

Abstract
Nephrotoxicity is a major side effect of cisplatin, a widely used cancer therapy drug. However, the mechanism of cisplatin nephrotoxicity remains unclear and no effective kidney protective strategies are available. Here, we report the induction of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) in both in vitro cell culture and in vivo mouse models of cisplatin nephrotoxicity. Notably, PFKFB3 was mainly induced in the nucleus of kidney tubular cells, suggesting a novel function other than its canonical role in glycolysis. Both pharmacological inhibition and genetic silencing of PFKFB3 led to the suppression of cisplatin-induced apoptosis in cultured renal proximal tubular cells (RPTCs). Moreover, cisplatin-induced kidney injury or nephrotoxicity was ameliorated in renal proximal tubule-specific PFKFB3 knockout mice. Mechanistically, we demonstrated the interaction of PFKFB3 with cyclin-dependent kinase 4 (CDK4) during cisplatin treatment, resulting in CDK4 activation and consequent phosphorylation and inactivation of retinoblastoma tumor suppressor (Rb). Inhibition of CDK4 reduced cisplatin-induced apoptosis in RPTCs and kidney injury in mice. Collectively, this study unveils a novel pathological role of PFKFB3 in cisplatin nephrotoxicity through the activation of the CDK4/Rb pathway, suggesting a new kidney protective strategy for cancer patients by blocking PFKFB3.
Introduction
Cisplatin is a widely used drug for chemotherapy of a variety of solid tumors. However, the clinical use and chemotherapy efficacy of cisplatin are limited by its side effects in normal tissues, especially nephrotoxicity manifested as acute kidney injury (AKI) and chronic kidney disease (CKD). Almost a third of cancer patients receiving cisplatin experience AKI, while chronic kidney problem develops in a significant portion of the patients.1–5 A major pathological feature of cisplatin nephrotoxicity is cell injury and death in kidney tubules, which involves multiple signaling pathways.1–4,6 Despite these studies, the underlying mechanism remains incompletely understood and, as a result, effective kidney protective strategies are not available.
6-Phosphofructo-2-kinase/fructose-2,6-Biphosphatase 3 (PFKFB3) is one of the bifunctional enzymes in the PFKFB family that catalyzes the synthesis of fructose 2,6-bisphosphate (F2,6P2), a potent allosteric activator of the rate-limiting enzyme 6-phosphofructo-1-kinase (PFK-1) in glycolysis.7–9 In mammals, there are four isoforms of PFKFB enzymes, which have different expression, regulation, and enzyme kinetics. Among them, PFKFB3 has the highest kinase to phosphatase activity ratio. Therefore, it produces high levels of F2,6P2 and promotes glycolysis.10 In addition to this well-recognized function in cytoplasm, scattered reports suggested that PFKFB3 may localize in the nucleus in cancer cells,11–13 where it may promote cell proliferation without affecting glucose metabolism,13 suggesting a non-canonical role of PFKFB3. In kidneys, Chen et al. recently reported that IL-22 may protect against cisplatin-induced nephrotoxicity and diabetic nephropathy by inducing PFKFB3 and associated metabolic reprogramming.14 However, the function and regulation of PFKFB3 in kidney diseases remains largely unknown.
The present study was designed to investigate the role and regulation of PFKFB3 in cisplatin nephrotoxicity. We showed that inhibition of PFKFB3 pharmacologically or genetically ameliorated cisplatin-induced tubular cell apoptosis in vitro and nephrotoxicity or AKI in mice. Interestingly, we found that PFKFB3 was mainly induced in the nucleus of kidney tubular cells in both cell culture and mouse models of cisplatin nephrotoxicity. To follow this lead, we demonstrated that PFKFB3 may directly interact with and activate cyclin-dependent kinase 4 (CDK4), which has been implicated in cisplatin nephrotoxicity recently.15–17 Together, these results demonstrate the first evidence of a pathogenic role of PFKFB3 in cisplatin nephrotoxicity by activating CDK4, suggesting a new strategy of kidney protection during chemotherapy.
Materials and Methods
Antibodies and special reagents
Primary antibodies were from the following sources: anti- poly (ADP-ribose) polymerase (PARP) (9532S), anti−cleaved caspase-3 (9664S), anti−cyclophilin B (43603S), anti-Phospho-RIP1 (Ser166) (31122S), anti-Phospho-RIP3 (Thr231/Ser232) (57220S), anti-Phospho-MLKL (Ser345) (37333S), anti-Rb (9313S) and anti-Phospho-Rb (8180S) from Cell Signaling Technology (Danvers, MA);anti-mTIM-1 (Kim-1) (AF1817) from R&D system (Minneapolis, MN); anti-PFKFB3 (ab181861 and ab218121) from Abcam (Waltham, MA); anti-CDK4 (A0366) from Abclonal (Woburn, MA) and (sc-23896) from Santa Cruz Biotechnology (Dallas, TX); anti−β-actin (A5441) Sigma-Aldrich (St. Louis, MO). Cisplatin was purchased from Sigma-Aldrich and West-Ward Pharmaceuticals (Berkeley Heights, NJ, USA). 1-(4pyridinyl)-3-(2-quinolinyl)-2-propen-1-one (PFK15, an inhibitor of PFKFB3) was purchased from Selleck Chemicals (Houston, TX). Ribociclib (an inhibitor of CDK4/6) was purchased from Chemietek (Indianapolis, IN).
Cell culture and treatments
Rat kidney proximal tubular cells (RPTCs) were originally obtained from Dr. Ulrich Hopfer (Case Western Reserve University). RPTCs were cultured in Ham’s F-12-DMEM supplemented with 10% FBS as previously described18. RPTCs were seeded in 35-mm collagen-coated dishes to reach more than 90% confluence by the next day for treatment with 20μM cisplatin for 24 hours in the presence or absence of 10μM PFK15. Cells were analyzed by morphology for apoptosis and cell lysate was collected for biochemical analyses.
shRNA-mediated gene silencing in cells
Stable knockdown of Pfkfb3 in RPTCs was achieved by transduction at 33°C with 5 different lentiviral short hairpin RNA (shRNA) constructs that were purchased from Sigma-Aldrich (MISSION Lentiviral shRNA: TRCN0000322299 (Sh#1), TRCN0000361974 (Sh#2), TRCN0000361967, TRCN0000322296, TRCN0000361975; all in pLKO-puro vector) following the manufacturer’s protocol. Virus control cells were generated by transducing lentivirus with TRC2 pLKO-puro empty vector control plasmid DNA. For selection of transfected colonies, 2.5 μg/mL of puromycin was added to the medium. Knockdown efficiency was confirmed by Western blot analysis for protein expression. Best knockdown was obtained with TRCN0000322299 (Sh#1). These cells were named Pfkfb3-ShRNA RPTCs and used for further experiments.
Animals and cisplatin treatment
All animals used in this study were housed with a 12:12-h light-dark cycle and fed with a standard rodent diet and water in the pathogen-free animal facility at Charlie Norwood VA Medical Center. Animal experiments were conducted with a protocol approved by the Institutional Animal Care and Use Committee of Charlie Nor-wood VA Medical Center. Male C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, ME). The proximal tubule-specific Pfkfb3 knockout mice were generated by crossing Pfkfb3flox/flox mice19 with PEPCK-Cre mice20 as previously described.21 For cisplatin treatment, mice were administered with a dose of cisplatin (30 mg/kg i.p.) as previously described.22,23 For CDK4 inhibition, Ribociclib (Chemietek, Indianapolis, IN) or vehicle solution were administered by oral gavage (150 mg/kg, dissolved in citrate buffer) four hours prior to cisplatin injection as described.15
Measurement of renal function
The decline of renal function was revealed by serum creatinine and BUN as previously described.24 Briefly, blood samples were clotted at room temperature and centrifuged to collect serum. For BUN measurement, the Urea Nitrogen Detection kit from Stanbio Laboratory (Boerne, TX) was used following the manufacturer’s instructions. For serum creatinine measurement, serum samples were assessed based on Jaffe reaction with reaction mixture of NaOH and picric. BUN and serum creatinine levels were calculated based on standard curves.
Renal histopathology
Mouse kidneys were fixed with 4% paraformaldehyde and embedded with paraffin. Kidney sections of 4 μm were subjected to a standard hematoxylin-eosin(H&E) staining. Histological damage was indicated by tubular dilation/flattening, loss of brush border, sloughing of cells into tubular lumen, formation of tubular casts, tubular degeneration, and vacuolization. Tissue injury was scored by the percentage of damaged renal tubules (0, no damage; 1, <25%; 2, 25−50%; 3, 50−75%; and 4, >75%.)
