

Abstract
Background and Purpose:
Macrophage-rich atherosclerotic arteries are highly active in glycolysis. PFKFB3, a key glycolytic enzyme, has emerged as a potential therapeutic target in atherosclerosis. Small-molecule inhibitors of PFKFB3, such as 3PO and PFK158, have demonstrated efficacy in hampering atherogenesis in preclinical models. However, genetic studies elucidating the role of Pfkfb3 in atherogenesis need to be conducted to validate pharmacological findings and to unveil potential pharmacological side effects.
Experimental Approach:
Apoe−/− mice with global heterozygous or myeloid cell-specific Pfkfb3 deficiency were fed a Western diet (WD), after which atherosclerosis development was determined. Monocyte subsets in atherosclerotic mice and patients were examined by flow cytometry. Monocyte infiltration was assayed by a Ly6Chi monocyte-specific latex labelling procedure. In situ efferocytosis was assessed on mouse aortic root sections. Additionally, metabolic status, macrophage motility, efferocytosis, and involved mechanisms were analysed in peritoneal macrophages.
Key Results:
Global heterozygous or myeloid cell-specific Pfkfb3 deficiency reduced atherogenesis in Apoe−/− mice. Mechanistic studies showed that PFKFB3 controlled the proliferation and infiltration of proinflammatory monocytes. Moreover, PFKFB3 expression was associated with inflammatory monocyte expansion in patients with atherosclerotic coronary artery disease. Surprisingly, homozygous loss of Pfkfb3 impaired macrophage efferocytosis and exacerbated atherosclerosis in Apoe−/− mice. Mechanistically, PFKFB3-driven glycolysis was shown to be essential for actin polymerization, thus aiding the efferocytotic function of macrophages.
Conclusion and Implications:
Collectively, these findings suggest the existence of a double-edged sword effect of myeloid PFKFB3 on the pathogenesis of atherosclerosis and highlight the need for caution in developing anti-atherosclerotic strategies that target PFKFB3.
Keywords: atherosclerosis, efferocytosis, glycolysis, macrophage, PFKFB3
1 |. INTRODUCTION
Atherosclerosis is initiated by monocyte–macrophage recruitment followed by foam cell formation, which results in the deposition of cholesterol-rich lipoproteins and chronic inflammation in the arterial wall (Hansson & Libby, 2006; Libby, 2002). Hypercholesterolaemia has been documented to induce monocytosis, which profoundly expands the number of circulating inflammatory Ly-6Chi monocytes (Murphy et al., 2011; Swirski et al., 2007). Circulating monocytes, upon atherogenic stimulation, binds to and migrates across the endothelium of the vessel wall to gain access to atherosclerotic lesions (Swirski et al., 2007). The monocyte count and its infiltrative capacity, particularly for the Ly-6Chi subset in mice and CD14+CD16+ subset in humans, independently predict cardiovascular events after adjustment for conventional risk factors (Rogacev et al., 2012).
In recent years, accumulating evidence demonstrates that cellular metabolic reprogramming accompanies the development of atherosclerosis (Ali et al., 2018; Shirai et al., 2016). In particular, atherosclerosis-related factors modify metabolic profiles in multiple cell types to fit their specific needs, including monocytes/macrophages and endothelial cells that exhibit high levels of glycolysis for their energy metabolism to regulate cellular function (Dolfi et al., 2021; Schnitzler et al., 2020). The shift to glycolysis is due, at least in part, to up-regulation of enzymes in the glycolytic pathway, including 6-phosphofructo-2-kinase/fructose-2,6-bisphophatase isoform 3 (PFKFB3), which catalyses the synthesis of fructose-2, 6-bisphophate (F2,6P2), leading to the activation of 6-phosphofructo-1-kinase (PFK-1), a potent stimulator of glycolysis (Sarrazy et al., 2016; Xu et al., 2014). In the context of atherosclerosis, it becomes evident that endothelial cells, upon lipoprotein(a) exposure, reprogram their glucose metabolism toward glycolysis through PFKFB3 induction to fuel inflammatory activation (Schnitzler et al., 2020). Additionally, when encountering inflammatory cues, monocytes differentiate into classical activated macrophages that exhibit a high level of PFKFB3-mediated glycolysis (Kelly & O’Neill, 2015; Rodríguez-Prados et al., 2010).
Since the initial reports, researchers have corroborated the efficacy of PFKFB3 inhibitors in preclinical models of atherosclerosis (Perrotta et al., 2020; Poels et al., 2020) and confirmed their athero-protective role; however, genetic intervention targeting PFKFB3 in atherosclerosis is lacking, and the precise role of PFKFB3 protein in monocyte/macrophage biology has not been thoroughly examined.
In the present study, through engineering global heterozygous and myeloid cell-specific Pfkfb3 knockout models, we directly investigated the role of PFKFB3 in atherosclerosis and the underlying cellular and molecular processes. Our results revealed the dual action of myeloid PFKFB3 in atherosclerosis. Heterozygous Pfkfb3 deletion reduced the severity of atherosclerosis in Apoe−/− mice. In contrast, mice with homozygous Pfkfb3 deletion in macrophages surprisingly showed more severe atherosclerosis. Our findings not only dissect the pro-atherogenic roles of PFKFB3, but also provide insight into the normal physiological role of macrophage PFKFB3 and bring to light the potential adverse side effects of PFKFB3 targeting in atherosclerotic vascular diseases.
2 |. METHODS
2.1 |. Animal procedures
The use of mice was approved by the Institutional Animal Care and Use Committee at Guangzhou Medical University (Guangzhou, China) and Augusta University (Augusta, GA, USA) in accordance with NIH guidelines. Animal studies are reported in compliance with the ARRIVE guidelines (Percie du Sert et al., 2020) and with the recommendations made by the British Journal of Pharmacology (Lilley et al., 2020). Global heterozygous Pfkfb3 (Pfkfb3+/−) knockout mice were generated as previously described (Chesney et al., 2005). The floxed Pfkfb3 (Pfkfb3flox/flox) mice were generated by Xenogen Biosciences Corporation (Cranbury, NJ, USA) (Xu et al., 2014). Haplodeficiency or homozygous deficiency of Pfkfb3 specific to myeloid cells (Pfkfb3flox/+/LysMcre/+ and Pfkfb3flox/flox/LysMcre/+; hereinafter referred as Mye-Pfkfb3+/− and Mye-Pfkfb3−/−) was achieved by cross-breeding LysMcre/+ transgenic mice (Stock Number: 004718, The Jackson Laboratory) with Pfkfb3flox/+ or Pfkfb3flox/flox mice, respectively, all on a C57BL/6 background. For the atherosclerosis study, Pfkfb3+/− mice were bred with Apoe−/− (Stock Number: 002052, The Jackson Laboratory) mice to generate Apoe−/−/Pfkfb3+/− mice as well as their littermate control Apoe−/−/Pfkfb3+/+ mice; Mye-Pfkfb3+/− and Mye-Pfkfb3−/− mice were bred with Apoe−/− mice to generate Apoe−/−/Mye-Pfkfb3+/− and Apoe−/−/Mye-Pfkfb3−/− mice as well as their littermate control Apoe−/−/Mye-Pfkfb3+/+ mice. All mice were housed on a 12:12-h light–dark cycle. For anaesthetising mice, ketamine (100 mg·kg−1 body weight) and xylazine (10 mg·kg−1) (Phoenix Scientific, Inc., St. Joseph, MO, USA) were intraperitoneally injected. For killing mice, CO2 asphyxiation was used followed by cervical dislocation.
2.2 |. Materials
The chemicals or reagents of Oil Red O (O0625), Haematoxylin (H9627), Eosin (861006), Brewer’s thioglycolate solution (T0632), glucose (G8270), oligomycin (495455), 2DG (D8375), Crystal violet (C0775), and staurosporine (569396) were purchased from Sigma-Aldrich (St. Louis, MO, USA). The fluorescent latex beads (18860) and phalloidin conjugated with rhodamine (ab235138) were purchased from Polysciences, Inc (Warrington, PA, USA) and Abcam (Cambridge, UK), respectively. The antibodies of anti-Mac-2 (ACL8942F), anti-PFKFB3 (ab181861), and anti-cleaved caspase 3 (9661) were obtained from Accurate Chemical & Scientific Corporation (Westbury, NY, USA), Abcam, and Cell Signaling Technology (Danvers, MA, USA), respectively. The antibodies of anti-CD11b-FITC (11-0112-82), anti-CD115-Percp-eFluor 710 (46-1152-82), anti-Ly6C-APC (17-5932-82), anti-CD45-PE (12-0451-82), anti-CD16- Percp-eFluor 710 (46-0168-42), and CD16/32 blocking antibody (14-0161-82) were purchased from eBioscience (San Diego, CA, USA). The antibodies of anti-CD14-FITC (557153) and anti-BrdU-PE (556029) were obtained from BD Biosciences (San Jose, CA, USA). VECTASTAIN® ABC kit (PK-4001), Total plasma cholesterol measurement kit (10007640), RNeasy Mini kit (74106), and iScriptTM cDNA synthesis kit (1708891) were purchased from Vector Labs (Malvern, PA, USA), Cayman (Ann Arbor, MI, USA), Qiagen (Shenzhen, China), and Bio-Rad (Hercules, CA, USA), respectively.
2.3 |. Patients and controls
All procedures involving human subjects were carried out in accordance with the Declaration of Helsinki and were approved by the Institutional Review Board at Guangzhou University of Chinese Medicine. Written informed consent was obtained from all participants prior to the inclusion of subjects. Twelve patients with coronary arterial disease (CAD) and 12 age- and sex-matched healthy individuals were recruited from the First Affiliated Hospital of Guangzhou University of Chinese Medicine. Patients had unequivocal CAD with at least one documented myocardial infarction and were enrolled more than 90 days after an ischaemic event. Healthy controls had no personal or family health history of autoimmune diseases, cancer, chronic viral infection, or any other inflammatory syndromes. Demographic characteristics of the patients and controls are given in Table S1.
2.4 |. Preparation of mouse aortas and quantification of atherosclerosis
To induce atherosclerosis, male Apoe−/−/Pfkfb3+/+, Apoe−/−/Pfkfb3+/−, Apoe−/−/Mye-Pfkfb3+/+, Apoe−/−/Mye-Pfkfb3+/−, and Apoe−/−/Mye-Pfkfb3−/− mice at 6 weeks of age were fed a high-fat Western diet (20% protein, 50% carbohydrate, 21% fat, 0.21% cholesterol; Research Diets, New Brunswick, NJ, USA) for 3 months. Mice were anaesthetised with ketamine (100 mg·kg−1) and xylazine (10 mg·kg−1) and then perfused through the left ventricle with 10 ml of ice-cold PBS. Aortas of atherosclerotic mice were collected, and both en face preparations of whole aortas and cross-sections of aortic sinuses were processed for Oil Red O staining. Each section of aortic sinus is 8 μm thick and sections taken for examination were 48 μm apart. Therefore, six slides over a length of 288 μm of aortic sinus were analysed. For each group, seven mice were included. Images obtained from the above experiments were scanned into a Macintosh computer and analysed with Image-Pro Plus version 5.0 software (Media Cybernetics, Inc., Rockville, MD, USA). The analysis of the en face Oil Red O staining was performed in a blinded manner.
