

Abstract
Collagen vascular disorders (CVDs), also known as connective tissue diseases (CTDs), are a heterogeneous group of entities that affect the connective tissues and are capable of causing end-organ damage to multiple systems, primarily cardiopulmonary and musculoskeletal. However, the occurrence and severity are highly variable among patients. Ocular involvement occurs in a significant number of these disorders and may precede the onset of other extraocular features, thereby serving as an important marker in the diagnosis of these diseases. A timely and accurate diagnosis enables the management of complications. CTDs are primarily immune-mediated inflammatory diseases; however, classifications have encompassed heritable disorders affecting collagen-containing structures and disorders of vascular development. A review of literature published until 25 January 2022 and collected from various databases using the relevant keywords was conducted. All publications (original articles, review articles, as well as case reports) describing the ocular features in CTDs were studied in detail. The objective of this review is to recognize the common ophthalmic presentations of various autoimmune and heritable CTDs, distinguish them from overlapping diseases, elaborate on the prognosis and management of these varied eye presentations, and deliberate on their impact on other ophthalmic surgeries.
Keywords: Autoimmune diseases, collagen disorder, connective tissue, heritable disorders, Marfan syndrome, ocular abnormalities, rheumatoid arthritis
Collagen vascular disorders (CVDs) or connective tissue diseases (CTDs) are primarily multisystem disorders affecting the synthesis of proteins that create the connective tissue framework of the body. Their diagnosis and management are often an interdisciplinary challenge. Ophthalmic presentations may be the primary presenting clinical feature.[1] These disorders have characteristic symptoms, signs, and autoantibodies to clinch the diagnosis. The common autoimmune CTDs are rheumatoid arthritis (RA), scleroderma, systemic lupus erythematosus (SLE), inflammatory myopathies (polymyositis [PM]/dermatomyositis [DM]), and Sjögren’s syndrome.[2] Certain heritable disorders that affect the production of extracellular matrix (ECM) like collagen, elastin, fibrillin, fibulin, and mucopolysaccharides are also included under the umbrella of CVDs. They affect the bones, joints, ligaments, skin, blood vessels, and valves. The commonly included disorders are Marfan syndrome (MFS), Ehler–Danlos syndrome (EDS), osteogenesis imperfecta (OI), Alport syndrome, Stickler syndrome (SS), and a few others.[3]
This review aims to summarize the ophthalmic manifestations and management of various CTDs, their systemic associations, and recent updates.
Autoimmune Disorders
The most frequently encountered CVDs are SLE, scleroderma, RA, inflammatory myopathies (PM and DM), and Sjögren’s syndrome
Systemic lupus erythematosus
SLE is a chronic, immune-mediated, multisystem condition that might affect various parts of the eye. Of the affected individuals, 90% are usually women in the age group of 15–45 years.[4] Fever, headache, malaise, arthralgias, myalgias, and loss of appetite are some constitutional features. Renal disease is usually the primary cause of death. Skin, neurologic, vascular, and hematologic systems are a few other commonly involved systems. SLE may be triggered by stress, infection, sunlight, pregnancy, and a few drugs such as oral contraceptive pills (OCPs) and sulfonamides.
B-cell lymphocyte hyperactivity leading to the production of numerous antibodies and immune complexes causing specific tissue damage is the primary pathogenesis; however, T-cell abnormalities are seen as well. The recent diagnostic criteria by the SLE International Collaborating Clinics (SLICC)[5] have been summarized in Table 1. Laboratory findings include the presence of antinuclear antibodies (ANA) not specific to SLE. Anti-ds DNA and anti-Sm antibodies are quite specific to SLE.
Table 1.
SLICC diagnostic criteria
| Clinical criteria | |
|---|---|
| 1 | Acute cutaneous lupus, including: Malar rash/bullous lupus/toxic epidermal necrolysis variant/maculopapular lupus rash/photosensitive lupus rash In the absence of dermatomyositis OR subacute cutaneous lupus |
| 2 | Chronic cutaneous lupus, including: Classic discoid rash/hypertrophic lupus/lupus panniculitis/mucosal lupus/lupus erythematosus tumidus/chilblains lupus/discoid lupus/lichen planus overlap |
| 3 | Oral, palate (buccal, tongue) OR nasal ulcers In the absence of other causes |
| 4 | Nonscarring alopecia In the absence of other causes |
| 5 | Synovitis- including >2 joints, characterized by swelling or effusion OR tenderness in >2 joints and at least 30 min morning stiffness |
| 6 | Serositis- typical pleurisy for more than 1 day OR pleural effusions OR pleural rub Typical pericardial pain OR pericardial effusion OR pericardial rub OR pericarditis by ECG |
| 7 | Renal- urine protein-to-creatinine ratio representing 500 mg protein/24 h OR red blood cell casts |
| 8 | Neurologic- seizures/psychosis/mononeuritis multiplex/myelitis/peripheral or cranial neuropathy/acute confusional state In the absence of other causes |
| 9 | Hemolytic anemia |
| 10 | Leukopenia (<4000/mm3 at least once) OR lymphopenia (<1000/mm3 at least once) In the absence of other causes |
| 11 | Thrombocytopenia (<100,000/mm3 at least once) |
|
| |
| Immunologic criteria | |
|
| |
| 1 | ANA level- above the laboratory reference range |
| 2 | Anti-ds DNA antibody level- above the laboratory reference range |
| 3 | Anti-Sm- the presence of antibody to Sm nuclear antigen |
| 4 | Antiphospholipid antibody positivity as determined by any of the following: Positive test result for lupus anticoagulant/false-positive test result for rapid plasma reagin/medium or high titer anticardiolipin antibody level/positive test result for anti-b2-glycoprotein 1 |
| 5 | Low complement (C3/C4/CH50) |
| 6 | Direct Coombs test In the absence of hemolytic anemia |
For a diagnosis of SLE, at least four criteria, including at least one clinical and one immunologic criterion, must be satisfied or the patient must have biopsy-proven lupus nephritis in the presence of antinuclear antibodies or anti–double-stranded DNA antibodies. ANA=antinuclear antibodies, SLE=systemic lupus erythematosus, SLEICC=SLE International Collaborating Clinics
Ocular manifestations
Though ocular involvement is not included in the SLICC criteria, they might present well in advance of a definitive diagnosis. Various ocular manifestations have been summarized in Table 2. Jawahar et al.[6] found that the prevalence of each ocular manifestation, except for retinal vaso-occlusive disease, was consistently less than 5%. The overall prevalence of dry eye disease (DED) was 16% among patients with SLE in a recent meta-analysis, where it was found that at least two positive findings were present in them among the subjective symptoms, tear function test, and vital staining test.[7] Studies show that up to 29% of patients with active SLE manifest retinal disease.[8] The exact prevalence of choroidal disease is unknown, but it is thought to be less common than retinopathy due to underdiagnosis. Optic nerve disease, represented by optic neuritis and anterior/posterior ischemic optic neuropathy, affects approximately 1% of SLE patients.[7] These ocular manifestations have been associated with neurologic flares, antiphospholipid antibodies, nephropathy, and increased mortality.
Table 2.
Ocular manifestations of SLE
| Ocular Structure | Common Ocular Manifestations |
|---|---|
| Eyelids | Discoid lupus of the eyelids |
| Cornea and conjunctiva | Commonly- KCS, conjunctivitis Others include superficial punctate and interstitial keratitis, PUK |
| Sclera | Episcleritis/scleritis |
| Uvea | Uveitis, ischemic choroidopathy, choroidal vasculitis |
| Retina | Commonly- vascular occlusions (venous and arterial) Others include cotton wool spots, hemorrhages, vasculitis, proliferative retinopathy, pseudo-RP–like retinopathy, serous detachments |
| Neuro-ophthalmic | VI nerve palsy, optic neuritis, ischemic optic neuropathy, INO, pupil and oculomotor abnormalities, hemianopia and amaurosis, visual hallucinations, geniculocalcarine blindness |
| Orbit | Myositis, pseudotumor, trochlearitis, vasculitis, paniculitis |
INO=internuclear ophthalmoplegia, KCS=keratoconjunctivitis sicca, PUK=peripheral ulcerative keratitis, RP=retinitis pigmentosa, SLE=systemic lupus erythematosus
Dry eye symptoms and abnormal Schirmer’s test were found in 26% and 24% of patients with SLE, respectively, and 12% of patients also met the criteria of secondary Sjögren’s syndrome.[6] Retinal vasculitis, uveitis, and isolated cotton wool spots were associated with more active SLE disease. Retinopathy in SLE is suggestive of high disease activity, and it is a marker of poor visual outcome and prognosis for survival.[9]
Ocular investigations
Corneal confocal microscopy identifies distal corneal nerve fiber loss and increased immune cell density in patients with SLE, and corneal nerve loss is associated with disease activity.[10] Patients suspected of SLE should receive optical coherence tomography angiography (OCTA) examination in a comprehensive eye examination to detect changes in ocular microcirculation at an early stage.[11] Liu et al.[12] demonstrated decreased inner retinal thickness and superior vascular in OCTA.
Management
Adequate systemic treatment is required for disease control using various drugs as follows: immunosuppressants (hydroxychloroquine, azathioprine, mycophenolate, cyclophosphamide, and glucocorticoids) and biologicals (rituximab, belimumab, epratuzumab). Anticoagulants are required in patients with antiphospholipid syndrome. Ophthalmic manifestations may be an indicator that the underlying SLE is not adequately managed. They are managed symptomatically; for instance, DED may be managed based on Dry Eye WorkShop (DEWS) recommendations. Few reports have highlighted the role of intravenous corticosteroid (methylprednisolone pulse therapy) along with a tapering regimen of oral prednisolone despite treatment with systemic azathioprine in vision-threatening conditions.[13] Rituximab, in particular, has been found beneficial in SLE retinal vasculitis and orbital pseudotumor that is refractory to steroids and cyclophosphamide.[14]
Scleroderma
Scleroderma is s chronic, acquired CTD that results in progressive thickening of the connective tissues.[15] It could be classified into two major forms – localized scleroderma (involving only skin) and systemic sclerosis (SSc; multisystem involvement). Localized scleroderma is subdivided into limited morphea, generalized morphea, and linear scleroderma. SSc is subdivided into limited and diffuse based on the extent of skin involvement.[16]
The primary site of pathogenesis is the small vessels such as arterioles and capillaries. Microvascular endothelial cell damage leads to initial increase in permeability and interstitial edema, followed by fibroblast and myofibroblast stimulation that results in fibrosis and ischemic injury. Trophic factors such as Platelet Derived Growth Factor (PDGF) play a critical role in the regulation of collagen synthesis. Helper T cells play a vital role in pathogenesis as a treatment against them helps in skin tightness in these patients.[17,18] The diagnostic criteria were laid by the American College of Rheumatology/European League Against Rheumatism, which included skin thickening proximal to metacarpophalangeal joints, fingertip lesions like ulcers or pitting scars, telangiectasia, abnormal nailfold capillaries, Raynaud phenomenon, pulmonary artery hypertension with/without interstitial lung disease, and SSc-related antibodies (anticentromere, anti-topoisomerase I [anti–Scl-70], anti-RNA polymerase III); however, no ocular features were included in the diagnostic criteria.[19]
Raynaud’s phenomenon is the most common finding in scleroderma patients (90%). Skin and gastrointestinal tract are the commonly involved organs in SSc.[20] Cardiac (myocardial fibrosis, dysrhythmias, myocardial ischemia, and heart failure) and pulmonary (interstitial fibrosis, pulmonary hypertension, pleuritis) involvement must be looked for in these patients. Renal failure is the leading cause of death in patients with SSc.[21]
ANA can be detected in >90% of the patients. Other major antibodies present are anticentromere antibodies (almost exclusively in limited cutaneous SSc), anti-topoisomerase, anti-RNA polymerase, and antihistone antibodies (in diffuse cutaneous SSc).
