

Abstract
The CC-chemokine receptor CCR5 mediates fusion and entry of the most commonly transmitted human immunodeficiency virus type 1 (HIV-1) strains. We have isolated six new anti-CCR5 murine monoclonal antibodies (MAbs), designated PA8, PA9, PA10, PA11, PA12, and PA14. A panel of CCR5 alanine point mutants was used to map the epitopes of these MAbs and the previously described MAb 2D7 to specific amino acid residues in the N terminus and/or second extracellular loop regions of CCR5. This structural information was correlated with the MAbs’ abilities to inhibit (i) HIV-1 entry, (ii) HIV-1 envelope glycoprotein-mediated membrane fusion, (iii) gp120 binding to CCR5, and (iv) CC-chemokine activity. Surprisingly, there was no correlation between the ability of a MAb to inhibit HIV-1 fusion-entry and its ability to inhibit either the binding of a gp120-soluble CD4 complex to CCR5 or CC-chemokine activity. MAbs PA9 to PA12, whose epitopes include residues in the CCR5 N terminus, strongly inhibited gp120 binding but only moderately inhibited HIV-1 fusion and entry and had no effect on RANTES-induced calcium mobilization. MAbs PA14 and 2D7, the most potent inhibitors of HIV-1 entry and fusion, were less effective at inhibiting gp120 binding and were variably potent at inhibiting RANTES-induced signaling. With respect to inhibiting HIV-1 entry and fusion, PA12 but not PA14 was potently synergistic when used in combination with 2D7, RANTES, and CD4-immunoglobulin G2, which inhibits HIV-1 attachment. The data support a model wherein HIV-1 entry occurs in three stages: receptor (CD4) binding, coreceptor (CCR5) binding, and coreceptor-mediated membrane fusion. The antibodies described will be useful for further dissecting these events.
Human immunodeficiency virus type 1 (HIV-1) induces viral-to-cell membrane fusion to gain entry into target cells (9, 15, 63). The first high-affinity interaction between the virion and the cell surface is the binding of the viral surface glycoprotein gp120 to the CD4 antigen (13, 28, 37, 38). This in turn induces conformational changes in gp120, which enable it to interact with one of several chemokine receptors (5, 6, 19, 33). The CC-chemokine receptor CCR5 is the major coreceptor for macrophage-tropic (R5) strains and plays a crucial role in the transmission of HIV-1 (5, 6, 19, 33). T-cell line-tropic (X4) viruses use CXCR4 to enter target cells and usually, but not always, emerge late in disease progression or as a consequence of virus propagation in tissue culture (5, 6, 19, 33). Some primary HIV-1 isolates are dualtropic (R5X4) since they can use both coreceptors, though not always with the same efficiency (12, 53). Mutagenesis studies coupled with the resolution of the gp120 core crystal structure have demonstrated that the coreceptor-binding site on gp120 includes several highly conserved residues (30, 49, 62).
We and others have demonstrated that tyrosines and negatively charged residues in the amino-terminal domain (Nt) of CCR5 are essential for gp120 binding to the coreceptor and for HIV-1 fusion and entry (7, 16, 18, 20, 25, 29, 48, 50). Residues in the extracellular loops (ECLs) 1 to 3 of CCR5 were dispensable for coreceptor function, and yet the CCR5 interdomain configuration had to be maintained for optimal viral fusion and entry (22). This led us to conclude either that gp120 forms interactions with a diffuse surface on the ECLs or that the Nt is maintained in a functional conformation by bonds with residues in the ECLs. Studies with chimeric coreceptors and anti-CCR5 monoclonal antibodies (MAbs) have also shown the importance of the ECLs for viral entry (6, 50, 60).
Molecules that specifically bind to CCR5 and block interactions with its ligands are a powerful tool to further probe the structure-function relationships of this coreceptor. Characterizing such compounds could also assist in designing effective therapeutic agents that target coreceptor-mediated steps of viral entry. Inhibitors of CCR5 or CXCR4 coreceptor function identified to date are diverse in nature and include small molecules, peptides, chemokines and their derivatives, and MAbs. No small molecule that specifically inhibits only CCR5-mediated fusion has been described, although a distamycin analogue has been reported to inhibit HIV-1 entry and to bind CCR5, CXCR4, and other chemokine receptors (26). Inhibition of HIV-1 entry by CC-chemokines is mediated by at least two distinct mechanisms: blockage of the gp120-coreceptor interaction and internalization of the chemokine-receptor complex (1, 4, 24, 55, 59). The variant AOP-RANTES also inhibits recycling of CCR5 to the cell surface (36, 52). Variants such as RANTES 9-68 and Met-RANTES only prevent the gp120-CCR5 interaction and do not down-regulate CCR5 (64). Three sets of anti-CCR5 MAbs have been previously described (25, 46, 60, 61). Of the approximately 25 MAbs generated, only 2D7 has been shown to inhibit efficiently HIV-1 entry and CC-chemokine-induced calcium mobilization (60). The 2D7 epitope is located in ECL2, which also contains the CC-chemokine binding site (51). Several anti-CCR5 MAbs were used to probe differences in epitope presentation when CCR5 is expressed on different cell types or mutated in its Nt region. The patterns of reactivity observed suggested cell type-specific alterations in CCR5 structure (25). Only one anti-CXCR4 MAb, 12G5, has been extensively characterized for its antiviral properties. The efficiency of 12G5 inhibition of viral entry has been reported to be both cell and isolate dependent (39, 54). This MAb binds to ECL2 of CXCR4, but the mechanism by which it inhibits entry is unknown (8).
Using a novel screening procedure that selects for HIV-1 inhibitory activity, we have isolated and characterized a panel of six murine MAbs, designated PA8, PA9, PA10, PA11, PA12, and PA14. All six MAbs specifically bound to CCR5+ cells but with different efficiencies that were cell type dependent. Epitope mapping studies identified the residues that are important for MAb binding and also revealed information about the folding and interactions of the CCR5 extracellular domains. Surprisingly, MAb inhibition of HIV-1 entry and fusion did not correlate with inhibition of either the binding of a gp120-soluble CD4 (sCD4) complex to CCR5 or CC-chemokine activity. Potent synergistic inhibition of HIV-1 fusion and entry was observed when certain CCR5 MAbs were used in combination with other HIV-1 attachment and fusion inhibitors. The results are consistent with a model for HIV-1 entry that involves three distinct stages: receptor binding, coreceptor binding, and coreceptor-mediated membrane fusion.
MATERIALS AND METHODS
Reagents.
MAb 2D7 was purchased from Pharmingen (San Diego, Calif.), and CC- and CXC-chemokines were obtained from R&D Systems (Minneapolis, Minn.). CD4-immunoglobulin G2 (IgG2) (2), sCD4 (3), and recombinant HIV-1JR-FL gp120 and HIV-1LAI gp120 (55) were produced by Progenics Pharmaceuticals, Inc. CD4-IgG2 is an antibody-like recombinant fusion protein in which the D1 and D2 domains of human CD4 are linked to the heavy and light chain constant regions of human IgG2. sCD4 contains the extracellular domains D1 to D4 of CD4.
Isolation and purification of anti-CCR5 MAbs.
Murine L1.2-CCR5+ cells (59) were incubated for 16 h in the presence of 5 mM sodium butyrate, which activates transcription from the cytomegalovirus promoter that controls CCR5 expression, resulting in a 10-fold increase in cell surface coreceptor density. Female BALB/c mice were immunized intraperitoneally with 107 L1.2-CCR5+ cells at 3-week intervals and administered an intravenous boost of 107 L1.2-CCR5+ cells 3 days prior to splenectomy. Hybridomas were generated by standard methods whereby splenocytes were fused with the Sp2/0 cell line and selected in hypoxanthine-, aminopterin-, and thymidine-supplemented medium. In the primary screen, supernatants were tested for the ability to inhibit HIV-1 envelope-mediated fusion between PM1 cells (11), which naturally express CCR5 and CD4, and HeLa-EnvJR-FL+ cells in a resonance energy transfer (RET) assay, as previously described (17, 35). Of the 10,000 hybridoma supernatants screened, 120 were further tested by flow cytometry for specific binding to CCR5+ L1.2 cells. Hybridomas that produced the most potent supernatants were then cloned by limiting dilution. Ascites fluids were prepared by Harlan Bioproducts for Science, Inc. (Indianapolis, Ind.), from BALB/c mice that were injected with hybridomas producing the anti-CCR5 MAbs PA8, PA9, PA10, PA11, PA12, and PA14. The MAbs were individually purified to >95% homogeneity by precipitation with ammonium sulfate followed by protein A chromatography. All MAbs were resuspended in phosphate-buffered saline (PBS) at a final concentration of 5 mg/ml.