2.5 |. Histological procedures
Mac-2 immunohistochemistry (IHC) staining: As previously reported (Xu, Wang, Yan, Yang, et al., 2017), 8 μm frozen sections were cut through the aortic root in the heart. Sections were fixed in cold acetone; blocked with 0.1% triton X-100, normal serum of the same species as the secondary antibody, Avidin/Biotin and quenched with 3% hydrogen peroxide. After blocking, sections were incubated with anti-Mac-2 antibody (3 μg·ml−1, ACL8942F, Accurate Chemical & Scientific Corporation). Biotinylated secondary antibodies were applied, and sections were stained with VECTASTAIN® ABC kit (1:200, Vector Labs) according to the manufacturer’s instructions. Control sections were incubated with IgG2a isotype antibody (eBioscience). Sections were counterstained with haematoxylin.
Haematoxylin and eosin (H&E) staining: H&E staining was performed to show necrotic areas on cross-sections of atherosclerotic lesions. Quantification of % Mac 2 staining area and necrotic area were performed using the Image-Pro Plus version 5.0 software (Media Cybernetics, Inc., MD). Data are reported as a mean of eight sections of aortic root per heart.
Immunofluorescent (IF) staining: For cleaved caspase 3 and Mac 2 co-IF staining in aortic root, 8 μm frozen sections were cut through the aortic root in the heart. Sections were heated at 98 °C for 8 min in citric acid buffer for antigen retrieval and incubated with anti-cleaved caspase 3 (1:200, 9661, Cell Signaling) and anti-Mac-2 (3 μg·ml−1, ACL8942F, Accurate Chemical & Scientific Corporation) antibodies followed by incubation with an Alexa Fluor 594-labelled secondary antibody (1:400, Molecular Probes, Eugene, OR, USA) and/or Alexa Fluor 488-labelled secondary antibody (1:400, Molecular Probes). The slides were then immersed in ProLong Gold mounting medium with DAPI (Invitrogen, Shanghai, China) to visualize the nuclei. For PFKFB3 and F-actin co-IF staining in cultured cells, peritoneal macrophages were treated with 50 mg·m−1 oxLDL for 6 h. Then, cells were fixed with 4% paraformaldehyde (PFA) at 37 °C for 5 min, washed, and processed for immunostaining with the following antibodies and reagents; anti-PFKFB3 antibody (1:100, ab181861, Abcam), anti-Rabbit-Alexa 488 (1:500, #A-11008, Invitrogen), and phalloidin conjugated with rhodamine (ab235138, Abcam). The slides were then immersed in ProLong Gold mounting medium with DAPI (Invitrogen) to visualize the nuclei. Images were acquired using a SP8 inverted confocal microscope (TCS SP8; Leica, Morrisville, USA) with a ×63 oil objective lens (HCX PL APO 63×/1.40–0.60 oil CS lens; Leica) and HyD detectors (Leica).
2.6 |. Measurement of 18F-FDG uptake
Male Apoe−/−/Mye-Pfkfb3+/+, Apoe−/−/Mye-Pfkfb3+/−, and Apoe−/−/Mye-Pfkfb3−/− mice at 6 weeks of age were fed a high-fat Western diet (20% protein, 50% carbohydrate, 21% fat, 0.21% cholesterol; Research Diets) for 6 weeks to induce atherosclerosis. Mice were fasted overnight before receiving a tail vein injection of 100 μCi 18F-FDG. Two hours later, mice were killed with CO2 asphyxiation followed by cervical dislocation, and the thoracic aortas were collected and weighed. The tissues were immediately fixed in a solution containing L-(+)-lysine hydrochloride (75 mmol·L−1) and 4% PFA in phosphate buffer (37.5 mmol·L−1; pH 7.4). Radioactivity was measured with a well-type γ-counter (1480 WIZARD3; Wallac Co. Ltd., Finland). The results were expressed as (% ID/g) × kg, which was calculated as (tissue activity/tissue weight)/(injected radiotracer activity/animal body weight).
2.7 |. Flow cytometry
For analysis of mouse monocyte subsets: Single-cell suspensions of blood, spleen, and bone marrow were harvested when mice were killed. After lysis of erythrocytes in ice-cold lysis buffer (8.4 g·L−1 NH4Cl, 1 g·L−1 NaHCO3, 37 mg·L−1 EDTA) for 3 min, Fc receptors in cell suspensions were blocked with CD16/32 blocking antibody (0.5 μg/100 μl, #14-0161-82, eBioscience) and then were incubated with anti-CD11b-FITC (1 μg/100 μl, 11-0112-82, eBioscience), anti-CD115- Percp-eFluor 710 (0.06 μg/100 μl, #46-1152-82, eBioscience), anti-Ly6C-APC (0.125 μg/100 μl, #17-5932-82, eBioscience) or anti-CD45-PE (12-0451-82, 0.125 μg·μl−1, eBioscience) antibodies. Stained cells were washed and resuspended in 400 μl of FACS Flow (BD Pharmingen, San Jose, CA, USA), and the whole suspension was analysed using a FACSCalibur flow cytometer with CellQuest software (BD Pharmingen). Isotype controls (BD Pharmingen) were used in parallel. Monocytes were identified as CD45+CD11b+CD115+ cells and further gated as Ly6Chi or Ly6Clo.
For analysis of human monocyte subsets: Erythrocytes in single-cell suspensions of human blood were lysed in ice cold lysis buffer (8.4 g·L−1 NH4Cl, 1 g·L−1 NaHCO3, 37 mg·L−1 EDTA) for 5 min. The resulting lysates were then stained with anti-CD14-FITC (20 μl/100 μl, #557153, BD Biosciences) and anti-CD16- Percp-eFluor 710 (0.06 μg/100 μl, #46-0168-42, eBioscience) antibodies. Stained cells were washed and resuspended in 400 μl of FACS Flow (BD Pharmingen), and the whole suspension was analysed using a FACSCalibur flow cytometer with CellQuest software (BD Pharmingen).
2.8 |. Cholesterol level assay
Blood samples were taken by tail-vein bleeding 1 day before introduction of the Western type of diet and at death. Briefly, the mouse was placed in a restraint cone allowing its tail to hang freely, and the tip of tail was wiped with alcohol. Then, the tail tip was cut off with the sterile scalpel blade, and 30 μl blood sample was collected into EDTA-treated blood collection tube. Total plasma cholesterol was determined by use of kits from Cayman Chemicals (No. 10007640) according to the manufacturer’s instructions.
2.9 |. Cell culture
Bone marrow macrophages derived from non-adherent precursors, and peritoneal macrophages were cultured in RPMI 1640 supplemented with L929 conditioned medium or mouse recombinant M-CSF (20 ng·ml−1, ImmunoTools, GmbH) as previously described.
2.10 |. In vivo migration assay
Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3+/− mice were injected intraperitoneally with 1 ml of 3% Brewer’s thioglycolate solution (Sigma-Aldrich). After 4 days, when most recruited cells are macrophages, mice were killed with CO2 asphyxiation followed by cervical dislocation, and peritoneal cells were collected for microscopic quantification by Giemsa staining.
2.11 |. Latex bead labelling and histochemical analysis
To specifically label Ly6chi monocytes, as described previously (Tacke et al., 2007), mice were intravenously injected with fluorescent latex beads (Polysciences) 18 h after receiving 250 μl chlodronate-contained liposomes (Liposome, Beijing) to transiently deplete monocytes. Mice were killed with CO2 asphyxiation followed by cervical dislocation 24 h after labelling. Hearts were isolated for cryosectioning and mounted with DAPI to analyse the number of latex bead-positive cells per aortic sinus section.
2.12 |. Real time PCR
The total RNA from peritoneal macrophages or from mice aortas was extracted with a RNeasy Mini Kit (Qiagen, Venlo, The Netherlands), and qRT-PCR was performed as described previously (Xu, Wang, Yan, Zhou, et al., 2017). Briefly, a 0.5–1 μg sample of RNA was used as a template for reverse transcription using the iScriptTM cDNA synthesis kit (Bio-Rad). qRT-PCR was performed on an ABI 7500 Real Time PCR System (Applied Biosystems) with the respective gene-specific primers listed in Table S2. All qRT-PCR experiments were performed in biological triplicates that were repeated at least twice independently. Relative gene expression was converted using the 2−ΔΔCt method against the internal control β-actin. The data from each of these experimental groups were analysed independently and revealed statistically significant differences between the indicated groups. Due to inherent variation in experiments performed at significantly different times, we limited the statistical analysis to data sets comprised of biological triplicates from one experiment performed at the same time.
2.13 |. Protein extraction and Western blotting
Peritoneal macrophages were lysed with RIPA buffer (Sigma) with 1% proteinase inhibitor cocktail (Roche, Basel, Switzerland) and 1% PMSF. After centrifugation of the cell lysates at 12000 g for 15 min at 4°C, proteins were quantified with a BCA assay and then loaded in 8%–12% sodium dodecyl sulfate polyacrylamide gel electrophoresis gels at 20 μg per lane. Antibodies used in this study were as follows: anti-PFKFB3 (1:2000, ab181861, Abcam) and β-actin (1:2000, 3700, Cell Signaling Technology). Images were taken with the ChemiDoc MP system (Bio-Rad), and band densities were quantified using Image Lab software (Bio-Rad). The immuno-related procedures used comply with the recommendations made by the British Journal of Pharmacology (Alexander et al., 2018).
2.14 |. BrdU labelling and flow cytometric analysis
Mice were injected intraperitoneally with 200 ul of 2 mg BrdU. Eighteen hours later, mice were killed with CO2 asphyxiation, and blood was drawn for the analysis of circulating monocytes. Single-cell suspensions of blood were lysed of erythrocytes in ice-cold lysis buffer (8.4 g·L−1 NH4Cl, 1 g·L−1 NaHCO3, 37 mg·L−1 EDTA) for 3 min and then were centrifuged at 500 g for 5 min at room temperature. The cell pellets were resuspended in 100 μL flow cytometry staining buffer (BD Biosciences) and then were stained using anti-CD11b-FITC (0.5 μg/100 μl, 11-0112-82, eBioscience, San Diego, CA, USA), anti-CD115- Percp-eFluor 710 (0.06 μg/100 μl, #46-1152-82, eBioscience), anti-Ly6C-APC (0.125 μg/100 μl, #17-5932-82, eBioscience) and anti-BrdU-PE (20 μl/100 μl, #556029, BD Biosciences) antibodies. Stained cells were washed and resuspended in 400 μl of FACS Flow (NC0022088, BD Biosciences), and the whole suspension was analysed using a FACSCalibur flow cytometer with CellQuest software (BD Biosciences). Isotype controls (BD Biosciences) were used in parallel.
