

Syk blockade induces macrophage polarization to an immunostimulatory phenotype, which enhances CD8+ T-cell responses and improves gemcitabine efficacy in pancreatic ductal adenocarcinoma, a clinically challenging malignancy.
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is an insidious disease with a low 5-year survival rate. PDAC is characterized by infiltration of abundant tumor-associated macrophages (TAM), which promote immune tolerance and immunotherapeutic resistance. Here we report that macrophage spleen tyrosine kinase (Syk) promotes PDAC growth and metastasis. In orthotopic PDAC mouse models, genetic deletion of myeloid Syk reprogrammed macrophages into immunostimulatory phenotype, increased the infiltration, proliferation, and cytotoxicity of CD8+ T cells, and repressed PDAC growth and metastasis. Furthermore, gemcitabine (Gem) treatment induced an immunosuppressive microenvironment in PDAC by promoting protumorigenic polarization of macrophages. In contrast, treatment with the FDA-approved Syk inhibitor R788 (fostamatinib) remodeled the tumor immune microenvironment, “re-educated” protumorigenic macrophages towards an immunostimulatory phenotype and boosted CD8+ T-cell responses in Gem-treated PDAC in orthotopic mouse models and an ex vivo human pancreatic slice culture model. These findings illustrate the potential of Syk inhibition for enhancing the antitumor immune responses in PDAC and support the clinical evaluation of R788 either alone or together with Gem as a potential treatment strategy for PDAC.
Significance:
Syk blockade induces macrophage polarization to an immunostimulatory phenotype, which enhances CD8+ T-cell responses and improves gemcitabine efficacy in pancreatic ductal adenocarcinoma, a clinically challenging malignancy.
Graphical Abstract
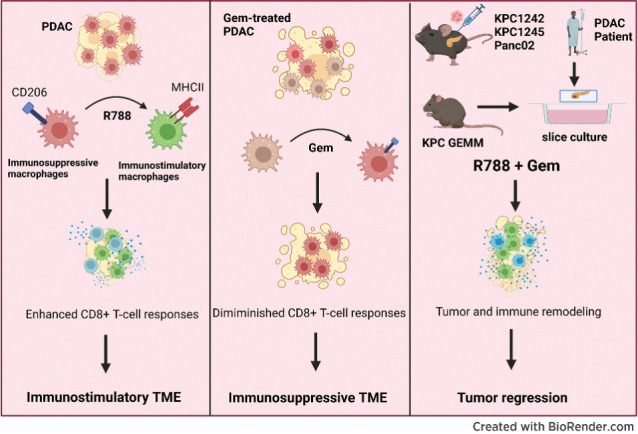
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive lethal malignancy with a mortality rate closely paralleling its incidence rates (1, 2). With a 5-year survival rate of 11%, PDAC represents the third leading cause of cancer-related deaths in the United States (3). Current standard therapies are not curative for advanced disease and minimally improve survival (4, 5); hence novel therapies are urgently required to combat this devastating malignancy.
The characteristic features of PDAC are the presence of a highly desmoplastic stroma and extreme intratumoral heterogeneity, which drives immune and drug resistance in these tumors (6, 7). Tumor-infiltrating immune cells of the myeloid lineage, including tumor-associated macrophages (TAM), and myeloid-derived suppressor cells (MDSC), play crucial roles in generating an immunosuppressive microenvironment that diminishes effective antitumor immune responses in pancreatic cancer (8–11). Among these myeloid cells, CD206+MHCII− TAMs are abundantly infiltrated in the PDAC tumors and promote PDAC growth (12). Emerging studies have shown that TAMs interact with the CD8+ T cells in the TME to reduce the mobility of T cells in the tumor stroma and limit their entry into the tumors (13). TAMs can also inhibit the function of CD8+ T cells by secreting immunosuppressive cytokines like TGFβ (14) or limiting the metabolites required for T-cell proliferation (L-arginine) by expressing arginase-1 enzyme (15). Recent studies have highlighted that gemcitabine (Gem) chemotherapy, the single most commonly used agent in PDAC, also promotes the polarization of macrophages to an immunosuppressive phenotype in the PDAC tumor microenvironment (TME), ultimately leading to Gem resistance (16, 17). Consequently, approaches that modulate the protumorigenic functions of TAMs have demonstrated that targeting these immune cells can unleash effector T-cell responses and enhance responses to Gem treatment in PDAC (10, 11, 18, 19).
We have recently shown that spleen tyrosine kinase (Syk) signaling downstream of α4β1 integrin promotes stabilization of hypoxia-inducible factor (HIF1α) to limit CD8+ T-cell responses in lung adenocarcinoma (20, 21). Moreover, using Syk inhibitor, R788 (fostamatinib), which is an FDA-approved drug for the treatment of patients with chronic immune thrombocytopenia (22), we have shown that Syk inhibition induces immunostimulatory transcriptional programming in macrophages to suppress tumor growth (20). The role of Syk in PDAC progression has not been previously explored in detail and one study reported that it acts as a tumor suppressor (23). For this reason, we sought to study the role of Syk in PDAC growth and metastasis.
Here we report that Syk is abundantly present in PDAC-associated macrophages and promotes polarization of macrophages into a protumorigenic type, leading to immunosuppression, tumor growth, and metastasis in mouse models of PDAC. Using orthotopic mouse models and an organotypic PDAC slice culture model, we demonstrated that pharmacologic inhibition of Syk using FDA-approved drug R788, reprogrammed TAMs towards immunostimulatory phenotype that promoted CD8+ T-cell activation and improved responses to Gem. These results suggest the therapeutic potential of R788 alone or in combination with chemotherapy to augment antitumor immune responses in this cancer.
Materials and Methods
Human samples
Human tissues from deidentified patient samples were received from UC San Diego Moores Cancer Center Biorepository under IRB-approved protocol no. 181755.
Cell lines
The murine KPC1245 and KPC1242 pancreatic adenocarcinoma cell lines derived from KrasG12D/+, p53R172H/+, Pdx1-Cre+ (KPC) mice (24) were a kind gift from Dr. David Tuveson (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY). The Panc02 cell line derived from C57BL/6 mice is described previously (25). All cell lines were grown in DMEM media supplemented with 10% FBS and 1% penicillin and streptomycin (Panc02) or supplemented with sodium pyruvate, nonessential amino acids for KPC-derived cells. All cell lines were maintained at low passage numbers and were cultured at 37°C and 5% CO2 in a humidified incubator. Cells were routinely tested for mycoplasma contamination and no cell manipulations were performed for this study.
Animals and in vivo experiments
All procedures involving animals were approved by the UCSD Animal Care Committee, which serves to ensure that all federal guidelines concerning animal experimentation are met. Floxed Syk mice, lysozyme M (LysM) Cre and CD19 cre recombinase transgenic mice were purchased from the Jackson laboratories. Sykfl/fl mice were crossed with LysM cre or CD19 cre mouse models to specifically delete Syk from monocyte and macrophages (SykMC-KO mice; ref. 20) or from B cells (SykBC-KOmice), respectively. C57BL/6 mice used for drug treatment studies were obtained from Charles River Laboratories. Fifty thousand KPC1245 or KPC1242 cells and one million Panc02 cells were implanted orthotopically into the pancreata of 6- to 8-week-old SykMC-WT (aka SykBC-WT), SykBC-KO and SykMC-KO mice, and C57BL/6 mice (25, 26). The metastatic nodules on mesenteric lymph nodes and liver were counted under dissecting microscope. In some studies, C57BL/6 mice bearing PDAC tumors were treated with 50 mg/kg R788 (five times/week) either alone or in combination with 15 mg/kg Gem (3 times/week) or in combination with 200 μg anti-PDL1 mAb (clone 10F.9G2, BioXCell), or isotype control LTF2 (BioXCell). R788 was obtained from Selleck Chemicals and Gemcitabine-HCL was purchased from Biotang. CD11b+F4/80+ macrophages were isolated using CD11b and F4/80 magnetic beads (Miltenyi Biotec) according to manufacturer's instructions.