Flow cytometric analysis and epitope mapping of anti-CCR5 MAbs.
Flow cytometry was used to detect cell surface reactivity of MAbs PA8 to PA12 and PA14 with CCR5. Sodium butyrate-treated L1.2-CCR5+ cells (106) were incubated with 5 μg of antibody per ml for 20 min at 4°C in Dulbecco’s PBS (DPBS) containing 0.1% sodium azide (NaN3). The CCR5 MAb 2D7 was used as a positive control; nonspecific murine IgG1 or IgG2a was used as a negative control. The cells were spun down, washed, and incubated with phycoerythrin (PE)-labeled goat anti-mouse IgG (Caltag, Burlingame, Calif.) diluted 1:100, under the same conditions as the first antibody incubation. Finally, cells were analyzed by flow cytometry. Peripheral blood mononuclear cells (PBMC) were isolated and stimulated as previously described (56). After 3 days of stimulation, CD4+ lymphocytes were purified as previously described (56) and incubated for another 4 days in the presence of 200 U of interleukin-2. Staining was performed as described for the L1.2 cells. Mean fluorescence intensities (MFI), referred to below, pertain to the gated CCR5+ cell population, not the whole cell population.
A similar procedure was used for epitope mapping of the anti-CCR5 MAbs. A panel of 70 CCR5 point mutants has been described elsewhere (18, 22, 48). The coding sequences of these proteins are subcloned into the pcDNA3.1 vector (Stratagene, La Jolla, Calif.) from which transcription can be driven by a 5′ T7 polymerase promoter. The CCR5 mutants carry a 9-residue hemagglutinin (HA) tag at the C terminus for detection of protein in cell lysates or by flow cytometry. HeLa cells (2 × 106) were incubated for 5 h with 20 μg of Lipofectin per ml and an equal amount of wild-type or mutant CCR5-expressing plasmid in OPTI-MEM (Life Technologies, Gaithersburg, Md.). The cells were then infected for 12 h with 2 × 107 PFU of vTF7-3, which encodes the T7 RNA polymerase (21); detached with 2 mM EDTA in PBS; and washed once with binding buffer (1% bovine serum albumin–0.05% NaN3 in DPBS). Cells (106) were surface labeled with MAbs as described in the above paragraph, washed once with the incubation buffer, and resuspended in 1 ml of 1× FACSlyse in water (Becton Dickinson, Franklin Lakes, N.J.) for 30 min at room temperature, to permeabilize the cell membranes. The cells were then spun down, washed with the incubation buffer, and incubated for 1 h at 37°C with 4 μg of a fluorescein isothiocyanate (FITC)-labeled mouse anti-HA MAb (BabCo, Richmond, Calif.) per ml for intracellular labeling. Finally, cells were washed once with binding buffer and once with DPBS, resuspended in 1% formaldehyde in PBS, and analyzed by flow cytometry. The extent of binding of a MAb to mutant CCR5 was determined by the equation (mutant CCR5 PE MFI/wild-type CCR5 PE MFI)/(mutant CCR5 FITC MFI/wild-type CCR5 FITC MFI) × 100%. This normalizes MAb binding for mutant coreceptor expression levels.
gp120-sCD4-binding assay.
gp120 and sCD4 were biotinylated with N-hydroxysuccinate–biotin (Pierce, Rockford, Ill.) according to the manufacturer’s instructions, and uncoupled biotin was removed by diafiltration. Sodium butyrate-treated L1.2-CCR5+ cells were incubated with varying dilutions of the individual biotinylated proteins, an equimolar mixture of sCD4 and biotinylated gp120, or 1.25 μg of sCD4 per ml and 2.5 μg of biotinylated gp120 per ml in the presence of varying concentrations of anti-CCR5 MAbs PA8 to PA12, PA14, or 2D7 or a nonspecific murine IgG1, for 1 h at room temperature in 0.1% NaN3 in DPBS. Cells were washed with the incubation buffer and incubated with streptavidin-PE (Becton Dickinson) diluted 1:50, for 1 h at room temperature. Finally, cells were washed with binding buffer and analyzed with a fluorescence plate reader (PerSeptive Biosystems, Framingham, Mass.).
Inhibition of envelope-mediated membrane fusion and HIV-1 entry by anti-CCR5 MAbs.
HIV-1 envelope-mediated fusion between HeLa-EnvJR-FL+ and PM1 cells was detected by the RET assay. Equal numbers (2 × 104) of fluorescein octadecyl ester (F18)-labeled envelope-expressing cells and octadecyl rhodamine (R18)-labeled PM1 cells were plated in 96-well plates in 15% fetal calf serum in DPBS and incubated for 4 h at 37°C in the presence of varying concentrations of the anti-CCR5 MAbs PA8 to PA12, PA14, or 2D7 or a nonspecific murine IgG1. Fluorescence RET was measured with a Cytofluor plate reader (PerSeptive Biosystems), and percent RET was determined as previously described (35).
NLLuc+ Env− viruses complemented in trans by envelope glycoproteins from JR-FL or Gun-1 were produced as previously described (18). U87MG-CD4+ CCR5+ cells (14) were infected with chimeric, reporter viruses containing 50 to 100 ng of p24 per ml in the presence of varying concentrations of the individual MAbs. After 2 h at 37°C, virus-containing media were replaced by fresh, MAb-containing media. Fresh media, without antibodies, were added again after 12 h. After 72 h, 100 μl of lysis buffer (Promega) was added to the cells, and luciferase activity (relative light units [RLU]) was measured as described elsewhere (18). The percent inhibition of HIV-1 infection is defined as [1 − (RLU in the presence of antibody/RLU in the absence of antibody)] × 100%.
Calcium signaling assays.
The fluorochrome Indo-1AM (Molecular Probes, Eugene, Oreg.) was added to sodium butyrate-treated L1.2-CCR5+ cells at a final concentration of 5 μM. After incubation at 37°C for 30 min, the cells were washed once and resuspended in Hanks’ buffered saline. Cells (106) were stimulated sequentially with an anti-CCR5 MAb or PBS, followed 60 s later with RANTES. MAbs PA8 to PA12 and PA14 were used at a concentration of 100 μg/ml, 2D7 was used at 20 μg/ml, and RANTES was used at 250 ng/ml. Calcium flux inhibition by PA14 and 2D7 was also tested for a wide range of MAb concentrations, ranging from 0 to 100 μg/ml. Intracellular calcium levels were monitored with a Perkin-Elmer LS-50S fluorescence spectrophotometer by measuring the ratio of fluorescence emissions at 402 nm (bound dye) to that at 486 nm (free dye) following excitation at 358 nm.
RESULTS
Isolating anti-CCR5 MAbs PA8, PA9, PA10, PA11, PA12, and PA14.
We have found that peptides corresponding to the extracellular domains of CCR5 are inefficient at raising specific, high-titer antibody against the native, cell surface receptor (45, 46). BALB/c mice were immunized, therefore, with murine L1.2-CCR5+ cells, and hybridoma culture supernatants were tested for their ability to inhibit JR-FL envelope-mediated membrane fusion with CD4+ CCR5+ PM1 cells in the RET assay (17, 35). Of 10,000 hybridoma supernatants screened, well over 100 inhibited fusion by >50%, but only 6—designated PA8, PA9, PA10, PA11, PA12, and PA14—specifically and intensely stained L1.2-CCR5+ but not the parental L1.2 cells, as demonstrated by flow cytometry (data not shown). Based on previous experience, we assume that the other MAbs capable of inhibiting fusion were probably directed against cell surface adhesion molecules such as LFA-1 (34). Hybridomas PA8 to PA12 and PA14 were determined by isotyping enzyme-linked immunosorbent assay (Cappel, Durham, N.C.) to secrete IgG1 MAbs. Ascites fluids were prepared from BALB/c mice that were injected with the six hybridomas, and the IgG1 fractions were purified. PA8, PA9, PA11, PA12, and PA14 exhibited distinct isoelectric focusing profiles, whereas PA10 had a profile very similar to that of PA9 and therefore may be a second isolate of the same MAb (data not shown).
MAb binding to CCR5 transfectants and CD4+ lymphocytes.