2.15 |. Metabolic measurements
FACS-sorted Ly6Chi and Ly6Clo monocytes from spleens were seeded in quintuplicate in Seahorse XF96 polystyrene tissue culture plates (2 × 105 monocytes per well) (Seahorse Bioscience). The metabolic rates of monocytes were analysed in four consecutive measurements in XF Base Medium (unbuffered DMEM with 5.5 mM glucose and 2 mM L-glutamine, pH adjusted to 7.4). After three basal measurements, three consecutive measurements were taken following the addition of 10 mM glucose, 2 μM oligomycin, and 50 mM 2-DG to determine basal and maximum ECAR. All compounds used during the Seahorse runs were from Sigma-Aldrich.
2.16 |. Macrophage function assays (invasion, migration, efferocytosis, and apoptosis)
Invasion and migration: Thioglycollate-elicited macrophages were loaded (2 × 105 cells per well) into the upper portion of a modified Boyden chamber (PVP-free, pore size 8 μM, Corning Incorporated, Corning, NY, USA), coated with matrigel (invasion assays) or uncoated (migration assays; BD Biosciences). As a chemoattractant, MCP-1 (10 ng·m−1; R&D Systems) was added to the lower chamber for both invasion and migration assays. Chambers were incubated for 24 h at 37°C in a 5% CO2 atmosphere. Then, cells on the underside of the membrane were stained with crystal violet (Sigma-Aldrich) and quantitated by dissolving the cell-bound crystal violet in 10% acetic acid for 5 min and subsequent spectrophotometric analysis at 450 nm.
Wound healing assay: Thioglycollate-elicited macrophage monolayers on 0.2% gelatin-coated 24-well plates (5 × 105 cells per well) were serum-starved for 24 h, scraped with a pipette tip, incubated 48 h in complete medium and either photomicrographed for quantification of cell migration or collected for assessment of proliferation.
Efferocytosis: Standard in vitro phagocytosis assays were performed as previously reported (Kojima et al., 2016). Apoptosis was induced in cell tracker CMTMR (5 μM, Molecular Probes)-labelled Jurkat T cells by incubation with staurosporine (1 μM, Sigma) for 2 h. The degree of apoptosis was ≥70% as judged by Annexin V and Propidium Iodide staining (BD Biosciences) and flow cytometry. Anti-F4/80-PE labelled Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3−/− BMDM were added to each well and co-incubated for 2 h in serum-free medium. Double-positive cells were analysed by flow cytometry. Efferocytosis was evaluated by the percentage of F4/80-positive cells that were also cell tracker CMTMR-positive.
Apoptosis: Bone-marrow-derived macrophages were serum-starved for 8 h to remove the effect of any exogenous growth factors. Thereafter, cells were stimulated with 100 μg·ml−1 oxLDL. 48 h later the cells were washed 2 times with annexin-V buffer, incubated with annexin V-APC (550474, BD Pharmingen) and F4/80-FITC (clone BM8, eBioscience) for 20 min, washed further and fixed. Apoptotic macrophages (Annexin V+F4/80+) were analysed with flow cytometry. At least 10,000 events were collected. Data were analysed with CellQuest v3.3 software (BD Biosciences) as instructed.
2.17 |. Macrophage morphology
Cell morphology and polarization: The elongated or rounded peritoneal macrophages were judged by Elongation Index (EI, defined as the ratio of cell length to breadth) as greater than 2 or lower than 1.2, respectively, 36 h after collection and culture. Cells displaying more than three filopodial extensions were considered to have filopodia.
2.18 |. In situ efferocytosis
Aortic root sections were co-stained with cleaved caspase 3 (1:200, #9661, Cell Signaling Technology) and Mac-2 (3 μg·ml−1, ACL8942F, Accurate Chemical & Scientific Corporation) to label apoptotic bodies and lesional macrophages. Apoptotic cells were then determined to be either macrophage-associated (colocalizing or juxtaposed with macrophages) or free (not associated with macrophages). Data were plotted as a ratio of associated to free cells to represent efferocytosis efficiency. Images were captured using a Zeiss fluorescence microscope and analysed using the Image-Pro Plus version 5.0 software (Media Cybernetics, Inc., MD) by an observer blinded to the group assignment of each sample.
2.19 |. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology. (Curtis et al., 2018). All studies were designed, where possible, to generate groups of equal size using randomization and blinded analysis. The minimum number of animals and sample sizes required to achieve statistical significance were determined by power analysis and prior experience, assuming 80% power at a significance level of 0.05. The data were analysed with GraphPad Prism Software version 9.1.10. For continuous variables, data normality was assessed by Shapiro–Wilk test and homogeneity of variance was assessed by Levene’s test. Two group comparisons were performed by two-sided unpaired Student’s t test, and multiple comparisons were made by one-way analysis of variance (ANOVA) with Tukey’s post hoc test or two-way ANOVA with Bonferroni’s post hoc test. Post hoc tests were only conducted when F in ANOVA achieved P < 0.05 and there was no significant inhomogeneity of variance. The number of experiments performed is provided in the figure legends (biological replicates). Statistical analysis was only undertaken for studies where each group size n was at least 5. Data are expressed as the mean ± standard error of the mean, and the null hypothesis was rejected at P ≤ 0.05.
2.20 |. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, and are permanently archived in the Concise Guide to PHARMACOLOGY 2021/22 (Alexander et al., 2021).
3 |. RESULTS
3.1 |. Anti-atherosclerotic effect by heterozygous disruption of Pfkfb3 and underlying monocyte-related mechanisms
Because homozygous disruption of Pfkfb3 is embryonically lethal (Chesney et al., 2005), heterozygous Pfkfb3 knockout mice on the Apoe−/− background were used for these experiments. Haplodeficiency of Pfkfb3 globally did not affect body weight or plasma cholesterol levels before (185.6 mg·dl−1 vs. 192.8 mg·dl−1 for Apoe−/−/Pfkfb3+/+) or after (991.3 mg·dl−1 vs. 962.5 mg·dl−1 for Apoe−/−/Pfkfb3+/+) introduction of a Western diet (WD) for 3 months. Oil Red O staining revealed significantly decreased lesion sizes in the whole aorta and aortic sinus of Apoe−/−/Pfkfb3+/− mice in comparison with Apoe−/− controls (Figure 1a–d). Haematoxylin–eosin (H&E) and cleaved caspase 3 staining showed no differences in necrotic areas (Figure 1e,f) and the number of apoptotic cells (Figure S1A) in aortic sinus between the two groups. Additionally, heterozygous deletion of Pfkfb3 markedly decreased the macrophage content in atherosclerotic lesions (Figure 1g,h). Haematological analysis showed no differences in circulating white blood cell counts or total monocytes in blood derived from Apoe−/−/Pfkfb3+/− mice compared with Apoe−/−/Pfkfb3+/+ controls (Table S3). However, flow cytometric analysis showed that Apoe−/−/Pfkfb3+/− mice exhibited lower percentages and numbers of Ly-6Chi monocytes relative to total (CD11b+CD115+) monocytes in the blood (Figure 2a,b), spleen (Figure 2c,d), and bone marrow (Figure 2e,f) in comparison with Apoe−/−/Pfkfb3+/+ mice, whereas the frequency of Ly-6Clo monocytes was increased. Through a Ly6Chi monocyte-specific latex labelling procedure described by Tacke et al. (2007), we observed a significant decrease of in vivo monocyte recruitment and infiltration into the vascular wall in Apoe−/−/Pfkfb3+/− mutants compared with in Apoe−/−/Pfkfb3+/+ mice as shown by reduced amounts of latex bead-contained macrophages in lesions (Figure 2g). Collectively, these data imply that the alleviated atherosclerosis in Pfkfb3 heterozygous mice may be related, at least partially, to the impaired systemic monocytosis and reduced monocyte infiltration into the lesions.
FIGURE 1.

Global heterozygous pfkfb3 deficiency prevents atherogenesis. (a, b) Oil red O-stained en face aortic preparations from Apoe−/−/Pfkfb3+/+ and Apoe−/−/Pfkfb3+/− mice fed a Western diet (WD) for 3 months (a), and quantification of the oil red O-stained areas (b) (n = 12–13 mice per group). (c, d) Oil red O staining of aortic roots (c), and quantification of lesion areas (d) in aortic roots of the 3-month-WD-fed Apoe−/−/Pfkfb3+/+ and Apoe−/−/Pfkfb3+/− mice (n = 7 mice per group). (e, f) H&E staining of aortic roots (e), and quantification of necrotic areas (f ) in aortic roots of 3-month-WD-fed Apoe−/−/Pfkfb3+/+ and Apoe−/−/Pfkfb3+/− mice (n = 7 mice per group). Red lines show the boundaries of necrotic cores. (g, h) Immunohistochemical (IHC) staining of Mac2 (g), and quantification of Mac2+ areas (h) in aortic roots of 3-month-WD-fed Apoe−/−/Pfkfb3+/+ and Apoe−/−/Pfkfb3+/− mice (n = 6 mice per group). All images are representative. For all bar graphs, data are mean ± SEM, *P < 0.05 (unpaired, two-tailed Student’s t-test).
FIGURE 2.

Global heterozygous Pfkfb3 deficiency inhibits Ly6Chi monocytosis and monocyte infiltration into the atherosclerotic lesions. (a) Relative frequency of Ly6Chi (II) and Ly6Clo (I) monocyte subsets in the blood of 3-month-WD-fed Apoe−/−/Pfkfb3+/+ and Apoe−/−/Pfkfb3+/− mice (n = 6 mice per group). (b) Total Ly6Chi and Ly6Clo monocytes in the blood of 3-month-WD-fed Apoe−/−/Pfkfb3+/+ and Apoe−/−/Pfkfb3+/− mice (n = 6 mice per group). (c) Relative frequency of Ly6Chi (II) and Ly6Clo (I) monocyte subsets in the spleen of 3-month-WD-fed Apoe−/−/Pfkfb3+/+ and Apoe−/−/Pfkfb3+/− mice (n = 6 for Apoe−/−/Pfkfb3+/+ group and n = 5 for Apoe−/−/Pfkfb3+/− group). (d) Total Ly6Chi and Ly6Clo monocytes in the spleen of 3-month-WD-fed Apoe−/−/Pfkfb3+/+ and Apoe−/−/Pfkfb3+/− mice (n = 6 for Apoe−/−/Pfkfb3+/+ group and n = 5 for Apoe−/−/Pfkfb3+/− group). (e) Relative frequency of Ly6Chi (II) and Ly6Clo (I) monocyte subsets in the bone marrow of 3-month-WD-fed Apoe−/−/Pfkfb3+/+ and Apoe−/−/Pfkfb3+/− mice (n = 6 mice per group). (f) Total Ly6Chi and Ly6Clo monocytes in the bone marrow of 3-month-WD-fed Apoe−/−/Pfkfb3+/+ and Apoe−/−/Pfkfb3+/− mice (n = 6 mice per group). (g) in vivo monocyte infiltration into lesions of aortic root in Apoe−/−/Pfkfb3+/+ and Apoe−/−/Pfkfb3+/− mice after tail vein injection of Ly6Chi monocyte-specific latex beads, with representative images at left and quantitative data at right (n = 8 mice per group). Arrows indicate FITC+ latex bead-containing F4/80+ macrophages in lesions. All images are representative. For all bar graphs, data are mean ± SEM, *P < 0.05 (unpaired, two-tailed Student’s t-test).