Macrophage-depletion and CD8+ T-cell depletion experiments
For macrophage depletion experiments, mice bearing KPC1245 tumors were treated with 50 mg/kg of anti-CSF1R antibody (clone AFS 98, BioXcell), administered intraperitoneally, every alternate day, until tumors were harvested on day 15. For CD8 depletion experiments, mice with KPC1245 tumors were treated with 200 μg of anti-CD8 (clone YTS169.4) or an isotype rat IgG2b control (LTF-2) from Bio-X-Cell administered intraperitoneally on day −3, 0, 3, 6, and 10 of tumor inoculation as described previously (20).
Cell proliferation assay
KPC1245 cells were plated at 4 × 104 cells/well in 96-well plates in media containing DMEM + 10% FBS. Cells were incubated overnight and treated with DMSO or R788 for 48 hours. Cell viability assay was performed using AlamarBlue (Roche) according to the manufacturer's protocol.
Single-cell preparation for flow cytometry
Single-cell suspensions from tumors were prepared as described previously (20). Briefly, tumors were isolated, minced, and incubated for 30 to 45 minutes at 37°C in a dissociation solution containing Hanks Balanced Salt Solution supplemented with 0.5 mg/mL collagenase IV (Sigma), 0.1 mg/mL hyaluronidase V (Sigma), and 0.005 MU/ml DNAse I (Sigma). The undigested tissues were removed by passing through 70 μm nylon mesh and centrifuged at 1,500 rpm for 5 minutes. The red blood cells were lysed using RBC lysis buffer (Pharm Lyse; BD Biosciences) and cells were prepared for flow cytometry.
Flow cytometry
For flow cytometry staining, dissociated cells were incubated with a fixable viability stain 510 (BD Biosciences) followed by staining with primary antibodies directed against CD45 (30-F11), CD11b (M1/70), Gr1 (RB6–8C5), F4/80 (BM8), CD206 (C068C2), MHCII (AF6–120.1), CD44 (IM7), PDL1 (MIH5), TIGIT (1G9), and CD69 (H1.2F3) obtained from BD Biosciences; CD3 (145–2C11), CD4 (GK1.5), CD8 (53–6.7), and pSykY348 (moch1ct) from eBioscience, PD1 (29F.1A12), and CD62 L (MEL-14) from BioLegend. Samples were acquired on BD Fortessa or LSRII and data were analyzed using FlowJo software version 10 (TreeStar).
IHC and immunofluorescence
Formalin-fixed paraffin embedded (FFPE) tissue sections (4 μm) of human PDAC were obtained from UC San Diego Moores Cancer Center Biorepository. FFPE tissue sections (4 μm) of human PDAC or mouse tumors were used for IHC or immunofluorescence (IF) studies. Slides were baked overnight and then either stained for H and E staining or Trichome staining or processed for IHC and IF staining. For IHC studies, human samples were stained with the following antibodies: anti-CD68 (MA5–12407, 1:50; Invitrogen), anti-CD206 (ab64693, 1:100; Abcam), and anti-CD8 (70306S, 1:50; Cell Signaling Technology). Mouse tumors were stained with the following antibodies: anti-SMA (ab5694, 1:1,000; Abcam), anti-CD3 (ab16669, 1:500; Abcam), and anti-CD8 (14–0195–82, 1:50; Invitrogen). Antigen retrieval was carried out in citrate buffer (pH 6.0, Vector Laboratories) at 95°C for 30 minutes. After antigen retrieval, tissue sections were incubated with BLOXALL (Vector Laboratories) for 10 minutes followed by blocking Blotto (Thermo Fisher Scientific) for 10 minutes. Sections were stained with primary antibodies at recommended dilutions in Blotto for 1 hour at room temperature. After washing primary antibodies, the samples were stained with anti-mouse or anti-rabbit secondary antibodies HRP Polymer (Cell IDX) for 30 minutes at room temperature, followed by DAB staining using DAB chromogen (VWR, 95041–478) for 5 minutes. The images with positive staining were captured using Olympus inverted microscope. The whole area of the tumor was selected as a field of interest, and the area with immunohistochemically positive staining within the field of interest was calculated by the ImageJ software after setting the thresholds. The results are expressed as the percentage of the positively immunolabeled area within the total area of the tumor. For immunofluorescence staining, antigen retrieval was carried out as described above followed by blocking with Blotto for 10 minutes. The tissues were then incubated with Syk (clone EP573Y, ab40781, 1:1,000; Abcam) and CD68 (MA5–12407, 1:50; Invitrogen) antibodies in Blotto for 1 hour at room temperature. After two washings with 1× TBST buffer, samples were stained with antimouse or anti-rabbit HRP polymer-conjugated secondary antibodies (Cell IDX) for 30 minutes at room temperature. Tissues were then incubated with Alexa Fluor 488 and Alexa Fluor 594 Tyramide reagents (Thermo Fisher Scientific) for 10 minutes. DAPI was used to counterstain the nuclei. For some studies, the slides were deparaffinized, and rehydrated, followed by antigen retrieval in citrate buffer. The slides were bleached with 4.5% H2O2 and 24 mmol/L NaOH in PBS with exposure to light. After 45 minutes the bleaching solution was refreshed, and bleaching was continued for 45 more minutes. The slides were then blocked, followed by incubation with the following antibodies: anti-CD8a (14–0808–82, 1:400; Invitrogen) and anti-Ki67 (ab16667, 1:200; Abcam) detected with donkey anti-rat AF647 (A48272, 1:500; Invitrogen) and donkey anti-rabbit (A10042, 1:500; Invitrogen), anti-CD31 (DIA-310, 1:200; Dianova) detected with donkey anti-rat AF647 (A48272, 1:500; Invitrogen), and anti-F4/80 (70076S, 1:200; Cell Signaling Technology) detected with goat anti-rabbit AF647 (A32733, 1:500; Invitrogen).The images of fluorescent staining were captured using Keyence BZX-700 fluorescent microscope.
Macrophage polarization studies
Bone marrow derived macrophages (BMDM) were generated from C57BL/6 mice as described previously (21). Tumor conditioned media (TCM) was prepared from confluent KPC1245 cells. For this, cells were grown to 80% confluence, washed with PBS and media changed to DMEM without FBS for another 48 hours. TCM was collected after 48 hours. and used for coculture experiments. For inhibitor studies, BMDMs stimulated with 20 ng/mL IL4, or WT BMDMs incubated with TCM from KPC1245 cells were treated with 500 nmol/L R788 for 24 hours followed by RNA isolation.
Syk activation studies
TAMs were isolated from Panc02 PDAC tumors based on FACS sorting of cells stained with CD11b and F4/80. TAMs were stimulated for Fc gamma ligation or LPS or α4β1 integrin stimulation. For Fc gamma ligation, cells were incubated with purified rat antimouse CD16/CD32 (BD Pharmingen) at 1.0 μg/106 cells for 10 minutes 4°C and then incubated with goat anti-rat F(ab’)2 (25 μg/mL; Jackson ImmunoResearch) for 10 minutes, followed by lysate preparation. For TLR4 signaling, TAMs were stimulated with 1 μg/mL of LPS for 10 minutes, followed by lysate preparation. For α4β1 integrin studies, nontissue culture petri dishes were coated with 10 μg/mL of H296 (ligand for α4β1 integrin) in PBS for 1 hour, followed by washing with PBS and plating of cells for 10 minutes and lysate preparation.