Ocular manifestations
A systematic review revealed limited proven associations and association with eye, mainly in terms of DED and choroidal thickness.[22] Vision-threatening occlusive retinal vasculitis may develop in selected patients with SSc.[23] The presence of elevated anti-phospholipid antibody titers may confer increased risk for this vision-threatening complication.[23] SSc itself may affect retinal and choroidal microvasculature, independent of hypertension.[24] Various ocular manifestations are compiled in Table 3.
Table 3.
Ocular manifestations of scleroderma
| Ocular Structure | Common Ocular Manifestations |
|---|---|
| Eyelids | Periorbital edema and fibrosis leading to blepharophimosis, lid telangiectasia, lid induration, cicatricial ectropion, MGD |
| Cornea and conjunctiva | KCS, subepithelial fibrosis, and progressive forniceal shortening are most common. Congestion and telangiectasia, exposure keratopathy (secondary to lid changes), decreased corneal sensation, keratoconus |
| Sclera | Scleral ischemia, scleral pit |
| Uvea | Iritis, iris sector atrophy and heterochromia, iris sphincter dysfunction, choroidal hypoperfusion, and ischemic choroidopathy |
| Retina | Cotton wool spots, retinal hemorrhages, hard exudates, retinal vascular occlusions, macular ischemia, parafoveal telangiectasia |
| Neuro-ophthalmic | Optic disk neovascularization, perineuritis |
| Orbit | Acquired Brown syndrome, painful retrobulbar fibrosis, ocular myopathy (superior rectus commonly) |
KCS=keratoconjunctivitis sicca, MGD=meibomian gland dysfunction
Ocular investigations
In vivo confocal microscopy of the cornea in patients with SSc demonstrated lower keratocyte cell densities in the anterior stroma and significantly lower sub-basal nerve fiber parameters.[25] Multimodal evaluation in SSc demonstrated reduced anterior chamber depth (ACD), corneal elasticity, and endothelial density before there are any detectable changes in corneal thickness or intraocular pressure (IOP).[26] Nagy et al.[27] demonstrated that all pachymetric values, corneal volume, and ACD were significantly lower among SSc patients. Corneas were found to be steeper in these patients.[28]
Decrease in radial peripapillary capillary network and macular microvascular vessel densities and changes in Optic nerve Head (ONH) parameters were found in OCTA.[29,30] However, this difference was not found with limited scleroderma.[31] OCTA can disclose early ocular vascular abnormalities, which could have a potential role in diagnosis, monitoring, and prognosis stratification in SSc.[32]
Management
Scleroderma is difficult to treat, and the management is directed toward specific organ involvement. D-Penicillamine is useful to reduce interstitial fibrosis. Immunosuppressive drugs are necessary when there is life-threatening renal and pulmonary involvement. Antiplatelet drugs and calcium channel blockers are required to tackle Raynaud’s phenomenon. Ocular management involves copious lubricants to treat keratoconjunctivitis sicca (KCS). Periocular skin involvement benefits from systemic colchicine and D-penicillamine.[33] Severe vision-threatening cases of retinopathy and choroidopathy benefit from systemic steroids, chlorambucil, azathioprine, and cyclophosphamide.
Rheumatoid arthritis
RA is a chronic, debilitating, autoimmune CTD affecting primarily the joints with multisystem involvement. Initial inflammation of the microvasculature (postcapillary venules) facilitates recruitment of various inflammatory cells (T and B cells, macrophages) and an array of cytokines (tumor necrosis factor [TNF] and interleukin [IL]-1, IL-6, gamma-interferon [γ-IFN], and matrix metalloproteinases), resulting in chronic synovial inflammation and destruction of the connective tissue. IL-17 has especially been found to play a crucial role in pathogenesis, which gives an opening for new targeted treatment strategies.[34] Similarly, collagen and proteoglycans present in the cornea and sclera get involved.
Patients predominantly present with symmetric involvement of the proximal interphalangeal, metacarpophalangeal, and wrist joints. They usually complain of pain, joint swelling, and morning stiffness (lasting >1 h) along with deformities in chronic cases.[35] Various extra-articular manifestations include subcutaneous nodules (periarticular and pressure areas), pulmonary manifestations (interstitial fibrosis, pleuritis, pneumonitis), cardiac involvement (pericarditis, myocarditis, endocardial and valvular nodules), rheumatoid vasculitis, and hematological and neurological manifestations.
Rheumatoid factor (RF) is usually present in 30% of the RA patients; however, this is nonspecific. Anticitrullated peptide antibodies have enhanced the diagnostic specificity and are also especially useful as a prognostic marker. Recent RA classification criteria given by American College of Rheumatology/European League Against Rheumatism have been compiled in Table 4.[36]
Table 4.
Rheumatoid arthritis classification criteria: American College of Rheumatology/European League Against Rheumatism[36]
| Ocular Structure | Common Ocular Manifestations |
|---|---|
| Joint involvement | |
| One large joint | 0 |
| 2–10 large joints | 1 |
| One to three small joints, ±large joint | 3 |
| >10 joints (at least one small joint) | 5 |
| Serology (need at least one) | |
| Negative RF, negative anti-CCP | 0 |
| Low positive RF or low positive anti-CCP | 2 |
| High positive RF or high positive anti-CCP | 3 |
| Acute phase reactants (need at least one) | |
| Normal CRP, normal ESR | 0 |
| Abnormal CRP or abnormal ESR | 1 |
| Duration of symptoms | |
| <6 weeks | 0 |
| >6 weeks | 1 |
>6/10- definitive RA. CRP=C-reactive protein, ESR=erythrocyte sedimentation rate, RA=rheumatoid arthritis, RF=rheumatoid factor
Ocular manifestations
In a large study, DED was found to be the most common ocular manifestation in RA (28%).[37] Other manifestations include episcleritis (3%), scleritis (2%), peripheral ulcerative keratitis (PUK) (1%), and sclerosing keratitis (1%). There is a strong association between the presence of Anti-cyclic citrullinated peptide (anti-CCP) antibodies and ocular manifestations of RA. RA is a major cause of secondary Sjögren’s syndrome; hence, DED has to be considered regardless of RA disease activity/severity.
DED can result from meibomian gland dysfunction (MGD) or lacrimal gland or goblet cell dysfunction. Abd-Allah et al.[38] found the severity of DED is not correlated with the activity of RA, but with its duration. The cornea in these patients is found to be thinner and steeper compared to healthy individuals.[39] PUK is a serious, vision-threatening complication presenting with peripheral thinning, neovascularization, and epithelial defect. PUK could be a sign of impending systemic vasculitis similar to scleritis.[40,41] Various ocular manifestations in RA are summarized in Table 5.
Table 5.
Ocular manifestations of rheumatoid arthritis
| Ocular Structure | Common Ocular Manifestations |
|---|---|
| Cornea and conjunctiva | KCS (most common), filamentary keratitis, peripheral ulcerative keratitis (contact lens cornea), sterile central ulceration/keratolysis, sclerosing keratitis, secondary microbial keratitis |
| Sclera | Episcleritis, scleritis (diffuse/nodular/scleromalacia perforans/necrotizing with inflammation) |
| Others | Subcutaneous rheumatoid nodules on eyelid (especially at the outer canthus of upper lids), venous stasis retinopathy, orbital apex syndrome (due to orbital nodules), cranial nerve palsies, geniculocortical blindness, vertebrobasilar insufficiency, Brown syndrome (inflammation of trochlea) |
KCS=keratoconjunctivitis sicca
Ocular investigations
Detailed DED evaluation including Schirmer’s test, tear film break-up time, tear osmolarity, and staining with rose bengal, fluorescein, or lissamine green should be done.[40] Noninvasive break-up time using interferometry, topography, confocal microscopy, and aberrometry should also be considered.[42]
Ultrasonography (USG) reveals T-sign or any associated exudative retinal detachment in cases with posterior scleritis. Meibography or LipiView can be used to analyze the meibomian glands and conjunctival impression cytology to analyze the goblet cells.[43]
Management
Early initiation of the conventional synthetic disease-modifying antirheumatic drug (csDMARD) helps in reducing further disability. Commonly used csDMARDs include methotrexate, leflunomide, sulfasalazine, and hydroxychloroquine. A short course of systemic glucocorticoid therapy should be considered during initiation or changing treatment. Biologicals such as infliximab, adalimumab, and rituximab should also be considered. TNF inhibitors improve tear production and promptly recover the goblet cells, which could also be used as a biomarker to look for the response to treatment.[44]
Ocular management for dry eye includes lubricating drops, topical cyclosporine, and oral pilocarpine (secretagogues). Autologous serum eye drops can also be considered. Varenicline solution (0.03 mg) was the first approved nasal spray for the treatment of DED.[45] Lifitegrast, a novel T-cell inhibitor (leukocyte function-associated antigen [LFA-1] and intracellular adhesion molecule-1 [ICAM-1]), could also be considered.[46] Punctal occlusion, amniotic membrane transplantation, and tarsorrhaphy can be performed in severe cases, along with medical management.
Systemic steroids with or without immunologic agents are the mainstay treatment for PUK. Additionally, topical medroxyprogesterone and acetylcysteine (collagenase inhibitors) and oral tetracycline (Matrix metalloproteinase Inhibitors (MMP) inhibitors) should be considered. Surgical interventions for tectonic support, such as tissue adhesives, Amniotic Membrane Graft (AMG), and keratoplasty (lamellar/full-thickness patch grafts), should be considered in cases of severe thinning.[47]
Episcleritis is usually self-limiting, and a short course of low potent topical steroids or nonsteroidal anti-inflammatory drugs (NSAIDs) can be considered for symptomatic relief. Scleritis, however, is a painful, vision-threatening condition. Topical therapies are generally ineffective in scleritis, and systemic therapy with NSAIDs or corticosteroids is mandatory. In refractory cases, infliximab and adalimumab are currently recommended.[41] Various other agents such as subconjunctival sirolimus, gevokizumab, rituximab, and tofacitinib are under trial. Tectonic patch graft might be necessary in cases of necrotizing scleritis with severe thinning.
