65(1):104400. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available upon request to corresponding authors.