Western blot analysis
BMDMs, CD90.2+ T cells, and CD19+ B cells isolated from splenocytes of PDAC-bearing mice, in vitro cultured Panc02, KPC1245 cells, and CD11b+F4/80+ TAMs stimulated with LPS or by Fc gamma ligation or α4β1 integrin ligation were solubilized in RIPA buffer containing protease and phosphatase inhibitors. Proteins were run on SDS-PAGE gels, electro-transferred to nitrocellulose membranes and were immunoblotted with antibodies from pSykY348 (Abcam), Syk (Santa Cruz Biotechnology), and β-actin (Santa Cruz Biotechnology) as described previously (21).
Real-time PCR
Total RNA was isolated from BMDMs or murine or human PDAC slices using the Qiagen RNAeasy Kit (Qiagen) and cDNA was prepared using iscript cDNA Synthesis Kit (Bio-Rad). cDNA was amplified by RT-PCR reactions with 1× SYBR Green Supermix (Bio-Rad) in 96-well plates on an CFX96 real-time system (Bio-Rad), using the program and the custom designed primers obtained from Integrated DNA Technologies. Relative expression levels were normalized to Gapdh expression as described previously (20).
Organotypic slice culture
Human PDAC slices and slices derived from 8 to 9-weeks-old KPC mice—a genetically engineered mouse model (GEMM) were cultured as described previously (27, 28). The tissues were first sectioned into fragments (approximately 5–10 mL cubed) and embedded in 4% low-gelling temperature agarose dissolved in PBS. Tissue slices were then cut off 200 μm on a vibroslicer (VT1000P; Leica Biosystems). The slices were placed on top of 0.4 μm transwell cell culture inserts (catalog no. CLS3450–24EA) containing RPMI media. These slices were incubated overnight at 37°C. The RPMI media was switched to RPMI media containing either 250 nmol/L Gem or 2.5 μmol/L R788 or combination of Gem and R788. These slices were incubated for another 3 days, supernatants were used for cytokine ELISA, and tissue slices were used for RNA isolation or IHC.
ELISA assays
Tissue culture supernatants from PDAC slices were assayed for total TGFβ1 or MCP1 (CCL2) by using commercially available Legend Max ELISA Kits from BioLegend according to manufacturer's instructions.
Gene expression analysis
Gene expression microarray data and sample information of matched tumor and normal samples from 36 patients with pancreatic adenocarcinoma were downloaded from Gene Expression Omnibus (GEO) accession GSE15471 (29). Sequence analysis on IL4 polarized BMDMs from SykMC-WT and SykMC-KO mice was performed as described previously (20). Briefly, RNA-sequencing expression data of IL4 polarized BMDMs from SykMC-WT and SykMC-KO were downloaded from GEO accession GSE115109 (20) and aligned using a STAR (30) to the mm10 mouse reference genome with reads quantified by Salmon (31) in a standard RNA-seq BCBio pipeline workflow. Differential gene expression in the IL4-exposed SykMC-WT and SykMC-KO BMDMs were compared using DESeq2 (32) with apeglm being used for empirical shrinkage effect size estimation (33). P values were multiple hypotheses corrected using the Benjamini–Hochberg method in the python stats models package. The heatmap of relevant differentially expressed genes was made using the Python seaborn package. The results are available at GSE115109.
Single-cell RNA-seq data acquisition, integration, and analyses
Single-cell RNA-seq (scRNA-seq) data from 24 human PDAC tumor samples and 11 normal pancreases were downloaded from GSA accession CRA001160 along with sample information and cell identity metadata, available at https://ngdc.cncb.ac.cn/gsa/browse/CRA001160. The cells for each sample were separated, and counts of each gene from cells of the same type were averaged (sum of gene counts/number of cells of type in sample); nonimmune cells were grouped together in the nonimmune category for clarity. Finally, a box plot of SYK expression across the different cell types was generated and P values between each cell type were computed using the Kruskal–Wallace paired sample test due to the paired nature of the data.
Statistical analysis
For all in vitro studies, experiments were performed three or more times with three or more biological replicates per group. All in vivo experiments were performed at least twice with 8 to 10 animals assigned/group. Data were normalized to standard where applicable. Significance testing was performed by one-way ANOVA with Tukey post hoc testing for multiple pair-wise testing with more than two groups and by Student t test when only two groups were compared. Statistical analysis was performed using GraphPad Prism V.9 software. Unless otherwise stated, data are shown as mean ± SEM. In all cases, *, P < 0.05, **, P < 0.01, ***, P < 0.001, ****, P < 0.0001, and ns, not significant.
Data availability
Gene expression microarray data of matched tumor and normal samples from Pancreatic Adenocarcinoma patients were downloaded from GEO accession GSE15471. RNA-seq expression data of IL4 polarized BMDMs from SykMC-WT and SykMC-KO were downloaded from GEO accession GSE115109. scRNA-seq data from human PDAC tumor samples and normal pancreases were downloaded from accession CRA001160 available at https://ngdc.cncb.ac.cn/gsa/browse/CRA001160. All other raw data are available upon request from the corresponding author. Data and code used for analysis are available as a Code Ocean capsule (https://codeocean.com/capsule/5100346/tree/v1).
Results
Syk-positive macrophages accumulate in murine and human PDAC
TAMs are principal regulators of tumor progression and chemo- and immunotherapeutic resistance in PDAC (8–10, 34). Similar to previous reports (11, 35) demonstrating the increased infiltration of TAMs into pancreatic tumors, we also observed an increase in the presence of CD68+ macrophages in human PDAC compared with healthy pancreatic tissue (Fig. 1A). To examine the accumulation of macrophages in murine PDAC, we orthotopically implanted KPC1245 and KPC1242 cells in C57BL/6 mice. Immunostaining of pancreata from normal and tumor-bearing mice revealed that F4/80+ macrophages are abundantly infiltrated in KPC1245 and KPC1242 tumors (Supplementary Fig. S1A and S1B). In addition, we found that macrophages are crucial regulators of PDAC growth as administration of antimouse anti-CSF1R suppressed tumor growth in mice bearing KPC1245 tumors (Supplementary Fig. S2A–S2C).
Figure 1.

Syk-positive macrophages accumulate in murine and human PDAC. A and B, Left, IHC or IF staining of human invasive PDAC patient sample and normal pancreas for CD68 (A) and SYK (B). Scale bar, 20 μm. Right, quantification of CD68+ macrophages or SYK+ cells/40× microscopic field in the tissue sections (n = 3). The tissue sections were stained with DAPI to detect nuclei in B. Statistical significance was determined by Student t test. C and D, Relative SYK mRNA expression in human PDAC as compared with normal pancreas tissue in data set GSE15471 (C) and scRNA-seq data set (CRA001160; D). Statistical significance was determined by Wilcoxin-signed rank test. E, Western blot images showing expression of Syk and β-actin in cell lines, B cells, T cells, BMDMs, and TAMs. F, Syk expression in macrophages, B cells, and nonimmune cells in human PDAC, using scRNA-seq data set (CRA001160). G, Figure shows IF staining of Syk (red) and CD68 (aqua) in a tissue section from human invasive PDAC. The dual-positive cells are indicated by arrows. Scale bar, 10 μm. H, FACS quantification of pSyk348+ CD11b+F4/80+ cells in normal pancreas and KPC1245 PDAC (n = 3). I, Immunoblot showing pSyk348 activation in Panc02 PDAC TAMs stimulated with Fc gamma ligation, 1 μg/mL LPS, or adhered to H296 (ligand for α4β1). **, P < 0.01; ****, P < 0.0001. N.S., no stimulation.