Immunohistochemical analysis
Immunohistochemical staining was performed as described in our recent studies.25,26 Briefly, paraffin-embedded kidney sections were sequentially treated through deparaffinization, hydration, and antigen retrieval by incubation with 0.1M Tris-EDTA (pH 9.0) at 100°C. After subsequent incubation in 3% H2O2, 2% normal goat serum, 0.2% non-fat milk and 0.8% Triton X-100 to reduce nonspecific binding, tissue sections were exposed to anti-PFKFB3 (Abcam, Ab181861) or anti-CDK4 (Abclonal, A0366) at 4°C overnight, followed by exposure to horseradish peroxidase (HRP) -conjugated secondary antibody for 1 h at room temperature. Signals of the antigen−antibody complexes were developed with a DAB Peroxidase Substrate Kit (Vector Laboratories) following the manufacturer’s introduction.
Immunofluorescence staining
For RPTCs, the cells were fixed in 4% paraformaldehyde for 10 min. Fixed cells were incubated with 2% BSA for 1h at room temperature. The cells were then incubated with anti-PFKFB3 (ab218121, 1:200) primary antibody at 4°C overnight followed by a complete wash. The cells were further incubated with diluted secondary antibodies for 1 h. After washes, the cells were mounted on slides with a solution containing DAPI. For immunofluorescence in kidney tissues, paraffin-embedded kidney tissue sections underwent deparaffinization, rehydration, and antigen retrieval by incubation with 0.1M Tris-EDTA (pH 9.0) at 95−100°C for 1 h. After subsequent incubations with3% H2O2, 2% normal goat serum, 0.2% nonfat milk and 0.8% Triton X-100, tissue sections were exposed to primary antibodies at 4°C overnight and then Alexa-conjugated secondary antibodies. Fluorescence images were collected by fluorescence confocal microscopy (Zeiss).
Analysis of apoptosis
For kidney tissues, TUNEL assay was done with the In Situ Cell Death Detection Kit (Roche Applied Science) following the manufacturer’s instructions. Positive staining was identified by fluorescence microscopy. Ten representative fields were randomly selected in each tissue section, and the TUNEL-positive cells per mm2 was counted.
For cultured cells, apoptosis was analyzed by cell morphology, nuclear staining with Hoechst 33342, and caspase activity. Apoptotic cells showed a round-up and shrunken morphology in phase contrast images with fragmented, condensed nuclei in
Hoechst 33342 staining. The activity of caspases was measured by an enzymatic assay as described in our previous studies24, 27. Briefly, cytosolic lysate was collected with a buffer containing 1% Triton X-100. Twenty micrograms of protein were used for the enzymatic reaction to detect the cleavage of DEVD-AFC to generate free AFC. The fluorescence of free AFC was measured at excitation 360 nm/emission 530 nm before and after 1-hour reaction with a GENios plate reader (Tecan US). The caspase activity was calculated based on the standard curve as the production of nanomoles free AFC/hour/mg protein.
Immunoprecipitation and Immunoblot analysis
Immunoprecipitation analysis were performed using the Direct Magnetic IP/Co-IP Kit (Thermo Fisher, 88828) according to the manual. Briefly, PFKFB3 antibody or normal rabbit IgG (as negative control) were incubated with magnetic beads (25 μL) for 60 min. RPTCs or kidney cortex tissues were washed twice with cold PBS, and then incubated on ice with the lysis buffer from the Direct Magnetic IP/Co-IP Kit. Super-natants were collected by centrifugation. After protein measurement, 1μg/μL protein were added into the tube with antibody-coupled magnetic beads and incubated for 2 h at room temperature on a rotator. Precipitated proteins were eluted from beads and prepared for Western blot analysis. For Western blot analysis, proteins were separated by 10% SDS-PAGE and analyzed by immunoblotting using a standard method.
Statistical analysis
Statistical analysis was performed with GraphPad Prism software. The significance of the differences was determined by one-way or two-way ANOVA test. All quantitative data results are expressed as mean ± SD. P < 0.05 was considered significant. All biological experiments were repeated at least three times using independent cell cultures or individual animals (biological replications).
Results
PFKFB3 is induced during cisplatin nephrotoxicity in mice and renal tubular cells
First, we examined the expression of PFKFB3 after cisplatin treatment of cultured RPTCs in vitro and mice in vivo by immunoblot analysis. RPTCs showed two protein bands of PFKFB3, which were later confirmed by knockdown experiments. Compared with the control groups, the lower band of PFKFB3 started to increase at 6 hours of cisplatin treatment before noticeable apoptosis (data not shown) and remained elevated till 24 hours of cisplatin treatment (Fig 1A). In control mouse kidneys, PFKFB3 was barely detectable in immunoblot. Following a single injection of cisplatin (30 mg/kg), PFKFB3 began to increase at day 1 when no obvious renal function loss was detected (Supplementary Fig 1C). This induction was time-dependent and reached a higher level at day 3 after cisplatin injection (Fig 1B, C) when there was obvious renal tubular damage (Fig 1D). We further examined the localization of PFKFB3 induction in kidneys and in RPTCs by immunofluorescence (Fig 1E and F). Control RPTCs showed PFKFB3 staining in both cytosol and the nucleus. With cisplatin treatment, there was an obvious increase of nuclear PFKFB3, accompanied by a decrease of cytosolic PFKFB3 signal (Fig 1F). In mouse kidneys, PFKFB3 staining was mainly detected in the cytosol as well as the nucleus of renal tubules, and the staining was markedly increased in cisplatin-treated kidneys, especially in tubular cell nuclei (Fig 1E and Supplementary Fig 1D). Notably, PFKFB3 induction occurred in kidney tubules that were co-stained with kidney injury molecule-1 (Kim-1), a specific marker of injured proximal tubules,28 suggesting the possible involvement of PFKFB3 in tubular damage in cisplatin nephrotoxicity.
Fig 1.
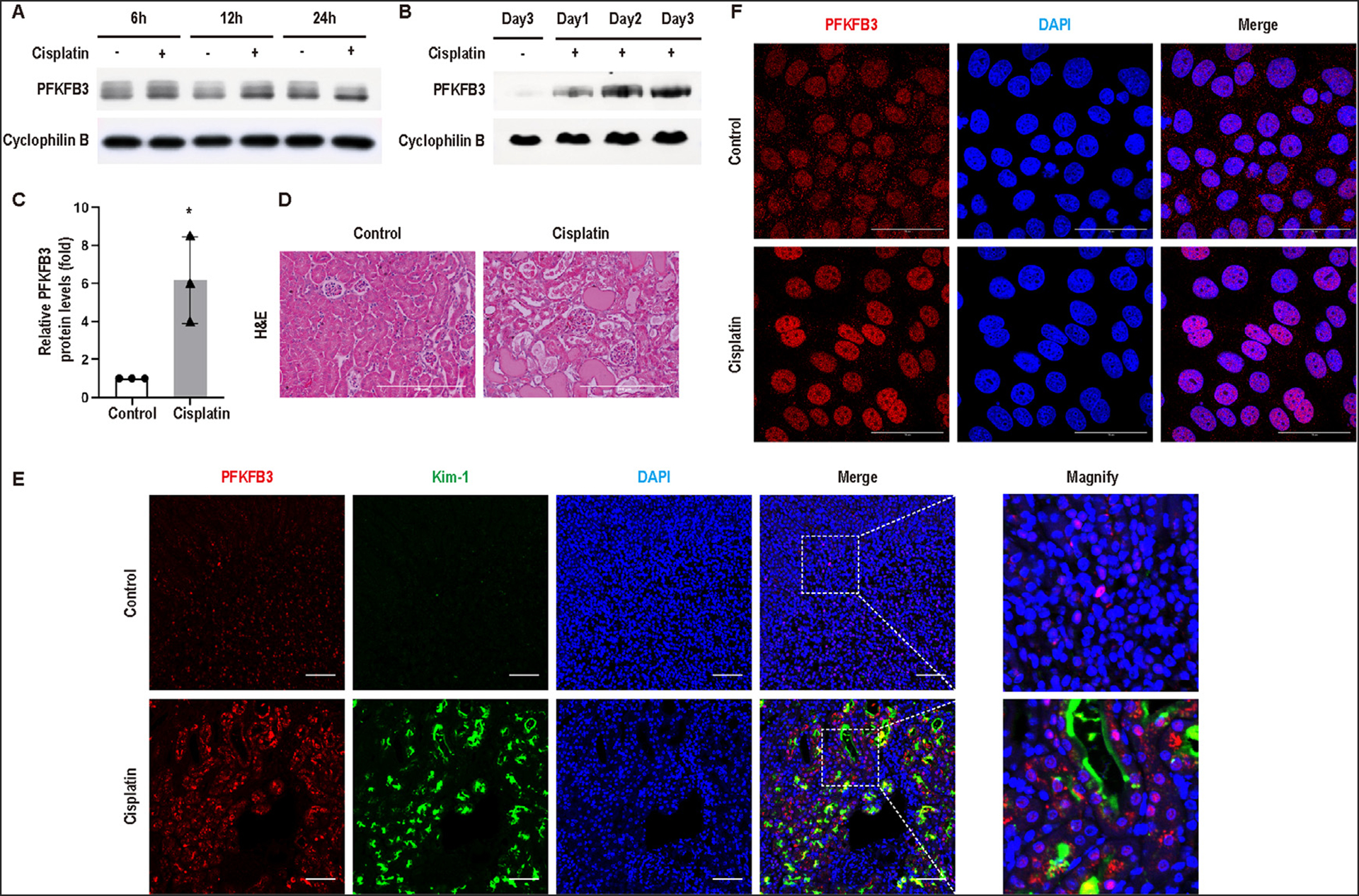
PFKFB3 is induced during cisplatin nephrotoxicity in mice and renal tubular cells. (A) Rat renal proximal tubular cells (RPTCs) were incubated with vehicle (Control) or 20μM cisplatin for 6, 12, or 24 hours. Whole cell lysates were collected for immunoblot analysis of PFKFB3. Cyclophilin B was detected as internal control. (B-E) C57BL/6 mice were injected with saline (Sham) or 30mg/kg cisplatin. Kidneys were harvested at day 1, 2, or 3 for immunoblot analysis, hematoxylin and eosin (H&E) staining, and immunofluorescence. (B) Time course of PFKFB3 induction in kidney cortex after cisplatin injection. (C) Densitometry of PFKFB3 induction at day 3 after cisplatin injection with cyclophilin B as the internal loading control. (D) Representative images of H&E staining of kidney tissues at day 3 after cisplatin injection. Scale bar = 200μm. (E) Representative images of immunofluorescence for PFKFB3 (red), Kim-1 (green) and DAPI (blue) in kidney tissues at day 3 after cisplatin injection. Scale bar = 50μm. (F) Representative images of immunofluorescence for PFKFB3 (red) and DAPI (blue) in RPTCs. Scale bar = 50μm.