3.2 |. Haplodeficiency of Pfkfb3 in myeloid cells confers athero-protective role in Apoe−/− mice
We then determined whether mere PFKFB3 deficiency in myeloid cells is enough to protect against atherogenesis. For this purpose, Pfkfb3f/+/LysMcre/+ (hereinafter referred to as Mye-Pfkfb3+/−) mice with haplodeficiency of Pfkfb3 in myeloid cells and their wild-type control Pfkfb3+/+/LysMcre/+ (hereinafter referred to as Mye-Pfkfb3+/+) mice were constructed and bred with Apoe−/− mice. Measuring the mRNA and protein levels of PFKFB3 in isolated peritoneal macrophages confirmed the partial deletion of myeloid cell PFKFB3 in Apoe−/−/Mye-Pfkfb3+/− mice (Figure S2A and S2B). Accordingly, 18F-FDG uptake assay demonstrated partially reduced glycolysis in arteriosclerotic vessel walls in Apoe−/−/Mye-Pfkfb3+/− mice compared with that in Apoe−/−/Mye-Pfkfb3+/+ mice (Figure S2C). Myeloid haplodeficiency of Pfkfb3 did not affect body weight or plasma cholesterol levels before (202.3 mg·dl−1 vs. 215.8 mg·dl−1 for Apoe−/−/Mye-Pfkfb3+/+) or after (956.3 mg·dl−1 vs. 988.9 mg·dl−1 for Apoe−/−/Mye-Pfkfb3+/+) introduction of a WD for 3 months. The circulating white blood cell counts or total monocytes in blood were comparable between the two groups with 3 months of WD feeding (Table S4). Nevertheless, the Apoe−/−/Mye-Pfkfb3+/− mice exhibited a significant reduction in atherosclerotic lesion sizes (Figure 3a–d). No differences in necrotic areas (Figure 3e,f) and the number of apoptotic cells (Figure S1B) were observed in aortic sinus between the two groups. Quantification of MAC2-positive areas at the level of the aortic sinus revealed a significant decrease in macrophage accumulation in Apoe−/−/Mye-Pfkfb3+/− mice compared with that in Apoe−/−/Mye-Pfkfb3+/+ controls (Figure 3g,h). Decreased percentages of Ly-6Chi and increased percentages of Ly-6Clo monocytes were observed in the blood, spleen, and bone marrow of Apoe−/−/Mye-Pfkfb3+/− mice compared with controls (Figure 3i). In addition, haplodeficiency of Pfkfb3 resulted in a significant reduction of the Ly6Chi monocyte infiltration into the lesions in Apoe−/−/Mye-Pfkfb3+/− compared to Apoe−/−/Mye-Pfkfb3+/+ mice (Figure 3j). These data demonstrate that haplodeficiency of Pfkfb3 in myeloid cells phenocopies the global heterozygous deletion of this enzyme regarding the pathogenesis of atherosclerosis.
FIGURE 3.

Haplodeficiency of Pfkfb3 in myeloid cells inhibits atherosclerosis. (a, b) Oil red O-stained en face aortic preparations from 3-month-WD-fed Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3+/− mice (a), and quantification of the oil red O-stained areas (b) (n = 15 mice per group). (c, d) Oil red O staining of aortic roots (c), and quantification of lesion areas (d) in aortic roots of 3-month-WD-fed Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3+/− mice (n = 7 mice per group). (e, f) H&E staining of aortic roots (e), and quantification of necrotic areas (f) in aortic roots of 3-month-WD-fed Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3+/− mice (n = 7 mice per group). Red lines show the boundaries of necrotic cores. (g, h) IHC staining of Mac2 (g), and quantification of Mac2+ areas (h) in aortic roots of 3-month-WD-fed Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3+/− mice (n = 7 mice per group). (i) Relative frequency of Ly6Chi and Ly6Clo monocyte subsets in the blood, spleen, and bone marrow of 3-month-WD-fed Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3+/− mice (n = 6 mice per group). (j) In vivo monocyte infiltration into lesions of aortic roots in Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3+/− mice after tail vein injection of Ly6Chi monocyte-specific latex beads, with representative images at left and quantitative data at right (n = 8 mice per group). All images are representative. For all bar graphs, data are mean ± SEM, *P < 0.05 (unpaired, two-tailed Student’s t-test).
3.3 |. PFKFB3 is predominantly expressed in Ly6Chi monocytes and regulates monocytosis
To determine how Pfkfb3 deficiency reduces the expansion of Ly6Chi monocytes, we analysed the PFKFB3 expression level and found that the mRNA level of Pfkfb3 was about two times higher in FACS-sorted Ly6Chi than in Ly6Clo monocytes (Figure 4a). The extracellular acidification rate (ECAR) was then assessed to test the glucose metabolic differences between monocyte subsets. Analysis of the mean data indicated that the basal glycolysis, maximal glycolytic capacity, and glycolytic reserve were significantly higher in Ly6Chi than those in Ly6Clo monocytes (Figure 4b,c), suggesting that Ly6Chi monocytes are more glycolytic. This was due, at least partially, to the high expression level of PFKFB3, because haplo-insufficiency of Pfkfb3 modestly decreased the basal glycolysis, the maximal glycolytic capacity, and the glycolytic reserve in Ly6Chi monocytes (Figure 4b,c) as well as glucose uptake in peritoneal macrophages (Figure S3A). In addition, we observed that the expression level of PFKFB3 in Ly6Chi monocytes was up-regulated by high cholesterol diet feeding, which was accompanied with higher levels of basal glycolysis, maximal glycolytic capacity, and glycolytic reserve (Figure 4d,e).
FIGURE 4.
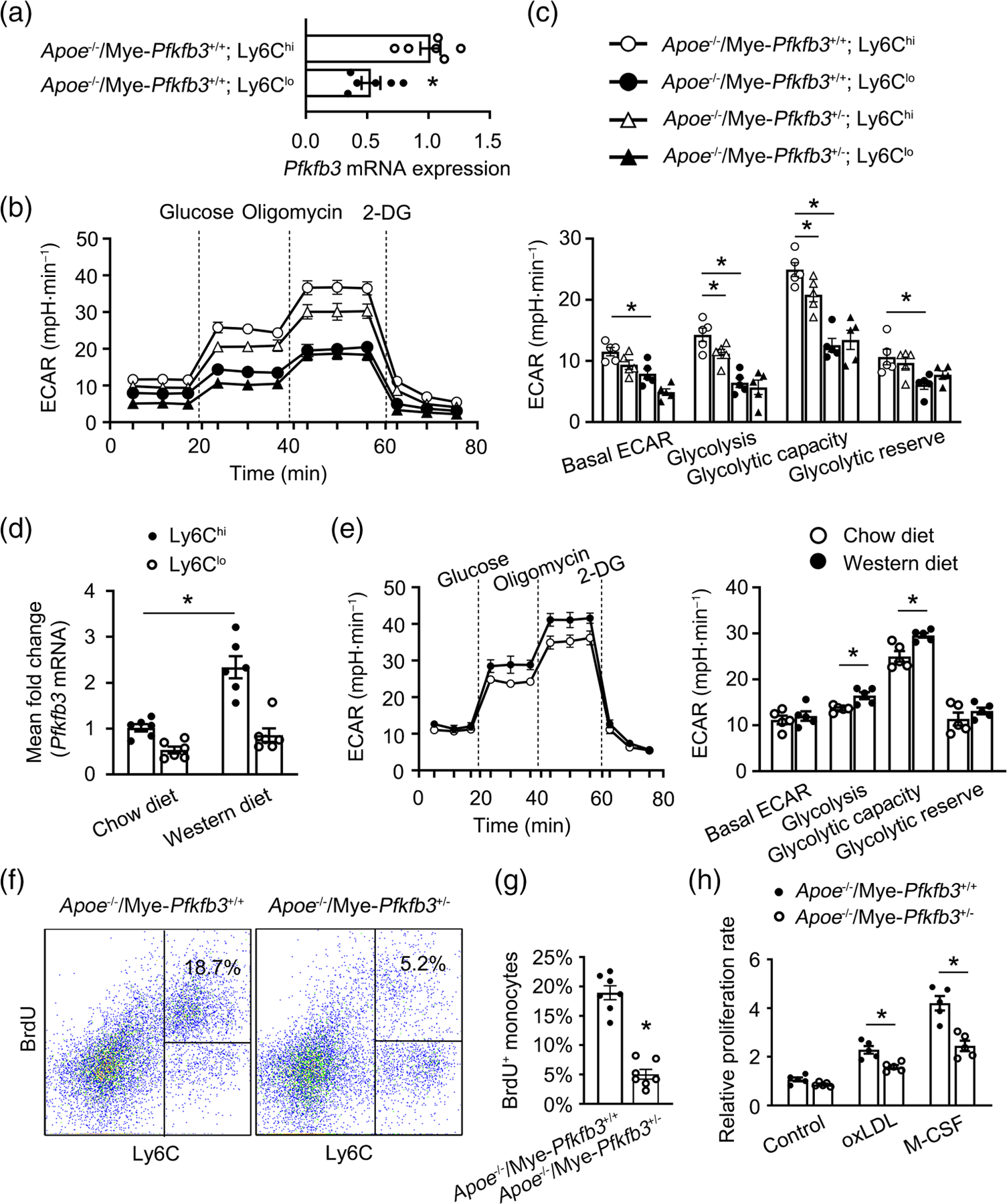
PFKFB3 regulates hypercholesterolaemia-associated monocytosis of Ly6Chi monocytes. (a) Real-time PCR analysis of Pfkfb3 mRNA expression in Ly6Chi and Ly6Clo monocytes sorted by flow cytometry from the spleen of Apoe−/− mice (n = 6 mice per group). (b) Extracellular acidification rate (ECAR) profile showing glycolytic function in Ly6Chi and Ly6Clo monocytes sorted by flow cytometry from the spleen of Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3+/− mice (n = 5). Vertical lines indicate the time of addition of glucose (10 mmol·L−1), oligomycin (2 μmol L−1), and 2-DG (50 mmol L−1). (c) Quantification of glycolytic function parameters from b (n = 5). (d) Real-time PCR analysis of Pfkfb3 mRNA expression in Ly6Chi and Ly6Clo monocytes sorted by flow cytometry from the spleen of Apoe−/− mice fed chow diet or WD for 3 months (n = 6 mice per group). (e) ECAR profile showing glycolytic function and quantification of glycolytic function parameters in Ly6Chi monocytes sorted by flow cytometry from the spleen of 3-month-chow diet or WD-fed Apoe−/− mice (n = 5). (f) Flow cytometry analysis of proliferating CD11b+CD115+ monocytes in the blood from 8-week-WD-fed Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3+/− mice. Cells were gated as Ly6Chi 5-bromo-2′-deoxyuridine (BrdU)+ (n = 7 mice per group). (f) Quantification of BrdU+ cells among total CD11b+CD115+ monocytes (n = 7 mice per group). (h) The relative proliferative activity of Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3+/− macrophages incubated with M-CSF or oxLDL (n = 5). All images are representative. For all bar graphs, data are mean ± SEM, *P < 0.05 (unpaired, two-tailed Student’s t-test for a, c, and e–h; one-way ANOVA with Tukey’s post hoc test for d).