On the basis of the abundance of macrophages in PDAC, we sought to test if Syk is expressed in PDAC and if therapeutic targeting of this kinase is efficacious in treating this recalcitrant neoplasm. We found an abundant increase in SYK+ cells in the tissue sections of invasive human PDAC as compared with normal tissue using immunofluorescence microscopy (Fig. 1B). Correspondingly, analysis of the publicly available dataset, GSE15471, and scRNA-seq data set (accession no.: CRA001160) revealed high expression of SYK in human PDAC as compared with normal pancreas (Fig. 1C and D).
Like human PDAC, Syk levels were high in murine PDAC tumors compared with normal pancreatic tissue (Supplementary Fig. S3A). Syk was expressed specifically in CD45+ immune cells isolated from orthotopically implanted KPC1245 tumors and minimally in CD45− tumor cells as revealed by RTPCR (Supplementary Fig. S3B). Most notably, we observed the presence of Syk in CD11b+F4/80+ TAMs isolated from murine PDAC tumors (Fig. 1E). Syk is also expressed in CD19+ B cells isolated from spleens of mice bearing PDAC tumors and to a lower extent in BMDMs. However, Syk is not expressed in murine PDAC cell lines, Panc02 and KPC1245 and CD90.2+ T cells from spleens of mice bearing PDAC tumors (Fig. 1E). Likewise, the analysis of scRNA-seq data (CRA001160) revealed that Syk is maximally expressed in macrophages, followed by B cells and nonimmune cells whereas not in T cells from human PDAC (Fig. 1F). Immunofluorescence microscopy also confirmed immunoreactivity of Syk in CD68+ TAMs in the tissue sections of human PDAC specimens (Fig. 1G).
In macrophages, Syk is known to be activated upon binding to phosphorylated ITAMs of Fc gamma receptor, or by binding to the cytoplasmic domains of integrin adhesion receptors, most notably β1 and β3 integrin or by TLR4 (36, 37). We have previously shown that α4β1 integrin can maximally activate Syk at Y348 site in TAMs isolated from the lung adenocarcinoma tumors (20). Here, we found that macrophages in PDAC tumors showed higher expression of pSYK348 as compared with macrophages from normal pancreas (Fig. 1H). In addition, TAMs are activated downstream of α4β1 integrin and Fc gamma receptors in the PDAC TME, with no activation by TLR4-mediated signaling (Fig. 1I). Taken together, these results clearly indicate that Syk is abundantly present in PDAC-associated macrophages and is activated downstream of α4β1 integrin or/and Fc gamma receptors in the PDAC TME and may function to modulate PDAC growth and metastasis.
Macrophage Syk promotes PDAC growth and metastasis and limits infiltration, proliferation, and activation of CD8+ T cells in the PDAC TME
As Syk is abundantly present in macrophages and to some extent in B cells (Fig. 1E and F), we next investigated if deletion of Syk in macrophages or B cells can impede PDAC growth. For this study, we used our previously established conditional myeloid Syk KO mice (SykMC-KO; ref. 20) and recently generated B cell Syk KO (SykBC-KO) mice and examined tumor growth of two syngeneic murine PDAC cell lines: KPC1245 and Panc02 according to the depicted schema (Fig. 2A). We found that CD19+ B cells were significantly reduced in SykBC-KO mice, but tumor growth is similar in SykBC-WT and SykBC-KO mice implanted with orthotopic KPC1245 tumors (Supplementary Fig. S4A–S4C), suggesting that Syk deletion in B cells is not responsible for PDAC growth. However, we found that both orthotopic Panc02 and KPC1245 pancreatic tumors from SykMC-KO mice were significantly smaller than pancreatic tumors from SykMC-WT mice (Fig. 2B and C). Remarkably, the prevalence of metastases in colonic lymph nodes and liver was also substantially reduced in pancreatic tumors from SykMC-KO mice, indicating that myeloid Syk promotes PDAC growth and metastatic progression (Fig. 2B and C). Analysis of pancreata from KPC1245-bearing mice by immunofluorescence or IHC reveals that SykMC-WT animals exhibited abundant expression of smooth muscle actin (SMA) and Trichrome, as well as increased CD31 staining (Fig. 2D), suggesting a role for myeloid Syk in promoting desmoplasia.
Figure 2.

Macrophage Syk regulates immunosuppression, PDAC growth, and metastasis. A, Panc02 and KPC1245 tumors were orthotopically implanted into SykMC-WT and SykMC-KO mice according to the depicted schema. B and C, Left, weights of pancreata containing Panc02 (B) or KPC1245 (C) tumors from SykMC-WT and SykMC-KO mice. Right panels of B and C show quantification of metastatic nodules in colonic lymph nodes and liver. Significance testing was performed by nonparametric t tests. D, Left, representative images showing trichrome, αSMA, and CD31 staining in KPC1245 PDAC tumors. Scale bar, 50 μm. Right, CD31 quantification. E, FACS quantification of CD3+, CD4+, and CD8+ T cells in Panc02 PDAC tumors. F, Left, IHC of KPC1245-PDAC tumors for CD3, CD4+, and CD8. Scale bar, 20 μm. Right, immunodetection of T cells/microscopic field (n = 3). G, Top, IF staining of CD8 (yellow), DAPI (blue), and Ki67 (red) in KPC1245-PDAC tumors. Bottom, quantification data. Scale bar, 10 μm. H, mRNA expression of Ifng, Gzm, and Prf in orthotopic SykMC-WT and SykMC-KO Panc02 tumors. Statistical significance was determined using the Student t test or one-way ANOVA with Tukey post hoc multiple pairwise testing when analyzing more than two groups. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant.
As myeloid Syk deletion has significant impact on PDAC growth (Fig. 2B and C), we analyzed alterations in immune cells in tissues from PDAC-bearing SykMC-WT and SykMC-KO mice by flow cytometry. We found that Syk deletion has no significant impact on the recruitment of CD19+ B cells, CD11b+Gr1− monocytes, CD11b+Gr1+ granulocytes, and CD11b+Gr1−F4/80+ TAMs into PDAC tumors (Supplementary Fig. S5A–S5D). Importantly, both Panc02 and KPC1245 PDACs from SykMC-KO mice exhibited significantly more CD3+ and CD8+ T cells than PDACs from SykMC-WT mice (Fig. 2E and F), with no significant impact on the systemic infiltration of T cells in the spleens of tumor bearing SykMC-WT and SykMC-KO mice (Supplementary Fig. S6A–S6D). We found a significant increase in the T-cell chemo-attractants CXCL5, CXCL9, and CXCL10 in the SykMC-KO-treated tumors (Supplementary Fig. S6E). In addition to increased infiltration, CD8+ T cells proliferate in the SykMC-KO-treated tumors (Fig. 2G). Accordingly, T cells isolated from the PDAC-bearing SykMC-KO mice showed higher expression of Ifng, Gzmb, and Prf (Fig. 2H). These results clearly suggest that myeloid Syk promotes immunosuppression, PDAC growth, and metastasis.