Nonspecific mouse IgG1 and IgG2a did not stain either L1.2 or L1.2-CCR5+ cells (Fig. 1a and data not shown). MAbs PA9 to PA12 and PA14 stained >90%, and PA8 stained ∼70%, of L1.2-CCR5+ cells, but not the parental L1.2 cells, as determined by flow cytometry, showing that they specifically recognized CCR5 (Fig. 1b to g). The anti-CCR5 MAb 2D7, which was a positive control in our experiments, also stained >90% of L1.2-CCR5+ cells (Fig. 1h). PA8 to PA12 and PA14 are all IgG1 and react equally with goat anti-mouse IgG, whereas 2D7 is IgG2a and may react slightly differently with the reporter antibody. Only MFI measured with MAbs PA8 to PA12 and PA14 therefore are directly comparable. The rank order of MFI was PA12 ∼ PA11 > (2D7 =) PA14 ∼ PA10 ∼ PA9 > PA8. The difference between PA12 MFI and PA8 MFI was threefold. The fluorescence shifts observed for PA8 and other MAbs remained constant over a wide range of antibody concentrations (data not shown), suggesting that the relative shifts do not reflect differences in MAb affinities for CCR5 as expressed on these cells.
FIG. 1.

Detection of CCR5 expression by flow cytometry on L1.2-CCR5+ cells and CD4+ lymphocytes. Flow cytometry was used to detect CCR5 protein expression on the surface of L1.2-CCR5+ cells, CD4+ lymphocytes purified from phytohemagglutinin–interleukin-2-stimulated PBMC, and PM1 cells. Cells were incubated with saturating concentrations of each MAb, which were detected with a PE-labeled anti-mouse IgG reporter antibody. Flow cytometry histograms from a representative experiment are shown: staining of L1.2 and L1.2-CCR5+ cells with a murine IgG1 isotype control antibody (a) or anti-CCR5 MAbs (b to h); staining of CD4+ lymphocytes (i to o) with anti-CCR5 MAbs (solid lines) or isotype control antibody (dotted lines); staining of PM1 cells (p) by anti-CCR5 MAb PA12 or isotype control antibody. The x axis is the number of cells × 102; the y axis is MFI.
Compared with L1.2-CCR5+ cells, 7-day-old CD4+ lymphocytes purified from mitogen-stimulated PBMC exhibited different patterns of staining by the anti-CCR5 MAbs. PA8 did not stain CD4+ lymphocytes whereas 2D7 and PA14 stained >25%, and PA9 to PA12 stained 6 to 12% (Fig. 1i to o). The MFI of the stained CD4+ lymphocytes were low but still higher than those for the PM1 cell line (Fig. 1p). Their rank order was (2D7 >) PA14 > PA12 ∼ PA11 ∼ PA10 > PA9 > PA8. This differed somewhat from the order of reactivities observed on CCR5 transfectants. The difference between PA9 MFI and PA14 MFI was fivefold. Other groups have observed similar differences in the ability of anti-CCR5 MAbs to stain stable, CCR5+ cell lines versus PBMC (25). This may be due to cell-specific differences in CCR5 conformation, posttranslational modification, or oligomerization. Alternatively, association with other cell surface molecules may differ between cells. Since an obvious choice for such a molecule would be the CD4 cell surface antigen, which is absent from L1.2-CCR5+ cells and present on PBMC, we also tested the ability of PA8 to PA12, PA14, and 2D7 to stain HeLa cells transiently expressing CCR5 alone or with CD4. We observed no differences in the ability of any of the MAbs to stain cell surface CCR5 in the presence of CD4 (data not shown). If there is an association between these two proteins, it does not involve epitopes recognized by the anti-CCR5 MAbs available to us. Alternatively, an association between CCR5 and CD4 might occur only on primary lymphocytes.
Epitope mapping of the MAbs with CCR5 alanine mutants.
None of the antibodies was able to detect reduced and denatured CCR5 protein by Western blotting, indicating that they recognize conformationally sensitive epitopes (data not shown). MAb epitope mapping studies were performed with a panel of 70 alanine point mutants of residues in the Nt and ECLs of CCR5. HeLa cells were transfected with mutant or wild-type CCR5 coding sequences appended with C-terminal HA tags and infected with vTF7-3 to boost coreceptor expression. The cells were then incubated with the anti-CCR5 MAbs, binding of which was revealed by a PE-labeled goat anti-mouse IgG (21). A second, intracellular stain was performed with an FITC-labeled anti-HA MAb (BabCo). This internal control allowed us to directly normalize staining by the anti-CCR5 MAbs for mutant coreceptor expression levels on the cell surface. MAb binding to each mutant is expressed as a percentage of binding to wild-type CCR5 (Fig. 2). The expression levels of the CCR5 mutants measured by this technique are equivalent to those detected by dot blotting of plasma membrane extracts of CCR5-expressing cells (18, 22, 48).
FIG. 2.

Epitope mapping of anti-CCR5 MAbs. A two-color staining protocol was used to assess binding of MAbs to mutant CCR5 proteins, tagged at the C terminus with the HA peptide. HeLa cells expressing CCR5 point mutants were incubated with saturating concentrations of each MAb followed by detection with a PE-labeled anti-mouse IgG. Cell surface coreceptor expression was measured by double staining of the cells with an FITC-labeled anti-HA MAb. The four grids correspond to the four extracellular domains of CCR5. The first row of every grid indicates the amino acid sequence of the corresponding CCR5 extracellular domain. Binding of anti-CCR5 MAbs to the alanine mutant of each residue is expressed as a percentage of binding to wild-type CCR5, as described in Materials and Methods.
Certain point mutations reduced by >50% the binding of most of the antibodies to CCR5. In general, PA8 to PA12 were the most affected and PA14 and 2D7 were the least affected by this class of mutants, which included the cysteine pair C101A and C178A; the Nt mutants Y10A, D11A, and K25A; the ECL1 mutant D95A; the ECL2 mutants K171A/E172A, Q188A, and K191A/N192A; and the ECL3 mutants F263A and F264A (Fig. 2). These mutations probably cause conformational perturbations that have a common effect on the binding of all MAbs. However, we cannot exclude that some of these residues are part of the epitopes of some of the MAbs. We assumed that if a mutation lowered binding of an individual MAb by >75% and did not also lower binding of most of the other antibodies, the residue was probably a direct contributor to the epitope recognized by the MAb. Using these stringent guidelines, we concluded that the seven anti-CCR5 MAbs recognize overlapping but distinct epitopes (Fig. 2). MAb PA8 binding to CCR5 depended on N13 and Y15 in the Nt. MAbs PA9 and PA10 required D2, Y3, Q4, P8, and N13 in the Nt and Y176 and T177 in ECL2. MAb PA9 also required S7 in the Nt. MAb PA11 and PA12 binding depended on Q4 in the Nt. PA14 required D2 in the Nt and R168 and Y176 in ECL2. Finally, MAb 2D7 required Q170 and K171/E172 in ECL2 in order to bind to CCR5.
Chemokine signaling in the presence of anti-CCR5 MAbs.
Chemokine receptor-binding agents can be antagonists or, more rarely, agonists of receptor-mediated intracellular signaling. Alternatively, they could have no effect on signaling. CCR5 is able to bind three CC-chemokines, RANTES, MIP-1α, and MIP-1β, and transduce a signal that modulates cytosolic calcium levels. We therefore tested the agonist-antagonist activity of various concentrations of MAbs PA8 to PA12, PA14, and 2D7. Changes in intracellular calcium concentrations, [Ca2+]i, were measured in Indo-1-loaded L1.2-CCR5+ cells. None of the MAbs stimulated a change in [Ca2+]i, indicating that they are not agonists for CCR5. PA8 to PA12 were also unable to inhibit Ca2+ fluxes induced by RANTES (Fig. 3a and data not shown), even at concentrations as high as 100 μg/ml, showing that they are not antagonists either. These concentrations provide saturating binding of the MAbs to L1.2-CCR5+ cells, as shown by flow cytometry and the gp120-CCR5 binding assay (Fig. 3d and data not shown). MAbs PA14 and 2D7, however, blocked calcium mobilization induced by RANTES, although with different potencies (Fig. 3a and b). The 50% inhibitory concentration (IC50) for PA14 calcium influx inhibition was 45 μg/ml, which was approximately eightfold higher than the IC50 for 2D7 (Fig. 3b). RANTES-, MIP-1α-, and MIP-1β-induced calcium fluxes were each inhibited by similar concentrations of PA14 (data not shown). None of the MAbs affected SDF-1-induced calcium mobilization in L1.2-CCR5+ cells, which endogenously express murine CXCR4 (data not shown). Finally, neither MAbs nor CC-chemokines affected cytosolic calcium levels in parental L1.2 cells (data not shown).
FIG. 3.

Inhibition of calcium mobilization into CCR5+ cells by anti-CCR5 MAbs. L1.2-CCR5+ cells were loaded with Indo-1AM and stimulated sequentially with an anti-CCR5 MAb or PBS, followed by RANTES (a). Fluorescence changes were measured with a spectrofluorometer, and the tracings are from a representative experiment. Calcium flux inhibition by PA14 and 2D7 was tested for a wide range of MAb concentrations (b). Results are plotted as percent inhibition of calcium influx = [1 − (peak relative fluorescence in the presence of MAb/peak relative fluorescence in the absence of MAb)] × 100% and are means of values from three independent experiments.