Aerobic glycolysis has been shown to support cell proliferation, pushing us to test whether PFKFB3 affects monocyte/macrophage proliferation, and thus contributes to the expansion of Ly6Chi monocytes. Mice were injected with 5-bromo-2′-deoxyuridine (BrdU) before death at 8 weeks upon WD feeding. Apoe−/−/Mye-Pfkfb3+/− mice exhibited a decreased percentage of BrdU+ monocytes compared with Apoe−/−/Mye-Pfkfb3−/− mice (Figure 4f,g). Moreover, the decrease in BrdU incorporation occurred mainly in the Ly6Chi subset (Figure 4f). These data suggest that Ly6Chi monocyte proliferation regulated by PFKFB3 contributes to monocytosis in WD-fed Apoe−/− mice. A BrdU-based chemiluminescence assay was then applied to further prove the potential role of PFKFB3 in monocyte/macrophage proliferation. Oxidized low density lipoprotein (oxLDL) or macrophage colony-stimulating factor (M-CSF) both significantly induced the proliferation of macrophages from Apoe−/−/Mye-Pfkfb3+/− and Apoe−/−/Mye-Pfkfb3+/+ mice, respectively (Figure 4h); however, macrophages from Apoe−/−/Mye-Pfkfb3+/− mice showed significantly less BrdU incorporation than those from control mice (Figure 4h).
3.4 |. PFKFB3 expression associates with inflammatory monocyte expansion in patients with atherosclerotic coronary artery disease
While our data suggest that PFKFB3 expression level influences monocytosis in an animal model of atherosclerosis, whether a similar phenomenon is operational in human monocytes is not yet known. Therefore, we quantified PFKFB3 expression in sorted classical CD14+CD16−, intermediate CD14+CD16+, and nonclassical CD14−CD16+ monocytes from patients suffering from coronary artery disease (CAD) or age-matched individuals. We observed that PFKFB3 was highly expressed in CD14+CD16− and CD14+CD16+ monocyte subsets (Figure 5a). Furthermore, the PFKFB3 level was much higher in CD14+CD16+ monocytes from CAD patients than that in the same cell subset from healthy individuals (Figure 5a). The distribution of monocyte subsets was then analysed. The ratio of the relatively small population of CD14−CD16+ monocytes was similar in both groups (Figure 5b,c). While the frequencies of classical CD14+CD16− monocytes was reduced, the intermediate CD14+CD16+ monocyte subset was expanded in CAD patients (Figure 5b,c). Intermediate CD14+CD16+ monocytes have been proposed to possess inflammatory properties reminiscent of the murine Ly6Chi monocytes (also termed “inflammatory” monocytes) and independently predict cardiovascular events. Collectively, these data suggest that inflammatory monocyte expansion is associated with high PFKFB3 expression level in patients with CAD.
FIGURE 5.
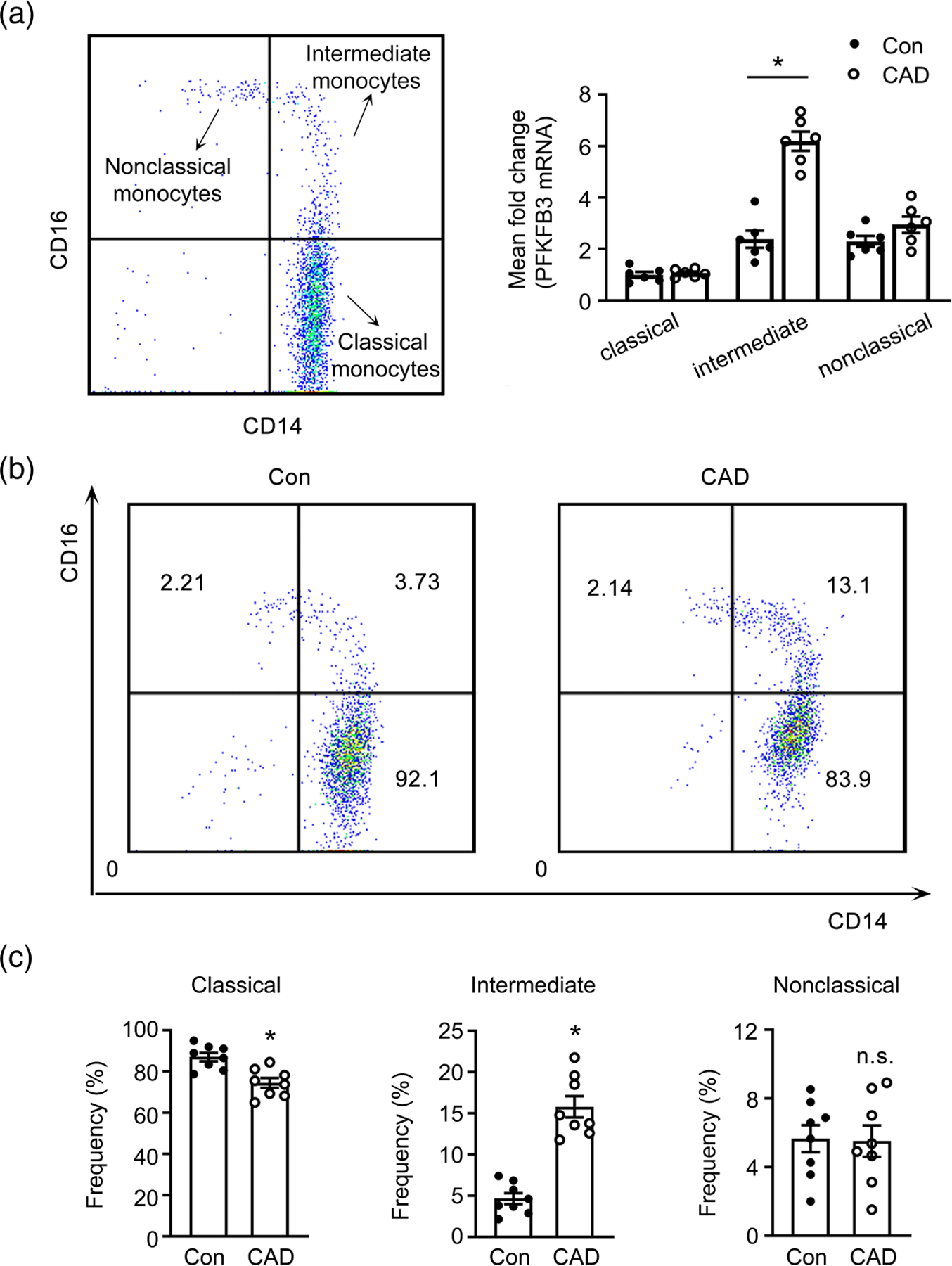
PFKFB3 expression associates with inflammatory monocyte expansion in patients with coronary artery disease. (a) Real-time PCR analysis of Pfkfb3 mRNA expression in classical (CD14+CD16−), intermediate (CD14+CD16+), and nonclassical (CD14−CD16+) monocytes sorted by flow cytometry from blood of patients with coronary artery disease (CAD) and age-matched healthy individuals (Con) (n = 6). (b, c) representative dot plots (b) and frequencies of the three monocyte subpopulations (c) are shown (n = 8). For all bar graphs, data are mean ± SEM, *P < 0.05 (unpaired, two-tailed Student’s t-test).
3.5 |. Loss of PFKFB3 significantly impairs myeloid cell aggregation, invasion, and motility
Ly6Chi monocytes are known to be hypermigratory and could selectively enter atherosclerotic lesions. Since PFKFB3 could affect migration of other cell types, we determined how the loss of PFKFB3 in monocytes/macrophages affect the processes of aggregation, invasion, and motility, which are essential for the tissue infiltration of monocytes and macrophages. We found that peritoneal macrophages from Apoe−/−/Mye-Pfkfb3+/+ mice reply to plating on growth factor-reduced matrigel by rapid and obvious homotypic adhesion, a process widely used as an in vitro assay to mirror leukocyte-endothelial cell interactions; nevertheless, PFKFB3-deficient macrophages showed impaired homotypic adhesion (Figure 6a). In addition, the capacity of PFKFB3-deficient macrophages to invade matrigels toward a chemotactic agent, MCP-1, is severely damaged (Figure 6a,b). Deficiency of PFKFB3 also inhibited directed migration in the absence of matrigel by 50% (Figure 6a,c). Furthermore, the inhibition of PFKFB3 by its inhibitor 3PO at multiple concentrations prevented MCP-1-induced THP-1 cell chemotaxis (Figure 6d). Even in vivo, 40% fewer PFKFB3-deficient macrophages migrated into the peritoneal cavity following thioglycolate treatment compared with WT macrophages (Figure 6e). In a wound healing assay, PFKFB3-haplodeficient macrophages exhibited significantly ablated wound migration (Figure 6f). In addition, PFKFB3-haplodeficient macrophages presented a changed morphology in vitro characterized by lack of elongation (Figure 6g) and morphological polarization (Figure 6h), while the cells also exhibited defects in the formation of filopodia and lamellipodia (Figure 6i–k). To seek the underlying mechanisms, we analysed and found no differences for the expression of Psgl-1, L-selectin, CD11b, Ccr2, and Cxcr2 in macrophages isolated from Apoe−/−/Mye-Pfkfb3+/− mice and Apoe−/−/Mye-Pfkfb3+/+ controls (Figure S3B), suggesting that mechanisms other than altered chemokine or chemokine receptor signalling mediate the proinvasive/promotile capabilities of PFKFB3. A previous study has shown that PFKFB3 colocalizes with F-actin in lamellipodia to create an assembly line of glycolysis, facilitating efficient and rapid local ATP production to fuel migration for endothelial cells (De Bock et al., 2013). Therefore, we examined the intracellular localization of PFKFB3 in macrophages. Immunocytochemical analysis revealed that PFKFB3 co-localizes with F-actin in filopodia and lamellipodia (Figure 6l). These results imply the critical role of glycolytic metabolism in monocyte/macrophage migration through fuelling filopodia and lamellipodia formation.
FIGURE 6.