Syk enhances an immunosuppressive phenotype of macrophages in vitro and in vivo
Because we observed that Syk deletion promotes recruitment and activation of T cells in PDAC, we postulated that Syk promotes immunosuppressive programming of macrophages to impede CD8+ T-cell responses in PDAC. We found that Syk deletion reprogramed TAMs towards immunostimulatory phenotype, as we observed an increase in the frequency of MHCII+ TAMs with a concomitant reduction in CD206+ immunosuppressive TAMs in PDAC tumors from SykMC-KO mice (Fig. 3A and B). Similarly, we found higher expression of immunosuppressive genes in TAMs isolated from KPC1245 tumors implanted in SykMC-WT mice whereas TAMs sorted from SykMC-KO mice showed higher expression of proinflammatory genes like Il12, Ifng, Nos2, as shown by RT-PCR (Fig. 3C). We next determined if Syk can regulate the transcriptional programming in macrophages in vitro. It is well-documented that incubation with IL4 or TCM promotes immune suppressive polarization of macrophages in vitro (38). RNA-seq data revealed that IL4-stimulated SykMC-WT BMDMs exhibited higher expression of genes related to immune suppression such as Ido1, Vegfd, Arg1, and Ccl2 as compared with IL4-stimulated SykMC-KO BMDMs (Fig. 3D). Interestingly, we also found higher expression of genes related to metastasis, such as Pdgfa, Pdgfb (39), in IL4-stimulated SykMC-WT BMDMs, which may help explain the increased metastasis observed in SYKMC-WT mice (Fig. 2B and C). We next asked if pharmacologic inhibition of Syk can reduce protumorigenic polarization of macrophages in vitro. For this, we used a commercially available Syk inhibitor, R788 has an IC50 of 41 nmol/L for Syk with no inhibition of ZAP-70 and Lyn kinases (22). We observed that R788 prevented IL4-induced expression of immunosuppressive genes (Fig. 3E). Although coculture of BMDMs with TCM from KPC-1245 tumor cells increased the gene expression of Il6, Il10, Ccl2, Arg, Tgfb, Mmp9, and Vegf, R788 downregulated the expression of these genes (Fig. 3F). Taken together, these results indicate that Syk inhibition reduces expression of immune suppressive genes and stimulates expression of proinflammatory genes in macrophages, which likely contribute to enhanced CD8+ T-cell recruitment, proliferation, and activation in PDAC tumors.
Figure 3.

Syk promotes immunosuppressive programing of macrophages in vitro and in vivo. A and B, Representative FACS plots (left) and FACS quantification (right) of MHCII+ TAMs (A) and CD206+ TAMs (B) in representative KPC1245 tumors from SykMC-WT and SykMC-KO mice (n = 4). Cells were gated on CD11b+F4/80+Gr1− TAMs. C, Relative mRNA expression of genes in TAMs isolated from orthotopic SykMC-WT and SykMC-KO Panc02 tumors. D, Relative mRNA expression of genes in nonstimulated (N.S.) and IL4-stimulated SykMC-WT and SykMC-KO BMDMs as determined by RNA sequencing. E and F, Relative mRNA expression of genes in R788-treated macrophages polarized with IL4 in vitro (E) or KPC1245-TCM induced TAMs in vitro (F). G, Weight of R788-treated and control-treated orthotopic tumors from SykMC-WT and SykMC-KO mice (n = 5). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant.
To investigate if deletion of Syk specifically in macrophages is sufficient to block PDAC growth, we treated SykMC-WT and SykMC-KO mice bearing KPC1245 tumors with R788. We found that administration of R788 significantly reduced tumor growth in SykMC-WT mice (Fig. 3G). However, R788 further did not significantly reduce tumor growth in SykMC-KO mice (Fig. 3G). As Syk is predominantly expressed in CD45+ immune cells in PDAC tumors (Supplementary Fig. S2B) with no contribution of B-cell Syk in PDAC growth (Supplementary Fig. S4C) and minimal effect of Syk inhibitors on PDAC cells in vitro (Supplementary Fig. S7), we conclude that macrophage Syk promotes immunosuppression and PDAC growth.
Syk inhibition with FDA-approved drug R788 synergizes with Gem to suppress PDAC growth and metastasis
As Syk controls immunosuppressive programming of macrophages, we speculated that Syk inhibitors either alone or together with immunotherapy or chemotherapy will improve the outcomes of pancreas cancer. To test this hypothesis, Panc02, and KPC1245 cells were orthotopically injected into the pancreata of WT mice, and mice were then treated with R788 either alone or in combination with anti-PDL1 mAb or Gem, according to the schema as depicted in Fig. 4A. We observed that vehicle mice developed large pancreatic tumors with numerous metastatic nodules in liver and colon, whereas R788-treated tumors were significantly smaller with rare metastatic nodules (Fig. 4B–D). We found that antibodies targeted against programmed death ligand 1 (anti-PDL1 mAb) either alone or in combination with R788 significantly reduced tumor growth in the Panc02 PDAC (Fig. 4B). However, no significant synergy was observed between anti-PDL1 mAb and R788 in the KPC1245 PDAC (Fig. 4C and D). We found that PDL1 is expressed both on CD45− tumor cells and CD45+ TAMs, and R788 didn't impact the expression of PDL1 either on macrophages or on TAMs (Supplementary Fig. S8), which explains the lack of synergy between R788 and anti-PDL1 mAb in KPC1245-treated tumors. However, we found that R788 synergizes with Gem to reduce tumor growth, and metastasis in Panc02, KPC1245, and in another KPC-derived cell line, KPC1242 (Fig. 4B–E). In addition, histological evaluation of KPC-1245 tumors from Gem and R788-treated mice showed reduced desmoplasia as revealed by Trichome staining (Fig. 4F).
Figure 4.

Syk inhibition reduces tumor growth and sensitizes PDAC tumors to gemcitabine. A, Schemas for the administration of R788 or anti-PDL1 mAb or Gem in Panc02 or KPC1245 tumors. B and C, Weights of pancreata from Panc02 (B) and KPC1245 (C) tumors treated with drugs (n = 5) as depicted in schema in A. D, Metastatic nodules in treated KPC1245 (n = 5) PDAC tumors. Significance testing was performed by one-way ANOVA with Tukey post hoc multiple pairwise testing. E, Weights of pancreata from KPC1242 tumors treated with drugs (n = 5) as depicted in schema. F, Images showing Trichrome staining in PDAC tumors. Scale bar, 50 μm. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant.
Gem endows TAMs with protumorigenic properties but R788 reprograms TAMs to overcome Gem-induced immunosuppression in PDAC in a CD8+ T-cell–dependent manner
Several studies have shown that macrophage infiltration and macrophage-induced immunosuppression both contribute to Gem resistance in PDAC (16, 17). As Syk inhibition reduced immune suppressive polarization of macrophages in vitro and in vivo (Fig. 3), we investigated if R788 can overcome Gem-induced immunosuppression and can boost CD8+ T cells responses in these tumors. We found that treatment of KPC1245 tumors with R788 or Gem did not significantly reduce infiltration of total CD45+ cells, whereas there is significant decrease in CD11b+ cells on R788 treatment (Fig. 5A and B). Most notably, Gem treatment reduced infiltration of TAMs but has no impact on the infiltration of CD206+ immunosuppressive TAMs in the PDAC tumors while R788 alone or together with Gem significantly reduced the infiltration of CD206+ TAMs in the tumor bed (Fig. 5C and D). In addition, Gem-treated tumors show increased gene expression of Arg, Il10, Tgfb, and Ccl2 as compared with vehicle tumors and R788 reduced Gem-induced immunosuppressive transcription in these tumors (Fig. 5E).
Figure 5.