Inhibition of CCR5 coreceptor function by the MAbs.
MAbs PA8 to PA12 and PA14 were initially selected on the basis of their ability to inhibit HIV-1 envelope-mediated membrane fusion. This activity was confirmed and quantified for the purified MAbs. As expected, all six MAbs, as well as MAb 2D7, blocked fusion between CD4+ CCR5+ PM1 cells and HeLa-EnvJR-FL+ cells in the RET assay. The rank order of potency was 2D7 ∼ PA14 > PA12 > PA11 > PA10 ∼ PA9 ∼ PA8 (Fig. 4a). IC50 for PA14 and 2D7 were 1.7 and 1.6 μg/ml, respectively; for PA11 and PA12, these were 25.5 and 10.0 μg/ml, respectively (Table 1). PA8, PA9, and PA10 inhibited fusion by only 10 to 15% at 300 μg/ml. None of the MAbs affected fusion between PM1 cells and HeLa-EnvLAI+ cells, which express the full-length envelope protein from an X4 virus (data not shown).
FIG. 4.

Inhibition of CCR5 coreceptor function by anti-CCR5 MAbs. Inhibition of cell-cell fusion by anti-CCR5 MAbs was tested in the RET assay (a). A total of 0 to 250 μg of PA8 to PA12 per ml or 0 to 25 μg of PA14 or 2D7 per ml was added to a mix of HeLa-EnvJR-FL+ and PM1 cells. Results are mean RET values from three independent experiments and are expressed as percent inhibition of fusion = [1 − (% RET in the presence of MAb/% RET in the absence of MAb)] × 100%. Inhibition of HIV-1 entry by anti-CCR5 MAbs was tested in a single-round replication luciferase-based entry assay (b). U87-CD4+ CCR5+ cells were infected with NLLuc+ Env− reporter virus carrying the JR-LF envelope in the presence of 0 to 250 μg of PA8 to PA12 per ml or 0 to 25 μg of PA14 or 2D7 per ml. Luciferase activity (RLU) was measured in cell lysates 72 h postinfection. Results are from a representative experiment and are expressed as percent inhibition of entry = [1 − (RLU in the presence of MAb/RLU in the absence of MAb)] × 100%. Shown is binding of biotinylated (b) gp120, sCD4, and b-gp120-CD4 complexes to L1.2-CCR5+ cells (c). Strong binding is observed when gp120 derived from the R5 virus HIV-1JR-LF is complexed with an equimolar amount of sCD4. No binding is observed in the absence of sCD4 or for gp120 derived from the X4 virus HIV-1LAI. Background binding to CCR5-L1.2 cells has been subtracted from all curves. Inhibition of gp120-sCD4 binding to L1.2-CCR5+ cells was tested in the presence of varying concentrations of each antibody (d). Cells were preincubated in 96-well plates with an anti-CCR5 MAb followed by an incubation with a saturating concentration of biotinylated gp120-sCD4. Finally, binding of PE-labeled streptavidin to cells was measured with a fluorescence plate reader. Results are from a representative experiment and are expressed as percent inhibition of gp120-sCD4 binding = [1 − (MFI in the presence of MAb/MFI in the absence of MAb)] × 100%.
TABLE 1.
Potency of anti-CCR5 MAbs in inhibiting HIV-1 envelope-mediated membrane fusion, viral entry, gp120-CCR5 binding, and chemokine signalinga
| MAb | Epitope(s) | IC50 (μg/ml) for inhibition of:
|
|||
|---|---|---|---|---|---|
| Membrane fusion | Viral entry | gp120 binding | Calcium flux | ||
| PA8 | Nt | ||||
| PA9 | Nt-ECL2 | 0.24 | |||
| PA10 | Nt-ECL2 | 0.13 | |||
| PA11 | Nt | 25.5 | 0.33 | ||
| PA12 | Nt | 10.0 | 0.24 | ||
| PA14 | Nt-ECL2 | 1.7 | 0.024 | 1.58 | 45 |
| 2D7 | ECL2 | 1.6 | 0.026 | 1.38 | 6.4 |
For comparative purposes, we have summarized the IC50 obtained for the anti-CCR5 MAbs in the various assays. IC50 are reported only for MAbs that could inhibit >90% of fusion, entry, or binding.
We also tested the ability of the different anti-CCR5 MAbs to inhibit entry of an R5 virus, JR-FL, and an R5X4 virus, Gun-1, in a single-round replication, luciferase-based entry assay. We typically measured 10,000 to 20,000 RLU in the absence of antibody and 1 to 5 RLU in the absence of virus. The rank order of potency in the entry assay was similar to the one determined in the fusion assay (Fig. 4b). We were unable to obtain >50% inhibition of JR-FL or Gun-1 entry with PA8 to PA11. The IC50 for PA12 was 2.5 μg/ml; however, we were unable to inhibit entry by >60% with this MAb. The discrepancies between fusion and entry data are probably due to cell type-specific differences such as coreceptor density. The IC50 for PA14 and 2D7 inhibition of JR-FL entry were determined to be 0.024 and 0.026 μg/ml, respectively (Table 2), and were 60-fold lower than those obtained in the fusion assay. Entry of dualtropic Gun-1 was two- to threefold more sensitive to inhibition by anti-CCR5 MAbs than was JR-FL entry (data not shown).
TABLE 2.
CI values for different combinations of MAbs and viral inhibitorsa
| Inhibitor combination | Concn ratio | Assay | CI
|
|
|---|---|---|---|---|
| 90% inhibition | 50% inhibition | |||
| PA12-2D7 | 10:1 | Entry | 0.043 | 0.291 |
| 2:1 | Fusion | 0.017 | 0.019 | |
| 10:1 | Fusion | 0.087 | 0.067 | |
| 50:1 | Fusion | 0.158 | 0.046 | |
| PA12-PA14 | 10:1 | Entry | 0.437 | 0.535 |
| 10:1 | Fusion | 0.22 | 0.263 | |
| PA14-2D7 | 1:1 | Entry | 2.85 | 1.85 |
| 1:1 | Fusion | 1.34 | 1.74 | |
| PA12-PA11 | 1:1 | Entry | 0.707 | 0.641 |
| PA12-RANTES | 1,000:1 | Fusion | 0.331 | 0.156 |
| PA14-RANTES | 100:1 | Fusion | 1.6 | 1.37 |
| 2D7-RANTES | 100:1 | Fusion | 0.972 | 0.962 |
| PA12-CD4-IgG2 | 10:1 | Fusion | 0.3 | 0.31 |
| PA14-CD4-IgG2 | 1:1 | Fusion | 0.957 | 0.566 |
| 2D7-CD4-IgG2 | 1:1 | Fusion | 1.127 | 0.302 |
Experiments like those described in the legend to Fig. 5 were performed for different combinations of viral entry inhibitors. Anti-CCR5 MAbs were tested in combination with each other, CC-chemokines, and CD4-IgG2, which inhibits HIV-1 attachment to target cells. The concentration ranges were as follows: PA11 and PA12, 0 to 250 μg/ml; 207 and PA14, 0 to 25 μg/ml; RANTES, 0 to 250 ng/ml; and CD4-IgG2, 0 to 25 μg/ml. The concentrations of single agents or their mixtures required to produce 50% and 90% inhibition of fusion or entry were quantitatively compared in a term known as the CI.
Anti-coreceptor MAbs might inhibit envelope-mediated fusion either by directly affecting the gp120-CCR5 interaction or by impeding postbinding steps involved in the formation of an active fusion complex. To examine the mechanism of inhibition of viral fusion and entry by PA8 to PA12 and PA14, we tested the ability of the different MAbs to block the gp120-CCR5 interaction. For this, we used an assay that detects binding to L1.2-CCR5+ cells of biotinylated HIV-1JR-FL gp120 complexed with sCD4. No binding of biotinylated gp120 was observed in the absence of sCD4 or CCR5 or when HIV-1LAI gp120 was used (Fig. 4c). Similarly, binding of biotinylated sCD4 was gp120 and CCR5 dependent (Fig. 4c and data not shown).