Inhibition of PFKFB3 by PFK15 decreases cisplatin-induced apoptosis in renal tubular cells
To understand the role of PFKFB3 in cisplatin-induced renal tubular injury, we tested the effect of PFK15, a small molecule inhibitor of PFKFB3, on cisplatin-treated RPTCs. Twenty-four hours of treatment with 20 μM cisplatin led to massive apoptosis, which was shown by typical apoptotic morphology (cellular shrinkage and formation of apoptotic bodies, nuclear condensation and fragmentation) and caspase activation (Fig 2A, B, C). PFK15 treatment suppressed both apoptotic morphology and caspase activation during cisplatin treatment (Fig 2A, B, C). The protective effect of PFK15 was further verified by immunoblot analysis of PARP cleavage and active/cleaved caspase-3 (Fig 2D and E).
Fig 2.
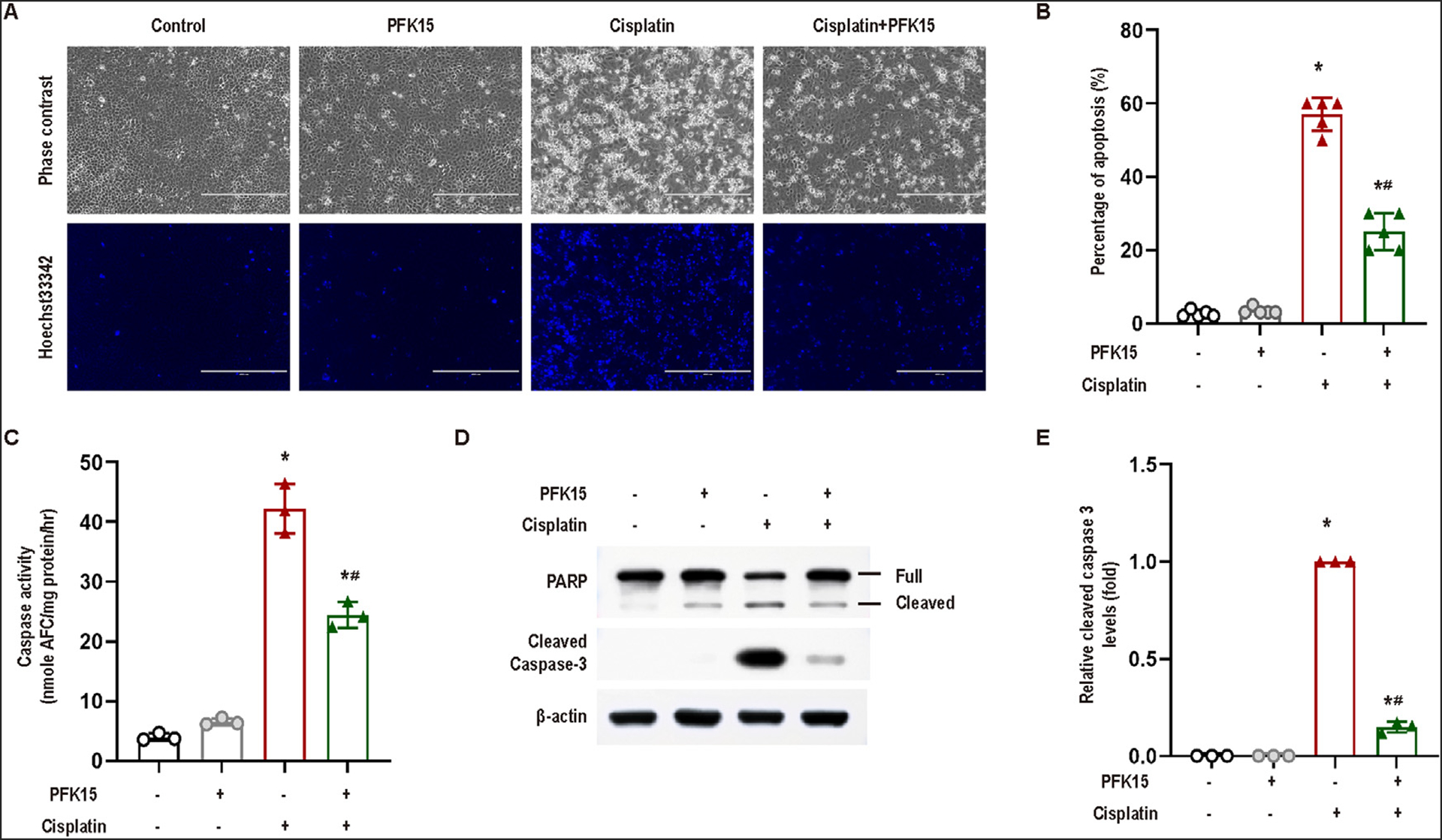
Inhibition of PFKFB3 by PFK15 decreases cisplatin-induced apoptosis in RPTCs. RPTCs were incubated with 20 μM cisplatin in the absence or presence of 10μM PFK15 for 24 hours. (A) Representative cell and nuclear images after Hoechst 33342 staining. Scale bar = 200 μm. (B) Percentage of apoptosis assessed by the cells with typical apoptotic morphologies. Data are expressed as means±SD (n = 5). (C) Caspase activity. Data are expressed as means ± SD (n = 3). (D) Immunoblots for PFKFB3, cleaved caspase-3 and poly (ADP-ribose) polymerase (PARP). β-Actin was used as the internal loading control. (E) Densitometry analysis of cleaved caspase-3, normalized with β-actin. Immunoblots are representative of at least 3 independent experiments. Quantitative data are expressed as mean ± SD; *, P < 0.05 vs control groups; #, P < 0.05 vs the cisplatin-treated group without PFK15.
Knockdown of PFKFB3 in renal tubular cells alleviates cisplatin-induced apoptosis
To further confirm the role of PFKFB3, we established stable PFKFB3 knockdown RPTC cell lines with lentiviral particles containing shRNAs against Pfkfb3, together with a negative control (NC-ShRNA) RPTC cell line as control. PFKFB3 depletion was verified in two knockdown cell lines (sh1, sh2) (Fig 3A). Following cisplatin treatment, these PFKFB3 knockdown cells showed obviously less caspase 3 cleavage or activation than negative control vector transfected cells (NC) (Fig 3B, C). Morphological analysis also showed that cisplatin induced less apoptosis in knockdown cells (Pfkfb3-ShRNA) than in negative control transfected cells (Fig 3D, E). Consistently, PFKFB3 knockdown cells had lower caspase activity after cisplatin treatment (Fig 3F).
Fig 3.