DM and PM
These are idiopathic inflammatory CTDs, characterized by progressive proximal muscle weakness predominantly, due to autoantibodies against the striated muscles. DM is also associated with cutaneous involvement.[48] Pathognomonic cutaneous manifestations include Gottron papules (violaceous papules over dorsal interphalangeal and metacarpophalangeal joints) and heliotrope rash. Other cutaneous findings include Gottron sign (erythematous papules or macules over elbows/knees), facial erythema, poikiloderma, shawl, and V-sign (erythema over the posterior and anterior aspects of the neck, respectively). Various other systems such as cardiac, respiratory, and esophageal are involved.[49] Approximately one-third of patients with DM may be associated with malignancy (paraneoplastic). Ocular involvement other than eyelids is quite rare, and various individual case reports have been described, which are compiled in Table 6.
Table 6.
Ocular manifestations of dermatomyositis
| Ocular Structure | Common Ocular Manifestations |
|---|---|
| Eyelids | Heliotrope rash (periorbital edema along with violaceous discoloration of the eyelids)- characteristic, telangiectatic vessels at the eyelid margin,[15] urticarial patches with fine scaling on eyelids,[15] calcified subcutaneous nodules (calcinosis) with secondary ulceration and cellulitis, poikiloderma, plaque-like atrophic patches, vitiligo or hyperpigmentation[50] |
| Cornea and conjunctiva | Dry eye[51] Conjunctival and episcleral vessel tortuosity[52] |
| Uvea and retina | Chorioretinopathy with paracentral acute middle maculopathy,[53,54] CRVO,[55,56] Purtscher-like retinopathy,[57] frosted branch angiitis,[58] retinal venous stasis[59] |
| Neuro-ophthalmic | Diplopia[60] Papillitis[59] |
| Orbit | Myositis, orbital inflammatory pseudotumor[61] |
CRVO=central retinal vein occlusion
Similar to other CTDs, DM patients have thinner corneas, and decreased corneal parameters are significantly associated with the occurrence of some extramuscular manifestations.[51]
Investigations
Blood investigations include muscle enzymes such as creatine kinase, lactate dehydrogenase, aspartate and alanine aminotransferases, and aldolase. Autoantibodies such as ANA, antisynthetase antibody (most commonly, myositis-specific antibody), and anti-Jo should be looked for. Muscle biopsy of the weak (involved) muscle is the most accurate way of diagnosis. Electromyography of the affected muscle is a noninvasive method for diagnosis.
It is important to rule out any underlying malignancy; colonoscopy, fecal occult blood, urine analysis, mammography, pap smears, and computed tomography (CT) chest, abdomen, and pelvis should be considered.[48]
Management
The first line of treatment is systemic corticosteroids. They are initially started at high doses till the muscle enzyme levels decline and the strength is regained and later tapered over 9–12 months. Immunosuppressants alleviate the long-term side effects of steroids; hence, they should be initiated along with corticosteroids to decrease the need for it. Azathioprine and methotrexate are the usually preferred agents. In resistant cases, mycophenolate mofetil, calcineurin inhibitors, or intravenous immunoglobulin should be administered. Calcinosis could be managed with calcium channel blockers (diltiazem). Physiotherapy and general sun-protective measures should be advised.[48]
Heritable Connective Tissue Disorders
Heritable disorders of the connective tissue (HDCTs) are syndromes that disrupt connective tissue integrity. They include MFS, EDS, Alport syndrome, SS, OI, and a few other disorders. Many patients with HDCTs have refractive errors, commonly myopia, and often seek refractive surgery.
Marfan syndrome
MFS, first described by Antoine Bernard-Jean Marfan, is an autosomal inherited multisystem CTD caused by a mutation in the FBN-1 gene on chromosome 15 encoding for fibrillin-1. This protein is involved in the formation of connective tissue in the cardiovascular, musculoskeletal, ophthalmologic, and pulmonary systems.[62]
The Ghent nosology criteria for diagnosis of MFS stressed the importance of a positive genetic finding to differentiate it from other syndromes which may clinically overlap with MFS.[63] Ectopia lentis (EL) was a major criterion based on this nosology, and other ocular features such as abnormally flat cornea, increased axial length, and hypoplastic iris or ciliary body are included as minor criteria.
These criteria were further revised to give more importance to the characteristic clinical features of MFS, aortic root aneurysm, and EL.[64] The revised Ghent’s criteria emphasized that in the absence of positive family history, the presence of these two cardinal features is enough to establish a diagnosis of MFS. If either of them is absent, the presence of FBN-1 mutation or a combination of systemic associations is required [Table 7a]. The revised criteria offered the advantage of making genetic testing not mandatory; however, at the same time, they gave adequate importance to genetic testing. The only ocular feature included besides EL is myopia >3 D. The revised classification also included a systemic scoring, thereby elaborating on the various phenotypic features [Table 7b].
Table 7a.
Revised Ghent criteria for diagnosing Marfan syndrome and related conditions
| Criteria | Diagnosis |
|---|---|
| Family history of Marfan syndrome absent | |
| AO (Z-score >2) and ectopia lentisa | Marfan syndrome |
| AO (Z-score >2) and FBN1 gene mutation | Marfan syndrome |
| AO (Z-score >2) and systemic scoreb >7a | Marfan syndrome |
| Ectopia lentis and FBN1 gene mutation with known AOc | Marfan syndrome |
| Ectopia lentis with or without systemic score >7b and FBN1 gene mutation Unknown with AOd or no FBN1 gene mutation | Ectopia lentis syndrome |
| AO (Z-score <2) and systemic scoreb >5, with at least one skeletal feature, without ectopia lentis | MASS |
| Mitral valve prolapse, AO (Z-score <2), and systemic scoreb <5, without ectopia lentis | Mitral valve prolapse syndrome |
| Family history of Marfan syndrome present | |
| Ectopia lentis | Marfan syndrome |
| Systemic score>7a | Marfan syndrome |
| AO (Z-score >2) in patients aged >20 years, (Z-score >3) in patients aged <20 yearsa | Marfan syndrome |
aWithout discriminating features of Shprintzen–Goldberg syndrome, Loeys–Dietz syndrome, or vascular form of Ehlers–Danlos syndrome and after collagen biochemistry and TGFBR1 and 2 and COL3A1 gene testing, if indicated. bAs described in Table 7b. cFBN1 gene mutation that has been identified in a patient with aortic aneurysm. dFBN1 gene mutation that has not been associated with aortic root aneurysm/dissection. AO=aortic diameter at the sinuses of Valsalva with indicated Z-score or aortic root dissection; FBN1=fibrillin-1 gene; MASS=mitral valve prolapse, aorta root diameter at the upper limit of normal, striae, and skeletal features similar to Marfan syndrome
Table 7b.
Systemic scoring system for the diagnosis of Marfan syndrome
| Feature (points) | Maximum possible score |
|---|---|
| Chest deformity | 2 |
| Pectus carinatum deformity (2) | |
| Pectus excavatum (1) | |
| Chest asymmetry (1) | |
| Dural ectasia (2) | 2 |
| Facial featuresa (1) | 1 |
| Foot deformity | 2 |
| Hindfoot deformity (2) | |
| Plain pes planus (1) | |
| Mitral valve prolapse, all types (1) | 1 |
| Myopia >3 D (1) | 1 |
| Pneumothorax (2) | 2 |
| Protrusio acetabuli (2) | 2 |
| Reduced elbow extension (1) | 1 |
| Reduced US/LS ratio, bincreased arm/height, and no severe scoliosis (1) | 1 |
| Skin striae (1) | 1 |
| Spine deformity | 1 |
| Scoliosis (1) | |
| Thoracolumbar kyphosis (1) | |
| Wrist and thumb deformities | 3 |
| Wrist sign (1) | |
| Thumb sign (1) | |
| Wrist and thumb signs (3) |
Maximum total score 20. aPresence of three of the following features: dolichocephaly, enophthalmos, downslanting palpebral fissures, malar hypoplasia, retrognathia. bUS length is the total arm span from each finger. LS is measured from the top of the symphysis pubis to the floor. LS=lower segment, US=upper segment
The most devastating feature of MFS is aortic root dissection. However, with the advent of aortic root surgery, the life expectancy of MFS patients has increased.
Ocular manifestations
Ocular features may be the initial presentation even before the cardiovascular manifestations. The most prominent features are superotemporal subluxation of the crystalline lens, myopia, glaucoma, cataract, keratoconus, and retinal detachment.[65]
EL can be present in 50% of eyes and may develop both in childhood as well as in adults. (Fig 1) Studies in different age groups have reported varied frequencies in children; however, there might be a progression of EL after childhood, and most dislocations develop before 18 years of age.[66,67]
Figure 1.
Temporal subluxation of crystalline lens in a patient of Marfan syndrome
Since increased axial length (altered fibrillin allows for increased stretching of sclera) induces myopia and flattened cornea decreases this effect, the refractive error depends on their net effect. However, myopia >3 D tends to be stable over time in adult patients.[68,69]
Cataracts develop at an earlier age than in the general population.[63] The risk of retinal detachment due to increased axial length is between 4% and 15%.[67] Due to the changes in elastic fibers, there is also a higher risk of keratoconus reported in a few studies.[69]
Management
Correction of refractive error with spectacles may suffice in mild cases. If the subluxated lens bisects the pupil, aphakic correction may provide better vision by overcoming lenticular astigmatism. In a few patients, contact lenses provide an acceptable alternative.[70]
Lens extraction is indicated in patients unable to achieve good corrected vision and in those with impending or complete dislocation, secondary glaucoma, or cataract. All efforts should be made to preserve the capsular bag.[71] In cases where the zonular weakness is not extensive, the capsular bag can be preserved and low-flow phacoemulsification is feasible. In some cases, surgery can be performed with the aid of retractors, enabling intralenticular lens aspiration and “in-the-bag fixation of IOL” with appropriate capsular fixation devices. If the lens subluxation is severe, the capsular bag may have to be sacrificed by performing lensectomy and vitrectomy. The IOL can then be fixated to the iris or scleral fixated (sutured and nonsutured).[72] The creation of a well-sized and centered capsulorrhexis is critical and can be achieved manually or with the aid of femtosecond laser-assisted cataract surgery (FLACS).[73] Postoperative complications include IOL decentration, cystoid macular edema, and the risk of retinal detachment.