PFKFB3 regulates monocyte/macrophage aggregation, invasion, and motility. (a) Representative images of aggregation (upper), invasion (middle), and migration (lower) of Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3+/− macrophages. For aggregation (upper), peritoneal macrophages plated at a concentration of 106 ml−1 on growth factor-reduced matrigel were allowed to aggregate for 24 h. For invasion (middle), peritoneal macrophages were seeded to the upper wells of the modified Boyden chambers with cell culture inserts coated with matrigel, and were allowed to invade toward 5% FCS in the lower wells for 24 h. For migration (lower), peritoneal macrophages were seeded to the upper wells of the modified Boyden chambers without added matrigel and were allowed to migrate for 24 h. (b) Quantification of the invaded macrophages in a (n = 5). (c) Quantification of the migrated macrophages in part a (n = 5). (d) Quantification of chemotaxis of THP-1 monocytes induced by MCP-1 (n = 5). THP-1 cells were preincubated with 3PO at concentrations of 2.5, 5, or 10 μM for 30 min, and then were treated with 50 ng·ml−1 MCP-1. (e) Quantification of the number of Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3+/− macrophages migrated to the thioglycolate-elicited peritoneal cavity (n = 5 mice per group). (f) Scratch wound healing analysis of migration of Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3+/− macrophages 24 h after wound scratch (n = 5). (g, h) Morphology of Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3+/− peritoneal macrophages cultured for 36 h (n = 5, 60 cells per replicate). (G) Mean elongation index (EI) defined as the ratio of cell length to cell breadth. (h) Percentage of rounded (EI < 1.2), partially elongated (1.2 ≤ EI ≤ 2), and elongated (EI > 2) macrophages. (i) Percentage of Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3+/− peritoneal macrophages cultured for 16 h, forming filopodia (n = 5, 60 cells per replicate). (j) Percentage of Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3+/− peritoneal macrophages cultured for 16 h, forming lamellipodia (n = 5, 60 cells per replicate). (k) Representative images showing morphology of Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3+/− peritoneal macrophages. (l) Representative immunofluorescent staining of PFKFB3 (red) and Phalloidin (green) in peritoneal macrophages under oxLDL treatment is shown. F-actin and PFKFB3 were co-localized in lamellipodia and filopodia. For all bar graphs, data are mean ± SEM, *P < 0.05 (unpaired, two-tailed Student’s t-test for b, c, and e–j; one-way ANOVA with Tukey’s post hoc test for d).
3.6 |. Homozygous deletion of Pfkfb3 in myeloid cell exacerbates atherosclerosis in Apoe−/− mice
To further confirm the role of myeloid cell PFKFB3 in atherogenesis, we aimed to test how homozygous deletion of Pfkfb3 affects atherosclerosis. To this aim, Pfkfb3f/f/LysMcre/+ (hereinafter referred to as Mye-Pfkfb3−/−) mice with homozygous deletion of Pfkfb3 in myeloid cells were constructed and bred with Apoe−/− mice to generate Apoe−/−/Mye-Pfkfb3−/− mice. The mRNA and protein levels of PFKFB3 were significantly decreased in peritoneal macrophages from Apoe−/−/Mye-Pfkfb3−/− mice compared with those from Apoe−/−/Mye-Pfkfb3+/+ controls (Figure S2A and S2B). Meanwhile, homozygous deletion of myeloid Pfkfb3 induced a substantial attenuation of glycolysis in the arterial wall (Figure S2C) and in the peritoneal macrophages (Figure S2D). Hematological analysis showed a nonsignificant trend toward reduced circulating monocytes in Apoe−/−/Mye-Pfkfb3−/− mice compared with those in Apoe−/−/Mye-Pfkfb3+/+ mice (Table S4). However, flow cytometric analysis demonstrated significantly decreased percentages of Ly6Chi and increased percentages of Ly6Clo subsets in the blood, spleen, and bone marrow of Apoe−/−/Mye-Pfkfb3−/− mice in comparison with Apoe−/−/Mye-Pfkfb3+/+ mice on being fed with WD (Figure S4A). Additionally, homozygous deletion of pfkfb3 resulted in a significant decrease of in vivo Ly6Chi monocyte recruitment and infiltration into the vascular wall (Figure S4B) as well as a dramatical decrease in macrophage aggregation, invasion, and motility ex vivo (Figure S4C–S4E). These findings in Apoe−/−/Mye-Pfkfb3−/− mice were consistent with those observed in Apoe−/−/Mye-Pfkfb3+/− mice, further confirming a major cell-autonomous role of PFKFB3 in controlling monocyte expansion and its entry into lesions. Thus, we hypothesized a prominent athero-protective role of homozygous deletion of Pfkfb3 in myeloid cells. Unexpectedly, despite a lower baseline weight, Apoe−/−/Mye-Pfkfb3−/− mice gained more weight after 3 months WD feeding (Figure S4F), and the blood cholesterol content was slightly higher in Apoe−/−/Mye-Pfkfb3−/− mice than in Apoe−/−/Mye-Pfkfb3+/+ controls (Figure S4G). Oil-red O staining exhibited a slight increase in atherosclerotic lesion size in Apoe−/−/Mye-Pfkfb3−/− mice (Figure 7a–d). However, the Mac2+ area was significantly decreased in vascular lesions in Apoe−/−/Mye-Pfkfb3−/− mice compared with Apoe−/−/Mye-Pfkfb3+/+ controls (Figure 7e,f), which might be due to impaired Ly6Chi monocyte infiltration into lesions (Figure S4B). In addition, the atherosclerotic lesions in aortas from Apoe−/−/Mye-Pfkfb3−/− mice contained an increased necrotic core area (Figure 7g,h) and elevated number of cells showing activation of caspase 3 (Figure 7i,j) compared with lesions from Apoe−/−/Mye-Pfkfb3+/+ mice, suggesting that increased lesion instability could contribute to the development of more severe atherosclerosis in Apoe−/−/Mye-Pfkfb3−/− mice.
FIGURE 7.

Homozygous deletion of Pfkfb3 in myeloid cell exacerbates atherosclerosis. (a, b) Oil red O-stained en face aortic preparations from 3-month-WD-fed Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3−/− mice (a), and quantification of the oil red O-stained areas (b) (n = 12 mice per group). (c, d) Oil red O staining of aortic roots (c), and quantification of lesion areas (d) in aortic roots of 3-month-WD-fed Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3−/− mice (n = 7 mice per group). (e, f) IHC staining of Mac2 (e), and quantification of Mac2+ areas (f) in aortic roots of 3-month-WD-fed Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3−/− mice (n = 6 mice per group). (g, h) H&E staining of aortic roots (g), and quantification of necrotic areas (h) in aortic roots of 3-month-WD-fed Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3−/− mice (n = 7 mice per group). Red lines show the boundaries of necrotic cores. (i, j) cleaved caspase 3 staining (i) and quantification of apoptotic bodies (j) in aortic roots of 3-month-WD-fed Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3−/− mice (n = 7 mice per group). (k, l) representative flow cytometry phagocytosis plots (l), and quantificative analysis of efferocytosis (k). Peritoneal macrophages from Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3−/− mice were incubated for 45 min with apoptotic Jurkat T cells, and then were assessed for efferocytosis by flow cytometry (n = 6). (m) Representative immunofluorescent staining of cleaved caspase 3 (red) and Mac2 (green) in aortic roots of 3-month-WD-fed Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3−/− mice (n = 6 mice per group). (n) the number of “free” apoptotic bodies not associated with Mac2+ phagocytic macrophages in aortic roots of 3-month-WD-fed Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3−/− mice (n = 6 mice per group). (o) The number of caspase 3 and Mac2 double-positive macrophages in aortic roots of 3-month-WD-fed Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3−/− mice (n = 6 mice per group). (p) F-actin formation during efferocytosis in Apoe−/−/Mye-Pfkfb3+/+ and Apoe−/−/Mye-Pfkfb3−/− macrophages. Peritoneal macrophages were co-incubated with CFSE-labelled apoptotic Jurkat T cells for 30 min and stained with phalloidin. Actin polymerization was measured by flow cytometry (n = 5). MFI, mean fluorescence intensity; rel., relative; MΦ, macrophage. For all bar graphs, data are mean ± SEM, *P < 0.05 (unpaired, two-tailed Student’s t-test for a–o; one-way ANOVA with Tukey’s post hoc test for p).
3.7 |. Homozygous deletion of Pfkfb3 in myeloid cells impairs efferocytosis in macrophages
One major cause of necrotic core formation in advanced atherosclerosis is impaired phagocytic clearance of apoptotic cells (efferocytosis) within lesions (Schrijvers et al., 2005). Therefore, we investigated whether homozygous deletion of Pfkfb3 had affected macrophage phagocytic ability. With an established in vitro phagocytosis assay, we found that peritoneal macrophages isolated from Apoe−/−/Mye-Pfkfb3−/− mice cleared less apoptotic Jurkat T cells than those from Apoe−/−/Mye-Pfkfb3+/+ mice (Figure 7k,l), indicating that homozygous deletion of Pfkfb3 compromised macrophage phagocytic ability in vitro. In addition, cleaved caspase 3 and Mac2 co-staining of the aortic sinus demonstrated that the number of “free” apoptotic cells that were not associated with Mac2+ macrophages (indicative of defective efferocytosis) was higher in atherosclerotic lesions from Apoe−/−/Mye-Pfkfb3−/− mice (Figure 7m,n), further confirming the pro-efferocytotic effect of PFKFB3. However, we observed that the number of caspase 3+ macrophages also increased in Apoe−/−/Mye-Pfkfb3−/− mice (Figure 7m,o), implying increased sensitivity of PFKFB3-deficient macrophages to apoptosis. Indeed, oxLDL stimulation induced more apoptosis in peritoneal macrophages from Apoe−/−/Mye-Pfkfb3−/− mice compared with Apoe−/−/Mye-Pfkfb3+/+ cells (Figure S4H).
Aerobic glycolysis has been demonstrated to fuel active actin filament assembly, which is essential for efferocytosis (Morioka et al., 2018). We then tested whether PFKFB3 supports actin polymerization in macrophages during efferocytosis. Increased actin polymerization was observed in apoptotic Jurkat T cell-engulfing macrophages, as demonstrated by phalloidin staining (Figure 7p). However, actin polymerization in efferocytic macrophages from Apoe−/−/Mye-Pfkfb3−/− mice was compromised compared with that in Apoe−/−/Mye-Pfkfb3+/+ efferocytic macrophages (Figure 7p), indicating that PFKFB3-driven glycolysis provides a metabolic basis for optimal efferocytosis.
4 |. DISCUSSION
Atherosclerotic plaque formation and progression are a result of a chronic non-resolving inflammatory response involving a constant expansion and infiltration of monocytes and impaired clearance of dying macrophages within the plaque (Libby, 2002). Therefore, the mechanisms regulating the expansion and recruitment of monocytes as well as the death and elimination of lipid-laden macrophages in lesions are crucial for atherosclerosis. Our results presented here showed that myeloid PFKFB3 exerts a double-edged sword effect on atherosclerosis. It not only promotes monocytosis and Ly6Chi monocyte infiltration into lesions to accelerate atherosclerosis, but also maintains a physiological role for macrophages to clear apoptotic cellular debris to prevent the progress of atherosclerosis. The impairment of the former dominates in Apoe−/−/Mye-Pfkfb3+/− mice and sensitizes these mice to WD-induced atherosclerosis. Nevertheless, the latter is compromised in Apoe−/−/Mye-Pfkfb3−/− mice, thus advancing atherosclerosis and inducing plaque instability. This study indicates a gene dosage effect of PFKFB3 on atherosclerosis.
Glycolysis in myeloid cells plays an important role in the development of atherosclerosis. In the present study, partial inhibition of PFKFB3 via global heterozygous gene deletion dramatically suppressed the development of atherosclerosis. As a hallmark of PFKFB3 deficiency, atherosclerotic lesions displayed significantly reduced amounts of macrophages, which might be due to the attenuated monocyte infiltration. In addition, a marked decrease of Ly6Chi monocytes, a subset known for its hypermigratory profile, was observed in Pfkfb3 haplodeficient mice. These changes in monocytes are likely caused by the cell-autonomous roles of PFKFB3, as myeloid cell haplodeficiency of PFKFB3 also suppressed the expansion of Ly6Chi monocytes and their infiltration into atherosclerotic lesions. Despite this strength, our study has limitations. PFKFB3 is expressed by other vascular and immune cell types, including endothelial cells, vascular smooth muscle cells, neutrophils, and T cells (Telang et al., 2012; Xiao et al., 2021), which are present in atherosclerotic lesions, and all of which are recognized to contribute to disease progression. Thus, the attenuated atherosclerosis in global heterozygous Pfkfb3-deficient mice might also be due to the decreased glycolysis in these cell types.