With the exception of PA8, all MAbs abrogated gp120-sCD4 binding to L1.2-CCR5+ (Fig. 3d). MAbs PA9, PA10, PA11, and PA12 inhibited binding with IC50 of 0.24, 0.13, 0.33, and 0.24 μg/ml, respectively (Table 1). Surprisingly, MAbs PA14 and 2D7 were among the least efficient inhibitors of gp120-sCD4 binding, with IC50 of 1.58 and 1.38 μg/ml, respectively (Table 1). Therefore, there was no correlation between the ability of a MAb to inhibit CCR5-mediated membrane fusion and entry and its ability to block gp120-sCD4 binding to the coreceptor. Inhibition by PA8 saturated at ∼40%. Taken together with the flow cytometry data of Fig. 1, this result suggests that PA8 binds to only a subset of CCR5 molecules as expressed on L1.2 transfectants, although other interpretations are possible.
Synergistic inhibition of HIV-1 fusion by combinations of anti-CCR5 MAbs and other viral entry inhibitors.
Coreceptor-specific agents may act at multiple stages of the entry process and exhibit nonadditive effects when used in combination. From a clinical perspective, it is important to determine the interactions of coreceptor-specific drug candidates with endogenous chemokines, which may afford some level of protection against disease progression. CCR5 MAbs were therefore tested in combination with each other or with RANTES, or with CD4-IgG2, which binds to gp120 to inhibit HIV-1 attachment to target cells. Dose-response curves were obtained for the agents used individually and in combination in viral fusion and entry assays. Data were analyzed by the median effect principle (10). The concentrations of single agents or their mixtures required to produce a given effect were quantitatively compared in a term known as the combination index (CI). A CI value greater than 1 indicates antagonism, CI ∼ 1 indicates an additive effect, and CI < 1 indicates a synergistic effect wherein the presence of one agent enhances the effect of another.
Combinations of PA12 and 2D7 were the most potently synergistic, with CI values ranging between 0.02 and 0.29, depending on the ratio of the antibodies (Fig. 5 and Table 2). The degree of synergy is known to vary with the stoichiometry of the agents. The viral entry and fusion assays were generally consistent in identifying MAb combinations that are highly synergistic (PA12 and 2D7), moderately synergistic (PA12 and PA14), additive (PA11 and PA12), and weakly antagonistic (PA14 and 2D7). The lack of synergy between PA14 and 2D7 is not surprising given that these MAbs cross-compete for binding to CCR5+ cells as determined by flow cytometry (data not shown). The additive effect observed for the combination of PA11 and PA12 is also consistent with their binding to similar epitopes in CCR5, including a shared dependency on residue Q4 in the Nt.
FIG. 5.

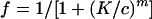 |
1 |
We also tested the ability of MAbs PA12, PA14, and 2D7 to synergize with RANTES in blocking cell-cell fusion. PA12 and RANTES combinations exhibited moderate synergy (Table 2). PA14 and 2D7 exhibited no synergy with RANTES, which is consistent with these MAbs being inhibitory of RANTES binding and signaling (Fig. 3a and b). Finally, we tested synergy between MAbs PA12, PA14, and 2D7 and CD4-IgG2, which interacts with gp120. We observed moderate synergy between PA12 and CD4-IgG2 over a broad range of concentrations but no synergy between PA14 or 2D7 and CD4-IgG2 when used at concentrations near their IC90 (Table 2).
DISCUSSION
We have isolated and characterized six murine anti-CCR5 IgG1 MAbs. Whereas PA8, PA9, PA11, PA12, and PA14 are distinct molecular species, PA9 and PA10 are practically indistinguishable by our analyses and therefore are probably the same MAb. All of the MAbs that we isolated recognize complex conformational epitopes, as is often the case with MAbs raised against native, cell surface proteins. Epitope mapping was performed with a panel of CCR5 alanine point mutants. Mutations that affected binding of all MAbs similarly were assumed to cause conformational perturbations in the coreceptor, though we cannot formally exclude that they participate in some of the MAb epitopes. The latter would be especially true if some of these residues were immunodominant. Only two of these residues, Y10 and D11, have been shown to affect HIV-1 entry (18, 48). We assumed that a residue was part of a MAb epitope if its substitution for an alanine inhibited MAb binding by >75%. This stringent guideline may have excluded some residues that interact weakly with the MAbs. According to our criteria, the PA8, PA11, and PA12 epitopes are located in the Nt domain. Since only the Q4→A4 mutation significantly affects PA11 and PA12 binding, these MAbs also might interact with residues that we have not mutated or bind peptide backbone atoms whose presentation may be unchanged by mutagenesis. MAbs PA9, PA10, and PA14 recognize epitopes that include residues in both the Nt and the ECL2 domains of CCR5, whereas the 2D7 epitope is located in ECL2. The PA14 epitope comprises both D2 in the Nt and R168 in ECL2, indicating that these two residues are proximal to one another within the context of a MAb footprint. They may even directly interact with one another through their opposite charges.
MAbs PA8 to PA12 and PA14 stained CCR5+ cells with different intensities and in a cell type-dependent manner. All MAbs except PA8 stained >90% L1.2-CCR5+ cells, the highest MFI being observed with PA11 and PA12. However, PA14 and 2D7 stained the highest percentage of CD4+ lymphocytes and also yielded the highest MFI on these cells. Hill et al. (25) have recently characterized a panel of anti-CCR5 MAbs that similarly stained transfected cells, but only two of eight stained PBMC, and none stained primary monocytes. A low affinity for CCR5 probably accounted for the nonreactivity of two of the MAbs with primary cells, but this was unlikely to be the explanation for the failure of the other four to react. In our MAb panel, we observe the most intense staining of CD4+ lymphocytes by MAbs 2D7 and PA14, which have epitopes located entirely or partially in the first 10 residues of ECL2. Hill et al. report, however, that MAbs specific for the Nt and ECL1 stain PBMC, while MAbs to ECL2 and ECL3 do not stain PBMC, and so a consistent pattern of reactivity has not been identified. One explanation for cell type-specific staining by MAbs would be that activated PBMC (and monocytes) secrete CC-chemokines that bind to cell surface CCR5, masking some MAb epitopes. However, one would expect this to be especially true for PA14 and 2D7, which are antagonists of chemokine-induced calcium mobilization and presumably compete with CC-chemokines for binding to CCR5. Yet these MAbs stain CD4+ lymphocytes the most intensely. Alternatively, differential CCR5 epitope exposure may reflect cell type-specific receptor oligomerization, association with other cell surface molecules, or different posttranslational modifications such as glycosylation. We have shown that differences in MAb binding probably do not reflect cell type-specific differences in CD4-CCR5 interactions.
A >90% inhibition of fusion could be attained with PA11, PA12, and PA14, and >90% inhibition of entry could be attained with PA14. The most potent of the six MAbs in blocking fusion and entry was PA14, which was slightly more effective than 2D7. Surprisingly, PA14 and 2D7 were among the least potent inhibitors of gp120-sCD4 binding to L1.2-CCR5+ cells, whereas PA9 to PA12 blocked with similar potencies, and PA8 was unable to block >40% of gp120-sCD4 binding.
However, even at antibody concentrations of 300 μg/ml, PA8, PA9, and PA10 blocked cell-cell fusion by <15% and viral entry by <40%. It is thus somewhat puzzling that these hybridomas came to be selected during primary screening, which employed the identical cell-cell fusion assay. One possibility is that the secreted MAbs acted synergistically with chemokines or other factors in the hybridoma supernatants. Another possibility is that their potency was diminished during subcloning and purification.
Inhibition of cell-cell fusion required in some cases almost 2 orders of magnitude more antibody than what was needed to block viral entry. Presumably, more gp120-CD4-CCR5 interactions as well as interactions between adhesion molecules are established and act cooperatively during cell-cell fusion, compared to virus-cell fusion, making it more difficult to inhibit. This is commonly observed with antibodies to LFA-1 or to the HIV-1 envelope glycoprotein (41, 47).
The low staining of CD4+ lymphocytes and the partial inhibition of fusion and entry by some of our MAbs suggest that they are able to bind to only a subset of CCR5 molecules expressed on primary CD4+ lymphocytes, PM1 and U87MG-CD4+ CCR5+ cell lines. Yet, other than PA8, all MAbs are able to stain >90% of L1.2-CCR5+ cells and to completely block binding of the gp120-sCD4 complex to these cells. At least one difference between L1.2-CCR5+ cells and the other cells that we have used is the density of coreceptor protein on the cell surface. Indeed, we estimate that the L1.2-CCR5+ cells express 10- to 100-fold more cell surface coreceptor than do PM1 and U87MG-CD4+ CCR5+ cells. But when HeLa cells are engineered to transiently express as much coreceptor as the L1.2-CCR5+ cell line, as determined by 2D7 staining, we are still unable to detect gp120-sCD4 binding to them (data not shown). Overexpression of CCR5 on L1.2, along with other cell-specific factors, therefore might favor a coreceptor conformation that prominently exposes the Nt, making it more accessible to both MAbs and gp120. Such a conformation might be induced by receptor oligomerization, by diminished or altered associations with cell surface proteins, or by receptor interactions with G proteins (23, 58). Do multiple conformations of CCR5 coexist on the cell surface, and are they permissive for viral entry? The patterns of MAb reactivity would suggest so, since HIV-1 entry and fusion can occur, albeit at reduced levels, in the presence of MAb concentrations that saturate epitopes required for gp120 binding to L1.2-CCR5+ cells. We favor the hypothesis that the coreceptor molecules present on L1.2-CCR5+ cells possess one HIV-1 entry-competent conformation whereas CCR5 molecules on PBMC, PM1, and CCR5+ U87MG exist in multiple entry-competent states that display different MAb reactivities. Whereas PA14 and 2D7 may recognize all conformations, other MAbs may not. Why L1.2 cells are conducive to a particular coreceptor conformation remains to be determined.