R788 overcomes Gem-induced immunosuppression and increases cytotoxic T cells in PDAC. A–D, FACS quantification of intratumoral CD45+ cells (A), CD11b+ cells (B), CD11b+Gr1−F4/80+ TAMs (C), and CD206+ TAMs (D) in end-stage KPC1245 tumors (n = 4) from Fig. 4C. E, Relative mRNA expression data of immunosuppressive genes in KPC1245 tumors from Fig. 4C (n = 3). F, IHC of KPC1245 PDAC tumors for CD8 (scale bar, 20 μm; top) and IF staining of CD8 (yellow) and Ki67 (red) in KPC1245-PDAC tumors (bottom). G, Quantification of CD8+ cells/40× (n = 3; top) and quantification of Ki67+CD8+ cells/40× (bottom) in tissue sections. Scale bar, 20 μm H. FACS plots (right) and quantification (left) of CD3+, CD4+ CD8+ T cells, CD44+CD62L−, CD69+ T cells in KPC1245 PDAC. I, Relative mRNA expression of Ifnγ and Gzmb in KPC1245 tumors (n = 3). J, Top, schema showing administration of different drugs together with anti-CD8 depleting antibodies. Bottom, weight of pancreata containing KPC1245 tumors. Significance testing was performed by one-way ANOVA with Tukey post hoc multiple pairwise testing. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant.
Remarkably, we observed that R788 treatment increased infiltration of CD8+ T cells as compared with vehicle tumors (Fig. 5F–H). Furthermore, these CD8+ T cells are more proliferative in the R788-treated group or Gem + R788-treated group as compared with vehicle or Gem (Fig. 5F and G). In addition, PDAC infiltrating CD8+ T cells were highly activated in R788 and Gem + R788-treated group as they exhibit increased infiltration of CD44+CD62L− effector T cells and CD69+ T cells as well as enhanced gene expression of Ifnγ and Gzm in treated tumors (Fig. 5H and I). Similarly, we found higher infiltration of CD69+ CD8+ T cells in the R788-treated KPC1242 tumors (Supplementary Fig. S9A–S9B). Most notably, we did not observe any significant changes in the expression of exhaustion markers PD1 and TIGIT on tumor infiltrating CD8+ T cells in the R788-treated tumors (Supplementary Fig. S9C–S9D). We found that depletion of CD8+ T cells in R788-treated tumors or Gem + R788-treated tumors restored the tumor growth in both groups, with no changes in tumor growth in the Gem-treated group (Fig. 5J), elucidating that synergy between Gem and R788 treatment is CD8+ T-cell dependent. Altogether these results indicate that Gem treatment promotes immune suppressive polarization of macrophages (this is actually a wound-healing response), but when combined with Syk inhibitors it remodels the TME towards immune stimulation, boosted T-cell proliferation, and activation to improve antitumor immune responses in PDAC.
R788 remodels the immune microenvironment in ex vivo murine and human PDAC slice cultures
To determine whether R788 together with Gem can improve antitumor immune responses in spontaneous murine models and human PDAC, we employed a tumor slice culture model as shown in Fig. 6A. Similar to the results observed in mouse orthotopic tumor models, we found that slices derived from KPC GEMM mouse tumors and treated with Gem showed increased expression of Arg, Vegf, Tgfβ, Ccl2, and Il10 as compared with vehicle slices, showing immunosuppressive TME in Gem-treated tumor slices (Fig. 6B–F). However, the treatment of these slices with R788 downregulated the expression of these immunosuppressive genes (Fig. 6B–F) and upregulated the expression of Ifng, and Gzmb (Fig. 6G and H). R788 and Gem treatment also reduced the protein expression of MCP1 (CCL2; Fig. 6I).
Figure 6.

R788 remodels immune microenvironment in KPC GEMM PDAC slice culture. A, Schematic representation of slice culture technique used for generating human and KPC GEMM PDAC slices. B–H, Relative mRNA expression of immune-response genes in the KPC GEMM PDAC slices treated with either 250 nmol/L Gem or 2.5 μmol/L R788 or a combination of both drugs. I, Concentration of MCP-1 (CCL2) in Gem and/or R788-treated slices. Significance testing was performed by one-way ANOVA with Tukey post hoc multiple pairwise testing. ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Similarly, treatment of human PDAC slices with Gem and R788 led to the reduction in the mRNA expression of ARG1, VEGF, IL10, and TGFB in three different patient PDAC samples (Fig. 7A–C; Supplementary Table S1) and decreased the secretion of TGFβ (Supplementary Fig. S10). We also found that R788 treatment alone was able to reduce the CD206+ TAMs without any significant changes in the number of CD68+ macrophages as shown by IHC (Fig. 7D and E). Most notably, we also observed an increase in the number of CD8+ T cells in R788-treated slices as well as Gem + R788-treated slices, suggesting an effect on T-cell proliferation (Fig. 7D–F).
Figure 7.

R788 reduces the expression of markers associated with immunosuppressive TAMs and augments immunostimulatory responses in human PDAC slice culture ex vivo. A–C, Relative mRNA expression of immunosuppressive genes in the human PDAC slices treated with either 250 nmol/L Gem and/or 2.5 μmol/L R788 from patient 1 (A), patient 2 (B), and patient 3 (C). Significance testing was performed by one-way ANOVA with Tukey post hoc multiple pairwise testing. D and E, IHC (D) of human PDAC slices derived from donor 1 for CD206+, CD68+ macrophages, and CD8+ T cells (scale bar, 20 μm) and quantification (E) of CD68+, CD206+, and CD8+ cells/40× field. F, IHC of human PDAC slices derived from patient 4 for CD8+ T cells. Scale bar, 20 μm. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant.
Discussion
PDAC tumors exhibit profound infiltration of macrophages, which promote immunosuppression, and relapse from therapy (12, 18). It is now well accepted that relieving the immunosuppressive activities of these cells to enhance cytotoxic T-cell activity and to mobilize CD8+ T cells in the tumor stroma, can prove useful for mounting durable antitumor immune responses (10, 11). Here, we have identified Syk as a potential regulator of immunosuppression and metastasis in pancreatic cancer. Contrary to a previous study by Layton and colleagues (23), we found higher expression and activation of Syk in CD68+ TAMs infiltrating in mouse and human PDAC, with no expression of Syk in pancreatic cell lines. We found that macrophage Syk plays a critical role in regulating PDAC growth and metastasis. Our studies reveal that Syk promotes immunosuppressive transcriptional programming of macrophages by stimulating the expression of Arg, Ccl2, Il10, Mmp9, and Tgfβ. In contrast, inhibition of this kinase increased the expression of Il12, Ifng, and Nos2 and skewed the macrophages toward an immunostimulatory phenotype in vitro and in vivo. This reprogramming of macrophages towards an immunostimulatory phenotype in Syk-inhibitor treated tumors was accompanied by increased CD8+ T-cell recruitment to PDAC tumors, enhanced CD8+ T-cell proliferation, increased T-cell expression of Ifng, Gzm, and Prf and bolstered CD8+ T-cell responses in the PDAC tumors. We found that Syk inhibitors synergize with Gem to slow PDAC growth in a T-cell-dependent manner. Finally, using slice cultures established from resected human PDAC tumor specimens, we show that Syk inhibitors, either alone or in combination with Gem, decrease the expression of markers associated with an immunosuppressive phenotype.
Syk can be activated downstream of various receptors and play diverse biological functions (36). Similar to our observation in lung adenocarcinoma tumors (20), macrophage Syk is activated downstream of α4β1 integrin in the PDAC tumors. In contrast to lung tumors, Syk is also activated downstream of Fc gamma receptors in the PDAC TME. In a related study, BTK-mediated signaling in B cells and FcRγ-positive macrophages promotes PDAC growth (10). Although, we have not evaluated the Syk-mediated cross-talk between B cells and FcRγ-positive macrophages in PDAC, we observed that Syk signaling in B cells has no role in PDAC growth. Although B cells, are reported to play a protumorigenic role in PDAC progression (10, 40), there are discrepancies in the literature regarding their role, and a recent study has shown that B cells do not favor PDAC progression (41). However, the role of regulatory B cells (Breg) in promoting PDAC progression and immunosuppression has recently been reported by several groups (40, 42). It might be feasible that similar to BTK, Syk has a role in regulating Breg-mediated immunosuppression in PDAC. In our studies, we used CD19 cre mice that depletes all mature and immature B cells and not specifically the Breg population. Hence, future studies will be focused on using specific strategies to deplete Bregs to study the role of Syk in Breg differentiation and Breg-mediated immunosuppression in PDAC.