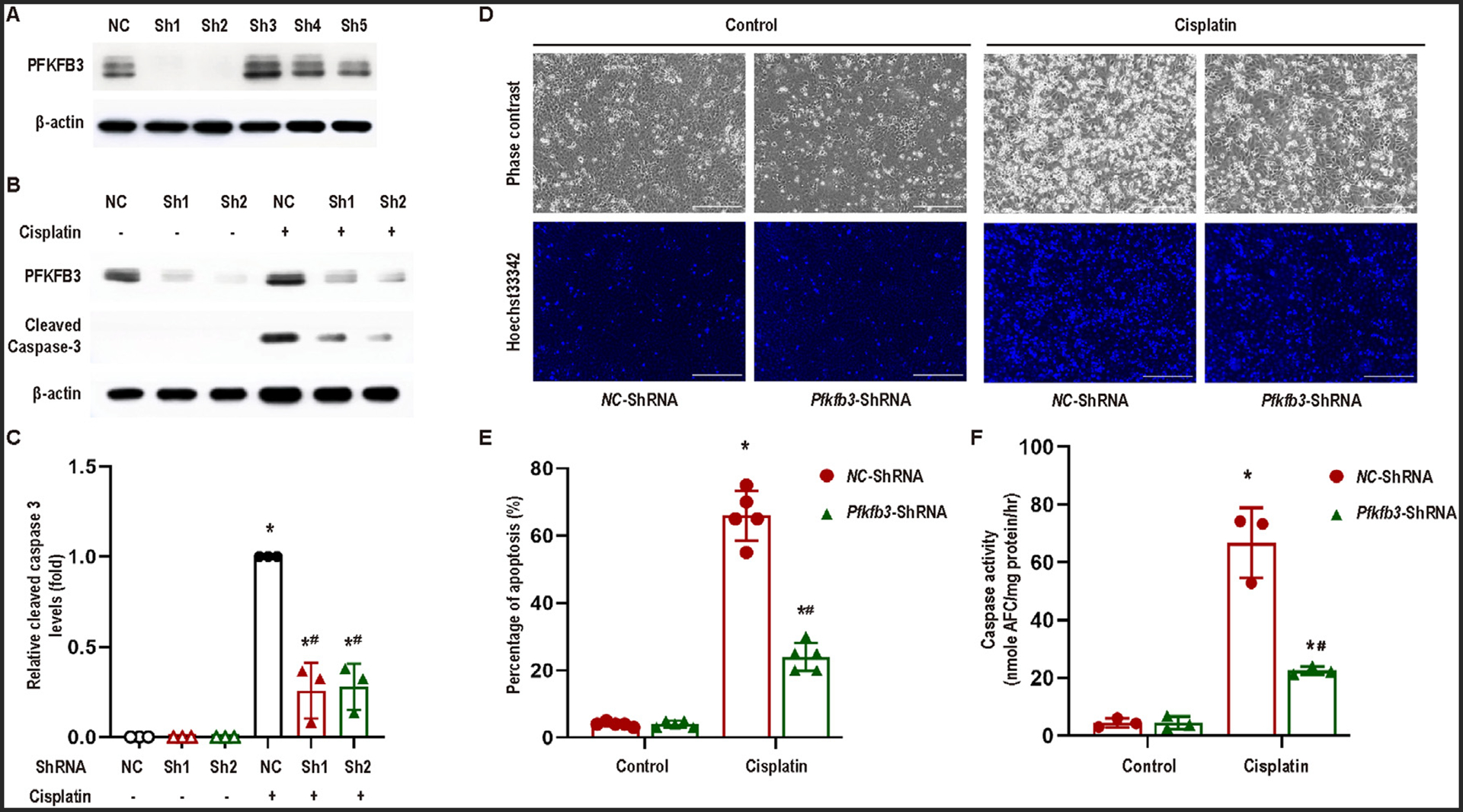
Knockdown of PFKFB3 alleviates cisplatin-induced apoptosis in RPTCs. (A) RPTCs were transfected with different Pfkfb3-ShRNAs (Sh1–5) or negative control shRNA (NC), and then selected with puromycin for 2 weeks. Whole cell lysates were collected for immunoblot analysis of PFKFB3 and β-actin. The cells stably transfected with Pfkfb3-ShRNA1 were used for subsequent experiments. (B)−(F) Pfkfb3-ShRNA and negative control (NC) RPTCs were incubated with or without 20 μM cisplatin for 24 hours. (B) Immunoblot analysis for PFKFB3 and cleaved caspase-3 in Pfkfb3-knockdown and negative control RPTCs. β-Actin was used as the internal loading control. (C) Densitometry analysis of cleaved caspase-3 normalized by β-Actin signals (n = 3). (D) Representative cell and nuclear images after Hoechst 33342 staining. Scale bar = 200 μm. (E) Percentage of apoptosis assessed morphologically (n = 5). (F) Caspase activity. Data are expressed as means ± SD, n = 3. *, P < 0.05 vs control groups; #, P < 0.05 vs. cisplatin-treated NC-ShRNA RPTCs.
Knockout of proximal tubule PFKFB3 ameliorates cisplatin-induced AKI in mice
To verify the pathological role of PFKFB3 in cisplatin-induced AKI in vivo, we established a mouse model with PFKFB3 knockout in kidney proximal tubular cells (Pfkfb3-KO) by crossing PEPCK-Cre20 mice with Pfkfb3flox/flox mice19 (Supplementary Fig A). The deletion of Pfkfb3 in kidney was verified by PCR-based genotyping (Supplementary Fig B) and immunoblotting (Fig 4A). We further confirmed the depletion of PFKFB3 in proximal tubules in Pfkfb3-KO kidney by immunohistochemical staining. As shown in Fig 4B, PFKFB3 staining was observed in the nucleus and cytosol of renal tubular cells in Pfkfb3-WT mouse kidney, which was mainly induced in tubular cell nucleus following cisplatin treatment. However, PFKFB3 was absent in the majority of kidney tubular cells in Pfkfb3-KO mice (Fig 4B).
Fig 4.

Knockout of kidney proximal tubule PFKFB3 ameliorates cisplatin induced AKI in mice. Pfkfb3-WT and Pfkfb3-KO littermate mice were intraperitoneally injected with 30 mg/kg cisplatin or saline. Mice were euthanized at day 3 after injection to collect kidney tissues and blood samples. (A) Immunoblots of PFKFB3 in kidney cortex with cyclophilin B as internal loading control. (B) Immunohistochemical staining of PFKFB3 in renal cortex. Scale bar = 200 μm. (C) Representative immunoblot of cleaved caspase-3 in kidney cortical lysates from Pfkfb3-WT and Pfkfb3-KO mice with or without cisplatin treatment. Cyclophilin B was detected as internal loading control. (D) Representative H&E staining of kidney tissues. Scale bar = 200 μm. (E) Densitometry of cleaved caspase-3. The signal was normalized to the internal loading control. (F) Blood urea nitrogen. (G) Representative images of TUNEL staining. Scale bar = 200 μm. (H) Serum creatinine. (I) Histology scores of tubular injury. (J) Quantification of TUNEL-positive cells in kidney tissues. Data are expressed as means ±SD; n =5 mice for each group. *, P < 0.05 vs wild-type control groups; #, P < 0.05 vs cisplatin-treated wild-type group.
Under control conditions, Pfkfb3-KO mice and Pfkfb3-WT mice had similarly low levels of blood urea nitrogen (BUN) and serum creatinine (SCr) at the experiment age of 2−3 months. After cisplatin treatment, Pfkfb3-WT mice showed increases in both BUN and SCr, which were significantly suppressed in Pfkfb3-KO mice (Fig 4F and H). Consistently, cisplatin induced obvious renal tubular damage in wild-type mice showing loss of brush boarder, tubular dilation, protein cast formation, and totally lysed tubules, which was partially ameliorated in Pfkfb3-KO mice (Fig 4D). This conclusion is supported by semiquantification of tubular damage in these animals (Fig 4I). We further examined renal apoptosis by transferase dUTP nick-end labeling (TUNEL) assay (Fig 4G) and immunoblotting of caspase-3 (Fig 4C). Pfkfb3-KO mice showed significantly fewer TUNEL-positive cells in kidneys (Fig 4G and J) and less active/cleaved caspase-3 (Fig 4C and E) after cisplatin treatment than their wild-type littermates. For regulated necrosis, we analyzed the phosphorylation of RIP1, RIP3, and MLKL, molecular hallmarks of necroptosis in cisplatin nephrotoxicity.29 As shown in Supplementary Fig 2, cisplatin treatment led to a remarkable phosphorylation of RIP3 and MLKL in kidneys at day 3, but the phosphorylation of RIP1 decreased. Cispaltin induced RIP1 at earlier time-points, eg. 6 hours (unshown data). Regardless, Pfkfb3-KO in proximal tubules did not affect the phosphorylation or activation of these necroptosis mediators. We further monitored animal death after cisplatin treatment (Supplementary Fig 3). Wild-type mice had 10% death at day 3 after cisplatin injection that increased to 40% at day 4 and to 100% at day 5. Animal death in Pfkfb3-KO mice started at day 4 but also reached 100% at day 5. Thus, Pfkfb3-KO in proximal tubules delayed, but did not prevent, animal death. The limited effect of Pfkfb3-KO on animal survival was most likely due to the severe injury induced by a high dose of cisplatin in this study.