No obvious correlation was observed between the HIV-1 and CC-chemokine inhibitory activities of the MAbs. PA8 to PA12 did not inhibit CC-chemokine induced calcium mobilization in L1.2-CCR5+ cells, nor did they mediate signaling through CCR5. Compared with 2D7, PA14 is equipotent at inhibiting HIV-1 fusion and entry but 10-fold less potent at inhibiting RANTES-induced signaling. Whereas 2D7 and RANTES binding maps primarily to ECL2, the PA14 epitope maps to both ECL2 and the Nt domain and may have less potential for steric overlap. These data demonstrate the feasibility of developing chemokine receptor-specific HIV-1 inhibitors that do not block normal receptor activity, an observation with considerable therapeutic implications.
Synergy between CCR5 MAbs and other viral entry inhibitors may reflect their interactions with distinct epitopes that are involved in interdependent, consecutive steps of HIV-1 entry. The degree of synergy observed between PA12 and 2D7 (CI < 0.1 under many circumstances) is extraordinary since CI values <0.2 are rarely observed for combinations of anti-HIV-1 antibodies (31, 32, 57), reverse transcriptase inhibitors (27), or protease inhibitors (40). Because of its potency, the PA12-2D7 combination was examined in multiple assay formats and concentration ratios, for which consistently high levels of synergy were observed. The PA12-2D7 combination did not act as a CCR5 agonist. Moreover, the presence of PA12 had no effect on the ability of 2D7 to block RANTES signaling in L1.2-CCR5+ cells (data not shown), indicating that the mechanism of synergy does not involve receptor down-regulation.
Important synergies were also observed when PA12 was used in combination with RANTES, but we do not know to what extent RANTES-induced CCR5 down-regulation contributed to this phenomenon. Moderate synergy was observed for PA12 combined with PA14. Agents that bind similar regions on CCR5 (PA11-PA12 or PA14-2D7-RANTES) generally displayed additive or antagonistic effects when used in combination. We also observed synergy between PA12 and CD4-IgG2. The CD4-gp120 complex is metastable, and if it is unable to interact with a coreceptor, it decays into a nonfusogenic state (41–44). Synergy would be expected if PA12 prevented the activated virus from interacting with CCR5 prior to decay. The lack of synergy between MAb PA14 and CD4-IgG2 suggests that they act on two nonconsecutive and independent steps of viral entry.
We and others have recently demonstrated that determinants of gp120 binding reside in the first 20 residues of the CCR5 Nt domain and center on a region spanning Y10 to E18 (18, 20, 22, 48). MAbs that map to this region potently block the gp120-CCR5 interaction but are not nearly as efficient at inhibiting HIV-1 fusion and entry into target cells as are PA14 and 2D7, whose epitopes lie outside this region. PA14 recognizes the tip of the Nt and residues in ECL2, whereas the 2D7 epitope seems to be located exclusively in ECL2. At present, we can only speculate about the mechanism of action of these MAbs. It may be that their binding to the first few residues of ECL2 induces conformational changes in the coreceptor that prevent membrane fusion. Alternatively, obstruction of ECL2 epitopes might impede coreceptor oligomerization and the formation of a fusion-competent protein complex. Yet another possibility is that residues in ECL2 line the fusion pore and binding of the MAbs impedes gp41 from inserting the fusion peptide into the plasma membrane. In contrast, MAbs PA8 to PA12 probably inhibit fusion and entry only by directly competing for binding with gp120-CD4 complexes. We do not know if parameters other than epitope exposure and affinity for CCR5 determine the antiviral activity of these MAbs. It is unclear why inhibiting steps subsequent to the gp120-coreceptor interaction would be more efficient than directly blocking that interaction. One possibility is that the off rate of gp120 binding to CCR5 is much lower than the on rate of MAb binding to CCR5. Thus, every time a MAb detaches itself from a coreceptor molecule, a virion-associated gp120 molecule replaces it in a quasi-irreversible fashion since this interaction leads to membrane fusion. Another possibility is that the interactions of CCR5 with oligomeric gp120-gp41 are more complex than that with monomeric gp120.
Our observations are consistent with a model wherein HIV-1 entry occurs in three distinct, sequential steps involving receptor binding, coreceptor binding, and coreceptor-mediated membrane fusion. Separate coreceptor binding and fusion events are suggested by the lack of correlation between the MAbs’ abilities to block gp120 binding and HIV-1 entry-fusion. The chronology of events during fusion is further suggested by the patterns of synergies observed. MAbs that potently inhibit the middle step of the process, namely, gp120 binding, act synergistically with inhibitors of prior and subsequent steps. In keeping with this model, no or weak synergies were observed between agents, such as 2D7 and CD4-IgG2, that act at the first and third steps. The antibodies described herein provide tools for probing these hypotheses.
ACKNOWLEDGMENTS
This work was supported by grants AI43847-01, AI41420-01, and AI40810-01.
We thank Alexandra Trkola and Tom Sakmar for helpful advice on experiments.
REFERENCES
- 1.Alkhatib G, Locati M, Kennedy P E, Murphy P M, Berger E A. HIV-1 co-receptor activity of CCR5 and its inhibition by chemokines: independence from G protein signaling and importance of coreceptor downmodulation. Virology. 1997;234:340–348. doi: 10.1006/viro.1997.8673. [DOI] [PubMed] [Google Scholar]
- 2.Allaway G P, Davis-Bruno K L, Beaudry B A, Garcia E B, Wong E L, Ryder A M, Hasel K W, Gauduin M C, Koup R A, McDougal J S, Maddon P J. Expression and characterization of CD4-IgG2, a novel heterotetramer that neutralizes primary HIV type 1 isolates. AIDS Res Hum Retroviruses. 1995;11:533–539. doi: 10.1089/aid.1995.11.533. [DOI] [PubMed] [Google Scholar]
- 3.Allaway G P, Ryder A M, Beaudry G A, Maddon P J. Synergistic inhibition of HIV-1 envelope-mediated cell fusion by CD4-based molecules in combination with antibodies to gp120 or gp41. AIDS Res Hum Retroviruses. 1993;9:581–587. doi: 10.1089/aid.1993.9.581. [DOI] [PubMed] [Google Scholar]
- 4.Amara A, Gall S L, Schwartz O, Salamero J, Montes M, Loetscher P, Baggiolini M, Virelizier J L, Arenzana-Seisdedos F. HIV coreceptor down-regulation as antiviral principle: SDF-1a-dependent internalization of the chemokine receptor CXCR4 contributes to inhibition of HIV replication. J Exp Med. 1997;186:139–146. doi: 10.1084/jem.186.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berger E A. HIV entry and tropism: the chemokine receptor connection. AIDS. 1997;11(Suppl. A):S3–S16. [PubMed] [Google Scholar]
- 6.Bieniasz P D, Cullen B R. Chemokine receptors and human immunodeficiency virus infection. Front Biosci. 1998;3:44–58. doi: 10.2741/a265. [DOI] [PubMed] [Google Scholar]