The present study advances our understanding of the mechanisms underlying monocytosis and Ly6Chi monocyte recruitment ability. The relationship between monocytosis and atherosclerosis has been well documented in atherosclerosis (Swirski et al., 2007); however, detailed mechanisms responsible for monocytosis are lacking. Our study showed that PFKFB3 is highly expressed in Ly6Chi monocytes and controls Ly6Chi monocyte proliferation during hypercholesterolaemia. These findings suggest that dominating myeloid cell proliferation during hypercholesterolaemia is a pivotal factor determining the magnitude of monocytosis and the atherogenic response. Ly6Chi monocytes are known to be hypermigratory and could selectively enter atherosclerotic lesions, compared with the Ly6Clo subset. We demonstrated here that a high level of glycolysis governed by PFKFB3 allows myeloid cells to meet the energy demands for dynamic rapid changes in motility, and this could explain, at least in part, the hypermigratory feature of Ly6Chi monocytes.
Macrophages might need glycolysis to sustain viability and remain energetic for exerting their efferocytotic effect. We observed in this study that homozygous deletion of myeloid Pfkfb3 impaired plaque stability in Apoe−/− mice. This is likely due to the following reasons. On the one hand, the decreased cellular energetics upon Pfkfb3 homozygous deficiency might sensitize macrophages to cell death. Previous studies have demonstrated the pro-survival role of PFKFB3-driven glycolysis in macrophages (Rodríguez-Prados et al., 2010; Tawakol et al., 2015). In the current study, although heterozygous deletion of Pfkfb3 did not sensitize macrophages to oxLDL-induced apoptosis (Figure S5A), homozygous deletion did do so (Figure 7p). Thus, increased apoptosis of Pfkfb3-deficient macrophages is expected to facilitate the formation of necrotic cores. On the other hand, the impaired efferocytosis could be a vital contributor to the unstable plaque in Apoe−/−/Mye-Pfkfb3−/− mice. The pro-efferocytotic effect of PFKFB3-driven glycolysis is supported by a previous study showing that PFKFB3 promotes the efferocytosis-dependent antiviral capacity of macrophages by metabolically sustaining the uptake and elimination of virus-infected cells (Jiang et al., 2016). The efferocytosis process involves active actin filament assembly, whereas PFKFB3-driven glycolysis is involved to supply the required ATP for this process, similar to the model observed in this study for macrophage migration and to an earlier model for endothelial tip cell migration (De Bock et al., 2013). This working hypothesis is further supported by a finding demonstrating that glycolysis within phagocytes contributes to actin polymerization and the continued uptake of garbage (Morioka et al., 2018). However, haplodeficiency of PFKFB3 induced a nonsignificant trend toward decreased actin polymerization in macrophages during efferocytosis (Figure S5B) and did not increase necrotic cores within plaques (Figure 3e,f). These suggest a gene-dose-dependent effect of PFKFB3 on macrophage efferocytosis and plaque stability.
Genetic deletion of Pfkfb3 results in an outcome inconsistent with effects observed with PFKFB3 inhibitors. A previous study showed that either a preventive regimen (50 mg·kg−1, 2×/week, 10 weeks, starting after 4 weeks of WD, ip) or a curative regimen (50 mg·kg−1, 4×/week, 4 weeks, starting after 16 weeks of WD, ip) of treatment with 3PO, a PFKFB3 inhibitor, only decreased plaque formation, but had no effect on plaque stability (Perrotta et al., 2020). However, a curative regimen (2 mg·kg−1, 3×/week, 5 weeks, starting after 16 weeks of WD, ip) of treatment with PFK158, a 3PO analogue, was reported to improve plaque stability. In our study, we did observe a decreased lesion size but not the improved plaque stability in Apoe−/− mice with global heterozygous deletion of Pfkfb3. Several reasons may explain these inconsistent results. First, a time-dependent effect of PFKFB3 inhibition might exist in atherosclerosis. Second, a dose-dependent effect of PFKFB3 inhibition might exist. Third, the PFKFB3 inhibitors may have “off-target” effects, that have already been demonstrated (Emini Veseli et al., 2021; Wik et al., 2020). Further investigations are needed to verify these possibilities.
Very recently, Tillie et al., exploiting Ldlr−/− mice with myeloid cell homozygous deletion of Pfkfb3 (Ldlr−/−/Mye-Pfkfb3−/−), claimed that partial inhibition of Pfkfb3 in myeloid cells has no effect on atherosclerotic lesion size and plaque stability (Tillie et al., 2021), which seemingly conflicts with our findings that partial inhibition of PFKFB3 in Apoe−/−/Mye-Pfkfb3+/− myeloid cells decreases lesion size. The inherent differences between Ldlr−/− and Apoe−/− models may account for the inconsistences. Monocytosis is shown to be much more prominent in WD-fed Apoe−/− mice than Ldlr−/− mice (Murphy et al., 2011). Therefore, the dramatically decreased monocytosis and monocyte accumulation in lesions by myeloid Pfkfb3 haplodeficiency in Apoe−/− mice may be a major contributor to the attenuated atherosclerosis in this mouse model. In addition, considering the gene-dose-dependent effect of PFKFB3 on atherosclerosis, the discrepant knockdown efficiency of Pfkfb3 gene could explain the different phenotypes observed in the two studies. Tillie et al. showed that myeloid Pfkfb3 knockdown efficiency in Ldlr−/−/Mye-Pfkfb3−/− mice is about 50% at the RNA level; yet 40% and 75% knockdown of PFKFB3 at the protein level was seen in Apoe−/−/Mye-Pfkfb3+/− and Apoe−/−/Mye-Pfkfb3−/− macrophages, respectively, in our study. The different efficiencies for Cre-mediated recombination may depend on the distinct inflammatory status in Ldlr−/− and Apoe−/− macrophages that might affect activity of the Lyz2 promoter, since Apoe has been shown to exhibit anti-inflammatory properties (Getz & Reardon, 2016). However, this discrepancy warrants further clarification.
In summary, we have unveiled here, using multiple genetic animal models, the double-edged sword effect of PFKFB3 on the pathogenesis of atherosclerosis: aiding monocytosis and monocyte recruitment to lesions to accelerate atherogenesis and potentiating macrophage efferocytosis to hamper the progress of atherosclerosis. The net effect of PFKFB3 inhibition or deletion on atherosclerosis depends on the balance of these two effects. Caution should be warranted with PFKFB3 inhibitor-based therapeutics, highlighting the importance of monitoring cardiovascular function in developing therapies that target PFKFB3, an inhibitor of which, PFK158, has already been included in a phase I clinical trial (clinicaltrials.gov # NCT02044861) for the treatment of cancer.
Supplementary Material
What is already known
Macrophage-rich atherosclerotic arteries are highly active in glycolysis.
Small molecule inhibitors of PFKFB3 have demonstrated efficacy in hampering atherogenesis in preclinical models.
What does this study add
Heterozygous Pfkfb3 deletion in myeloid cells reduces the severity of atherosclerosis in Apoe−/− mice.
Homozygous loss of Pfkfb3 in myeloid cells impairs macrophage efferocytotic function in Apoe−/− mice.
What is the clinical significance
Caution should be warranted with PFKFB3 inhibitor-based therapeutics.
It is very important to monitor cardiovascular function in developing therapies that target PFKFB3.
ACKNOWLEDGMENTS
This work was supported by the open research funds from the Sixth Affiliated Hospital of Guangzhou Medical University, Qingyuan People’s Hospital, and grants from the Natural Science Foundation of China (Grant number: 81870217 and 81700395), the Key Project of Department of Education of Guangdong Province (Grant number: 2018KZDXM053), the Key Project of Guangzhou Science and Technology Bureau (Grant number: 202102010425), and American Heart Association (Grant number: 15POST22810024).
Funding information
Guangdong Medical Research Foundation, Grant/Award Number: 2018KZDXM053; Guangzhou Municipal Science and Technology Bureau, Grant/Award Number: 202102010425; National Natural Science Foundation of China, Grant/Award Numbers: 81700395, 81870217; American Heart Association, Grant/Award Number: 15POST22810024; Key Project of Guangzhou Science and Technology Bureau; Key Project of Department of Education of Guangdong Province; Natural Science Foundation of China; Sixth Affiliated Hospital of Guangzhou Medical University, Qingyuan People’s Hospital
Abbreviations:
- 3PO
3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one
- Apoe
apolipoprotein E
- BrdU
5-bromo-2′-deoxyuridine
- CAD
coronary arterial disease
- CCR2
C-C chemokine receptor type 2
- Cxcr2
C-X-C chemokine receptor type 2
- ECAR
extracellular acidification rate
- FDG
fluorodeoxyglucose
- H&E staining
haematoxylin and eosin staining
- Ly6C
lymphocyte antigen 6 complex
- LysM
lysozyme C type M
- M-CSF
macrophage-colony-stimulating factor
- oxLDL
oxidized low density lipoprotein
- PFK-1
6-phosphofructo-1-kinase-1
- PFK158
(E)-1-pyridin-4-yl-3-[7-(trifluoromethyl)quinolin-2-yl]prop-2-en-1-one
- PFKFB3
6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3
- PSGL1
P-selectin glycoprotein ligand 1
- WD
Western diet
Footnotes
CONFLICTS OF INTERESTS
The authors declare no conflict of interests.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design and Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers and other organizations engaged with supporting research.