CDK4/Rb signaling in cisplatin-induced apoptosis in RPTCs
After cisplatin treatment, PFKFB3 was remarkably induced in cell nuclei in renal tubules (Fig 1E), suggesting a novel role of PFKFB3 in the nucleus other than a glycolysis enzyme in the cytoplasm. Interestingly, there is evidence that PFKFB3 may promote cell cycle via cyclin-dependent kinases (CDKs) in the nucleus in cancer cells.12,13 Moreover, CDK4/6 have been implicated in cisplatin-induced AKI or nephrotoxicity.15–17 Thus, we hypothesized that PFKFB3 may promote kidney tubular cell death during cisplatin treatment by regulating CKD4 in the nucleus. To test this, we initially examined the change of CDK4 protein in cisplatin-treated RPTCs. As shown in Fig 5A, cisplatin induced CDK4 expression at 6 and 12 hours, and this induction decreased at 24 hours. Rb is an important downstream target of CDK4/6, which is inactivated following CDK4/6-medaited phosphorylation.30 In RPTCs, phosphorylated Rb (p-Rb) increased after 12 and 24 hours of cisplatin treatment. To verify the role of CDK4 in cisplatin-induced cell death in RPTCs, we tested the effect of Ribociclib, a CDK4/6 inhibitor. As shown inFig 5B and C, Ribociclib significantly reduced apoptosis during cisplatin treatment. Consistently, Ribociclib suppressed cisplatin-induced caspase activation as shown by the appearance of active/cleaved caspase-3 (Fig 5D and E). These results support a role of CKD4/Rb signaling in cisplatin-induced cell death in renal proximal tubular cells.
Fig 5.

CDK4/Rb signaling in cisplatin-induced apoptosis in RPTCs. Whole cell lysates were collected for immunoblotting. (A) RPTCs were incubated with or without 20 μM cisplatin for 6, 12, or 24 hours to collect whole cell lysate for immunoblot analysis of CDK4, phosphorylated-Rb, and cyclophilin B (internal control). (B −E) RPTCs were incubated with 20 μM cisplatin in the absence or presence of Ribociclib for 24 hours. (B) Representative cell and nuclear images after Hoechst 33342 staining. Scale bar = 200μm. (C) Percentage of apoptosis (n = 3). (D) Representative immunoblot of cleaved caspase-3 in RPTCs. β-Actin was used as the internal loading control. (E) Densitometry of cleaved caspase-3 (n=3). The signal of cleaved caspase-3 was normalized with β-Actin. Quantitative data are expressed as means ±SD; *, P < 0.05 vs control groups; #, P < 0.05 vs cisplatin-treated group without Ribociclib.
Role of CDK4 signaling in cisplatin-induced AKI in mice
In mice, CDK4 started to rise at day 1 after cisplatin injection, peaked at day 2, and then decreased at day 3. p-Rb showed a mild induction at day 2 following cisplatin treatment and a marked increase at day 3 (Fig 6A). Immunohistochemistry demonstrated a remarkable CDK4 increase and accumulation in the nucleus of renal tubular cells after cisplatin treatment of mice (Fig 6B), indicative of the activation of CDK4.31
Fig 6.

Role of CDK4 signaling in cisplatin-induced AKI in mice. (A) & (B) C57BL/6 mice were injected with saline (Control) or 30 mg/kg cisplatin to collect kidneys at day 1, 2, or 3. (A) Immunoblot analysis for CDK4 and phosphorylated-Rb in kidney cortical lysates with cyclophilin B as internal control. (B-G) Pfkfb3-WT mice were given 150 mg/kg Ribociclib 4 hours before intraperitoneal injection of 30 mg/kg cisplatin or saline. Mice were euthanized at day 3 after injection to collect kidney tissues for histology and immunoblot analysis. (B) Immunohistochemical staining of CDK4 in kidneys following cisplatin or saline injection. The lower panels were ampli-fied from the boxed areas of the upper panels. Scale bar = 200 μm. (C) Immunoblot analysis for phosphorylated-Rb, cleaved caspase-3 and β-actin. (D) Blood urea nitrogen. (E) Serum creatinine. (F) Representative histology of H&E staining. Scale bar = 200 μm. (G) Histology scores of tubular injury. Qantitative data are expressed as means ±SD; *, P < 0.05 vs control groups; #, P < 0.05 vs only cisplatin-treated groups.
We then tested the effect of Ribociclib on cisplatin nephrotoxicity in mice. Ribociclib suppressed p-Rb expression during cisplatin treatment, indicating the inhibitory effect of Ribociclib on CDK4 (Fig 6C). Importantly, Ribociclib showed protective effects against cisplatin-induced AKI as indicated by better renal function (Fig 6D and E), less tubular damage (Fig 6F and G) and less active/cleaved caspase 3 (Fig 6C). These results indicate that CDK4 activation plays a significant role in cisplatin-induced AKI.
PFKFB3 mediates CDK4 activation in cisplatin-induced AKI
To investigate whether the activation of CDK4 signaling is dependent on PFKFB3, we examined the effect of PFKFB3 knockdown. In negative control vector-transfected RPTCs, CDK4 was induced at 24 hours of cisplatin treatment, while this induction was obviously suppressed in PFKFB3 knockdown cells (Fig 7A). Meanwhile, p-Rb was increased at 24 hours of cisplatin treatment and its induction was suppressed in PFKFB3 knockdown cells (Fig 7A). In vivo, we detected a remarkable p-Rb induction in Pfkfb3-WT mice kidneys at day 3 of cisplatin treatment, but not in KO mice (Fig 7B), further indicating the regulation of CDK4/Rb pathway by PFKFB3.
Fig 7.
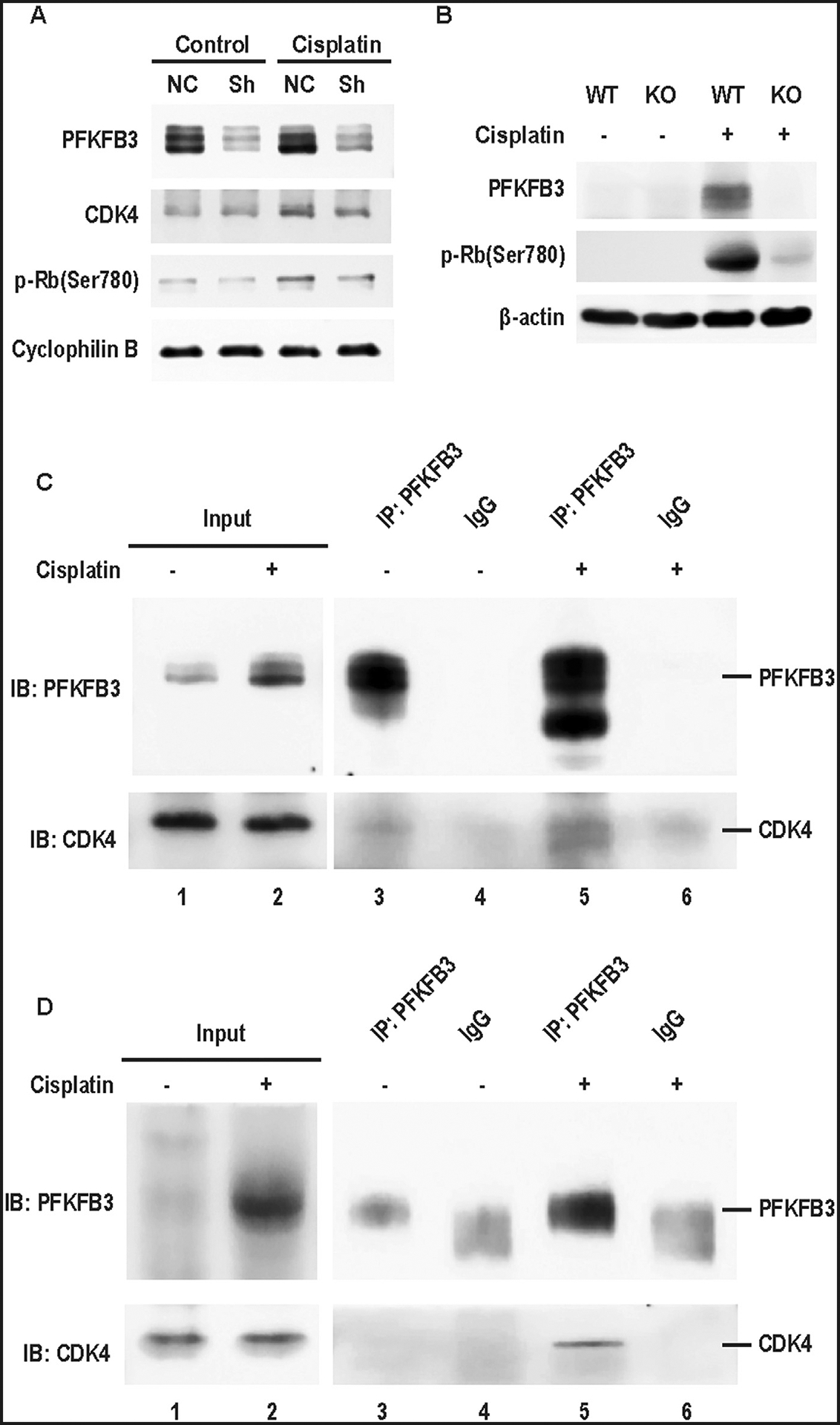
CDK4 activation is dependent on PFKFB3 in cisplatin-induced AKI. (A) Pfkfb3-knockdown (Sh) and negative control (NC) RPTCs were incubated with or without 20μM cisplatin for 24 hours. Whole cell lysates were collected for immunoblotting of PFKFB3, CDK4 and phosphorylated-Rb, and cyclophilin B (internal control). (B and D) Pfkfb3-WT and Pfkfb3-KO littermate mice were intraperitoneally injected with 30 mg/kg cisplatin or saline. Mice were euthanized at day 3 after cisplatin injection to collect kidney cortex tissues for immunoblot and immunoprecipitation analysis. (B) Immunoblots of phosphorylated-Rb and β-actin in Pfkfb3-WT and Pfkfb3-KO littermate mice with or without cisplatin treatment. (C) RPTCs were incubated with or without 20 μM cisplatin for 24 hours to collect whole cell lysate for immunoprecipitation with anti-PFKFB3 antibody or IgG (negative control). Immunoprecipitates were analyzed by SDS-PAGE and immunoblotting with indicated antibodies. (D) Lysates of mouse kidney cortex tissues were immunoprecipitated with anti-PFKFB3 antibody or IgG (negative control). Immunoprecipitates were analyzed by SDS-PAGE and immunoblotting with indicated antibodies.