Management of other complications
Corneal refractive surgery is not recommended for most patients with MFS as the cornea is flat in these cases. Retinal detachment surgery in MFS is challenging because of a thin sclera and the possibility of multiple breaks. Preoperative retinal screening before lens surgery is critical. Retinal screening in the fellow eye should also be done. Patients should be advised to seek consultation in case of flashes, floaters, or loss of visual acuity.[74]
Ehler–Danlos syndrome
The hallmark of EDS is defective connective tissue synthesis, manifesting principally as skin hyperelasticity, joint hypermobility, easy bruising, and tissue fragility. The genetic mutations affect type I, III, and V collagen and are commonly inherited in an autosomal dominant manner. The overall prevalence varies between 1 in 2500 and 1 in 5000.[75] There are at least 13 subtypes identified based on their varying clinical presentations: classical, classical-like, cardiac-valvular, vascular, hypermobile, arthrochalasia, dermatosparaxis, kyphoscoliotic, spondylodysplastic, musculocontractural, myopathic, periodontal, and brittle cornea syndrome (BCS).[76] Classical, kyphoscoliotic, and BCS subtypes are associated with significant ophthalmologic findings (i.e., globe rupture). The other subtypes present with minor ocular presentations.
Ocular manifestations
Ophthalmic features are commonly minor, though not infrequent in EDS. A prospective, cross-sectional study of 44 eyes reported that the most consistent association in EDS included xerophthalmia, steeper corneas, pathologic myopia, vitreous abnormalities, and minor lens opacities.[77] Various ocular manifestations are summarized in Table 8.
Table 8.
Ocular manifestations of Ehler–Danlos syndrome
| Ocular Structure | Common Ocular Manifestations |
|---|---|
| Eyelid and conjunctival abnormalities[78] | Epicanthal folds, telecanthus, floppy eyelids, conjunctivochalasis[79] |
| Strabismus | |
| Cornea[80] | Thin cornea, keratoconus, keratoglobus, damage on minor trauma, brittle cornea,[81] microcornea |
| Globe perforation[82] | Blue sclera, abnormal scleral fragility |
| Xerophthalmia[77] | Higher OSDI scores, lower Schirmer’s and TBUT values |
| Pathologic myopia[77] | |
| Lens opacities[77] | Minor lens opacities |
| Retinal detachment[83] |
OSDI=ocular surface disease index, TBUT=tear break-up time
Importance in ophthalmic surgery
A large case series of EDS reported that 24% of patients required some form of ophthalmic surgery, of which 45.5% had at least one complication, which is an unusually high rate compared to the general population.[84] The complications included regression, dry eye, and corneal ectasia following refractive surgery. After cataract surgery, the reported complications included wound leak, dry eye, retinal detachment, and pigment dispersion. Most patients had undergone primary surgery before their diagnosis of EDS. This highlighted the importance of preoperative screening to look for EDS before surgery, especially refractive surgery.[85]
Refractive errors prompt patients with EDS to seek refractive surgery; however, it is considered as a contraindication to refractive surgery, given the collagen abnormality and reported ectasias after surgery. For kyphoscoliotic EDS and BCS, refractive surgery is not advocated due to corneal thinning and ocular fragility. Corneal perforation has also been reported following crosslinking in these eyes. Those with normal corneal tomography, central corneal thickness (CCT) >500 μm, and no dry eye may receive refractive surgery with appropriate counseling regarding the postoperative complications.[86]
Alport syndrome
Alport syndrome is a heterogeneous disorder that results from mutations in collagen IV genes, thereby involving the glomerular, cochlear, and ocular basement membranes.[87] It was described initially in 1927 as a syndrome of hereditary nephritis and deafness.[88] Ocular features were subsequently described in 1956.[89] It affects one in 5000 newborns, and males are more likely to be affected than females.
Diagnosis is based on genetic testing, the presence of typical clinical features (renal failure, hearing loss, lenticonus), or renal biopsy. The individuals may present with hematuria, proteinuria, and eventually progress to renal failure. Early diagnosis is important as it facilitates close observation of the affected individuals and relatives, thereby enabling early initiation of appropriate treatment.
Collagen IV is found in various basement membranes of the eye, including cornea, lens capsule, and retina. The genes mentioned above code for collagen IV α5, α4, and α3 chains. Mutations occur in the COL4A3, COL4A4, and COL4A5 variants of the gene. The COL4A5 gene is located on the X chromosome, while the other two variants are located on chromosome 2.[90] Hence, the disease can be inherited by the following mechanisms: X-linked, autosomal recessive, and autosomal dominant, resulting in the variants mentioned below:
X-linked form of Alport syndrome (XLAS)- 80%
Autosomal recessive Alport syndrome (ARAS)- 15%
Autosomal dominant Alport syndrome (ADAS)- rare
Ocular manifestations
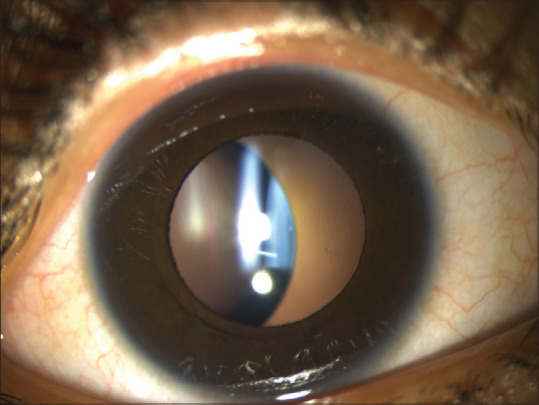
Identifying the cardinal ocular features in Alport syndrome can help in diagnosis, to detect treatable complications such as a macular hole, and to identify the risk of early-onset renal failure. Lenticonus is pathognomonic of Alport syndrome, occurring in one-fourth of these patients. It may result in progressive lenticular myopia and significant astigmatism. It is evident clinically as an “oil droplet” on direct ophthalmoscopy. Anterior lenticonus is absent at birth, but usually develops by the second or third decade of life.[91] Examination on slit lamp reveals a bulge in the anterior lens surface. The lenticonus may be easily missed on an undilated examination. (Fig 2a and b) Small ruptures in the lens capsule can lead to cataract formation.
Figure 2.

(a, b) Retroillumination images of bilateral anterior lenticonus in a patient with Alport syndrome. (c, d) Slit images of anterior lenticonus in the same patient
A recent cross-sectional study described the importance of temporal retinal thinning in this disorder and indicated that the presence of thinning as evidenced on optical coherence tomography (OCT) distinguished Alport syndrome from the more benign thin membrane nephropathy. It is also indicative of a clinically worse disease.[92]
The macular holes in Alport syndrome differ from idiopathic macular holes as they have an earlier age of onset and are larger.[93] Other ocular manifestations are summarized in Table 9.
Table 9.
Ocular manifestations and their management in Alport syndrome
| Abnormality | Description | Symptoms | Treatment | |
|---|---|---|---|---|
| Common presenting features | Anterior lenticonus | Central portion of the lens protrudes forward into the anterior chamber Mostly bilateral | Difficulty in focusing | Corrective spectacles/contact lenses/IOL surgery |
| Dot fleck retinopathy | Whitish or yellowish flecks in the central retina sparing the fovea OR Peripheral coalescent flecks | Do not affect vision | No treatment | |
| Retinal thinning[94] | Thinning temporal macular “staircase pattern” on OCT | -do- Associated with early-onset renal failure | -do- | |
| Other reported abnormalities | Posterior lenticonus | Cone shaped protrusion/bowing on posterior lens surface | Astigmatism | Refractive correction/lens surgery |
| Subcapsular cataracts | Surgery | |||
| Posterior polymorphous dystrophy[95] | Visible on slit-lamp/specular microscopy- thickened DM, endothelial vesicles in isolation or band like clusters. typically, bilateral | Commonly asymptomatic. rarely corneal edema | May require posterior lamellar keratoplasty | |
| Recurrent corneal erosions[95] | Commonly bilateral | Episodes of pain, photophobia, watering | Lubricants, patching, avoidance of triggers | |
| Keratoconus[96] | Refractive correction, crosslinking for progressive disease | |||
| Giant macular hole | Reduced vision | Surgery | ||
| Bull’s eye maculopathy[94] | Choroidal thinning | |||
| Macular retinoschisis[94] | Impairment of basement membrane of RPE- Bruch’s membrane causes passage of fluid into the retina | |||
| Loss of choriocapillaris[121] | Evident on swept-source OCT angiography |
DM=Descemet’s membrane; IOL=intraocular lens; OCT=optical coherence tomography; RPE=retinal pigment epithelium
Ocular investigations
The investigative modalities for ocular features include slit-lamp biomicroscopy, corneal topography, endothelial microscope analysis, OCT, and fundus photography.
Management
Management of refractive errors due to the abnormal lens curvature includes corrective glasses. The threats to visual acuity include anterior lenticonus and retinal holes and require surgical intervention.
The challenges of performing phacoemulsification surgery in anterior lenticonus include accurate IOL power calculation in patients with irregular astigmatism and a fragile anterior capsule, leading to a difficult capsulorrhexis. The use of trypan blue dye has been advocated to increase capsular stiffness. Moreover, FLACS, intraoperative aberrometry, and toric IOLs have been described as useful in these eyes.[97]
Ocular manifestations of Alport syndrome can help in the right diagnosis of this multisystemic disease. A nephrologist consultation is mandatory to monitor renal abnormalities. The patient’s close relatives should also be examined, and genetic counseling should be offered.
Stickler syndrome
SS is a heritable CTD caused by mutations in the COL2A1 and COL11A1 genes. It was initially described by Stickler et al.[98] in 1965 as a hereditary progressive arthro-ophthalmopathy. The estimated incidence is between 1 in 7000 and 9000. Though four subtypes have been described, STL1 and STL2 are the most common and are autosomal dominant. The mutations affect collagen I and II. Clinical features include ocular, auditory (hearing loss), craniofacial (micrognathia, cleft palate), and articular (early-onset arthritis and joint mobility) manifestations.
Ocular manifestations
The overall prevalence of childhood myopia among SS is reported to be 86%, with the mean refractive error ranging between −8.70 and −14.50 D. The risk of developing a retinal detachment varies between 14% and 65%, and it usually has an earlier onset (first two decades of life) and bilateral involvement. Presenile cataract (30%) and glaucoma (11%) are seen less frequently.[99] Various ocular manifestations of SS have been summarized in Table 10.
Table 10.