- 7.Bieniasz P D, Fridell R A, Aramori I, Ferguson S S G, Caron M C, Cullen B R. HIV-1-induced cell fusion is mediated by multiple regions within both the viral envelope and the CCR5 co-receptor. EMBO J. 1997;16:2599–2609. doi: 10.1093/emboj/16.10.2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brelot A, Heveker N, Pleskoff O, Sol N, Alizon M. Role of the first and third extracellular domains of CXCR4 in human immunodeficiency virus coreceptor activity. J Virol. 1997;71:4744–4751. doi: 10.1128/jvi.71.6.4744-4751.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan D C, Kim P S. HIV entry and its inhibition. Cell. 1998;93:681–684. doi: 10.1016/s0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- 10.Chou T C, Rideout D C. Synergism and antagonism in chemotherapy. New York, N.Y: Academic Press; 1991. [Google Scholar]
- 11.Cocchi F, DeVico A L, Garzino-Derno A, Arya S K, Gallo R C, Lusso P. Identification of RANTES, MIP-1α and MIP-1β as the major HIV-suppressive factors produced by CD8 T-cells. Science. 1995;270:1811–1815. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- 12.Connor R I, Sheridan K E, Ceradini D, Choe S, Landau N R. Change in co-receptor use correlates with disease progression in HIV-1 infected individuals. J Exp Med. 1997;185:621–628. doi: 10.1084/jem.185.4.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dalgleish A G, Beverly P C L, Clapham P R, Crawford D H, Greaves M F, Weiss R A. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature. 1984;312:763–766. doi: 10.1038/312763a0. [DOI] [PubMed] [Google Scholar]
- 14.Deng H K, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, DiMarizio P, Marmon S, Sutton R E, Hill C M, Peiper S C, Schall T J, Littman D R, Landau N R. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 15.Dimitrov D S. How do viruses enter cells? The HIV co-receptors teach us a lesson of complexity. Cell. 1997;91:721–730. doi: 10.1016/s0092-8674(00)80460-2. [DOI] [PubMed] [Google Scholar]
- 16.Doranz B J, Lu Z-H, Rucker J, Zhang T-Y, Sharron M, Cen Y-H, Wang Z-X, Guo H-H, Du J-G, Accavitti M A, Doms R W, Peiper S C. Two distinct CCR5 domains can mediate coreceptor usage by human immunodeficiency virus type 1. J Virol. 1997;71:6305–6314. doi: 10.1128/jvi.71.9.6305-6314.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dragic T, Litwin V, Allaway G P, Martin S R, Huanh Y, Nagashima K A, Cayanan C, Maddon P J, Koup R A, Moore J P, Paxton W A. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature. 1996;381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 18.Dragic T, Trkola A, Lin S W, Nagashima K A, Kajumo F, Zhao L, Olson W C, Wu L, Mackay C R, Allaway G P, Sakmar T P, Moore J P, Maddon P J. Amino-terminal substitutions in the CCR5 coreceptor impair gp120 binding and human immunodeficiency virus type 1 entry. J Virol. 1998;72:279–285. doi: 10.1128/jvi.72.1.279-285.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dragic T, Trkola A, Moore J P. HIV co-receptors: gateways to the cell. Adv Res Ther. 1997;7:2–13. [Google Scholar]
- 20.Farzan M, Choe H, Vaca L, Martin K, Sun Y, Desjardins E, Ruffing N, Wu L, Wyatt R, Gerard N, Gerard C, Sodroski J. A tyrosine-rich region in the N terminus of CCR5 is important for human immunodeficiency virus type 1 entry and mediates an association between gp120 and CCR5. J Virol. 1998;72:1160–1164. doi: 10.1128/jvi.72.2.1160-1164.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fuerst T R, Niles E G, Studier F W, Moss B. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc Natl Acad Sci USA. 1986;83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Genoud S, Kajumo F, Guo Y, Thompson D A D, Dragic T. CCR5-mediated human immunodeficiency virus entry depends on an amino-terminal domain gp120-binding site and on the conformational integrity of all four extracellular domains. J Virol. 1999;73:1645–1648. doi: 10.1128/jvi.73.2.1645-1648.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gether U, Kobilka B K. G protein-coupled receptors. J Biol Chem. 1998;273:17979–17982. doi: 10.1074/jbc.273.29.17979. [DOI] [PubMed] [Google Scholar]
- 24.Gordon C J, Muesing M A, Proudfoot A E I, Power C A, Moore J P, Trkola A. Enhancement of human immunodeficiency virus type 1 infection by the CC-chemokine RANTES is independent of the mechanism of virus-cell fusion. J Virol. 1999;73:684–694. doi: 10.1128/jvi.73.1.684-694.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hill C M, Kwon D, Jones M, Davis C B, Marmon S, Daugherty B L, DeMartino J A, Springer M S, Unutmaz D, Littman D R. The amino terminus of human CCR5 is required for its function as a receptor for diverse human and simian immunodeficiency virus envelope glycoproteins. Virology. 1998;248:357–371. doi: 10.1006/viro.1998.9283. [DOI] [PubMed] [Google Scholar]
- 26.Howard O M Z, Oppenheim J J, Hollingshead M G, Covey J M, Bigelow J, McCormack J J, Buckheit J R W, Clanton D J, Tupin J A, Rice W G. Inhibition of in vitro and in vivo HIV replication by a distamycin analogue that interferes with chemokine receptor function: a candidate for chemotherapeutic and microbicidal application. J Med Chem. 1998;41:2184–2193. doi: 10.1021/jm9801253. [DOI] [PubMed] [Google Scholar]
- 27.Johnson V A, Merrill D P, Videler J A, Chou T C, Byington R E, Eron J J, D’Aquila R T, Hirsch M S. Two-drug combinations of zidovudine, didanosine, and recombinant interferon-alpha A inhibit replication of zidovudine-resistant human immunodeficiency virus type 1 synergistically in vitro. J Infect Dis. 1991;164:646–655. doi: 10.1093/infdis/164.4.646. [DOI] [PubMed] [Google Scholar]
- 28.Klatzmann D, Champagne E, Chamaret S, Gruest J M, Guetard D, Hercend T, Gluckman J C, Montagnier L. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature. 1984;312:382–385. doi: 10.1038/312767a0. [DOI] [PubMed] [Google Scholar]
- 29.Kuhmann S E, Platt E J, Kozak S L, Kabat D. Polymorphisms in the CCR5 genes of African green monkeys and mice implicate specific amino acids in infections by simian and human immunodeficiency viruses. J Virol. 1997;71:8642–8656. doi: 10.1128/jvi.71.11.8642-8656.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kwong P D, Wyatt R, Robinson J, Sweet R W, Sodroski J, Hendrickson W A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laal S, Burda S, Gorny M K, Karwowska S, Buchbinder A, Zolla-Pazner S. Synergistic neutralization of human immunodeficiency virus type 1 by combinations of human monoclonal antibodies. J Virol. 1994;68:4001–4008. doi: 10.1128/jvi.68.6.4001-4008.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li A, Katinger H, Posner M R, Cavacini L, Zolla-Pazner S, Gorny M K, Sodroski J, Chou T C, Baba T W, Ruprecht R M. Synergistic neutralization of simian-human immunodeficiency virus SHIV-vpu+ by triple and quadruple combinations of human monoclonal antibodies and high-titer anti-human immunodeficiency virus type 1 immunoglobulins. J Virol. 1998;72:3235–3240. doi: 10.1128/jvi.72.4.3235-3240.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Littman D R. Chemokine receptors: keys to AIDS pathogenesis. Cell. 1998;93:677–680. doi: 10.1016/s0092-8674(00)81429-4. [DOI] [PubMed] [Google Scholar]
- 34.Litwin, V. Unpublished results.