PFKFB3 was reported to interact with CDK4 in cancer cells.12 To investigate the mechanism of PFKFB3 regulation of CDK4 in cisplatin nephrotoxicity, we analyzed their interaction by co-immunoprecipitation experiments. In control RPTCs, PFKFB3 antibodies pulled down a small amount of CDK4, but it pulled down much more CKD4 from the lysate of cisplatin-treated cells (Fig 7C: lanes 3, 5). Similarly, PFKFB3/CDK4 co-IP was minimal in control kidney lysate, but it became obvious in cisplatin-treated kidney lysate (Fig 7D: lanes 3, 5). Thus, PFKFB3 may directly interact with CDK4 for its activation in cisplatin-induced AKI.
Discussion
In this study, we have demonstrated the first evidence of a role of PFKFB3 in cisplatin-induced AKI or nephrotoxicity using both in vitro cell culture and in vivo mouse models. In these models, inhibition of PFKFB3 pharmacologically and genetically protected renal tubular cells and kidneys against cisplatin toxicity, indicating an injurious function of PFKFB3. Mechanistically, although PFKFB3 is known as an enzyme in glycolysis in the cytosol, our results show that PFKFB3 is mainly induced in the nucleus in renal tubular cells during cisplatin treatment. Upon induction in cell nucleus, PFKFB3 may interact with CDK4 to promote its activation, leading to the phosphorylation and inactivation of Rb with ensuing tubular cell injury and death. Therapeutically, we proved the protective effects of the PFKFB3 inhibitor PFK15 and the CDK4 inhibitor Ribociclib, suggesting a kidney protective potential of these inhibitors in cisplatin chemotherapy.
Kidney injury and repair is associated with changes in metabolism in renal tubular cells.32–37 Especially, proximal tubules, the main injury and repair site, normally have none or very limited capacity of glycolysis, but after injury, they lose functional mitochondria and may regain the ability of glycolysis for bioenergetics and cell viability.33,37 In view of these findings, we initially speculated that PFKFB3, well known as an enzyme in glycolysis, might contribute to the metabolic shift in kidney injury and repair. However, in our study PFKFB3 was mainly induced in the nucleus during cisplatin treatment of renal tubular cells in vitro and mouse in vivo, suggesting a non-canonical function of PFKFB3 that is separate from glycolysis in the cytosol. While this was surprising, PFKFB3 was recently implicated in cancer cell proliferation.38 Our subsequent experiments showed that PFKFB3 may interact with and activate CDK4, a key cyclin-dependent protein kinase for G1/S progression in the cell cycle, resulting in Rb phosphorylation and inactivation both in vitro and in vivo (Fig 7). Interestingly, previous studies demonstrated the protective effects of CDK inhibitors against cisplatin nephrotoxicity15–17,39,40 as well as other forms of AKI.17,41,42 Consistently, the CDK4 inhibitor Ribociclib ameliorated cisplatin-induced tubular cell death and kidney injury in our present study (Fig. 5 and 6). Collectively, the results indicate PFKFB3 is induced mainly in the nucleus of renal tubular cells during cisplatin nephrotoxicity, where it interacts with and activates CDK4 to promote tubular cell death and AKI.
The mechanism underlying the protective effects of CDK inhibitors in AKI is not fully understood, but it may involve the induction of G1/S cell cycle arrest and therefore allow more time to repair damaged DNA before progressing into S phase and subsequent G2/M phase for mitosis.15–17,39,40 In this connection, p21, a cell-cycle inhibitory protein, was induced in renal tubular cells in AKI, and p21-knockout mice developed much more severe kidney damage than wild-type mice after cisplatin injection, which may involve cell cycle progression via CDK2/E2F1 activation.40,41,43,44 In our study, we detected PFKFB3 induction in tubular cell nucleus, CDK4 activation, and the interaction between PFKFB3 and CKD4 during cisplatin treatment. In addition, inhibition of either PFKFB3 or CDK4 reduced cisplatin injury, suggesting that PFKFB3 may act upstream of CDK4 to promote cell cycle activation and sensitize tubular cells to injury. It is unclear why both cell cycle inhibitors (eg, p21) and activators (eg, PFKFB3/CDK4) are induced in renal tubular cells during cisplatin nephrotoxicity. One possibility is that p21 is induced in the cells that survive the injury whereas PFKFB3/CDK4 is induced in those that die. Regardless, these observations support the idea that cell cycle status is a determinant in tubular cell death during AKI.
In the cell cycle, CDK4 mainly phosphorylates Rb to prevent its inhibitory binding to E2F, a family of transcription factors for G1/S transition. In the absence of Rb binding, E2F induces the expression of various genes including those for DNA replication.45–47 In the present study, we detected Rb phosphorylation following PFKFB3 induction and CDK4 activation during cisplatin treatment (Fig. 5A, 6A, C, and 7A). Currently, the role of Rb in AKI is not very clear. In this regard, Kim and colleagues recently reported that ablation of Rb from renal tubule cells did not change the severity of cisplatin-induced kidney injury, but it diminished the protective effect of Ribociclib.15 While these results support a critical role of Rb in the protective effect of CDK4 inhibition, CDK4 may also induce apoptosis through Rb-independent pathways. For example, CDK4 activity is closely related to mitochondrial biogenesis in Drosophila, and excess CDK4 activity results in inefficient oxidative phosphorylation (OXPHOS) coupling and increased mitochondrial superoxide production, cumulating in cell injury and death.48
In our study, specific knockout of PFKFB3 from kidney proximal tubules protected mice from cisplatin nephrotoxicity. Similar protective effect was shown for the CDK4 inhibitor Ribociclib. Together, these results suggest a therapeutic strategy of kidney protection by targeting the PFKFB3/CDK4 pathway. As we proposed in 2008,1 an effective strategy of kidney protection in cisplatin chemotherapy should not diminish the anti-cancer efficacy of cisplatin. In this regard, it is well-recognized that cancer cells tend to use glycolysis for energy production even in the presence of oxygen, a phenomenon called Warburg effect. Glycolysis promotes cell proliferation, tumorigenesis and malignancy progression.13,49,50 Therefore, inhibition of PFKFB3 may suppress glycolysis and associated cell proliferation in cancers. Indeed, there are reports about the anti-cancer effects of PFKFB3 inhibitors.51–53 More over, PFKFB3 inhibitors may enhance the chemotherapeutic efficacy of cisplatin in tumor treatment.54,55 In our study, PFK15 (PFKFB3 inhibitor) protected renal tubular cells during cisplatin treatment in vitro (Fig 2), but it didn’t show significant protective effects against cisplatin-induced AKI in mice (data not shown). The possible reasons of the lack of effect in mice may include the nonspecific side effect of PFK15 in addition to PFKFB3 suppression and the potential toxicity of PFKFB3 inhibition in other renal cells. For example, some cells in kidneys, such as distal tubule cells56 and macrophage,57 rely mostly on glycolysis for energy production. By inhibiting PFKFB3 and therefore blocking glycolysis, PFK15 may induce toxicity in these cells causing kidney damage. Therefore, it is important to identify PFKFB3 inhibitors that may suppress its effect on CKD4 but do not block its enzymatic activity for glycolysis. For this purpose, it would be interesting to further investigate the interaction between PFKFB3 and CKD4, which may lead to the discovery of chemicals or peptides that block the molecular interaction and therefore CDK4 activation without affecting the enzymatic activity of PFKFB3 in glycolysis.