SUPPORTING INFORMATION
Additional supporting information can be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
REFERENCES
- Alexander SP, Fabbro D, Kelly E, Mathie A, Peters JA, Veale EL, Armstrong JF, Faccenda E, Harding SD, Pawson AJ, Southan C, Davies JA, Annett S, Boison D, Burns KE, Dessauer C, Gertsch J, Helsby NA, Izzo AA, … Wong SS (2021). THE CONCISE GUIDE TO PHARMACOLOGY 2021/22: Enzymes. British Journal of Pharmacology, 178(Suppl 1), S313–S411. 10.1111/bph.15542 [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Roberts RE, Broughton BRS, Sobey CG, George CH, Stanford SC, Cirino G, Docherty JR, Giembycz MA, Hoyer D, Insel PA, Izzo AA, Ji Y, MacEwan DJ, Mangum J, Wonnacott S, & Ahluwalia A (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology. British Journal of Pharmacology, 175(3), 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali L, Schnitzler JG, & Kroon J (2018). Metabolism: The road to inflammation and atherosclerosis. Current Opinion in Lipidology, 29(6), 474–480. 10.1097/MOL.0000000000000550 [DOI] [PubMed] [Google Scholar]
- Chesney J, Telang S, Yalcin A, Clem A, Wallis N, & Bucala R (2005). Targeted disruption of inducible 6-phosphofructo-2-kinase results in embryonic lethality. Biochemical and Biophysical Research Communications, 331(1), 139–146. 10.1016/j.bbrc.2005.02.193 [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA, Hoyer D, Insel PA, Izzo AA, Ji Y, MacEwan DJ, Sobey CG, Stanford SC, Teixeira MM, Wonnacott S, & Ahluwalia A (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175(7), 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquière B, Cauwenberghs S, Eelen G, Phng LK, Betz I, Tembuyser B, Brepoels K, Welti J, Geudens I, Segura I, Cruys B, Bifari F, … Carmeliet P (2013). Role of PFKFB3-driven glycolysis in vessel sprouting. Cell, 154(3), 651–663. 10.1016/j.cell.2013.06.037 [DOI] [PubMed] [Google Scholar]
- Dolfi B, Gallerand A, Haschemi A, Guinamard RR, & Ivanov S (2021). Macrophage metabolic regulation in atherosclerotic plaque. Atherosclerosis, 334, 1–8. 10.1016/j.atherosclerosis.2021.08.010 [DOI] [PubMed] [Google Scholar]
- Emini Veseli B, Van Wielendaele P, Delibegovic M, Martinet W, & De Meyer GRY (2021). The PFKFB3 inhibitor AZ67 inhibits angiogenesis independently of glycolysis inhibition. International Journal of Molecular Sciences, 22(11), 5970. 10.3390/ijms22115970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getz GS, & Reardon CA (2016). Do the Apoe−/− and Ldlr−/− mice yield the same insight on Atherogenesis? Arteriosclerosis, Thrombosis, and Vascular Biology, 36(9), 1734–1741. 10.1161/ATVBAHA.116.306874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson GK, & Libby P (2006). The immune response in atherosclerosis: A double-edged sword. Nature Reviews. Immunology, 6(7), 508–519. 10.1038/nri1882 [DOI] [PubMed] [Google Scholar]
- Jiang H, Shi H, Sun M, Wang Y, Meng Q, Guo P, Cao Y, Chen J, Gao X, Li E, & Liu J (2016). PFKFB3-driven macrophage glycolytic metabolism is a crucial component of innate antiviral defense. Journal of Immunology, 197(7), 2880–2890. 10.4049/jimmunol.1600474 [DOI] [PubMed] [Google Scholar]
- Kelly B, & O’Neill LA (2015). Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Research, 25(7), 771–784. 10.1038/cr.2015.68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, Direnzo D, Nanda V, Ye J, Connolly AJ, Schadt EE, Quertermous T, Betancur P, Maegdefessel L, Matic LP, Hedin U, Weissman IL, & Leeper NJ (2016). CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature, 536(7614), 86–90. 10.1038/nature18935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P (2002). Inflammation in atherosclerosis. Nature, 420(6917), 868–874. 10.1038/nature01323 [DOI] [PubMed] [Google Scholar]
- Lilley E, Stanford SC, Kendall DE, Alexander SPH, Cirino G, Docherty JR, George CH, Insel PA, Izzo AA, Ji Y, Panettieri RA, Sobey CG, Stefanska B, Stephens G, Teixeira M, & Ahluwalia A (2020). ARRIVE 2.0 and the British Journal of Pharmacology: Updated guidance for 2020. British Journal of Pharmacology, 177(16), 3611–3616. 10.1111/bph.15178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morioka S, Perry JSA, Raymond MH, Medina CB, Zhu Y, Zhao L, Serbulea V, Onengut-Gumuscu S, Leitinger N, Kucenas S, Rathmell JC, Makowski L, & Ravichandran KS (2018). Efferocytosis induces a novel SLC program to promote glucose uptake and lactate release. Nature, 563(7733), 714–718. 10.1038/s41586-018-0735-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy AJ, Akhtari M, Tolani S, Pagler T, Bijl N, Kuo CL, Wang M, Sanson M, Abramowicz S, Welch C, Bochem AE, Kuivenhoven JA, Yvan-Charvet L, & Tall AR (2011). ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. The Journal of Clinical Investigation, 121(10), 4138–4149. 10.1172/JCI57559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl U, Emerson M, Garner P, Holgate ST, Howells DW, Karp NA, Lazic SE, Lidster K, MacCallum CJ, Macleod M, … Würbel H (2020). The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biology, 18(7), e3000410. 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrotta P, van der Veken B, van der Veken P, Pintelon I, Roosens L, Adriaenssens E, Timmerman V, Guns PJ, de Meyer GRY, & Martinet W (2020). Partial inhibition of glycolysis reduces Atherogenesis independent of Intraplaque neovascularization in mice. Arteriosclerosis, Thrombosis, and Vascular Biology, 40(5), 1168–1181. 10.1161/ATVBAHA.119.313692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poels K, Schnitzler JG, Waissi F, Levels JHM, Stroes ESG, Daemen M, Lutgens E, Pennekamp AM, de Kleijn DPV, Seijkens TTP, & Kroon J (2020). Inhibition of PFKFB3 hampers the progression of atherosclerosis and promotes plaque stability. Frontiers in Cell and Development Biology, 8, 581641. 10.3389/fcell.2020.581641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Prados JC, Través PG, Cuenca J, Rico D, Aragonés J, Martín-Sanz P, Cascante M, & Boscá L (2010). Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. Journal of Immunology, 185(1), 605–614. 10.4049/jimmunol.0901698 [DOI] [PubMed] [Google Scholar]
- Rogacev KS, Cremers B, Zawada AM, Seiler S, Binder N, Ege P, Große-Dunker G, Heisel I, Hornof F, Jeken J, Rebling NM, Ulrich C, Scheller B, Böhm M, Fliser D, & Heine GH (2012). CD14++CD16+ monocytes independently predict cardiovascular events: A cohort study of 951 patients referred for elective coronary angiography. Journal of the American College of Cardiology, 60(16), 1512–1520. 10.1016/j.jacc.2012.07.019 [DOI] [PubMed] [Google Scholar]
- Sarrazy V, Viaud M, Westerterp M, Ivanov S, Giorgetti-Peraldi S, Guinamard R, Gautier EL, Thorp EB, de Vivo DC, & Yvan-Charvet L (2016). Disruption of Glut1 in hematopoietic stem cells prevents Myelopoiesis and enhanced glucose flux in atheromatous plaques of ApoE(−/−) mice. Circulation Research, 118(7), 1062–1077. 10.1161/CIRCRESAHA.115.307599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnitzler JG, Hoogeveen RM, Ali L, Prange KHM, Waissi F, van Weeghel M, Bachmann JC, Versloot M, Borrelli MJ, Yeang C, de Kleijn DPV, Houtkooper RH, Koschinsky ML, de Winther MPJ, Groen AK, Witztum JL, Tsimikas S, Stroes ESG, & Kroon J (2020). Atherogenic lipoprotein(a) increases vascular glycolysis, thereby facilitating inflammation and leukocyte extravasation. Circulation Research, 126(10), 1346–1359. 10.1161/CIRCRESAHA.119.316206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrijvers DM, De Meyer GR, Kockx MM, Herman AG, & Martinet W (2005). Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology, 25(6), 1256–1261. 10.1161/01.ATV.0000166517.18801.a7 [DOI] [PubMed] [Google Scholar]
- Shirai T, Nazarewicz RR, Wallis BB, Yanes RE, Watanabe R, Hilhorst M, Tian L, Harrison DG, Giacomini JC, Assimes TL, Goronzy JJ, & Weyand CM (2016). The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. The Journal of Experimental Medicine, 213(3), 337–354. 10.1084/jem.20150900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, & Pittet MJ (2007). Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. The Journal of Clinical Investigation, 117(1), 195–205. 10.1172/JCI29950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, Lira SA, Habenicht AJ, & Randolph GJ (2007). Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. The Journal of Clinical Investigation, 117(1), 185–194. 10.1172/JCI28549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawakol A, Singh P, Mojena M, Pimentel-Santillana M, Emami H, MacNabb M, Rudd JHF, Narula J, Enriquez JA, Través PG, Fernández-Velasco M, Bartrons R, Martín-Sanz P, Fayad ZA, Tejedor A, & Boscá L (2015). HIF-1α and PFKFB3 mediate a tight relationship between Proinflammatory activation and Anerobic metabolism in atherosclerotic macrophages. Arteriosclerosis, Thrombosis, and Vascular Biology, 35(6), 1463–1471. 10.1161/ATVBAHA.115.305551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telang S, Clem BF, Klarer AC, Clem AL, Trent JO, Bucala R, & Chesney J (2012). Small molecule inhibition of 6-phosphofructo-2-kinase suppresses t cell activation. Journal of Translational Medicine, 10, 95. 10.1186/1479-5876-10-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillie R, de Bruijn J, Perales-Patón J, Temmerman L, Ghosheh Y, van Kuijk K, Gijbels MJ, Carmeliet P, Ley K, Saez-Rodriguez J, & Sluimer JC (2021). Partial inhibition of the 6-Phosphofructo-2-kinase/Fructose-2,6-Bisphosphatase-3 (PFKFB3) enzyme in myeloid cells does not affect atherosclerosis. Frontiers in Cell and Development Biology, 9, 695684. 10.3389/fcell.2021.695684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wik JA, Lundback P, la Cour Poulsen L, Haraldsen G, Skalhegg BS, & Hol J (2020). 3PO inhibits inflammatory NFkappaB and stress-activated kinase signaling in primary human endothelial cells independently of its target PFKFB3. PLoS ONE, 15(3), e0229395. 10.1371/journal.pone.0229395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Oldham WM, Priolo C, Pandey AK, & Loscalzo J (2021). Immunometabolic endothelial phenotypes: Integrating inflammation and glucose metabolism. Circulation Research, 129(1), 9–29. 10.1161/CIRCRESAHA.120.318805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, An X, Guo X, Habtetsion TG, Wang Y, Xu X, Kandala S, Li Q, Li H, Zhang C, Caldwell RB, Fulton DJ, Su Y, Hoda MN, Zhou G, Wu C, & Huo Y (2014). Endothelial PFKFB3 plays a critical role in angiogenesis. Arteriosclerosis, Thrombosis, and Vascular Biology, 34(6), 1231–1239. 10.1161/ATVBAHA.113.303041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Wang Y, Yan S, Yang Q, Zhou Y, Zeng X, Liu Z, An X, Toque HA, Dong Z, Jiang X, Fulton DJ, Weintraub NL, Li Q, Bagi Z, Hong M, Boison D, Wu C, & Huo Y (2017). Regulation of endothelial intracellular adenosine via adenosine kinase epigenetically modulates vascular inflammation. Nature Communications, 8(1), 943. 10.1038/s41467-017-00986-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Wang Y, Yan S, Zhou Y, Yang Q, Pan Y, Zeng X, An X, Liu Z, Wang L, Xu J, Cao Y, Fulton DJ, Weintraub NL, Bagi Z, Hoda MN, Wang X, Li Q, Hong M, … Huo Y (2017). Intracellular adenosine regulates epigenetic programming in endothelial cells to promote angiogenesis. EMBO Molecular Medicine, 9(9), 1263–1278. 10.15252/emmm.201607066 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.