We demonstrated that administration of anti-PDL1 mAb alone reduced tumor growth in orthotopic mouse models as reported before (43–45). However, we did not observe additive effects of targeting Syk and PDL1 together in PDAC. These findings are similar to our studies in the B16 melanoma model, where we demonstrated that Syk inhibitors and anti-PDL1 mAb alone reduced tumor growth but combination of R788 and anti-PDL1 showed no significant additive effect (20). Interestingly, in related studies, PI3Kγ inhibitors or BTK inhibitor ibrutinib reprogrammed the macrophages and slowed PDAC growth but showed no additive effects in combination with immunotherapies (10, 11).
Notably, our studies have shown that Syk inhibitors can be used as a potential agent to reduce Gem-induced immunosuppression in the PDAC TME. It has been reported before that multiple doses of clinical Gem treatment regimen can induce an immunosuppressive TME and hampers CD8+ T-cell activity in PDAC tumors (46). This study has shown that treatment of KPC GEMM mice with 100 mg/kg dose of Gem for 2 weeks, lead to an increase in PDL1 expression and an increase in the expression of IL1, CCL2, and TGFβ (46). Similarly, Wu and colleagues have shown that multiple doses of clinical Gem treatment regimen induce immunosuppressive TME, although the tumor growth was delayed in the E0771-tumor bearing mice (47). In our studies, we used a low dose of Gem as reported previously (10) and found that Gem reduced tumor growth but induced immunosuppressive transcriptional programming in macrophages, leading to diminished cytotoxic T-cell activity in PDAC tumors. These observations are similar to previous studies demonstrating that PI3Kγ and BTK inhibitors suppress macrophage polarization and improve responses to gemcitabine in PDAC (10, 11).
Interestingly, we also observed that Gem treatment reduced the infiltration and activation of CD8+ T cells. Although the effect of Gem on T cells has not been widely explored (48), one study has shown that Gem treatment reduced CD4+, CD8+ T cells in a non–small lung cancer mouse model (49) and another study has shown that CD3+ T cells and memory T cells decrease in Gem-treated patients with PDAC after second and third infusions (50). Our studies on orthotopic mouse models showed that R788 reinvigorates cytotoxic T cells by reducing macrophage-mediated immunosuppression, and combining R788 with Gem eliminated the inhibitory effects of Gem in T-cell-dependent manner. Surprisingly, we also observed increased CD8+ T cells in R788-treated slices ex vivo. Similar to our observations in in vivo models, R788 might have increased the proliferation and reactivation of endogenous tumor reactive CD8+ T cells.
Taken together these studies establish a paradigm that Syk has a crucial role in regulating macrophage-mediated immunosuppression in PDAC tumors and concomitantly in calibrating the effective antitumor immune responses in these tumors. Syk inhibitors have long been used in the treatment of B-cell malignancies. However, Syk inhibitors including FDA approved drug fostamatinib (R788) have never been tested in patients with PDAC. The data presented in this manuscript prompt the hypothesis that fostamatinib in combination with gemcitabine may be an effective therapeutic strategy for PDAC that could be rapidly tested in clinical trials.
Supplementary Material
Supplementary table
Acknowledgments
The authors would like to acknowledge UCSD histology core and UCSD microscopy core. This work was supported by NIH grants K22 CA229594, R01NS122835 to Shweta Joshi and Pediatric Padres Pedal award to Shweta Joshi as well as The Fund to Cure Pancreatic Cancer and Ride the Point to Andrew M. Lowy. This work was also supported by NIH grants: R01CA154480, R01CA121941, R01CA247551, U01CA176058, R01DE026870, U24CA220341, U24CA248457, R01CA226803, U01CA217885, and R01CA109467, a State of California Initiative to Advance Precision Medicine award (OPR18112) to Pablo Tamayo, and the GCBSR shared resources at the UCSD Moores Cancer Center P30CA023100.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Disclosures
T.V. Pham reports grants from NIH, State of California Initiative to Advance Precision Medicine award, and GCBSR shared resources at the UCSD Moores Cancer Center during the conduct of the study. A.M. Lowy reports personal fees from Kinnate, Steba Biotech, Rafael, Bluestar Genomics, Merck, Pfizer, and Corcept outside the submitted work. No disclosures were reported by the other authors.
Authors' Contributions
D. Rohila: Conceptualization, data curation, formal analysis, investigation, visualization, methodology, writing–original draft, writing–review and editing. I.H. Park: Conceptualization, data curation, formal analysis, visualization, methodology, writing–review and editing. T.V. Pham: Data curation, software, formal analysis, visualization, methodology, writing–review and editing. J. Weitz: Conceptualization, investigation, methodology, writing–original draft, writing–review and editing. T. Hurtado de Mendoza: Data curation, formal analysis, methodology, writing–review and editing. S. Madheswaran: Data curation, formal analysis, methodology, writing–review and editing. M. Ishfaq: Data curation, formal analysis, methodology, writing–review and editing. C. Beaman: Data curation, formal analysis, methodology, writing–review and editing. E. Tapia: Data curation, formal analysis, methodology, writing–review and editing. S. Sun: Data curation, formal analysis, methodology, writing–review and editing. J. Patel: Data curation, methodology, writing–review and editing. P. Tamayo: Conceptualization, resources, data curation, supervision, validation, investigation, writing–original draft, writing–review and editing. A.M. Lowy: Conceptualization, resources, supervision, funding acquisition, validation, investigation, writing–original draft, writing–review and editing. S. Joshi: Conceptualization, resources, data curation, software, formal analysis, supervision, funding acquisition, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Rawla P, Sunkara T, Gaduputi V. Epidemiology of pancreatic cancer: global trends, etiology and risk factors. World J Oncol 2019;10:10–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McGuigan A, Kelly P, Turkington RC, Jones C, Coleman HG, McCain RS. Pancreatic cancer: a review of clinical diagnosis, epidemiology, treatment and outcomes. World J Gastroenterol 2018;24:4846–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin 2022;72:7–33. [DOI] [PubMed] [Google Scholar]
- 4. Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25. [DOI] [PubMed] [Google Scholar]
- 5. Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol 2011;29:4548–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Watt J, Kocher HM. The desmoplastic stroma of pancreatic cancer is a barrier to immune cell infiltration. Oncoimmunology 2013;2:e26788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schober M, Jesenofsky R, Faissner R, Weidenauer C, Hagmann W, Michl P, et al. Desmoplasia and chemoresistance in pancreatic cancer. Cancers (Basel) 2014;6:2137–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res 2014;74:5057–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 2014;63:1769–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, et al. Bruton tyrosine kinase-dependent immune cell cross-talk drives pancreas cancer. Cancer Discov 2016;6:270–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kaneda MM, Cappello P, Nguyen AV, Ralainirina N, Hardamon CR, Foubert P, et al. Macrophage PI3Kgamma drives pancreatic ductal adenocarcinoma progression. Cancer Discov 2016;6:870–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang S, Liu Q, Liao Q. Tumor-associated macrophages in pancreatic ductal adenocarcinoma: origin, polarization, function, and reprogramming. Front Cell Dev Biol 2020;8:607209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci U S A 2018;115:E4041–E50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol 2017;14:399–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res 2004;64:5839–49. [DOI] [PubMed] [Google Scholar]