The other therapeutic strategy is to directly inhibit CDK4/6. Previous studies reported the involvement of CDK4/6 in kidney injury.15–17,58 Notably, inhibitors of CDK4/6 showed remarkable protective effects in both cisplatin nephrotoxicity15–17 and renal ischemia-reperfusion injury.17 Consistently, in our study the CDK4/6 inhibitor Ribociclib ameliorated cisplatin-induced apoptosis in RPTCs and attenuated cisplatin nephrotoxicity in mice. CDK4/6 inhibitors, including Ribociclib, have been used for chemotherapy of various tumor types, such as breast cancer,59–61 non−small cell lung cancer,62 renal cell carcinoma,63 and metastatic castration-resistant prostate cancer.64 Thus, CDK4/6 inhibitors, when used in combination with cisplatin, may protect kidneys during chemotherapy while enhancing the anticancer efficacy of cisplatin.
In summary, we have demonstrated the first evidence of the involvement of PFKFB3 in acute nephrotoxicity of cisplatin. Mechanistically, PFKFB3 is mainly induced in the nucleus in kidney tubule cells during cisplatin treatment, where it interacts with and activates CDK4, resulting in Rb phosphorylation and inactivation and tubular cell death. The work highlights PFKFB3/CDK4 as a potential therapeutic target for kidney protection in cisplatin-mediated chemotherapy.
Supplementary Material
At A Glance Commentary.
Wen L, et al.
Background.
Cisplatin is a widely used chemotherapy drug, but it induces nephrotoxicity. The underlying mechanism is not well-understood, and effective kidney protective therapeutics are not available. PFKFB3 is known as a glycolysis enzyme, and its involvement in kidney diseases is unclear.
Translational Significance.
We demonstrate that PFKFB3 is induced in cell nucleus of kidney tubules during cisplatin treatment, where it interacts with and activates CDK4, leading to Rb phosphorylation and inactivation with ensuing tubular cell death. Therapeutically, the work highlights PFKFB3/CDK4 as a potential target for kidney protection during chemotherapy in cancer patients.
Acknowledgments
We thank Dr. Yuqing Huo for providing Pfkfb3-floxed mice for establishing the Pfkfb3-KO mouse model. Z.D. is a recipient of a Senior Research Career Scientist award of the Department of Veterans Administration of USA.
This study was supported partially by grants from the National Institutes of Health of USA (DK087843, DK058831), and Department of Veterans Administration of USA (BX000319).
Abbreviations:
- AKI
acute kidney injury
- BUN
blood urea nitrogen
- CKD
chronic kidney disease
- CDKs
cyclin-dependent kinases
- CDK4
cyclin-dependent kinase 4
- F2,6P2
fructose-2,6-bisphosphate
- Kim-1
kidney injury molecule-1
- OXPHOS
oxidative phosphorylation
- PFK-1
6-phosphofructo-1-kinase
- PFK15
1-(4pyridinyl)-3-(2-quinolinyl)-2-propen-1-one
- PFKFB3
6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3
- p-Rb
phosphorylated Rb
- Rb
retinoblastoma tumor suppressor
- RPTCs
rat renal proximal tubular cells
- SCr
serum creatinine
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling
Footnotes
All authors have read the journal’s policy on disclosure of potential conflicts of interest and declared no conflict of interest. L.W. and Z.D. contributed to the conceptualization, design and outline of this study; Q. W., M.L., and Y.H. provided mice, L.W. performed experiments; L.W., Q. W., M.L., and Z.D. interpreted results of experiments; L.W. and Q.W. prepared the original draft with figures. L.W., Q.W., S.L., X.H., G.D., Y.L. and Z.D. contributed to the revision and editing. All authors have read the journal’s authorship agreement, reviewed and approved the manuscript.
The data reported in this study are available from the corresponding author upon request. Source data are provided with this paper.
Supplementary materials
Supplementary material associated with this article can be found in the online version at doi:10.1016/j.trsl.2022.10.001.
References
- 1.Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int 2008;73:994–1007. [DOI] [PubMed] [Google Scholar]
- 2.Holditch SJ, Brown CN, Lombardi AM, Nguyen KN, Edelstein CL. Recent advances in models, mechanisms, biomarkers, and interventions in cisplatin-induced acute kidney injury. Int J Mol Sci 2019;20:3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sears S, Siskind L. Potential therapeutic targets for cisplatin-induced kidney injury: lessons from other models of AKI and fibrosis. J Am Soc Nephrol 2021;32:1559–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang C, Cai J, Yin XM, Weinberg JM, Venkatachalam MA, Dong Z. Mitochondrial quality control in kidney injury and repair. Nat Rev Nephrol 2021;17:299–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Curry JN, McCormick JA. Cisplatin-induced kidney injury: delivering the goods. J Am Soc Nephrol 2022;33:255–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller R, Tadagavadi R, Ramesh G, Reeves W. Mechanisms of cisplatin nephrotoxicity. Toxins 2010;2:2490–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inhibition of 6-phosphofructo-2-kinase (PFKFB3) induces autophagy as a survival mechanism, Cancer Metab, 2, 2014, 2.https://cancerandmetabolism.biomedcentral.com/articles/10.1186/2049-3002-2-2 Accessed January 23, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alvarez R, Mandal D, Chittiboina P. Canonical and non-canonical roles of PFKFB3 in brain tumors. Cells 2021;10:2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song C, Wang S, Fu Z, et al. IGFBP5 promotes diabetic kidney disease progression by enhancing PFKFB3-mediated endothelial glycolysis. Cell Death Dis 2022;13:340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakakibara R, Kato M, Okamura N, et al. Characterization of a human placental fructose-6-phosphate, 2-kinase/fructose-2,6-bisphosphatase. J Biochem 1997;122:122–8. [DOI] [PubMed] [Google Scholar]
- 11.Shi WK, Zhu XD, Wang CH, et al. PFKFB3 blockade inhibits hepatocellular carcinoma growth by impairing DNA repair through AKT. Cell Death Dis 2018;9:428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jia W, Zhao X, Zhao L, et al. Non-canonical roles of PFKFB3 in regulation of cell cycle through binding to CDK4. Oncogene 2018;37:1685–98. [DOI] [PubMed] [Google Scholar]
- 13.Yalcin A, Clem BF, Simmons A, et al. Nuclear targeting of 6-phosphofructo-2-kinase (PFKFB3) increases proliferation via cyclin-dependent kinases. J Biol Chem 2009;284:24223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen W, Shen Y, Fan J, et al. IL-22-mediated renal metabolic reprogramming via PFKFB3 to treat kidney injury. Clin Transl Med 2021;11:e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim JY, Jayne LA, Bai Y, et al. Ribociclib mitigates cisplatin-associated kidney injury through retinoblastoma-1 dependent mechanisms. Biochem Pharmacol 2020;177:113939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pabla N, Gibson AA, Buege M, et al. Mitigation of acute kidney injury by cell-cycle inhibitors that suppress both CDK4/6 and OCT2 functions. Proc Natl Acad Sci U S A 2015;112:5231–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DiRocco DP, Bisi J, Roberts P, et al. CDK4/6 inhibition induces epithelial cell cycle arrest and ameliorates acute kidney injury. Am J Physiol Renal Physiol 2014;306: F379–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woost PG, Orosz DE, Jin W, et al. Immortalization and characterization of proximal tubule cells derived from kidneys of spontaneously hypertensive and normotensive rats. Kidney Int 1996;50:125–34. [DOI] [PubMed] [Google Scholar]
- 19.Xu Y, An X, Guo X, et al. Endothelial PFKFB3 plays a critical role in angiogenesis. Arterioscler Thromb Vasc Biol 2014;34:1231–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rankin EB, Tomaszewski JE, Haase VH. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res 2006;66:2576–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao Y, Zhang X, Wang L, et al. PFKFB3-mediated endothelial glycolysis promotes pulmonary hypertension. Proc Natl Acad Sci U S A 2019;116:13394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pabla N, Dong G, Jiang M, et al. Inhibition of PKCdelta reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer. J Clin Invest 2011;121:2709–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei Q, Dong G, Yang T, Megyesi J, Price PM, Dong Z. Activation and involvement of p53 in cisplatin-induced nephrotoxicity. Am J Physiol Renal Physiol 2007;293: F1282–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei Q, Sun H, Song S, et al. MicroRNA-668 represses MTP18 to preserve mitochondrial dynamics in ischemic acute kidney injury. J Clin Invest 2018;128:5448–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu S, Hu X, Ma Z, et al. p53 in proximal tubules mediates chronic kidney problems after cisplatin treatment. Cells 2022;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Livingston MJ, Shu S, Fan Y, et al. Tubular cells produce FGF2 via autophagy after acute kidney injury leading to fibroblast activation and renal fibrosis. Autophagy 2022: 1–22. 10.1080/15548627.2022.2072054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song Z, Zhu J, Wei Q, Dong G, Dong Z. Canagliflozin reduces cisplatin uptake and activates Akt to protect against cisplatin-induced nephrotoxicity. Am J Physiol Renal Physiol 2020;318:F1041–F52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han WK, Bailly V, Abichandani R, Thadhani R, Bonventre JV. Kidney Injury Molecule-1 (KIM-1): a novel biomarker for human renal proximal tubule injury. Kidney Int 2002;62(1):237–44. [DOI] [PubMed] [Google Scholar]