Ocular manifestations of Stickler syndrome
| Ocular Structure | Common Ocular Manifestations |
|---|---|
| Refractive | Congenital nonprogressive high myopia[100] |
| Retina[101] | Retinal detachment |
| Foveal hypoplasia | |
| Multiple retinal tears | |
| Giant retinal tear | |
| Vitreous | Vitreous anomalies |
| Pigment dispersion | |
| Glaucoma | Infantile/secondary[102] |
| Cataract[103] | Nuclear sclerotic |
| Lamellar | |
| Quadrantic | |
| Cortical |
Retinal detachment can be potentially devastating to the visual acuity. Prophylactic treatment using laser or cryotherapy reduces the risk of developing a detachment.[104] A recent case series concluded that cryopexy combined with scleral buckling significantly reduced the risk of fellow eye Retinal Detachment (RD) in SS patients who had already lost one eye to RD.[105]
Osteogenesis imperfecta
OI is a heritable CTD caused by mutations in the COL1A1 and COL1A2 genes affecting the production of type I collagen, which is found in cornea and lens.[106] It is commonly inherited as an autosomal dominant disorder with an incidence of around 1 in 15,000 births. It is divided into subtypes from Type I to Type VII based on clinical and radiographic findings. Common manifestations include blue sclera, hearing loss, bone deformities and fractures, short stature, dental anomalies, and cardiovascular disorders.[107]
Ocular manifestations
The literature on ocular features in OI is mostly limited to case reports. However, few case series have documented the common ocular manifestations.[106,108,109] The ocular features in OI have been summarized in Table 11.
Table 11.
Ocular manifestations of osteogenesis imperfecta
| Ocular Structure | Common Ocular Manifestations |
|---|---|
| Cornea | Reduced thickness |
| Keratoconus[110] | |
| Corneal rupture due to minor trauma[111] | |
| Microcornea | |
| Bowman membrane agenesis | |
| Sclera | Low ocular rigidity |
| Blue sclera[111] | |
| Glaucoma | Congenital glaucoma |
| Primary open-angle glaucoma[107] | |
| Lens | Ectopia lentis |
| Vitreous/retina | Subhyaloid hemorrhage |
| Retinal detachment[112] (fragile sclera) | |
| Optic atrophy | |
| Refractive errors | Myopia due to altered corneal and scleral biomechanics[107] |
A recent systematic review suggested that the most significant association is globe rupture following minor trauma and complications of routine ophthalmic surgeries.[107]
Reduced CCT, corneal hysteresis (CH), and Corneal resistance factor (CRF) lead to underestimation of IOP.[108] This, coupled with the altered structure of the trabecular meshwork, contributes to the development of glaucoma.
Blue sclera is a common finding that occurs due to reduced scleral thickness and increased visibility of underlying choroid. In patients with corneal or scleral rupture following minor trauma, EDS, OI, and BCS should be excluded.[111] Performing a patch graft in these eyes could be challenging due to thinned-out host tissue. It has been recommended that patients with OI should wear protective eyeglasses to shield them from accidental trauma.
Ophthalmic surgeries that might result in complications such as scleral perforation include strabismus surgery, trabeculectomy, and scleral buckling. Hence, any ophthalmic surgery in a patient with OI should be approached cautiously.[107]
Loeys–Dietz Syndrome
Loeys–Dietz syndrome (LDS) is an autosomal dominant CTD caused by mutations in the genes coding for components of the transforming growth factor-beta (TGF-β) signal pathway. The estimated prevalence is <1 in 100,000.[113] There is an overlap of clinical features with MFS. The overlapping features are vascular aneurysms, scoliosis, pes planus, anterior chest deformity, spontaneous pneumothorax, and craniofacial anomalies. However, the distinguishing features of LDS include hypertelorism, bifid uvula, cleft palate, and aortic and arterial tortuosity. The cardiovascular malformations are more severe, with an increased propensity of aneurysm dissection.[114]
Ocular manifestations include myopia blue sclera, RD, cataracts, retinal tortuosity, and strabismus. Characteristically, there is no association with EL, the myopia is less severe, and the association with blue sclera is present. Given the more aggressive aortic root dissections, these patients need careful monitoring and distinction from MFS.
Epidermolysis bullosa
Epidermolysis bullosa (EB) can be inherited in an Autosomal dominant (AD) or Autosomal Recessive (AR) manner with mutations involving keratin, laminin, or collagen. It is characterized by fragile epithelium that results in blisters and erosions with minor trauma. There are four main types: EB simplex, dystrophic EB, junctional EB, and Kindler syndrome.[115]
Ocular features include corneal erosions, conjunctival injection, bullous keratopathy, DED and its sequelae like pannus, scarring, symblepharon, and ectropion.[116]
Wagner syndrome
Wagner syndrome is a vitreoretinopathy caused by an autosomally dominant inherited mutation of the VCAN gene on chromosome 5q that codes for an ECM responsible for the integrity of the vitreous. It is a rare disorder with an estimated prevalence of 1 in 1,000,000.[117] Ophthalmic features include an optically empty vitreous with avascular strands, membranes, or veils. The other features are myopia, chorioretinal atrophy causing night blindness, retinal detachment due to peripheral vitreoretinal adhesions, and a presenile cataract. Detachment can be rhegmatogenous or tractional and has an earlier onset. However, there are no associated systemic features, which distinguish it from SS.[118]
These patients require periodic screening by a retina specialist. It is also important to screen family members to recognize asymptomatic RD or those at risk for RD.
Pseudoxanthoma elasticum
Pseudoxanthoma elasticum (PXE) is caused by an autosomal recessive inherited mutation in the ABCC6 gene on chromosome 6 that affects cell membrane transport and buildup of abnormal calcification in the elastic tissues of the skin, eyes, and blood vessels. The prevalence is between 1 in 25,000 and 100,000.[119] Clinical features include yellow skin plaques on the neck and flexural areas, wrinkled loose skin, atherosclerotic cardiovascular disease, angioid streaks, and gastrointestinal bleed in a few patients.
The most common ocular feature is angioid streaks (85%), followed by optic disk drusen, chorioretinal atrophy, and macular degeneration with choroidal neovascular membrane (CNVM). Angioid streaks appear as jagged lines radiating from the optic disk and are crack-like dehiscences in the Bruch’s membrane. They are mostly asymptomatic. Macular CNVM can lead to irreversible visual loss. Periodic follow-up is recommended to look for and treat CNVM.[120]
Refractive Surgery in Connective Tissue Disorders
A recent review by Moshirfar et al.[86] suggested that since myopia is more prevalent in patients with CTD and the presentation between patients is quite diverse, it is not appropriate to consider them as a blanket contraindication to refractive surgery. Certain subtypes of CTD may be amenable to surgery.[86] Chen et al. suggested that among the autoimmune disorders, except for primary Sjögren’s syndrome, other patients who are well controlled with no recent flare-up of the disease process and not on multidrug regimens may undergo refractive surgery with appropriate counseling regarding postoperative recovery and complications.[122]
A review of literature was conducted by searching the following databases: PubMed (United States National Library of Medicine), Embase (Reed Elsevier Properties SA), Web of Science (Thomson Reuters), and Scopus (Elsevier BV). Only peer-reviewed scientific reports were included. Articles in languages other than English were included if the abstract was available in English or if the translated version of the article was available. The last electronic search was conducted on 25 Jan 2022. The literature search used various combinations of the following keywords: ONE from connective tissue disorders (CTDs), collagen vascular disorders (CVDs), rheumatoid arthritis, systemic lupus, Marfan, Ehler Danlos, heritable, autoimmune and ONE from ocular features, ophthalmic manifestations. Prospective studies, randomized controlled trials (RCTs), relevant retrospective studies, and case reports were included. Full text and reference lists of all studies were screened to ensure that any relevant study was not excluded.
Conclusion
To conclude, CVDs (both autoimmune and heritable) frequently present with various ophthalmic manifestations, with specific disorders having different common presentations. A comprehensive ocular evaluation by the ophthalmologist enables early detection of disease, appropriate referral to other subspecialities, initiation of immunomodulatory therapy, limitation of end-organ damage involving other systems, arresting the progression of disease, and management of the sight-threatening ocular complications.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Jennings BJ. Connective tissue diseases. Optom Clin Off Publ Prentice Soc. 1994;3:45–85. [PubMed] [Google Scholar]
- 2.Streifler JY, Molad Y. Connective tissue disorders: Systemic lupus erythematosus, Sjögren's syndrome, and scleroderma. Handb Clin Neurol. 2014;119:463–73. doi: 10.1016/B978-0-7020-4086-3.00030-8. [DOI] [PubMed] [Google Scholar]
- 3.Hsu R-H, Chien Y-H, Hwu W-L, Lee N-C. Diversity in heritable disorders of connective tissue at a single center. Connect Tissue Res. 2021;62:580–5. doi: 10.1080/03008207.2020.1816994. [DOI] [PubMed] [Google Scholar]