- 35.Litwin V, Nagashima K, Ryder A M, Chang C H, Carver J M, Olson W C, Alizon M, Hasel K W, Maddon P J, Allaway G P. Human immunodeficiency virus type 1 membrane fusion mediated by a laboratory-adapted strain and a primary isolate analyzed by resonance energy transfer. J Virol. 1996;70:6437–6441. doi: 10.1128/jvi.70.9.6437-6441.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mack M, Luckow B, Nelson P J, Cihak J, Simmons G, Clapham P R, Signoret N, Marsh M, Stangassinger M, Borlat F, Wells T N C, Schlondorff D, Proudfoot A E I. Aminooxypentane-RANTES induces CCR5 internalization but inhibits recycling: a novel inhibitory mechanism of HIV infectivity. J Exp Med. 1998;187:1215–1224. doi: 10.1084/jem.187.8.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maddon P J, Dalgleish A G, McDougal J S, Clapham P R, Weiss R A, Axel R. The T4 gene encodes the AIDS virus receptor and is expressed in the immune system and the brain. Cell. 1986;47:333–348. doi: 10.1016/0092-8674(86)90590-8. [DOI] [PubMed] [Google Scholar]
- 38.McDougal J S, Kennedy M S, Sligh J M, Cort S P, Mawle A, Nicholson J K A. Binding of HTLVIII/LAV to T4+ T cells by a complex of the 110K viral protein and the T4 molecule. Science. 1986;231:382–385. doi: 10.1126/science.3001934. [DOI] [PubMed] [Google Scholar]
- 39.McKnight A, Wilkinson D, Simmons G, Talbot S, Picard L, Ahuja M, Marsh M, Hoxie J A, Clapham P R. Inhibition of human immunodeficiency virus fusion by a monoclonal antibody to a coreceptor (CXCR4) is both cell type and virus strain dependent. J Virol. 1997;71:1692–1696. doi: 10.1128/jvi.71.2.1692-1696.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Merrill D P, Manion D J, Chou T C, Hirsch M S. Antagonism between human immunodeficiency virus type 1 protease inhibitors indinavir and saquinavir in vitro. J Infect Dis. 1997;176:265–268. doi: 10.1086/517263. [DOI] [PubMed] [Google Scholar]
- 41.Moore J P, Cao Y, Qing L, Sattentau Q J, Pyati J, Koduri R, Robinson J, Barbas III C F, Burton D R, Ho D D. Primary isolates of human immunodeficiency virus type 1 are relatively resistant to neutralization by monoclonal antibodies to gp120, and their neutralization is not predicted by studies with monomeric gp120. J Virol. 1995;69:101–109. doi: 10.1128/jvi.69.1.101-109.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moore J P, Jameson B A, Weiss R A, Sattentau Q J. The HIV-cell fusion reaction. In: Bentz J, editor. Viral fusion mechanisms. Boca Raton, Fla: CRC Press, Inc.; 1993. pp. 233–289. [Google Scholar]
- 43.Moore J P, McKeating J A, Huang Y, Ashkenazi A, Ho D D. Virions of primary human immunodeficiency virus type 1 isolates resistant to soluble CD4 (sCD4) neutralization differ in sCD4 binding and glycoprotein gp120 retention from sCD4-sensitive isolates. J Virol. 1992;66:235–243. doi: 10.1128/jvi.66.1.235-243.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore J P, Sweet R W. The HIV gp120-CD4 interaction: a target for pharmacological and immunological intervention. Prospect Drug Discovery Design. 1993;1:235–250. [Google Scholar]
- 45.Olson, W. C. Unpublished results.
- 46.Osbourn J K, Earnshaw J G, Johnson K S, Parmentier M, Timmermans V, McCafferty J. Directed selection of MIP-1a neutralizing CCR5 antibodies from a phage display human antibody library. Nat Biotechnol. 1998;16:778–781. doi: 10.1038/nbt0898-778. [DOI] [PubMed] [Google Scholar]
- 47.Pantaleo G, Poli G, Butini L, Fox C, Dayton A I, Fauci A S. Dissociation between syncytia formation and HIV spreading. Expression of syncytia does not necessarily reflect inhibition of HIV infection. Eur J Immunol. 1991;21:1771–1774. doi: 10.1002/eji.1830210730. [DOI] [PubMed] [Google Scholar]
- 48.Rabut G E E, Konner J A, Kajumo F, Moore J P, Dragic T. Alanine substitutions of polar and nonpolar residues in the amino-terminal domain of CCR5 differently impair entry of macrophage- and dualtropic isolates of human immunodeficiency virus type 1. J Virol. 1998;72:3464–3468. doi: 10.1128/jvi.72.4.3464-3468.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rizzuto C, Wyatt R, Hernandez-Ramos N, Sun Y, Kwong P, Hendrickson W, Sodroski J. Identification of a conserved human immunodeficiency virus gp120 glycoprotein structure important for chemokine receptor binding. Science. 1998;280:1949–1953. doi: 10.1126/science.280.5371.1949. [DOI] [PubMed] [Google Scholar]
- 50.Rucker J, Samson M, Doranz B J, Libert F, Berson J F, Yi Y, Smyth R J, Collman R G, Broder C C, Vassart G, Doms R W, Parmentier M. Regions in the β-chemokine receptors CCR-5 and CCR-2b that determine HIV-1 cofactor specificity. Cell. 1996;87:437–446. doi: 10.1016/s0092-8674(00)81364-1. [DOI] [PubMed] [Google Scholar]
- 51.Samson M, LaRosa G, Libert F, Paindavoine P, Detheux M, Vassart G, Parmentier M. The second extracellular loop of CCR5 is the major determinant of ligand specificity. J Biol Chem. 1997;272:24934–24941. doi: 10.1074/jbc.272.40.24934. [DOI] [PubMed] [Google Scholar]
- 52.Simmons G, Clapham P R, Picard L, Offord R E, Rosenkilde M M, Schwartz T W, Buser R, Wells T N C, Proudfoot A E I. Potent inhibition of HIV-1 infectivity in macrophages and lymphocytes by a novel CCR5 antagonist. Science. 1997;276:276–279. doi: 10.1126/science.276.5310.276. [DOI] [PubMed] [Google Scholar]
- 53.Simmons G, Wilkinson D, Reeves J D, Dittmar M T, Beddows S, Weber J, Carnegie G, Desselberger U, Gray P W, Weiss R A, Clapham P R. Primary, syncytium-inducing human immunodeficiency virus type 1 isolates are dual-tropic and most can use either Lestr or CCR5 as coreceptors for virus entry. J Virol. 1996;70:8355–8360. doi: 10.1128/jvi.70.12.8355-8360.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Strizki J M, Davis Turner J, Collman R G, Hoxie J, Gonzalez-Scarano F. A monoclonal antibody (12G5) directed against CXCR-4 inhibits infection with the dual-tropic human immunodeficiency virus type 1 isolate HIV-189.6 but not the T-tropic isolate HIV-1HxB. J Virol. 1997;71:5678–5683. doi: 10.1128/jvi.71.7.5678-5683.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trkola A, Dragic T, Arthos J, Binley J, Olson W C, Allaway G P, Cheng-Mayer C, Robinson J, Maddon P J, Moore J P. CD4-dependent, antibody sensitive interactions between HIV-1 and its co-receptor CCR-5. Nature. 1996;384:184–187. doi: 10.1038/384184a0. [DOI] [PubMed] [Google Scholar]
- 56.Trkola A, Paxton W A, Monard S P, Hoxie J A, Siani M A, Thompson D A, Wu L, Mackay C R, Horuk R, Moore J P. Genetic subtype-independent inhibition of human immunodeficiency virus type 1 replication by CC- and CXC chemokines. J Virol. 1997;72:396–404. doi: 10.1128/jvi.72.1.396-404.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vijh-Warrier S, Pinter A, Honnen W J, Tilley S A. Synergistic neutralization of human immunodeficiency virus type 1 by a chimpanzee monoclonal antibody against the V2 domain of gp120 in combination with monoclonal antibodies against the V3 loop and the CD4-binding site. J Virol. 1996;70:4466–4473. doi: 10.1128/jvi.70.7.4466-4473.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ward S G, Bacon K, Westwick J. Chemokines and lymphocytes: more than an attraction. Immunity. 1998;9:1–11. doi: 10.1016/s1074-7613(00)80583-x. [DOI] [PubMed] [Google Scholar]
- 59.Wu L, Gerard N P, Wyatt R, Choe H, Parolin C, Ruffing N, Borsetti A, Cardoso A A, Desjardin E, Newman W, Gerard C, Sodroski J. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature. 1996;384:179–183. doi: 10.1038/384179a0. [DOI] [PubMed] [Google Scholar]
- 60.Wu L, LaRosa G, Kassam N, Gordon C J, Heath H, Ruffing N, Chen H, Humblias J, Samson M, Parmentier M, Moore J P, Mackay C R. Interaction of chemokine receptor CCR5 with its ligands: multiple domains for HIV-1 gp120 binding and a single domain for chemokine binding. J Exp Med. 1997;186:1373–1381. doi: 10.1084/jem.186.8.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu L, Paxton W, Kassam N, Ruffing N, Rittman J B, Sullivan N, Choe H, Sodroski J, Newman W, Koup R A, Mackay C. CCR5 levels and expression patterns correlate with infectability by macrophage-tropic HIV-1, in vitro. J Exp Med. 1997;185:1681–1691. doi: 10.1084/jem.185.9.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wyatt R, Kwong P D, Desjardins E, Sweet R, Robinson J, Hendrickson W, Sodroski J. The antigenic structure of the human immunodeficiency virus gp120 envelope glycoprotein. Nature. 1998;393:705–711. doi: 10.1038/31514. [DOI] [PubMed] [Google Scholar]
- 63.Wyatt R, Sodroski J. The HIV-1 envelope glycoproteins: fusogens, antigens and immunogens. Science. 1998;280:1884–1888. doi: 10.1126/science.280.5371.1884. [DOI] [PubMed] [Google Scholar]
- 64.Ylisastigui L, Vizzavona J J, Drakopoulou E, Paindavoine P, Calvo C F, Parmentier M, Gluckman J C, Vita C, Benjouad A. Synthetic full length and truncated RANTES inhibit HIV-1 infection of primary macrophages. AIDS. 1998;12:977–984. [PubMed] [Google Scholar]