- 29.Landau SI, Guo X, Velazquez H, et al. Regulated necrosis and failed repair in cisplatin-induced chronic kidney disease. Kidney Int 2019;95:797–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Topacio BR, Zatulovskiy E, Cristea S, et al. Cyclin D-Cdk4,6 drives cell-cycle progression via the retinoblastoma protein’s C-terminal helix. Mol Cell 2019;74:758–70. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tong W, Pollard JW. Progesterone inhibits estrogen-induced cyclin D1 and cdk4 nuclear translocation, cyclin E- and cyclin A-cdk2 kinase activation, and cell proliferation in uterine epithelial cells in mice. Mol Cell Biol 1999;19:2251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou HL, Zhang R, Anand P, et al. Metabolic reprogramming by the S-nitroso-CoA reductase system protects against kidney injury. Nature 2019;565:96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lan R, Geng H, Singha PK, et al. Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J Am Soc Nephrol 2016;27:3356–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wen L, Li Y, Li S, Hu X, Wei Q, Dong Z. Glucose metabolism in acute kidney injury and kidney repair. Front Med (Lausanne) 2021;8:744122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Verissimo T, Faivre A, Rinaldi A, et al. Decreased renal gluconeogenesis is a hallmark of chronic kidney disease. J Am Soc Nephrol 2022;33:810–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Osada Y, Nakagawa S, Ishibe K, et al. Antibiotic-induced microbiome depletion alters renal glucose metabolism and exacerbates renal injury after ischemia-reperfusion injury in mice. Am J Physiol Renal Physiol 2021;321:F455–F65. [DOI] [PubMed] [Google Scholar]
- 37.van der Rijt S, Leemans JC, Florquin S, Houtkooper RH, Tammaro A. Immunometabolic rewiring of tubular epithelial cells in kidney disease. Nat Rev Nephrol 2022;18:588–603. [DOI] [PubMed] [Google Scholar]
- 38.Kotowski K, Rosik J, Machaj F, et al. Role of PFKFB3 and PFKFB4 in cancer: genetic basis, impact on disease development/progression, and potential as therapeutic targets. Cancers (Basel) 2021;13:909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang L, Zhang Y, Yuan L, Liu C, Fu L, Mei C. Cyclin-dependent kinase inhibitor p18INK4c is involved in protective roles of heme oxygenase-1 in cisplatin-induced acute kidney injury. Int J Mol Med 2014;34:911–7. [DOI] [PubMed] [Google Scholar]
- 40.Price PM, Yu F, Kaldis P, et al. Dependence of cisplatin-induced cell death in vitro and in vivo on cyclin-dependent kinase 2. J Am Soc Nephrol 2006;17:2434–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Megyesi J, Andrade L, Vieira JM Jr. , Safirstein RL, Price PM. Positive effect of the induction of p21WAF1/CIP1 on the course of ischemic acute renal failure. Kidney Int 2001;60:2164–72. [DOI] [PubMed] [Google Scholar]
- 42.Bhatt K, Wei Q, Pabla N, et al. MicroRNA-687 induced by hypoxia-inducible factor-1 targets Phosphatase and Tensin homolog in renal ischemia-reperfusion injury. J Am Soc Nephrol 2015;26:1588–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu F, Megyesi J, Safirstein RL, Price PM. Involvement of the CDK2-E2F1 pathway in cisplatin cytotoxicity in vitro and in vivo. Am J Physiol Renal Physiol 2007;293:F52–9. [DOI] [PubMed] [Google Scholar]
- 44.Hodeify R, Tarcsafalvi A, Megyesi J, Safirstein RL, Price PM. Cdk2-dependent phosphorylation of p21 regulates the role of Cdk2 in cisplatin cytotoxicity. Am J Physiol Renal Physiol 2011;300(5):F1171–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rubin SM, Sage J, Skotheim JM. Integrating Old and New Paradigms of G1/S Control. Mol Cell 2020;80:183–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goel S, Bergholz JS, Zhao JJ. Targeting CDK4 and CDK6 in cancer. Nat Rev Cancer 2022;22:356–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.VanArsdale T, Boshoff C, Arndt KT, Abraham RT. Molecular pathways: targeting the cyclin D-CDK4/6 axis for cancer treatment. Clin Cancer Res 2015;21:2905–10. [DOI] [PubMed] [Google Scholar]
- 48.Icreverzi A, de la Cruz AF, Van Voorhies WA, Edgar BA. Drosophila cyclin D/Cdk4 regulates mitochondrial biogenesis and aging and sensitizes animals to hypoxic stress. Cell Cycle 2012;11:554–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yalcin A, Clem BF, Imbert-Fernandez Y, et al. 6-Phosphofructo-2-kinase (PFKFB3) promotes cell cycle progression and suppresses apoptosis via Cdk1-mediated phosphorylation of p27. Cell Death Dis 2014;5:e1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bartrons R, Rodriguez-Garcia A, Simon-Molas H, Castano E, Manzano A, Navarro-Sabate A. The potential utility of PFKFB3 as a therapeutic target. Expert Opin Ther Targets 2018;22:659–74. [DOI] [PubMed] [Google Scholar]
- 51.Li HM, Yang JG, Liu ZJ, et al. Blockage of glycolysis by targeting PFKFB3 suppresses tumor growth and metastasis in head and neck squamous cell carcinoma. J Exp Clin Cancer Res 2017;36:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clem BF, O’Neal J, Tapolsky G, et al. Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol Cancer Ther 2013;12:1461–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sarkar Bhattacharya S, Thirusangu P, Jin L, Staub J, Shridhar V, Molina JR. PFKFB3 works on the FAK-STAT3-SOX2 axis to regulate the stemness in MPM. Br J Cancer 2022;127:1352–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ninou AH, Lehto J, Chioureas D, et al. PFKFB3 Inhibition sensitizes DNA cross-linking chemotherapies by suppressing Fanconi anemia repair. Cancers (Basel) 2021;13:3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li FL, Liu JP, Bao RX, et al. Acetylation accumulates PFKFB3 in cytoplasm to promote glycolysis and protects cells from cisplatin-induced apoptosis. Nat Commun 2018;9:508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee M, Harley G, Katerelos M, et al. Mutation of regulatory phosphorylation sites in PFKFB2 worsens renal fibrosis. Sci Rep 2020;10:14531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jing C, Castro-Dopico T, Richoz N, et al. Macrophage metabolic reprogramming presents a therapeutic target in lupus nephritis. Proc Natl Acad Sci U S A 2020;117:15160–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feliers D, Frank MA, Riley DJ. Activation of cyclin D1-Cdk4 and Cdk4-directed phosphorylation of RB protein in diabetic mesangial hypertrophy. Diabetes 2002;51 (11):3290–9. [DOI] [PubMed] [Google Scholar]
- 59.Hortobagyi GN, Stemmer SM, Burris HA, et al. Overall survival with Ribociclib plus Letrozole in advanced breast cancer. N Engl J Med 2022;386:942–50. [DOI] [PubMed] [Google Scholar]
- 60.Spring LM, Clark SL, Li T, et al. Phase 1b clinical trial of ado-trastuzumab emtansine and ribociclib for HER2-positive metastatic breast cancer. NPJ Breast Cancer 2021;7:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alves CL, Ehmsen S, Terp MG, et al. Co-targeting CDK4/6 and AKT with endocrine therapy prevents progression in CDK4/6 inhibitor and endocrine therapy-resistant breast cancer. Nat Commun 2021;12:5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Santoro A, Su WC, Navarro A, et al. Phase Ib/II study of ceritinib in combination with ribociclib in patients with ALK-rearranged non-small cell lung cancer. Lung Cancer 2022;166:170–7. [DOI] [PubMed] [Google Scholar]
- 63.Sager RA, Backe SJ, Ahanin E, et al. Therapeutic potential of CDK4/6 inhibitors in renal cell carcinoma. Nat Rev Urol 2022;19:305–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.de Kouchkovsky I, Rao A, Carneiro BA, et al. A phase Ib/II study of the CDK4/6 inhibitor ribociclib in combination with docetaxel plus prednisone in metastatic castration-resistant prostate cancer. Clin Cancer Res 2022;28:1531–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.