- 4.Cunningham ET, Tabbara KF, Zierhut M. Systemic lupus erythematosus and the eye. Ocul Immunol Inflamm. 2018;26:1143–5. doi: 10.1080/09273948.2018.1539589. [DOI] [PubMed] [Google Scholar]
- 5.Petri M, Orbai A-M, Alarcón GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the systemic lupus international collaborating clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64:2677–86. doi: 10.1002/art.34473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jawahar N, Walker JK, Murray PI, Gordon C, Reynolds JA. Epidemiology of disease-activity related ophthalmological manifestations in systemic lupus erythematosus: A systematic review. Lupus. 2021;30:2191–203. doi: 10.1177/09612033211050337. [DOI] [PubMed] [Google Scholar]
- 7.Wang L, Xie Y, Deng Y. Prevalence of dry eye in patients with systemic lupus erythematosus: A meta-analysis. BMJ Open. 2021;11:e047081. doi: 10.1136/bmjopen-2020-047081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Andrade FA, Guimarães Moreira Balbi G, Bortoloti de Azevedo LG, Provenzano SáG, Vieira de Moraes Junior H, Mendes Klumb E, et al. Neuro-ophthalmologic manifestations in systemic lupus erythematosus. Lupus. 2017;26:522–8. doi: 10.1177/0961203316683265. [DOI] [PubMed] [Google Scholar]
- 9.Mimier-Janczak M, Kaczmarek D, Janczak D, Kaczmarek R. Optical coherence tomography angiography as a new tool for evaluation of the subclinical retinal involvement in patients with systemic lupus erythematosus-A review. J Clin Med. 2021;10:2887. doi: 10.3390/jcm10132887. doi:10.3390/jcm10132887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bitirgen G, Kucuk A, Ergun MC, Baloglu R, Gharib MH, Al Emadi S, et al. Subclinical corneal nerve fiber damage and immune cell activation in systemic lupus erythematosus: A corneal confocal microscopy study. Transl Vis Sci Technol. 2021;10:10. doi: 10.1167/tvst.10.14.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi W-Q, Han T, Liu R, Xia Q, Xu T, Wang Y, et al. Retinal microvasculature and conjunctival vessel alterations in patients with systemic lupus erythematosus-An optical coherence tomography angiography study. Front Med. 2021;8:724283. doi: 10.3389/fmed.2021.724283. doi:10.3389/fmed. 2021.724283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu R, Wang Y, Xia Q, Xu T, Han T, Cai S, et al. Retinal thickness and microvascular alterations in the diagnosis of systemic lupus erythematosus: A new approach. Quant Imaging Med Surg. 2022;12:823–37. doi: 10.21037/qims-21-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hernandez-Da Mota SE, Arellanes-Garcia L, Recillas-Gispert C, Cornejo-Ballesteros H, Melgoza-del-Angel C, Teran-Estrada L, et al. Lupus relapse presented as frosted branch retinal angiitis: Case report. Ocul Immunol Inflamm. 2011;19:367–9. doi: 10.3109/09273948.2011.603879. [DOI] [PubMed] [Google Scholar]
- 14.Escudero González CM, Rodríguez Montero S, Martínez Pérez R, Pastor Mañosa C, Velloso Feijoo ML, Marenco de la Fuente JL. [Resistant orbital pseudotumor treated with rituximab in a patient with systemic lupus erythematosus. A case presentation] Reumatol Clin. 2010;6:214–6. doi: 10.1016/j.reuma.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 15.James WD, Berger TG, Elston DM, Odom RB. Andrews'Diseases of the Skin: Clinical Dermatology. Philadelphia: Saunders Elsevier; 2006. [Google Scholar]
- 16.Penmetsa GK, Sapra A. StatPearls. Treasure Island (FL): StatPearls Publishing; 2022. [Last accessed on 2022 Mar 28]. Morphea. Available from: http://www.ncbi.nlm.nih.gov/books/NBK559010/ [Google Scholar]
- 17.Varga J, Jimenez SA. Modulation of collagen gene expression: Its relation to fibrosis in systemic sclerosis and other disorders. Ann Intern Med. 1995;122:60–2. doi: 10.7326/0003-4819-122-1-199501010-00010. [DOI] [PubMed] [Google Scholar]
- 18.Sakkas LI, Platsoucas CD. Is systemic sclerosis an antigen-driven T cell disease?Arthritis Rheum. 2004;50:1721–33. doi: 10.1002/art.20315. [DOI] [PubMed] [Google Scholar]
- 19.van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: An American college of rheumatology/European league against rheumatism collaborative initiative. Ann Rheum Dis. 2013;72:1747–55. doi: 10.1136/annrheumdis-2013-204424. [DOI] [PubMed] [Google Scholar]
- 20.Herrick AL, Wigley FM. Raynaud's phenomenon. Best Pract Res Clin Rheumatol. 2020;34:101474. doi: 10.1016/j.berh.2019.101474. doi:10.1016/j.berh. 2019.101474. [DOI] [PubMed] [Google Scholar]
- 21.Hudson M, Ghossein C, Steen V. Scleroderma renal crisis. Presse Medicale Paris Fr 1983. 2021;50:104063. doi: 10.1016/j.lpm.2021.104063. [DOI] [PubMed] [Google Scholar]
- 22.Kreps EO, Carton C, Cutolo M, Cutolo CA, Vanhaecke A, Leroy BP, et al. Ocular involvement in systemic sclerosis: A systematic literature review, it's not all scleroderma that meets the eye. Semin Arthritis Rheum. 2019;49:119–25. doi: 10.1016/j.semarthrit.2018.12.007. [DOI] [PubMed] [Google Scholar]
- 23.Ng CC, Suresh S, Rosenbaum JT, McDonald HR, Cunningham ET. Occlusive retinal vasculitis associated with systemic sclerosis and antiphospholipid antibodies. Am J Ophthalmol Case Rep. 2021;24:101206. doi: 10.1016/j.ajoc.2021.101206. doi:10.1016/j.ajoc. 2021.101206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kök M, Ayan A, Fatih Küçük M, Erol MK, Yaprak L. Evaluation of the direct effects on retinal and choroidal microvascularity of systemic scleroderma. Microvasc Res. 2021;136:104166. doi: 10.1016/j.mvr.2021.104166. doi:10.1016/j.mvr. 2021.104166. [DOI] [PubMed] [Google Scholar]
- 25.Szalai E, Szucs G, Szamosi S, Aszalos Z, Afra I, Kemeny-Beke A. An in vivo confocal microscopy study of corneal changes in patients with systemic sclerosis. Sci Rep. 2021;11:11111. doi: 10.1038/s41598-021-90594-9. doi:10.1038/s41598-021-90594-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mayali H, Altinisik M, Sencan S, Pirildar T, Kurt E. A multimodal ophthalmic analysis in patients with systemic sclerosis using ocular response analyzer, corneal topography and specular microscopy. Int Ophthalmol. 2020;40:287–96. doi: 10.1007/s10792-019-01173-x. [DOI] [PubMed] [Google Scholar]
- 27.Nagy A, Rentka A, Nemeth G, Ziad H, Szücs G, Szekanecz Z, et al. Corneal manifestations of systemic sclerosis. Ocul Immunol Inflamm. 2019;27:968–77. doi: 10.1080/09273948.2018.1489556. [DOI] [PubMed] [Google Scholar]
- 28.Gomes BF, Santhiago MR, Kara-Junior N, Moraes HV. Evaluation of corneal parameters with dual scheimpflug imaging in patients with systemic sclerosis. Curr Eye Res. 2018;43:451–4. doi: 10.1080/02713683.2017.1414855. [DOI] [PubMed] [Google Scholar]
- 29.Küçük MF, Yaprak L, Erol MK, Ayan A, Kök M. Evaluations of the radial peripapillary, macular and choriocapillaris microvasculature using optical coherence tomography angiography in patients with systemic sclerosis. J Fr Ophtalmol. 2022;45:81–92. doi: 10.1016/j.jfo.2021.06.009. [DOI] [PubMed] [Google Scholar]
- 30.Rommel F, Prangel D, Prasuhn M, Grisanti S, Ranjbar M. Correlation of retinal and choroidal microvascular impairment in systemic sclerosis. Orphanet J Rare Dis. 2021;16:27. doi: 10.1186/s13023-020-01649-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.KılınçHekimsoy H, Şekeroğlu AM, Koçer AM, Hekimsoy V, Akdoğan A. Evaluation of the optic nerve head vessel density in patients with limited scleroderma. Ther Adv Ophthalmol. 2021;13:2515841421995387. doi: 10.1177/2515841421995387. doi:10.1177/2515841421995387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carnevali A, Giannaccare G, Gatti V, Battaglia C, Randazzo G, Yu AC, et al. Retinal microcirculation abnormalities in patients with systemic sclerosis: An explorative optical coherence tomography angiography study. Rheumatol Oxf Engl. 2021;60:5827–32. doi: 10.1093/rheumatology/keab258. [DOI] [PubMed] [Google Scholar]
- 33.Zhao M, Wu J, Wu H, Sawalha AH, Lu Q. Clinical treatment options in scleroderma: Recommendations and comprehensive review. Clin Rev Allergy Immunol. 2022;62:273–91. doi: 10.1007/s12016-020-08831-4. [DOI] [PubMed] [Google Scholar]
- 34.Ruiz de Morales JMG, Puig L, Daudén E, Cañete JD, Pablos JL, Martín AO, et al. Critical role of interleukin (IL)-17 in inflammatory and immune disorders: An updated review of the evidence focusing in controversies. Autoimmun Rev. 2020;19:102429. doi: 10.1016/j.autrev.2019.102429. [DOI] [PubMed] [Google Scholar]
- 35.Littlejohn EA, Monrad SU. Early diagnosis and treatment of rheumatoid arthritis. Prim Care. 2018;45:237–55. doi: 10.1016/j.pop.2018.02.010. [DOI] [PubMed] [Google Scholar]
- 36.Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO, et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62:2569–81. doi: 10.1002/art.27584. [DOI] [PubMed] [Google Scholar]
- 37.Vignesh APP, Srinivasan R. Ocular manifestations of rheumatoid arthritis and their correlation with anti-cyclic citrullinated peptide antibodies. Clin Ophthalmol Auckl NZ. 2015;9:393–7. doi: 10.2147/OPTH.S77210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abd-Allah NM, Hassan AA, Omar G, Hamdy M, Abdelaziz STA, Abd El Hamid WM, et al. Dry eye in rheumatoid arthritis: Relation to disease activity. Immunol Med. 2020;43:92–7. doi: 10.1080/25785826.2020.1729597. [DOI] [PubMed] [Google Scholar]
- 39.Gurlevik U, Karakoyun A, Yasar E. When rheumatoid arthritis is mentioned, should only dryness come to mind?Clin Rheumatol. 2020;39:3317–21. doi: 10.1007/s10067-020-05124-1. [DOI] [PubMed] [Google Scholar]
- 40.Artifoni M, Rothschild P-R, Brézin A, Guillevin L, Puéchal X. Ocular inflammatory diseases associated with rheumatoid arthritis. Nat Rev Rheumatol. 2014;10:108–16. doi: 10.1038/nrrheum.2013.185. [DOI] [PubMed] [Google Scholar]