- 16. Deshmukh SK, Tyagi N, Khan MA, Srivastava SK, Al-Ghadhban A, Dugger K, et al. Gemcitabine treatment promotes immunosuppressive microenvironment in pancreatic tumors by supporting the infiltration, growth, and polarization of macrophages. Sci Rep 2018;8:12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bulle A, Dekervel J, Deschuttere L, Nittner D, Libbrecht L, Janky R, et al. Gemcitabine recruits M2-Type tumor-associated macrophages into the stroma of pancreatic cancer. Transl Oncol 2020;13:100743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lankadasari MB, Mukhopadhyay P, Mohammed S, Harikumar KB. TAMing pancreatic cancer: combat with a double edged sword. Mol Cancer 2019;18:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang W, Marinis JM, Beal AM, Savadkar S, Wu Y, Khan M, et al. RIP1 kinase drives macrophage-mediated adaptive immune tolerance in pancreatic cancer. Cancer Cell 2018;34:757–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Joshi S, Liu KX, Zulcic M, Singh AR, Skola D, Glass CK, et al. Macrophage Syk-PI3Kgamma inhibits antitumor immunity: SRX3207, a novel dual Syk-PI3K inhibitory chemotype relieves tumor immunosuppression. Mol Cancer Ther 2020;19:755–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Joshi S, Singh AR, Zulcic M, Bao L, Messer K, Ideker T, et al. Rac2 controls tumor growth, metastasis and M1-M2 macrophage differentiation in vivo. PLoS One 2014;9:e95893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Newland A, Lee EJ, McDonald V, Bussel JB. Fostamatinib for persistent/chronic adult immune thrombocytopenia. Immunotherapy 2018;10:9–25. [DOI] [PubMed] [Google Scholar]
- 23. Layton T, Stalens C, Gunderson F, Goodison S, Silletti S. Syk tyrosine kinase acts as a pancreatic adenocarcinoma tumor suppressor by regulating cellular growth and invasion. Am J Pathol 2009;175:2625–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005;7:469–83. [DOI] [PubMed] [Google Scholar]
- 25. Andrews FH, Singh AR, Joshi S, Smith CA, Morales GA, Garlich JR, et al. Dual-activity PI3K-BRD4 inhibitor for the orthogonal inhibition of MYC to block tumor growth and metastasis. Proc Natl Acad Sci U S A 2017;114:E1072–E80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Joshi S, Singh AR, Liu KX, Pham TV, Zulcic M, Skola D, et al. SF2523: dual PI3K/BRD4 inhibitor blocks tumor immunosuppression and promotes adaptive immune responses in cancer. Mol Cancer Ther 2019;18:1036–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Seo YD, Jiang X, Sullivan KM, Jalikis FG, Smythe KS, Abbasi A, et al. Mobilization of CD8(+) T cells via CXCR4 blockade facilitates PD-1 checkpoint therapy in human pancreatic cancer. Clin Cancer Res 2019;25:3934–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weitz J, Hurtado de Mendoza T, Tiriac H, Lee J, Sun S, Garg B, et al. An ex-vivo organotypic culture platform for functional interrogation of human appendiceal cancer reveals a prominent and heterogenous immunological landscape. Clin Cancer Res 2022;28:4793–806. [DOI] [PubMed] [Google Scholar]
- 29. Badea L, Herlea V, Dima SO, Dumitrascu T, Popescu I. Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepatogastroenterology 2008;55:2016–27. [PubMed] [Google Scholar]
- 30. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 2017;14:417–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhu A, Ibrahim JG, Love MI. Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences Bioinformatics 2019;35:2084–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov 2018;17:887–904. [DOI] [PubMed] [Google Scholar]
- 35. Nielsen SR, Quaranta V, Linford A, Emeagi P, Rainer C, Santos A, et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat Cell Biol 2016;18:549–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mocsai A, Ruland J, Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol 2010;10:387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yi YS, Son YJ, Ryou C, Sung GH, Kim JH, Cho JY. Functional roles of Syk in macrophage-mediated inflammatory responses. Mediators Inflamm 2014;2014:270302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen SJ, Lian GD, Li JJ, Zhang QB, Zeng LJ, Yang KG, et al. Tumor-driven like macrophages induced by conditioned media from pancreatic ductal adenocarcinoma promote tumor metastasis via secreting IL-8. Cancer Med 2018;7:5679–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ding M, He SJ, Yang J. MCP-1/CCL2 mediated by autocrine loop of PDGF-BB promotes invasion of lung cancer cell by recruitment of macrophages via CCL2-CCR2 axis. J Interferon Cytokine Res 2019;39:224–32. [DOI] [PubMed] [Google Scholar]
- 40. Pylayeva-Gupta Y, Das S, Handler JS, Hajdu CH, Coffre M, Koralov SB, et al. IL35-producing B cells promote the development of pancreatic neoplasia. Cancer Discov 2016;6:247–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Spear S, Candido JB, McDermott JR, Ghirelli C, Maniati E, Beers SA, et al. Discrepancies in the tumor microenvironment of spontaneous and orthotopic murine models of pancreatic cancer uncover a new immunostimulatory phenotype for B cells. Front Immunol 2019;10:542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Das S, Bar-Sagi D. BTK signaling drives CD1d(hi)CD5(+) regulatory B-cell differentiation to promote pancreatic carcinogenesis. Oncogene 2019;38:3316–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mace TA, Shakya R, Pitarresi JR, Swanson B, McQuinn CW, Loftus S, et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 2018;67:320–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pan Y, Fei Q, Xiong P, Yang J, Zhang Z, Lin X, et al. Synergistic inhibition of pancreatic cancer with anti-PD-L1 and c-Myc inhibitor JQ1. Oncoimmunology 2019;8:e1581529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li E, Huang X, Zhang G, Liang T. Combinational blockade of MET and PD-L1 improves pancreatic cancer immunotherapeutic efficacy. J Exp Clin Cancer Res 2021;40:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Principe DR, Narbutis M, Kumar S, Park A, Viswakarma N, Dorman MJ, et al. Long-term gemcitabine treatment reshapes the pancreatic tumor microenvironment and sensitizes murine carcinoma to combination immunotherapy. Cancer Res 2020;80:3101–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu C, Tan X, Hu X, Zhou M, Yan J, Ding C. Tumor microenvironment following gemcitabine treatment favors differentiation of immunosuppressive Ly6C(high) myeloid cells. J Immunol 2020;204:212–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mandili G, Curcio C, Bulfamante S, Follia L, Ferrero G, Mazza E, et al. In pancreatic cancer, chemotherapy increases antitumor responses to tumor-associated antigens and potentiates DNA vaccination. J Immunother Cancer 2020;8:e001071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nowak AK, Robinson BW, Lake RA. Gemcitabine exerts a selective effect on the humoral immune response: implications for combination chemo-immunotherapy. Cancer Res 2002;62:2353–8. [PubMed] [Google Scholar]
- 50. Plate JM, Plate AE, Shott S, Bograd S, Harris JE. Effect of gemcitabine on immune cells in subjects with adenocarcinoma of the pancreas. Cancer Immunol Immunother 2005;54:915–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table
Data Availability Statement
Gene expression microarray data of matched tumor and normal samples from Pancreatic Adenocarcinoma patients were downloaded from GEO accession GSE15471. RNA-seq expression data of IL4 polarized BMDMs from SykMC-WT and SykMC-KO were downloaded from GEO accession GSE115109. scRNA-seq data from human PDAC tumor samples and normal pancreases were downloaded from accession CRA001160 available at https://ngdc.cncb.ac.cn/gsa/browse/CRA001160. All other raw data are available upon request from the corresponding author. Data and code used for analysis are available as a Code Ocean capsule (https://codeocean.com/capsule/5100346/tree/v1).