- 41.Promelle V, Goeb V, Gueudry J. Rheumatoid arthritis associated episcleritis and scleritis: An update on treatment perspectives. J Clin Med. 2021;10:2118. doi: 10.3390/jcm10102118. doi:10.3390/jcm10102118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McGinnigle S, Naroo SA, Eperjesi F. Evaluation of dry eye. Surv Ophthalmol. 2012;57:293–316. doi: 10.1016/j.survophthal.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 43.Noh SR, Chung JL, Lee JM, Seo KY, Koh K. Meibomian gland atrophy with duration of Sjogren's syndrome in adult females. Int Ophthalmol. 2022;42:191–200. doi: 10.1007/s10792-021-02013-7. [DOI] [PubMed] [Google Scholar]
- 44.Usuba FS, de Medeiros-Ribeiro AC, Novaes P, Aikawa NE, Bonfiglioli K, Santo RM, et al. Dry eye in rheumatoid arthritis patients under TNF-inhibitors: Conjunctival goblet cell as an early ocular biomarker. Sci Rep. 2020;10:14054. doi: 10.1038/s41598-020-70944-9. doi:10.1038/s41598-020-70944-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quiroz-Mercado H, Hernandez-Quintela E, Chiu KH, Henry E, Nau JA. A phase II randomized trial to evaluate the long-term (12-week) efficacy and safety of OC-01 (varenicline solution) nasal spray for dry eye disease: The MYSTIC study. Ocul Surf. 2021;24:15–21. doi: 10.1016/j.jtos.2021.12.007. [DOI] [PubMed] [Google Scholar]
- 46.Abidi A, Shukla P, Ahmad A. Lifitegrast: A novel drug for treatment of dry eye disease. J Pharmacol Pharmacother. 2016;7:194–8. doi: 10.4103/0976-500X.195920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cao Y, Zhang W, Wu J, Zhang H, Zhou H. Peripheral ulcerative keratitis associated with autoimmune disease: Pathogenesis and treatment. J Ophthalmol. 2017;2017:7298026. doi: 10.1155/2017/7298026. doi:10.1155/2017/7298026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sasaki H, Kohsaka H. Current diagnosis and treatment of polymyositis and dermatomyositis. Mod Rheumatol. 2018;28:913–21. doi: 10.1080/14397595.2018.1467257. [DOI] [PubMed] [Google Scholar]
- 49.Volc-Platzer B. [Dermatomyositis-update. Hautarzt Z Dermatol Venerol Verwandte Geb. 2015;66:604–10. doi: 10.1007/s00105-015-3659-0. [DOI] [PubMed] [Google Scholar]
- 50.Wolff K, Goldsmith L, Katz S, Gilchrest B, Paller A, Leffell D. Fitzpatrick's Dermatology in General Medicine. 7th ed. New York: McGraw-Hill; 2008. [Google Scholar]
- 51.Griger Z, Danko K, Nemeth G, Hassan Z, Aszalos Z, Szabo K, et al. Anterior segment parameters associated with extramuscular manifestations in polymyositis and dermatomyositis. Int J Ophthalmol. 2020;13:1443–50. doi: 10.18240/ijo.2020.09.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Young TA, Al-Mayouf S, Feldman BM, Levin AV. Clinical assessment of conjunctival and episcleral vessel tortuosity in juvenile dermatomyositis. J AAPOS Off Publ Am Assoc Pediatr Ophthalmol Strabismus. 2002;6:238–40. doi: 10.1067/mpa.2002.123428. [DOI] [PubMed] [Google Scholar]
- 53.Choi RY, Swan RJ, Hersh A, Vitale AT. Retinal manifestations of juvenile dermatomyositis: Case report of bilateral diffuse chorioretinopathy with paracentral acute middle maculopathy and review of the literature. Ocul Immunol Inflamm. 2018;26:929–33. doi: 10.1080/09273948.2017.1305421. [DOI] [PubMed] [Google Scholar]
- 54.Starr MR, Softing Hataye AL, Bakri SJ. Asymptomatic multifocal paracentral acute middle maculopathy associated with juvenile dermatomyositis: Optical coherence angiography findings. Retin Cases Brief Rep. 2021;15:500–3. doi: 10.1097/ICB.0000000000000849. [DOI] [PubMed] [Google Scholar]
- 55.Wang Y, Morgan ML, Espino Barros Palau A, Lee AG, Foroozan R. Dermatomyositis-related nonischemic central retinal vein occlusion. J Neuroophthalmol. 2015;35:289–92. doi: 10.1097/WNO.0000000000000235. [DOI] [PubMed] [Google Scholar]
- 56.Sharma M, Prashar A, Tuli R, Sharma RK, Mahajan VK. Atypical central retinal artery occlusion: An uncommon cause of retinopathy and visual loss in dermatomyositis. Int J Rheum Dis. 2019;22:325–30. doi: 10.1111/1756-185X.12750. [DOI] [PubMed] [Google Scholar]
- 57.Yan Y, Shen X. Purtscher-like retinopathy associated with dermatomyositis. BMC Ophthalmol. 2013;13:36. doi: 10.1186/1471-2415-13-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee MY, Kim HH, Kim KS. Frosted branch angiitis, presumably related to dermatomyositis. Ocul Immunol Inflamm. 2011;19:129–31. doi: 10.3109/09273948.2010.531895. [DOI] [PubMed] [Google Scholar]
- 59.Wu CM, Dunn JP, Sergott RC. Papillitis with retinal venous congestion and intraocular inflammation. Am J Ophthalmol Case Rep. 2020;20:100913. doi: 10.1016/j.ajoc.2020.100913. doi:10.1016/j.ajoc. 2020.100913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rosés Sáiz R, Debono C, Hernández Pardines F, Piñero Cutillas C, Del Olmo Diaz L. [Bilateral dermatomyositis and diplopia of unknown origin: A case report. J Fr Ophtalmol. 2021;44:e373–5. doi: 10.1016/j.jfo.2020.09.036. [DOI] [PubMed] [Google Scholar]
- 61.Espinoza GM. Orbital inflammatory pseudotumors: Etiology, differential diagnosis, and management. Curr Rheumatol Rep. 2010;12:443–7. doi: 10.1007/s11926-010-0128-8. [DOI] [PubMed] [Google Scholar]
- 62.Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–9. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 63.De Paepe A, Devereux RB, Dietz HC, Hennekam RC, Pyeritz RE. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet. 1996;62:417–26. doi: 10.1002/(SICI)1096-8628(19960424)62:4<417::AID-AJMG15>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 64.Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47:476–85. doi: 10.1136/jmg.2009.072785. [DOI] [PubMed] [Google Scholar]
- 65.Bitterman AD, Sponseller PD. Marfan syndrome: A clinical update. J Am Acad Orthop Surg. 2017;25:603–9. doi: 10.5435/JAAOS-D-16-00143. [DOI] [PubMed] [Google Scholar]
- 66.Salchow DJ, Gehle P. Ocular manifestations of Marfan syndrome in children and adolescents. Eur J Ophthalmol. 2019;29:38–43. doi: 10.1177/1120672118761333. [DOI] [PubMed] [Google Scholar]
- 67.Chandra A, Ekwalla V, Child A, Charteris D. Prevalence of ectopia lentis and retinal detachment in Marfan syndrome. Acta Ophthalmol (Copenh) 2014;92:e82–83. doi: 10.1111/aos.12175. [DOI] [PubMed] [Google Scholar]
- 68.Konradsen TR, Zetterström C. A descriptive study of ocular characteristics in Marfan syndrome. Acta Ophthalmol (Copenh) 2013;91:751–5. doi: 10.1111/aos.12068. [DOI] [PubMed] [Google Scholar]
- 69.Gehle P, Goergen B, Pilger D, Ruokonen P, Robinson PN, Salchow DJ. Biometric and structural ocular manifestations of Marfan syndrome. PloS One. 2017;12:e0183370. doi: 10.1371/journal.pone.0183370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nelson LB, Maumenee IH. Ectopia lentis. Surv Ophthalmol. 1982;27:143–60. doi: 10.1016/0039-6257(82)90069-8. [DOI] [PubMed] [Google Scholar]
- 71.Chee S-P, Ti S-E, Chan NS-W. Management of the subluxated crystalline lens: A review. Clin Experiment Ophthalmol. 2021;49:1091–101. doi: 10.1111/ceo.13975. [DOI] [PubMed] [Google Scholar]
- 72.Hoffman RS, Snyder ME, Devgan U, Allen QB, Yeoh R, Braga-Mele R, et al. Management of the subluxated crystalline lens. J Cataract Refract Surg. 2013;39:1904–15. doi: 10.1016/j.jcrs.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 73.Chee S-P, Wong MHY, Jap A. Management of severely subluxated cataracts using femtosecond laser-assisted cataract surgery. Am J Ophthalmol. 2017;173:7–15. doi: 10.1016/j.ajo.2016.09.021. [DOI] [PubMed] [Google Scholar]
- 74.Esfandiari H, Ansari S, Mohammad-Rabei H, Mets MB. Management strategies of ocular abnormalities in patients with Marfan syndrome: Current perspective. J Ophthalmic Vis Res. 2019;14:71–7. doi: 10.4103/jovr.jovr_29_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Malfait F, Kariminejad A, Van Damme T, Gauche C, Syx D, Merhi-Soussi F, et al. Defective initiation of glycosaminoglycan synthesis due to B3GALT6 mutations causes a pleiotropic Ehlers-Danlos-syndrome-like connective tissue disorder. Am J Hum Genet. 2013;92:935–45. doi: 10.1016/j.ajhg.2013.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.De Paepe A, Malfait F. The Ehlers-Danlos syndrome, a disorder with many faces. Clin Genet. 2012;82:1–11. doi: 10.1111/j.1399-0004.2012.01858.x. [DOI] [PubMed] [Google Scholar]
- 77.Gharbiya M, Moramarco A, Castori M, Parisi F, Celletti C, Marenco M, et al. Ocular features in joint hypermobility syndrome/ehlers-danlos syndrome hypermobility type: A clinical and in vivo confocal microscopy study. Am J Ophthalmol. 2012;154:593–600.e1. doi: 10.1016/j.ajo.2012.03.023. [DOI] [PubMed] [Google Scholar]
- 78.Joseph AW, Joseph SS, Francomano CA, Kontis TC. Characteristics, diagnosis, and management of Ehlers-Danlos syndromes: A review. JAMA Facial Plast Surg. 2018;20:70–5. doi: 10.1001/jamafacial.2017.0793. [DOI] [PubMed] [Google Scholar]
- 79.Whitaker JK, Alexander P, Chau DYS, Tint NL. Severe conjunctivochalasis in association with classic type Ehlers-Danlos syndrome. BMC Ophthalmol. 2012;12:47. doi: 10.1186/1471-2415-12-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cameron JA. Corneal abnormalities in Ehlers-Danlos syndrome type VI. Cornea. 1993;12:54–9. doi: 10.1097/00003226-199301000-00009. [DOI] [PubMed] [Google Scholar]
- 81.Beighton P. Serious ophthalmological complications in the Ehlers-Danlos syndrome. Br J Ophthalmol. 1970;54:263–8. doi: 10.1136/bjo.54.4.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lozada R, Amaral C, Alvarez-Falcón S, Izquierdo NJ, Oliver AL. Successful repair of a spontaneous scleral rupture in a patient with type VI Ehlers-Danlos syndrome. Am J Ophthalmol Case Rep. 2020;20:100961. doi: 10.1016/j.ajoc.2020.100961. doi:10.1016/j.ajoc. 2020.100961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lumi X, Bergant G, Lumi A, Mahnic M. Outcomes of vitrectomy for retinal detachment in a patient with Ehlers-Danlos syndrome type IV: A case report. J Med Case Reports. 2021;15:249. doi: 10.1186/s13256-021-02855-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Louie A, Meyerle C, Francomano C, Srikumaran D, Merali F, Doyle JJ, et al. Survey of Ehlers-Danlos Patients'ophthalmic surgery experiences. Mol Genet Genomic Med. 2020;8:e1155. doi: 10.1002/mgg3.1155. doi:10.1002/mgg3.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Galperin G, Berra M, Berra A. Keratectasia following laser in situ keratomileusis in a low-risk patient with benign joint hypermobility syndrome. Arq Bras Oftalmol. 2014;77:119–21. doi: 10.5935/0004-2749.20140030. [DOI] [PubMed] [Google Scholar]
- 86.Moshirfar M, Barke MR, Huynh R, Waite AJ, Ply B, Ronquillo YC, et al. Controversy and consideration of refractive surgery in patients with heritable disorders of connective tissue. J Clin Med. 2021;10:3769. doi: 10.3390/jcm10173769. doi:10.3390/jcm10173769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kashtan CE. Alport syndrome: Achieving early diagnosis and treatment. Am J Kidney Dis Off J Natl Kidney Found. 2021;77:272–9. doi: 10.1053/j.ajkd.2020.03.026. [DOI] [PubMed] [Google Scholar]
- 88.Watson S, Padala SA, Bush JS. StatPearls. Treasure Island (FL): StatPearls Publishing; 2022. [Last accessed on 2022 Jan 23]. Alport syndrome. Available from: http://www.ncbi.nlm.nih.gov/books/NBK470419/ [Google Scholar]
- 89.Sohar E. Renal disease, inner ear deafness, and ocular changes;a new heredofamilial syndrome. AMA Arch Intern Med. 1956;97:627–30. doi: 10.1001/archinte.1956.00250230121013. [DOI] [PubMed] [Google Scholar]
- 90.Kashtan C. Multidisciplinary management of Alport syndrome: Current perspectives. J Multidiscip Healthc. 2021;14:1169–80. doi: 10.2147/JMDH.S284784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Junk AK, Stefani FH, Ludwig K. Bilateral anterior lenticonus: Scheimpflug imaging system documentation and ultrastructural confirmation of Alport syndrome in the lens capsule. Arch Ophthalmol Chic Ill 1960. 2000;118:895–7. [PubMed] [Google Scholar]
- 92.Chen Y, Colville D, Ierino F, Symons A, Savige J. Temporal retinal thinning and the diagnosis of Alport syndrome and Thin basement membrane nephropathy. Ophthalmic Genet. 2018;39:208–14. doi: 10.1080/13816810.2017.1401088. [DOI] [PubMed] [Google Scholar]
- 93.Shah SN, Weinberg DV. Giant macular hole in Alport syndrome. Ophthalmic Genet. 2010;31:94–7. doi: 10.3109/13816811003767128. [DOI] [PubMed] [Google Scholar]
- 94.Ghadiri NJ, Stanojcic N, Raja M, Burton BJ. A triad of retinal signs in Alport syndrome: The 'stair-case'fovea, choroidal thinning and peripheral schisis. Eur J Ophthalmol. 2019;29(1_suppl):10–4. doi: 10.1177/1120672119841002. [DOI] [PubMed] [Google Scholar]
- 95.Nicklason E, Mack H, Beltz J, Jacob J, Farahani M, Colville D, et al. Corneal endothelial cell abnormalities in X-linked Alport syndrome. Ophthalmic Genet. 2020;41:13–9. doi: 10.1080/13816810.2019.1709126. [DOI] [PubMed] [Google Scholar]
- 96.Chugh KS, Sakhuja V, Agarwal A, Jha V, Joshi K, Datta BN, et al. Hereditary nephritis (Alport's syndrome)--clinical profile and inheritance in 28 kindreds. Nephrol Dial Transplant Off Publ Eur Dial Transpl Assoc-Eur Ren Assoc. 1993;8:690–5. doi: 10.1093/ndt/8.8.690. [DOI] [PubMed] [Google Scholar]
- 97.Orts-Vila P, Amparo F, Rodríguez-Prats JL, Tañá-Rivero P. Alport syndrome and femtosecond laser-assisted cataract surgery. J Ophthalmic Vis Res. 2020;15:264–9. doi: 10.18502/jovr.v15i2.6748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stickler GB, Belau PG, Farrell FJ, Jones JD, Pugh DG, Steinberg AG, et al. Hereditary progressive arthro-ophthalmopathy. Mayo Clin Proc. 1965;40:433–55. [PubMed] [Google Scholar]
- 99.Boysen KB, La Cour M, Kessel L. Ocular complications and prophylactic strategies in Stickler syndrome: A systematic literature review. Ophthalmic Genet. 2020;41:223–34. doi: 10.1080/13816810.2020.1747092. [DOI] [PubMed] [Google Scholar]
- 100.Wilson MC, McDonald-McGinn DM, Quinn GE, Markowitz GD, LaRossa D, Pacuraru AD, et al. Long-term follow-up of ocular findings in children with Stickler's syndrome. Am J Ophthalmol. 1996;122:727–8. doi: 10.1016/s0002-9394(14)70494-5. [DOI] [PubMed] [Google Scholar]
- 101.Ang A, Poulson AV, Goodburn SF, Richards AJ, Scott JD, Snead MP. Retinal detachment and prophylaxis in type 1 Stickler syndrome. Ophthalmology. 2008;115:164–8. doi: 10.1016/j.ophtha.2007.03.059. [DOI] [PubMed] [Google Scholar]
- 102.Wubben TJ, Branham KH, Besirli CG, Bohnsack BL. Retinal detachment and infantile-onset glaucoma in Stickler syndrome associated with known and novel COL2A1 mutations. Ophthalmic Genet. 2018;39:615–8. doi: 10.1080/13816810.2018.1509355. [DOI] [PubMed] [Google Scholar]
- 103.Seery CM, Pruett RC, Liberfarb RM, Cohen BZ. Distinctive cataract in the Stickler syndrome. Am J Ophthalmol. 1990;110:143–8. doi: 10.1016/s0002-9394(14)76982-x. [DOI] [PubMed] [Google Scholar]
- 104.Naravane AV, Belin PJ, Pierce B, Quiram PA. Risk and prevention of retinal detachments in patients with Stickler syndrome. Ophthalmic Surg Lasers Imaging Retina. 2022;53:7–11. doi: 10.3928/23258160-20211213-02. [DOI] [PubMed] [Google Scholar]
- 105.Ripandelli G, Rossi T, Pesci FR, Cecere M, Stirpe M. The prophylaxis of fellow-eye retinal detachment in Stickler syndrome: A retrospective series. Retina Phila Pa. 2022;42:250–5. doi: 10.1097/IAE.0000000000003304. [DOI] [PubMed] [Google Scholar]
- 106.Keleş A, Doğuizi S, Şahin NM, Koç M, Aycan Z. Anterior segment findings in patients with osteogenesis imperfecta: A case-control study. Cornea. 2020;39:935–9. doi: 10.1097/ICO.0000000000002345. [DOI] [PubMed] [Google Scholar]
- 107.Treurniet S, Burger P, Ghyczy EAE, Verbraak FD, Curro-Tafili KR, Micha D, et al. Ocular characteristics and complications in patients with osteogenesis imperfecta: A systematic review. Acta Ophthalmol (Copenh) 2022;100:e16–28. doi: 10.1111/aos.14882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lagrou LM, Gilbert J, Hannibal M, Caird MS, Thomas I, Moroi SE, et al. Altered corneal biomechanical properties in children with osteogenesis imperfecta. J AAPOS Off Publ Am Assoc Pediatr Ophthalmol Strabismus. 2018;22:183–7.e1. doi: 10.1016/j.jaapos.2017.12.015. [DOI] [PubMed] [Google Scholar]
- 109.Lyster ML, Hald JD, Rasmussen ML, Grauslund J, Folkestad L. Risk of eye diseases in osteogenesis imperfecta - A nationwide, register-based cohort study. Bone. 2022;154:116249. doi: 10.1016/j.bone.2021.116249. doi:10.1016/j.bone. 2021.116249. [DOI] [PubMed] [Google Scholar]
- 110.Zeri F, Swann PG, Naroo S. Osteogenesis imperfecta and keratoconus in an Italian family. Clin Exp Optom. 2018;101:400–3. doi: 10.1111/cxo.12617. [DOI] [PubMed] [Google Scholar]
- 111.Campagna G, Al-Mohtaseb Z, Khandelwal S, Chang E. Sequential traumatic corneal open globe rupture in a patient with osteogenesis imperfecta type I. Am J Ophthalmol Case Rep. 2018;11:35–6. doi: 10.1016/j.ajoc.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fleissig E, Barak A. Surgical management of retinal detachment in osteogenesis imperfecta: Case report and review of the literature. Retin Cases Brief Rep. 2019;13:43–6. doi: 10.1097/ICB.0000000000000527. [DOI] [PubMed] [Google Scholar]
- 113.Meester JAN, Verstraeten A, Schepers D, Alaerts M, Van Laer L, Loeys BL. Differences in manifestations of Marfan syndrome, Ehlers-Danlos syndrome, and Loeys-Dietz syndrome. Ann Cardiothorac Surg. 2017;6:582–94. doi: 10.21037/acs.2017.11.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Busch C, Voitl R, Goergen B, Zemojtel T, Gehle P, Salchow DJ. Ocular findings in Loeys-Dietz syndrome. Br J Ophthalmol. 2018;102:1036–40. doi: 10.1136/bjophthalmol-2017-311254. [DOI] [PubMed] [Google Scholar]
- 115.Hon KL, Chu S, Leung AKC. Epidermolysis bullosa: Pediatric perspectives. Curr Pediatr Rev. 2021 doi: 10.2174/1573396317666210525161252. [DOI] [PubMed] [Google Scholar]
- 116.Rousseau A, Prost-Squarcioni C, Doan S, Leroux-Villet C, Caux F, Hoang-Xuan T, et al. Ocular involvement in epidermolysis bullosa acquisita with long-term follow-up. Br J Ophthalmol. 2020;104:235–40. doi: 10.1136/bjophthalmol-2019-313960. [DOI] [PubMed] [Google Scholar]
- 117.Araújo JR, Tavares-Ferreira J, Estrela-Silva S, Rocha P, Brandão E, Faria PA, et al. WAGNER syndrome: Anatomic, functional and genetic characterization of a Portuguese family. Graefes Arch Clin Exp Ophthalmol. 2018;256:163–71. doi: 10.1007/s00417-017-3800-0. [DOI] [PubMed] [Google Scholar]
- 118.Acón D, Hussain RM, Yannuzzi NA, Berrocal AM. Complex combined tractional and rhegmatogenous retinal detachment in a twenty-three-year-old male with Wagner syndrome. Ophthalmic Surg Lasers Imaging Retina. 2020;51:467–71. doi: 10.3928/23258160-20200804-07. [DOI] [PubMed] [Google Scholar]
- 119.Germain DP. Pseudoxanthoma elasticum. Orphanet J Rare Dis. 2017;12:85. doi: 10.1186/s13023-017-0639-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Choudhary S, Srivastava A, Gupta S. Pseudoxanthoma elasticum and angioid streaks. QJM Mon J Assoc Physicians. 2021;114:274. doi: 10.1093/qjmed/hcaa190. [DOI] [PubMed] [Google Scholar]
- 121.Swaminathan SS, Shah P, Zheng F, Gregori G, Rosenfeld PJ. Detection of choriocapillaris loss in Alport syndrome with swept-source OCT angiography. Ophthalmic Surg Lasers Imaging Retina. 2018;49:138–41. doi: 10.3928/23258160-20180129-10. [DOI] [PubMed] [Google Scholar]
- 122.Chen TY, Chu DS. Refractive surgery for the patient with autoimmune diseases. Curr Opin Ophthalmol. 2020;31:247–52. doi: 10.1097/ICU.0000000000000668. [DOI] [PubMed] [Google Scholar]